

Abstract
Background:
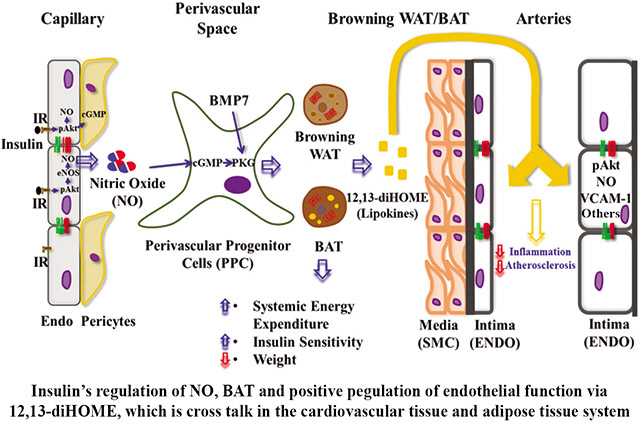
Insulin resistance (IR) can increase atherosclerotic and cardiovascular risk by inducing endothelial dysfunction (ED), decreasing nitric oxide (NO) production and accelerating arterial inflammation. The aim is to determine the mechanism by which insulin action and NO production in EC can improve systemic bioenergetics and decrease atherosclerosis via differentiation of perivascular progenitor cells (PPCs) into brown adipocytes (BAT).
Methods:
Studies used various endothelial transgenic and deletion mutant ApoE−/− mice of insulin receptors, eNOS and ETB receptors for assessments of atherosclerosis.
Cells were isolated from perivascular fat and micro-vessels for studies on differentiation and signaling mechanisms in responses to NO, insulin and lipokines from BAT.
Results:
Enhancing insulin’s actions on EC and NO production in ECIRS1 transgenic mice reduced body weight and increased systemic energy expenditure and BAT mass and activity by inducing differentiation of PPCs into beige/BAT even with high fat diet. However, positive changes in bioenergetics, BAT differentiation from PPCs and weight loss were inhibited by L-NAME, an inhibitor of endothelial NO synthase (eNOS), in ECIRS1 mice and eNOSKO mice. The mechanism mediating NO’s action on PPC differentiation into BAT was identified as the activation of solubilized guanylate cyclase/cGMP protein-dependent kinase Iα/GSK3β pathways. Plasma lipidomics from ECIRS1 mice with NO-induced increased BAT mass revealed elevated 12,13-diHOME production. Infusion of 12,13-diHOME improved ED and decreased atherosclerosis, whereas its reduction had opposite effects in ApoE−/− mice.
Conclusion:
Activation of eNOS and EC by insulin enhanced the differentiation of PPC to BAT and its lipokines and improved systemic bioenergetics and atherosclerosis, suggesting that ED is a major contributor of energy disequilibrium in obesity.
Keywords: Basic Science Research, Cell Signaling/Signal Transduction, Animal Models of Human Disease, Vascular biology
Graphical Abstract
Introduction
Insulin’s actions on endothelial cells (EC) have important direct effects on the vascular wall and systemic circulation1-4. Loss of insulin actions in EC causes endothelial dysfunction (ED), increasing cardiovascular disease risk5, 6. A major cause of ED is insulin signaling inhibition in the EC with selective diminution of PI3K/Akt pathway activation and decreased endothelial nitric oxide synthase (eNOS) phosphorylation to increase pro-atherogenic adhesion molecules such as VCAM-I, and accelerate atherosclerosis7-10. A major question is how insulin’s modest effect on eNOS activation by Akt can significantly decrease atherosclerosis and improve insulin sensitivity11-13. We and others have shown that free fatty acids and hyperglycemia can activate protein kinase C and p38/MAPK to increase serine/threonine phosphorylation of IRS1/2 and PI3K and inhibit their activation by insulin in EC 14-16.
This study explores whether insulin’s activation of eNOS/NO can regulate perivascular progenitor cells (PPC) differentiation into BAT, weight reduction, and systemic bioenergetics, and ameliorate high fat diet- and diabetes-induced ED and atherosclerosis. Previous reports showed that mice with eNOS deletion had decreased BAT and weight gain, 17, 18 and that BAT activation was associated with decreased atherosclerosis19, 20. However, these reports suggested that BAT decreased atherosclerosis by improving systemic insulin resistance rather than specific insulin actions on EC and BAT metabolites. Further, vascular endothelial growth factor (VEGF)-induced neovascularization in adipose tissue has been linked to increased white adipose tissue (WAT) and BAT, with conflicting changes in body weight21, 22. We have reported that enhancing insulin action by overexpressing IRS1 targeted to EC in ApoE−/− (ECIRS1) mice decreased atherosclerosis severity with enhanced NO production11. This report showed that insulin’s activation of eNOS in EC has the novel action to enhance BAT differentiation from PPCs, leading to decreased weight gain and increased caloric expenditure and levels of a BAT lipokine, 12,13-diHOME, which may decrease atherosclerosis.
Methods
Data Availability
Data supporting findings of this study are available from corresponding authors upon reasonable request. Detailed Expanded Materials and Methods including a Major Resources Table are presented in the Supplemental Materials.
ECIRS1 and ETBRKO mice were generated as described11, and ApoE−/− and eNOSKO mice purchased from Jackson Laboratories. Animal use in all study experiments is described in Supplemental Materials and was performed according to the Animal Care and Use Committee guidelines at Joslin Diabetes Center.
Statistical analysis.
Data were analyzed using GraphPad Prism v.9.0.2 (GraphPad Software, San Diego, CA) and presented as means ± SD. For normally distributed data (Shapiro-Wilk test P>0.05), differences between means of groups were evaluated by unpaired t tests for two groups, or One-way analysis of variance (ANOVA) for more than two groups. For the ANOVA tests, Tukey’s post-hoc procedures were additionally employed to identify specific between-group differences while adjusting for multiple comparisons. If data showed evidence of significant deviations from a normal distribution (Shapiro-Wilk P<0.05), or had a small sample size (n<6), non-parametric procedures were used, including, the Mann-Whitney U test for two, or Kruskal-Wallis test (with Dunn’s multiple comparisons test) for more than 2 groups . P<0.05 was considered significant for all comparisons.
Results
Insulin’s endothelial functions on NO production and systemic metabolism.
After 10 weeks of residual (RD) or high fat diet (HFD), body weight (BW) gained by transgenic ECIRS1 mice were 8.7±1.5% (RD, P=0.07) and 15.4±2.6% (HFD, P<0.05) less, respectively, compared to wild type (Wt, C57BL6/J) mice (Figure1A). Body composition analysis (DEXA) showed that total and subcutaneous white adipose tissue (scWAT) mass were reduced by 15±2.7% and 12±2.1%, respectively (both P<0.05) in HFD-ECIRS1 mice compared to HFD-Wt mice. Addition of L-NAME, a selective inhibitor of NOS, in drinking water (Figure1A) eliminated differences in BW and total and subcutaneous fat mass between HFD-Wt and HFD-ECIRS1 mice and reduced systemic insulin sensitivity in HFD-ECIRS1 mice compared to HFD-Wt mice (Figure1B and Figure S1A&B). Although L-NAME treatment increased BW in both RD-Wt and RD-ECIRS1 mice, no statistical differences in BW and fat mass were observed between RD-Wt and RD-ECIRS1 mice (Figure 1A and Figure S1C). Assessment of energy expenditure and food intake by comprehensive laboratory animal monitoring system (CLAMS) showed that respiratory exchange ratio (RER, VCO2/VO2) significantly improved in ECIRS1 mice compared to Wt mice (Figure 1C). No statistical differences in motor activity or food intake were observed (Figure S1D&E), suggesting elevation of fatty acid (FA) oxidation in HFD-ECIRS1 mice (Figure 1C). Increased energy expenditure was confirmed by elevated heat generation (Figure1E) and interscapular BAT (iBAT) weight (3.3 fold; p<0.05, Figure 1E) in ECIRS1 mice compared to Wt mice. Histologically, iBAT from ECIRS1 mice showed reduced intracellular lipid droplet size and increased immunostaining for CD31, a marker of EC, compared to Wt mice, in parallel with improved insulin action via PI3K/Akt pathway activation in iBAT, scWAT and skeletal muscle, but no statistical difference was observed for epididymal WAT (eWAT) (Figure 1F and Figure S1F-H). L-NAME treatment partially reversed iBAT lipid changes in HFD-ECIRS1 mice (Figure 1J), but did not reduce elevated VEGF mRNA expression or CD31 levels in iBAT of HFD-ECIRS1 mice (Figure 1F). These results suggest that NO loss did not affect neovascularization but reduced BAT whitening. We reported that increased NO production in ECIRS1 mice, with reduction of atherosclerosis, was due to enhanced endothelin receptor type B (ETBR) expression and Ca2+/calmodulin-dependent protein kinase (CAMK) activation in EC 11. Thus, using ETBR knockout (ETBRKO) and control (ETBR (fl/fl)) mice, we investigated whether ETBR deletion in EC would also increase BW and fat mass and decrease iBAT activation. After HFD, ETRBKO mice exhibited increased BW (18±3.1%) and intracellular lipid droplet size in iBAT, and lower RER, compared to HFD-control mice (Figure S2A-C).
Figure 1. Effect of enhancing endothelial functions and NO production on systemic metabolism.

(A&B) Body weight (A) and subcutaneous fat mass (B) in Wt and ECIRS1 mice in the absence or presence of L-NAME. (C&D) Representative and quantification of respiratory exchange rate (C) and heat generation (D) assessed by CLAMS analysis. (E) Representative and quantification of BAT mass. (F) Histological analysis of lipid droplet by H&E staining (J) and quantitation of capillary density (K) using anti-CD31antibody staining in BAT. (L) Vegf mRNA levels in BAT of all mice groups. Biological replicates n=8 for each group. P values were determined by Two-way ANOVA with Tukey’s multiple comparisons test in A, B, C, D, E and F. Data presented as mean ± SD; n=mice/group as indicated.
Endothelial function on BAT activity.
Analysis of gene expressions involved in FA oxidation (Mcad, Lcad, and Pgc1α), electron transfer (Etfa and Etfb) and electron transport/mitochondrial biogenesis (Ndufv1, Uqcrc1, Tfam, Nrf1 and Ucp1) showed significant iBAT elevation in HFD-ECIRS1 mice, compared to HFD-Wt mice (Figure 2A). However, increased gene expression of FA oxidation, electron transfer and mitochondrial biogenesis were inhibited in iBAT of L-NAME-treated HFD-ECIRS1 mice, compared to HFD-ECIRS1 mice (Figure 2A). Mitochondrial gene expression in iBAT showed that proteins of mitochondrial complex I, II, III and IV of the electron transport chain were increased in iBAT of HFD-ECIRS1 mice compared to HFD-Wt mice, which were inhibited in L-NAME treated HFD-ECIRS1 mice (Figure 2B). Morphometric histological analysis of iBAT via electron microscopy showed that mitochondrial volume and density per cell were greater in iBAT from HFD-ECIRS1 mice compared to HFD-Wt mice (P=0.033, Figure 2C). Mitochondrial function as assessed by oxygen consumption rate (OCR),revealed that both basal and maximal respiratory capacity were increased in the iBAT mitochondria of HFD-ECIRS1 mice compared to HFD-Wt mice, which were inhibited by L-NAME (Figure 2D). To assess mitochondrial fusion-fission, mRNA expression of Mfn2 and Opa, not Dnm, were significantly elevated in iBAT from ECIRS1 mice on RD and HFD, compared to Wt mice (Figure 2E). ETBRKO mice also exhibited the expected increases in BW and impaired RER compared to control mice (Figure S2A-C), and had decreased mitochondrial volume, expression of complexes I-V, and mitochondrial activity by Seahorse analysis, compared to control mice (Figure S2D-G).
Figure 2. Endothelial function on mitochondrial function and BAT activity.

(A) Mitochondrial mRNA levels including fatty acid oxidation, electron transfer, electron transport and biogenesis in BAT from Wt and ECIRS1 mice. (B) Immunoblotting (Top) and quantification (Bottom) of Complex I-V from BAT of Wt and ECIRS1 mice fed on RD or HFD. (C) Representative electron microscopy (Top) and quantification of mitochondria (Bottom) inBAT (X 40,000, Scale Bar 0.5um) and mitochondrial volume density. (D) Oxygen consumption rate (OCR) (Top) and quantification (Bottom) of mitochondria isolated from BAT of Wt and ECIRS1 mice by Seahorse assay. (E) Expression of mitochondrial fission (Mfn2 and Opa) and fusion (Dnm1) mRNA in the BAT of Wt and ECIRS1 mice. Biological replicates n=3 for each group. P values were determined by Kruskal-Wallis with Dunn’s multiple comparison test in A. Biological replicates n=6 for each group. P values were determined by Two-way ANOVA with Tukey’s multiple comparisons test in B, C, D and E. Data presented as mean ± SD; n=mice/group as indicated.
UCP1 expression in scWAT of ECIRS1 mice by cold challenge.
Increased mitochondrial activity and mass in iBAT of ECIRS1 mice indicated protection against cold-induced lowering of body temperature. After exposure to 5°C for one week, ECIRS1 mice were protected from decreased body temperature compared to Wt mice (Figure 3A). Immunohistochemical analysis showed elevated UCP1 expression in scWAT, but not in eWAT, of ECIRS1 mice compared to Wt mice (Figure S3A-C). L-NAME diminished cold resistance and UCP1 expression in both Wt and ECIRS1 mice (Figure 3A and Figure S3D). Expression of thermogenic genes Prdm16 and Cidea were increased in scWAT of ECIRS1 mice after cold exposure, which were inhibited by L-NAME (Figure 3B). UCP1 gene expression was not elevated in eNOSKO mice during cold exposure (Figure 3C&D). Among non-metabolic changes, Angiotensin II (AngIl)-infused mice had elevated blood pressure and loss of cold protection, with significantly lower BAT mass and decreased UCP1 expression as compared to Wt mice (Figure 3E and Figure S3E-G). Ucp1 and Prdm16 mRNA were decreased in scWAT of AngII-infused mice compared to Wt mice (Figure S3H). When treated with hydralazine, a vasodilator, AngII’s effects on body temperature, blood pressure, and protein and mRNA of UCP1 expression and mitochondrial complexes I-V levels were reversed, but no significant differences in insulin and glucose sensitivity were observed (Figure S4A-D). In ECIRS1 mice, AngII-mediated ED reversed the protective effect on body temperature against cold-induced thermogenesis and expression of Ucp1 and mitochondrial complexes I-V (Figure 3F, Figure S4E&F). Interestingly, no significant difference in Ucp1 expression was observed in scWAT of mice with insulin receptor deletion targeted to the endothelium (VEIRKO mice), compared to control (IR (fl/fl)) mice, during cold challenge (Figure S4G&H).
Figure 3. Enhancement of UCP1 expression in scWAT of ECIRS1 mice by cold challenge.

(A) Rectal body temperature of Wt and ECIRS1 mice in the presence of L-NAME or PBS after cold challenge for 7 days. (B) Real-time PCR analysis of thermogenic gene expression from scWAT of all mice. (C) Histology of scWAT of Wt and eNOSKO mice after exposure to room temperature (23°C) or cold (5°C) by H&E staining and UCP1 immunostaining (scale bar, 0.5mm). (D) Representative and quantification of immunoblot of UCP1 expression in scWAT of control and eNOSKO mice after exposure to room temperature (23°C) or cold (5°C). (E) Representative and quantification of immunoblot of UCP1 expression in scWAT of AngII-treated mice. (F) Cold-induced Ucp1, Cidea and Prdm16 mRNA expression in scWAT of ECIRS1 mice treated without or with AngII. Biological replicates n=6 for each group. P values were determined by unpaired t tests in D and E, or by Two-way ANOVA with Tukey’s multiple comparisons test in A, B and F. Data presented as mean ± SD; n=mice/group as indicated.
Characterization of perivascular progenitor cells for beige/BAT in vivo.
To determine the cells targeted by NO for increase in beige/BAT mass, PPCs from control and ECIRS1 mice were characterized by FACS analysis to estimate cell transition from stromal vascular fraction (SVF) into differentiated white or brown adipocytes. EC (CD31) and hematopoietic cell (Lin) markers were depleted from SVF to enrich for PPCs (CD31(−):Lin(−) cells). These CD31(−):Lin(−) cells (65.6% of total cells) were further selected by positive immunostaining for stem cell markers CD34(+) and Sca1(+). Then, the CD31(−):Lin(−):CD34(+):Sca1(+) subpopulation (31.8±5.5% of total cells) was enriched for BAT-like cells using markers CD140b(+) and CD146(+), and to obtain PPCs from BAT (CD31(−):Lin(−):CD34(+):Sca1(+):CD140b(+):CD146(+)), comprising 1.2% of the total original cells (Figure S5A&B and Figure 4A) 23, 24. After inducing adipogenesis25, 26, PPC differentiation into adipocytes was marked by lipids stained with O red oil (Figure S5C). Expression of adipogenic and adipokine genes, including C/EBPα, Leptin, Adpn and Adipsin mRNA, were significantly increased, but not of the Pref-1 gene, a marker of WAT (Figure S5D)27. Ucp1, Pparγ and Zfp423 were increased in sodium nitroprusside (SNP)-treated PPCs, compared to untreated PPCs (Figure 4B). Ucp1 mRNA expression was elevated 2.3-fold from day 4-7 (0-100nM), when BMP7 was added (Figure 4C and Figure S5E).
Figure 4. Identification and characterization of perivascular progenitors cells of beige/brown adipocytes.

(A) Population of PPCs in scWAT of Wt and ECIRS1 mice after 23°C or 5°C exposure by FACS. (B) Gene expression of white adipogenesis in PPCs of ECIRS1 mice treated without or with SNP. (C) UCP1 expression levels in the differentiated beige/brown adipocytes from PPCs in time dependent manner of NO using SNP (100nM) and PBS. (D) Synergetic effect of BMP7 and NO on induction of thermogenic genes including Prdm16 and Ucp1 mRNA levels in the differentiated beige/brown adipocytes. (E) UCP1 expression in different subpopulations after treated with a combination of BMP7 and NO. (F) Confocal images of whole-mounted scWAT from PDGFα and PDGFα-ECIRS1 mice (red: membrane-targeted dTomato; green: membrane-targeted eGFP, indicating Cre excision of dTomato) after exposure to 5°C in the presence or absence of L-NAME (scale bar, 50um). Biological replicates n=6 for each group. P values were determined by unpaired t tests in B and C or by Two-way ANOVA with Tukey’s multiple comparisons test in A, D, E and F. Data presented as mean ± SD; n=mice/group as indicated.
To determine the NO-responsive steps in PPC differentiation, SNP and BMP7, a known BAT differentiation factor, were added to PPCs, resulting in increased Ucp1 and Prdm16 mRNA expression above levels induced by SNP or BMP7 alone (Figure 4D). Next, we evaluated whether cells at each PPC differentiation step (Figure S5A&B) could respond to NO and synergistically with BMP7. Analysis of Ucp1 mRNA expression at each step showed significantly increased Ucp1 expression in Lin(−), Lin(−):CD31(−), Lin(−):CD31(−):CD34(+), Lin(−):CD31(−):CD34(+):CD140b(+), Lin(−):CD31(−):CD34(+):CD140b(+):CD146(+), and Lin(−):CD31(−):CD34(+):CD140b(+):CD146(+) cell populations with SNP or BMP7 alone or in combination (Figure 4E).
The origin of BAT was determined by lineage tracing analysis of PPCs by crossing mice with a fluorescent-membrane tdTomato/eGFP (mT/mG), reporter to PDGFRα-Cre mice (PDGFRα-Cre:mT/mG mice). PPCs that expressed PDGFRα were marked by tamoxifen treatment and then followed-up to trace their differentiation into mature beige/BAT. PDGFRα-ECIRS1 mice were the result of crossbreeding ECIRS1 mice and PDGFRα-Cre:mT/mG mice (Figure S5F). Whole mount confocal microscopy of scWAT from PDGFRα and IRS1-PDGFRα mice showed that cold exposure induced PDGFRα-derived GFP(+) cells in the scWAT of both mouse types, but there were more PDGFRα-derived GFP(+) cells in PDGFRα-ECIRS1 mice than in PDGFRα mice. The increased GFP(+) cells (PPCs) in the scWAT of both mice were blocked by L-NAME (Figure 4F). These findings were confirmed by Oil Red O staining (Figure S5G), indicating that PPCs at early differentiation steps were already responsive to NO and BMP7.
To identify the mechanism for the additive effect of NO and BMP7 on PPC differentiation, the role of soluble guanylate cyclase (sGC), a known receptor for NO, was studied using a specific inhibitor of sGC, 1H-[1,2,4] oxadiazolo [4,3,-a]quinoxalin-1-one (ODQ). SNP and BMP7 increased PRDM16 and UCP1 mRNA expression, individually, but ODQ only inhibited the effect of SNP and not of BMP7. The combination of BMP7 and SNP exhibited additive effects on Prdm16 and Ucp1 mRNA expression, which were inhibited by 47±8.1% and 55±9.2%, respectively, using ODQ (Figure 5A, P<0.01). The role of cGMP protein-dependent kinase Iα (PKGIα) was studied using small molecule inhibitor of PKGIα (KT5823), a target of NO/cGMP signaling18, 28. As expected, ODQ decreased cGMP levels in SNP (100nM)-treated PPCs, which was not significantly inhibited by KT5823 (100nM) (Figure S6A). However, both SNP and BMP7 significantly increased PKGIα activity in PPCs, both individually and additively (Figure 5B), which was in turn inhibited by KT5823. Similarly, the addition of KT5823 inhibited BMP7 and SNP’s additive effects on Ucp1 mRNA expression (Figure 5C), indicating that PKGIα is a common mediator of these BAT inducers.
Figure 5. Synergistic mechanism of NO/cGMP and BMP3 signaling on differentiation of perivascular progenitor cells.
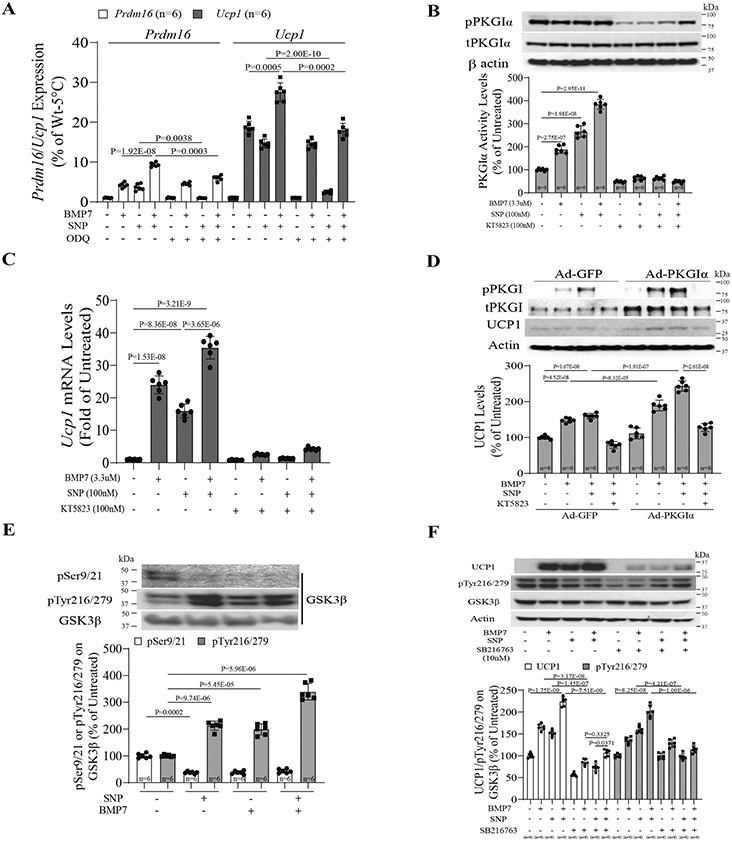
(A) Effect of NO/cGMP on Ucp1 and Prdm16 mRNA levels in the treated PPCs with combination of SNP and BMP7 using the soluble guanylyl cyclase inhibitor, ODQ. (B&C) Effect of PKGIα activity (B) and Ucp1 mRNA expression (C) in the treated PPCs with SNP and BMP7 in the absence or presence of KT5823. (D) PKGIα activity is required to induce UCP1 expression in the PPCs by overexpressing adenoviral PKGIα as a gain of PKGIα function. (E) Tyrosine (216/279) and serine (9/21) phosphorylation of GSK3β in the PPC with combination of SNP and BMP7. (F) Synergistic effect of NO/cGMP and BMP7 signaling on differentiation of PPCs and UCP1 expression via tyrosine (216/279) phosphorylation of GSK3β using the GSK3β inhibitor, SB216763 in the PPCs. Biological replicates n=6 for each group. P values were determined by Two-way ANOVA with Tukey’s multiple comparisons test in E, and by Three-way ANOVA with Tukey’s multiple comparisons test in A, B, C, D and F. Data presented as mean ± SD; n=mice/group as indicated.
PKGIα/GSK3β Signaling of NO on PPCs.
To demonstrate that PKGIα mediates beige/BAT differentiation with BMP7 and NO, PPCs overexpressed with PKGIα elevated UCP1 protein expression by 2.1-fold, compared to PPCs infected with adeno-GFP in the presence of BMP7 and SNP (Figure 5D). Again, the additive effects of BMP7 and SNP on UCP1 expression was inhibited by KT5823 in PPCs with or without PKGIα overexpression. The activation of PKGIα on UCP1 expression was inhibited by small interfering RNA (siRNA), which decreased PKGIα and UCP1 expression by 64% and 74%, respectively, compared to siScramble-treated PPCs in the presence of SNP and BMP7 (Figure S6B). GSK3β is known to be activated by PKGIα via tyrosine phosphorylation at 216 and 279 positions29. SNP or BMP7 individually increased GSK3β tyrosine phosphorylation (pTyr216/279) (Figure 5E) and decreased GSK3β serine phosphorylation (pSer9/21) in PPCs. These GSK3β-pTyr and pSer changes were additive with SNP and BMP7 in parallel with PKGIα activity (pSer126) and UCP1 induction (Figure 5C). To demonstrate interactions between PKGIα and GSK3β, PPC extracts were immunoprecipitated for GSK3β and blotted for PKGIα phosphorylation in cells overexpressing GFP or dominant-negative PKGIα. Results showed that BMP7 increased the association of PKGIα and GSK3β; however, overexpression of dominant-negative PKGIα in PPCs reduced the amount of BMP7-mediated pPKGIα /GSK3β protein complex through inhibition of pTyr216/279 on GSK3β (Figure S6C). Direct association of PKGIα and GSK3β induced by SNP and BMP7 was inhibited by a specific inhibitor of GSK3β phosphorylation at Ser21/9, SB216763 (10nM) (Figure S6D). To assess whether the complex of PKGIα and GSK3β required pTyr216/279 on GSK3β to induce UCP1 expression in PPCs, we observed that UCP1 expression and pTyr 216/279 on GSK3β induced by SNP and BMP7 addition were inhibited by SB216763 (Figure 5F). GSK3β is a negative regulator of β-catenin by phosphorylation at serine 55230. Thus, we determined whether SNP and BMP7-mediated pTyr 216/279 on GSK3β can alter β-catenin activation. Results showed that nuclear β-catenin was significantly increased in SNP- and BMP7-treated differentiated PPCs, whereas its level was inhibited by SB216763 (Figure S6E). These data suggested that NO and BMP7 signaling converge at the pPKGIα step, which induces GSK3β activation through pTyr 216/279, resulting in β-catenin stabilization to the nucleus, where it enhances Ucp1 and Prdm16 gene expression (Figure S6F).
12,13-diHOME and its anti-atherogenic actions in EC.
Previously, BAT effects in decreasing atherosclerosis were attributed to improvements in systemic metabolism19, 20. Plasma lipidomic analysis of these mouse strains showed that the levels of two diols including 9,10- and 12,13-diHOME were elevated in ECIRS1 mice as compared to Wt mice and were decreased by L-NAME treatment (Figure S7A). Tseng’s group has reported that BAT-induced plasma levels of 12,13-diHOME were decreased in insulin-resistant rodents and patients with elevated BMI31, 32. Thus, we propose that enhanced insulin action on the endothelium will elevate 12,13-diHOME production due to increased BAT differentiation from PPCs, which can recirculate and have direct anti-atherogenic actions on EC. Cold exposure increased several lipokines including 9,10-diHOME (3.0-fold on RD and 2.8-fold on HFD) and 12,13-diHOME (1.9-fold on RD and 1.87-fold on HFD) in ECIRS1 mice compared to Wt mice (Figure 6A and Figure S4A). L-NAME decreased 9,10-diHOME by 54±10.7% and 12,13-diHOME by 87±14.3% (Figure 6A). However, the epoxide precursors of 9,10- or 12,13-diHOME were not significantly changed in HFD-Wt and ECIRS1 mice, compared to untreated mice.
Figure 6. Levels of plasma lipokines in Wt and ECIRS1 mice and effects of exogenous 12,13-diHOME on EC and severity of atherosclerosis.

(A) Plasma 9,10-diHOME (Left) 12,13-diHOME (Right) levels from lipidomic analysis data of the plasma from Wt and ECIRS1 mice on RD or HFD with or without L-NAME. (B) Representative immunoblot analysis (Left) and quantification (Right) of VCAM-1 and ICAM-1 protein levels in BAEC after treatment with TNFα, OA, Ox-LDL or PA with or without 12,13-diHOME (100uM). (C) Measurement of Ca2+ influx by Fluor-4 in BAEC after being treated with different doses of 12,13-diHOME (1nM-1uM). (D) en face stained aorta and quantification of aortas from HFD-ApoE−/− mice treated with 12,13-diHOME (scale bar, 1mm). (E) Representative images and quantification for collagen and macrophages stained in cryo-sections of the aortic sinus (scale bar, 500um). (F) en face stained aorta and quantification of aortas from WD-ApoE−/− mice exposure at 23°C or 30°C (scale bar, 1mm). Biological replicates n=5 for each group. P values were determined by Mann-Whitney U test in C, D, E, and F. Biological replicates n=5-6 for each group. P values were determined by Kruskal-Wallis with Dunn’s multiple comparison test in A and B. Data presented as mean ± SD; n=mice/group as indicated.
In bovine EC (BAEC), 12,13-diHOME significantly decreased protein levels of VCAM-1, but not of ICAM-1, without decreasing their mRNA levels in the presence of oxidized LDL (ox-LDL), which increased Vcam-1 and Icam-1 mRNA by 3.4- and 4.2-fold, respectively (Figure 6B, and Figure S7B&C). Functionally, 12,13-diHOME reduced ox-LDL and palmitate-induced adhesion of fluorescent-labeled monocytes to BAEC, which were inhibited by antibodies against VCAM-1 and not by control IgG (Figure S7D). 12,13-diHOME (1nM-1uM), but not 9,10-diHOME, increased NO production by 2.5-fold, as measured by nitrite/nitrate in BAEC media, and did not significantly increase reactive oxygen species in cell lysates (Figure S7E&F). NO production can be induced by Akt activation with either phosphorylation of eNOS or Ca++/calmodulin activation 33. Addition of 12,13-diHOME, not 9, 10-diHOME, induced pAkt activity from 1nM-1uM by 4.1-fold, but did not increase peNOS levels (Figure S7G&H). However, intracellular Ca++ levels measured by Fura 2 were increased in BAEC by 12,13-diHOME from 1-100nM, indicating that activation of Ca++/CAMKII pathways (Figure 6C) increases NO production. Using myography, acetylcholine (ACh)-induced endothelium-dependent vasodilation was increased in 12,13-diHOME-treated carotid arteries, compared to vehicle-treated arteries, from ApoE−/− mice, (Figure S8A). However, no statistical difference of Ach-induced vasodilation was observed between vehicle-treated and 12,13-diHOME-treated carotid arteries from eNOSKO mice (Figure S8B).
To demonstrate whether 12,13-diHOME can directly affect vascular function and severity of atherosclerosis in vivo, ApoE−/− mice were treated with 10ug/kg BW of 12,13-diHOME or vehicle in drinking water, and atherosclerosis severity assessed after three months. Plasma 12,13-diHOME was 27.7nM and 7.1nM in ApoE−/− mice on RD and HFD, respectively, compared to 53.1nM in those on HFD and fed with 12,13-diHOME (Figure S8C). After three months, the extent of Sudan IV stained aortic root and descending aorta decreased by 52±8.5% in 12,13-diHOME-treatedApoE−/− mice, compared to vehicle-treated ApoE−/− mice (Figure 6D). Analysis of atherosclerosis complexity revealed that 12,13-diHOME feeding reduced extracellular matrix (Masson Trichrome stains) and macrophage accumulation (MAC2 staining) in aortic roots of HFD-ApoE−/− mice (n=5) by 32±5.7% and 45±6.3% (P=0.003 and P=0.004, respectively) compared to vehicle-treated HFD-ApoE−/− mice (Figure 6E).
While 12,13-diHOME treatment did not significantly decrease plasma triglycerides (TG), FA or total cholesterol levels in HFD-ApoE−/− mice, they exhibited lower levels of very low-density (VLDL) and low-density lipoprotein (LDL) without blood pressure changes in these mice (Figure S8D-H). No detectable UCP1 protein levels were observed in scWAT, and no significant differences in CD36 and UCP1 expression were observed in iBAT of ApoE−/− mice with or without 12,13-diHOME (Figure S9A-C). Moreover, while immunoblot and gene expression analysis of iBAT and scWAT showed that eNOSKO mice have less UCP1 expression in both iBAT and scWAT compared to Wt mice, UCP1 expression was not significantly affected in 12,13-diHOME treated eNOSKO mice after cold exposure for a week, compared to Wt mice (Figure S9D-F).
The effect of lowering circulating 12,13-diHOME on atherosclerosis was evaluated by increasing environmental ambient temperature from 23°C to 30°C, commonly referred as thermo-neutral temperature (TN)34. In WD-ApoE−/− mice reared at 30°C (TN), compared to those at 23°C, circulating 12, 13-diHOME levels decreased by 25±4.3% (Figure S10A). The extent of atherosclerosis, measured by Sudan IV staining, showed that lipid deposition and macrophage composition in the plaques of the aortic sinuses (n=5) were increased by 172±33.1% and 187±25.5% (P=0.011 and P=0.024), respectively, without significant changes in total cholesterol and TG levels (Figure 6F and Figure S10B-E). However, TN-WD-ApoE−/− mice experienced significantly higher average daily RER compared with 23°C-WD-ApoE−/− mice, indicating higher dependence on carbohydrates without statistically significant changes in glucose tolerance and insulin levels (Figure S10F-H), and supporting the hypothesis that TN inhibited BAT activation and decreased 12,13-diHOME expression.
Correlation of plasma 12,13-diHOME levels with hemodynamic and thermogenic changes and NO production.
To test whether lowering NO production caused by non-metabolic factors could decrease BAT mass and 12,13-diHOME levels, Wt mice were treated with AngII for 7 days, followed by cold exposure for another 7 days (Figure 3E and Figure S3E). Parallel to elevation of blood pressure and impaired protection against cold exposure, plasma 12,13-diHOME levels and Ucp1 and Prdm16 mRNA expression were decreased significantly in AngII-treated mice compared to vehicle-treated mice(Figure 7A and Figure S3F-H). Normalizing blood pressure by hydralazine reversed AngII action on decreasing plasma 12,13-diHOME (Figure S4C). We studied the effect of exposure to 5°C and 23°C on circulating 12,13-diHOME in ETBRKO mice and eNOSKO mice, which exhibited defects in thermogenesis and BAT development (Figure 3C&D). Plasma 12,13-diHOME was significantly reduced in both mice at 23°C and were dramatically reduced after 7 days at 5°C, compared to their respective control mice (Figure 7B). Furthermore, increased plasma 12,13-diHOME levels in ECIRS1 mice were inhibited by L-NAME and AngII administration (Figure 7C). To determine whether the 12,13-diHOME changes depended on eNOS activity, NO production or both, we performed ex vivo assays of NOS activity, which measured conversion of L-arginine to L-citrulline in aortas from ECIRS1 mice treated with AngII or eNOSKO mice treated with 12,13-diHOME after cold exposure at either 23°C or 5°C for 10 days. Results showed that eNOS activity decreased in the aorta of ECIRS1-AngII mice as compared to ECIRS1 mice, and 12,13-diHOME did not significantly change NOS activity in eNOSKO mice (Figure S11A&B). Results from eNOSKO mice showed that eNOS activity was not significantly increased even in EC with IRS1 overexpression (Figure S11C). To evaluate whether NO changes correlated with circulating 12,13-diHOME levels, nitrite/nitrate levels were measured with 12,13-diHOME levels in the plasma of the same mice. As shown in Figure 7A-C, levels of 12,13-diHOME correlated with nitrite /nitrate ratios. The nitrite/nitrate levels were significantly lower in AngII-treated mice, ETBRKO mice, and eNOSKO mice at 23°C or 5°C, compared to age-matched Wt or their respective control mice (Figure 7D&E). ECIRS1 mice exhibited higher levels of nitrite/nitrate compared to Wt mice both at 23°C and at 5°C, but AngII and L-NAME treatment decreased these nitrite/nitrate levels (Figure 7F). Nitrite/nitrate levels and ex vivo NOS activities of the aorta were not significantly different in RD or WD-ApoE mice exposed at 23°C versus those exposed to 30°C (Figure S11D&E). To evaluate whether 12,13-diHOME-induced NO production may be derived from iNOS and nNOS in WAT, we found that iNOS protein and mRNA levels were increased in WAT from WD-ApoE−/− mice treated with vehicle compared to mice on RD, but no significant difference in iNOS levels was observed in WAT from WD-ApoE−/− mice treated with 12,13-diHOME (Figure S11F&G).
Figure 7. Effect of inhibiting endothelial function and NO production by non-metabolic changes on circulating 12,13-diHOME.

(A) Circulating 12,13-diHOME levels in plasma of vehicle-treated or AngII-treated mice after 7 days of exposure at 23°C or 5°C for 7 days in the absence or the presence of hydralazine. (B) Plasma 12,13-diHOME concentrations in ETBRKO mice (Left) and eNOSKO mice (Right) at 23°C or 5°C for 7 days. (C) Plasma 12,13-diHOME levels in L-NAME treated mice or AngII treated mice after cold exposure 7days. (D) Plasma Nitrite/Nitrate levels in vehicle-treated or AngII-treated mice after cold exposure. (E) Plasma Nitrite/Nitrate levels in ETBRKO mice (Left) and eNOSKO mice (Right) at 23°C or 5°C for 7 days. (F) Plasma Nitrite/Nitrate levels in L-NAME treated mice or AngII treated mice after cold exposure. Biological replicates n=3-4 for each group. P values were determined by Kruskal-Wallis with Dunn’s multiple comparison test in A, B, C, D, E and F. Data presented as mean ± SD; n=mice/group as indicated.
Discussion
This study identified a new pathway where insulin signaling on EC can increase BAT differentiation from PPCs and alter whole body bioenergetics. The increase in BAT differentiation, with resultant elevation of 12,13-diHOME, uncovered a feedback circuit that targeted EC, possibly, to decrease VCAM-1 expression and monocyte recruitment and thus ameliorating arterial wall inflammation and atherosclerosis. We showed that eNOS activation in EC, whether induced by insulin or inhibited by hemodynamic changes such as angiotensin, has significant effects on the arterial wall and affected systemic bioenergetics since L-NAME infusion removed the protection of weight gain in ECIRS1 mice on HFD. Enhancing IRS1, specifically to EC, as in ECIRS1/ApoE−/− mice11, increased NO production and decreased atherosclerosis, as reported. Insulin action on EC and eNOS activation affected BW, as supported by ETBR deletion and ETBRKO mice, which exhibited loss of insulin action on eNOS activation and decreased 12,13-diHOME levels.
The importance of NO in BAT development and systemic bioenergetics is clear in eNOSKO mice which had reduced BAT mass, nitrite/nitrate ratio, and 12,13-diHOME levels with cold stress. These findings suggest that ED, rather than being a secondary effect of systemic insulin resistance, can be an initiating event that induces systemic bioenergetics changes, increasing cardiovascular disease risk and weight gain as part of metabolic syndrome. The mechanism that protects ECIRS1 mice against weight gain and cold stress is related to elevated NO production and action on PPC differentiation into BAT, with corresponding increased energy expenditure. This new finding is supported by increased subcutaneous beige/BAT and less temperature decline when mice were subjected to stress at 5°C, in parallel with elevated nitrite/nitrate levels. The increased beige/BAT mass is also functionally active since ECIRS1 mice generated more heat, had more mitochondria mass, and exhibited greater oxidative phosphorylation than Wt mice. However, the significance of NO in regulating weight gain with extreme food intake is unclear since L-NAME failed to impact body weight in HFD-Wt mice.
Previous reports have suggested that NO may also elevate VEGF expression to increase WAT35,36. However, our results indicated that VEGF expression in BAT is not directly due to NO since L-NAME did not significantly decrease VEGF expression in BAT. Thus, the elevation of VEGF expression associated with BAT increase is secondarily due to increased metabolic demand of enlarged BAT mass. The target of NO on PPC is selective with specific differentiation into BAT. This novel idea was determined by lineage tracing studies using ECIRS1-PDGFRα mice exhibiting cold-induced adipocyte hyperplasia with increased thermogenic genes for beige/BAT from PPCs, which were abrogated by L-NAME. Effects of NO on PPC differentiation to BAT are also selective for cell type since PPCs have the potential to develop into WAT, fibroblasts, smooth muscle cells (SMC), osteoclasts and pericytes 35. Selective actions of NO on PPCs for BAT differentiation were supported by findings in eNOSKO mice, which manifested significant losses in BAT but not in WAT.
Signaling studies indicated that these differentiation factors activate separate signaling pathways at the beginning of their cascade for inducing BAT from PPCs. The mechanism of NO induction is initiated by sGC activation with cGMP elevation, an established pathway for NO in SMC 36. However, BMP7’s lack of any statistically observable effect on cGMP elevation supports the idea that BMP7 and NO are activating different signaling pathways. Our results demonstrated that NO/cGMP and BMP7 signaling converges at PKGIα in PPCs. NO signaling is known to activate PKGIα via cGMP, whereas the known signaling pathway for BMP7 is by SMAD phosphorylation28. Our results indicated that BMP7’s effect on PPCs was mediated by tyrosine phosphorylation at the 216/279 positions of PKGIα. Further, SMAD4 interacted with PKGIα after BMP7 induction which enhanced NO activation at pTyr (216/279 positions) of PKGIα. Once activated by NO and BMP7, additively, PKGIα increased pGSK3β and activated nuclear translocation of β-catenin, a novel additive signaling pathway for BMP7 and NO to enhance PPC differentiation into beige/BAT.
The association of BAT mass elevation with decreased atherosclerosis, as found in ECIRS1 mice, is consistent with multiple reports19, 20, 37. Recently, Cao’s group reported that BAT activation by systemic treatment with a β3-adrenergic activator, mirabegron, led to worsening of atherosclerosis in ApoE−/− mice in parallel with elevations of plasma LDL, VLDL or reduction of adiponectin38. The findings observed after mirabegron treatment are difficult to interpret since this also involved changes in the induction of hepatic genes for lipid metabolism in ApoE−/− mice 39. In ECIRS1/ApoE−/− mice, increased BAT did not significantly affect lipoprotein profile, which allowed the interpretation of BAT effects on atherosclerosis without being confounded by systemic changes. Furthermore, data from the feedback mechanism of 12,13-diHOME on EC also supported the idea that BAT activation can directly decrease atherosclerosis 31. Our studies demonstrated that elevating circulating 12,13-diHOME levels by feeding can directly decrease the complexity of atherosclerosis. In addition, 12,13-diHOME did not significantly change UCP1 expression in both BAT and scWAT, while increasing Ach-endothelium-dependent vasodilation in the mesenteric arteries of HFD-ApoE−/− mice, indicating that 12,13-diHOME can potentially decrease atherosclerosis by enhancing eNOS activity in HFD-ApoE−/− mice. The direct actions of 12,13-diHOME on EC also suggested that its induction of NO production is through activation of eNOS via Ca++/calmodulin enhancement, although circulatory nitrite/nitrate remained unchanged. Future studies will need to be performed in female mice and also involve clinical subjects in order to confirm whether these findings are applicable to both genders and in people. Thus, these findings have identified that the endothelium, via NO, can regulate systemic bioenergetics by facilitating the selective differentiation of PPCs into BAT with a positive feedback action on itself due to specific BAT lipokines, thus delaying atherosclerosis.
Supplementary Material
Novelty and Significance section.
What Is Known?
Obesity is associated with imbalance and dysfunction of white and brown adipose tissue (WAT and BAT), which are strongly associated with insulin resistance, type 2 diabetes, endothelial dysfunction and atherosclerosis.
Targeting angiogenesis in WAT and BAT affects thermogenesis, and may even ameliorate obesity and severity of atherosclerosis.
Circulating lipokine levels from BAT are decreased in insulin resistant rodents and in people with T2DM and obesity.
What New Information Does This Article Contribute?
Insulin induced nitric oxide (NO) production from endothelial cells (EC) can regulate whole body’s bioenergetics by increasing the differentiation perivascular progenitor cells (PPCs) to BAT, resulting in protection against obesity and atherosclerosis.
This novel action of NO is mediated by the activation of solubilized guanylate cyclase/cGMP protein-dependent kinase Iα(PKGIα)/GSK3β pathways in the PPCs to induce BAT and the production of lipokine, 12,13-diHOME, which has direct actions on EC to reduce inflammation and atherosclerosis.
Systemic vascular diseases such as hypertension and its associated endothelial dysfunction may initiate defects in BAT differentiation and vasoactive lipokines, 12,13-diHOME to affect whole body bioenergetics, weight gains and atherosclerosis. This also enhances insulin’s actions on EC and NO production as ECIRS1 transgenic mice had reduced body weight and increased systemic energy expenditure and brown adipose tissue (BAT) mass and activity by inducing differentiation of PPCs into beige/BAT even with high fat diet. However, positive changes in bioenergetics, BAT differentiation from PPCs, and weight loss were inhibited by L-NAME, an inhibitor of endothelial NO synthase (eNOS), or in eNOSKO mice. The mechanism mediating NO’s action on PPC differentiation into BAT was activation of the solubilized guanylate cyclase/ PKGIα/GSK3β pathways. Plasma lipidomics analysis from ECIRS1 mice with NO-induced increased BAT mass, revealed elevated 12,13-diHOME production. Infusion of 12,13-diHOME improved endothelial dysfunction (ED) and decreased atherosclerosis, whereas, reducing 12,13-diHOME had opposite effects in ApoE−/− mice. Activation of eNOS and EC by insulin enhanced the differentiation of PPC to BAT and its related lipokine and improved systemic bioenergetics and atherosclerosis, suggesting ED is a major contributor of energy disequilibrium in obesity.
Acknowledgements
Plasma total cholesterol, free fatty acids and lipid profiles were measured at the Lipid Cores of Vanderbilt University Medical Center. Plasma lipidomic analysis was performed by Berg Inc. (Framingham, MA).
Sources of Funding
This study was supported by NIH grants DK053105, DK036836, and NIDDK Diabetes Research Center award, 5P30DK036836-35.
Non-standard Abbreviations and Acronyms
- Ach
Acetylcholine
- Akt
Protein kinase B
- AngII
Angiotensin II
- ApoE−/−
Apolipoprotein E Null
- BAEC
Bovine Aortic Endotelial Cells
- BAT
Brown Adipose Tissue
- BMI
Body Mass Index
- BMP7
Bone Morphogenetic Protein 7
- BW
Body Weight
- CAMK
Ca2+/Calmodulin-dependent Protein Kinase
- CLAMS
Comprehensive Laboratory Animal Monitoring System
- DEXA
Dual-Energy X-ray Absorptiometry
- EC
Endothelial Cells
- ED
Endothelial Cells Dysfunction
- eNOS
Endothelial Nitric Oxide Synthase
- ETBR
Endothelin Receptor Type B
- eWAT
Epididymal White Adipose Tissue
- FA
Fatty Acid
- GSK3β
Glycogen synthase kinase 3 beta
- HFD
High Fat Diet
- iBAT
Interscapular Brown Adipose Tissue
- ICAM-1
Intercellular Adhesion Molecule 1
- iNOS
Induced Nitric Oxide Synthase
- IRS1
Insulin Receptor Substrate 1
- LDL
Low Density Lipoprotein
- L-NAME
N(gamma)-nitro-L-arginine methyl ester
- MAPK
Mitogen-Activated Protein Kinase
- NO
Nitric Oxide
- ODQ
1H-[1,2,4] oxadiazolo [4,3,-a]quinoxalin-1-one
- Ox-LDL
Oxidized Low Density Lipoprotein
- PI3K
Phosphoinositide 3-Kinase
- PKGIα
cGMP protein-dependent kinase Iα
- PPCs
Perivascular Progenitor Cells
- RER
Respiratory Exchange Ratio
- scWAT
Subcutaneous white adipose tissue
- sGC
Soluble guanylate cyclase
- siRNA
Small interfering RNA
- SMC
Smooth Muscle Cells
- SNP
Sodium Nitroprusside
- TG
Triglycerides
- TN
Thermo-Neutral Temperature
- UCP1
Uncoupling Protein 1
- VCAM-I
Vascular Adhesion Molecule 1
- VEGF
Vascular endothelial growth factor
- VLDL
Very Low Density Lipoprotein
- Wt
Wild Type
Footnotes
Disclosures
None
References
- 1.Abe H, Yamada N, Kamata K, Kuwaki T, Shimada M, Osuga J, Shionoiri F, Yahagi N, Kadowaki T, Tamemoto H, Ishibashi S, Yazaki Y and Makuuchi M. Hypertension, hypertriglyceridemia, and impaired endothelium-dependent vascular relaxation in mice lacking insulin receptor substrate-1. J Clin Invest. 1998;101:1784–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W and Liu Z. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia. 2009;52:752–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King GL and Johnson SM. Receptor-mediated transport of insulin across endothelial cells. Science. 1985;227:1583–6. [DOI] [PubMed] [Google Scholar]
- 4.King GL, Park K and Li Q. Selective Insulin Resistance and the Development of Cardiovascular Diseases in Diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes. 2016;65:1462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeFronzo RA and Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–94. [DOI] [PubMed] [Google Scholar]
- 6.Wong ND, Zhao Y, Patel R, Patao C, Malik S, Bertoni AG, Correa A, Folsom AR, Kachroo S, Mukherjee J, Taylor H and Selvin E. Cardiovascular Risk Factor Targets and Cardiovascular Disease Event Risk in Diabetes: A Pooling Project of the Atherosclerosis Risk in Communities Study, Multi-Ethnic Study of Atherosclerosis, and Jackson Heart Study. Diabetes Care. 2016;39:668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ and Sessa WC. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang ZY, He Z, King BL, Kuroki T, Opland DM, Suzuma K, Suzuma I, Ueki K, Kulkarni RN, Kahn CR and King GL. Characterization of multiple signaling pathways of insulin in the regulation of vascular endothelial growth factor expression in vascular cells and angiogenesis. J Biol Chem. 2003;278:31964–71. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH and Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–54. [DOI] [PubMed] [Google Scholar]
- 10.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, Clermont A, Geraldes P, Dall'Osso C, Wagers AJ, Huang PL, Rekhter M, Scalia R, Kahn CR and King GL. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park K, Mima A, Li Q, Rask-Madsen C, He P, Mizutani K, Katagiri S, Maeda Y, Wu IH, Khamaisi M, Preil SR, Maddaloni E, Sorensen D, Rasmussen LM, Huang PL and King GL. Insulin decreases atherosclerosis by inducing endothelin receptor B expression. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R and Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. [DOI] [PubMed] [Google Scholar]
- 13.Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE and Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9:845–8. [DOI] [PubMed] [Google Scholar]
- 14.Maeno Y, Li Q, Park K, Rask-Madsen C, Gao B, Matsumoto M, Liu Y, Wu IH, White MF, Feener EP and King GL. Inhibition of insulin signaling in endothelial cells by protein kinase C-induced phosphorylation of p85 subunit of phosphatidylinositol 3-kinase (PI3K). J Biol Chem. 2012;287:4518–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park K, Li Q, Rask-Madsen C, Mima A, Mizutani K, Winnay J, Maeda Y, D'Aquino K, White MF, Feener EP and King GL. Serine phosphorylation sites on IRS2 activated by angiotensin II and protein kinase C to induce selective insulin resistance in endothelial cells. Mol Cell Biol. 2013;33:3227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun XJ, Goldberg JL, Qiao LY and Mitchell JJ. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999;48:1359–64. [DOI] [PubMed] [Google Scholar]
- 17.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S and Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–9. [DOI] [PubMed] [Google Scholar]
- 18.Roberts LD, Ashmore T, Kotwica AO, Murfitt SA, Fernandez BO, Feelisch M, Murray AJ and Griffin JL. Inorganic nitrate promotes the browning of white adipose tissue through the nitrate-nitrite-nitric oxide pathway. Diabetes. 2015;64:471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berbee JF, Boon MR, Khedoe PP, Bartelt A, Schlein C, Worthmann A, Kooijman S, Hoeke G, Mol IM, John C, Jung C, Vazirpanah N, Brouwers LP, Gordts PL, Esko JD, Hiemstra PS, Havekes LM, Scheja L, Heeren J and Rensen PC. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nature communications. 2015;6:6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoeke G, Kooijman S, Boon MR, Rensen PC and Berbee JF. Role of Brown Fat in Lipoprotein Metabolism and Atherosclerosis. Circulation research. 2016;118:173–82. [DOI] [PubMed] [Google Scholar]
- 21.Kolonin MG, Saha PK, Chan L, Pasqualini R and Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nature medicine. 2004;10:625–32. [DOI] [PubMed] [Google Scholar]
- 22.Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R and Folkman MJ. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99:10730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nayak RC, Berman AB, George KL, Eisenbarth GS and King GL. A monoclonal antibody (3G5)-defined ganglioside antigen is expressed on the cell surface of microvascular pericytes. J Exp Med. 1988;167:1003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodeheffer MS, Birsoy K and Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–9. [DOI] [PubMed] [Google Scholar]
- 25.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF and Prockop DJ. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. 2004;103:1662–8. [DOI] [PubMed] [Google Scholar]
- 26.Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y, Ahrens MJ, Dudley AT, Norris AW, Kulkarni RN and Kahn CR. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD and Graff JM. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwappacher R, Weiske J, Heining E, Ezerski V, Marom B, Henis YI, Huber O and Knaus P. Novel crosstalk to BMP signalling: cGMP-dependent kinase I modulates BMP receptor and Smad activity. EMBO J. 2009;28:1537–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasaki Y, Kugimiya F, Chikuda H, Kamekura S, Ikeda T, Kawamura N, Saito T, Shinoda Y, Higashikawa A, Yano F, Ogasawara T, Ogata N, Hoshi K, Hofmann F, Woodgett JR, Nakamura K, Chung UI and Kawaguchi H. Phosphorylation of GSK-3beta by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes. The Journal of clinical investigation. 2008;118:2506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegfried E, Chou TB and Perrimon N. wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell. 1992;71:1167–79. [DOI] [PubMed] [Google Scholar]
- 31.Lynes MD, Leiria LO, Lundh M, Bartelt A, Shamsi F, Huang TL, Takahashi H, Hirshman MF, Schlein C, Lee A, Baer LA, May FJ, Gao F, Narain NR, Chen EY, Kiebish MA, Cypess AM, Bluher M, Goodyear LJ, Hotamisligil GS, Stanford KI and Tseng YH. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nature medicine. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasan SK, Noordam R, Gowri MS, Neville MJ, Karpe F and Christodoulides C. The proposed systemic thermogenic metabolites succinate and 12,13-diHOME are inversely associated with adiposity and related metabolic traits: evidence from a large human cross-sectional study. Diabetologia. 2019;62:2079–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang PL. Endothelial nitric oxide synthase and endothelial dysfunction. Curr Hypertens Rep. 2003;5:473–80. [DOI] [PubMed] [Google Scholar]
- 34.Giles DA, Ramkhelawon B, Donelan EM, Stankiewicz TE, Hutchison SB, Mukherjee R, Cappelletti M, Karns R, Karp CL, Moore KJ and Divanovic S. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol Metab. 2016;5:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armulik A, Genove G and Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. [DOI] [PubMed] [Google Scholar]
- 36.Beavo JA and Brunton LL. Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–8. [DOI] [PubMed] [Google Scholar]
- 37.Kikai M, Yamada H, Wakana N, Terada K, Yamamoto K, Wada N, Motoyama S, Saburi M, Sugimoto T, Irie D, Kato T, Kawahito H, Ogata T and Matoba S. Adrenergic receptor-mediated activation of FGF-21-adiponectin axis exerts atheroprotective effects in brown adipose tissue-transplanted apoE(−/−) mice. Biochemical and biophysical research communications. 2018;497:1097–1103. [DOI] [PubMed] [Google Scholar]
- 38.Sui W, Li H, Yang Y, Jing X, Xue F, Cheng J, Dong M, Zhang M, Pan H, Chen Y, Zhang Y, Zhou Q, Shi W, Wang X, Zhang H, Zhang C, Zhang Y and Cao Y. Bladder drug mirabegron exacerbates atherosclerosis through activation of brown fat-mediated lipolysis. Proc Natl Acad Sci U S A. 2019;116:10937–10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang SH, Reddick RL, Piedrahita JA and Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–71. [DOI] [PubMed] [Google Scholar]
- 40.Sun K, Kusminski CM, Luby-Phelps K, Spurgin SB, An YA, Wang QA, Holland WL and Scherer PE. Brown adipose tissue derived VEGF-A modulates cold tolerance and energy expenditure. Mol Metab. 2014;3:474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batista TM, Jayavelu AK, Wewer Albrechtsen NJ, Iovino S, Lebastchi J, Pan H, Dreyfuss JM, Krook A, Zierath JR, Mann M and Kahn CR. A Cell-Autonomous Signature of Dysregulated Protein Phosphorylation Underlies Muscle Insulin Resistance in Type 2 Diabetes. Cell Metab. 2020;32:844–859 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmittgen TD and Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Park K, Li C, Rask-Madsen C, Mima A, Qi W, Mizutani K, Huang P and King GL. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase C-beta isoform in the endothelium. Circ Res. 2013;113:418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park K, Li Q, Evcimen ND, Rask-Madsen C, Maeda Y, Maddaloni E, Yokomizo H, Shinjo T, St-Louis R, Fu J, Gordin D, Khamaisi M, Pober D, Keenan H and King GL. Exogenous Insulin Infusion Can Decrease Atherosclerosis in Diabetic Rodents by Improving Lipids, Inflammation, and Endothelial Function. Arterioscler Thromb Vasc Biol. 2018;38:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting findings of this study are available from corresponding authors upon reasonable request. Detailed Expanded Materials and Methods including a Major Resources Table are presented in the Supplemental Materials.
ECIRS1 and ETBRKO mice were generated as described11, and ApoE−/− and eNOSKO mice purchased from Jackson Laboratories. Animal use in all study experiments is described in Supplemental Materials and was performed according to the Animal Care and Use Committee guidelines at Joslin Diabetes Center.