

Abstract
Background:
Establishment of the myocardial wall requires proper growth cues from nonmyocardial tissues. During heart development, the epicardium and epicardium-derived cell (EPDC)s instruct myocardial growth by secreting essential factors including fibroblast growth factor 9 (FGF9) and insulin-like growth factor 2 (IGF2). However, it is poorly understood how the epicardial secreted factors are regulated, in particular by chromatin modifications for myocardial formation. The current study is to investigate whether and how histone deacetylase 3 (HDAC3) in the developing epicardium regulates myocardial growth.
Methods:
Various cellular and mouse models in conjunction with biochemical and molecular tools were employed to study the role of HDAC3 in the developing epicardium.
Results:
We deleted Hdac3 in the developing murine epicardium and mutant hearts showed ventricular myocardial wall hypoplasia with reduction of EPDCs. The cultured embryonic cardiomyocytes with supernatants from Hdac3 knockout (KO) mouse epicardial cells (MECs) also showed decreased proliferation. Genome-wide transcriptomic analysis revealed that Fgf9 and Igf2 were significantly downregulated in Hdac3 KO MECs. We further found that Fgf9 and Igf2 expression is dependent on HDAC3 deacetylase activity. The supplementation of FGF9 or IGF2 can rescue the myocardial proliferation defects treated by Hdac3 KO supernatant. Mechanistically, we identified that microRNA (miR)-322 and miR-503 were upregulated in Hdac3 KO MECs and Hdac3 epicardial KO hearts. Overexpression of miR-322 or miR-503 repressed FGF9 and IGF2 expression, while knockdown of miR-322 or miR-503 restored FGF9 and IGF2 expression in Hdac3 KO MECs.
Conclusions:
Our findings reveal a critical signaling pathway in which epicardial HDAC3 promotes compact myocardial growth by stimulating FGF9 and IGF2 through repressing miR-322/miR-503, providing novel insights in elucidating etiology of congenital heart defects, and conceptual strategies to promote myocardial regeneration.
Keywords: Basic Science Research, Developmental Biology, Mechanisms
Graphical Abstract
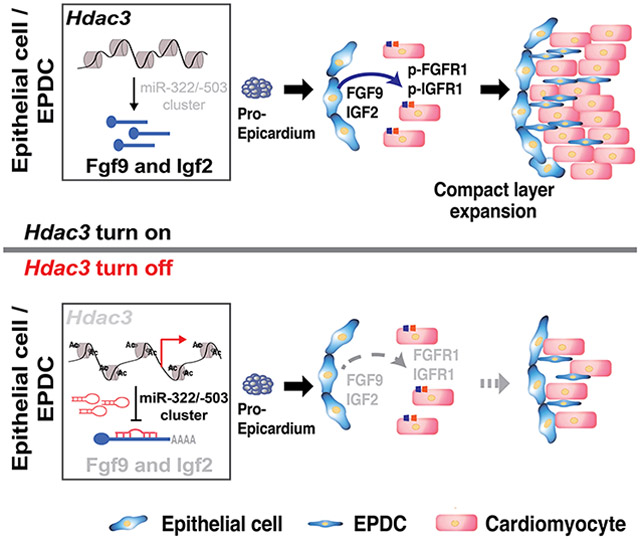
Introduction
Congenital heart disease (CHD) is still the most common birth defect worldwide1. Myogenic defects are often associated with many forms of CHD. In the past, most research has focused on identifying the intrinsic factors in the myocardium to understand the potential causes, whereas the contributions from nonmyocytes through intercellular communications or regulations between cardiomyocyte (CM)s and nonmyocytes have been largely overlooked2. The nonmyocyte compartments such as the epicardium and epicardium-derived cell (EPDC)s are capable of regulating the development of adjacent tissues such as compact myocardium via paracrine signaling crosstalk3. Thus, it is possible that the disrupted signaling communications other than myocardium itself account for the myocardial malformations. Further, many of these paracrine signaling pathways are reinvested during heart repair and/or regeneration. For instance, the epicardium acts as a pivotal hub for mediating heart regeneration3-6. Studies in both adult zebrafish hearts and neonatal mouse hearts have revealed that the epicardium plays a critical role for heart regeneration by providing paracrine growth factors, such as fibroblast growth factor (FGF)s and insulin-like growth factor (IGF)s to proliferating CMs6, 7. Understanding how these paracrine signals are regulated may provide important insights for both the pathogenesis of CHD and novel strategies for promoting heart regeneration.
The epicardium is composed of a single layer of mesothelial cells. It covers the outermost layer of the heart. The epicardium originates from a cluster of progenitor cells, which are located at the venous pole of the developing heart, known as the proepicardium (PE)8. The PE is initiated around embryonic day (E) 8.5 in mice9. The EPDCs emancipate from the epicardium through an epithelial-to-mesenchymal transition (EMT) event and give rise to several cardiac cell lineages including interstitial cardiac fibroblasts and coronary smooth muscle cells, which constitute cardiac stroma and provide oxygen and nutrients to heart muscle10, 11. During heart development, the epicardium also plays an important role by nurturing the underlying myocardium through secreting paracrine trophic factors such as FGF9 and IGF23.
FGF9 belongs to the FGF super family12. The binding of FGF9 to FGF receptors (FGFRs) triggers phosphorylation of FGFRs, and then subsequently activates the PI3K/AKT pathway and the MEK/ERK signaling cascade to drive cell proliferation and tissue morphogenesis12, 13. Either global deletion of FGF9 or conditional knockout of FGFR1/2 in the myocardium leads to ventricular hypoplasia14, suggesting that FGF9 and its downstream signaling is important for myocardial growth. IGF2 is another major paracrine growth signal released from epicardial cells and converges to the same downstream AKT and ERK signaling axis3, 15. Conditional knockout of either IGF2 in the epicardium or its major receptor, IGFR1 in cardiac progenitors (Nkx2-5+) resulted in reduced CM proliferation and ventricular wall hypoplasia16-18. In contrast, conditional deletion of IGF2 in the endocardium or myocardium did not give rise to any apparent cardiac phenotypes18. In the developing heart, FGF9 and IGF2 are mainly secreted by the epicardium and its EPDCs, with minor contributions from cardiac endothelial cells3, 19. Most previous research has focused on understanding the function and/or the downstream signaling of these growth factors12, 15. How the expression of these growth factors in the developing epicardium is regulated is still poorly understood.
Gene transcription can be heavily influenced by chromatin’s accessibility, which can be regulated by post-translational modifications of histones, including acetylation/deacetylation, methylation/demethylation, phosphorylation/dephosphorylation and ubiquitination/deubiquitination20. By switching between any of those two states, the associated genes can be dynamically programmed to be transcriptionally active or repressed. Histone deacetylase 3 (HDAC3), a member of the class I HDAC family that catalyzes the removal of acetyl groups from lysine residues in histone tails, has been implicated in many biological processes by modulating gene expression21. Mesodermal or global knockout of Hdac3 resulted in myogenic defects and early embryonic lethality22, 23. Interestingly, ablation of Hdac3 specifically in the myocardium does not give rise to cardiac morphological phenotypes during early heart development, but rather compromises cardiac function at later postnatal stages23, 24. These findings suggest that the function of HDAC3 in nonmyocyte compartments may be critical for early myocardial development.
MicroRNAs (miRs) are small non-coding RNAs (about 22 nucleotides in length) that post-transcriptionally regulate gene expression, and are greatly implicated in heart development, disease, and regeneration25, 26. MiRs can modulate the expression of a wide range of genes including growth factors. For instance, the expression of FGF2 and IGF2 in neural stem cells, cancer, or skeletal muscle is subject to the regulation by miR-1275, miR-483, and others27-30. It is not clear whether and how the expression of FGF9 and IGF2 is epigenetically (e.g., via HDACs or miRs) regulated in the developing epicardium.
In the present study, we investigated the role of HDAC3 in the epicardium during early heart development.
Methods
Data Availability
A detailed description of all experimental procedures and statistical tests can be found in the Supplemental Materials & Methods section in the Supplemental Materials.
Results
Specific inactivation of Hdac3 in the developing epicardium results in ventricular wall hypoplasia
To study the potential role of Hdac3 in the epicardium during heart development, we specifically ablated Hdac3 using Wt1CreERT2 10, a tamoxifen inducible epicardial Cre mouse, and Hdac3 floxed mice31. As expected, after tamoxifen induction at E8.5, Hdac3 was effectively and specifically deleted in the epicardium, whereas its expression in non-epicardial cells, such as CMs, was unaffected (Fig. 1A). We did not observe any leakiness of Cre in the Wt1CreERT2 allele (Fig. S1). To evaluate potential impact of Hdac3 deletion on epicardial development, we performed epicardial lineage tracing in both epicardial Hdac3 deficient (Hdac3f/f, Wt1CreERT2/+, R26eGFP/+) (Hdac3eko) hearts and their littermate control (Hdac3f/+, Wt1CreERT2/+, R26eGFP/+) (CTL) hearts. At E10.5 and E12.5, the epicardium was similarly traced in Hdac3eko and CTL hearts (Fig. S2A and S2B), suggesting proepicardial development and epicardial migration to the heart is not affected by epicardial Hdac3 deletion. From E12.5 to E16.5, EPDCs undergo EMT and migrate into the compact myocardium to become fibroblasts and coronary vascular cells32, 33. Interestingly, we found that Hdac3 deficient EPDCs were significantly fewer in E14.5 Hdac3eko hearts as compared to CTL hearts (Fig. S2A and S2B), although the contributions to each epicardial cell lineage including fibroblasts, smooth muscle cells appeared to be unaffected (Fig. S2C). Furthermore, the percentage of EPDCs that migrate into the compact myocardium was significantly lower in E14.5 Hdac3eko hearts as compared to CTL hearts (Fig. S2D), suggesting a potential EMT/migration defect in Hdac3 deficient EPDCs. Consistently, epicardial EMT markers such as Snail2 and Twist1 were significantly downregulated in Hdac3eko hearts (Fig. S2E). The epicardium is an important source for contributing to embryonic coronary development by both lineage contribution and providing paracrine patterning cues3, 34. Sox17 has been widely used as an early coronary vascular cell marker35-37. We found that Sox17+ cells were significantly fewer in Hdac3eko hearts as compared to CTL hearts (Fig. S3), suggesting that coronary vascular development is compromised in Hdac3eko hearts.
Figure 1. Epicardial deletion of Hdac3 resulted in hypoplasia of ventricular compact wall.

(A) Representative immunofluorescence staining of HDAC3 and WT1 in Hdac3eko (Hdac3f/f; Wt1CreERT2/+) hearts and control (CTL; Hdac3f/+; Wt1CreERT2/+) hearts. Tamoxifen was given to dams intraperitoneally (150 mg/kg body weight) at E8.5. Scale bars: 25 μm. (B) Representative H&E staining of Hdac3eko and CTL hearts. Scale bars: 250 μm. Quantification is shown on the right. CTL: n=6, Hdac3eko: n=7. The relative thickness of compact wall and the relative area of trabeculae for each sample was derived by dividing its actual measured thickness or trabecular area by the mean measured thickness or area value of the CTL group. (C) Representative immunofluorescence staining of BrdU and p-H3 in Hdac3eko hearts and CTL hearts. Scale bars: 250 μm. Quantification is shown on the right. For BrdU, CTL: n=6, Hdac3eko: n=6; For P-H3, CTL: n=7, Hdac3eko: n=7. P-values were determined by unpaired two tailed Student’s t-test for (B) and (C).
Both epicardium and EPDCs are major contributors to compact myocardial growth through cell-cell crosstalk3. Thus, we assessed the myocardia of epicardial Hdac3 deficient (Hdac3eko) hearts at several embryonic stages. The ventricular free wall morphogenesis was similar between Hdac3eko and their littermate CTL hearts at E9.5, E11.5 and E12.5 (Fig. S4). However, the compact layer in Hdac3eko hearts was significantly thinner than in the littermate CTL hearts at E13.5 and E14.5 (Fig. S4 and Fig. 1B), whereas there was no apparent morphological phenotypic difference in the Hdac3eko trabecular myocardia as compared to the littermate CTL hearts.
Next, we investigated whether altered cell proliferation and/or apoptosis contributes to the ventricular wall hypoplastic phenotypes in Hdac3eko hearts. At E13.5, we found that the percentage of p-H3+ or BrdU+ myocytes was significantly lower in Hdac3eko compact myocardia as compared to the littermate CTL hearts (Fig. 1C). In contrast, there was no significant difference in cell apoptosis between Hdac3eko and the littermate CTL hearts (Fig. S5). These findings are consistent with the hypoplastic cardiac phenotypes seen in Hdac3eko hearts (Fig. S4 and Fig. 1B).
Reduced expression of FGF9 and IGF2 in Hdac3 deficient epicardial cells contributes to deceased CM proliferation
To further understand the potential molecular mechanisms contributing to the hypoplastic ventricular wall phenotypes in Hdac3eko hearts, we effectively deleted Hdac3 in immortalized MECs using CRISPR/Cas9 technology (Fig. 2A). Hdac3 KO MECs grow slower than Hdac3 EV MECs. By performing bulk RNA sequencing and gene ontology (GO) analyses on Hdac3 KO and EV MECs, we found 1,681 downregulated genes and 1,549 upregulated genes. GO pathway analyses identified that the top 14 out of 20 significantly affected pathways are all involved in cell division (Fig. 2B). In a search for EPDC secreted growth factors in downregulated genes, we identified Fgf9 and Igf2 as our top candidates (volcano plot in Fig. 2C). We further validated the decrease of FGF9 and IGF2 at both mRNA and protein level in Hdac3 KO MECs (Fig. 2D and E). Consistently, FGF9 and IGF2 were also significantly decreased in Hdac3eko hearts (Fig. S6). These results are consistent with the cardiac growth defects seen in Hdac3eko hearts.
Figure 2. Hdac3 deletion resulted in downregulation of FGF9 and IGF2.

(A) Generation of Hdac3 knockout (KO) and empty vector control (EV) MECs by CRISPR/Cas9. Deletion of Hdac3 was verified by western blot. n=4 in each group. (B) Gene ontology (GO) pathway analyses and (C) Volcano plot of RNA Sequencing in Hdac3 KO and EV MECs. n=3 in each group. Log2 fold changes were calculated by RPKM (Reads per kilo base of transcript per million mapped reads) per gene in the KO group divided by the mean RPKM per gene in the EV group then followed by calculation of Log (fold change, 2). Significantly downregulated genes are shown in light blue, and significantly up-regulated genes are shown in red. Cut-off criteria: adjusted P-value<0.01. (D) Quantification of Fgf9 and Igf2 in Hdac3 KO MECs by qRT-PCR. Gapdh was used as cDNA loading control. n=4 in each group. (E) Quantification of FGF9 and IGF2 in Hdac3 KO and EV MECs by western blot. GAPDH was used as protein loading control. n=6 in each group. P-values were determined by the Mann-Whitney U test for (A), (D) and unpaired two tailed Student’s t-test for (E).
Next, we sought to determine whether reduction of FGF9 and IGF2 accounts for decreased CM proliferation. First, we found that FGF9 and IGF2 were significantly decreased in the supernatants from Hdac3 KO MECs as compared to that from Hdac3 EV MECs (Fig. 3A). Then, we treated cultured primary embryonic CMs isolated from E13.5 Tnnt2nGFP/+38 hearts with supernatants from either Hdac3 KO MECs or Hdac3 EV MECs, respectively. Hdac3 KO supernatant treatment resulted in significant decrease of the percentage of p-H3+ CMs and the total number of CMs, as compared to Hdac3 EV supernatant treatment (Fig. 3B and C). Consistently, Hdac3 KO supernatant attenuated the activation of downstream signaling pathway for CM proliferation, such as p-ERK (Fig. S7A). Lastly, to determine whether the reduction of FGF9 and IGF2 in Hdac3 KO supernatants is a major contributor to the decreased CM proliferation, we supplemented Hdac3 KO supernatants with recombinant mouse FGF9 or IGF2 protein, respectively. Strikingly, supplementation with FGF9 or IGF2 successfully rescued CM proliferation defects by Hdac3 KO supernatant treatment (Fig. 3B and C). We further confirmed the reactivation of p-FGFR1 or p-IGFR1 by FGF9 or IGF2 supplementation in Hdac3 KO supernatant treated CMs (Fig. S7B and S7C). Altogether, these results suggest that FGF9 and IGF2 secreted from epicardial cells provide important cues for driving CM proliferation.
Figure 3. Supplementation of FGF9 or IGF2 rescues CM proliferation defects.

(A) Secretion of FGF9 and IGF2 from Hdac3 KO and EV MECs. Coomassie brilliant blue staining of total extracted proteins from supernatants served as protein loading controls. FGF9 and IGF2 in the MEC supernatants were detected by western blot. Arrows point to the target bands. Quantifications are shown on the right. n=5 in each group. (B and C) The effects of MEC supernatants and/or recombinant FGF9 or IGF2 on E13.5 Tnnt2nGFP/+ CM proliferation. Representative immunofluorescence micrographs are shown. Scale bars: 275 μm. Percentage of p-H3+ CMs and total number of CMs were quantitated. Independent samples: FGF9, n=6 in each group; IGF2, n=12 in each group. P-values were determined by the Mann-Whitney U test for (A) and One-way ANOVA followed by the Tukey post hoc test for (B) and (C).
HDAC3 induces FGF9 and IGF2 expression dependent on its deacetylase enzymatic activities
To determine whether the deacetylase activities are required for HDAC3 to induce the expression of FGF9 and IGF2, we treated MECs with RGFP966, a selective HDAC3 deacetylase inhibitor39. Interestingly, RGFP966 treatment significantly decreased FGF9 and IGF2 (Fig. 4A and B). To further confirm that HDAC3 regulates FGF9 and IGF2 dependent on its enzymatic activities, we performed a set of genetic rescue experiments. As expected, re-expression of lentiviral wildtype Hdac3 in Hdac3 KO MECs successfully restored the expression of Fgf9 and Igf2 (Fig. 4C). In contrast, re-expression of lentiviral Hdac3 Y298H, a deacetylase dead mutant Hdac340, failed to rescue the expression of Fgf9 and Igf2 (Fig. 4C).
Figure 4. HDAC3 induces the expression of FGF9 and IGF2 dependent on its deacetylase activity.

(A) Decease of Fgf9 and Igf2 mRNAs after RGFP966 (a selective Hdac3 inhibitor) treatment. MECs were treated with 10 uM RGFP966 or vehicle for 24 hours. mRNA levels were quantified by qRT-PCR. Gapdh was used as a cDNA loading control. Fgf9, n=4 in each group; Igf2, n=5 in each group. (B) Decease of FGF9 and IGF2 protein levels after RGFP966 treatment. MECs were treated with 2.5 uM (n=6), 5 uM (n=6) or 10 uM (n=6) RGFP966 or vehicle (n=9) for 24 hours. FGF9 and IGF2 were quantified by western blot. GAPDH was used as a protein loading control. (C) mRNA levels of Fgf9 and Igf2 in Hdac3 KO and EV MECs after 24 hours treatment with Hdac3 WT (wild type), Y298H mutant, or mCherry (CTL) lentivirus. The expression of HDAC3 was quantified by western blot. n=6 in each group. The mRNA levels of Fgf9 and Igf2 were quantified by qRT-PCR. n=6 in each group. P-values were determined by the Mann-Whitney U test for (A) and One-way ANOVA followed by the Tukey post hoc test for (B) and (C).
HDAC3 induces the expression of FGF9 and IGF2 through repressing miR-322 and miR-503
The expression of FGFs and IGFs can be modulated by miRs41, 42, which can be downstream targets of HDACs in certain biological contexts43. To identify the potential HDAC3 downstream miR targets, we performed miR sequencing in Hdac3 KO and EV MECs. Through differential expression analyses, 42 miRs were significantly upregulated in Hdac3 KO MECs (Fig. 5A). Among the top 20 hits (fold change equal to or greater than 1.5 and P<0.01), we identified 11 miRs that have putative binding sites on either Fgf9 or Igf2 using the “DIANA MicroT-CDS” analyses tool44. We treated MECs with these 11 miR mimics and found that the treatment of miR-322 mimics or miR-503 mimics significantly inhibited the expression of both Fgf9 and Igf2 (Fig. 5B). miR-322 and miR-503 are encoded as one cluster by H19X, which is located in chromosome X, and they share the same “AGCAGC” sequences within the seed region at the 5’ end. The 3’ UTRs of both Fgf9 and Igf2 harbor putative binding sites for miR-322 and miR-503 (Fig. 5C). We further validated the significant inhibitory effects of miR-322 mimics or miR-503 mimics treatment on the expression of FGF9 and IGF2 by western blot (Fig. 5D). These results indicate that miR-322/miR-503 represses the expression of Fgf9 and Igf2. Further, we treated cultured E13.5 CMs with either miR-322 or miR-503 mimics and found that these treatments significantly decreased the percentage of p-H3+ CMs (Fig. 5E), suggesting that miR-322 and miR-503 inhibit CM proliferation.
Figure 5. miR-322 and miR-503 repress the expression of FGF9 and IGF2 and CM proliferation.

(A) Volcano plot of miR sequencing of Hdac3 KO and EV MECs. n=3 in each group. MiRNAs with reads less than 100 were discarded and miRNA expression levels were normalized by TPM (transcript per million) values (TPM = (miRNA total reads/total clean reads) × 106). Log2 fold changes were calculated by TPM per miR in the KO group divided by the mean TPM per miR in the EV group then followed by calculation of Log (fold change, 2). Significantly downregulated miRs are shown in light blue, and significantly up-regulated miRs are shown in red. Cut-off criteria: adjusted P-value<0.01. (B) Quantification of Fgf9 and Igf2 expression in MECs after miR mimics treatment (final concentration: 10 nM) by qRT-PCR. Gapdh was used as a cDNA loading control. Fgf9, n=7 in each group; Igf2, n=6 in each group. Fold changes were compared to the miR scrambles (SCR) group. (C) miR-322 and miR-503 share high similarity of their seed binding motifs to 3’UTRs of Fgf9 and Igf2. Binding motifs or complementary bases are in red. (D) Quantification of the expression of FGF9 and IGF2 after miR-322 or miR-503 mimics treatment by western blot. n=4 in each group. (E) The effects of supernatants from miR-322 and miR-503 mimics treated MECs on E13.5 CM proliferation. Representative immunofluorescence micrographs are presented. Scale bars: 275 um (upper images); 50 um (lower images). Percentage of p-H3+ CMs were quantified. Independent samples: n=12 in each group. (F) Quantification of the expression of miR-322 and miR-503 in Hdac3 KO MECs, RGFP966-treated MECs (10 uM), or E13.5 Hdac3eko hearts by qRT-PCR. U6 snRNA was used for normalization. miR-322 or miR-503 expression in EV or KO MECs: n=4 in each group; miR-322 expression after vehicle or RGFP966 treatment: n=5 in each group; miR-503 expression after vehicle or RGFP966 treatment: n=4 in each group; miR-322 expression in E13.5 hearts: CTL (n=6), Hdac3eko (n=8); miR-503 expression in E13.5 hearts: CTL (n=5), Hdac3eko (n=8). P-values were determined by One-way ANOVA followed by the Dunnett post hoc test for (B), One-way ANOVA followed by the Tukey post hoc test for (E), and the Mann-Whitney U test for (D) and (F).
We further validated the upregulation of miR-322 and miR-503 in Hdac3 KO MECs (Fig. 5F). Interestingly, miR-322 and miR-503 were similarly significantly upregulated when the deacetylase activity of HDAC3 is inhibited by RGFP966 treatment (Fig. 5F). The significant upregulation of miR-322 and miR-503 was also observed in E13.5 Hdac3eko hearts as compared to the littermate CTL hearts (Fig. 5F).
To determine whether the upregulation of miR-322 and miR-503 has causal effects on the reduction of FGF9 and IGF2 when Hdac3 is knocked out or inhibited, we knocked down the expression of miR-322 or miR-503 by miRZip lentivirus in Hdac3 KO MECs (Fig. 6A). Remarkably, knockdown of miR-322 or miR-503 significantly restored the expression of FGF9 and IGF2 in Hdac3 KO MECs (Fig. 6B). These results suggest that HDAC3 promotes the expression of FGF9 and IGF2 through repressing miR-322 and miR-503.
Figure 6. Knockdown of miR-322 or miR-503 restores the expression of FGF9 and IGF2.

(A) Quantification of miR-322 and miR-503 after Hdac3 KO or EV MECs were infected with LentimiRa-GFP-miRZip (miR-322 or miR-503) or pGreenPuro Scramble Hairpin control lentivirus (SCR) respectively. miR levels were quantified by qRT-PCR. n=6 in each group. (B) Quantification of the expression of FGF9 and IGF2 after miRZip lentiviral treatment by western blot. n=8 in each group. P-values were determined by One-way ANOVA followed by the Tukey post hoc test for (A) and (B).
HDAC3 represses miR-322/miR-503 promoter activity
H3K27Ac is a marker for active promoters and a direct downstream target of HDAC321. As expected, Hdac3 deletion resulted in significant increase of H3K27Ac in MECs (Fig. 7A). Identified by the ENCODE project, the promoter region of miR-322/miR-503 is subject to epigenetic regulations such as H3K27Ac (Fig. 7B). To determine whether Hdac3 deletion would affect the chromatin accessibility of the miR-322/miR-503 promoter, we surveyed H3K27Ac binding affinity in Hdac3 KO MECs by ChIP-qPCR. We found that Hdac3 deletion significantly increased the binding of H3K27Ac to the miR-322/miR-503 promoter (Fig. 7C), suggesting that Hdac3 deletion renders the miR-322/miR-503 promoter more accessible to transcriptional factors. As expected, HDAC3 binds to this promoter region, as determined by HDAC3 ChIP-qPCR (Fig. 7D). To further test whether this increased chromatin accessibility would affect the miR-322/miR-503 promoter activities, we performed an miR-322/miR-503 promoter luciferase reporter assay. We found the luciferase activity was significantly increased in Hdac3 KO MECs as compared to Hdac3 EV MECs (Fig. 7E). Strikingly, inhibition of HDAC3 by RGFP966 replicated this result (Fig. 7E), suggesting that the regulation of HDAC3 on the miR-322/miR-503 promoter is dependent on its deacetylase activities.
Figure 7. HDAC3 represses miR-322/miR-503 promoter activity.

(A) Quantification of H3K27Ac in Hdac3 EV and KO MECs by western blot. n=5 in each group. (B) Schematic diagram of the miR-322/miR-503 locus from the UCSC Genome Browser. In the upstream regulatory regions as well as gene bodies, active epigenetic marker H3K27Ac was identified by the ENCODE project. (C) Quantification of H3K27Ac binding affinity in the miR-322/miR-503 promoter region in Hdac3 KO and EV MECs by ChIP-qPCR. Primers targeting a gene desert region were used as a negative control. n=6 in each group. (D) Quantification of binding of HDAC3 to the miR-322/miR-503 promoter region by ChIP-qPCR in MECs. n=5 in each group. (E) Dual luciferase reporter assays on the −1.5 kb miR-322/miR-503 promoter when Hdac3 is either knocked out or inhibited by RGFP966 (10 uM) treatment. The ratio of firefly:Renilla luciferase light units (RLU) was determined 48 hours after transfection. n=6 in each group. (F) The schematics of the working model. In the developing epicardium, HDAC3 represses the expression of miR-322/miR-503 to release their suppression on the expression of FGF9 and IGF2. When Hdac3 is deleted, the expression of miR-322/miR-503 is increased, which subsequently suppresses the expression of FGF9 and IGF2 to a stronger extent, and the decrease of FGF9 and IGF2 leads to ventricular wall hypoplasia. P-values were determined by the Mann-Whitney U test for (A) and (D), the one-way ANOVA followed by the Tukey post hoc test for (C) and unpaired two tailed Student’s t-test for (E).
Discussion
Early elegant studies in avian embryos have revealed that delay or blockade of the epicardium formation leads to decreased CM proliferation in the ventricular free wall without affecting trabecular myocardial development, which is more dependent on the support from the endocardium45, 46. Subsequent studies found that epicardium stimulates ventricular wall expansion by providing mitogens to CMs such as retinoic acid, FGFs, and IGFs16, 47, 48. However, it is unclear how these mitogens are initially induced in the epicardium. Epigenetics has been increasingly recognized as an important regulator of gene transcription in a variety of physiological/pathological processes including cardiac development and congenital diseases49. Our current study suggests that HDAC3 induces the expression of FGF9 and IGF2 in the epicardium, and thus stimulates ventricular myocardial wall expansion through paracrine signaling. Our study provides strong evidence that the epigenetics in the epicardium regulates ventricular wall morphogenesis, a new perspective on the mechanisms of cardiac development.
HDAC1, HDAC2 and HDAC3 are categorized in the same family (Class I HDACs), and global knockout in each case is lethal although their cardiovascular phenotypes are all dinstinct50, suggesting that they all have unique functions. HDAC1 and HDAC2 are almost identical and they are often found in same repressive complexes such as Sin3 and NuRD51, while HDAC3 usually partners with Nuclear receptor co-repressor 1 (NCoR1 or NCoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT)52. The difference in these biochemical characteristics may underlie their distinct functions. Knockout of Hdac3 alone in the developing epicardium resulted in cardiac defects, indicating that other HDACs are not functionally redundant with HDAC3/unable to compensate for the loss of HDAC3. Meanwhile, it awaits investigation whether deletion of other Hdacs in the developing epicardium will elicit cardiac phenotypes.
The molecular action of HDAC3 is complex. NCoR and SMRT are both nuclear receptor co-repressors. The NCoR/SMRT complex stoichiometrically recruits HDAC3 and activates its intrinsic deacetylase activity21. HDAC3 mainly deacetylates H3K9Ac and H3K27Ac. Deacetylation of these two sites renders chromatin structure unfavorable for the recruitment of transcriptional factors, and thus dampens gene transcription. This is the mechanism by which HDAC3 epigenetically suppresses gene transcription in regulation of skeletal muscle metabolism53, 54. However, when HDAC3 determines CM fate, it acts as a recruiter to tether genes to the nuclear periphery for silencing and its deacetylase enzymatic activity is dispensable55. Interestingly, several recent studies highlight a critical function for HDAC3 as a gene activator, rather than a gene repressor55-58. Whether HDAC3 acts as a repressor or an activator might depend on the nature of its interacting partners and cell context. Our current study demonstrates that in the developing epicardium, HDAC3 works as a gene repressor to suppress the expression of miR-322/miR-503 through its deacetylase activities.
To identify the potential targets of HDAC3 in the epicardium, we narrowed down to FGF9 and IGF2 by both unbiased screen (RNA-Seq) and candidate (known growth factors) approaches. It is possible that other unaccounted dysregulated genes caused by Hdac3 deletion might contribute to the hypoplastic ventricular wall phenotype. We will explore those possibilities in future studies.
miR-322 (ortholog of human miR-424) and miR-503 belong to the miR-15/107 family. They are encoded as one cluster by H19X. miR-322 and miR-503 regulate many fundamental processes such as cell proliferation, death, differentiation, metabolism, and stress response. Thus, they are widely implicated in many disorders such as cardiovascular disease, neural disease, and cancer59, 60. During heart development, miR-322/miR-503 has been shown to drive early cardiac progenitor cells toward CM lineage, although the underlying mechanism is undetermined61. Our current study demonstrates that HDAC3 represses miR-322 and miR-503 expression to induce the expression of several major growth ligands including FGF9 and IGF2 for ventricular wall CM proliferation (Fig. 7F). This is the first line of study to demonstrate that the myocardial growth cues from the epicardium are tightly controlled by double inhibitory epigenetic regulation with HDACs and microRNAs. HDAC3 modulates the accessibility of miR-322/miR-503 promoter (Fig. 7C). Using transcription factor search bioinformatic tools, we identified over 300 putative transcription factors that bind to the 1.5kb miR-322/miR-503 promoter region. These transcription factors include C/EBPß, RXRα, Smad, ETS-1 and LEF-1/TCF-1, which have been shown to regulate epicardial development62-66. Some of these transcription factors may work with HDAC3 to regulate miR-322/503 expression. We will explore it more in future studies.
Many forms of CHD, such as hypoplastic left heart syndrome, are associated with defective cardiac myogenesis, which further compromises cardiac contractile function67. Proper cardiac myogenesis requires precise coordination among multiple cell types in the developing heart, rather than just requiring CMs. Nonmyocytes including EPDCs provide key growth signals to the adjacent compact myocardium in a paracrine fashion2. Our findings suggest that disrupted epicardial signaling (FGF9 and IGF2) significantly compromises the compact wall expansion in Hdac3eko hearts. Epicardium also plays important roles during heart regeneration and repair, such as providing paracrine signals and stimulating neovascularization3-6, 68. It’s very common that some growth signaling pathways employed during heart development are reactivated upon myocardial injuries/stresses and contribute to heart regeneration/repair. In future studies, it will be interesting to determine whether this HDAC3—miR-322/miR-503—FGF9/IGF2 axis is reutilized during regenerative responses and manipulation of this pathway affects the outcome of neonatal/adult heart repair after injuries.
During heart development, EPDCs mainly give rise to interstitial cardiac fibroblasts and coronary smooth muscle cells11, maybe to CMs and endothelial cells with controversial results10, 69-71. We found that the total number of EPDCs is significantly decreased in Hdac3eko hearts, whereas the percentage of contributions to each cell type was not affected (Fig. S2). The reduction of EPDC population in Hdac3eko hearts likely also contributes to the ventricular hypoplasia phenotypes since the total amount of FGF9 and IGF2 will be decreased as a result of EPDC population reduction. Further, Hdac3 deletion appeared to specifically affect EPDC invasion to the compact myocardium (Fig. S2). It is not well understood how the lineage contribution by EPDCs is regulated. Growth factors (e.g., FGF10) can promote this process by either inducing EMT or stimulating the proliferation of epicardium-derived terminal cells48, 72. It is possible that the reduction of FGF9 and IGF2 in Hdac3eko hearts accounts for decreased derivation of epicardial lineages.
In summary, we demonstrated that epicardial HDAC3 orchestrates ventricular wall expansion by inducing FGF9 and IGF2 expression through repressing miR-322/miR-503. Our study provides strong evidence that epigenetic factors such as HDAC3 play pivotal roles for the expression of these paracrine growth signals in the epicardium. Our findings strengthen the importance of epicardial paracrine signaling for myocardial development, which is implicated in the pathogenesis of CHDs and adult heart regeneration.
Supplementary Material
Novelty and Significance.
WHAT IS KNOWN?
Paracrine growth signals including fibroblast growth factors (FGFs) and insulin-like growth factors (IGFs) from the developing epicardium and epicardium-derived cells (EPDCs) promote the expansion of compact myocardium. However, it is unclear how these signals are transcriptionally regulated.
Histone deacetylase 3 (HDAC3) is essential for heart development in general, but its function in the developing epicardium has not been studied.
microRNA (miR)-322 and miR-503 are important for cardiac progenitor cell proliferation and differentiation. Their potential roles in other cardiac cells including EPDCs have been elusive.
WHAT NEW INFORMATION DOES THIS ARTICLE CONTRIBUTE?
The expression of Hdac3 in the developing epicardium is critical for compact myocardial growth.
HDAC3 is important for EPDC derivation and migration.
miR-322 and miR-503 suppress the transcription of Fgf9 and Igf2.
HDAC3 promotes the transcription of Fgf9 and Igf2 by repressing the expression of miR-322 and miR-503.
In the present study, we discovered that deletion of Hdac3 in epicardial cells compromises developing CM proliferation both in vitro and in vivo. Through lineage tracing at several developmental stages, we found that deletion of Hdac3 in the epicardium resulted in reduced EPDC derivation and migration. Hdac3 deletion led to downregulation of FGF9 and IGF2 both in vitro and in vivo. Further, we showed that HDAC3 induces the expression of FGF9 and IGF2 dependent on its deacetylase activity. We found that miR-322 and miR-503 were significantly upregulated in Hdac3 deficient epicardial cells both in vitro and in vivo, whereas ectopic expression of these miRs in epicardial cells resulted in the reduced expression of FGF9 and IGF2, and subsequently led to decreased CM proliferation. Importantly, inhibition of miR-322 or miR-503 restored the expression of FGF9 and IGF2 in Hdac3 deleted epicardial cells. Finally, we demonstrated that HDAC3 physically binds to the promoter of miR-322/miR-503 and suppresses their expression. Overall, we conclude that epicardial HDAC3 regulates compact myocardial growth by inducing FGF9 and IGF2 expression through repressing miR-322 and miR-503. Our study exemplifies how the expression of growth factors is regulated by a dual epigenetic mechanism (histone modifiers and microRNAs), and dysregulation of such signaling axis among distinct cell types may elicit congenital heart defects. On the other hand, this mechanism may well be invested to induce cardiac regeneration and repair.
Acknowledgments
We gratefully acknowledge Dr. Mitchell Lazar (University of Pennsylvania, Philadelphia, Pennsylvania, USA) for providing Hdac3flox/+ mice. We are also very grateful to Dr. Zheng Sun at Baylor College of Medicine for providing Hdac3 plasmids. We also thank Dr. Sari D. Holmes at the Johns Hopkins University School of Medicine for her critical reading and editing of the manuscript.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute R01 grant HL153406 (D.L.) and startup funds from Department of Surgery, University of Maryland School of Medicine.
Abbreviations
- CHD
Congenital Heart Disease
- EPDC
Epicardium-Derived Cell
- CM
Cardiomyocyte
- EMT
Epithelial-to-Mesenchymal Transition
- HDAC3
Histone Deacetylase 3
- FGF9
Fibroblast Growth Factor 9
- IGF2
Insulin-like Growth Factor 2
- miR
MicroRNA
- BrdU
5-bromo-2'-deoxyuridine
Footnotes
Disclosure
None.
References
- 1.Zaidi S and Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circulation research. 2017;120:923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian Y and Morrisey EE. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circulation research. 2012;110:1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quijada P, Trembley MA and Small EM. The Role of the Epicardium During Heart Development and Repair. Circulation research. 2020;126:377–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlueter J and Brand T. Epicardial progenitor cells in cardiac development and regeneration. J Cardiovasc Transl Res. 2012;5:641–53. [DOI] [PubMed] [Google Scholar]
- 5.Saifi O, Ghandour B, Jaalouk D, Refaat M and Mahfouz R. Myocardial regeneration: role of epicardium and implicated genes. Mol Biol Rep. 2019;46:6661–6674. [DOI] [PubMed] [Google Scholar]
- 6.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN and Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao J and Poss KD. The epicardium as a hub for heart regeneration. Nat Rev Cardiol. 2018;15:631–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manner J The development of pericardial villi in the chick embryo. Anat Embryol (Berl). 1992;186:379–85. [DOI] [PubMed] [Google Scholar]
- 9.Niderla-BieliNska J, Jankowska-Steifer E, Flaht-Zabost A, Gula G, Czarnowska E and Ratajska A. Proepicardium: Current Understanding of its Structure, Induction, and Fate. Anat Rec (Hoboken). 2019;302:893–903. [DOI] [PubMed] [Google Scholar]
- 10.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR and Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamora M, Manner J and Ruiz-Lozano P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc Natl Acad Sci U S A. 2007;104:18109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh N, Ohta H, Nakayama Y and Konishi M. Roles of FGF Signals in Heart Development, Health, and Disease. Front Cell Dev Biol. 2016;4:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soszynska A, Klimczewska K and Suwinska A. FGF/ERK signaling pathway: how it operates in mammalian preimplantation embryos and embryo-derived stem cells. Int J Dev Biol. 2019;63:171–186. [DOI] [PubMed] [Google Scholar]
- 14.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J and Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. [DOI] [PubMed] [Google Scholar]
- 15.Iosef Husted C and Valencik M. Insulin-like growth factors and their potential role in cardiac epigenetics. J Cell Mol Med. 2016;20:1589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Bruning JC, Pashmforoush M and Sucov HM. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development. 2011;138:1795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Shen H, Gan P, Cavallero S, Kumar SR, Lien CL and Sucov HM. Differential roles of insulin like growth factor 1 receptor and insulin receptor during embryonic heart development. BMC Dev Biol. 2019;19:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen H, Cavallero S, Estrada KD, Sandovici I, Kumar SR, Makita T, Lien CL, Constancia M and Sucov HM. Extracardiac control of embryonic cardiomyocyte proliferation and ventricular wall expansion. Cardiovasc Res. 2015;105:271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barak Y, Hemberger M and Sucov HM. Phases and Mechanisms of Embryonic Cardiomyocyte Proliferation and Ventricular Wall Morphogenesis. Pediatric cardiology. 2019;40:1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar R, Deivendran S, Santhoshkumar TR and Pillai MR. Signaling coupled epigenomic regulation of gene expression. Oncogene. 2017;36:5917–5926. [DOI] [PubMed] [Google Scholar]
- 21.Emmett MJ and Lazar MA. Integrative regulation of physiology by histone deacetylase 3. Nature reviews Molecular cell biology. 2019;20:102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW and Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewandowski SL, Janardhan HP and Trivedi CM. Histone Deacetylase 3 Coordinates Deacetylase-independent Epigenetic Silencing of Transforming Growth Factor-beta1 (TGF-beta1) to Orchestrate Second Heart Field Development. The Journal of biological chemistry. 2015;290:27067–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R and Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quiat D and Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med. 2014;6:239ps3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novak J, Vinklarek J, Bienertova-Vasku J and Slaby O. MicroRNAs involved in skeletal muscle development and their roles in rhabdomyosarcoma pathogenesis. Pediatr Blood Cancer. 2013;60:1739–46. [DOI] [PubMed] [Google Scholar]
- 28.Fawzy IO, Hamza MT, Hosny KA, Esmat G, El Tayebi HM and Abdelaziz AI. miR-1275: A single microRNA that targets the three IGF2-mRNA-binding proteins hindering tumor growth in hepatocellular carcinoma. FEBS Lett. 2015;589:2257–65. [DOI] [PubMed] [Google Scholar]
- 29.Liu M, Roth A, Yu M, Morris R, Bersani F, Rivera MN, Lu J, Shioda T, Vasudevan S, Ramaswamy S, Maheswaran S, Diederichs S and Haber DA. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes & development. 2013;27:2543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Barkho BZ, Luo Y, Smrt RD, Santistevan NJ, Liu C, Kuwabara T, Gage FH and Zhao X. Epigenetic regulation of the stem cell mitogen Fgf-2 by Mbd1 in adult neural stem/progenitor cells. The Journal of biological chemistry. 2008;283:27644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, Feng D, Steger DJ, Schug J, Artis D and Lazar MA. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes & development. 2011;25:2480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quijada P, Trembley MA, Misra A, Myers JA, Baker CD, Perez-Hernandez M, Myers JR, Dirkx RA Jr., Cohen ED, Delmar M, Ashton JM and Small EM. Coordination of endothelial cell positioning and fate specification by the epicardium. Nature communications. 2021;12:4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su T, Stanley G, Sinha R, D'Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT, Roper WA, Li G, Quinn KE, Caron KM, Wu S, Miquerol L, Butcher EC, Weissman I, Quake S and Red-Horse K. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. 2018;559:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma B, Chang A and Red-Horse K. Coronary Artery Development: Progenitor Cells and Differentiation Pathways. Annu Rev Physiol. 2017;79:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma B, Ho L, Ford GH, Chen HI, Goldstone AB, Woo YJ, Quertermous T, Reversade B and Red-Horse K. Alternative Progenitor Cells Compensate to Rebuild the Coronary Vasculature in Elabela- and Apj-Deficient Hearts. Dev Cell. 2017;42:655–666 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Hernandez S, Gomez MJ, Sanchez-Cabo F, Mendez-Ferrer S, Munoz-Canoves P and Isern J. Sox17 Controls Emergence and Remodeling of Nestin-Expressing Coronary Vessels. Circulation research. 2020;127:e252–e270. [DOI] [PubMed] [Google Scholar]
- 37.Han M and Zhou B. Sox17 and Coronary Arteriogenesis in Development. Circulation research. 2020;127:1381–1383. [DOI] [PubMed] [Google Scholar]
- 38.Yan J, Zhang L, Sultana N, Oh JG, Wu B, Hajjar RJ, Zhou B and Cai CL. A series of robust genetic indicators for definitive identification of cardiomyocytes. J Mol Cell Cardiol. 2016;97:278–85. [DOI] [PubMed] [Google Scholar]
- 39.Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR and Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, Koch U, De Francesco R, Steinkuhler C and Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katoh Y and Katoh M. FGFR2-related pathogenesis and FGFR2-targeted therapeutics (Review). Int J Mol Med. 2009;23:307–11. [DOI] [PubMed] [Google Scholar]
- 42.van de Worp W, Theys J, van Helvoort A and Langen RCJ. Regulation of muscle atrophy by microRNAs: 'AtromiRs' as potential target in cachexia. Curr Opin Clin Nutr Metab Care. 2018;21:423–429. [DOI] [PubMed] [Google Scholar]
- 43.Tchio Mantho CI, Harbuzariu A and Gonzalez-Perez RR. Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J Clin Oncol. 2017;8:178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T and Hatzigeorgiou AG. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manner J Experimental study on the formation of the epicardium in chick embryos. Anat Embryol (Berl). 1993;187:281–9. [DOI] [PubMed] [Google Scholar]
- 46.Gittenberger-de Groot AC, Vrancken Peeters MP, Bergwerff M, Mentink MM and Poelmann RE. Epicardial outgrowth inhibition leads to compensatory mesothelial outflow tract collar and abnormal cardiac septation and coronary formation. Circulation research. 2000;87:969–71. [DOI] [PubMed] [Google Scholar]
- 47.Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR and Evans RM. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes & development. 1994;8:1007–18. [DOI] [PubMed] [Google Scholar]
- 48.Vega-Hernandez M, Kovacs A, De Langhe S and Ornitz DM. FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development. 2011;138:3331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moore-Morris T, van Vliet PP, Andelfinger G and Puceat M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol Rev. 2018;98:2453–2475. [DOI] [PubMed] [Google Scholar]
- 50.Haberland M, Montgomery RL and Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews Genetics. 2009;10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang XJ and Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–53. [DOI] [PubMed] [Google Scholar]
- 52.Yang XJ and Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nature reviews Molecular cell biology. 2008;9:206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong MM, Guo C and Zhang J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol. 2014;2:169–87. [PMC free article] [PubMed] [Google Scholar]
- 54.Song S, Wen Y, Tong H, Loro E, Gong Y, Liu J, Hong S, Li L, Khurana TS, Chu M and Sun Z. The HDAC3 enzymatic activity regulates skeletal muscle fuel metabolism. J Mol Cell Biol. 2019;11:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagan JE, Sun Z, Wang Q, Li L, Dubois NC, Morrisey EE, Lazar MA, Smith CL, Epstein JA and Jain R. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell. 2017;171:573–587 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, Baur JA, Pei L, Won KJ, Seale P, Gerhart-Hines Z and Lazar MA. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. 2017;546:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janardhan HP, Milstone ZJ, Shin M, Lawson ND, Keaney JF Jr. and Trivedi CM. Hdac3 regulates lymphovenous and lymphatic valve formation. J Clin Invest. 2017;127:4193–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang L, He X, Liu L, Jiang M, Zhao C, Wang H, He D, Zheng T, Zhou X, Hassan A, Ma Z, Xin M, Sun Z, Lazar MA, Goldman SA, Olson EN and Lu QR. Hdac3 Interaction with p300 Histone Acetyltransferase Regulates the Oligodendrocyte and Astrocyte Lineage Fate Switch. Dev Cell. 2016;36:316–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang F, Liang R, Tandon N, Matthews ER, Shrestha S, Yang J, Soibam B, Yang J and Liu Y. H19X-encoded miR-424(322)/-503 cluster: emerging roles in cell differentiation, proliferation, plasticity and metabolism. Cell Mol Life Sci. 2019;76:903–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Wu Y, Zhang J, Sun H and Wang X. Role of miRNA-424 in Cancers. Onco Targets Ther. 2020;13:9611–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen X, Soibam B, Benham A, Xu X, Chopra M, Peng X, Yu W, Bao W, Liang R, Azares A, Liu P, Gunaratne PH, Mercola M, Cooney AJ, Schwartz RJ and Liu Y. miR-322/-503 cluster is expressed in the earliest cardiac progenitor cells and drives cardiomyocyte specification. Proc Natl Acad Sci U S A. 2016;113:9551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang GN, Thatcher JE, McAnally J, Kong Y, Qi X, Tan W, DiMaio JM, Amatruda JF, Gerard RD, Hill JA, Bassel-Duby R and Olson EN. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science. 2012;338:1599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dronkers E, Wauters MMM, Goumans MJ and Smits AM. Epicardial TGFbeta and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart? Biomolecules. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, Chien KR and Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005;102:18455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macias D, Perez-Pomares JM, Garcia-Garrido L, Carmona R and Munoz-Chapuli R. Immunoreactivity of the ets-1 transcription factor correlates with areas of epithelial-mesenchymal transition in the developing avian heart. Anat Embryol (Berl). 1998;198:307–15. [DOI] [PubMed] [Google Scholar]
- 66.von Gise A, Zhou B, Honor LB, Ma Q, Petryk A and Pu WT. WT1 regulates epicardial epithelial to mesenchymal transition through beta-catenin and retinoic acid signaling pathways. Dev Biol. 2011;356:421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yester JW and Kuhn B. Mechanisms of Cardiomyocyte Proliferation and Differentiation in Development and Regeneration. Curr Cardiol Rep. 2017;19:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pellegrini L, Foglio E, Pontemezzo E, Germani A, Russo MA and Limana F. Cardiac Repair: The Intricate Crosstalk between the Epicardium and the Myocardium. Curr Stem Cell Res Ther. 2020;15:661–673. [DOI] [PubMed] [Google Scholar]
- 69.Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J and Evans SM. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudat C and Kispert A. Wt1 and epicardial fate mapping. Circulation research. 2012;111:165–9. [DOI] [PubMed] [Google Scholar]
- 71.Carmona R, Barrena S, Lopez Gambero AJ, Rojas A and Munoz-Chapuli R. Epicardial cell lineages and the origin of the coronary endothelium. FASEB J. 2020;34:5223–5239. [DOI] [PubMed] [Google Scholar]
- 72.Cao Y, Duca S and Cao J. Epicardium in Heart Development. Cold Spring Harb Perspect Biol. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwartz RJ, Field LJ, Atkinson SJ and Shou W. Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development. 2011;138:303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Bruning JC, Pashmforoush M and Sucov HM. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development. 2011;138:1795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, Won KJ and Lazar MA. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L, Feng Z, Wang X, Wang X and Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8. [DOI] [PubMed] [Google Scholar]
- 77.Schmittgen TD and Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of all experimental procedures and statistical tests can be found in the Supplemental Materials & Methods section in the Supplemental Materials.