

Abstract
Histone deacetylases (HDACs) are important epigenetic regulators involved in many diseases, especially cancer. Five HDAC inhibitors have been approved for anticancer therapy and many are in clinical trials. Among the 11 zinc‐dependent HDACs, HDAC10 has received relatively little attention by drug discovery campaigns, despite its involvement, e. g., in the pathogenesis of neuroblastoma. This is due in part to a lack of robust enzymatic conversion assays. In contrast to the protein lysine deacetylase and deacylase activity of most other HDAC subtypes, it has recently been shown that HDAC10 has strong preferences for deacetylation of oligoamine substrates like acetyl‐putrescine or ‐spermidine. Hence, it is also termed a polyamine deacetylase (PDAC). Here, we present the first fluorescent enzymatic conversion assay for HDAC10 using an aminocoumarin‐labelled acetyl‐spermidine derivative to measure its PDAC activity, which is suitable for high‐throughput screening. Using this assay, we identified potent inhibitors of HDAC10‐mediated spermidine deacetylation in vitro. Based on the oligoamine preference of HDAC10, we also designed inhibitors with a basic moiety in appropriate distance to the zinc binding hydroxamate that showed potent inhibition of HDAC10 with high selectivity, and we solved a HDAC10‐inhibitor structure using X‐ray crystallography. We could demonstrate selective cellular target engagement for HDAC10 but a lysosomal phenotype in neuroblastoma cells that was previously associated with HDAC10 inhibition was not observed. Thus, we have developed new chemical probes for HDAC10 that allow further clarification of the biological role of this enzyme.
Keywords: HDAC, histone deacetylases, HDAC10, medicinal chemistry, polyamines
HDAC10 is as a polyamine deacetylase from the histone deacetylase (HDAC) family. Using a fluorescent acetylspermidine derivative, we developed the first enzymatic conversion assay for HDAC10. We designed HDAC10 selective inhibitors and demonstrated that they do not cause a lysosomal phenotype previously associated with HDAC10 inhibition in cancer cells.

Introduction
Histone deacetylases are important players in epigenetic regulation. [1] Besides their eponymous deacetylase activity on histones, histone deacetylases (HDACs) have been found to be active on an increasing number of non‐histone proteins. One of the most prominent examples is the tumor suppressor protein p53. [2] Other examples include the cytoskeleton protein α‐tubulin and proteins such as SMC3, HSP90 and ERRα. [3] HDACs are also known as lysine deacetylases (KDACs), a name that better reflects their broad substrate specificity. The 18 known human KDACs are divided into two groups ‐ the classical zinc‐dependent enzymes (class I, IIa/b, IV) and the NAD+‐dependent sirtuins (class III, Sirt1‐7). The zinc‐dependent enzymes are subdivided into four classes based on phylogenetic analysis: class I consists of HDAC1, 2, 3 and 8; class IIa consists of HDAC4, 5, 7 and 9; class IIb consists of HDAC6 and 10; and class IV consists of only HDAC11. [4] Recently, the substrate specificities of HDAC10 and 11 were redefined: HDAC11 was discovered to be a protein‐lysine fatty‐acid deacylase [5] and HDAC10 was discovered to be a polyamine deacetylase (PDAC). [6] Thus, HDAC10 has important non‐protein, non‐lysine deacetylase activity.
Regarding their broad range of interaction partners HDACs are involved in many physiological and pathological processes, such as regulation of metabolism, [7] aging, [8] gene transcription [9] and homologous recombination. [10] Furthermore, a prominent role in neurodegenerative diseases, [11] metabolic disorders [12] and cancer [13] was reported for this enzyme class. Since only limited treatment options are available for these diseases, HDACs are emerging targets for new therapeutic approaches.
As previously mentioned, the substrate specificity of HDAC10 stands out compared to the other family members. Hai et al. demonstrated that acetylated polyamines are preferred substrates. [6] The highest catalytic activity was measured for N8‐acetylspermidine (1) (Figure 1). Furthermore, acetylputrescine (2) and N1,N8‐diacetylspermidine (3) were deacetylated as well. In contrast, N1‐acetylspermidine (4) was converted to a much lesser extent.
Figure 1.

Polyamine substrates of HDAC10.
Critical for substrate recognition is a negatively charged glutamate (Glu272 hHDAC10 resp. Glu274 in drHDAC10) at the entrance of the active site, which acts as a gatekeeper to favor the binding of protonated and hence positively charged polyamine substrates. The crystal structure of inactivated drHDAC10 complexed with N8‐acetylspermidine shows that Glu274 engages the protonated secondary amino group of the substrate with two water‐mediated hydrogen bonds. [14] The preferential binding of N8‐acetylspermidine versus N1‐acetylspermidine is explained by the position and orientation of the secondary amino group. A distance of four carbons between the amide moiety and the secondary amino group is favorable for the substrate recognition. [6]
In recent years, HDAC10 has been linked to tumor development and proliferation. [15] The development of potential drugs that block HDAC10 has emerged as a potential new therapeutic strategy for the treatment of cancer, e. g. neuroblastoma,[ 13a , 16 ] lung cancer, [17] ovarian cancer [18] and leukemia. [19]
The first potent HDAC inhibitors (HDACis) trichostatin A (TSA, 5) (Figure 2) and trapoxin were reported many years ago. [20] Since vorinostat (6) was approved as the first HDACi for treatment of cutaneous T‐cell lymphoma by the FDA three more HDAC inhibitors (romidepsin (7), belinostat (8), panobinostat (9)) received FDA approval for cancer treatment. [21] In China one more substance is approved ‐ tucidinostat (chidamide, 10). [22] Additional drug candidates are in clinical trials for cancer treatment. Examples are quisinostat (11), abexinostat (12) or mocetinostat (13). [23]
Figure 2.

Structures of selected HDAC inhibitors.
Many HDAC inhibitors show unselective activity over a wider range of HDAC subtypes. This broad target range has been suggested to be the source of unwanted side effects [24] and hence an increasing number of subtype selective HDAC inhibitors has been developed as chemical tools and drug candidates [25] (reviewed in ref. [26]). While hydroxamates and benzamides dominate the literature in general and the clinically approved inhibitors, there are also other scaffolds with thiols and hydrazides as notable zinc binding groups. [27]
Suitable assay systems must be available to enable the development of isozyme‐selective HDAC inhibitors, but certain isozymes currently lack a facile activity assay. Specifically, due to the newly discovered substrate specificity of HDAC10, the standard HDAC activity assay based on the hydrolysis of an acetyllysine substrate is inappropriate. The fixed‐point acetylpolyamine assay developed by Hai and colleagues[ 6 , 28 ] employs a polyamine substrate but is not well‐suited for high‐throughput screening. Only weak lysine deacetylase activity was observed for HDAC10, which increases the risk of false positive results if cell‐derived enzyme samples are contaminated with other HDAC isozymes.[ 6 , 29 ] As an alternative to a substrate conversion assay, two inhibitor based binding assay systems for HDAC10 have been reported in literature. A time resolved fluorescence energy transfer (TR‐FRET) in vitro assay with recombinant HDAC10 and a bioluminescence energy transfer (BRET) cellular assay were presented. [30] In both cases the displacement of a fluorescent HDAC10 probe by a competing binder leads to a change of the measured signal. Using these displacement assay systems, Géraldy and colleagues discovered potent HDAC10 hits by testing a set of inhibitors thought to be HDAC6‐selective, including tubastatin A (14), HPOB and nexturastat, [31] which was not unexpected in the end since both HDAC6 and HDAC10 are class IIb enzymes. Further investigations on tubastatin A (14) and derivatives (15‐17) identified the basic amine next to the indole structure as crucial for HDAC10 binding, which is consistent with the specificity for polyamine substrates based on the gatekeeper glutamate (see Figure 3). Compound 14 and 16 were bound strongly by HDAC10, while removing basic properties by substitution of the amine by oxygen (15) or by Boc‐protection (17) led to a strongly diminished binding affinity. A salt bridge between the basic amine structure and the gatekeeper residue was postulated for the HDAC10 binders. Géraldy and colleagues assumed an additional flexibility in the L1 loop structure of HDAC10 to be necessary to bind more bulky molecules, such as tubastatin A. [32] While the interaction with the gatekeeper is also reported by Uba and colleagues, the change of the conformation of the L1 loop is not proposed by them. [33]
Figure 3.

HDAC6 (selective) inhibitor tubastatin A and analogs. Only compounds with a basic nitrogen near the heterocyclic core (14, 16) exhibit strong HDAC10 binding.
For further HDAC10 inhibitor optimization campaigns, we aimed to develop a HDAC10 activity assay suitable for high‐throughput screening of inhibitors. Due to its ease of preparation and its similarity to the human enzyme,[ 6 , 34 ] we used HDAC10 from Danio rerio (zebrafish, drHDAC10) in our assay development studies. Inspired by the discovery that HDAC10 is a polyamine deacetylase, [6] we developed a new polyamine‐based assay substrate suitable for high‐throughput activity assays and characterized available inhibitors for the inhibition of polyamine deacetylation activity. We also developed selective HDAC10 inhibitors with a basic nitrogen and demonstrated that they do not induce the lysosomal phenotype resulting from HDAC10 previously thought to be dependent on HDAC10 enzymatic inhibition.
Results and Discussion
Substrate synthesis
As outlined above, we set out to synthesize a new HDAC10 substrate based on a polyamine structure and containing a fluorescent moiety. We decided to develop an assay based on an acetylated spermidine derivative. Since N8‐acetylspermidine and N1,N8‐diacetylspermidine were reported to be very well recognized and N1‐acetylspermidine showed a lower affinity to HDAC10, the N8‐site was chosen to be acetylated while the N1‐site was selected to be modified by a fluorescent reporter group. In Scheme 1 the synthesis route is depicted. First a building block (20) was obtained by an amide coupling of Boc‐β‐alanine with 7‐amino‐4‐methylcoumarin, followed by Boc deprotection of 18 and nosyl protection of 19 via 2‐nitrobenzenesulfonyl chloride. To obtain Ac‐spermidine‐AMC (23), the building block (20) was alkylated with N‐(4‐bromobutyl)acetamide (21) to obtain 22. The nosyl group of 22 was cleaved off by a nucleophilic aromatic substitution with subsequent elimination of SO2 using thiophenol resulting in the desired substrate 23. Spermidine‐AMC (26), the deacetylated substrate, was synthesized in three steps from the building block 20; an alkylation of 20 with 4‐(Boc‐amino)butylbromide to 24, followed by Boc deprotection to 25, the nosyl deprotection of 25 leads finally to the Spermidine‐AMC (26).
Scheme 1.

Synthesis of a protected aminopropionyl aminocumarin (20) (a), Ac‐spermidine‐AMC (23) (b) and Spermidine‐AMC (26) (c). Reagents and conditions: (a) BOP−Cl, Et3N, DCM, r.t., overnight; (b) TFA, Et3SiH, DCM, 40 °C, 2 h; (c) Et3N, THF, 0 °C to r.t., 4 h; (d) K2CO3, DMF, 45 °C, 4 h, then r.t., overnight; (e) K2CO3, MeCN, 35 °C, 3 h.
Substrate validation
With a potential substrate in hand, we aimed to develop a homogeneous assay. To initially confirm deacetylation, we monitored conversion of the Ac‐spermidine‐AMC (23) by HDAC10 using HPLC. Direct detection of the enzymatic product 26 via HPLC was not possible due to low sensitivity in this assay. Therefore, a derivatization of 26 with fluorescamine which would only react with the deacetylated product was performed and the amount of the product‐fluorescamine adduct was quantified. Retention times of 23 (11.5 min, method see experimental), the fluorescamine adduct (16.8 min) and fluorescamine (22.3 min) were determined (see Figure S1). A dilution series of the substrate 23 and the expected metabolite 26 was balanced to a concentration of 100 μM and fluorescamine was added and a calibration curve was generated (Figure 4a, Table S1). The linear calibration curve demonstrated the potential to measure deacetylation in the desired concentration range.
Figure 4.

Proof of substrate conversion by HPLC (UV, 210 nm): a) Calibration curve of Spermidine‐AMC, detection via derivatization with fluorescamine; b) Conversion of Ac‐spermidine‐AMC by drHDAC10: incubation for 0–60 min in buffer (20 mM Na2HPO4, pH 7.9, 10 mM NaCl, 0.25 mM EDTA). Reaction was stopped and product was detected by adding fluorescamine in acetonitrile.
To monitor enzymatic substrate conversion, Ac‐spermidine‐AMC was incubated with drHDAC10 (0.027 mg/mL) in buffer (20 mM Na2HPO4, pH 7.9, 10 mM NaCl, 0.25 mM EDTA). The reaction was stopped at different time points (0, 10, 15, 20, 30, 45, 60 min) by adding fluorescamine in acetonitrile. From time point “10 min” a new peak with a retention time of 16.8 min, the expected derivatized deacetylation product, appeared. An increase of the product was observed until time point 45 min and conversion was maximal around a level of about 50 % (Figure 4b). Thus, we demonstrated that 23 is a suitable substrate of drHDAC10, and that it is converted to 26.
Homogeneous assay format
In a next step, we monitored the enzymatic conversion in a microplate‐based assay design. Measurement in a plate reader format enables high‐throughput screening. We wanted to use naphthalene‐2,3‐dialdehyde (NDA) as a derivatization reagent to quantify conversion. This treatment of the deacetylated substrate leads to a benzisoindole formation on the amine which in turn quenches the fluorescence signal of the aminocoumarin intramolecularly, a strategy that we have previously used successfully for the development of homogeneous assays for AMC‐lysine derivatives. [35] Therefore, we investigated the stability and linearity of the fluorescence signal and the general possibility to quench the signal of the deacetylated metabolite by benzisoindole formation in a microplate based assay design (see Figure S2).
We observed that the fluorescence of both spermidine derivatives, Ac‐spermidine‐AMC (23) and Spermidine‐AMC (26), increased in a linear fashion with increasing concentration. Linearity (regression coefficient R2=0.999) was excellent in both cases (see Figures S2a+b). In a follow‐up experiment, the enzymatic conversion of Ac‐spermidine‐AMC was simulated. A dilution of Ac‐spermidine‐AMC (23), complemented to an initial total concentration of 10.5 μM with spermidine‐AMC (26) as the deacetylation product was prepared. A stable fluorescence signal for all dilution points was observed (see Figure S2c). With addition of an NDA containing stop solution the fluorescence signal of spermidine‐AMC (26) was quenched and a linear increase of the signal with the increasing concentration of acetylated substrate was observed (see Figure S2d, Table S2) as desired.
We designed an assay set‐up suitable for a high throughput as depicted in Figure 5. In the first assay step, the synthesized substrate (23) is deacetylated by HDAC10. For the second step, a stop solution containing NDA is added. In the presence of a nucleophile (here Mesna) NDA forms benzisoindoles with primary amines. [36] As mentioned above, the benzisoindole intramolecularly quenches the fluorescence signal of the aminocoumarin. This allows for the quantification of the remaining acetylated substrate by measuring the fluorescence signal. HDAC10 inhibitors decrease the deacetylation of the substrate by HDAC10 which leads to an increase of the measured fluorescence signal.
Figure 5.

Setup of the homogeneous microplate based assay system. 23 is incubated with drHDAC10. After adding stop solution containing naphthalene‐2,3‐dialdehyde (NDA) the fluorescence is measured (λex=330 nm, λem=390 nm): a) Ac‐spermidine‐AMC is not able to react with NDA; fluorescence is still high at 390 nm; b) Ac‐spermidine‐AMC is deacetylated by HDAC10; reaction of NDA with Spermidine‐AMC in the presence of a nucleophile (here Mesna) leads to formation of a substituted benzisoindole which quenches the fluorescence at 390 nm intramolecularly. Thus, inhibitors of PDAC activity lead to a high fluorescence signal at 390 nm.
We then determined the robustness of the system in the microplate format. The variability of an assay system can be described by using the Z'‐factor which designates the separation band between positive and negative controls. A value between 1 and 0.50 indicates an excellent assay quality. [37] According to the determined values (Z'‐factors≥0.50, Table S3) we demonstrated that our assay is capable of generating robust results.
Furthermore, the developed substrate was shown to be selectively converted by drHDAC10. This is not a prerequisite for in vitro screening as such but if, e. g., contamination with other deacetylases are present from the expression system, this reduces background signals not stemming from HDAC10 activity. For this purpose the activities of the investigated isotypes (hHDAC1, 6 and 8 and drHDAC10) were determined using Z‐(ϵ‐trifluoroacetyl)lysine‐7‐amino‐4‐methylcoumarin (ZMTFAL) as a substrate to normalize deacetylation efficacy. Trifluoracetylated lysines are described as good HDAC substrates in literature. [38] To determine activity on Ac‐Spermidine‐AMC, the amount of enzyme was normalized to approximately 15 % conversion of ZMTFAL (for hHDAC6 this activity was not reached). For drHDAC10 an activity was shown for the polyamine substrate that is between 16 and 27 times higher than for the other subtypes (see Tables S4 and S5).
To demonstrate the suitability for determination of HDAC10 inhibition, we measured the IC50 value of a reported HDAC10 inhibitor. For this purpose, we chose quisinostat which was recently determined to bind strongly to hHDAC10 with an EC50 of 10 nM in time‐resolved fluorescence resonance energy transfer experiments. [32] Using our new assay substrate Ac‐spermidine‐AMC (23), we observed an IC50 value of 50±5 nM (Figure 6). The similarity of the IC50 values determined in our assay and the EC50 values reported from the ligand displacement assay underlines the validity of our assay to measure inhibition of HDAC10.
Figure 6.

IC50 value of quisinostat on drHDAC10; one experiment performed in quadruplicate, error bars represent standard deviation of the mean (error of the IC50 value is shown as SEM of the non‐linear regression).
Screening for HDAC10 inhibitors
With a validated assay in hand we tested four sets of compounds for inhibition of HDAC10. The first compound set was compiled from reported HDAC inhibitors (see Table 1). This set contained unselective inhibitors (quisinostat (11), panobinostat (9), abexinostat (12) and vorinostat (6)), HDAC6‐selective inhibitors tubastatin A (also HDAC10, 14), bufexamac (27) [39] and BRD9757 (28), [40] the HDAC8‐selective compound PCI‐34051 (29) [41] and mocetinostat (13), a class I selective HDAC inhibitor. [42] We added further compounds from an in‐house library to the set (30–34). Table 2 depicts the inhibitory data of cinnamic acid derivatives. This compound class was previously described as inhibitors of hHDAC6 and Schistosoma mansoni histone deacetylase 8 (SmHDAC8). [43] Further, a selection of benzhydroxamates was tested (see Table 3). Benzhydroxamates were reported as potent HDAC8 inhibitors. [44] Since for tubastatin A, a “selective” HDAC6 inhibitor, effects against HDAC10 were shown, a set of oxazole compounds was included which had been reported by us as selective HDAC6 inhibitors (see Table 4). [45]
Table 1.
Structures and results of drHDAC10 testing for known HDAC inhibitors.
|
Compound |
NDA assay drHDAC10 |
|
|---|---|---|
|
|
% inhibition |
@ c [μM] |
|
quisinostat (11) |
91 % 60 % |
@ 1 @ 0.1 |
|
|
IC50: 0.05±0.005 μM |
|
|
panobinostat (9) |
>95 % 65 % |
@ 1 @ 0.1 |
|
|
|
|
|
abexinostat (12) |
>95 % 45 % |
@ 1 @ 0.1 |
|
|
|
|
|
vorinostat (6) |
43 % <10 % |
@ 1 @ 0.1 |
|
|
|
|
|
tubastatin A (14) |
>95 % 82 % |
@ 1 @ 0.1 |
|
|
|
|
|
bufexamac (27) |
64 % 26 % |
@ 1 @ 0.1 |
|
|
|
|
|
BRD9757 (28) |
93 % 45 % |
@ 1 @ 0.1 |
|
|
|
|
|
mocetinostat (13) |
<10 % <10 % |
@ 1 @ 0.1 |
|
|
|
|
|
PCI‐34051 (29) |
17 % 14 % |
@ 1 @ 0.1 |
|
|
|
|
|
ST70 (30) [46] |
51 % 11 % |
@ 1 @ 0.1 |
|
|
|
|
|
ST71 (31) [46] |
64 % <10 % |
@ 1 @ 0.1 |
|
|
|
|
|
TB5 (32) [43] |
56 % 11 % |
@ 1 @ 0.1 |
|
|
|
|
|
AW12 (33) [47] |
42 % <10 % |
@ 1 @ 0.1 |
|
|
|
|
|
AW19 (34) [48] |
19 % 11 % |
@ 1 @ 0.1 |
|
|
|
|
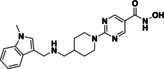
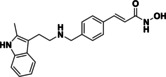
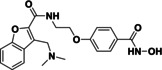
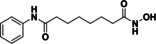
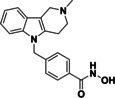
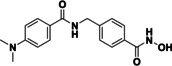
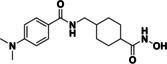
Table 2.
Structures and results of drHDAC10 testing of cinnamic acid derivatives.
|
Compound |
X |
Y |
Z |
NDA assay drHDAC10 |
|
|---|---|---|---|---|---|
|
|
|
|
|
% inhibition |
@ c [μM] |
|
TB8 (35 a) |
−Cl |
H |
H |
73 % 37 % |
@ 1 @ 0.1 |
|
TB51 (35 b) |
−Cl |
−Cl |
H |
>95 % 53 % |
@ 1 @ 0.1 |
|
TB53 (35 c) |
−Br |
−H |
−F |
50 % 12 % |
@ 1 @ 0.1 |
|
TB54 (35 d) |
−Br |
−H |
−OCH3 |
39 % 11 % |
@ 1 @ 0.1 |
|
TB76 (35 e) |
−Br |
−H |
−H |
85 % 23 % |
@ 1 @ 0.1 |
|
TB77 (35 f) |
−H |
−H |
−Cl |
69 % 18 % |
@ 1 @ 0.1 |
|
TB27 (35 g) |
|
−H |
−H |
24 % 18 % |
@ 1 @ 0.1 |
|
TB38 (35 h) |
−H |
|
−OCH3 |
30 % 17 % |
@ 1 @ 0.1 |
|
TB73 (36) |
|
54 % <10 % |
@ 1 @ 0.1 |
||
|
TB75 (37) |
|
64 % 46 % |
@ 1 @ 0.1 |
||
Table 3.
Structures and results of drHDAC10 testing of benzhydroxamate compounds.
|
Compounds |
R |
X |
NDA assay drHDAC10 |
|
|---|---|---|---|---|
|
|
|
|
% inhibition |
@ c [μM] |
|
TH65 (38 a) |
|
−OCH3 |
<10 % <10 % |
@ 1 @ 0.1 |
|
TH68 (38 b) |
|
−OCH3 |
31 % 14 % |
@ 1 @ 0.1 |
|
TH70 (38 c) |
|
−OCH3 |
<10 % <10 % |
@ 1 @ 0.1 |
|
TH77 (38 d) |
|
−Cl |
20 % 11 % |
@ 1 @ 0.1 |
|
TH95 (38 e) |
|
−OCH3 |
<10 % <10 % |
@ 1 @ 0.1 |
|
TH149 (38 f) |
|
−OCH3 |
19 % <10 % |
@ 1 @ 0.1 |
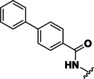
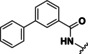
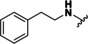
Table 4.
Structures and results of drHDAC10 testing of oxazole compounds.
|
Compounds |
R |
NDA assay drHDAC10 |
|
|---|---|---|---|
|
|
|
% inhibition |
@ c [μM] |
|
JS18 (39 a) |
|
45 % <10 % |
@ 1 @ 0.1 |
|
JS28 (39 b) |
|
>95 % 33 % |
@ 1 @ 0.1 |
|
JS35 (39 c) |
|
37 % <10 % |
@ 1 @ 0.1 |
|
JS41 (39 d) |
|
34 % <10 % |
@ 1 @ 0.1 |
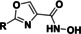
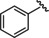
All compounds were tested in a first screening at two concentrations (1 and 0.1 μM). For further investigations we set a cut‐off of more than 30 % inhibition at 0.1 μM. Besides quisinostat, four of the unselective inhibitors, three cinnamic acid derivatives and one oxazole compound exceeded this potency. For these nine hits and vorinostat as a reference inhibitor IC50 values were determined (Table 5, Figure S3).
Table 5.
IC50 values against HDAC1, 6, 8 and 10 of selected hits.
|
Compound |
drHDAC10 (Ac‐spermidine‐AMC) |
HDAC10 binding assay |
hHDAC1 (ZMAL) |
hHDAC6 (ZMAL) |
hHDAC8 (FDL) |
|---|---|---|---|---|---|
|
quisinostat (11) |
50 ±5 nM |
10 nM[a] |
3 ±0.3 nM |
182 ±22 nM |
64 ±3 nM |
|
panobinostat (9) |
51 ±7 nM |
2 nM |
2 ±0.1 nM |
4 ±0.4 nM |
89 ±6 nM |
|
abexinostat (12) |
134 ±26 nM |
4 nM[a] |
24 ±2 nM |
9 ±0.3 nM |
820 ±149 nM |
|
vorinostat (6) |
2000 ±240 nM |
200 nM[a] |
117 ±6 nM |
104 ±9 nM |
400 ±100 nM |
|
tubastatin A (14) |
30 ±3 nM |
19 nM |
1916 ±420 nM |
34 ±17 nM |
1440 ±120 nM |
|
BRD9757 (28) |
147 ±15 nM |
32 nM |
4800 ±1300 nM |
455 ±75 nM |
<10 % @ 100 μM |
|
TB8 (35 a) |
185 ±47 nM |
2 nM |
1454 ±470 nM |
95 ±21 nM |
54 ±9 nM |
|
TB51 (35 b) |
112 ±19 nM |
6 nM |
3630 ±190 nM |
710 ±88 nM |
705 ±120 nM |
|
TB75 (37) |
273 ±58 nM |
24 nM |
2700 ±200 nM |
225 ±33 nM |
205 ±32 nM |
|
JS28 (39 b) |
400 ±43 nM |
34 nM |
14470 ±1100 nM |
59 ±9 nM |
14370 ±2950 nM |
[a] From Géraldy et al. [32]
We compared the activity based potency with the recently published HDAC10 binding assay as mentioned above. This assay system was already used to show HDAC10 binding for tubastatin A, quisinostat and abexinostat. [32] On the one hand we were able to confirm the previously presented binding data for these compounds in our activity based system, on the other hand we verified the drHDAC10 inhibition by showing strong hHDAC10 binding for all the other hits (see Table 5).
Furthermore, the activities of the hit compounds against hHDAC1, 6 and 8 was measured (see Table 5) to analyze subtype selectivity. As expected, a strong effect on HDAC1 as well as HDAC8 was observed for the unselective inhibitors (6, 9, 11, 12). For the remaining substances only moderate to weak inhibition against HDAC1 and 8 was observed. Only TB8 (35 a) and TB75 (37) stood out with an IC50 of 54 nM and 205 nM against HDAC8. However, all compounds showed strong inhibition of hHDAC6. Since HDAC6 and HDAC10 are both members of class IIb and share a high similarity in their amino acid sequence, this was not surprising. The data also matched the results for the strong HDAC10 binding of the HDAC6 inhibitor tubastatin A and its derivatives.
We observed that the interaction between a basic part of the molecule and the gatekeeper was not mandatory for HDAC10 inhibition. While many of the most active compounds 9, 11, 12 and 14 contain a basic amine capable of interacting with the gatekeeper, we also see strong inhibition for other structures (TB8 (35 a) and TB51 (35 b)) that lack a basic moiety. Indeed, TB8 and TB51 gave the strongest binding in the FRET‐assay. Vorinostat was presented as a strong HDAC10 binder in previous studies (0.2 μM [32] ). In contrast, our activity assay indicated weak inhibition with an IC50 value of 2 μM. The discrepancy between the two assay systems might result from the use of different enzymes: recombinant human HDAC10 was used for the binding assay, whereas recombinant zebrafish HDAC10 was used for the activity assay. Further, some smaller discrepancies were also noted for other compounds. Overall, a good general agreement between the two different setups was noted. While all identified inhibitors showed binding affinities in a similar range, their inhibitory activities differed more substantially. For the compounds with a basic moiety, binding affinity and inhibition differed less than for the others. The cinnamic acid moiety seems to be favorable for HDAC10 inhibition. Besides panobinostat (9), some new HDAC10 inhibitors (TB8 (35 a), TB51 (35 b) and TB75 (37)) were identified within this set. No hit was identified among our set of simple benzhydroxamate compounds. However, for the more complex compounds abexinostat and tubastatin A, both containing a benzhydroxamate moiety, strong inhibition of HDAC10 was shown. Thus, we conclude that the benzhydroxamate moiety in combination with an appropriate cap group may lead to potent HDAC10 inhibition. The data from the oxazole set demonstrated that it is possible to diverge between HDAC6 and 10 inhibition. JS28 (39 b) turned out to be a good HDAC10 inhibitor, whereas the other oxazole compounds (39 a, 39 c and 39 d) had just a moderate inhibitory effect on HDAC10.
In order to rationalize the obtained biochemical data, docking studies were carried out using available crystal structures of drHDAC10 (PDB ID 6UHU) [34] as well as human HDAC6 (PDB ID 5EDU), [28] HDAC1 (PDB ID 5ICN) [49] and HDAC8 (PDB ID 2 V5X) [50] to account for the observed selectivity profile of some hits. Of note, crystal structures of drHDAC6 in complex with hydroxamic acid derivatives have shown that the inhibitors can chelate the catalytic zinc ion in either mono‐ or bidentate fashion.[ 28 , 51 ] Hence, two different settings were used for docking of the hits into HDAC6 structure to investigate plausible binding modes.
The pan HDAC inhibitors abexinostat (12), quisinostat (11) and panobinostat (9), which all bear a basic moiety in the capping group, were among the most active compounds tested against HDAC10. The derived docking results, reveal that the capping group of these inhibitors (Figure 7) is able to undergo salt bridge interactions between the protonated amine and the gatekeeper residue Glu274 [32] as well as hydrophobic interactions with Phe204 or Trp205. Additionally, the benzhydroxamate moiety shows the classically observed interactions in the lysine binding tunnel that include a bidentate chelation of the zinc ion, three hydrogen bond interactions with His136, His137 and Tyr307, and aromatic interactions with residues lining the tunnel.
Figure 7.

Predicted binding mode in drHDAC10 (PDB ID 6UHU) of pan‐inhibitors abexinostat (12), quisinostat (11) and panobinostat (9): a) Abexinostat (colored salmon), b) quisinostat (colored teal), and c) panobinostat (colored green). Side chains of binding site residues are shown as white sticks and the catalytic zinc ion as orange spheres. Hydrogen bonds and salt bridge interactions are depicted as blue‐dashed lines and coordination of the zinc ion by the ligand as yellow‐dashed lines.
As previously discussed, several reported potent and “selective” HDAC6 inhibitors also exhibited potent inhibition of HDAC10. This comes as no surprise, since HDAC6 and 10 are the only class IIb HDAC members and share a sequence identity of >45 % (sequence identity of the ligand binding site is around 68 %). Tubastatin A (14), for instance, showed equal nanomolar potency against HDAC6 and HDAC10 and weak activity against HDAC1 and HDAC8. The derived docking studies in drHDAC10 reveal that, as previously proposed, [32] the piperidine‐NH of tubastatin A is able to undergo salt bridge interactions with Glu274, while the indole ring shows hydrophobic interactions with Ile27 and Trp205 (Figure 8a). In HDAC6, we obtained a docking pose similar to that described in the literature. [52] Here, tubastatin A chelates the zinc ion in a monodentate fashion, the phenyl ring of the linker is embedded in the hydrophobic lysine tunnel, while the tetrahydro‐γ‐carboline moiety is embedded against a hydrophobic patch formed by Phe620, Pro501, His500 and Leu749 (Figure 8b). In the case of HDAC8, although the docking pose displays a bidentate coordination of the zinc ion, the hydrophobic cap group is significantly solvent‐exposed at the protein surface, which might account for the weak activity of tubastatin A against HDAC8 (Figure 8c). Meanwhile, in the obtained docking pose in HDAC1 proper chelation of the zinc ion is not achieved (Figure 8d).
Figure 8.

Predicted binding modes of tubastatin A (14) in different HDAC isoforms: a) Tubastatin A (yellow sticks) in drHDAC10 (PDB ID 6UHU), b) tubastatin A (teal sticks) in HDAC6 (PDB ID 5EDU), c) tubastatin A (orange sticks) in HDAC8 (PDB ID 2 V5X), d) tubastatin A (magenta sticks) in HDAC1 (PDB ID 5ICN). The surface of the protein is colored according to lipophilicity; green for hydrophobic and magenta for hydrophilic. Side chains of binding site residues are shown as white sticks and the catalytic zinc ion as orange spheres. Hydrogen bonds and salt bridge interactions are depicted as blue‐dashed lines and coordination of the zinc ion by the ligand as yellow‐dashed lines. Distances are shown as black lines.
Docking studies offer little explanation for the selectivity of other previously reported inhibitors towards HDAC6 and HDAC10, e. g., BRD9757 (28). As seen in the case of BRD9757, the inhibitor seems to bind almost identically in the different HDAC isoforms and only interacts with residues lining the highly conserved lysine tunnel (Figure S4). Here it is important to note that studies have shown that the selectivity of some HDAC6 inhibitors is driven by entropic factors and that the binding of the linker in the lysine tunnel of HDAC6 may be driven by desolvation. [53] The binding of BRD9757 to HDAC6 is accompanied by entropic gain, whereas in HDAC8 it is accompanied by entropic loss. [53a] Hence, the selectivity for the compounds towards HDAC10 over HDAC1 and HDAC8 might also be entropically driven.
Regarding the cinnamic acid based derivative TB75 (37), docking into the active site of drHDAC10 shows that the compound is able to coordinate the zinc ion in a bidentate manner and undergo the three common hydrogen bond interactions with the conserved histidine and tyrosine residues at the bottom of the tunnel. The naphthyl capping group is embedded in the lysine tunnel where it undergoes π‐π stacking interactions with Trp205 and Phe146 (Figure 9a). A similar binding mode is observed for TB75 (37) in HDAC6 and HDAC8 (Figure 9b and Figure 9c, respectively). Meanwhile for HDAC1, where TB75 (37) only shows weak inhibitory activity, our docking studies show that the ligand is not able to properly chelate the zinc ion (Figure 9d). A similar observation was obtained for the docking of TB8 (35 a) and TB51 (35 b) in the various HDAC isoforms, as exemplified in Figure 10. In both drHDAC10 and HDAC6, a bidentate coordination of the zinc ion is observed and the chlorophenyl moiety is nicely accommodated in the hydrophobic lysine tunnel (Figure 10a and Figure 10b, respectively). On the other hand, the bulky linker cannot be properly embedded into the lysine tunnel of HDAC1, hence, no proper chelation of the zinc ion is observed (Figure 10c).
Figure 9.

Predicted binding modes of TB75 (37) in different HDAC isoforms: a) TB75 (yellow sticks) in drHDAC10 (PDB ID 6UHU), b) TB75 (teal sticks) in HDAC6 (PDB ID 5EDU), c) TB75 (orange sticks) in HDAC8 (PDB ID 2V5X), d) TB75 (magenta sticks) in HDAC1 (PDB ID 5ICN). The surface of the proteins is colored according to lipophilicity; green for hydrophobic and magenta for hydrophilic. Side chains of binding site residues are shown as white sticks and the catalytic zinc ion as orange spheres. Hydrogen bonds and salt bridge interactions are depicted as blue‐dashed lines and coordination of the zinc ion by the ligand as yellow‐dashed lines. Distances are shown as black lines.
Figure 10.

Predicted binding modes of TB51 (35 b) in different HDAC isoforms: a) TB51 (yellow sticks) in drHDAC10 (PDB ID 6UHU), b) TB51 (teal sticks) in HDAC6 (PDB ID 5EDU), c) TB51 (magenta sticks) in HDAC1 (PDB ID 5ICN). The surface of the proteins is colored according to lipophilicity; green for hydrophobic and magenta for hydrophilic. Side chains of binding site residues are shown as white sticks and the catalytic zinc ion as orange spheres. Hydrogen bonds and salt bridge interactions are depicted as blue‐dashed lines and coordination of the zinc ion by the ligand as yellow‐dashed lines. Distances are shown as black lines.
In summary, the obtained docking results can partly explain the experimentally observed inhibitory activity and selectivity profile of the herein reported hits. Nevertheless, one has to bear in mind that entropic factors also play essential roles in the binding of the ligands. Regarding benzhydroxamate derivatives, bulky capping groups at the p‐position can be well accommodated in the binding cleft of both HDAC6 and HDAC10 and usually results in selectivity against other HDAC subtypes. A basic moiety in the capping group which is able to undergo salt bridge interaction with the gatekeeper Glu274 seems to be not essential for HDAC10 inhibition, albeit it might contribute to increased potency.
However, m‐substituted benzhydroxamate derivatives, previously reported as selective HDAC8 inhibitors,[ 44 , 54 ] were proven to show little inhibitory activity against HDAC10. The docking studies clearly show that the m‐substitution pattern is not suitable for binding to HDAC10. In the obtained docking poses we observe clashes between the p‐substituent (−OCH3 or −Cl) and Trp205/Asp94 as well as electrostatic clashes between the capping phenyl group and the gatekeeper Glu274 (Figure S5).
On the other hand, cinnamic acid derivatives seem to represent good starting points for the development of HDAC10 inhibitors; bulkier groups like o‐chlorophenyl and naphthyl moieties are still well accommodated in the lysine tunnel and the mouth of the active site cleft. These bulky groups are less suitable for binding to HDAC1, which leads to selectivity over this HDAC isoform. Further modifications of the capping group can be exploited to develop more potent and selective inhibitors.
Design and synthesis of selective HDAC10 inhibitors
Based on our SAR data and the crystal structure of an N8‐acetylspermidine analogue inhibitor determined by Herbst‐Gervasoni and colleagues [34] we designed piperidine‐4‐hydroxamates as potential HDAC10‐selective inhibitors. To mimic the oligoamine structure, a basic amino group was introduced into the molecule with an ethylene bridge to realize a proper spacing of the basic center from the piperidine. The distal amine was endcapped with a lipophilic benzyl group. This was exemplified by the benzyl compound 48 a and its 4'‐bromo congener 48 b (see Figure 11 and Scheme 2).
Figure 11.

Obtained docking poses of 48 b in different HDAC isoforms: a) 48 b (yellow sticks) in drHDAC10 (PDB ID 6UHU), b) 48 b (teal sticks) in HDAC6 (PDB ID 5EDU), c) 48 b (orange sticks) in HDAC8 (PDB ID 2V5X), d) 48 b (magenta sticks) in HDAC1 (PDB ID 5ICN); the surface of HDAC1 binding site is colored according to lipophilicity; green for hydrophobic and magenta for hydrophilic. Binding site residues are shown as white sticks and the catalytic zinc ions as orange spheres. Hydrogen bonds interactions are depicted as blue‐dashed lines, salt bridge interactions as magenta‐dashed lines, cation‐π interactions as teal‐dashed lines, and coordination of the zinc ion by the ligand as yellow‐dashed lines.
Scheme 2.

(a) K2CO3, MeCN, r.t., overnight; (b) TFA, Et3SiH, DCM, 40 °C, 2 h; (c) Et3N, THF, 0 °C to r.t., 4 h; (d) X−Ph‐CH2‐Br, K2CO3, DMF, r.t., overnight; (e) LiOH, THF, 40 °C, 4 h; (f) BOP−Cl, Et3N, DCM, r.t., overnight; (g) K2CO3, MeCN, 35 °C, 3 h, (h) TFA, Et3SiH, DCM, r.t.
Good interactions with drHDAC10 and an expected selectivity over the other subtypes (HDAC1, 6 and 8) were predicted for the two model compounds in a docking study (see Figure S6). Docking of these derivatives into the crystal structure of HDAC10 yielded a similar proposed binding orientation as observed for the N8‐acetylspermidine analogue inhibitor and an overlap of their basic amino moieties (Figure S6a). Both derivatives were able to undergo extensive interactions in the HDAC10 binding pocket, which explains their strong inhibitory activity on this isoform (Figure S6b and Figure 11a). The hydroxamate moiety of 48 a and 48 b chelates the catalytic zinc ion in bidentate fashion and shows the typical hydrogen bond interactions with the neighboring histidine and tyrosine residues. Meanwhile, the capping group shows extensive interactions with the amino residues at the rim of the binding pocket. The protonated amine is placed between Asp94 and the gatekeeper residue Glu274 exhibiting two salt bridge interactions and additionally a cation‐π interaction with Trp205. The phenyl group undergoes π‐π stacking interactions with Phe204 and the p‐bromo substituent of 48 b displays an additional halogen bond with the backbone of Ser203; the latter interaction might explain the increased activity of 48 b with respect to 48 a (see below).
In HDAC6 (Figure 11b), the compounds showed a bidentate chelation of the zinc ion, however, the capping group did not show any major stabilizing interactions, except a hydrogen bond interaction between the protonated amine and Ser568. The phenyl moiety of the capping group was significantly solvent exposed. In HDAC8 (Figure 11c), we similarly observed a bidentate chelation of the zinc ion. Meanwhile the capping group showed a salt bridge interaction between the protonated amine and Asp101 as well as a π‐π stacking interaction between the phenyl group and Phe152. This might explain the relatively higher inhibitory activity for HDAC8 as compared to HDAC6. In contrast to the previously discussed inhibitors, docking of 48 a and 48 b into the crystal structure of HDAC1 yielded docking poses (Figure 11d) where the hydroxamate moiety chelated the zinc ion in bidentate fashion. Similar to the obtained docking poses in HDAC8, the protonated amine showed a salt bridge interaction with Asp99. However, we observed that the phenyl group is embedded in a hydrophilic subpocket at the rim of HDAC1 binding site, which may indicate that this binding orientation is not favored. Since the acetyllysine binding pocket of HDAC1 is narrower, the interaction of the bulky piperidine moiety might be less favorable compared to HDAC8 and 10.
Compounds 48 a and 48 b were hence synthesized according to following synthesis route (see Scheme 2). A nucleophilic substitution was used to obtain 40 from methyl piperidine‐4‐carboxylate and 2‐(Boc‐amino)‐ethyl bromide, followed by Boc deprotection to 41 and nosyl protection with 2‐nitrobenzenesulfonyl chloride resulting in 42. 42 was alkylated with benzyl bromide (a) respectively 4‐bromobenzyl bromide (b) to obtain the methyl ester compounds (44 a/b). After hydrolysis under basic conditions the carboxylates (45 a/b) were converted to the nosyl and trityl protected hydroxamic acids (46 a/b). The nosyl group was cleaved off by a nucleophilic aromatic substitution reaction with subsequent elimination of SO2 using thiophenol resulting in 47 a/b. Final deprotection under acidic conditions finally led to the inhibitors 48 a and 48 b.
Compounds 48 a and 48 b were tested for their inhibition against drHDAC10 and hHDAC1, hHDAC6 and hHDAC8. Further, the affinity to hHDAC10 was determined in the binding assay (Table 6). Both compounds showed inhibition of drHDAC10 in the two‐digit nanomolar range and binding in the low nanomolar region. The 4'‐bromo derivative 48 b is somewhat more potent than the unsubstituted 48 a, which might be explained by a halogen bond between S203 and the bromine atom of 48 b in the docked complex (Figure 11a). Both compounds are essentially inactive on HDAC1 and the other class IIb enzyme, HDAC6 while they show inhibition around 1 μM for HDAC8 (30‐fold selectivity for both inhibitors), Thus, both compounds are highly potent and selective inhibitors of HDAC10.
Table 6.
Inhibition of HDAC1, 6, 8 and 10 by 48 a and 48 b.
|
Compound |
drHDAC10 (IC50 Ac‐sperm.‐AMC) |
HDAC10 binding assay (FRET) |
hHDAC1 (ZMAL) |
hHDAC6 (ZMAL) |
hHDAC8 (IC50 FDL) |
|---|---|---|---|---|---|
|
48 a |
64 ±7 nM |
5 nM |
20 % @ 10 <10 % @ 1 |
<10 % @ 10 <10 % @ 1 |
1973 ±309 nM |
|
48 b |
37 ±5 nM |
2 nM |
11 % @ 10 <10 % @ 1 |
11 % @ 10 <10 % @ 1 |
694 ±52 nM |
Structure of the HDAC10‐48 a complex
To provide experimental validation for our computational modeling approach, we solved the crystal structure of the HDAC10‐48 a complex at 2.18 Å resolution (Figure 12). Inhibitor binding does not cause any major long‐range structural changes in the deacetylase domain, and the root‐mean‐square deviation of 345 Cα atoms is 0.10 Å in comparison with the structure of the HDAC10‐acetate complex (PDB 7KUV). The P(E,A)CE motif helix sterically constricts the active site to confer specificity for long, slender polyamine substrates, but occasionally exhibits conformational flexibility in the binding of certain bulky inhibitors. [55] Here, the P(E,A)CE motif helix shifts slightly to accommodate the binding of 48 a (maximum Cα shift of 0.9 Å).
Figure 12.

Stereoview of a Polder omit map of 48 a (contoured at 5.0σ) bound in the active site of HDAC10. Atoms are color‐coded as follows: C=light gray (HDAC10) or orange (48 a), N=blue, O=red, and Zn2+=gray sphere. Metal coordination interactions are shown as solid black lines and hydrogen bonds are represented as dashed black lines.
The crystal structure of the HDAC10‐48 a complex reveals bidentate hydroxamate coordination to the catalytic Zn2+ ion (C=O⋅⋅⋅Zn2+ and N−O−⋅⋅⋅Zn2+ separations of 2.3 Å and 2.2 Å, respectively). The hydroxamate carbonyl oxygen accepts a hydrogen bond from Y307, the hydroxamate NH group donates a hydrogen bond to H137, and the hydroxamate N−O− group accepts a hydrogen bond from H136.
The linker between the hydroxamate moiety and the phenyl capping group of the inhibitor consists of a piperidine ring with a tertiary amino group, presumed to bind as a positively charged ammonium cation based on typical pKa values for tertiary amines. The NH moiety of the tertiary ammonium cation donates a hydrogen bond to a water molecule, which in turn accepts a hydrogen bond from zinc ligand H176 and donates a hydrogen bond to E274. This water‐mediated hydrogen bond with E274 mimics one of those observed for the binding of the preferred substrate N8‐acetylspermidine. [14] The tertiary ammonium cation of the piperidine ring also appears to make a cation‐π interaction with W205. The secondary ammonium cation of 48 a donates hydrogen bonds to E24 and another water molecule; the hydrogen bond with E24 may be facilitated by the slight shift of the P(E,A)CE motif helix. The phenyl capping group of 48 a is characterized by strong electron density, indicating that it is well ordered, but it does not make any intermolecular interactions in the HDAC10 active site. Indeed, the phenyl group extends into a solvent‐filled region of the crystal lattice and does not interact with any residues on the protein surface.
Cellular target engagement, selectivity and phenotypic evaluation
To show cellular activity/binding of the piperidine based selective inhibitors 48 a–b, a BRET‐assay system was used. BRET data had already been presented by Géraldy and colleagues for the control compound vorinostat (6) as well for the highly active compounds 9, 11, 12 and 14. [32] For both new inhibitors, IC50 values in the in vitro conversion (Table 6) and in vitro binding (see Table 7) assays as well as EC50 values in the cellular BRET assay were determined to be in the low nanomolar range. The observed activity is similar or superior to the activities of the highly potent reference inhibitors. The BRET data confirm the data from the HDAC10 binding assay and the new activity assay. Furthermore, these data demonstrate cell permeability and cellular activity for the selective piperidine inhibitors.
Table 7.
In‐vitro (FRET) and in‐cellulo (BRET) binding data of selected inhibitors.
|
Compound |
FRET assay (IC50) [nM] |
BRET assay (EC50) [nM] |
|---|---|---|
|
vorinostat (6) |
200[a] |
630[a] |
|
quisinostat (11) |
10 |
40[a] |
|
abexinostat (12) |
4 |
8[a] |
|
tubastatin A (14) |
19 |
13[a] |
|
48 a |
5 |
26 |
|
48 b |
2 |
10 |
[a] From Géraldy et al. [32]
To validate the selectivity observed in vitro, we probed the inhibitors 48 a/b for their effect on the classical HDAC substrates acetyl Histone H3 (class I, representative HDAC1) and acetyl tubulin (HDAC6). In concentrations of 10 μM there was no increase of either substrate whereas the control inhibitor vorinostat (6) showed robust hyperacetylation for both proteins (Figure S7).
Further, screening hits were also investigated in a cellular LysoTracker‐Assay in neuroblastoma cells. Oehme and colleagues showed that doxorubicin treatment in a neuroblastoma cell model induces autophagic flux as a major resistance mechanism. The control of lysosomal activity was linked to HDAC10 activity. HDAC10 inhibition by unselective inhibitors as well as its depletion by knockout induced accumulation of lysosomes, which also affected autophagy and sensitized for drug‐induced cell death. HDAC10 was identified as a promising target in advanced stage 4 neuroblastoma. [16a] HDAC10 inhibition mediated accumulation of lysosomes is detected by the LysoTracker‐Assay. This assay system allows us to determine cellular effects of our HDAC10 inhibitors. [16b]
We studied the impact of our screening hits on the accumulation of lysosomes in neuroblastoma cells. Accumulation was monitored by fluorescence microscopy and quantified via flow cytometry analysis. Via fluorescence microcopy an increase of the LysoTracker signal was monitored for all of our hits (see Figure 13). While for JS28 (39 b) and BRD9757 (28) high concentrations were necessary to get a signal, a significant increase of the accumulation of lysosomes at moderate concentrations (1.0–7.5 μM) was observed for the rest of the hits. For the highly potent and unselective inhibitors quisinostat (11), panobinostat (9) and abexinostat (12) a very strong response was monitored. Tubacin, a selective HDAC6 inhibitor, was included as negative control compound. [56] No influence of HDAC6 inhibition on the accumulation of lysosomes was seen. Surprisingly, our selective HDAC10 inhibitor 48 b did not show an accumulation of lysosomes.
Figure 13.

Fluorescence microscopy analysis of LysoTracker DND‐99 staining 24 h after treatment of SK‐N‐BE(2)‐C cells with 7.5 μM tubacin (HDAC6i), 7.5 μM tubastatin A (14), 0.5 μM quisinostat (11), 5 μM TB51 (35 b) and 5.0 μM TB75 (37). Nuclei were stained with DAPI (4′,6‐diamidino‐2‐phenylindole).
Quantification via flow cytometry confirmed the tendency of fluorescence microscopy analysis (see Table 8 and Figure 14). LysoTracker fluorescence was normalized against DMSO control. The majority of the inhibitors that also target HDAC10 (6, 14, 35 a, 35 b, 37) showed a significant effect, between a 1.5‐ and 2.0‐fold increase. Under treatment with unselective inhibitors (9, 11 and 12) already at nanomolar concentrations, a strong increase of the measured signal up to 3‐fold occurred. While for HDAC class IIb inhibitors a weak signal was observed, selective HDAC6 inhibition did not result in a significant LysoTracker signal. For tubacin (7.5 μM) a 1.1‐fold change of the intensity of the LysoTracker signal was detected. Effects of BRD9757 (28) and JS28 (39 b) were in the same range (1.2–1.3 @ 20 μM). We observe that the more pronounced the HDAC class I activity of the compound is, the higher is the accumulation of lysosomes in neuroblastoma cells. However, the most selective HDAC10 inhibitor, 48 b, did not show an increase in signal.
Table 8.
Quantification of accumulation of lysosomes.
|
Compound |
Tested concentration |
LysoTracker effect (relative signal intensity as compared to control) |
|---|---|---|
|
quisinostat (11) n=3 |
0.5 μM |
3.0±0.65 |
|
panobinostat (9) 0.01 μM n=2 0.004 μM n=6 |
0.01 μM 0.004 μM |
2.0±0.15 1.5±0.2 |
|
abexinostat (12) [16b] n=5 |
0.1 μM |
1.7±0.23 |
|
vorinostat (6) n=2 |
1 μM |
2.0±0.03 |
|
tubastatin A (14) [16b] n=4 |
7.5 μM |
1.6±0.08 |
|
BRD9757 (28) n=3 |
20.0 μM |
1.3±0.07 |
|
JS28 (39 b) n=3 |
20.0 μM |
1.2±0.07 |
|
TB8 (35 a) n=3 |
5.0 μM |
1.5±0.24 |
|
TB51 (35 b) n=3 |
5.0 μM |
1.5±0.27 |
|
TB75 (37) n=3 |
5.0 μM |
1.7±0.04 |
|
tubacin [16b] n=4 |
7.5 μM |
1.1±0.07 |
|
48 b n=3 |
20.0 μM 10 μM |
1.04±0.05 1.07±0.15 |
Figure 14.

Quantitation of LysoTracker accumulation by flow cytometry for selective HDAC10 inhibitor 48 b. Tubastatin A (TubA) and Panobinostat (Pano) were used as positive controls.
Exemplarily, we show the underlying data for the analysis of the HDAC10 inhibitor 48 b by FACS where we did not observe any significant accumulation of lysosomes (Figure 14).
Conclusion
We designed and synthesized the first fluorescent polyamine substrate for HDAC10 which can be used in a validated conversion assay suitable for high‐throughput screening. By screening a set of HDAC inhibitors, the assay confirmed reported potent HDAC10 binders as strong HDAC10 inhibitors. Furthermore, we used the assay system to identify new HDAC10 inhibitors. It turned out that inhibition of HDAC10 tends to correlate with HDAC6 inhibition. Among hydroxamates, a strong affinity to HDAC1 and 8 does not exclude HDAC10 inhibition, as exemplified by the unselective inhibitors 9, 11, and 12, but the HDAC8 selective inhibitor PCI‐34051 did not inhibit HDAC10. We also tested mocetinostat, a benzamide HDAC inhibitor, which is described as class I selective agent, and did not see any effects on HDAC10. The selectivity profiles of the compounds were rationalized by docking studies.
Based on this SAR data and crystal structures we were able to design and synthesize selective HDAC10 inhibitors 48 a and 48 b which employ an arylmethyl‐aminomethyl‐piperidine hydroxamate structure. We were able to solve the crystal structure of the inhibitor 48 a in complex with HDAC10 and both amino groups show interactions with one glutamate each (E24 and E274). In a parallel, yet independent study, it has been shown very recently that a benzoyl instead of a benzyl‐group at the “outer” nitrogen also leads to piperidine‐hydroxamate HDAC10 inhibitors but with decreased potency (93 nM in FRET assay) and selectivity as compared to our new inhibitors. [57] In this amide series, open chain analogues (aza‐vorinostats) showed higher potency and selectivity than the piperidines. We do not have the matched pairs for the amines in the open chain series, but judging from the comparison in the piperidine series and the specific additional interactions of our second basic center with E24 we would argue that this second amine function is beneficial for potency and selectivity.
Surprisingly, our new selective HDAC10 inhibitors did not show an accumulation of lysosomes in neuroblastoma cells, which had previously been tied to cellular HDAC10 inhibition. The same was observed for other new selective HDAC10 inhibitors mentioned above. [57] Thus, the exact HDAC subtype selectivity profile for the induction of the lysosomal phenotype remains elusive so far.
In conclusion, this study outlines the development of important and highly selective molecular tools for phenotypic cellular assays as well as a new HDAC10 substrate conversion assay. Our new selective HDAC10 inhibitors are valuable chemical probes that can be used to interrogate HDAC10 biology and identify potential HDAC10 dependent pathologies. Moreover, these compounds may serve as potential lead compounds for drug discovery campaigns targeting HDAC10.
Experimental Section
Chemistry
Starting materials and reagents were purchased from different suppliers. No further purification was done. For Rf‐determination thin‐layer plates from Merck (TLC Silica gel 60 F254 and TLC Silica gel 60 RP‐18 F254s) were used and analyzed under UV light (254 nm). Mass spectrometry (MS) was performed on an Advion expression CMS spectrometer using an APCI ion source or ESI. Spectra for final compounds were recorded with high resolution mass spectrometry (HRMS) on an Exactive device (Thermo Fisher Scientific) operating in ESI mode. Theoretical masses were calculated with the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu). 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III HD spectrometer at 400 and 100 MHz by using the signal of the deuterated solvent as internal standard. The following abbreviation were used to report the spectra: 1H: chemical shift δ (ppm), multiplicity (s=singlet, d=doublet, dd=doublet of doublets, t=triplet, q=quartet, m=multiplet, b=broad), integration, coupling constant (J in Hz). 13C, chemical shift δ (ppm). HMBC and HSQC experiments were applied for the assignment. The purity of the final compounds (>95 %) was determined by HPLC and UV detection (λ=210 nm). HPLC analysis was performed using the following conditions: Eluent A, H2O containing 0.05 % TFA; Eluent B, acetonitrile containing 0.05 % TFA, flow rate 1 mL/min, linear gradient conditions (0–4 min, A=90 %, B=10 %; 4–29 min, linear increase to 100 % of B; 29–31 min, B=100 %; 31–40 min, A=10 %, B=90 %), Phenomenex Kinetex 5 μm XB‐ C 18 (100 Å, 250×4.60 mm).
tert ‐Butyl (3‐((4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)amino)‐3‐oxopropyl)carbamate (18): Boc‐β‐alanine (1778 mg, 9.40 mmol, 1.5 eq) and BOP−Cl (2635 mg, 10.35 mmol, 1.7 eq) were suspended in dry DCM (15 mL). After adding triethylamine (1903 mg, 18.81 mmol, 3.0 eq) and stirring for 30 min at room temperature 7‐amino‐4‐methylcoumarin (1098 mg, 6.27 mmol, 1.0 eq) was added. Reaction was stirred overnight at room temperature. After removing solvent under reduced pressure, water was added and pH<5 was adjusted with HCl (2 M). The suspension was extracted with DCM. Organic layers were dried over magnesium sulfate, filtrated and solvent was removed. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 68 % of a white solid. Rf, 0.58 (DCM/MeOH 95 : 5 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.38 (s, 1H, CO−NH−AMC), 7.77 (d, J=2.0 Hz, 1H, AMC H8), 7.71 (d, J=8.4 Hz, 1H, AMC H5), 7.48 (dd, J=8.4, 2.0 Hz, 1H, AMC H6), 6.92 (t, J=5.6 Hz, 1H, CO−NH−CH2), 6.27–6.25 (m, 1H, AMC H3), 3.27–3.20 (m, 2H, NH‐CH 2 ‐CH2‐CO), 2.56–2.50 (overlapping with DMSO signal, m, 2H, HN‐CH2‐CH 2 ‐CO), 2.40 (d, J=1.2 Hz, 3H, CH3), 1.38 (s, 9H, (CH 3 ) 3 ‐CH2‐O). 13C NMR (DMSO‐d6, δ [ppm]): 170.6 (CH2‐CO−NH), 160.5 (CO−AMC), 156.0 (Boc−CO−NH), 154.1 (AMC C9), 153.6 (AMC C4), 142.9 (AMC C7), 126.3 (AMC C5), 115.5 (AMC C6), 115.3 (AMC C10), 112.6 (AMC C3), 105.9 (AMC C8), 78.1 ((CH3)3‐CH2‐O), 37.3 (NH‐CH2‐CH2‐CO), 36.7 (HN‐CH2‐CH2‐CO), 28.7 ((CH 3 ) 3 ‐CH2‐), 18.4 (CH3). MS (APCI, +): 346.2 [M+H]+.
3‐Amino‐N‐(4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)propanamide (19): Compound 18 (1487 mg, 4.30 mmol, 1.0 eq) was solved in DCM (10 mL). Trifluoroacetic acid (4903 mg, 43.00 mmol, 10.0 eq) and triethylsilane (5000 mg, 43.00 mmol, 10.0 eq) were added and the mixture was stirred for 2 h at 40 °C. Solvent was removed under reduced pressure. The residue was suspended via ultrasonication in 5 mL ethyl acetate. After adding 5 mL cyclohexane and cooling on ice the suspension was filtrated and washed with a solvent mixture (EE/CH, 50/50, 0 °C). The precipitant was dried and use without further purification. Yield, 95 % of a white solid. Rf, 0.40 (DCM/MeOH 95 : 5 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.63 (s, 1H, CO−NH−AMC), 7.85–7.77 (m, 4H, + H 3 N‐CH2+AMC H8), 7.75 (d, J=8.4 Hz, 1H, AMC H5), 7.47 (dd, J=8.4, 2.0 Hz, 1H, AMC H6), 6.30–6.27 (m, 1H, AMC H3), 3.17–3.07 (m, 2H, +H3N‐CH 2 ‐CH2‐CO), 2.77 (t, J=6.6 Hz, 2H, +H3N‐CH2‐CH 2 ‐CO), 2.41 (d, J=1.2 Hz, 3H, CH3). 13C NMR (DMSO‐d6, δ [ppm]): 169.6 (HN−CO‐CH2), 160.4 (CO−AMC), 158.3 (q, 3 J=31 Hz, CO TFA) 154.1 (AMC C9), 153.5 (AMC C4), 142.6 (AMC C7), 126.5 (AMC C5), 115.5 (AMC C6+C10), 112.8 (AMC C3), 106.0 (AMC C8), 35.1 (+H3N‐CH2‐CH2‐CO), 33.9 (+H3N‐CH2‐CH2‐CO), 18.4 (CH3); CF3 of TFA not visible. MS (APCI, +): 247.2 [M+H]+.
N ‐(4‐Methyl‐2‐oxo‐2H‐chromen‐7‐yl)‐3‐((2‐nitrophenyl)sulfonamido)propanamide (20): Compound 19 (728 mg, 2.02 mmol, 1.0 eq) and 2‐nitrobenzenesulfonyl chloride (535 mg, 2.42 mmol, 1.2 eq) were dissolved in THF (10 mL, 0 °C). After adding triethylamine (818 mg, 8.08 mmol, 4.0 eq) the reaction was stirred for 30 min at room temperature. The reaction mixture (white flakes) was filtrated and washed with a solvent mixture (EE/CH, 75/25, 15 mL, 0 °C). Precipitation was dried and use without further purification. Yield, 100 % of a white solid. Rf, 0.47 (EE/CH 75/25 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.41 (s, 1H, CO−NH−AMC), 8.24 (t, J=5.6 Hz, 1H, SO2‐NH‐CH2), 8.06–8.01 (m, 1H, Nosyl H6), 8.00–7.95 (m, 1H, Nosyl H3), 7.90–7.83 (m, 2H, Nosyl H4,5), 7.74–7.69 (m, 2H, AMC H5,8), 7.44 (dd, J=8.8, 2.0 Hz, 1H, AMC H6), 6.28–6.25 (m, 1H, AMC H3), 3.27–3.20 (m, 2H, HN‐CH 2 ‐CH2‐CO), 2.62 (t, J=6.8 Hz, 2H, HN‐CH2‐CH 2 ‐CO), 2.40 (d, J=1.2 Hz, 3H, CH3). 13C NMR (DMSO‐d6, δ [ppm]): 169.9 (HN−CO‐CH2), 160.5 (CO−AMC), 154.1 (AMC C9), 153.5 (AMC C4), 148.2 (Nosyl C2), 142.7 (AMC C7), 134.5 (Nosyl C4), 133.1 (Nosyl C5), 132.9 (Nosyl C1), 129.9 (Nosyl C6), 126.3 (AMC C5), 124.9 (Nosyl C3), 115.5 (AMC C6), 115.4 (AMC C10), 112.7 (AMC C3), 106.0 (AMC C8), 39.1 (HN‐CH2‐CH2‐CO), 37.0 (HN‐CH2‐CH2‐CO), 18.4 (CH3). MS (APCI, +): 432.2 [M+H]+.
N ‐(4‐Bromobutyl)acetamide (21): 4‐Bromobutene‐1‐amine hydrobromide (529 mg, 2.29 mmol, 1.0 eq) was suspended in dry THF (10 mL). Acetylchloride (1786 mg, 22.9 mmol, 10.0 eq) and cesium carbonate (2985 mg, 9.16 mmol, 4.0 eq) were added. After stirring the reaction for 5 h at 60 °C, the solvent was removed and water was added. The aqueous mixture was extracted with DCM. Organic layers were dried over magnesium sulfate, filtrated and solvent was removed. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 64 % of a colorless oil. Rf, 0.49 (DCM/MeOH 95 : 5 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 7.86 (bs, 1H, CH2‐NH‐CO), 3.54 (t, J=6.8 Hz, 2H, Br‐CH 2 ‐CH2), 3.08–2.99 (m, 2H, CH2‐CH 2 ‐NH), 1.84–1.72 (m, 5H, −CH3+Br−CH2‐CH 2 ‐CH2), 1.55–1.44 (m, 2H, −CH2‐CH 2 ‐CH2‐NH). 13C NMR (DMSO‐d6, δ [ppm]): 169.5 (−COCH3), 37.9 (CH2‐CH2‐NH), 35.3 (Br‐CH2−), 30.2 (−CH2‐CH2‐CH2‐NH), 28.2 Br‐CH2‐CH2‐CH2), 23.0 (−CH3). MS (APCI, +): 194.1+196.1 [M+H]+.
3‐((N‐(4‐Acetamidobutyl)‐2‐nitrophenyl)sulfonamido)‐N‐(4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)propanamide (22): Compound 20 (147 mg, 0.34 mmol, 1.0 eq), N‐(4‐bromobutyl)acetamide (21) (132 mg, 0.68 mmol, 2.0 eq), potassium carbonate (71 mg, 0.51 mmol, 1.5 eq) and potassium iodide (11 mg, 0.07 mmol, 0.2 eq) were suspended in DMF (5 mL). The reaction mixture was stirred at 45 °C for 4 h and overnight at room temperature. Solvent was removed under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 48 % of a colorless oil. Rf, 0.63 (DCM/MeOH 90/10 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.45 (s, 1H, CO−NH−AMC), 8.06–8.02 (m, 1H, Nosyl H6), 7.99–7.95 (m, 1H, Nosyl H3), 7.91–7.80 (m, 3H, Nosyl H4,5+CH2‐NH−CO), 7.74–7.70 (m, 2H, AMC H5,8), 7.43 (dd, J=8.8, 2.0 Hz, 1H, AMC H6), 6.28–6.26 (m, 1H, AMC H3), 3.63–3.56 (m, 2H, N‐CH 2 ‐CH2‐CO), 3.34–3.28 (m, 2H, Nosyl‐N‐CH 2 ‐CH2‐CH2), 3.04–2.96 (m, 2H, CH2‐CH 2 ‐NH−CO), 2.73–2.66 (m, 2H, N‐CH2‐CH 2 ‐CO), 2.40 (d, J=1.2 Hz, 3H, AMC CH3), 1.78 (s, 3H, CO‐CH3), 1.58–1.56 (m, 2H, CH 2 ‐CH2‐NH−CO), 1.40–1.29 (m, 2H, Nosyl‐N‐CH2‐CH 2 ‐CH2). 13C NMR (DMSO‐d6, δ [ppm]): 169.9 (CH2‐CO−NH), 169.4 (HN‐CO‐CH3), 160.4 (AMC CO), 154.1 (AMC C9), 153.5 (AMC C4), 148.0 (Nosyl C2), 142.6 (AMC C7), 134.9 (Nosyl C4), 133.0 (Nosyl C5), 132.1 (Nosyl C1), 130.2 (Nosyl C6), 126.4 (AMC C5), 124.8 (Nosyl C3), 115.5 (AMC C6), 115.4 (AMC C10), 112.7 (AMC C3), 105.9 (AMC C8), 48.2 (Nosyl‐N‐CH2‐CH2‐CH2), 43.9 (Nosyl‐N‐CH2‐CH2‐CO), 38.4 (CH2‐CH2‐NH‐CO), 36.4 (Nosyl‐N‐CH2‐CH2‐CO), 26.7 (Nosyl‐N‐CH2‐CH2‐CH2), 25.8 (CH2‐CH2‐NH‐CO), 23.0 (NH‐CO‐CH3), 18.4 (AMC CH3). MS (APCI, +): 545.2 [M+H]+.
3‐((4‐Acetamidobutyl)amino)‐N‐(4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)propanamide (23): Compound 22 (34 mg, 0.06 mmol, 1.0 eq) and potassium carbonate (17 mg, 0.13 mmol, 2.0 eq) were dissolved in MeCN (5 mL). After adding thiophenol (10 mg, 0.09 mmol, 1.5 eq) the reaction mixture was stirred for 4 h at 35 °C. Solvent was removed and crude product was purified via flash column chromatography (H2O/MeCN+0.1 % TFA). Yield, 53 % of a white solid. Rf, 0.30 (H2O/MeCN 50/50 (v/v)+0.05 % TFA). 1H NMR (DMSO‐d6, δ [ppm]): 10.65 (s, 1H, CO−NH−AMC), 8.48 (b s, 2H, CH2‐NH 2 +‐CH2), 7.90 (t, J=5.6 Hz, 1H, CONH‐CH2‐CH2), 7.80 (d, J=2.0 Hz, 1H, AMC H8), 7.75 (d, J=8.4 Hz, 1H, AMC H5), 7.48 (dd, J=8.4, 2.0 Hz, 1H, AMC H6), 6.31–6.27 (m, 1H, AMC H3), 3.28–3.18 (m, 2H, CH 2 ‐CH2‐CO), 3.06 (q, 6.8 Hz, 2H, CONH‐CH2‐CH2‐CH2‐CH 2 ‐NH2 +), 3.01–2.92 (m, 2H, CONH‐CH 2 ‐CH2‐CH2‐CH2‐NH2 +), 2.83 (t, J=6.8 Hz, 2H, CH2‐CH 2 ‐CO), 2.41 (d, J=1.2 Hz, 3H, AMC CH3), 1.81 (s, 3H, CO‐CH 3 ), 1.65–1.55 (m, 2H, CONH −CH2‐CH2‐CH 2 ‐CH2‐ NH2 +), 1.49–1.39 (m, 2H, m, 2H, CONH‐CH2‐CH 2 ‐CH2‐CH2‐NH2 +). 13C NMR (DMSO‐d6, δ [ppm]): 169.5 (H3C‐CONH), 169.3 (CH2‐CH2‐CONH), 160.4 (AMC C2), 158.4 (q, 3 J=32 Hz, CO TFA), 154.1 (AMC C10), 153.5 (AMC C4), 142.5 (AMC C7), 126.5 (AMC C5), 115.6 (AMC C6+C9), 112.8 (AMC C3), 106.1 (AMC C8), 47.2 (CONH‐CH2‐CH2‐CH2‐CH2‐ NH2 +), 42.8 (CH2‐CH2‐CO), 38.2 (CONH‐CH2‐CH2‐CH2‐CH2‐ NH2 +), 32.8 (CH2‐CH2‐CO), 26.7 (CONH‐CH2‐CH2‐CH2‐CH2‐ NH2 +), 23.4 (CONH −CH2‐CH2‐CH2‐CH2‐ NH2 +), 23.0 (COCH3), 18.4 (AMC CH3), CF3 of TFA not visible. HRMS (ESI, +): 360.1914 [M+H]+. Calculated mass: 360.1923 [M+H]+. Purity: 97 % (11.50 min).
tert ‐Butyl (4‐((N‐(3‐((4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)amino)‐3‐oxopropyl)‐2‐nitrophenyl)sulfonamido)butyl)carbamate (24): Compound 20 (200 mg, 0.47 mmol, 1.0 eq), 4‐(bocamino)butylbromide (141 mg, 0.56 mmol, 1.2 eq), potassium carbonate (96 mg, 0.70 mmol, 1.5 eq) and potassium iodide (15 mg, 0.09 mmol, 0.2 eq) were suspended in DMF (5 mL). The reaction mixture was stirred at 45 °C for 4 h and overnight at room temperature. Solvent was removed under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 31 % of a colorless solid. Rf, 0.59 (EE/CH 75/25 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.45 (s, 1H, CO−NH−AMC), 8.06–8.02 (m, 1H, Nosyl H6), 7.99–7.95 (m, 1H, Nosyl H3), 7.91–7.81 (m, 2H, Nosyl H4,5), 7.74–7.69 (m, 2H, AMC H5,8), 7.43 (dd, J=8.0, 2.0 Hz, 1H, AMC H6), 6.83 (t, J=5.5 Hz, 1H, CH2‐NH‐Boc), 6.27 (d, J=1.2 Hz, 1H, AMC H3), 3.63–3.56 (m, 2H, N‐CH 2 ‐CH2‐CO), 3.33–3.26 (m, 2H, Nosyl‐N‐CH 2 ‐CH2‐CH2), 2.93–2.83 (m, 2H, CH2‐CH 2 ‐NH‐Boc), 2.74–2.66 (m, 2H, N‐CH2‐CH 2 ‐CO), 2.40 (d, J=1.2 Hz, 3H, AMC CH3), 1.57–1.45 (m, 2H, CH 2 ‐CH2‐NH−Boc), 1.36 (s, 9H, (CH 3 ) 3 ‐C−O), 1.34–1.28 (m, 2H, Nosyl‐N‐CH2‐CH 2 ‐CH2). 13C NMR (DMSO‐d6, δ [ppm]): 169.9 (CH2‐CO−NH), 160.4 (AMC CO), 156.0 (Boc−CO), 154.1 (AMC C9), 153.5 (AMC C4), 148.0 (Nosyl C2), 142.6 (AMC C7), 134.9 (Nosyl C4), 133.0 (Nosyl C5), 132.1 (Nosyl C1), 130.1 (Nosyl C6), 126.4 (AMC C5), 124.8 (Nosyl C3), 115.5 (AMC C6), 115.4 (AMC C10), 112.7 (AMC C3), 105.9 (AMC C8), 77.8 ((CH3)3‐C−O), 48.2 (Nosyl‐N‐CH2‐CH2‐CH2), 43.9 (N‐CH2‐CH2‐CO), 39.8 (CH2‐NH−Boc (HMBC)), 36.4 (N‐CH2‐CH2‐CO), 28.7 ((CH 3 ) 3 ‐C−O), 27.0 (Nosyl‐N‐CH2‐CH2‐CH2), 25.7 (CH2‐CH2‐NH‐Boc), 18.4 (AMC CH3). MS (ESI, +): 624.9 [M+Na]+.
3‐((N‐(4‐Aminobutyl)‐2‐nitrophenyl)sulfonamido)‐N‐(4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)propanamide (25): Compound 24 (138 mg, 0.23 mmol, 1.0 eq) was dissolved in DCM (5 mL). Trifluoroacetic acid (392 mg, 3.44 mmol, 15.0 eq) and triethylsilane (266 mg, 2.29 mmol, 10.0 eq) were added and the mixture was stirred for 2 h at 40 °C. Solvent was removed under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 86 % of a colorless oil. Rf, 0.38 (DCM/MeOH 90/10 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.53 (s, 1H, CO−NH−AMC), 8.07–8.03 (m, 1H, Nosyl H6), 8.00–7.96 (m, 1H, Nosyl H3), 7.92–7.82 (m, 2H, Nosyl H4,5), 7.75–7.70 (m, 2H, AMC H5,8), 7.51–7.37 (m, 4H, AMC H6+NH3 +), 6.28 (d, J=1.2 Hz, 1H, AMC H3), 3.65–3.58 (m, 2H, N‐CH 2 ‐CH2‐CO), 3.41–3.27 (overlapping with H2O‐Peak, m, 2H, Nosyl‐N‐CH 2 ‐CH2‐CH2), 2.82–2.74 (m, 2H, CH2‐CH 2 ‐NH3 +), 2.73–2.66 (m, 2H, N‐CH2‐CH 2 ‐CO), 2.40 (d, J=1.2 Hz, 3H, AMC CH3), 1.67–1.56 (m, 2H, CH 2 ‐CH2‐NH3 +), 1.56–1.45 (m, 2H, Nosyl‐N‐CH2‐CH 2 ‐CH2). 13C NMR (DMSO‐d6, δ [ppm]): 169.9 (CH2‐CO−NH), 160.5 (AMC CO), 158.4+158.1 (TFA), 154.1 (AMC C9), 153.5 (AMC C4), 148.0 (Nosyl C2), 142.6 (AMC C7), 135.0 (Nosyl C4), 133.0 (Nosyl C5), 132.0 (Nosyl C1), 130.1 (Nosyl C6), 126.4 (AMC C5), 124.8 (Nosyl C3), 115.5 (AMC C6), 115.4 (AMC C10), 112.7 (AMC C3), 105.9 (AMC C8), 48.0 (Nosyl‐N‐CH2‐CH2‐CH2), 43.8 (N‐CH2‐CH2‐CO), 39.0 (CH2‐CH2‐NH3 +), 36.3 (N‐CH2‐CH2‐CO), 25.3 (CH2‐CH2‐NH3 +), 25.0 (Nosyl‐N‐CH2‐CH2‐CH2), 18.4 (AMC CH3). MS (APCI, +): 503.2 [M+H]+.
3‐((4‐Aminobutyl)amino)‐N‐(4‐methyl‐2‐oxo‐2H‐chromen‐7‐yl)propanamide (26): Compound 25 (122 mg, 0.20 mmol, 1.0 eq) and potassium carbonate (109 mg, 0.79 mmol, 4.0 eq) were dissolved in MeCN (8 mL). After adding thiophenol (65 mg, 0.59 mmol, 3.0 eq) the reaction mixture was stirred for 4 h at 35 °C. Crude product was purified via flash column chromatography (H2O/MeCN+0.1 % TFA). Yield, 80 % of a yellow solid. Rf, 0.50 (H2O/MeCN 50/50 (v/v)+0.1 % TFA). 1H NMR (DMSO‐d6, δ [ppm]): 10.70 (s, 1H, CO−NH−AMC), 8.64 (bs, 2H, CH2‐NH 2 +‐CH2), 7.92–7.77 (m, 4H, + H 3 N‐CH2+AMC H8), 7.75 (d, J=8.8 Hz, 1H, AMC H5), 7.47 (dd, J=8.8, 2.0 Hz, 1H, AMC H6), 6.29 (d, J=1.2 Hz, 1H, AMC H3), 3.29–3.19 (m, 2H, +H2N‐CH 2 ‐CH2‐CO), 3.05–2.93 (m, 2H, +H3N‐CH2‐CH2‐CH2‐CH 2 ‐NH2 +), 2.89–2.77 (m, 4H, +H3N‐CH 2 ‐CH2 CH2‐CH2‐NH2 ++CH 2 ‐CO), 2.41 (d, J=1.2 Hz, 3H, AMC CH3), 1.71–1.53 (m, 4H, +H3N‐CH2‐CH 2 ‐CH 2 ‐CH2‐NH2 +). 13C NMR (DMSO‐d6, δ [ppm]): 169.4 (CH2‐CO‐NH), 160.4 (AMC CO), 158.5 (q, 3 J=31 Hz, CO TFA), 154.1 (AMC C9), 153.6 (AMC C4), 142.5 (AMC C7), 126.5 (AMC C5), 115.6 (AMC C6+C10), 112.8 (AMC C3), 106.1 (AMC C8), 46.7 (+H3N‐CH2‐CH2‐CH2‐CH2‐NH2 +), 42.7 (CH2‐CH2‐CO), 38.7 (+H3N‐CH2‐CH2‐CH2‐CH2‐NH2 +), 32.7 (+H2N‐CH2‐CH2‐CO), 24.6 (+H3N‐CH2‐CH2‐CH2‐CH2‐NH2 +), 22.9 (+H3N‐CH2‐CH2‐CH2‐CH2‐NH2 +), 18.4 (AMC CH3), CF3 TFA not visible. HRMS (ESI, m/z): 318.1810 [M+H]+. Calculated mass: 318.1818 [M+H]+. Purity: 99 % (10.51 min).
Methyl 1‐(2‐((tert‐butoxycarbonyl)amino)ethyl)‐piperidine‐4‐carboxylate (40): Potassium carbonate (7246 mg, 52.43 mmol, 2.5 eq) was placed in a round‐bottom flask and suspended in acetonitrile. Methyl piperidine‐4‐carboxylate (3000 mg. 20.97 mmol, 1.0 eq) was added and stirred for 5 min. After adding 2‐(Boc‐amino)‐ethyl bromide (4677 mg, 20.97 mmol, 1.0 eq), the mixture was stirred overnight at room temperature. Product formation was observed by TLC analysis (EE/CH, 66 : 33 (v/v)). When full conversion was reached, solvent was evacuated under reduced pressure. The crude residue was resuspended in water. The aqueous suspension was extracted with ethyl acetate (3 times). Collected organic layers were washed with saturated NaHCO3 solution and NaCl solution before being dried over MgSO4, filtered off and the solvent evaporated under reduced pressure. Crude product was purified via flash column chromatography (EE/CH). Yield, 66 % of a yellow solid. Rf, 0.49 (DCM/MeOH 90/10 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 6.64 (t, J=5.6 Hz, 1H, OCO−NH‐CH2), 3.60 (s, 3H, O‐CH3), 3.01 (q, 6.4 Hz, 2H, NH‐CH 2 ‐CH2), 2.81–2.73 (m, 2H, Piperidine H2,6), 2.33–2.23 (m, 3H, CH2‐CH 2 ‐N‐(Piperidine)+Piperidine H4), 2.02–1.91 (m, 2H, Piperidine H2,6), 1.81–1.73 (m, 2H, Piperidine H3,5), 1.59–1.46 (m, 2H, Piperidine H3,5), 1.37 (s, 9H, ((CH 3 ) 3 ‐C−O). 13C NMR (DMSO‐d6, δ [ppm]): 175.3 (COOCH3), 155.9 (OCONH), 77.9 ((CH3)3‐C−O), 57.9 (CH2‐CH 2 ‐N‐(Piperidine)), 52.8 (Piperidine C2,6), 51.8 (O‐CH3), 40.6 (Piperidine C4), 37.9 (NH‐CH 2 ‐CH2), 28.7 ((CH 3 ) 3 ‐C−O), 28.4 (Piperidine C3,5). MS (APCI, +): 287.2 [M+H]+.
Methyl 1‐(2‐aminoethyl)‐piperidine‐4‐carboxylate (41): Compound 40 (2123 mg, 7.42 mmol, 1.0 eq) was dissolved in dichloromethane. Trifluoroacetic acid (8469 mg, 74.16 mmol, 10.0 eq) and triethylsilane (8623 mg, 74.16 mmol, 10.0 eq) were added and the mixture was stirred for 2 h at 40 °C. Solvent was removed under reduced pressure. The crude product was use without further purification. Yield, 100 % of a yellow oil. Rf, 0.72 (H2O/AcN 50/50 (v/v)+0.5 % TFA). 1H NMR (DMSO‐d6, δ [ppm]): 10.12 (s, 1H, CH2‐NH + ‐(Piperidine)), 8.20 (s, 3H, H 3 N + ‐CH2), 3.65 (s, 3H, O‐CH3), 3.62–3.46 (m, 2H, Piperidine C2,6), 3.34–3.18 (m, 4H, H3N+‐CH 2 ‐CH 2 ‐NH+‐(Piperidine)), 3.15–2.95 (m, 2H, Piperidine H2,6), 2.76–2.58 (m, 1H, Piperidine H4), 2.18–1.96 (m, 2H, Piperidine H3,5), 1.87–1.67 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 173.7 (COOCH3), 159.0 (q, 3 J=32 Hz, CO TFA), 117.1 (q, 1 J=296 Hz, CF3 TFA), 53.3 (H3N+‐CH2‐CH 2 ‐NH+‐(Piperidine)), 52.3 (O‐CH3), 51.8 (Piperidine C2,6), 37.9 (Piperidine C4), 33.9 (H3N+‐CH 2 ‐CH2‐NH+‐(Piperidine)), 25.8 (Piperidine C3,5). MS (APCI, +): 187.2 [M+H]+.
Methyl 1‐(2‐((2‐nitrophenyl)sulfonamido)ethyl)‐piperidine‐4‐carboxylate (42): Primary amine 41 (3072 mg, 7.42 mmol, 1 eq) and 2‐nitrobenzenesulfonyl chloride (1967 mg, 8.90 mmol, 1.2 eq) were dissolved in THF, under cooling with ice water. After adding of triethylamine (3001 mg, 29.66 mmol, 4 eq), the reaction mixture was stirred for 4 h at room temperature. The reaction was quenched by solvent evaporating and suspending the crude residue with water and dichloromethane (50 : 50). The aqueous layer was extracted with dichloromethane (5 times). Organic layers were dried over MgSO4, filtered off and the solvent was evaporated under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 82 % of a yellow oil. Rf, 0.95 (DMC/MeOH 90/10 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 8.09–8.03 (m, 1H, Nosyl H6), 8.02–7.96 (m, 1H, Nosyl H3), 7.90–7.77 (m, 3H, Nosyl H4,5+SO2 NH), 3.59 (s, 3H, O‐CH3), 3.03 (t, J=6.4 Hz, 2H, SO2NH‐CH 2 ‐CH2), 2.66–2.58 (m, 2H, Piperidine H2,6), 2.31 (t, J=6.4 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 2.28–2.19 (m, 1H, Piperidine H4), 1.95–1.85 (m, 2H, Piperidine H2,6), 1.74–1.65 (m, 2H, Piperidine H3,5), 1.48–1–35 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 175.2 (COOCH3), 148.0 (Nosyl C2) 134.4 (Nosyl C4), 133.4 (Nosyl C1), 133.1 (Nosyl C5), 130.0 (Nosyl C6), 124.9 (Nosyl C3), 57.2 (CH2‐CH 2 ‐N‐(Piperidine)), 52.5 (Piperidine C2,6), 51.8 (O‐CH3), 40.7 (SO2NH‐CH 2 ‐CH2),40.6 (Piperidine C4), 28.2 (Piperidine C3,5). MS (APCI, +): 372.6 [M+H]+.
Methyl 1‐(2‐((N‐benzyl‐2‐nitrophenyl)sulfonamido)ethyl)‐piperidine‐4‐carboxylate (44 a): Nosyl protected building block 43 (1002 mg, 2.70 mmol, 1.0 eq), benzyl bromide (923 mg, 5.40 mmol, 2.0 eq), potassium carbonate (559 mg, 4.05 mmol, 1.5 eq) and potassium iodide (90 mg, 0.54 mmol, 0.2 eq) were suspended in dimethylformamide. The reaction mixture was stirred overnight at room temperature. Solvent was removed under reduced pressure and the residue was suspended in water/dichloromethane. The aqueous layer was extracted with dichloromethane (3 times). The organic layer was dried over MgSO4 and then filtered off before the solvent was evaporated under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 69 % of a yellow solid. Rf, 0.52 (EE/CH 66/33 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 8.16 (dd, J=7.6, 1.6 Hz, 1H, Nosyl H6), 8.02 (dd, J=7.6, 1.6 Hz, 1H, Nosyl H3), 7.91 (td, J=7.6, 1.6 Hz, 1H, Nosyl H4), 7.85 (td, J=7.6, 1.6 Hz, 1H, Nosyl H5), 7.40–7.28 (m, 5H, Phenyl), 4.57 (s, 2H, Phenyl‐CH 2 ‐N), 3.58 (s, 3H, O‐CH3), 3.29 (t, J=6.6 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.62–2.53 (m, 2H, Piperidine H2,6), 2.26–2.16 (m, 3H, CH2‐CH 2 ‐N‐(Piperidine)+Piperidine H4), 1.88–1.77 (m, 2H, Piperidine H2,6), 1.72–1.62 (m, 2H, Piperidine H3,5), 1.45–1.33 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 175.2 (COO−), 148.0 (Nosyl C2), 136.8 (Phenyl C1), 134.8 (Nosyl C4), 132.9 (Nosyl C5), 132.8 (Nosyl C1), 130.1 (Nosyl C6), 129.0 (Phenyl C3,5), 128.3 (Phenyl C2,6), 128.2 (Phenyl C4), 124.8 (Nosyl C3), 56.1 (CH2‐CH 2 ‐N‐(Piperidine)), 52.6 (Piperidine C2,6), 51.9 (Phenyl‐CH 2 ‐N), 51.8 (O‐CH3), 44.9 (SO2N‐CH 2 ‐CH2), 40.5 (Piperidine C4), 28.2 (Piperidine C3,5). MS (APCI, +): 462.8 [M+H]+.
Lithium 1‐(2‐((N‐benzyl‐2‐nitrophenyl)sulfonamido)ethyl)‐piperidine‐4‐carboxylate (45 a): Compound 44 a (852 mg, 1.85 mmol, 1.0 eq) was dissolved in tetrahydrofuran and lithium hydroxide solution [1 M] (5.5 mL, 5.54 mmol, 1.5 eq) was added. The mixture was stirred for 4 h at 40 °C. After removing solvent, the crude residue was resuspended in water and washed with ethyl acetate. The aqueous layer was evaporated and product was used without further purification. Yield, 100 % of a white crystalline solid. 1H NMR (DMSO‐d6, δ [ppm]): 8.22 (dd, J=7.6, 1.6 Hz, 1H, Nosyl H6), 8.02 (dd, J=7.6, 1.6 Hz, 1H, Nosyl H3), 7.91 (td, J=7.6, 1.6 Hz, 1H, Nosyl H4), 7.85 (td, J=7.6, 1.6 Hz, 1H, Nosyl H4), 7.39–7.24 (m, 5H, 5× Phenyl), 4.56 (s, 2H, Phenyl‐CH 2 ‐NSO2), 3.29 (t, J=6.8 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.17 (t, J=6.8 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 1.78–1.66 (m, 2H, Piperidine C2,6), 1.65–1.55 (m, 3H, Piperidine H3,5+H4), 1.45–1.31 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 174.5 (−COO−), 148.0 (Nosyl C2), 136.8 (Phenyl C1), 134.9 (Nosyl C4), 132.9 (Nosyl C5), 132.8 (Nosyl C1), 130.2 (Nosyl C6), 129.0 (Phenyl C3,5), 128.3 (Phenyl C2,6), 128.2 (Phenyl C4), 124.8 (Nosyl C3), 56.7 (CH2‐CH 2 ‐N‐(Piperidine)), 54.0 (Piperidine C2,6), 51.9 (Phenyl‐CH 2 ‐NSO2), 44.7 (SO2N‐CH 2 ‐CH2), 44.1 (Piperidine C4), 29.9 (Piperidine C3,5). MS (APCI, +): 448.2 [M+H]+.
1‐(2‐((N‐Benzyl‐2‐nitrophenyl)sulfonamido)ethyl)‐N‐(trityloxy)‐piperidine‐4‐carboxamide (46 a): Compound 45 a (839 mg, 1.85 mmol, 1.0 eq) and BOP‐Cl (941 mg, 3.70 mmol, 2.0 eq) were placed in a round‐bottom flask and suspended in dichloromethane. After adding triethylamine (748 mg, 7.39 mmol, 4 eq), the mixture was stirred for 10 min at room temperature. O‐Tritylhydroxylamine (611 mg, 2.22 mmol, 1.2 eq) was added and the mixture was stirred overnight at room temperature. The reaction was quenched by removing the volatiles and followed by the resuspension of the crude material in saturated NaHCO3 solution. The aqueous suspension was extracted with ethyl acetate (3 times). Organic layers were washed with saturated NaHCO3 solution and NaCl solution and dried over MgSO4 before the solvent was evaporated under reduced pressure. The crude product was purified via flash column chromatography (DCM/MeOH). Yield, 41 % of a yellow solid. Rf, 0.40 (EE/CH 66/33 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.27 (s, 1H, CO−NH−O), 8.17 (dd, J=7.6, 1.2 Hz, 1H Nosyl H6), 8.00 (dd, J=7.6, 1.2 Hz, 1H, Nosyl H3), 7.89 (td, J=7.6, 1.2 Hz, 1H, Nosyl H4), 7.81 (td, J=7.6, 1.2 Hz, 1H, Nosyl H5), 7.38–7.21 (m, 20H, 5× Phenyl+15× Trityl), 4.52 (s, 2H, Phenyl‐CH 2 ‐N), 3.24 (t, J=6.4 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.48 (2H, Piperidine C2,6, HSQC, covert by DMSO), 2.12 (t, J=6.4 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 1.87–1.76 (m, 1H, Piperidine H4), 1.65–1.53 (m, 2H, Piperidine H2,6), 1.20–1.03 (m, 4H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 172.8 (CONH), 148.0 (Nosyl C2), 142.8 (3× Phenyl C1 (Trityl)), 136.7 (Phenyl C1), 134.9 (Nosyl C4), 132.8 (Nosyl C5), 132.7 (Nosyl C1), 130.2 (Nosyl C6), 129.4 (3× Phenyl C2,6 or C3,6 (Trityl)), 128.9 (Phenyl C3,5), 128.3 (Phenyl C2,6), 128.1 (Phenyl C4), 127.9 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.8 (3× Phenyl C4 (Trityl)), 124.7 (Nosyl C3), 92.2 (O−C‐Trityl), 56.2 (CH2‐CH 2 ‐N‐(Piperidine)), 52.9 (Piperidine C2,6), 51.7 (Phenyl‐CH 2 ‐N), 44.7 (SO2N‐CH 2 ‐CH2), 39.1 (Piperidine C4), 28.3 (Piperidine C3,5). MS (ESI, +): 705.3 [M+H]+.
1‐(2‐(Benzylamino)ethyl)‐N‐(trityloxy)‐piperidine‐4‐carboxamide (47 a): Compound 46 a (312 mg, 0.44 mmol, 1.0 eq) and potassium carbonate (245 mg, 1.77 mmol, 4.0 eq) were dissolved in acetonitrile. After adding thiophenol (136 μL, 1.32 mmol, 3.0 eq) the reaction mixture was stirred for 3 h at 35 °C. Solvent was removed and the crude product was purified via flash column chromatography (DCM/MeOH). Yield, 69 % of a colorless crystalline solid. Rf, 0.53 (DCM/MeOH 90/10 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.29 (s, 1H, CO−NH−O), 7.41–7.18 (m, 20H, 5x Phenyl+15x Trityl), 3.67 (s, 2H, Phenyl‐CH 2 ‐N), 2.72–2.63 (m, 2H, Piperidine H2,6), 2.49 (2H, N‐CH 2 ‐CH2‐N‐(Piperidine), HSQC, covert by DMSO), 2.28 (t, J=6.4 Hz, 2H, N‐CH2‐CH 2 ‐N‐(Piperidine)), 1.89 (q, J=8.0 Hz, 1H, Piperidine H4), 1.75–1.61 (m, 2H, Piperidine H2,6), 1.31–1.15 (m, 4H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 173.0 (CONH), 142.9 (3× Phenyl C1 (Trityl)), 141.1 (Phenyl C1), 129.4 (3× Phenyl C2,6 or 3,5 (Trityl)), 128.5 (Phenyl C3,5), 128.3 (Phenyl C2,6), 127.9 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.8 (3× Phenyl C4 (Trityl)), 127.0 (Phenyl C4), 92.2 (O−C‐Trityl), 58.0 (N‐CH2‐CH 2 ‐N‐(Piperidine)), 53.3 (Phenyl‐CH 2 ‐N), 53.2 (Piperidine C2,6), 45.9 (N‐CH 2 ‐CH2‐N‐(Piperidine)), 39.6 (Piperidine C4), 28.5 (Piperidine C3,5). MS (ESI, +): 520.3 [M+H]+.
1‐(2‐(Benzylamino)ethyl)‐N‐hydroxypiperidine‐4‐carboxamide (48 a): Compound 47 a (103 mg, 0.20 mmol, 1.0 eq) was dissolved in dry dichloromethane and trifluoroacetic acid (226 mg, 1.98 mmol, 10.0 eq) and triethylsilane (230 mg, 1.98 mmol, 10.0 eq) were added before the mixture was stirred at room temperature. Product formation was observed by TLC analysis (DCM/MeOH, 90 : 10 (v/v)). When full conversion was reached, solvent was removed. The crude product was dissolved in water. The aqueous solution was washed with cyclohexane (3 times) and solvent was removed under reduced pressure. Yield, 100 % of a colorless oil. 1H NMR (DMSO‐d6, δ [ppm]): 10.64 (s, 1H, CO−NH−OH), 9.62 (bs, 1H, CH2‐NH + ‐(Piperidine)), 9.28 (bs, 2H, CH2‐NH 2 +‐CH2), 7.58–7.41 (m, 5H, Phenyl), 4.22 (s, 2H, Phenyl‐CH 2 ‐NH2 +), 3.65–3.47 (m, 2H, Piperidine H2,6), 3.46–3.36 (m, 4H, NH2 +‐CH 2 ‐CH 2 ‐NH+‐(Piperidine)), 3.12–2.94 (m, 2H, Piperidine H2,6), 2.36–2.23 (m, 1H, Piperidine H4), 2.00–1.71 (m, 4H, Piperidine H3,5), CO−NH−OH not visible. 13C NMR (DMSO‐d6, δ [ppm]): 170.1 (CONH), 158.9 (q, 3 J=34 Hz, CO TFA), 132.1 (Phenyl C1), 130.3 (Phenyl C2,6), 129.6 (Phenyl C4), 129.3 (Phenyl C3,5), 116.8 (q, 1 J=294 Hz, CF3 TFA), 52.1 (pieridine C2,6, NH2 +‐CH2‐CH 2 ‐NH+‐(Piperidine), 50.8 (Phenyl‐CH 2 ‐NH2 +), 41.2 (NH2 +‐CH 2 ‐CH2‐NH+‐(Piperidine)), 36.5 (Piperidine C4), 26.3 (Piperidine C3,5). HRMS (ESI, m/z): 278.1864 [M+H]+. Calculated mass: 278.1869 [M+H]+. Purity: 96 % (8.66 min, modification of method: flow rate 0.5 mL/min).
Methyl 1‐(2‐((N‐(4‐bromobenzyl)‐2‐nitrophenyl)sulfonamido)ethyl)‐piperidine‐4‐carboxylate (44 b): Compound 43 (500 mg, 1.35 mmol. 1.0 eq), 4‐bromobenzyl bromide (672 mg, 2.69 mmol, 2.0 eq), potassium carbonate (279 mg, 2.02 mmol, 1.5 eq) and potassium iodide (45 mg, 0.27 mmol, 0.2 eq) were suspended in dimethylformamide. The reaction mixture was stirred overnight at room temperature. Solvent was removed under reduced pressure and the residue was suspended in water/dichloromethane. The aqueous layer was extracted with dichloromethane (3 times). The organic layer was dried over MgSO4 and then filtered off before the solvent was evaporated under reduced pressure. Crude product was purified via flash column chromatography (DCM/MeOH). Yield, 66 % of a green oil. Rf, 0.35 (EE/CH 66/33 (v/v)).1H NMR (DMSO‐d6, δ [ppm]): 8.16 (dd, J=8.0, 1.2 Hz, 1H, Nosyl H6), 8.02 (dd, J=8.0, 1.2 Hz, 1H, Nosyl H3), 7.91 (td, J=8.0, 1.2 Hz, 1H, Nosyl H4), 7.85 (td, J=8.0, 1.2 Hz, 1H, Nosyl H5), 7.60–7.54 (m, 2H, Phenyl H3,5), 7.32–7.26 (m, 2H, Phenyl H2,6), 4.54 (s, 2H, Phenyl‐CH 2 ‐NSO2), 3.59 (s, 3H, O−CH3), 3.31 (t, J=6.4 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.63–2.53 (m, 2H, Piperidine C2,6), 2.27–2.16 (m, 3H, CH2‐CH 2 ‐N‐(Piperidine)+Piperidine H4), 1.90–1.78 (m, 2H, Piperidine H2,6), 1.73–1.63 (m, 2H, Piperidine H3,5), 1.45–1.36 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 175.2 (COOCH3), 148.0 (Nosyl C2), 136.6 (Phenyl C1), 134.9 (Nosyl C4), 132.9 (Nosyl C5), 132.6 (Nosyl C1), 131.8 (Phenyl C3,5), 130.4 (Phenyl C2,6), 130.1 (Nosyl C6), 124.8 (Nosyl C3), 121.2 (Phenyl C4), 56.1 (CH2‐CH 2 ‐N‐(Piperidine)), 52.6 (Piperidine C2,6), 51.8 (O−CH 3 ), 51.3 (Phenyl‐CH 2 ‐NSO2), 45.2 (SO2N‐CH 2 ‐CH2), 40.5 (Piperidine C4, HSQC), 28.2 (Piperidine C3,5). MS (APCI, +): 540.1+542.1 [M+H]+.
Lithium 1‐(2‐((N‐(4‐bromobenzyl)‐2‐nitrophenyl)sulfonamido)ethyl)‐piperidine‐4‐carboxylate (45 b): Compound 44 b (468 mg, 0.87 mmol, 1.0 eq) was dissolved in tetrahydrofuran and lithium hydroxide solution [1 M] (2.6 mL, 2.61 mmol, 3.0 eq) was added. The mixture was stirred for 4 h at 40 °C. After removing solvent, the crude residue was resuspended in water and washed with ethyl acetate. The aqueous layer was evaporated and product was used without further purification. Yield, 90 % of a yellow solid. 1H NMR (DMSO‐d6, δ [ppm]): 8.21 (dd, J=8.0, 1.6 Hz, 1H, Nosyl H6), 8.01 (dd, J=8.0, 1.6 Hz, 1H, Nosyl H3), 7.91 (td, J=8.0, 1.6 Hz, 1H, Nosyl H4), 7.85 (td, J=8.0, 1.6 Hz, 1H, Nosyl H5), 7.58–7.53 (m, 2H, Phenyl H3,5), 7.31–7.25 (m, 2H, Phenyl H2,6), 4.54 (s, 2H, Phenyl‐CH 2 ‐N), 3.30 (t, J=6.4 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.60–5.53 (m, 2H, Piperidine H2,6), 2.19 (t, J=6.4 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 1.79–1.67 (m, 3H, Piperidine H2,6+H4), 1.63–1.58 (m, 2H, Piperidine H3,5), 1.45–1.32 (m, 2H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 179.3 (COO−), 148.0 (Nosyl C2), 136.5 (Phenyl C1), 135.0 (Nosyl C4), 133.0 (Nosyl C5), 132.6 (Nosyl C1), 131.8 (Phenyl C3,5), 130.4 (Phenyl C2,6), 130.2 (Nosyl C6), 124.8 (Nosyl C3), 121.2 (Phenyl C4), 56.6 (CH2‐CH 2 ‐N‐(Piperidine)), 53.9 (Piperidine C2,6), 51.2 (Phenyl‐CH 2 ‐N), 45.0 (SO2N‐CH 2 ‐CH2), 44.0 (Piperidine C4), 29.8 (Piperidine C3,5). MS (APCI, +): 526.0+528.0 [M+H]+.
1‐(2‐((N‐(4‐Bromobenzyl)‐2‐nitrophenyl)sulfonamido)ethyl)‐N‐(trityloxy)‐piperidine‐4‐carboxamide (46 b): Compound 45 b (135 mg, 0.26 mmol, 1.0 eq) and BOP‐Cl (130 mg, 0.51 mmol, 2.0 eq) were placed in a round‐bottom flask and suspended in dichloromethane. After adding triethylamine (104 mg, 1.02 mmol, 4 eq), the mixture was stirred for 10 min at room temperature. O‐Tritylhydroxylamine (85 mg, 0.31 mmol, 1.2 eq) was added and the mixture was stirred overnight at room temperature. The reaction was quenched by removing the volatiles and followed by the resuspension of the crude in saturated NaHCO3 solution. The aqueous suspension was extracted with ethyl acetate (3 times). Organic layers were washed with saturated NaHCO3 solution and NaCl solution and dried over MgSO4 before the solvent was evaporated under reduced pressure. The crude product was purified via flash column chromatography (DCM/MeOH). Yield, 68 % of a yellow oil. Rf, 0.52 (DCM/MeOH 95/5 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.27 (s, 1H, CO−NH−O), 8.19–8.13 (m, 1H, Nosyl H6), 8.00 (dd, J=8.0, 1.2 Hz, 1H, Nosyl H3), 7.94–7.86 (m, 1H, Nosyl H4), 7.86–7.78 (m, 1H, Nosyl H5), 7.57–7.52 (m, 2H, Phenyl H3,5), 7.41–7.22 (m, 17H, Phenyl H2,6+15x Trityl), 4.50 (s, 2H, Phenyl‐CH 2 ‐NSO2), 3.26 (t, J=6.4 Hz, 2H, SO2N‐CH 2 ‐CH2), 2.53 (m, 2H, Piperidine H2,6, HSQC, covert by DMSO), 2.14 (t, J=6.4 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 1.89–1.76 (m, 1H, Piperidine H4), 1.65–1.53 (m, 2H, Piperidine H2,6), 1.21–1.04 (m, 4H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 172.8 (CONH), 148.0 (Nosyl C2), 142.8 (3× Phenyl C1 (Trityl)), 136.5 (Phenyl C1), 134.9 (Nosyl C4), 132.9 (Nosyl C5), 132.5 (Nosyl C1), 131.8 (Phenyl C3,5), 130.4 (Phenyl C2,6), 130.2 (Nosyl C6), 129.4 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.9 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.8 (3× Phenyl C4 (Trityl)), 124.8 (Nosyl C3), 121.2 (Phenyl C4), 92.2 (O−C‐Trityl), 56.2 (CH2‐CH 2 ‐N‐(Piperidine)), 52.9 (Piperidine C2,6), 51.1 (Phenyl‐CH 2 ‐NSO2), 45.0 (SO2N‐CH 2 ‐CH2), 38.9 (Piperidine C4), 28.3 (Piperidine C3,5). MS (ESI, +): 783.2+785.2 [M+H]+.
1‐(2‐((4‐Bromobenzyl)amino)ethyl)‐N‐(trityloxy)‐piperidine‐4‐carboxamide (47 b): Compound 46 (128 mg, 0.16 mmol, 1.0 eq) and potassium carbonate (90 mg, 0.65 mmol, 4.0 eq) were dissolved in acetonitrile. After adding thiophenol (50 μL, 0.49 mmol, 3.0 eq) the reaction mixture was stirred for 3 h at 35 °C. Solvent was removed and the crude product was purified via flash column chromatography (DCM/MeOH). Yield, 73 % of a colorless oil. Rf, 0.20 (DCM/MeOH 95/5 (v/v)). 1H NMR (DMSO‐d6, δ [ppm]): 10.29 (s, 1H, CO−NH−O), 7.51–7.46 (m, 2H, Phenyl H3,5), 7.42–7.19 (m, 17H, Phenyl H2,6+15x Trityl), 3.64 (s, 2H, Phenyl‐CH 2 ‐NSO2), 2.71–2.62 (m, 2H, Piperidine H2,6), 2.49–2.45 (m, 2H, SO2N‐CH 2 ‐CH2), 2.27 (t, J=6.4 Hz, 2H, CH2‐CH 2 ‐N‐(Piperidine)), 1.95–1.82 (m, 1H, Piperidine H4), 1.73–1.60 (m, 2H, Piperidine H2,6), 1.32–1.14 (m, 4H, Piperidine H3,5). 13C NMR (DMSO‐d6, δ [ppm]): 173.0 (CONH), 142.9 (3× Phenyl C1 (Trityl)), 140.9 (Phenyl C1), 131.3 (Phenyl C3,5), 130.5 (Phenyl C2,6), 129.4 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.9 (3× Phenyl C2,6 or C3,5 (Trityl)), 127.8 (3× Phenyl C4 (Trityl)), 92.2 (O−C‐Trityl), 58.1 (CH2‐CH 2 ‐N‐(Piperidine)), 53.2 (Piperidine C2,6), 52.5 (Phenyl‐CH 2 ‐NSO2), 45.9 (SO2N‐CH 2 ‐CH2), 40.0 (Piperidine C4, HSQC), 28.5 (Piperidine C3,5). MS (APCI, +): 598.2+600.2 [M+H]+.
1‐(2‐((4‐Bromobenzyl)amino)ethyl)‐N‐hydroxypiperidine‐4‐carboxamide (48 b): Compound 47 b (66 mg, 0.11 mmol, 1.0 eq) was dissolved in dry dichloromethane and trifluoroacetic acid (208 mg, 1.10 mmol, 10.0 eq) and triethylsilane (128 mg, 1.10 mmol, 10.0 eq) were added before the mixture was stirred at room temperature. Product formation was observed by TLC analysis (DCM/MeOH, 90 : 10 (v/v)). When full conversion was reached, solvent was removed. The crude product was purified via flash column chromatography (H2O/MeCN+0.5 % TFA). Yield, 47 % of a brown oil. Rf, 0.63 (H2O/MeCN 50/50 (v/v)+0.5 % TFA). 1H NMR (DMSO‐d6, δ [ppm]): 10.65 (s, 1H, CO−NH−OH), 9.73 (s, 1H, CH2‐NH + ‐(Piperidine)), 9.37 (s, 2H, CH2‐NH 2 +‐CH2), 7.69 (d, J=8.4 Hz, 2H, Phenyl H3,5), 7.46 (d, J=8.4 Hz, 2H, Phenyl H2,6), 4.21 (s, 2H, Phenyl‐CH 2 ‐NH2), 3.63–3.48 (m, 2H, Piperidine H2,6), 3.39 (s, 4H, NH2 +‐CH 2 ‐CH 2 ‐NH+‐(Piperidine)), 3.12–2.93 (m, 2H, Piperidine H2,6), 2.36–2.22 (m, 1H, Piperidine H4), 1.98–1.70 (m, 4H, Piperidine H3,5), CO−NH−OH not visible. 13C NMR (DMSO‐d6, δ [ppm]): 170.1 (CONH), 158.9 (q, 3 J=34 Hz, CO TFA), 132.6 (Phenyl C2,6), 132.2 (Phenyl C3,5), 131.4 (Phenyl C1), 123.1 (Phenyl C4), 116.8 (q, 1 J=293 Hz, CF3 TFA), 52.1 (Piperidine C2,6, NH2 +‐CH2‐CH 2 ‐NH+‐(Piperidine)), 49.9 (Phenyl‐CH 2 ‐NH2 +), 41.1 (NH2 +‐CH 2 ‐CH2‐NH+‐(Piperidine)), 36.5 (Piperidine C4), 26.2 (Piperidine C3,5). HRMS (ESI, +): 356.0963+358.0948 [M+H]+. Calculated mass: 356.0974+358.1070 [M+H]+. Purity: 95 % (9.95 min).
Protein expression
HDAC10 from Danio rerio (zebrafish) (residues 2–676) was prepared and purified as previously described [6] with minor modifications. Briefly, protein was expressed in E. coli BL21 (DE3) (Agilent) cells in 2× YT media in the presence of 50 μg/mL kanamycin (Gold Bio). Expression was induced when OD600 reached 1.0 by addition of 150 μM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG, Gold Bio) and 500 μM ZnSO4 (Fisher Scientific), and cell cultures were grown for an additional 18–22 hours at 16 °C. Cells were harvested by centrifugation at 6,000 g and resuspended in lysis buffer containing 50 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) (pH 8.0), 300 mM NaCl, 2 mM tris‐(2‐carboxyethyl) phosphine (TCEP), 10 % glycerol (v/v), 10 μM ZnCl2, 30 mM imidazole, 2 mini‐protease inhibitor tablets (Roche), 0.5 mg/mL hen egg‐white lysozyme (Millipore‐Sigma), and 0.1 mg/mL DNaseI (Millipore‐Sigma). Cells were lysed by sonication and lysate was cleared by centrifugation at 26,000 g for 1 hour at 4 °C. The supernatant was loaded onto a 5‐mL pre‐packed His trap (Ni2+) affinity column (GE Healthcare Life Sciences) and HDAC10 was eluted with buffer containing 50 mM HEPES (pH 8.0), 300 mM NaCl, 2 mM TCEP, 10 % glycerol (v/v), 10 μM ZnCl2, and 500 mM imidazole.
Peak fractions were incubated with Tobacco Etch Virus (TEV) protease and extensively dialyzed overnight at 4 °C against a buffer containing 50 mM HEPES (pH 8.0), 300 mM NaCl, 2 mM TCEP, 10 % glycerol (v/v), 10 μM ZnCl2, and 30 mM imidazole. The free zebrafish HDAC10 protein was separated from free His‐MBP tag by loading the dialyzed protein sample onto a 5‐mL pre‐packed His trap (Ni2+) affinity column (GE Healthcare Life Sciences) and then passing the His trap column flow‐through fraction over a column containing amylose resin (NEB Biolabs). The flow‐through sample from the amylose column was then loaded onto a HiLoad Superdex S200 (26/600) size exclusion column (GE Healthcare Life Sciences) pre‐equilibrated with a buffer containing 50 mM HEPES (pH 7.5), 300 mM KCl, 1 mM TCEP and 5 % glycerol (v/v). Protein was concentrated to 2–8 mg/mL, flash‐cooled in liquid nitrogen, and stored at −80 °C for further use.
Proof of substrate conversion by HPLC
For the calibration curve, stock solutions of Ac‐spermidine‐AMC [200 μM] and Spermidine‐AMC [200 μM] in buffer (20 mM Na2HPO4, pH 7.9, 10 mM NaCl, 0.25 mM EDTA) were prepared. Fluorescamine solution [600 μM] was prepared in acetonitrile. Ac‐spermidine‐AMC and Spermidine‐AMC stocks solutions were mixed in nine different mixing ratios to get 50 μL solution (see Table S1). Substrate‐product‐mixtures were complemented with 50 μL of fluorescamine solution. Different samples were analyzed via HPLC (Gradient see Chemistry, 5 μL injected, Phenomenex Kinetex column, UV absorption 210 nm).
Stock solutions of Ac‐spermidine‐AMC [240 μM] and Spermidine‐AMC [240 μM] in buffer (20 mM Na2HPO4, pH 7.9, 10 mM NaCl, 0.25 mM EDTA) were prepared to prove substrate conversion by drHDAC10. Fluorescamine solution [600 μM] was prepared in acetonitrile. The enzyme was diluted with buffer to a final concentration of 0.027 mg/mL.
Samples were prepared in Eppendorf tubes; each containing 50 μL Ac‐spermidine‐AMC solution and 10 μL of enzyme solution. The tubes were incubated at 37 °C for 0–60 min. The reaction was stopped at several time points (0, 10, 15, 20, 30, 45, 60 min) by adding fluorescamine solution, mixing and centrifugation (5 min, 20000 rpm). For time point 0 min stop solution was added before enzyme solution. Supernatant was analyzed by HPLC (see calibration curve, n=1).
Z'‐factor
The following equation was used to calculate the Z' value: Z′=1‐(3σc++3σc–)/|μc+–μc–|. The standard deviation of the positive control (σc+) is represented by relative fluorescence units of conversion by enzyme (buffer, enzyme solution, DMSO, substrate), for standard deviation of the negative control (σc‐) the relative fluorescence units of no substrate conversion was used (buffer, DMSO, substrate). It was shown that there is no significant difference between no substrate conversion (buffer, DMSO) and 100 % inhibition (buffer, enzyme solution, 10 μM quisinostat, substrate). μc+ and μc‐ are the mean of positive control (no inhibition) and negative control (no conversion of substrate).
Measurements were performed on three different days each with 35 values for positive control and 35 values for negative control.
Selectivity of substrate
ZMTFAL‐Assay. Enzyme solutions were prepared in assay buffer (15 mM Tris, pH 7.5, 50 mM KH2PO4, 10 mM KCl, 3 mM MgSO4 ⋅ 7H2O). 22.5 μL of enzyme solution, 2.5 μL DMSO and 5 μL of ZMTFAL (Z‐L‐Lys(ϵ‐trifluoroacetyl)‐AMC) substrate solution [150 μM] were incubated for 90 min at 25 °C in AreaPlate‐96 F microplates (PerkinElmer). 30 μL of stop solution, containing 2.5 μL trichostatin A (TSA) (33 μM) and 5 μL trypsin (6 mg/mL) in trypsin buffer (Tris‐HCl 50 mM, pH 8.0, NaCl 100 mM), were added. After incubation (30 min at 37 °C) fluorescence signal (POLARstar plate reader, λex=390 nm, λem=460 nm) was measured.
NDA‐Assay. Enzyme solutions were prepared in assay buffer leading to final concentrations (hHDAC1 1 μL/well, hHDAC6 1 μL/well, hHDAC8 0.005 μL/well, zHDAC10 0.5 μL/well). Enzyme activity was determined according to NDA‐Assay for HDAC10.
In vitro testing
hHDAC1/6: Commercial available human recombinant HDAC1 (BPS Bioscience, catalog no. 50051) and human recombinant HDAC6 (BPS Bioscience, catalog no. 50006) were used. Activity assays were performed in OptiPlateTM‐96 F black microplates (PerkinElmer). The total assay volume of 60 μL contained 52 μL of enzyme solution in incubation buffer (50 mM Tris‐HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mg/mL bovine serum albumin), 3 μL of increasing concentrations of inhibitors in DMSO, and 5 μL of the fluorogenic substrate ZMAL (Z‐(Ac)Lys‐AMC) (126 μM). After the incubation step (90 min, 37 °C), 60 μL of stop solution, containing 5 μL Trichostatin A (TSA) (33 μM) and 10 μL trypsin (6 mg/mL) in trypsin buffer (Tris‐HCl 50 mM, pH 8.0, NaCl 100 mM), were added and the plate was incubated for 30 min at 37 °C. Fluorescence signal was measured on a BMG LABTECH POLARstar OPTIMA plate reader (BMG Labtechnologies, Germany) with an excitation wavelength of 390 nm and an emission wavelength of 460 nm. [58]
hHDAC8: For HDAC8 activity testing commercial available Fluor de Lys (FDL) drug discovery kit (BML‐KI178) was used. Enzyme was obtained as described previously. [59] The assay was performed according to the manufacturer's instructions. Enzyme solution (15 μL), increasing inhibitor concentrations (10 μL), and FDL substrate solution (25 μL) were incubated for 90 min at 37 °C in AreaPlate‐96 F microplates (PerkinElmer). Developer solution (50 μL) was added and the assay was incubated for 45 min at 30 °C. The fluorescence signal was determined as described for HDAC1/6.
HDAC10: All stock solutions were prepared in DMSO; quisinostat (1 mM), NDA (16 mM) and Ac‐spermidine‐AMC (10 mM). Compounds for testing were dissolved and diluted to 12‐fold higher than test concentration in DMSO. Ac‐spermidine‐AMC stock solutions were diluted with assay buffer (20 mM Na2HPO4, pH 7.9, 100 mM NaCl, 0.25 mM EDTA, 10 % (v/v) glycerol, 10 mM Mesna, 0.01 % TWEEN 20) to 126 μM. For assay determination stop solution was prepared, containing 5 μL NDA (16 mM), 5 μL quisinostat (1 mM) and 190 μL borate buffer (100 mM boric acid, pH 9.5) per well. Immediately before use, enzyme solution (0.0054 mg/mL) was prepared in assay buffer.
The assay was performed in black 96‐well plates (PerkinElmer, OptiPlateTM‐96 F). Assay buffer was presented in the plate, 55 μL for the blank, 45 μL for the blank containing enzyme solution, 50 μL for the negative control and 40 μL for the positive control and test compounds. 5 μL of DMSO were added to the wells of blanks, positive and negative control. 5 μL of increasing concentrations of inhibitors in DMSO were added to the relevant wells. After adding 10 μL of enzyme solution (12 nM final assay concentration) to blank containing enzyme, positive control and test compounds, 5 μL Ac‐spermidine‐AMC solution (10.5 μM final assay concentration) were added to negative control, positive control and test compounds. The plate was incubated for 25 min at 25 °C. Before measuring fluorescence (POLARstar plate reader, λex=330 nm, λem=390 nm) each well was filled with 200 μL stop solution.
IC50 calculation
Inhibition was measured at increasing concentration and IC50 was calculated by nonlinear regression with Origin 9.0G software.
FRET assay
The FRET‐assay was performed in white 384‐well ProxiPlates (PerkinElmer). Reagents were diluted in assay buffer (50 mM HEPES pH 8.0, 150 mM NaCl, 10 mM MgCl2, 1 mM EGTA, 0.01 % Brij‐35). Final assay volume (10 μL) contains 3 nM GST‐HDAC10 (Life Technologies), 30 nM Tubastatin‐Alexa647‐Tracer, [32] and 0.5 nM LanthaScreen Eu‐anti‐GST (Life Technologies). Compound stocks (10 mM DMSO) were diluted in assay buffer. An 11‐fold 1 : 3‐serial dilution of test compounds (1 μL) were presented in the plate and complemented by 9 μL of assay mix. After incubation (1 h, room temperature) TR‐FRET was measured with EnVision plate reader (Ex: 3 flashes of the TRF‐europiumlaser; Em: 620 and 665 nm (665/620 nm ratio). Inhibition was calculated by using negative control (2 % DMSO) and positive control (20 μM vorinostat). Dose‐response curves were fitted in ActivityBase (IDBS) using a four‐parameter logistic model and IC50‐values were calculated. [32]
X‐ray structure determination
The “humanized” variant of HDAC10 was expressed and purified as previously described. [55] To a solution containing 10 mg/mL HDAC10 in buffer [50 mM HEPES (pH 7.5), 300 mM KCl, 5 % glycerol, and 1 mM tris(2‐carboxyethyl)phosphine (TCEP)] 2 mM of 48 a was added and allowed to incubate on ice for 1 h. Trypsin was then added at a 1 : 1000 trypsin : HDAC10 molar ratio and allowed to digest at room temperature for 1 h. Immediately following trypsin digestion, the solution was filtered using a 0.22 μm centrifuge filter.
Crystals of the HDAC10‐48 a complex were formed by the sitting drop vapor diffusion method at 4 °C. Briefly, a 100 nL drop of protein solution was added to a 100 nL drop of precipitant buffer [0.100 M NaH2PO4, 0.100 M Na2HPO4, and 20 % (w/v) PEG3350] on a 96‐well crystallization plate using a Mosquito crystallization robot (TTP Labtech). To each drop, 25 nL of microseed crystals in precipitant buffer were added. The drop was equilibrated against 80 μL of precipitant buffer in the well reservoir. Crystals formed in approximately one day.
X‐ray diffraction data for the HDAC10‐48 a complex were collected on NE‐CAT beamline 24‐ID‐C at the Advanced Photon Source of Argonne National Laboratory (Argonne, Il). Diffraction data were integrated using iMosflm2 and scaled with Aimless in the CCP4 program suite. [60] The initial electron density map was phased by molecular replacement using the atomic coordinates of the Y307F HDAC10‐trifluoroketone inhibitor complex (PDB 5TD7) [6] using Phaser. [61] Model building was performed using COOT and the model was refined using PHENIX in an iterative process. [62] The atomic coordinates of 48 a were not added to the model until the final stages of refinement. MolProbity10 was used to assess the quality of the final model. [63] All data collection and refinement statistics are listed in Table S6.
BRET assay
BRET‐Assay for HDAC10 was performed as previously described. [32] According to manufacturer's instructions 1.9×104 cells per well were plated in a 96‐well plate (3600, Corning) and tracer was added with a concentration of 0.3 μM. Inhibitors were tested at ten 1 : 4 serial dilutions in triplicates ranging from 129 pM to 40 μM. DMSO concentrations were normalized to 0.5 % for all wells. After incubating for 2 h at 37 °C NanoLuc substrate was added. Luminescence was measured (λem=450 and 650 nm) with a CLARIOstar plate reader (BMG Labtechnologies) 2 min after NanoLuc substrate addition. BRET ratios were calculated from 650 nm/450 nm luminescence and normalized using negative controls treated with 50 μM vorinostat and uninhibited positive controls. EC50 values were calculated using nonlinear regression with GraphPad Prism version 7.04 (GraphPad Software) software.
Western blotting
HL60 cells (ATCC CCL‐240) were cultured in RPMI medium 1640 containing 10 % FBS gold, 2 mM L‐glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. 2.5×105 cells per well were seeded in a 12‐well plate and immediately incubated with different concentrations of compounds for 4 h. After incubation, cells were collected in Eppendorf tubes and centrifuged with 500 g for 5 minutes at room temperature. Cells were washed with PBS and lysed in 90 μL of SDS sample buffer (Cell Signaling, 62.5 mM Tris‐HCl (pH 6.8 at 25 °C), 2 % w/v SDS, 10 % v/v glycerol, 50 mM dithiothreitol, 0.01 % bromophenol blue). After sonicating for 5 min, to shear DNA and reduce sample viscosity, the samples were heated to 95 °C for 1 min. Cell extracts were used directly for SDS‐PAGE or kept frozen at −20 °C until usage. For the SDS‐PAGE an amount of 10 μL of cell extracts was loaded onto a 12.5 % SDS gel and run at 160 V followed by the transfer to a nitrocellulose membrane via Western blotting for antibody‐based detection. After transfer the nonspecific binding was blocked by incubating the membrane in 25 mL of blocking buffer (5 % nonfat dry milk in Tris‐buffered saline with 0.1 % Tween 20 (TBS‐T)) for 1 h at room temperature or at 4 °C overnight. After washing the membrane 3 times for 5 min with TBS‐T, the primary antibody (antiacetylated α‐tubulin (Sigma‐Aldrich T7451‐200UL, 1 : 1000)) was added in 3 % milk in TBS‐T for 3 h at room temperature or overnight at 4 °C. Before exposing the membrane with the secondary antibody, it was washed again three times for 5 min with TBS‐T to remove unbound primary antibody. The secondary antibody anti‐mouse‐IgG‐HRP (Sigma‐Aldrich, 1 : 2000) was added in 3 % milk in TBS‐T at room temperature for 1 h. Afterward the membrane was washed again. The detection was performed via enhanced chemiluminescence (ECL Prime) after incubation for 5 min in the dark with a FUSION‐SL (PEQLAB) and the FUSION‐CAPT software. After detection of acetylated tubulin the whole procedure was repeated with the primary antibody antiacetyl‐histone H3 (Millipore 06.599, 1 : 2000) and the secondary antibody anti‐rabbit IgG‐HRP (Sigma‐Aldrich, 1 : 5000) to detect the acetylation of histone H3 and again with the primary antibody antiGAPDH (Sigma‐Aldrich 69545‐200UL, 1 : 5000) and the secondary antibody antirabbit IgG‐HRP (Sigma‐Aldrich, 1 : 10 000) to control the loading amount.
Cellular testing (fluorescence microscopic and flow cytometric analysis of LysoTracker® red staining)
Analysis of LysoTracker staining via confocal fluorescence microscopy or flow cytometry was performed after overnight treatment of SK‐N‐BE(2)‐C cells as described previously. [16b]
Computational methods
Molecular docking: The ligands and protein‐ligand complexes used herein were prepared using a similar method as reported in our previous published paper. [54a]
Ligand preparation: MOE (version 2014.09, Chemical Computing Group, Montreal, Canada) was used to generate the molecular structures of all compounds.
The ligands were prepared for docking using the LigPrep tool as implemented in Schrödinger's software (version 2018‐1), where all possible tautomeric forms, as well as stereoisomers, were generated. They were subsequently energy minimized using the integrated Optimized Potentials for Liquid Simulations (OPLS_2005) force field. A maximum of 64 conformers of prepared ligands were calculated with ConfGen using the default settings (Schrödinger's software version 2018–1).
Protein preparation: The crystal structures of HDAC10 (drHDAC10; PDB ID: 6UHU), HDAC1 (hsHDAC1; PDB ID: 5ICN), HDAC6 (hsHDAC6; PDB ID: 5EDU) and HDAC8 (hsHDAC8; PDB ID: 2V5X) were downloaded from the Protein Databank (PDB; www.rcsb.org) [64] with the exception of water molecules occupying the catalytic pockets that were used for the docking procedures. Further preparations of the protein structures were done using the Protein Preparation Wizard of Schrödinger's software (version 2018–1). Bond orders were assigned and hydrogen atoms added, and the hydrogen bond network was subsequently optimized. The protonation states at pH 7.0 were predicted using the Epik‐tool. The structures were finally subjected to a restrained energy minimization step (rmsd of the atom displacement for terminating the minimization was 0.3 Å) using the OPLS2005 force field.
Docking in HDAC6 with monodentate zinc coordination: The co‐crystallized ligand and three water molecules (HOH921, HOH999 and HOH1011) from PDB ID 5EF7 were retrieved and inserted into the herein used crystal structure of HDAC6 (PDB ID 5EDU) prior the protein preparation and minimization steps mentioned above.
Docking in HDAC6 with bidentate zinc chelation: three water molecules (HOH1015, HOH1006 and HOH1083) were retrieved from PDB ID 6CSQ were retrieved and inserted into the herein used crystal structure of HDAC6 (PDB ID 5EDU) prior the protein preparation and minimization steps mentioned above.
Docking to HDACs: A docking protocol using Glide (Schrödinger's software version 2018‐1) was developed and validated by redocking the co‐crystallized HDAC inhibitors with the corresponding crystal structure.
Docking studies were done using Glide. [65] The receptor grid preparation for the docking procedure was carried out by assigning the co‐crystallized ligand as the centroid of the grid box. The generated 3D conformers of the ligands (refer to Section 3.1.1) were docked into the receptor grid using Glide and the Standard Precision (SP) mode as the scoring function. A total of 20 poses per ligand conformer were included in the post‐docking minimization step, and a maximum of 2 docking poses was generated for each ligand conformer.
PAINS analysis: All tested compounds were checked for structural features which could interfere with the assay system. Screening against PAINS was performed by using PAINS‐Remover. [66] All compounds passed the filter.
Supporting information
Additional data on compound purity/identity by HPLC and NMR spectra, assay validation and IC50‐determinations, molecular modelling (including pdb‐files of docked complexes, molecular string data file.
Accession code
Refined atomic coordinates and structure factor amplitudes for the HDAC10‐48 a complex have been deposited in the Protein Data Bank with accession code 7U59.
Abbreviations
AMC, aminocoumarin; APCI, atmospheric pressure chemical ionization; BOP−Cl, Bis(2‐oxo‐3‐oxazolidinyl)phosphinic chloride; BRET, bioluminescence energy transfer; c, concentration; CH, cyclohexane; DAPI, 4′,6‐Diamidino‐2‐phenylindole; DCM, dichloromethane; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; EC50, half maximal effective concentration; EDTA, ethylenediaminetetraacetic acid; EE, ethyl acetate; EGTA, Ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid; ERRα, estrogen‐related receptor alpha; ESI, electrospray ionization; Et3N, triethylamine; Et3SiH, triethylsilane; eq, equivalent; FDA, Food and Drug Administration; FDL, Fluor de Lys; HDACi(s), histone deacetylase inhibitor(s); HDACs, histone deacetylases; (h)/(z)HDAC10, (human)/(zebrafish) histone deacetylase 10; HMBC, Heteronuclear Multiple Bond Correlation; HEPES, (4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid; HPLC, High‐performance liquid chromatography; HRMS, high resolution mass spectrometry; HSP90, heat shock protein 90; HSQC, Heteronuclear Single Quantum Coherence; IC50, half maximal inhibitory concentration; KDACs, lysine deacetylase(s); LC‐MS, liquid chromatography‐mass spectrometry; MeCN, acetonitrile; MeOH, methanol; min, minute; NAD+, Nicotinamide adenine dinucleotide; NDA, naphthalene‐2,3‐dialdehyde; NMR, nuclear magnetic resonance; p53, tumor suppressor p53; rpm, revolutions per minute; PBS, physiological buffer solution; PDAC, polyamine deacetylase; r.t., room temperature; Sirt, sirtuin; SMC3, structural maintenance of chromosomes protein 3; SmHDAC8, Schistosoma mansoni histone deacetylase 8; TCEP, tris(2‐carboxyethyl)phosphine; TFA, trifluoroacetic acid; THF, tetrahydrofuran; TLC, Thin Layer Chromatography; TR‐FRET, time resolved fluorescence energy transfer; TSA, Trichostatin A; UV detection, ultraviolet detection; ZMAL, Z‐(Ac)Lys‐AMC.
Author Contributions
Daniel Herp: Substrate synthesis, assay design and validation, in vitro testing of HDAC10, preparation of manuscript. Johannes Ridinger: Performed LysoTracker‐Assay (FACS and confocal microscopy). Dina Robaa: Computational methods, preparation of manuscript. Stephen A. Shinsky: Expression and purification of drHDAC10. Corey J. Herbst‐Gervasoni: Structure determination of the HDAC10‐48 a complex. Karin Schmidtkunz: In vitro testing of HDAC1, 6 and 8. Talha Z. Yesiloglu: Carried out the docking studies. Theresa Bayer: Synthesis of some of the HDAC inhibitors and provided material for biological testing. Raphael R. Steimbach: FRET and BRET assay testing Christophe Romier: Enzyme preparation, preparation of manuscript. Peter Sehr: Development of FRET‐HDAC10 assay and testing of selected inhibitors. Nikolas Gunkel: Coordination of FRET and BRET testing. Aubry K. Miller: Coordination of the FRET and BRET assays assay, preparation of manuscript. Ina Oehme: Supervision of cellular experiments, preparation of manuscript. David W. Christianson: Coordination of HDAC10 expression and crystal structure determination of the HDAC10‐48 a complex, preparation of manuscript. Wolfgang Sippl: Supervision of molecular modelling, preparation of manuscript. Manfred Jung: Design of study, preparation of manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the Deutsche Forschungsgesellschaft (DFG, M.J. (Ju295/13‐1), W.S. (Si868/22‐1 project number 469954457), I.O. (Oe542/2‐1) and INST 39/931‐1 for co‐financing of the NMR console) for funding. The work of I.O. and J.R. was supported by the H.W.&J. Hector foundation (Grant reference number: M91). Thanks to the National Institute of Health (NIH) for funding D.W.C. (GM49758) and S.A.S. (F32 GM125141). C.R. was supported by institutional funds from the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Université de Strasbourg. This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357. We thank A.‐T. Hauser for support in manuscript finalization. Open Access funding enabled and organized by Projekt DEAL.
D. Herp, J. Ridinger, D. Robaa, S. A. Shinsky, K. Schmidtkunz, T. Z. Yesiloglu, T. Bayer, R. R. Steimbach, C. J. Herbst-Gervasoni, A. Merz, C. Romier, P. Sehr, N. Gunkel, A. K. Miller, D. W. Christianson, I. Oehme, W. Sippl, M. Jung, ChemBioChem 2022, 23, e202200180.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐pqhtn‐v4).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Taunton J., Hassig C. A., Schreiber S. L., Science 1996, 272, 408–411; [DOI] [PubMed] [Google Scholar]
- 1b. Kuo M. H., Allis C. D., BioEssays 1998, 20, 615–626; [DOI] [PubMed] [Google Scholar]
- 1c. Leipe D. D., Landsman D., Nucleic Acids Res. 1997, 25, 3693–3697; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Yang W. M., Inouye C., Zeng Y. Y., Bearss D., Seto E., Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luo J., Su F., Chen D., Shiloh A., Gu W., Nature 2000, 408, 377–381. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P., Nature 2002, 417, 455–458; [DOI] [PubMed] [Google Scholar]
- 3b. Chakrabarti A., Melesina J., Kolbinger F. R., Oehme I., Senger J., Witt O., Sippl W., Jung M., Future Med. Chem. 2016, 8, 1609–1634; [DOI] [PubMed] [Google Scholar]
- 3c. Kovacs J. J., Murphy P. J., Gaillard S., Zhao X., Wu J. T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B., Yao T. P., Mol. Cell 2005, 18, 601–607; [DOI] [PubMed] [Google Scholar]
- 3d. Wilson B. J., Tremblay A. M., Deblois G., Sylvain-Drolet G., Giguere V., Mol. Endocrinol. 2010, 24, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregoretti I. V., Lee Y. M., Goodson H. V., J. Mol. Biol. 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Kutil Z., Novakova Z., Meleshin M., Mikesova J., Schutkowski M., Barinka C., ACS Chem. Biol. 2018, 13, 685–693; [DOI] [PubMed] [Google Scholar]
- 5b. Moreno-Yruela C., Galleano I., Madsen A. S., Olsen C. A., Cell Chem. Biol. 2018, 25, 849–856. [DOI] [PubMed] [Google Scholar]
- 6. Hai Y., Shinsky S. A., Porter N. J., Christianson D. W., Nat. Commun. 2017, 8, 15386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., Shaw R. J., Cell 2011, 145, 607–621; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Qian H., Chen Y. Y., Nian Z. Q., Su L., Yu H. Y., Chen F. J., Zhang X. Q., Xu W. Y., Zhou L. K., Liu J. M., Yu J. H., Yu L. X., Gao Y., Zhang H. C., Zhang H. H., Zhao S. M., Yu L., Xiao R. P., Bao Y. Q., Hou S. C., Li P. P., Li J. D., Deng H. T., Jia W. P., Li P., J. Clin. Invest. 2017, 127, 1353–1369; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Song S. Y., Wen Y. F., Tong H., Loro E., Gong Y. Y., Liu J. D., Hong S. G., Li L., Khurana T. S., Chu M. P., Sun Z., J. Mol. Cell Biol. 2019, 11, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sen P., Shah P. P., Nativio R., Berger S. L., Cell 2016, 166, 822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Gallinari P., Di Marco S., Jones P., Pallaoro M., Steinkuhler C., Cell Res. 2007, 17, 195–211; [DOI] [PubMed] [Google Scholar]
- 9b. Lai I. L., Lin T. P., Yao Y. L., Lin C. Y., Hsieh M. J., Yang W. M., J. Biol. Chem. 2010, 285, 7187–7196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K., Cell 2009, 138, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Kotian S., Liyanarachchi S., Zelent A., Parvin J. D., J. Biol. Chem. 2011, 286, 7722–7726; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Robert T., Vanoli F., Chiolo I., Shubassi G., Bernstein K. A., Rothstein R., Botrugno O. A., Parazzoli D., Oldani A., Minucci S., Foiani M., Nature 2011, 471, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Zhang R. H., Lu J. Y., Kong X. Q., Jin L., Luo C., Cns Neurol. Disord. Dr. 2013, 12, 126–141; [DOI] [PubMed] [Google Scholar]
- 11b. Graff J., Rei D., Guan J. S., Wang W. Y., Seo J., Hennig K. M., Nieland T. J. F., Fass D. M., Kao P. F., Kahn M., Su S. C., Samiei A., Joseph N., Haggarty S. J., Delalle I., Tsai L. H., Nature 2012, 483, 222–U123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Miao F., Gonzalo I. G., Lanting L., Natarajan R., J. Biol. Chem. 2004, 279, 18091–18097; [DOI] [PubMed] [Google Scholar]
- 12b. Sun L., de Evsikova C. M., Bian K., Achille A., Telles E., Pei H. D., Seto E., Ebiomedicine 2018, 33, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Oehme I., Lodrini M., Brady N. R., Witt O., Autophagy 2013, 9, 2163–2165; [DOI] [PubMed] [Google Scholar]
- 13b. Nakagawa M., Oda Y., Eguchi T., Aishima S., Yao T., Hosoi F., Basaki Y., Ono M., Kuwano M., Tanaka M., Tsuneyoshi M., Oncol. Rep. 2007, 18, 769–774; [PubMed] [Google Scholar]
- 13c. Wilson A. J., Byun D. S., Popova N., Murray L. B., L′Italien K., Sowa Y., Arango D., Velcich A., Augenlicht L. H., Mariadason J. M., J. Biol. Chem. 2006, 281, 13548–13558. [DOI] [PubMed] [Google Scholar]
- 14. Herbst-Gervasoni C. J., Christianson D. W., Biochemistry 2021, 60, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Song C. L., Zhu S. C., Wu C. Y., Kang J. H., J. Biol. Chem. 2013, 288, 28021–28033; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Wu J., Du C., Lv Z., Ding C., Cheng J., Xie H., Zhou L., Zheng S., Dig. Dis. Sci. 2013, 58, 3545–3553; [DOI] [PubMed] [Google Scholar]
- 15c. Duan B. Y., Ye D., Zhu S. C., Jia W. W., Lu C. Q., Wang G. Y., Guo X. D., Yu Y. Y., Wu C. Y., Kang J. H., Oncotarget 2017, 8, 61338–61349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Oehme I., Linke J. P., Bock B. C., Milde T., Lodrini M., Hartenstein B., Wiegand I., Eckert C., Roth W., Kool M., Kaden S., Grone H. J., Schulte J. H., Lindner S., Hamacher-Brady A., Brady N. R., Deubzer H. E., Witt O., Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Ridinger J., Koeneke E., Kolbinger F. R., Koerholz K., Mahboobi S., Hellweg L., Gunkel N., Miller A. K., Peterziel H., Schmezer P., Hamacher-Brady A., Witt O., Oehme I., Sci. Rep. 2018, 8, 10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Yang Y. W., Huang Y. T., Wang Z. T., Wang H. T., Duan B. Y., Ye D., Wang C. X., Jing R. Q., Leng Y., Xi J. J., Chen W., Wang G. Y., Jia W. W., Zhu S. C., Kang J. H., Oncotarget 2016, 7, 59388–59401; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Shan C. L., Lu Z. L., Li Z., Sheng H., Fan J., Qi Q., Liu S. P., Zhang S., Cell Death Dis. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Islam M. M., Banerjee T., Packard C. Z., Kotian S., Selvendiran K., Cohn D. E., Parvin J. D., Gynecol. Oncol. 2017, 144, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeyen P., Zeyn Y., Herp D., Mahmoudi F., Yesiloglu T., Erdmann F., Schmidt M., Robaa D., Romier C., Ridinger J., Christianson D. W., Oehme I., Jung M., Kraemer H., Sippl W., Eur. J. Med. Chem. 2022, 234, 114272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Yoshida M., Kijima M., Akita M., Beppu T., J. Biol. Chem. 1990, 265, 17174–17179; [PubMed] [Google Scholar]
- 20b. Kijima M., Yoshida M., Sugita K., Horinouchi S., Beppu T., J. Biol. Chem. 1993, 268, 22429–22435. [PubMed] [Google Scholar]
- 21.
- 21a. Duvic M., Vu J., Expert Opin. Invest. Drugs 2007, 16, 1111–1120; [DOI] [PubMed] [Google Scholar]
- 21b. Furumai R., Matsuyama A., Kobashi N., Lee K. H., Nishiyama N., Nakajima I., Tanaka A., Komatsu Y., Nishino N., Yoshida M., Horinouchi S., Cancer Res. 2002, 62, 4916–4921; [PubMed] [Google Scholar]
- 21c. Lee H. Z., Kwitkowski V. E., Del Valle P. L., Ricci M. S., Saber H., Habtemariam B. A., Bullock J., Bloomquist E., Li Shen Y., Chen X. H., Brown J., Mehrotra N., Dorff S., Charlab R., Kane R. C., Kaminskas E., Justice R., Farrell A. T., Pazdur R., Clin. Cancer Res. 2015, 21, 2666–2670; [DOI] [PubMed] [Google Scholar]
- 21d. Raedler L. A., Am. Health Drug Benefits 2016, 9, 84–87; [PMC free article] [PubMed] [Google Scholar]
- 21e. Richon V. M., Emiliani S., Verdin E., Webb Y., Breslow R., Rifkind R. A., Marks P. A., Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu X., Ning Z., Li Z., Cao H., Wang X., Intractable Rare Dis. Res. 2016, 5, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Tjulandin S., Fedyanin M., Vladimirov V. I., Kostorov V., Lisyanskaya A. S., Krikunova L., Cakana A., Azarova V., Karavaeva O., Vostokova N., Baranovsky S., J. Clin. Oncol. 2017, 35; [Google Scholar]
- 23b. Aggarwal R. R., Thomas S., Hauke R. J., Nordquist L. T., Munster P. N., J. Clin. Oncol. 2019, 37; [Google Scholar]
- 23c. Batlevi C. L., Crump M., Andreadis C., Rizzieri D., Assouline S. E., Fox S., van der Jagt R. H. C., Copeland A., Potvin D., Chao R., Younes A., Br. J. Haematol. 2017, 178, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balasubramanian S., Verner E., Buggy J. J., Cancer Lett. 2009, 280, 211–221. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Marek L., Hamacher A., Hansen F. K., Kuna K., Gohlke H., Kassack M. U., Kurz T., J. Med. Chem. 2013, 56, 427–436; [DOI] [PubMed] [Google Scholar]
- 25b. Martin M. W., Lee J. Y., Lancia D. R., Ng P. Y., Han B. S., Thomason J. R., Lynes M. S., Marshall C. G., Conti C., Collis A., Morales M. A., Doshi K., Rudnitskaya A., Yao L. L., Zheng X. Z., Bioorg. Med. Chem. Lett. 2018, 28, 2143–2147; [DOI] [PubMed] [Google Scholar]
- 25c. Zhang Y., Yan J., Yao T. P., Eur. J. Med. Chem. 2017, 141, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roche J., Bertrand P., Eur. J. Med. Chem. 2016, 121, 451–483. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Wang Y., Stowe R. L., Pinello C. E., Tian G., Madoux F., Li D., Zhao L. Y., Li J. L., Wang Y., Wang Y., Ma H., Hodder P., Roush W. R., Liao D., Chem. Biol. 2015, 22, 273–284; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. McClure J. J., Zhang C., Inks E. S., Peterson Y. K., Li J., Chou C. J., J. Med. Chem. 2016, 59, 9942–9959; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Jiang Y., Xu J., Yue K., Huang C., Qin M., Chi D., Yu Q., Zhu Y., Hou X., Xu T., Li M., Chou C. J., Li X., J. Med. Chem. 2022, 65, 285–302; [DOI] [PubMed] [Google Scholar]
- 27d. Fruhauf A., Meyer-Almes F. J., Molecules 2021, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hai Y., Christianson D. W., Nat. Chem. Biol. 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Li J., Staver M. J., Curtin M. L., Holms J. H., Frey R. R., Edalji R., Smith R., Michaelides M. R., Davidsen S. K., Glaser K. B., Life Sci. 2004, 74, 2693–2705; [DOI] [PubMed] [Google Scholar]
- 29b. Jones P., Altamura S., De Francesco R., Gallinari P., Lahm A., Neddermann P., Rowley M., Serafini S., Steinkuhler C., Bioorg. Med. Chem. Lett. 2008, 18, 1814–1819. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Marks B. D., Fakhoury S. A., Frazee W. J., Eliason H. C., Riddle S. M., J. Biomol. Screening 2011, 16, 1247–1253; [DOI] [PubMed] [Google Scholar]
- 30b. Robers M. B., Dart M. L., Woodroofe C. C., Zimprich C. A., Kirkland T. A., Machleidt T., Kupcho K. R., Levin S., Hartnett J. R., Zimmerman K., Niles A. L., Ohana R. F., Daniels D. L., Slater M., Wood M. G., Cong M., Cheng Y. Q., Wood K. V., Nat. Commun. 2015, 6, 10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Vreese R., D′Hooghe M., Eur. J. Med. Chem. 2017, 135, 174–195. [DOI] [PubMed] [Google Scholar]
- 32. Geraldy M., Morgen M., Sehr P., Steimbach R. R., Moi D., Ridinger J., Oehme I., Witt O., Malz M., Nogueira M. S., Koch O., Gunkel N., Miller A. K., J. Med. Chem. 2019, 62, 4426–4443. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Uba A. I., Yelekci K., J. Biomol. Struct. Dyn. 2020, 38„ 4397–4406; [DOI] [PubMed] [Google Scholar]
- 33b. I. Uba A., Yelekci K., J. Biomol. Struct. Dyn. 2019, 37, 3627–3636. [DOI] [PubMed] [Google Scholar]
- 34. Herbst-Gervasoni C. J., Christianson D. W., Biochemistry 2019, 58, 4957–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heltweg B., Jung M., J. Biomol. Screening 2003, 8, 89–95. [DOI] [PubMed] [Google Scholar]
- 36. Demontigny P., Stobaugh J. F., Givens R. S., Carlson R. G., Srinivasachar K., Sternson L. A., Higuchi T., Anal. Chem. 1987, 59, 1096–1101. [Google Scholar]
- 37. Zhang J. H., Chung T. D. Y., Oldenburg K. R., J. Biomol. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Heltweg B., Dequiedt F., Marshall B. L., Brauch C., Yoshida M., Nishino N., Verdin E., Jung M., J. Med. Chem. 2004, 47, 5235–5243; [DOI] [PubMed] [Google Scholar]
- 38b. Riester D., Wegener D., Hildmann C., Schwienhorst A., Biochem. Biophys. Res. Commun. 2004, 324, 1116–1123. [DOI] [PubMed] [Google Scholar]
- 39. Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A. M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Dumpelfeld B., Eberhard D., Huthmacher C., Mathieson T., Poeckel D., Reader V., Strunk K., Sweetman G., Kruse U., Neubauer G., Ramsden N. G., Drewes G., Nat. Biotechnol. 2011, 29, 255–265. [DOI] [PubMed] [Google Scholar]
- 40. Wagner F. F., Olson D. E., Gale J. P., Kaya T., Weiwer M., Aidoud N., Thomas M., Davoine E. L., Lemercier B. C., Zhang Y. L., Holson E. B., J. Med. Chem. 2013, 56, 1772–1776. [DOI] [PubMed] [Google Scholar]
- 41. Balasubramanian S., Ramos J., Luo W., Sirisawad M., Verner E., Buggy J. J., Leukemia 2008, 22, 1026–1034. [DOI] [PubMed] [Google Scholar]
- 42. Fournel M., Bonfils C., Hou Y., Yan P. T., Trachy-Bourget M. C., Kalita A., Liu J., Lu A. H., Zhou N. Z., Robert M. F., Gillespie J., Wang J. J., Ste-Croix H., Rahil J., Lefebvre S., Moradei O., Delorme D., MacLeod A. R., Besterman J. M., Li Z. M., Mol. Cancer Ther. 2008, 7, 759–768. [DOI] [PubMed] [Google Scholar]
- 43. Bayer T., Chakrabarti A., Lancelot J., Shaik T. B., Hausmann K., Melesina J., Schmidtkunz K., Marek M., Erdmann F., Schmidt M., Robaa D., Romier C., Pierce R. J., Jung M., Sippl W., ChemMedChem 2018, 13, 1517–1529. [DOI] [PubMed] [Google Scholar]
- 44. Heimburg T., Kolbinger F. R., Zeyen P., Ghazy E., Herp D., Schmidtkunz K., Melesina J., Shaik T. B., Erdmann F., Schmidt M., Romier C., Robaa D., Witt O., Oehme I., Jung M., Sippl W., J. Med. Chem. 2017, 60, 10188–10204. [DOI] [PubMed] [Google Scholar]
- 45. Senger J., Melesina J., Marek M., Romier C., Oehme I., Witt O., Sippl W., Jung M., J. Med. Chem. 2016, 59, 1545–1555. [DOI] [PubMed] [Google Scholar]
- 46. Schaefer S., Saunders L., Schlimme S., Valkov V., Wagner J. M., Kratz F., Sippl W., Verdin E., Jung M., ChemMedChem 2009, 4, 283–290. [DOI] [PubMed] [Google Scholar]
- 47. Jung M., Brosch G., Kolle D., Scherf H., Gerhauser C., Loidl P., J. Med. Chem. 1999, 42, 4669–4679. [DOI] [PubMed] [Google Scholar]
- 48. Uesato S., Kitagawa M., Nagaoka Y., Maeda T., Kuwajima H., Yamori T., Bioorg. Med. Chem. Lett. 2002, 12, 1347–1349. [DOI] [PubMed] [Google Scholar]
- 49. Watson P. J., Millard C. J., Riley A. M., Robertson N. S., Wright L. C., Godage H. Y., Cowley S. M., Jamieson A. G., Potter B. V., Schwabe J. W., Nat. Commun. 2016, 7, 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vannini A., Volpari C., Gallinari P., Jones P., Mattu M., Carfi A., De Francesco R., Steinkuhler C., Di Marco S., EMBO Rep. 2007, 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.
- 51a. Miyake Y., Keusch J. J., Wang L., Saito M., Hess D., Wang X., Melancon B. J., Helquist P., Gut H., Matthias P., Nat. Chem. Biol. 2016, 12, 748–754; [DOI] [PubMed] [Google Scholar]
- 51b. Porter N. J., Osko J. D., Diedrich D., Kurz T., Hooker J. M., Hansen F. K., Christianson D. W., J. Med. Chem. 2018, 61, 8054–8060; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51c. Osko J. D., Porter N. J., Narayana Reddy P. A., Xiao Y. C., Rokka J., Jung M., Hooker J. M., Salvino J. M., Christianson D. W., J. Med. Chem. 2020, 63, 295–308; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51d. Porter N. J., Mahendran A., Breslow R., Christianson D. W., Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen S., Svoboda M., Zhang G., Cavasin M. A., Motlova L., McKinsey T. A., Eubanks J. H., Bařinka C., Kozikowski A. P., ACS Med. Chem. Lett. 2020, 11, 706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.
- 53a. Porter N. J., Wagner F. F., Christianson D. W., Biochemistry 2018, 57, 3916–3924; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53b. Osko J. D., Christianson D. W., Bioorg. Med. Chem. Lett. 2020, 30, 127023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.
- 54a. Heimburg T., Chakrabarti A., Lancelot J., Marek M., Melesina J., Hauser A. T., Shaik T. B., Duclaud S., Robaa D., Erdmann F., Schmidt M., Romier C., Pierce R. J., Jung M., Sippl W., J. Med. Chem. 2016, 59, 2423–2435; [DOI] [PubMed] [Google Scholar]
- 54b. Marek M., Shaik T. B., Heimburg T., Chakrabarti A., Lancelot J., Ramos-Morales E., Da Veiga C., Kalinin D., Melesina J., Robaa D., Schmidtkunz K., Suzuki T., Holl R., Ennifar E., Pierce R. J., Jung M., Sippl W., Romier C., J. Med. Chem. 2018, 61, 10000–10016. [DOI] [PubMed] [Google Scholar]
- 55. Herbst-Gervasoni C. J., Steimbach R. R., Morgen M., Miller A. K., Christianson D. W., ACS Chem. Biol. 2020, 15, 2154–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Butler K. V., Kalin J., Brochier C., Vistoli G., Langley B., Kozikowski A. P., J. Am. Chem. Soc. 2010, 132, 10842–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Steimbach R. R., Herbst-Gervasoni C. J., Klinke G., Géraldy M., Tihanyi G., Ridinger J., Poschet G., Oehme I., Christianson D. W., Gunkel N., Miller A. K., ChemRxiv. 2021. 10.26434/chemrxiv-2021-37shs-v2. [Google Scholar]
- 58. Heltweg B., Trapp J., Jung M., Methods 2005, 36, 332–337. [DOI] [PubMed] [Google Scholar]
- 59. Marek M., Kannan S., Hauser A. T., Moraes Mourao M., Caby S., Cura V., Stolfa D. A., Schmidtkunz K., Lancelot J., Andrade L., Renaud J. P., Oliveira G., Sippl W., Jung M., Cavarelli J., Pierce R. J., Romier C., PLoS Pathog. 2013, 9, e1003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.
- 60a. Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W., Acta Crystallogr. Sect. D 2011, 67, 271–281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60b. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235–242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60c. Evans P. R., Murshudov G. N., Acta Crystallogr. Sect. D 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.
- 61a. Mccoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61b. Evans P., Mccoy A., Acta Crystallogr. Sect. D 2008, 64, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.
- 62a. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62b. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.
- 63a. Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63b. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B., Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., Richardson D. C., Protein Sci. 2018, 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burley S. K., Berman H. M., Christie C., Duarte J. M., Feng Z., Westbrook J., Young J., Zardecki C., Protein Sci. 2018, 27, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.
- 65a. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749; [DOI] [PubMed] [Google Scholar]
- 65b. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 66. Baell J. B., Holloway G. A., J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.