

SUMMARY
This study underlines the importance of treadmill exercise in reducing α-synuclein (α-syn) spreading in the A53T brain and protecting nigral dopaminergic neurons. Preformed α-syn fibril (PFF) seeding in the internal capsule of young A53T α-syn mice leads to increased spreading of α-syn to substantia nigra and motor cortex and concomitant loss of nigral dopaminergic neurons. However, regular treadmill exercise decreases α-syn spreading in the brain and protects nigral dopaminergic neurons in PFF-seeded mice. Accordingly, treadmill exercise also mitigates α-synucleinopathy in aged A53T mice. While investigating this mechanism, we have observed that treadmill exercise induces the activation of peroxisome proliferator-activated receptor α (PPARα) in the brain to stimulate lysosomal biogenesis via TFEB. Accordingly, treadmill exercise remains unable to stimulate TFEB and reduce α-synucleinopathy in A53T mice lacking PPARα, and fenofibrate, a prototype PPARα agonist, reduces α-synucleinopathy. These results delineate a beneficial function of treadmill exercise in reducing α-syn spreading in the brain via PPARα.
Graphical Abstract
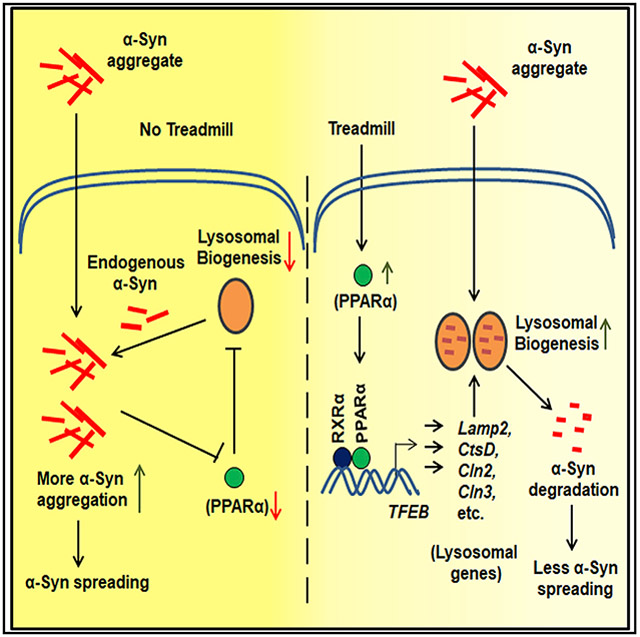
In brief
Dutta et al. demonstrate that regular treadmill exercise is capable of decreasing α-synuclein spreading in the brain and protecting nigral dopaminergic neurons in mice. However, treadmill running does not reduce α-synucleinopathy in mice lacking peroxisome proliferator-activated receptor α (PPARα), highlighting a role of PPARα in the beneficial effect of treadmill running.
INTRODUCTION
Accumulation of α-synuclein (α-syn), the major component of Lewy bodies (LBs) and glial cytoplasmic inclusions (GCIs), in multiple brain regions is the hallmark in all forms of α-synucleinopathies including Parkinson’s disease (PD), LB dementia (LBD), and multiple-system atrophy (MSA) (Goedert and Spillantini, 1998; Spillantini et al., 1997; Wakabayashi et al., 1998). Normal α-syn is required for vesicle trafficking and dopamine (DA) neurotransmission at the nerve terminals. However, overexpression of wild-type α-syn or the presence of mutated α-syn results in the generation of a higher molecular weight oligomeric form of the protein, which can evade the cellular degradation machineries, eventually causing neuronal death (Hofer et al., 2005; Mueller et al., 2005). Therefore, compromised cellular degradation machineries such as autophagy and lysosomal functions are important for α-syn aggregation in the brain (Bellomo et al., 2020; Lee et al., 2013). Eventually, neuron-secreted forms of α-syn progressively spread in selected brain regions of the CNS depending on the disease (Beach et al., 2009; Braak et al., 2003; Kantarci et al., 2010; Wenning and Jellinger, 2005). This prion-like propagation of α-syn has also been recapitulated in animals by multiple studies (Mao et al., 2016; Rey et al., 2019; Sorrentino et al., 2017). However, until now, no effective therapy has been available to prevent α-syn spreading in human brains of α-synucleinopathies.
Regular exercise has multiple benefits on human health. Since treadmills are easily accessible, it has been suggested that treadmill exercise may be an effective form of rehabilitation in patients with different neurological conditions for improving mobility. Accordingly, treadmill exercise is also currently being used by different experimental studies to improve locomotor activities in patients with PD (Jang et al., 2018; Minakaki et al., 2019). Here, we demonstrated that regular treadmill exercise not only reduced localized α-syn accumulation in brains of genetic A53T mice model but also inhibited the spreading of pathological phosphoserine 129 α-syn (pSyn129) in multiple brain regions of preformed α-syn fibril (PFF)-seeded A53T animals. It was also accompanied with protection of nigral dopaminergic (DAergic) neurons, normalization of striatal neurotransmitters, and attenuation of parkinsonian features. Furthermore, treadmill-mediated alleviation of α-syn spreading and accumulation was dependent on the activation of peroxisome proliferator and activated receptor α (PPARα) and consequent up-regulation of lysosomal biogenesis, as treadmill exercise remained unable to alleviate α-synucleinopathy in A53T mice lacking PPARα, and fenofibrate, a known agonist of PPARα, reduced α-synucleinopathy. These results suggest that treadmill exercise and/or FDA-approved lipid-lowering drug fenofibrate may be used for reducing α-syn spreading in PD, LBD, and MSA.
RESULTS
Treadmill exercise inhibits α-syn spreading in substantia nigra (SN) and cortex of PFF-seeded mice brain
Fibrils and PFF were validated by electron microscopy (EM), which showed a large fibril structure of the peptide formed by aggregation of full-length human α-syn monomers and the PFFs generated after sonication of fibrils (Figure 1A). The PFF was microinjected in the internal capsule (IC) region of 2-month-old A53T animals (Figure 1B), and following 2 months of surgery, mice were subjected to regular treadmill exercise for another 2 months (Figure 1C). The IC region of the forebrain contains corticospinal connections through which α-syn spreading was also found to happen in the motor cortex, rostral striatum, and caudal SN region following PFF injection at the IC (Sorrentino et al., 2017). Therefore, at first, the SN region was assessed for α-syn and pSyn129, respectively. Data revealed that PFF-injected A53T animals, which were not subjected to exercise (A53T + PFF NR), had significantly higher level of pSyn129 in the SN than the PBS-injected group, and it showed both somatic as well as neuritic pathology. However, in mice undergoing treadmill exercise (A53T + PFF R), pSyn129 content was significantly reduced in the SN, as shown by relative optical density analysis of pSyn129 in nigral sections (Figures 1D and 1E). The finding was further confirmed by immunoblot analysis, in which PFF-seeded mice had higher levels of total α-syn compared with the PBS-injected group. Treadmill exercise did not lower the detergent soluble content of α-syn, but the α-syn level in the detergent-insoluble fraction was significantly reduced (Figures 1F-1I). Moreover, the level of total α-syn was monitored in tyrosine hydroxylase (TH)-positive neurons in the SN by immunostaining. A53T + PFF NR mice exhibited more α-syn deposition in nigral TH neurons. A remarkable decrease of α-syn neuronal intensity in TH neurons of PFF-seeded animals was found following treadmill exercise in the A53T + PFF R group (Figures S1A and S1B). Furthermore, staining of α-syn in nigral sections following proteinase K (PK) digestion demonstrated a significant decrease in α-syn aggregates in PFF-seeded R mice compared with the NR group (Figures S1C and S1D). Reduction of PK-resistant α-syn species in the SN (Figures S1C and S1D) signifies that an insoluble and structurally complex form of α-syn is lowered by treadmill exercise, and this substantiates the finding obtained from western blot analysis (Figures 1F-1I). Next, we sought to monitor pSyn129 spreading in the rostral motor cortex and dorsal striatum region, where visible spreading and accumulation of pSyn129 were observed in the PFF-seeded NR mice. Parallel to the SN region, the level of pSyn129 was also significantly reduced in motor cortex as well as striatum following treadmill exercise (Figures 1J, 1K, S1E, and S1F).
Figure 1. Treadmill inhibits α-syn spreading in A53T mice brain.

(A) Preformed α-syn fibril (PFF) that was validated by electron microscopy.
(B and C) PFF was bilaterally injected into the internal capsule (IC) of 2-month-old A53T mice (B), and following 2 months of PFF injection, animals were run in a treadmill for the next 2 months (C).
(D–I) Spreading of α-syn in brains of PFF-seeded non-running (A53T + PFF NR) and running (A53T + PFF R) mice was evaluated by phosphoserine 129 (pSyn129) staining in midbrain sections (F3,36 = 166.2, p < 0.001, D and E) and immunoblotting of total Triton X-100 soluble and insoluble α-syn in the SN (F3,16 = 32.18, p < 0.01, F–I).
(J and K) Propagation of α-syn to motor cortex was evaluated by pSyn129 immunostaining followed by quantification (F3, 36 = 102.4, p < 0.01).
For immunostaining, at least two sections from each brain per group were stained, and 10–15 pSyn129-positive cells were quantified from each section by Fiji software. Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests. *p < 0.05, **p < 0.01, ***p < 0.001 indicate significance compared with respective groups; ns, not significant. Data are represented as mean ± SEM (n = 5 animals per each group).
Treadmill exercise protects nigral DAergic neurons and reduces parkinsonian pathology in PFF-seeded mice
Potential spreading of pathological α-syn resulted in an almost 40% loss of nigral DAergic neurons in PFF-seeded NR mice (Figures 2A and 2B), and the loss of neurons is corroborated with diminished TH protein levels in nigral tissues (Figures 2C and 2D). However, DAergic neurons, as well as the TH level, were markedly protected in PFF-seeded A53T R mice. In parallel to neuronal demise in the SN, PFF-injected NR mice exhibited a significant loss of DAergic terminals and TH protein levels in the striatum (Figures 2E-2G), resulting in the depletion of striatal DA and its metabolites, 3,4-dihyroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA) (Figures 2H-2J). Concomitant with nigral neuronal protection, regular treadmill exercise also prevented loss of DAergic terminals as well as the DA and its metabolites’ levels in striatum. Finally, significant motor behavioral impairment was evidenced by PFF-seeded A53T NR animals as demonstrated by reduced distance moved (Figures 2K and 2L), velocity (Figure 2M), and cumulative duration (Figure 2N) of moving in an open-field test and impaired feet movement in rotarod test (Figure 2O). The mice undergoing treadmill overcame the movement deficits and exhibited improved locomotor activities in both open-field and rotarod tests.
Figure 2. Attenuation of parkinsonian features by treadmill in PFF-seeded A53T mice.

(A–G) PFF-induced parkinsonian pathology was determined by tyrosine hydroxylase (TH) staining in nigral sections (A), stereological counting of TH-positive neurons (F3, 16 = 29.97, p < 0.01, B), total TH level in SN (F3, 16 = 13.67, p < 0.05, C and D), TH fiber staining (E), and total TH level (F3, 16 = 28.37, p < 0.01, F and G) in striatum of different groups of animals.
(H–J) Dopamine (DA) and its metabolites, 3,4-dihyroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA), were measured from striatal tissues by high-performance liquid chromatography electrochemical detection (HPLC-ECD; F3, 14 = 16.18, p < 0.05 for DA).
(M–Q) Behavioral deficit in PFF-seeded animals was monitored by open-field locomotor test (F3, 16 = 8.04, p < 0.01 for distance, F3, 16 = 8.04, p < 0.01 for velocity, and F3, 16 = 7.602, p < 0.05 for cumulative duration, M–P) and rotarod test (F3, 16 = 14.49, p < 0.01, Q).
Data were statistically analyzed by unpaired two-tailed t test for comparison between two samples and by one-way ANOVA followed by Tukey’s multiple comparison tests. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. Values are represented as mean ± SEM (n = 5 animals per each group).
Treadmill exercise also reduces α-syn aggregation in the brain of aged A53T mice
Next, we have evaluated the effect of treadmill exercise in the genetic and more progressive model of α-synucleinopathy. The A53T mice are known to develop significant localized neuron-specific α-syn aggregation in several brain regions and motor impairment starting from 7 to 8 months of age (Giasson et al., 2002). Therefore, aged A53T animals (8 months old) were subjected to treadmill exercise (Figure 1C). The data exhibited no change of α-syn in Triton X-100 soluble fraction isolated from midbrain (Figures 3A and 3C), but a significant reduction of Triton X-100 insoluble pathogenic forms of α-syn was observed in A53T R mice (Figures 3B and 3D). The data obtained from immunoblotting was further strengthened by immunostaining of α-syn in TH-positive DAergic neurons of the SN (Figure 3E). The area fraction (Figure 3F), average size (Figure 3G), and integrated density (Figure 3H) of α-syn aggregates in these neurons were found to be remarkably decreased following exercise. Consequently, the pathogenic pSyn129 level and intensity were also significantly down-regulated in the SN of A53T R mice brains (Figures 3I and 3J). To further corroborate the finding, α-syn staining coupled with PK digestion was performed in nigral sections. The results displayed a marked loss of α-syn mean fluorescence intensity (MFI) following PK treatment in both A53T NR and R animals. However, the MFI of α-syn aggregates in A53T R mouse brain sections was significantly less than that of the A53T NR, indicating that treadmill exercise caused a loss in structural rigidity of α-syn aggregates in the brain (Figures S2A and S2B). Other major brain regions including hippocampus and brain stem were analyzed for monitoring the content of pSyn129 (Figures S2C and S2F). The aged A53T mice showed significantly higher α-syn levels in hippocampus and brainstem compared with the non-transgenic (nTg) mice. Similar to the SN, the total α-syn level was also decreased in both the regions by exercise (Figures S2D, S2E, S2G, and S2H). Reduction of aggregated α-syn was accompanied with better behavioral performance of A53T R mice, as shown by locomotor tests and cognitive performance. To monitor movement abilities such as feet movement and forepaw used by experimental animals, rotarod analysis and rearing tests in the open-field arena were performed, respectively. The A53T NR group, at the age of 10 months, exhibited compromised movement compared with the age-matched nTg mice. However, regular exercise significantly improved the motor performance of these mice (Figures 3K and 3L). Importantly, spatial cognitive function also improved in A53T mice following treadmill exercise. A53T NR mice took more time and made more non-target visits prior to finding the goal box in Barnes maze compared with the nTg animals. A53T R mice performed better in Barnes maze than A53T NR mice, indicating the restoration of spatial memory following treadmill exercise (Figures 3M-3O). This also correlates well with α-syn reduction in hippocampus, as the depletion of total α-syn or pSyn129 levels in hippocampus might result in better cognitive function of A53T R mice.
Figure 3. Treadmill reduces α-synucleinopathy in aged A53T mice brain.

(A–H) Effect of regular treadmill on α-syn aggregates in aged (10 months old) A53T NR and R mice brain was demonstrated by assessing total Triton X-100 soluble and insoluble α-syn in midbrain tissues (F2, 11 = 29.44, p < 0.05 for insoluble α-syn, A–D, n = 5) and by double immunostaining of total α-syn in TH-positive neurons of SN coupled with quantification of size, area, and integrated density of α-syn intraneuronal puncta (**p < 0.01, E–H, n = 6). Two sections from each brain per group were used for staining purposes, where α-syn puncta was quantified in at least 10 TH-positive cells from each section.
(I and J) Level of pathological pSyn129 in nigral sections were shown by immunostaining followed by optical density measurement (***p < 0.001, n = 4).
(K and L) Effect of treadmill exercise on motor behavior was monitored by rotarod test and by counting the number of rearing in the open-field test (F2, 15 = 6.983, p < 0.05 for rotarod, F2, 15 = 24.58, p < 0.01 for rearing, n = 6).
(M–O) Spatial cognitive function of experimental animals was done by Barnes maze, where the latency time to reach goal box and number of errors made by each animal before reaching goal box were counted (F2, 12 = 12.35, p < 0.05, n = 5).
Statistical analyses were conducted by unpaired two-tailed t test for comparison between two samples and by one-way ANOVA followed by Tukey’s multiple comparison tests. *p < 0.05, **p < 0.01, ***p < 0.001. Data are represented as mean ± SEM.
PPARα is up-regulated in vivo in the brain following treadmill exercise
Reduction of α-syn aggregates in neurons by treadmill exercise indicated better clearance of these protein aggregates by the protein-degradation machineries. It is well manifested that the autophagy and lysosomal protein-degradation pathway is one of the prime mechanisms behind degradation of misfolded protein aggregates, and this pathway is reported to be severely affected in aged brains of A53T animals (Bellomo et al., 2020; Poehler et al., 2014). Therefore, to find out the core mechanism behind treadmill-exercise-induced reduction of α-syn, we primarily sought to monitor the level of different isoforms of PPAR proteins in brains of A53T animals due to their multifaceted roles covering fatty acid metabolism, inflammation, lysosomal biogenesis, and autophagy (Ghosh et al., 2012, 2015; Ghosh and Pahan, 2016; Roy and Pahan, 2009). It led us to carry out an in-depth investigation on the status of PPAR isoforms in the brain of A53T animals. Firstly, we started monitoring PPARα levels in different A53T brains, as this isoform has previously been shown to be intricately involved in up-regulating autophagy and lysosomal genes and the degradation of protein aggregates (Chandra et al., 2018; Ghosh et al., 2015; Raha et al., 2021b). Double immunostaining demonstrated the loss of PPARα isoform in TH-positive neurons of the SN of PFF-seeded A53T mice, and this level was significantly less than that of the PBS-injected mice. Treadmill exercise strikingly up-regulated the PPARα level in TH neurons of PFF-seeded mice (Figures S3A and S3B). In addition, a decreased level of PPARα was also found in SN DAergic neurons of aged A53T mice compared with nTg brains (Figures S4A and S4C). Down-regulation of PPARα was also observed in midbrain astrocytes of these animals (Figures S4B and S4D). Similar to the PFF-seeded A53T mice, PPARα was significantly up-regulated in both neurons and astrocytes of aged A53T brains by treadmill exercise. The increase in PPARα expression was further validated in midbrain tissues of mice by immunoblotting (Figures S4E and S4F).
To be more specific about treadmill-induced PPARα up-regulation, we also evaluated the expression of other PPAR isoforms including PPARγ and PPARβ. Immunostaining demonstrated significantly enhanced PPARγ expression in nigral TH neurons (Figures S5A and S5B), but the PPARb level remained unchanged following exercise (Figures S5C and S5D). Moreover, PPARβ expression in DAergic neurons was much less than other isoforms of the protein. Accordingly, whole-tissue expression of PPARγ increased remarkably in A53T R mice compared with the A53T NR group (Figures S4E and S4G), whereas the change in PPARβ level was statistically insignificant (Figures S4E and S4H). The expression of the transcriptional coactivator PGC1α, known to function along with PPAR isoforms to regulate gene expression, was also not changed in midbrain tissues of the A53T NR and R groups of mice (Figures S4E and S4I). To further confirm PPARα activation by treadmill exercise, we also monitored its level in the hippocampus of aged A53T brains, where A53T NR animals exhibited diminished levels of this protein, but in A53T R brains, the PPARα level was markedly up-regulated (Figures S5E and S5F). Since PPARα is a transcription factor, electrophoretic mobility shift assay (EMSA) was performed from midbrain tissues of aged A53T NR and R mice to monitor the DNA-binding activity of PPARα. Earlier, we demonstrated that the Tfeb promoter harbors a consensus peroxisome proliferator-response element (PPRE) for transcriptional activation of the Tfeb gene (Ghosh et al., 2015). Therefore, for EMSA, we selected a probe from the same Tfeb promoter containing the PPRE and found a marked increase in DNA-binding activity of PPARα in the midbrain of A53T R mice compared with the A53T NR group (Figure S4J).
Treadmill exercise up-regulates TFEB in the SN via PPARα
The TFEB promoter was found to contain the PPRE around 482 to 504 bp upstream of the transcription start site (Ghosh et al., 2015) (Figure S6A). Chromatin immunoprecipitation (ChIP) analysis followed by real-time PCR demonstrated increased recruitment of PPARα and its partner retinoid X receptor α (RXRα) to the PPRE of Tfeb promoter in the midbrain of A53T R mice (Figures S6B and S6C). This was specific, as treadmill running did not modulate the recruitment of either PPARβ or PPARγ to the Tfeb promoter in the midbrain of A53T mice (Figures S6B and S6C). Accordingly, PPARα-mediated transcriptional up-regulation resulted in higher Tfeb mRNA expression in A53T R mice brain (Figure S6D). The protein expression of TFEB also increased in midbrain tissues of A53T R mice with respect to A53T NR mice (Figures S6E and S6F). Since the level of PPARα increased in both neurons and astrocytes, we further monitored TFEB expression in TH-positive neurons and GFAP-positive astrocytes in the SN. The findings clearly exhibited a diminished level of TFEB in TH neurons as well as astrocytes of 10-month-old A53T NR mice compared with nTg mice. However, treadmill exercise enhanced the level of TFEB in these cells (Figures S6G and 4J).
Figure 4. Heterozygous deletion of TPP1 aggravates α-syn pathology in A53T mice.

(A) Homozygous A53T mice were crossed with homozygous null CLN2 mice (CLN2−/−) to produce mice carrying homozygous mutation of A53T form of α-syn and heterozygous mutation of CLN2 gene (A53TCLN2+/−).
(B–H) The α-Syn pathology was compared between A53T and A53TCLN2+/− mice by monitoring the level of total Triton X-100 soluble (B and D) and insoluble form of the protein (*p < 0.05, C, E, and F, n = 4) and by visualizing α-syn aggregation in nigral TH neurons through immunofluorescence analysis (*p < 0.05, G and H, n = 4). Two sections from each brain per group were used for staining purposes, where α-syn MFI was quantified in at least 10 TH-positive cells from each section. (I and J) TH immunohistochemistry (IHC) followed by stereological counting exhibited number of nigral dopaminergic neurons in SN of these two groups of mice (**p < 0.01, n = 4).
(K–P) Behavioral performance of experimental animals was assessed by open-field locomotor test to obtain parameters like distance (F2, 9 = 16.59, **p < 0.01), velocity (F2, 9 = 16.64, **p < 0.01), central frequency (F2, 9 = 5.394, *p < 0.05), and cumulative duration of moving (F2, 9 = 11.2, *p < 0.05) and also by rotarod test (F2, 9 = 12.94, **p < 0.01) (n = 4).
One-way ANOVA followed by Tukey’s multiple comparison tests was performed to determine statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001 indicate significance compared with respective groups; ns, not significant. Data are represented as mean ± SEM.
Treadmill exercise enhances autophagy-lysosome pathway in the SN
Next, we evaluated the mRNA expression of 22 genes that are involved in autophagy-lysosomal functioning (Figure S7A). The majority of these genes were previously reported to be directly regulated by TFEB (Martini-Stoica et al., 2016; Settembre et al., 2012), whereas CLN2 and NPC expression can be directly regulated by PPARα (Chakrabarti et al., 2019; Ghosh and Pahan, 2016). The dendrogram demonstrates that the gene cluster containing the genes LC3B, SQSTM1, cathepsin B, cathepsin D, and LAMP1 had the highest up-regulated genes in A53T R mice compared with the A53T NR group. The second gene cluster containing LAMP2, VPS8, and Beclin1 had moderately up-regulated genes, whereas VPS11, UVRAG, and TPP1 were found to be increased by a lesser fold in A53T R mice. Expression of other genes such as AMBRA1, NPC, GBA1, CLN3, ATP6v, and ATG family proteins were not significantly altered following treadmill exercise (Figure S7A). Based on the gene array data, we have selected the highly and moderately up-regulated genes along with tripeptidyl-peptidase 1 (TPP1; a less up-regulated gene), the protein product of CLN2 gene, in the string protein-protein interaction analysis to create the functional interactome among these proteins (Figure S7B). We found a strong interaction among the majority of these proteins except for VPS8, which is an endosomal membrane protein participating in the SNARE-mediated membrane fusion of endosomes. Interestingly, LAMP2 sits in the middle of the cluster with multiple interactions with other genes like LAMP1, GABARAP, cathepsin B, cathepsin D, and SQSTM1, suggesting an important role played by this molecule in mediating autophagy-lysosomal function (Figure S7B). Moreover, TPP1 was shown to have an interaction with another major lysosomal protease cathepsin D (CtsD), which is also known to cleave misfolded α-syn in the mature lysosome. Next, we confirmed the enhanced expression of certain candidate proteins such as LAMP2, CtsD, and TPP1 by immunoblotting. The expression of these proteins was significantly less in aged A53T brains compared with the nTg mice, whereas remarkable up-regulation of these proteins were observed in A53T R mice compared with the A53T NR mice (Figures S7C-S7F). Expression of LAMP2 (Figures S7G and S7H) and TPP1 (Figures S7I and S7J) was further validated by double immunostaining in TH-positive nigral neurons, where the level of these proteins was found to be significantly higher in A53T R mice brain compared with A53T NR mice.
Heterozygous knock down of CLN2 aggravates α-syn pathology and causes DAergic neuronal death in A53T mice
Is lysosomal function critical for the development of LB pathology and DAergic neuronal loss? To delineate this relationship, we have generated double-transgenic mice carrying homozygous A53T α-syn and heterozygous CLN2 gene (A53TCLN2+/−) (Figure 4A). Cln2/TPP1 deficiency is the major cause of late infantile neuronal ceroid lipofuscinosis (LINCL) (Sleat et al., 1997). We compared the α-syn pathology between only A53T and A53TCLN2+/− animals, both of which were 6 months old during the experiment. The monomeric α-syn level in either the detergent-soluble (Figures 4B and 4D) or -insoluble fraction (Figures 4C and 4E) of midbrain tissue of these two groups of animals exhibited no change. However, protein bands corresponding to higher molecular weight oligomeric α-syn (around 80 kDa) were seen to be increased significantly in detergent-insoluble fractions of A53TCLN2+/− animals compared with age-matched A53T animals (Figures 4C and 4F). Parallel to this, enhanced accumulation of total α-syn content was found in TH neurons of SN in double-transgenic animals (Figures 4G and 4H), indicating enhanced α-syn pathology due to decrease activity of TPP1. In addition, accumulation of more PK-resistant forms of α-syn aggregates was also confirmed in the SN of A53TCLN2+/− mice, and the α-syn content following PK digestion was significantly higher in these mice than the normal A53T animals (Figures S8A and S8B). Interestingly, although A53T animals at the age of 6 months do not show any loss of DAergic neurons in the SN, the age-matched A53TCLN2+/− animals exhibited 37% loss of nigral TH neurons compared with A53T animals (Figures 4I and 4J). The findings clearly indicate that a decrease in TPP1 increases the vulnerability DAergic neurons in A53T mice. As a result, impaired motor activity of A53TCLN2+/− animals was observed in the open-field locomotor test and rotarod test (Figures 4K-4P). Other locomotor parameters including velocity, distance moved, cumulative duration of moving, and central frequency were also significantly less in A53TCLN2+/− animals compared with the age-matched A53T animals.
Treadmill exercise is unable to reduce α-synucleinopathy in A53T mice lacking PPARα
Our experimental findings so far exhibited that regular treadmill exercise greatly elevates PPARα-mediated expression of TFEB and lysosomal biogenesis, ultimately leading to a decrease in α-syn burden. Therefore, to substantiate the role of PPARα in treadmill-exercise-mediated reduction of α-synucleinopathy, we generated mice carrying a homozygous mutation of the A53T form of α-syn and a homozygous null mutation of PPARα (A53TΔPPARα) (Figure 5A). As expected, treadmill-induced down-regulation in the pathological detergent-insoluble α-syn level was found in midbrain tissues of normal A53T animals (Figures 5C and 5E). However, no significant change in the total α-syn level (detergent soluble and insoluble) was observed in brains of A53TΔPPARα mice following treadmill exercise (Figures 5B-5E). Moreover, A53TΔPPARα mice exhibited the presence of higher molecular weight oligomeric forms of α-syn in Triton X-100 insoluble fractions. Neuronal specific α-syn content (Figures 5F and 5G), size (Figure 5H), and intensity (Figure 5I) of α-syn aggregates were also evaluated in TH neurons of SN, where a remarkable decrease in α-syn aggregates was clearly visible in A53T R brains as revealed by immunofluorescence analysis. In contrast, α-syn levels remained unchanged in nigral TH neurons of A53TΔPPARα mice after treadmill exercise. Therefore, the area, size, and integrated density of α-syn aggregates were found to be comparable in DAergic neurons of A53TDPPARα NR and A53TΔPPARα R mice brains (Figures 5G-5I). To further validate the molecular mechanism, TFEB expression was also monitored in nigral TH neurons by double immunostaining, which showed remarkable up-regulation in TFEB level in A53T R mice compared with the A53T NR group, whereas no change in TFEB was observed in TH neurons of A53TΔPPARα R mice compared with the A53TΔPPARα NR group (Figures S9A and S9B). The failure of exercise in mitigating α-syn pathology in A53TΔPPARα mice was also reflected in motor performance of these animals. The A53TΔPPARα NR mice showed poor performance on rotarod and pole tests, and there was no behavioral improvement of these mice after treadmill exercise (Figures 5J-5L).
Figure 5. The α-syn pathology is not reduced by treadmill exercise in PPARα-deficient A53T mice.

(A) Homozygous A53T mice were crossed with PPARα−/− mice to generate A53T mice deficient of functional PPARα (A53TΔPPARα).
(B–E) Total α-syn level in Triton X-100 soluble and insoluble fractions isolated from midbrain tissues of A53T and A53TΔPPARα mice was measured by immunoblotting (F1, 16 = 6.83, *p < 0.05 for insoluble fraction, n = 5).
(F) Deposition of aggregated α-syn in nigral TH neurons was evaluated by immunofluorescence followed by confocal imaging.
(G–L) Two sections from each brain per group were used for staining purpose, where α-syn puncta was quantified in at least 10 TH positive cells from each section.
(G–I) Average values of different α-syn parameters including area (F1,36 = 6.21, *p < 0.05), average size (F1,36 = 6.37, *p < 0.05), and MFI of protein aggregates (F1,36 = 4.18, *p < 0.05) obtained from each section of all the groups of mice are shown (n = 5).
(J–L) Movement behavior was examined by rotarod (F1,20 = 7.4, *p < 0.05, J, n = 6) and pole test, where pole turn (F1,20 = 6.30, *p < 0.05, n = 6) and climb-down time (F1,20 = 6.96, *p < 0.05, n = 6) by each mouse were recorded (K and L).
The significance of mean was compared using two-way ANOVA considering NR/R and genotype as two independent factors. F1,20 > Fc and p < 0.05 were considered the statistical significance, where *p < 0.05 and **p < 0.01 indicate significance compared with respective groups; ns, not significant. Data are represented as mean ± SEM.
Spreading of α-synucleinopathy is not reduced by treadmill exercise in A53TΔPPARα mice
Since in the absence of functional PPARα, treadmill exercise could not reduce α-syn aggregation in neurons of aged A53T mice, next, to strengthen the role of PPARα in alleviating α-syn spreading, we seeded α-syn PFF in the IC region of both A53T and A53TΔPPARα mice. To evaluate spreading of α-syn, SN sections were stained for pSyn129. The data demonstrated exaggerated accumulation of pSyn129 in nigral neurons of both PFF-seeded A53T and A53TΔPPARα mice. Similar to what was mentioned above, pSyn129 accumulation was greatly reduced in SN neurons of A53T mice undergoing regular treadmill exercise (Figures 6A and 6B). However, in the case of A53TΔPPARα mice, no reduction of pSyn129 pathology was observed in nigral neurons following treadmill exercise (Figures 6A and 6B). PBS-injected A53T and A53TΔPPARα mice showed similar pSyn129 intensity in neurons without having any significant difference between these two groups. We further confirmed this finding by immunoblotting of total α-syn in midbrain tissues. The level of α-syn in Triton X-100 soluble (Figures 6C and 6D) and insoluble (Figures 6E and 6F) fractions was significantly higher in PFF-seeded A53T NR animals than the PBS-injected group. As shown above, insoluble α-syn content was markedly decreased in PFF-seeded A53T R mice compared with NR mice (Figures 6E and 6F). PFF-seeded A53TΔPPARα NR mice also showed higher levels of soluble (Figures 6G and 6H) and insoluble (Figures 6I and 6J) α-syn than the PBS-injected animals. However, reduction of insoluble α-syn was not found in PFF-seeded A53TΔPPARα mice undergoing treadmill exercise (Figures 6I and 6J). These results indicate that treadmill exercise is unable to reduce α-syn spreading as well as aggregation in neurons in the absence of PPARα. In addition, the number of nigral DAergic neurons and the striatal DA level was monitored in the PFF-seeded A53T (NR and R) and A53TΔPPARα (NR and R) mice. Like the earlier findings, a significant protection of nigral DAergic neurons was observed in A53T + PFF R mice compared with the A53T + PFF NR mice (Figures S10A and S10B), and that parallels with the attenuation of DA loss in the striatum of the A53T + PFF R mice (Figure S10C). However, the level of DOPAC and HVA was not found to be significantly altered after treadmill exercise (Figures S10D and S10E). The PFF-seeded A53TΔPPARα+PFF NR mice also showed remarkable loss of DAergic neurons and striatal DA levels compared with the PBS-injected A53TΔPPARα mice (Figures S10A and S10B). Interestingly, following treadmill exercise, the DAergic neurons were found to be protected in these mice, resulting in an elevation of the striatal DA level (Figure S10C). These finding states that although PPARα is essential for hindering α-syn spreading by treadmill exercise in A53T animals, retention of DAergic system might be happening through another neuroprotective mechanism that is independent of PPARα activation.
Figure 6. Treadmill exercise does not reduce α-syn spreading in the brain in the absence of PPARα.

Age-matched A53T and A53T mice lacking functional PPARα (A53TΔPPARα) were injected with either PBS or PFF at the IC region of striatum, and following 2 months of seeding, animals were run in a treadmill daily for 2 months.
(A) IHC of pSyn129 in the SN followed by relative pSyn129 intensity analysis.For immunostaining, at least two sections from each brain per group were stained, and 10–15 pSyn129-positive cells were quantified in each section.
(B) Average intensity of pSyn129 obtained from each section of all groups of mice are shown (F6, 35 = 62.92, *p < 0.05, ***p < 0.001, n = 3).
(C, E, G, and I) The levels of total α-syn in the Triton X-100 soluble and insoluble fractions isolated from midbrain tissues (C and E) and A53TΔPPARα mice (G and I) were measured by immunoblotting.
(D, F, H, and J) Ratio of band intensities of α-syn to actin (D and H for soluble fractions and F and J for insoluble fractions, F4, 11 = 24.85, **p < 0.01 for A53T + PFF NR versus A53T + PFF R for insoluble fraction, n = 3).
The significance of mean was compared using one-way ANOVA followed by Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, and ***p < 0.001; ns, not significant. Data are represented as mean ± SEM.
Fenofibrate, a PPARα agonist, decreases α-syn spreading and protects TH neurons
So far, we have seen that PPARα is the major factor behind treadmill-exercise-mediated reduction of α-syn pathology in the brain. However, in addition to activating PPARα, regular treadmill exercise can modulate many other factors, which can also participate in the reduction of α-synucleinopathy. Therefore, we tested whether activation of PPARα alone was sufficient to hinder α-syn spreading and aggregation. Accordingly, PFF-seeded A53T mice were treated orally with canonical PPARα agonist fenofibrate via gavage for 1 month (Figure 7A). As expected, PFF-seeded mice showed a higher aggregation of pSyn129 in nigral neurons than the PBS-injected group. Interestingly, pSyn129 intensity in nigral neurons was found to be greatly reduced in PFF-seeded mice fed with fenofibrate (Figures 7B and 7C). Parallel to pSyn129 down-regulation, the total insoluble form of α-syn was also remarkably decreased in midbrain tissues by fenofibrate treatment (Figures 7F and 7G). However, no significant change in the soluble α-syn content was found in the fenofibrate-treated group compared with the PFF-seeded control animals (Figures 7D and 7E). As expected, a more considerable depletion of DAergic neurons (Figures 7H and 7I) and TH protein content (Figures 7J and 7K) was found in PFF-seeded mice than the PBS-injected group. Interestingly, fenofibrate treatment alone was sufficient to attenuate the loss of nigral TH in PFF-seeded A53T mice (Figures 7H-7K). This finding was also recapitulated at the DAergic terminals in the striatum, because PFF seeding simultaneously caused around 25% loss of DA level (Figure S11A), which was significantly protected by oral fenofibrate. However, no significant change in DOPAC and HVA level was found in these animals (Figures S11B and S11C). Next, the effect of fenofibrate on behavioral performance of PFF-seeded A53T mice was also monitored by open-field (Figure S11D) and rotarod tests. Parallel to DA depletion, PFF seeding also resulted in minute but significant retardation of locomotor parameters such as distance (Figure S11E), velocity (Figure S11F), cumulative duration of moving (Figure S11G), rearing (Figure S11H), and latency to fall in the rotarod test (Figure S11I). However, fenofibrate-treated animals exhibited significant improvement in the movement behaviors compared with the PFF-seeded control animals. Collectively, these findings clearly state that activation of PPARα solely by its agonist fenofibrate is sufficient to reduce α-syn pathology and attenuate parkinsonian features in mice.
Figure 7. Oral fenofibrate inhibits α-syn spreading and protects nigral DAergic neurons in PFF-seeded A53T mice brain.

(A) PFF-seeded A53T mice were fed with fenofibrate (2 mg/kg) daily via gavage for 1 month.
(B) Spreading of α-syn in SN was analyzed by pSyn129 IHC.
(C) Relative intensity of pSyn129 (F3, 20 = 109.7, ***p < 0.001, n = 3).
(D–G) The level of α-syn in Triton X-100 soluble (D and E) and insoluble tissue fractions (F3, 11 = 21.38 for A53T + PFF versus A53T + PFF + Feno, *p < 0.05, **p < 0.01, F and G, n = 3) was assessed by immunoblotting.
(H–K) Number of TH neurons in SN was shown by IHC of midbrain sections (H) followed by stereological counting in each hemisphere of mouse brain (*p < 0.05, **p < 0.01, I, n = 3), and the TH protein content in SN was measured by immunoblotting (*p < 0.05, J and K, n = 3).
The significance of mean was compared using one-way ANOVA followed by Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significance compared to respective groups; ns, not significant. Data are represented as mean ± SEM.
DISCUSSION
Spreading of α-syn and the propagation of pathological form of this protein coincide with progressive neuronal death in the brain (Chu et al., 2019; Rey et al., 2019; Yun et al., 2018). Moreover, progressive localized accumulation of α-syn aggregates is visible in several brain regions such as the SN, hippocampus, and brain stem in distinct α-synucleinopathies (Giasson et al., 2002; Rey et al., 2019; Tsika et al., 2010). Despite intense investigations, no effective therapy is yet available to control α-synucleinopathies in humans. In that respect, the significance of regular treadmill exercise for α-synucleinopathy is proven firstly by the fact that it mitigates striatonigral and striatum to cortical spreading of α-syn and prevents nigral DAergic cell death in a PFF-seeding model of PD. Secondly, a decrease in α-syn level in hippocampus and simultaneous improvement in spatial memory function of A53T mice confirms the impact of treadmill exercise in attenuating LBD-like pathology, where the limbic system is primarily affected in the early stage (Braak et al., 2003; Sanford, 2018). Therefore, treadmill exercise may be beneficial in patients with PD, MSA, and DLB.
How does treadmill exercise reduce α-syn pathology? Experimental studies executed up to date have strongly indicated that promoting degradation of intracellular α-syn aggregates can be one of the potential strategies against α-synucleinopathy (Brundin et al., 2017). This indication came from the findings that the autophagy-lysosomal pathway, which consists of macroautophagy and chaperone-mediated autophagy processes (Xicoy et al., 2019), is found to be deficient in almost all forms of α-synucleinopathies, resulting in the accumulation of more toxic forms of the protein (Dawson and Dawson, 2003; MacInnes et al., 2008; Poehler et al., 2014; Rubinsztein, 2006). PPARα is a transcription factor that regulates genes involved in fatty acid catabolism (Pahan, 2006; Roy and Pahan, 2015). Although liver is rich in PPARα, we have demonstrated that PPARα is constitutively expressed in different parts of the brain (Corbett et al., 2015; Roy et al., 2013, 2015; Roy and Pahan, 2015). While exploring the mechanism behind reduced α-syn pathology by treadmill, we investigated the level of different PPAR isoforms. We observed an increase in both PPARα and PPARγ in the midbrain following treadmill exercise, whereas PPARβ remained unaltered. In situ ChIP analysis convincingly established the specific involvement of PPARα in stimulating the transcription of TFEB, a master regulator of lysosomal biogenesis and autophagy, in vivo in the midbrain. Hence, it nullifies the involvement of PPARβ and PPARγ in up-regulating lysosomal biogenesis. It is noteworthy that PPARγ increase delineates activation of other pathways such as mitochondrial biogenesis, protein import, and anti-oxidant protein synthesis (Chaturvedi and Beal, 2008; Swanson et al., 2011) but not lysosomal biogenesis. Therefore, reduced level of PPARα in the SN and hippocampus might be responsible for deficiency of the autophagy-lysosome pathway resulting in chronic accumulation of α-syn in A53T brains. This finding was verified in A53TΔPPARα animals where, in the absence of functional PPARα, there is no up-regulation of TFEB expression, and therefore α-syn accumulation as well as spreading in the brain remains unaltered in these mice following treadmill exercise. This strongly indicates that activation of the PPARα-T-FEB axis is perhaps the underlying reason behind treadmill-exercise-induced reduction of α-syn pathology. The involvement of PPARα in α-syn clearance becomes more evident from the fact that the well-known PPARα agonist fenofibrate also remarkably hinders α-syn spreading and parkinsonian pathologies in mice. However, accumulation of α-syn aggregates and DAergic neuronal survival might not be well correlated with each other, as in absence of functional PPARα, DAergic neurons were yet protected by regular treadmill exercise. This fact might be due to the activation of PPARα-independent protective pathways such as upregulation of PPARγ by treadmill intervention, as found in the aged A53T animals, and possible induction of PPARγ-mediated mitochondrial biogenesis and inhibition of neuroinflammation in PFF-seeded mice (Makela et al., 2016; Pisanu et al., 2014; Schintu et al., 2009), although these possible mechanisms were not addressed in the present study.
It must be mentioned that TFEB down-regulation was also found in brains of PD models and in the brains of patients with MSA (Arotcarena et al., 2019), whereas TFEB overexpression rescued DAergic neurons from α-syn-mediated toxicity (Decressac et al., 2013), highlighting its functional relevance in the context of PD. As we also observed that PPARα and TFEB expression is significantly upregulated in brains following treadmill exercise, it further necessitated investigation on the PPARα-TFEB-mediated lysosomal biogenesis in greater detail. Conspicuously up-regulated proteins such as Lamp2 and CtsD have significant functional importance in α-syn breakdown via lysosome (Ambrosi et al., 2014; McGlinchey and Lee, 2015; Vidoni et al., 2016; Youn et al., 2018). Another up-regulated protein, TPP1, has never been reported to play any role in the catabolism of α-syn species. It is interesting that TPP1 down-regulation potentially leads to formation of higher order α-syn toxic forms in brain and simultaneous DAergic cell death in A53T animals, which do not exhibit any neuronal demise at the early age (6 months) of lifespan. This finding assures that TPP1 might be one crucial enzyme for α-syn degradation in cells. Most importantly, it also highlights the possibility that heterozygous human carriers of the CLN2 gene might be more prone to be affected with PD or other forms of α-synucleinopathies, although this assumption needs further in-depth investigations.
In summary, the present article made important revelations: First, treadmill exercise significantly mitigates both localized α-syn aggregation and spreading to different brain regions, resulting in prevention of neuronal demise. Second, treadmill exercise induces the activation of PPARα in neurons. Third, treadmill exercise up-regulates the autophagy-lysosomal pathway via PPARα-dependent transcription of TFEB. Fourth, heterozygous reduction of the CLN2 gene aggravates α-syn spreading and causes DA loss, emphasizing the importance of lysosome in α-syn pathology and indicating that the decrease in function of a pivotal lysosomal protein, TPP1, may increase the risk of α-synucleinopathy. Fifth, activation of PPARα by a prototype PPARα activator fenofibrate is also sufficient to reduce α-syn spreading and protect DAergic neurons. Therefore, either treadmill exercise or drug-mediated PPARα activation may be beneficial for preventing α-synucleinopathy and related neurodegeneration.
Limitations of the study
Recently, microglial activation and associated neuroinflammation has become a hallmark of different neurodegenerative disorders including α-synucleinopathy. However, this study does not describe whether treadmill exercise is capable of reducing microglial activation in PFF-seeded mice. We also do not know if PPARα is required for treadmill-mediated possible modulation of neuroinflammation in the brain of PFF-seeded mice. Moreover, although here we have delineated a beneficial function of treadmill exercise in halting α-syn spreading in the brain via PPARα, this molecule is known to partner with RXRα to exhibit transcriptional activity. For example, here, we have seen that treadmill exercise stimulates the recruitment of both PPARα and RXRα to the TFEB gene promoter in vivo in the midbrain of A53T mice. However, we do not know anything about the status of RXRα in PFF-seeded mouse brain before and after treadmill exercise. Therefore, future studies may be designed to address these issues.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Kalipada Pahan (Kalipada_Pahan@rush.edu).
Materials availability
Mouse lines generated in this study are available from the lead contact upon request with a completed Materials Transfer Agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does report any original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Adult C57BL6 mice were purchased from Envigo. PPARα−/− mice and A53T α-syn transgenic line M83 were purchased from Jackson Laboratories. CLN2−/− mice were kindly provided by Prof. Peter Lobel of Rutgers University. Mice of both sexes were used for treatment and analysis. While 2-month old A53T mice were used PFF seeding, 8-month old A53T mice were used for other experiments. Animal maintenance and experiments were performed in accordance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use committee of the Rush University Medical Center.
Generation of A53TΔPPARα and A53TCLN2+/− mice
Adult A53T mice were bred with PPARα−/− mice and the pups were genotyped using the primers as suggested by Jackson Laboratories. Mice, which were homozygous mutant for α-syn and PPARα−/−, were selected for further experiments. Similarly adult A53T mice were also bred with CLN2−/− mice and the litters were genotyped for selecting those animals, which were homozygous mutant of A53T and heterozygous mutant of CLN2 (CLN2+/−).
METHOD DETAILS
Preparation and validation of α-syn PFF
Recombinant monomeric endotoxin free α-syn was solubilized in 30 mM Tris-HCl (pH 7.4) at the concentration of 350 μM and rotated continuously in a rotary shaker at 250 rpm at 37°C for 7 days. Next, the fibrils were briefly sonicated (30 s) at 10% amplitude and were characterized by electron microscopy (EM) (Karampetsou et al., 2017; Mao et al., 2016). For EM, 1 μL of stock solution was diluted in 100 μL of phosphate buffer saline (PBS) and this solution was adsorbed to 300-mesh copper, Formvar-coated EM grid, washed, stained with 2% uranyl acetate and the grid was allowed to be dried for 15–20 min. Imaging was performed at 100,000X magnification using a JEOL JEM-1220 transmission electron microscope (operating at 80 kV). Digital micrographs were acquired using an Erlangshen ES1000W model 785 CCD camera and Digital Micrograph software (Version 1.7).
Mouse brain stereotaxy
Stereotaxic surgery was performed according to the procedure mentioned by Sorrentino and co-workers (Sorrentino et al., 2017). α-Syn PFF was injected into the internal capsule (IC) region of striatum of 2 months old A53T mice of both sexes bilaterally. Animals were injected with 5 μg of α-syn fibrils dissolved in 2.5 μL of PBS in the internal capsule region in both the hemispheres of brain (coordinates from Bregma: A/P −0.5, L - +/−1.5, D/V −3.0) (Dutta et al., 2021a). Similarly control animals received only PBS in both the hemispheres. The solution was injected at the rate of 0.4 μL/min and following the injection, the needle was kept for another 5 min on each side of the brain for proper diffusion of the protein solution.
Treadmill exercise
Aged A53T animals (8 months old) and 4 months old PFF-injected A53T animals (2 months post brain surgery) were subjected to treadmill exercise on 6 consecutive days per week for 2 months. Initial 2 weeks animals were acclimatized in the treadmill by keeping the speed at low magnitude (5m/min) for 30 min every day. After that, the speed was gradually increased to 8 m/min for 2 weeks, 10m/min for 2 weeks and 12 m/min for the last 2 weeks (Koo et al., 2017).
Fenofibrate treatment
Fenofibrate solution was prepared in 0.1% of methyl cellulose. Following 2 months of PFF seeding, A53T mice were fed with fenofibrate at the dose of 2 mg/kg via gavage daily for 30 days.
Behavioral tests
Behavioral tests for both the genetic and sporadic animal models were carried out after 6 weeks of initiation of treadmill exercise. For aged A53T mice, movement parameters were evaluated by conducting rotarod and pole tests, whereas memory deficits were measured by Barnes maze. In case of PFF-injected mice, motor performance was evaluated by open field and rotarod.
Open field test
Open field test was performed to analyze the locomotor activities of different groups of mice as described before (Patel et al., 2018, 2020; Raha et al., 2021b). Mice were put in the open field arena (40 × 40cm, 30 cm high walls) for 2 consecutive days, 10 min for each day per mice to acclimatize the animals. One day after the training, locomotor activity was monitored with a camera linked to Noldus system and EthoVision XT software (Netherlands). The instrument records the overall movement abilities of the animals including total distance moved, velocity, total moving time, resting time, center time and frequencies of movement and the number of rearing. After releasing the animal, data acquisition was started by the software for the next 5 min and the parameters related to the locomotor activities were collected by the software.
Rotarod test
Prior to the test, mice were placed on the rotarod instrument for 5–10 min daily for consecutive two days to train them. After one day of training, mice were placed on the rotating rod, which rotates with a gradual increasing speed of 4–40 rpm. The experiment was ended if the animals slips from the rotating rod to the base of the instrument or just grips the rod to turn reverse without rotating against the direction of the rod (Chandra et al., 2016, 2017; Dutta et al., 2021a; Yun et al., 2018).
Pole test
Pole test was performed as described earlier (Chandra et al., 2016). Briefly, the wooden pole (57 cm in height and 2 cm in width) was covered with gauze to make rough surface on the wooden rod. Animals were trained for 2 consecutive days prior to the test and each time 5 trials were given to each animal. During the test, animal was placed near the top of the pole facing upwards. The time taken by each animal to turn downwards was monitored as the pole turn time. Moreover, the time taken by each animal to reach the base of the pole was recorded as the pole climb down time. The maximum time for recording was set as 60 s. If any animal was found to stall for more than 60 s, the test was further performed for that particular animal (Matsuura et al., 1997).
Barnes maze
Barnes maze was performed for aged A53T animals to monitor the spatial memory deficits. The Barnes maze arena contained 20 holes, out of which only one hole contains the food pellet. During the training phase, mice were placed on the middle of the Barnes maze arena 10 min daily for consecutive 2 days. The arena was equipped with high wattage light to provide enough heat and motivation to the animals to find out the target hole containing the food pellet. Animals were given rest for one day and on the 4th day the experiment was performed. Animals were food-deprived for overnight and during the test each animal was placed in the arena for 5 min and the performance was captured using the Noldus system. Memory of animals was analyzed based on the parameters such as time taken to reach the goal box and number of errors made by each animal before reaching the goal box (Chandra et al., 2019; Rangasamy et al., 2018).
Real-time PCR
Midbrain tissues were isolated from mice brain and kept in Trizol solution (Sigma-Aldrich, St. Louis, MO) and stored at −20°C. Next day, total RNA was isolated from tissues following the protocol published before (Chandra et al., 2016, 2017; Dutta et al., 2021a). The isolated RNA was reverse transcribed into cDNA and real-time PCR was performed using the primers designed for lysosomal genes using Primer 3 software (Table S2). PCR reaction was carried out in ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) using the SYBR green real-time kit obtained from QuantaBio (Beverly, MA) as described before (Patel et al., 2020)and the data were evaluated using the method of Livak and Schmittgen (Livak and Schmittgen, 2001).Relative expression of genes was compared among experimental animals and the heatmap along with hierarchical clustering of differentially expressed genes were made using Morpheus (Broad institute, USA). Protein-protein interaction network of selected proteins were generated using String v11 software.
Tissue lysate preparation
Midbrain tissues were isolated from mice brain and homogenized in Triton X-100 soluble buffer containing 1% Triton X-100, 0.5 μM EDTA, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl and protease inhibitors and phosphatase inhibitor cocktails. The homogenate was centrifuged at 17500 xg for 15 min at 4°C. The supernatant obtained after centrifugation was the detergent soluble fraction of the tissue. The resultant pellet was dissolved in detergent insoluble buffer containing 50 mM Tris (pH 8.0), 1% Triton X-100, 2% SDS, 1% sodium deoxycholate, 1% NP-40,0.5 μM EDTA and protease, phosphatase inhibitor cocktails. The lysate was further centrifuged at 17,500 xg for 15 min at 4°C and the supernatant was collected as the Triton X-100 insoluble fraction (Dutta et al., 2021a; Yun et al., 2018). Total tissue lysate was obtained by dissolving the tissue in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 1% SDS, 0.5% sodium-deoxycholate) with complete protease and phosphatase inhibitor cocktails. Tissues were sonicated for 20–30 s and the homogenate was centrifuged at 17,500 xg for 15 min at 4°C, and the resulting supernatant was collected (Dutta et al., 2018, 2021a).
Western blotting
Western blotting was performed as described previously (Chandra et al., 2017; Dutta et al., 2021a). Equal amounts of proteins were electrophoresed in 10% or 12% SDS-PAGE and transferred onto nitrocellulose membrane. The blot was probed with primary antibodies overnight at 4°C. Next day primary antibodies were removed and the blots were washed with phosphate buffer saline containing 0.1% Tween-20 (PBST) and corresponding infrared fluorophore-tagged secondary antibodies (1:10,000; Jackson Immuno-Research) were added at room temperature (RT). Finally, blots were scanned with an Odyssey infrared scanner (Li-COR Biosciences, Lincoln, NE). Band intensities were quantified using ImageJ software (NIH, USA).
In situ chromatin immunoprecipitation
Animals were perfused with 4% paraformaldehyde and were kept in paraformaldehyde for overnight. Midbrain tissue was dissected out, washed with PBS and homogenized in Tris-EDTA (pH 7.6). The homogenate was kept at 52°C overnight until tissue fragments were dissolved. Next day, from that homogenate DNA was isolated following the phenol, chloroform, isopropanol (25:24:1) method. Aqueous phase from the final spin was collected and 0.1 vol of 3 M sodium acetate was added, vortexed followed by addition of I vol of isopropanol and incubated at −20°C overnight. The precipitated DNA was centrifuged at 12000 xg at 4°C for 15 min, washed by 75% ethanol and suspended in nuclease free Tris-EDTA buffer. Chromatin immunoprecipitation was performed as described previously with antibodies for PPARα, PPARβ, PPARγ, RXRα and IgG was used as the negative control. The PPRE-containing fragment of the mouse Tfeb promoter was amplified using the following primers: Sense: 5′-GAA CAT TCC AGG TGG AGG CA-3′ Antisense: 5′-CCC CCA ACA CAT GCT TCT CT-3′
For real-time PCR, data were normalized with the input and the fold change with respect to the untreated control was calculated (Dutta et al., 2021a; Ghosh et al., 2015).
Electrophoretic mobility shift assay (EMSA) for PPARα
EMSA was performed from nuclear extracts of midbrain using procedure described previously (Jana and Pahan, 2010; Paidi et al., 2021a, 2021b) using the peroxisome proliferator-response element (PPRE) of the Tfeb promoter. Briefly, IRDye infrared dye end-labeled oligonucleotide probes (Li-COR Biosciences) was incubated with 6 μg of nuclear extract with binding buffer and with infrared-labeled probe for 20 min. Subsequently, samples were separated on a 6% polyacrylamide gel in 0.25× TBE buffer (Tris borate-EDTA) and analyzed by the Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunostaining
Experimental animals were perfused with 4% paraformaldehyde, and the brains were kept in 30% sucrose solution at 4°C. Coronal sections (30 μm in width) were cut from the multiple brain regions including striatum, hippocampus, midbrain and brainstem and processed for immunostaining. Tissue sections were blocked with 3% BSA and 2% normal goat serum in PBS containing 0.5% Triton X-100 (Sigma-Aldrich) and 0.05% Tween 20 (Sigma-Aldrich) for 1 h. Then the samples were kept in primary antibodies for target proteins (Table S2) and incubated at 4°C temperature overnight under shaking conditions. Next day, sections were washed with PBS for 30 min and further incubated with Cy2- or Cy5-labeled secondary antibodies (all 1:500; Jackson Immuno-Research) for 2 h under similar shaking conditions. Following multiple washes with PBS, cells were incubated for 4–5 min with 4′,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma). For immunohistochemistry (IHC) of brain sections, samples were kept in solution containing biotin-tagged secondary antibodies for 2 h followed by incubation in Vectastain A and B mixture solution at RT. Sections were developed by 3,3′-diaminodenzidine (DAB) solution containing peroxide. Sections were mounted on coated slides, dried overnight and were run in an ethanol and xylene (Fisher) gradient and observed under either Olympus BX41 fluorescence microscope or imaged under Zeiss confocal microscope.
Mean fluorescence intensity (MFI) and counting of target proteins or cells were performed using ImageJ (Dutta et al., 2018, 2019; Raha et al., 2021a). Optical density of pSyn129 was measured using color deconvolution followed by analyze and measure options of the Fiji (ImageJ2 version) software. Briefly, each image was deconvoluted to achieve H-DAB stained images and then each cell was outlined and the intensity was measured by the Analyze-measure option provided in the software. At least 15 cells were selected from each section for this analysis and two random sections from nigral and motor cortex regions of each mouse were considered. The mean value of stained cells in A53T brains (including PBS and PFF-injected) was compared to that of cells of nTg brain. Darker staining indicates lower mean value of DAB staining. The mean value of white is considered to be the highest (255), and therefore the formula used for calculating relative O.D. is log10(255/mean of each cell) (Dutta et al., 2021a, 2021b). Analysis of α-syn aggregates in neurons was conducted using ImageJ. Briefly, each image was converted to 8-bit image and the background was subtracted using the software. Next, using the threshold option the α-syn puncta or aggregates were better visualized and each cell was selected with an outline provided in ImageJ. Then number, area occupied and the intensity of α-syn aggregates in each cell was obtained by using the Analyze Particle option of ImageJ. For quantifying the mean fluorescent intensity (MFI) of other proteins present in TH neurons, a contour was made around the area of the target neuron in substantia nigra (SN) and by using the Analyze-Measure option in the software the MFI was measured. Each MFI value was subtracted from the background fluorescence according to the equation given in ImageJ manual (NIH).
Proteinase K digestion
Proteinase K (PK) digestion of nigral sections was carried out during immunofluorescence staining of α-syn in dopaminergic neurons. The sections were kept in solution containing 10 μg/mL of PK made in PBS for 10 min at 37°C under constant rotation. Following the PK digestion, co-staining of α-syn and TH was performed using the protocol described above. Images were captured under Olympus BX41 fluorescence microscope and MFI was analyzed using ImageJ.
TH neuronal counting
TH IHC in both SN and striatum was performed according to the protocol mentioned earlier (Dutta et al., 2019, 2021a; Ghosh et al., 2007, 2009). Counting of TH neurons in SN of each hemisphere of brain was performed by using STEREO INVESTIGATOR software (MicroBrightfield, Williston, VT) having an optical fractionator. Every 6th section from the starting of nigra (around 2.8 mm from bregma) was considered for counting of TH neurons.
HPLC analysis for measurement of striatal DA and its metabolite levels
Striatal level of DA and its metabolites, 3,4-dihyroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA), was measured as mentioned earlier (Ghosh et al., 2007; Khasnavis et al., 2014; Mondal et al., 2012). In brief, after 7 days of MPTP treatment, mice were sacrificed by cervical dislocation and striatum was isolated from each mouse and sonicated in 0.2 M ice cold perchloric acid. The homogenate was centrifuged at 17500 xg for 15 min at 4°C. Supernatant was collected in a fresh tube and 10 μL of the supernatant was injected in an Eicompak SC-3ODS column (Complete Stand-Alone HPLC-ECD System EiCOMHTEC-500; JM Science, Grand Island, NY). The level of neurotransmitters was analyzed according to the manufacturer’s protocol (Chandra et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were performed using GraphPad Prism v7.02. Values are expressed as mean ± S.E. for all experiments. Statistical analyses for differences between two different samples were performed using unpaired two-tailed t test. One-way ANOVA followed by Tukey’s multiple comparisons was performed for statistical analyses among multiple groups. Two-way ANOVA was performed to determine statistical significance where two different parameters were involved. The criterion for statistical significance was p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Synuclein | Abcam | ab138501 (MJFR1) |
| α-Synuclein | BD Bioscience | 610787; AB_398107 |
| Phospho Ser129 α-synuclein | Abcam | ab51253 (EP1536Y) |
| Tyrosine hydroxylase (TH) | Pel-Freeze Biologicals | P40101 |
| PPARα | Santacruz | sc-398394 |
| PPARβ | Santacruz | sc-74517 |
| PPARγ | Santacruz | sc-7273 |
| PGC1α | Millipore | AB3242 |
| TFEB | Abcam | ab2636 |
| Lamp2 | Millipore | MABC40; AB_2662613 |
| Cathepsin D | Santacruz | sc-6487 |
| TPP1 | Abcam | ab54685 |
| Actin | Abcam | ab8226 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human α-syn | Anaspec | AS-1000 |
| Fenofibrate | Millipore Sigma | F6020-100G |
| SYBR green real-time kit | QuantaBio | K1070 |
| Proteinase K | Sigma | 3115828001 |
| IRDye infrared dye end-labeled oligonucleotide probe for PPAR | Li-COR Biosciences | Custom made |
| Deposited data | ||
| Raw and analyzed data | Figshare | https://doi.org/10.6084/m9.figshare.20032727 |
| Experimental models: Organisms/strains | ||
| M83 (A53T) | Jackson Laboratory | B6;C3-Tg(Prnp-SNCA*A53T)83Vle/J |
| CLN2−/− | Ghosh et al., 2017 | https://doi.org/10.1111/jnc.13987 |
| C57BL6 | Envigo | C57BL/6JOlaHsd |
| PPARα−/− | Jackson Laboratory | B6; 129S4-Pparatm1Gonz/J |
| Oligonucleotides | ||
| Primers for lysosomal genes, see Table S2 | This paper | NA |
| Primer corresponding to Tfeb promoter, sense: 5′-GAA CAT TCC AGG TGG AGG CA-3′ | This paper | NA |
| Primer corresponding to Tfeb promoter, antisense: 5′-CCC CCA ACA CAT GCT TCT CT-3′ | This paper | NA |
| Software and algorithms | ||
| Fiji (ImageJ2) | Schneider et al., 2012 | https://doi.org/10.1038/nmeth.2089 |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| GraphPad Prism v7.02 | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Treadmill exercise mitigates α-synuclein spreading in the brain
Striatal dopamine is protected in a PFF-seeding model by treadmill exercise
PPARα is activated in neurons by treadmill exercise
Activation of PPARα lowers α-synucleinopathy
ACKNOWLEDGMENTS
This study was supported by grants (NS108025, AG050431, and AG069229) from NIH and merit awards (BX003033 and BX005002) from the US Department of Veterans Affairs. Moreover, K.P. is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the US Department of Veterans Affairs.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111058.
DECLARATION OF INTERESTS
Authors declare no competing interests.
REFERENCES
- Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan-Gomez C, Bombardieri CR, Armentero MT, Zangaglia R, Pacchetti C, Mastroberardino PG, and Blandini F (2014). Bioenergetic and proteolytic defects in fibro-blasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta 1842, 1385–1394. 10.1016/j.bbadis.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Arotcarena ML, Bourdenx M, Dutheil N, Thiolat ML, Doudnikoff E, Dovero S, Ballabio A, Fernagut PO, Meissner WG, Bezard E, and Dehay B (2019). Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight 4, 129719. 10.1172/jci.insight.129719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, et al. (2009). Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 117, 613–634. 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo G, Paciotti S, Gatticchi L, and Parnetti L (2020). The vicious cycle between alpha-synuclein aggregation and autophagic-lysosomal dysfunction. Mov. Disord 35, 34–44. 10.1002/mds.27895. [DOI] [PubMed] [Google Scholar]
- Braak H, Tredici KD, Rüb U, de Vos RA, Jansen Steur EN, and Braak E (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Brundin P, Dave KD, and Kordower JH (2017). The rapeutic approaches to target alpha-synuclein pathology. Exp. Neurol 298, 225–235. 10.1016/j.expneurol.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Chandra S, Roy A, Dasarathi S, Kundu M, and Pahan K (2019). Upregulation of tripeptidyl-peptidase 1 by 3-hydroxy-(2, 2)-dimethyl butyrate, a brain endogenous ligand of PPARα: implications for late-infantile Batten disease therapy. Neurobiol. Dis 127, 362–373. 10.1016/j.nbd.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G, Rangasamy SB, Roy A, Kordower JH, and Pahan K (2016). Neutralization of RANTES and eotaxin prevents the loss of dopaminergic neurons in a mouse model of Parkinson disease. J. Biol. Chem 291, 15267–15281. 10.1074/jbc.M116.714824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chandra G, Roy A, Rangasamy SB, and Pahan K (2017). Induction of adaptive immunity leads to nigrostriatal disease progression in MPTP mouse model of Parkinson’s disease. J. Immunol 198, 4312–4326. 10.4049/jimmunol.1700149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Jana M, and Pahan K (2018). Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARα. J. Neurosci 38, 6682–6699. 10.1523/JNEUROSCI.0054-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Roy A, Jana M, and Pahan K (2019). Cinnamic acid activates PPARα to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol. Dis 124, 379–395. 10.1016/j.nbd.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, and Beal MF (2008). PPAR: a therapeutic target in Parkinson’s disease. J. Neurochem 106, 506–518. 10.1111/j.1471-4159.2008.05388.x. [DOI] [PubMed] [Google Scholar]
- Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J, Tamagnan G, Marek K, Luk KC, Trojanowski JQ, et al. (2019). Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142, 3565–3579. 10.1093/brain/awz296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett GT, Gonzalez FJ, and Pahan K (2015). Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 112, 8445–8450. 10.1073/pnas.1504890112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, and Dawson VL (2003). Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302, 819–822. 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, and Björklund A (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 110, E1817–E1826. 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Ali N, Banerjee E, Singh R, Naskar A, Paidi RK, and Mohanakumar KP (2018). Low levels of prohibitin in substantia nigra makes dopaminergic neurons vulnerable in Parkinson’s disease. Mol. Neurobiol 55, 804–821. 10.1007/s12035-016-0328-y. [DOI] [PubMed] [Google Scholar]
- Dutta D, Jana M, Majumder M, Mondal S, Roy A, and Pahan K (2021a). Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat. Commun 12, 5382. 10.1038/s41467-021-25767-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Kundu M, Mondal S, Roy A, Ruehl S, Hall DA, and Pahan K (2019). RANTES-induced invasion of Th17 cells into substantia nigra potentiates dopaminergic cell loss in MPTP mouse model of Parkinson’s disease. Neurobiol. Dis 132, 104575. 10.1016/j.nbd.2019.104575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Majumder M, Paidi RK, and Pahan K (2021b). Alleviation of Huntington pathology in mice by oral administration of food additive glyceryl tribenzoate. Neurobiol. Dis 153, 105318. 10.1016/j.nbd.2021.105318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Corbett GT, Gonzalez FJ, and Pahan K (2012). Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor alpha: implications for late infantile Batten disease therapy. J. Biol. Chem 287, 38922–38935. 10.1074/jbc.M112.365148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Jana M, Modi K, Gonzalez FJ, Sims KB, Berry-Kravis E, and Pahan K (2015). Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: implications for lysosomal storage disorders. J. Biol. Chem 290, 10309–10324. 10.1074/jbc.M114.610659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, and Pahan K (2016). PPARα in lysosomal biogenesis: a perspective. Pharmacol. Res 103, 144–148. 10.1016/j.phrs.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, and Pahan K (2007). Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 104, 18754–18759. 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, and Pahan K (2009). Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci 29, 13543–13556. 10.1523/JNEUROSCI.4144-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Rangasamy SB, Modi KK, and Pahan K (2017). Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis. J. Neurochem 141, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, and Lee VMY (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Goedert M, and Spillantini MG (1998). Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol. Psychiatr 3, 462–465. 10.1038/sj.mp.4000458. [DOI] [PubMed] [Google Scholar]
- Hofer A, Berg D, Asmus F, Niwar M, Ransmayr G, Riemenschneider M, Bonelli SB, Steffelbauer M, Ceballos-Baumann A, Haussermann P, et al. (2005). The role of alpha-synuclein gene multiplications in early-onset Parkinson’s disease and dementia with Lewy bodies. J. Neural. Transm 112, 1249–1254. 10.1007/s00702-004-0263-3. [DOI] [PubMed] [Google Scholar]
- Jana A, and Pahan K (2010). Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J. Neurosci 30, 12676–12689. 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Hwang DJ, Koo JH, Um HS, Lee NH, Yeom DC, Lee Y, and Cho JY (2018). Association of exercise-induced autophagy upregulation and apoptosis suppression with neuroprotection against pharmacologically induced Parkinson’s disease. J. Exerc. Nutr. Biochem 22, 1–8. 10.20463/jenb.2018.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Avula R, Senjem ML, Samikoglu AR, Zhang B, Weigand SD, Przybelski SA, Edmonson HA, Vemuri P, Knopman DS, et al. (2010). Dementia with Lewy bodies and Alzheimer disease: neurodegenerative patterns characterized by DTI. Neurology 74, 1814–1821. 10.1212/WNL.0b013e3181e0f7cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karampetsou M, Ardah MT, Semitekolou M, Polissidis A, Samiotaki M, Kalomoiri M, Majbour N, Xanthou G, El-Agnaf OMA, and Vekrellis K (2017). Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep 7, 16533. 10.1038/s41598-017-15813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasnavis S, Roy A, Ghosh S, Watson R, and Pahan K (2014). Protection of dopaminergic neurons in a mouse model of Parkinson’s disease by a physically-modified saline containing charge-stabilized nanobubbles. J. Neuroimmune Pharmacol 9, 218–232. 10.1007/s11481-013-9503-3. [DOI] [PubMed] [Google Scholar]
- Koo JH, Jang YC, Hwang DJ, Um HS, Lee NH, Jung JH, and Cho JY (2017). Treadmill exercise produces neuroprotective effects in a murine model of Parkinson’s disease by regulating the TLR2/MyD88/NF-κB signaling pathway. Neuroscience 356, 102–113. 10.1016/j.neuroscience.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, and Lee SJ (2013). Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp. Mol. Med 45, e22. 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- MacInnes N, Iravani MM, Perry E, Piggott M, Perry R, Jenner P, and Ballard C (2008). Proteasomal abnormalities in cortical Lewy body disease and the impact of proteasomal inhibition within cortical and cholinergic systems. J. Neural Transm 115, 869–878. 10.1007/s00702-008-0027-6. [DOI] [PubMed] [Google Scholar]
- Mäkelä J, Tselykh TV, Kukkonen JP, Eriksson O, Korhonen LT, and Lindholm D (2016). Peroxisome proliferator-activated receptor-γ (PPARγ) agonist is neuroprotective and stimulates PGC-1α expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 102, 266–275. 10.1016/j.neuropharm.2015.11.020. [DOI] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, et al. (2016). Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374. 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini-Stoica H, Xu Y, Ballabio A, and Zheng H (2016). The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 39, 221–234. 10.1016/j.tins.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Kabuto H, Makino H, and Ogawa N (1997). Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 73, 45–48. 10.1016/s0165-0270(96)02211-x. [DOI] [PubMed] [Google Scholar]
- McGlinchey RP, and Lee JC (2015). Cysteine cathepsins are essential in lysosomal degradation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 112, 9322–9327. 10.1073/pnas.1500937112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakaki G, Canneva F, Chevessier F, Bode F, Menges S, Timotius IK, Kalinichenko LS, Meixner H, Müller CP, Eskofier BM, et al. (2019). Treadmill exercise intervention improves gait and postural control in alpha-synuclein mouse models without inducing cerebral autophagy. Behav. Brain Res 363, 199–215. 10.1016/j.bbr.2018.11.035. [DOI] [PubMed] [Google Scholar]
- Mondal S, Roy A, Jana A, Ghosh S, Kordower JH, and Pahan K (2012). Testing NF-κB-based therapy in hemiparkinsonian monkeys. J. Neuroimmune Pharmacol 7, 544–556. 10.1007/s11481-012-9377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wüllner U, Meitinger T, and Gasser T (2005). Multiple regions of alpha-synucle in are associated with Parkinson’s disease. Ann. Neurol 57, 535–541. 10.1002/ana.20438. [DOI] [PubMed] [Google Scholar]
- Pahan K (2006). Lipid-lowering drugs. Cell. Mol. Life Sci 63, 1165–1178. 10.1007/s00018-005-5406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidi RK, Jana M, Mishra RK, Dutta D, and Pahan K (2021a). Selective inhibition of the interaction between SARS-CoV-2 spike S1 and ACE2 by SPIDAR peptide induces anti-inflammatory therapeutic responses. J. Immunol 207, 2521–2533. 10.4049/jimmunol.2100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidi RK, Jana M, Mishra RK, Dutta D, Raha S, and Pahan K (2021b). ACE-2-interacting domain of SARS-CoV-2 (AIDS) peptide suppresses inflammation to reduce fever and protect lungs and heart in mice: implications for COVID-19 therapy. J. Neuroimmune Pharmacol 16, 59–70. 10.1007/s11481-020-09979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Roy A, Kundu M, Jana M, Luan CH, Gonzalez FJ, and Pahan K (2018). Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc. Natl. Acad. Sci. USA 115, E7408–E7417. 10.1073/pnas.1802021115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Roy A, Raha S, Kundu M, Gonzalez FJ, and Pahan K (2020). Upregulation of BDNF and hippocampal functions by a hippocampal ligand of PPARα. JCI Insight 5, 136654. 10.1172/jci.insight.136654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, and Carta AR (2014). Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis 71, 280–291. 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Poehler AM, Xiang W, Spitzer P, May VEL, Meixner H, Rockenstein E, Chutna O, Outeiro TF, Winkler J, Masliah E, and Klucken J (2014). Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 10, 2171–2192. 10.4161/auto.36436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha S, Dutta D, Roy A, and Pahan K (2021a). Reduction of Lewy body pathology by oral cinnamon. J. Neuroimmune Pharmacol 16, 592–608. 10.1007/s11481-020-09955-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha S, Ghosh A, Dutta D, Patel DR, and Pahan K (2021b). Activation of PPARα enhances astroglial uptake and degradation of β-amyloid. Sci. Signal 14, eabg4747. 10.1126/scisignal.abg4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy SB, Jana M, Roy A, Corbett GT, Kundu M, Chandra S, Mondal S, Dasarathi S, Mufson EJ, Mishra RK, et al. (2018). Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J. Clin. Invest 128, 4297–4312. 10.1172/JCI96209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Bousset L, George S, Madaj Z, Meyerdirk L, Schulz E, Steiner JA, Melki R, and Brundin P (2019). α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the ol-factory bulb. Acta Neuropathol Commun 7, 221. 10.1186/s40478-019-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ, and Pahan K (2013). Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor α. Cell Rep. 4, 724–737. 10.1016/j.celrep.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Jana M, Kundu M, Corbett GT, Rangaswamy SB, Mishra RK, Luan CH, Gonzalez FJ, and Pahan K (2015). HMG-CoA reductase inhibitors bind to PPARα to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metabol. 22, 253–265. 10.1016/j.cmet.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, and Pahan K (2009). Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol. Immunotoxicol. 31, 339–351. 10.1080/08923970902785253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, and Pahan K (2015). PPARα signaling in the Hippocampus: crosstalk between fat and memory. J. Neuroimmune Pharmacol 10, 30–34. 10.1007/s11481-014-9582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sanford AM (2018). Lewy body dementia. Clin. Geriatr. Med 34, 603–615. 10.1016/j.cger.2018.06.007. [DOI] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E, and Carta AR (2009). PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci 29, 954–963. 10.1111/j.1460-9568.2009.06657.x. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108. 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, and Lobel P (1997). Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 277, 1802–1805. 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Sorrentino ZA, Brooks MMT, Hudson V 3rd, Rutherford NJ, Golde TE, Giasson BI, and Chakrabarty P (2017). Intrastriatal injection of alpha-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol. Neurodegener 12, 40. 10.1186/s13024-017-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, and Goedert M (1997). α-Synuclein in Lewy bodies. Nature 388, 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Swanson CR, Joers V, Bondarenko V, Brunner K, Simmons HA, Ziegler TE, Kemnitz JW, Johnson JA, and Emborg ME (2011). The PPAR-gamma agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflammation 8, 91. 10.1186/1742-2094-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsika E, Moysidou M, Guo J, Cushman M, Gannon P, Sandaltzopoulos R, Giasson BI, Krainc D, Ischiropoulos H, and Mazzulli JR (2010). Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J. Neurosci 30, 3409–3418. 10.1523/JNEUROSCI.4977-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidoni C, Follo C, Savino M, Melone MAB, and Isidoro C (2016). The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev 36, 845–870. 10.1002/med.21394. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S, and Takahashi H (1998). α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett 249, 180–182. 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- Wenning GK, and Jellinger KA (2005). The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol. 109, 129–140. 10.1007/s00401-004-0935-y. [DOI] [PubMed] [Google Scholar]
- Xicoy H, Penuelas N, Vila M, and Laguna A (2019). Autophagic- and lysosomal-related biomarkers for Parkinson’s disease: lights and shadows. Cells 8, E1317. 10.3390/cells8111317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J, Lee SB, Lee HS, Yang HO, Park J, Kim JS, Oh E, Park S, and Jang W (2018). Cerebrospinal fluid levels of autophagy-related proteins represent potentially novel biomarkers of early-stage Parkinson’s disease. Sci. Rep 8, 16866. 10.1038/s41598-018-35376-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med 24, 931–938. 10.1038/s41591-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does report any original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.