

Highlights
-
•
This review is part of the series on the clinical neurophysiology of movement disorders and focuses on Parkinson’s disease and parkinsonism.
-
•
The pathophysiology of cardinal parkinsonian motor symptoms and myoclonus are reviewed.
-
•
The recordings from microelectrode and deep brain stimulation electrodes are reported in detail.
Keywords: Tremor, Bradykinesia, Gait and balance, Electroencephalography, Electromyography, Long latency reflexes, Transcranial magnetic stimulation, Local field potentials, Microelectrode recording, Deep brain stimulation
Abstract
This review is part of the series on the clinical neurophysiology of movement disorders. It focuses on Parkinson’s disease and parkinsonism. The topics covered include the pathophysiology of tremor, rigidity and bradykinesia, balance and gait disturbance and myoclonus in Parkinson’s disease. The use of electroencephalography, electromyography, long latency reflexes, cutaneous silent period, studies of cortical excitability with single and paired transcranial magnetic stimulation, studies of plasticity, intraoperative microelectrode recordings and recording of local field potentials from deep brain stimulation, and electrocorticography are also reviewed. In addition to advancing knowledge of pathophysiology, neurophysiological studies can be useful in refining the diagnosis, localization of surgical targets, and help to develop novel therapies for Parkinson’s disease.
1. Introduction
The techniques for studying the pathophysiology of movement disorders were described in previous chapters of this series (Hallett et al., 2021, Rothwell et al., 2021). This chapter focuses on the pathophysiology of Parkinson’s disease (PD) and parkinsonism. We first discuss the pathophysiology of the cardinal motor signs of PD including tremor, rigidity, bradykinesia, gait disturbance and postural instability. This is followed by a review of results from non-invasive methods including electromyography (EMG), electroencephalography (EEG), transcranial magnetic stimulation (TMS), long-latency reflexes (LLR). The findings from invasive microelectrode recordings (MER) during deep brain stimulation (DBS) surgery, and local field potentials (LFP) recordings from MER, DBS and electrocorticographic (ECoG) electrodes are also addressed.
2. Tremor in Parkinson’s disease and parkinsonism
2.1. Clinical phenotype of parkinsonian tremor
Tremor is defined as an involuntary, rhythmic, oscillatory movement of a body part, and it is one of the cardinal motor signs of PD. The classic parkinsonian tremor occurs at rest, at a frequency of 3.5–7 Hz, often with a pill-rolling movement of the thumb across the fingers. Rest tremor is seen in roughly 75% of patients with PD, where it is an early sign that may decrease with disease progression (Helmich, 2018). The tremor usually has an asymmetric distribution, and may involve the arms, legs, and face. During posture holding such as with arms outstretched, rest tremor briefly disappears for a variable delay of several seconds to sometimes more than a minute. This phenomenon is called resetting, and it reliably distinguishes PD from essential tremor (ET) and dystonic tremor (Schwingenschuh et al., 2010, Papengut et al., 2013). After stable posturing, in two thirds of patients a “re-emergent tremor” returns (Fig. 1A,B) (Dirkx et al., 2018, Belvisi et al., 2017). This has led to the idea that PD tremors are expressions of “tremor of stability” (Hallett, 2014). Furthermore, about 15% of PD patients have a “pure postural tremor”, which starts without delay after posturing, and is at a higher frequency (±8 Hz; Fig. 1C)(Dirkx et al., 2018) than rest tremor. Finally, some PD patients have a kinetic tremor, which can have a broad frequency range of 6–15 Hz (Raethjen et al., 2005).
Fig. 1.

Tremor recording and time–frequency representations of Parkinson’s disease tremors. Panel A shows EMG and accelerometry recordings (left) and power spectral analysis (right) in a Parkinson’s disease (PD) patient with re-emergent tremor. The patient made a rapid wrist extension at the beginning of the trace, after which the tremor amplitude was transiently reduced (resetting). The extensor carpi radialis (ECR) and flexor carpi radialis (FCR) muscles show an alternating pattern of rhythmic EMG bursts at 5 Hz with harmonic at double tremor frequency (10 Hz). Panels B and C show time–frequency representations (TFR) of two different PD patients with re-emergent tremor (panel B) or pure postural tremor (panel C). The plots show EMG power (of a tremulous muscle, color coding) over the course of 70 s (x-axis) and as a function of frequency (y-axis). At 10 s, both patients extend their wrist. Panel B illustrates three key characteristics of PD tremor: (1) power at tremor frequency and double tremor frequency (first harmonic); (2) suppression of tremor power after wrist extension; (3) waxing and waning of tremor power over time. Panel C illustrates a patient with a typical “pure postural tremor”, which typically starts immediately after posturing and occurs at a relatively high frequency of ± 8 Hz. ECR = extensor carpi radialis muscle; EMG = electromyography; FCR = flexor carpi radialis muscle.
These various PD tremor types respond differently to dopaminergic medication: rest tremor has the best response, although in 39% of patients it is resistant to even high doses of levodopa (Zach et al., 2020). Re-emergent tremor also responds to levodopa, albeit slightly less than rest tremor, and “pure postural tremor” and kinetic tremor do not respond to levodopa (Dirkx et al., 2018, Belvisi et al., 2018). PD rest tremor typically increases during cognitive tasks or motor coactivation, e.g., counting or tapping with the contralateral hand, and this can be used by clinicians to bring out the tremor in the clinic (Dirkx et al., 2020). PD tremor is very sensitive to stress, and it can be reduced with stress-reducing interventions such as mindfulness (van der Heide et al., 2021) and relaxation exercises (Blakemore et al., 2019). Patients with PD may also manifest tremor in the cranio-cervical area, including chin, tongue, and voice tremor. Voice tremor in PD may resemble voice tremor in non-parkinsonian conditions such as essential tremor and laryngeal dystonia (Fabbri et al., 2017, Suppa et al., 2021, Suppa et al., 2020, Suppa et al., 2022b).
Rest tremor is much less common in patients with atypical parkinsonism: it was seen in only 27.5% of patients with (pathologically confirmed) multiple system atrophy (MSA), 30.8% in patients with progressive supranuclear palsy (PSP), and 53.8% of patients with dementia with Lewy body dementia (DLB)(Miki et al., 2019). Furthermore, if present in atypical parkinsonism, tremor usually lacks the typical pill-rolling aspect. Action tremor is slightly more common than rest tremor in atypical parkinsonism. If present, it usually has a jerkier aspect than tremor in PD, and it is sometimes difficult to distinguish from (minipoly)myoclonus.
2.2. Neurophysiology of PD tremor
PD rest tremor typically involves alternating EMG bursts in agonist and antagonist muscles. The tremor rhythm is not strictly sinusoidal, which leads to relatively high power at harmonics (e.g., at double tremor frequency) in the power spectrum (Fig. 1B), as compared to ET (Muthuraman et al., 2011) and tremor in MSA (Su et al., 2020). When observed over longer time periods, PD tremor power waxes and wanes spontaneously (Fig. 1B), and it may also shift between different muscle pairs. Compared to ET, PD tremor has a lower tremor stability index (TSI), which indicates that there are more cycle-to-cycle changes in tremor frequency (di Biase et al., 2017). The TSI measures how “jittery” the tremor frequency of a given individual is. More specifically, the TSI quantifies how much the tremor slows down or speeds up across tremor cycles, and how these frequency adaptations vary as a function of tremor frequency. The low TSI in PD suggest that there are larger spontaneous changes in tremor frequency in PD than in ET. This may have diagnostic value in distinguishing ET from PD tremor, although higher tremor power also leads to lower TSI. It is not clear how the TSI informs us about the nature of tremor oscillators.
Deep brain recordings in the ventral intermediate nucleus (VIM) of the thalamus, which receives cerebellar input, and in the subthalamic nucleus (STN) and internal globus pallidus (GPi), have shown cells firing at tremor frequency (Lenz et al., 1988, Hurtado et al., 1999, Levy et al., 2000, Hutchison et al., 1997). Within each of these regions, different cells are involved in tremor in different body parts (Pedrosa et al., 2012). The presence of multiple oscillators explains why there is no coherence between tremor in different body parts. Furthermore, EEG and magnetoencephalography (MEG) studies have shown oscillatory activity at tremor frequency in the entire cerebello-thalamo-cortical circuit (Timmermann et al., 2003, Muthuraman et al., 2018). Within this circuit, tremor rhythm and tremor amplitude may be controlled by different nodes, depending on the tremor type. Specifically, single-pulse transcranial magnetic stimulation (TMS) over the primary motor cortex (M1) can “reset” the rhythm of both rest tremor and re-emergent tremor, indicating that M1 is part of the tremor oscillator (Ni et al., 2010, Helmich et al., 2021, Leodori et al., 2020). In contrast, TMS over the cerebellum can only reset postural and re-emergent tremor, but not rest tremor (Ni et al., 2010, Helmich et al., 2021). Furthermore, single pulse TMS over M1, but not over the cerebellum, transiently reduces tremor amplitude (Helmich et al., 2021). This suggests that M1 controls tremor amplitude, while the tremor oscillator is more distributed and may depend on the context in which the tremor occurs.
2.3. Functional magnetic resonance imaging (fMRI) in PD tremor
Although fMRI does not have the temporal resolution to identify cycle-by-cycle tremor changes, the slow waxing-and-waning of tremor power (captured with EMG or accelerometry during scanning; Fig. 1B) can be used to localize tremor-related brain activity. Combined EMG-fMRI has revealed a specific and reproducible pattern of tremor-related brain activity involving the cerebello-thalamo-cortical circuit and basal ganglia (Helmich et al., 2011, Dirkx et al., 2016, Dirkx et al., 2019). Network analyses suggest that within this distributed network, activity starts within the GPi and is then relayed to the cerebello-thalamo-cortical circuit (Dirkx et al. 2016). This has resulted in the “dimmer-switch model”, which states that the basal ganglia initiate tremor episodes, while the cerebello-thalamo-cortical circuit amplifies the tremor (Helmich 2018). This hypothesis has recently been confirmed using intracranial recordings from the STN and sensorimotor cortex, where inter-regional connectivity between no-tremor, tremor onset, and sustained tremor episodes was compared. During tremor onset, there was a drive from STN to cortex, which was reversed during sustained tremor, and the drive was absent when there was no tremor (Lauro et al., 2021). Tremor-related somatosensory afferents to the cerebellum may have an additional role in stabilizing the tremor rhythm (Muthuraman et al. 2018). The neurophysiological changes in the GPi that lead to tremor onsets have not been established, and this deserves future research.
Tremor-related activity in the cerebello-thalamo-cortical network is sensitive to dopaminergic medication, which reduces tremor by specifically inhibiting the VIM (Dirkx et al. 2017). Furthermore, cognitive load was found to increase tremor through excitatory, noradrenergic influences to the VIM (Dirkx et al. 2020). There are also inter-individual differences in the architecture of the cerebello-thalamo-cortical tremor circuit: PD patients with dopamine-resistant rest tremor had increased activity in the cerebellum (interposed nuclei) compared to patients with dopamine-response tremor (Dirkx et al. 2019). This suggests that dopamine-resistant PD tremor may be treated by interventions that target the cerebellum.
2.4. Summary for PD tremor
PD harbors different types of tremor, each with their own neurophysiological signature. In the future, pathophysiological insights in individual patients may be used for targeted interventions to reduce tremor.
3. Rigidity and bradykinesia
Bradykinesia and rigidity are the two main symptoms of PD and other parkinsonisms (Berardelli et al., 2001, Postuma et al., 2015, Berardelli et al., 2013). From a pathophysiological point of view, clinical and experimental evidence suggests that these symptoms are primarily related to dysfunction of the basal ganglia resulting from dopaminergic denervation (Delong and Wichmann, 2007). However, recent evidence shows that both bradykinesia and rigidity in PD and parkinsonism are underpinned by a complex pathophysiology that includes other brain areas in addition to the basal ganglia, namely motor cortical areas, cerebellum, and brainstem (Linn-Evans et al., 2020, Bologna et al., 2020).
3.1. Pathophysiology of rigidity
Rigidity is a major motor sign of PD, particularly in the context of the so-called bradykinetic-rigid form, and in parkinsonian syndromes (Postuma et al., 2015, Berardelli et al., 2013). Rigidity is defined as an increase in muscle tone that can be appreciated as speed-independent resistance to passive mobilization of a body segment (Antonini et al., 2013, Postuma et al., 2015). Although the extent of rigidity is generally associated with the severity of PD, it can vary significantly from patient to patient (Bologna and Paparella, 2020).
Concerning the pathophysiology of rigidity, in addition to the failure to relax, neurophysiological studies have demonstrated excitability changes at the cortical and subcortical levels in animal models and in patients with PD (Delwaide et al., 1986, Bologna and Paparella, 2020). Studies based on EMG techniques applied to the stretch reflex have revealed that alterations in LLR (discussed further in section 6) may contribute to rigidity (Berardelli et al., 1983, Rothwell et al., 1983) in a different way compared to spasticity, in which increased short-latency reflexes (SLR) have been found (Dietz and Sinkjaer, 2007). It is believed that alterations in the stretch reflexes contribute to altered integration of afferent sensory stimuli (mediated by group II afferents in the lower extremities and by Ia muscle spindle afferent fibers in the upper extremities) at both the spinal level and at the transcortical circuits. The spinal component of the LLR is particularly relevant for the lower limbs, whereas the transcortical component has greater importance for the upper limbs. At the spinal level, alterations in stretch reflexes are likely implicated in the pathophysiology of rigidity with changes in interneuron (Ia and Ib) and spinal motoneuron excitability (Simonetta Moreau et al., 2002, Marchand-Pauvert et al., 2011, Pasquereau and Turner, 2013). The latter are thought to reflect, at least in part, neurodegeneration in the brainstem. For example, neurodegenerations in the reticulospinal system and various brainstem nuclei, particularly the locus coeruleus and raphe nucleus, result in the altered influence of descending noradrenergic and serotonergic systems on spinal circuits (Delwaide et al., 1993, Braak et al., 2006, Simonetta Moreau et al., 2002, Marchand-Pauvert et al., 2011, Pasquereau and Turner, 2013, Xia et al., 2016). Additional evidence has supported the possible role of brainstem dysfunction in the pathophysiology of rigidity in PD. The phenomenon of reduced muscle activation (atony), which is considered a physiological phenomenon during rapid eye movement (REM) sleep, has been specifically studied (Chahine et al., 2014). PD patients with REM sleep without atonia (RSWA), which likely reflects more severe pathology of the brainstem and non-dopaminergic systems (Postuma et al., 2019), have been shown to exhibit more severe and symmetrical stiffness than individuals without RSWA during wakefulness (Chahine et al., 2014, Linn-Evans et al., 2020).
From a broader perspective, studies on the pathophysiology of rigidity in PD support the hypothesis that this motor sign is not exclusively an expression of altered basal ganglia activity due to dopaminergic denervation, but rather reflects the dysfunction of various brain areas (including the brainstem and spinal cord) and the involvement of non-dopaminergic neurotransmitter systems.
3.2. Pathophysiology of bradykinesia
By definition, the term bradykinesia (slowness of movement) includes the phenomenon of the 'sequence effect’ which refers to the progressive decrease in speed and amplitude of repetitive and continuous movements (Agostino et al., 2003, Berardelli et al., 2013, Postuma et al., 2015). However, in some cases the term bradykinesia is used extensively to indicate not only movement slowness and sequence effect, but also movements of reduced amplitude (hypokinesia), motor blocks, hesitation, and the absence of movement (akinesia)(Schilder et al., 2017). In addition to clinical observations, bradykinesia in PD and parkinsonism has been characterized in recent years using neurophysiological techniques based on quantitative movement analysis (Hasan et al., 2017). These studies have shown that the characteristics of bradykinesia can vary considerably because of several factors, including disease severity and the use of dopaminergic drugs (Espay et al., 2009, Espay et al., 2011, Bologna et al., 2016a, Bologna et al., 2016b). In the latter regard, clinical and experimental observations demonstrated that levodopa therapy improves bradykinesia but the effects are variable (Espay et al., 2009, Espay et al., 2011) and does not significantly influence the sequence effect (Kang et al., 2010, Wu et al., 2016, Suppa et al., 2017a, Bologna et al., 2018, Bologna et al., 2020).
From a pathophysiological perspective, bradykinesia is traditionally considered a consequence of nigrostriatal dopaminergic depletion, altered basal ganglia activity, and subsequent excessive thalamic and M1 inhibition (Berardelli et al., 2001, Delong and Wichmann, 2007). Originally, changes in firing frequencies in the various basal ganglia nuclei (direct and indirect pathways) were emphasized. The basal ganglia are implicated in the process of movement selection and inhibition (through direct and indirect pathways, respectively) and in the encoding of movement speed and amplitude. From a motor perspective, altered basal ganglia activity results in increased reaction times and decreased movement speed and amplitude due to dopaminergic depletion (Berardelli et al., 2001, Delong and Wichmann, 2007, Bologna et al., 2020). Recent evidence has pointed to more complex phenomena, including abnormal oscillatory activity, with increased beta activity (13–35 Hz) that correlates with specific features of bradykinesia (e.g., sequence effect) in some cases (Steiner et al., 2017, Bologna et al., 2020), and abnormal plasticity at the level of the basal ganglia (Milosevic et al., 2019).
In addition to the basal ganglia, recent evidence suggests the possible involvement of motor cortical areas (particularly M1) and the cerebellum in the pathophysiology of bradykinesia (Bologna et al., 2020). Electrophysiological studies conducted in animal models and in humans with PD using non-invasive neurophysiological techniques have shown that excitability and plasticity alterations in M1 may correlate with specific features of bradykinesia (Pasquereau and Turner, 2011, Pasquereau et al., 2016, Bologna et al., 2018). Cerebellar dysfunction is now considered an additional potential mechanism in the pathophysiology of bradykinesia, a hypothesis supported by finding of reciprocal anatomical connections between the cerebellum and basal ganglia and by results obtained in neurophysiological and neuroimaging studies (Wu and Hallett, 2013, Bostan and Strick, 2018, Bologna et al., 2020). For example, a possible relationship between cerebellar involvement and bradykinesia of the upper limb, particularly in writing (micrographia), has been demonstrated in PD (Wu et al., 2016). Motor brain areas and the cerebellum could play a compensatory role in counteracting the appearance of sequence effect since they are involved in movement feedback, which is particularly important in the execution of repetitive and continuous movements. Finally, altered integration of sensory information at the cortico-subcortical level is an important element in the pathophysiology of bradykinesia, as demonstrated by the study of sensorimotor integration and tactile temporal discrimination threshold.
Overall, bradykinesia should be interpreted as the consequence of a dysfunctional network, where the basal ganglia, motor areas, cerebellum, and altered integration of sensory information at the cortical and subcortical levels play a critical role. From a network perspective, the various features of bradykinesia (e.g., movement slowness and sequence effect) seem to be mediated by involvement of different brain areas and mechanisms. The network prospective may also explain the observation of bradykinesia in other clinically and pathophysiologically heterogeneous conditions, including those not associated with parkinsonism (Paparella et al., 2021).
4. Gait and balance in PD
Gait and balance disturbance plays a major role in the impairment of function and quality of life of PD patients (Moore et al., 2007). Impairment in gait and balance lead to frequent falls and are associated with increased mortality (Fasano et al., 2017a). A growing body of literature has attempted to understand the clinical, pathophysiological and therapeutic aspects of PD-related gait and balance disorders.
From a pathophysiological standpoint, these axial motor problems can be divided into: 1. Dopaminergic appendicular issues, 2. Gait-specific issues, and 3. Balance and non-dopaminergic issues. This subdivision follows the temporal progression of the neurodegenerative process of PD.
4.1. Dopaminergic appendicular issues
Probably the earliest clinical sign of gait involvement in PD is the reduction of the synkinetic arm swinging associated with human locomotion. In addition, instrumental evaluation (gait analysis) can detect subtle bradykinetic signs of the lower limbs, such as the narrowing of the base of support during dual task in patients with idiopathic REM sleep behavioural disorder (Ehgoetz Martens et al., 2019).
During progression through the clinical stages of the disease, a variety of motor signs can affect the lower limbs, usually asymmetrically. Bradykinesia is the most common and causes step length reduction, swing phase shortening, narrowing of the base of support and reduced foot clearance (shuffling gait) (Fig. 2A) (Fasano and Bloem, 2013). Bradykinesia also contributes to ‘sequence effect’, recognized in this context as the progressive shortening of step length (Iansek et al., 2006) (Fig. 2A). Leg rigidity, even when severe, plays a minor role, especially when compared to bradykinesia.
Fig. 2.

Schematic illustrations of Parkinson’s disease gait. (A) Parkinson’s disease gait is characterized by step length reduction, swing phase shortening, narrowing of the base of support and reduced foot clearance (shuffling gait); bradykinesia also contributes to ‘sequence effect’, defined as the progressive shortening of step length (bottom row). The asymmetry of these gait features tends to persist during disease progression. (B) The pharmacokinetics of levodopa provides a useful framework to understand how axial motor problems can respond to dopaminergic treatment, be caused by or be resistant to levodopa.
Lower limb rest tremor does not usually impact gait as it typically disappears during locomotion. In rare cases, PD can be associated with a typical high-frequency orthostatic tremor (Mestre et al., 2012) or a ‘pseudo-orthostatic tremor’, which is a slow dopamine-responsive rest tremor of the leg and trunk (Thomas et al., 2007). These tremors do not necessarily affect gait as they are present during quiet standing but contribute to a subjective feeling of imbalance. Orthostatic myoclonus with 9–16 Hz non-rhythmic EMG bursts of 50–100 ms durations affecting the legs during standing or gait initiation can also contribute to unsteadiness. This has been described in neurodegenerative conditions, including advanced PD (Glass et al., 2007).
Dystonia of the foot, either inversion or plantar flexion mimicking foot drop, can be one of the earliest PD signs, especially in young onset patients (Elia et al., 2014). Interestingly, some of these patients might present with a paroxysmal exercise-induced dystonia for years before PD is formally diagnosed (Bozi and Bhatia, 2003).
With the exception of orthostatic tremor, these signs respond well to dopaminergic treatments. Dopaminergic treatments, and particularly levodopa, can cause dyskinesias. Peak-dose dyskinesias generally do not impair gait as they tend to involve mainly the upper body, which can still be a destabilizing factor when balance is impaired (see below). Biphasic dyskinesias are less common but are an important source of gait impairment as they involve the lower limbs (Fig. 2B), often producing bizarre phenomenology (Ruzicka et al., 2011).
4.2. Gait-specific issues
While the spatial features of locomotion are impaired by PD, the temporal properties are not and can compensate. The typical short-stepped gait of PD patients is indeed characterized by increased cadence (steps/minute), which results in a normal or slightly lower speed, at least in the initial stages of the disease (Morris et al., 1994). The further reduction of step length, the occurrence of sequence effect and the compensatory increase in cadence are the basis of festination. During festination, gait velocity increases while the patient bends forward chasing their own center of gravity. Festination can end spontaneously if the patient is able to gain control of stepping generation or can terminate abruptly, either with an episode of freezing of gait (FOG) or a forward fall (Fasano and Bloem, 2013).
FOG is the most typical gait abnormality seen in PD. Its prevalence increases with disease progression, up to 96% of patients in late stages of the disease (Perez-Lloret et al., 2014). It is defined as the episodic inability to generate an effective stepping (Nieuwboer and Giladi, 2013) and can present with a variety of clinical phenomena (Table 1) (Fasano and Lang, 2015). Probably the most characteristic clinical features of FOG are represented by the hesitation when initiating gait or when turning. These phenomena are not preceded by festination, which is seen only when FOG occurs during straight ongoing walking.
Table 1.
Classification of freezing of gait (modified from (Fasano and Lang 2015)).
| Phenomenological classification* | |
| Akinetic FOG (occurs at gait initiation) |
|
| Motor FOG (arrests during ongoing gait) |
|
| Pharmacological classification* | |
| Off-state FOG | Most frequent type, relieved by dopaminergic medications |
| Pseudo-on-state FOG | Seen during a seemingly optimum on-state, but which nevertheless improves with increased dopaminergic medication |
| On-state FOG | Rarest form, induced by dopaminergic medication |
| Resistant (or unresponsive) FOG | Indifferent to changes in dopaminergic medication, often seen in parkinsonian disorders other than PD, or in the late stages of PD |
Abbreviations: *: Different freezing of gait types can appear in one patient; **: can be triggered or worsened by external circumstances; FOG: freezing of gait; PD: Parkinson’s disease.
The pathophysiology of FOG is not fully established and several theories have been proposed (Nieuwboer and Giladi, 2013). The prevalent view is that FOG results from the combination of several motor and non-motor factors. The motor factors include: sequence effect (Iansek et al., 2006), abnormal cadence/step length relationship (Morris et al., 1994), asymmetry (Plotnik et al., 2005), ineffective anticipatory postural adjustments (Jacobs et al., 2009) and abnormal cerebellar function (Fasano et al., 2017b). Different attempts have been made to unify these motor components (Fasano et al., 2016). The aforementioned walking issues also increase gait variability, which is further worsened by motor or cognitive dual tasking, thus indicating a certain degree of cortical compensation (Fasano and Bloem, 2013). Accordingly, non-motor factors alone are not sufficient to produce FOG, but they contribute to its generation and severity (Ehgoetz Martens et al., 2018). These include: executive cognitive dysfunction (Amboni et al., 2008, Shine et al., 2013), visuo-spatial issues (Vercruysse et al., 2012), and anxiety (Ehgoetz Martens et al., 2014). Several neuroimaging studies have tried to identify the network(s) involved in FOG pathophysiology and most findings converge on the connections between the frontal lobe and the basal ganglia, the caudate and brainstem circuits including the pedunculopontine nucleus (PPN), especially in the right hemisphere (Fasano et al., 2015b, Bharti et al., 2019). More recent studies recording LFP in DBS patients during walking have identified that FOG is associated with a spike of beta oscillation followed by theta oscillation in the STN and substantia nigra pars reticulata (Chen et al., 2019, Georgiades et al., 2019).
An important feature of FOG patients is their ability to temporarily overcome the problem when presented with visual cues or other external stimuli, which can be adopted for therapeutic purposes (Muthukrishnan et al., 2019). In fact, dopaminergic therapies often provide disappointing results (Fig. 2B, Table 1) (Espay et al., 2012), in keeping with the notion that extra-dopaminergic pathways are involved (Bohnen et al., 2014). The effects of DBS of different targets can be disappointing and often detrimental, also when targeting cholinergic experimental targets such as the PPN (Fasano et al., 2015a). New therapeutic approaches for FOG include closed-loop systems involving wearable sensors for the automatic detection or prediction of FOG, and the real-time administration of sensory stimuli to improve gait (Suppa et al., 2017b, Borzì et al., 2022).
4.3. Balance and non-dopaminergic issues
PD causes an anterior-posterior instability, clinically manifesting with retropulsion when the disease progresses (Fasano et al., 2012). In fact, forward or lateral falls tend to be caused by FOG during straight walking or turning, respectively. The anterior-posterior instability fits with the clinical observation that PD patients can perform tandem gait and ride a bicycle. Accordingly, gait parameters show a narrow base of support but a longer time spent on double limb support (Fig. 2A) (Fasano and Bloem, 2013). Falls can respond to dopaminergic treatments when it is caused by FOG or other appendicular dopaminergic signs, such as slow compensatory stepping in case of perturbation. Conversely, axial motor dysfunction is usually refractory to levodopa, which can lead to abnormal postural control when turning even at early stages of PD (Zampogna et al., 2021).
The spreading of degeneration to non-dopaminergic areas of the brain, particularly the cholinergic system, is an important factor in causing balance impairment, as shown by the association with cognitive impairment and REM sleep behavioural disorder, neuroimaging studies (Bohnen et al., 2009), and the modest but positive outcome of trials using cholinergic drugs (Chung et al., 2010) or PPN DBS (Moro et al., 2010).
Balance is however a complex function, also involving higher cortical functions, such attention and risk awareness. Clinically this manifest with ‘reckless gait’ or features of ‘highest level gait disorders’ (Fasano and Bloem, 2013). By integrating kinematic and EMG recordings, posturography may be used to examine the specific components of the complex sensorimotor system underlying balance control, including short-, medium- and long-latency postural reflexes (Rogers and Mille, 2018). In PD, posturography disclosed abnormal central proprioceptive integration as well as impaired scaling of postural responses during external perturbations, possibly contributing to postural instability (Nonnekes et al., 2013, Zampogna et al., 2020). Finally, other factors can contribute to falls in PD patients, such as the side effects of sedating drugs, dyskinesias or dysautonomia causing orthostatic hypotension (Fasano et al., 2017a).
5. EEG and evoked potential studies in PD
Neurons, especially in thalamic nuclei and in the cerebral cortex, exhibit intrinsic oscillations, which form the basis for macroscopic rhythms, detectable with EEG and magnetoencephalography (MEG).
5.1. Cortical oscillations in PD
EEG has been used to investigate the pathophysiology of PD and related disorders. Oscillatory activities have been reported at a variety of frequencies between 4 and 60 Hz. The frequency range between 13 Hz and 30 Hz is labelled as beta frequency.
EEG and MEG studies suggest that advanced PD is associated with pathologically increased cortical beta power (Berendse and Stam, 2007). This association between cortical beta power and PD has also been demonstrated in animal models of PD with dopamine depletion (Mallet et al., 2008). However, increased cortical beta power has also been shown in early PD, especially in bilateral primary sensorimotor cortices (Pollok et al., 2012).
In line with these findings, stimulation at the beta frequency of cortical sites may cause slowing of movements as shown by studies using transcranial alternating current stimulation (tACS) in healthy subjects (Joundi et al., 2012). There are some discrepant findings in PD patients with STN DBS. A study found worsening of Unified Parkinson’s disease rating scale (UPDRS) motor scores with STN DBS at 10 Hz but not at 20 Hz (beta frequency) compared to no stimulation (Timmermann et al., 2004). Other studies reported that finger tapping rate decreased with STN DBS at 5 Hz and 20 Hz but not at 10 Hz (Eusebio et al., 2008), while the rising slope in a reaction time grip task decreased with 20 Hz but not with 5 or 10 Hz STN DBS compared to no stimulation, although 50 Hz appeared to have similar effect as 20 Hz (Chen et al., 2011). These studies did not measure motor UPDRS, and a study showed no worsening of motor UPDRS with beta frequency stimulation (Tsang et al., 2012a). These findings suggest that no single pathological frequency can reflect all parkinsonian motor symptoms and specific frequency bands may be correlated with specific motor performance parameters (Timmermann and Florin, 2012).
Other neurophysiological biomarker in PD have also been examined. A measure known as phase-amplitude coupling (PAC) examines the synchronization between phases of a low frequency and the amplitude of a higher frequency. The PAC between beta phase and broadband gamma amplitude (>50 Hz), which is thought to reflect the spiking activity of populations of neurons (Manning et al., 2009), has been examined. Elevated beta-gamma PAC in PD was originally found with invasive electrocorticography (ECoG) studies (de Hemptinne et al., 2013, de Hemptinne et al., 2015) and subsequently shown in EEG studies (Swann et al., 2015, Jackson et al., 2019). Beta-gamma PAC over sensorimotor cortex was elevated in PD patients off medications compared to those on medications and to healthy controls (Swann et al., 2015). Elevated PAC might reflect modulation of spiking activity by low-frequency rhythms (Canolty et al., 2006), and in PD there is excessive synchrony (de Hemptinne et al., 2013, de Hemptinne et al., 2015). Neurophysiological biomarkers that change with the disease state may be helpful to improve patient care with appropriate adjustments of pharmacological treatments as well as brain stimulation parameters such as in closed-loop DBS. The shape of the beta oscillations is another neurophysiological biomarker. PD patients are characterised by beta oscillations with non-sinusoidal shape (Jackson et al., 2019). Moreover, the shape changes with the medication state as greater sharpness asymmetry and steepness asymmetry of canonical beta oscillations over the sensorimotor cortex was found in PD patients off medications than on medications (Jackson et al., 2019). A novel neurophysiological biomarker in PD involves nonlinear cortical and subcortical signals has also been described (Ozkurt et al., 2020). Using LFP and MEG recordings in PD patients treated with STN-DBS, nonlinearity in the subcortical high beta band in off-medication state and in the cortical alpha (8–12 Hz) band in the on-medication state was found (Ozkurt et al., 2020). These measures are promising candidates for use in closed-loop DBS devices and to optimise pharmacological treatment.
5.2. Somatosensory evoked potentials studies in PD
Somatosensory evoked potentials (SEPs) are another neurophysiological parameter. PD patients showed reduction and in some cases absent frontal N30 SEPs to median nerve (MN) stimulation at rest (Rossini et al., 1989, Rossini et al., 1991, Cheron et al., 1994, Bostantjopoulou et al., 2000). However, some studies did not confirm this finding (Garcia et al., 1995, Drory et al., 1998). Several studies showed that dopaminergic treatments such as apomorphine or levodopa (Rossini et al., 1993, Cheron et al., 1994, Ulivelli et al., 1999) and bilateral STN or GPi DBS (Pierantozzi et al., 1999) increased the N30 SEP amplitude. Consequentially, the frontal N30 SEP has been suggested as a dopamine-dependent physiological marker of basal ganglia modulation of non-primary motor cortical generators of this SEP (Cheron, 1999). Abnormal sensory attenuation, also known as sensory gating, has been found in PD (Abbruzzese and Berardelli, 2003). A study found reduced sensory attenuation of the N20-P25 SEP component, measured as reduced SEP amplitudes at the onset of movement, in PD patients off dopaminergic treatment but improved in patients on treatment (Macerollo et al., 2016). These data support the hypothesis that a failure in sensory gating prior to movement onset in PD may contribute to difficulties in movement initiation in PD. More specifically, bradykinesia may be related to deficits in the modulation of the gain of the afferent signal.
Taken together these studies indicate the utility of SEPs as a neurophysiological biomarker in different phases of active movements. Indeed, SEPs at the onset of movement rather than in the later tonic phase of movement may reflect different aspects of sensorimotor integration and plasticity changes.
In conclusion, SEPs can evaluate pathological changes related to PD and parkinsonism in research studies. Moreover, they may be useful as clinical outcomes of rehabilitation protocols for neurodegenerative diseases in which somatosensory and motor systems are involved. For instance, a rehabilitation technique that interfere or influence somatosensory processing such as vibratory peripheral stimulation, brain stimulation and repetitive motor tasks may use SEPs pre- and post- treatment to develop a more thorough understanding of changes in the somatosensory system. Overall, advances in EEG processing and analysis as well as ability to combine with brain stimulation techniques such as TMS, transcranial direct current stimulation (tDCS) and DBS has expanded the possibility to develop valuable tools to investigate pathophysiology related to sensorimotor integration as well as neural plasticity.
6. Long latency reflex and cutaneous silent period
6.1. Long latency reflexes
LLR include the muscle stretch reflex (MSR), mixed nerve electrical stimulation evoked long latency reflex (eLLR), and cutaneomuscular reflex (CMR). These reflexes may be useful tools to assess the interactions between the peripheral and central nervous systems. The latencies of some long latency reflexes are in the range of 50 ∼ 80 ms, which are longer than the expected traveling time if they only involve the peripheral sensory-motor reflex circuits alone. Therefore, they could be mediated by a transcortical reflex pathway from peripheral sensory nerve to the posterior column in the spinal cord, ascend to the sensory cortex, and then to the motor cortex and descend back to alpha motor neuron pools through the pyramidal tract (Lourenço et al., 2006). Primate studies also suggested that spinal and brainstem structures such as the red nucleus may mediate the long latency responses (Soteropoulos and Baker, 2020, Herter et al., 2015).
6.2. Muscle stretch reflex
MSR is a commonly used LLR measure performed with rapid passive joint flexion or extension and can be measured in both the upper and lower limbs. The MSR consists of the M1, M2 and M3 responses. The M1 response, which is also termed the SLR, has onset latencies of 20–40 ms in forearm muscles and 35–40 ms in the soleus muscle, and represents spinal segmental reflexes. Both M2 and M3 are considered LLR. The M2, which is sometimes labeled as the medium latency reflex, has onset of around 50–60 ms in forearm muscles and 60–80 ms in the soleus muscle. In the upper limbs, the M2 is mainly mediated by Ia afferents from muscle spindles (Grey et al., 2001). There is evidence that it involves a transcortical pathway (Capaday et al., 1991, Palmer and Ashby, 1992, Thilmann et al., 1991). The pathways mediating the M2 is different in the lower limb. Since the MSR M2 response was unaffected by cold temperature and ischemia, it could be mediated mainly by group II afferents from the secondary spindle endings and convergent excitation from group I afferents onto the group II interneurons (Berardelli et al., 1983, Pierrot-Deseilligny and Burke, 2012). The M2 in the lower limb probably rely more on spinal pathways rather than supraspinal transcortical loops (Schieppati and Nardone, 1999). The M3 response in the upper limb has latency of about 80 ms and can be found in about 30% of normal subjects (Rothwell et al., 1983), but in some subjects may not be a clear peak distinct from M2 (Ahmadi-Pajouh et al., 2012). It may traverse through cerebellum before reaching the motor cortex (Lee and Tatton, 1975). In the lower limb, the M3 of the tibialis anterior muscle is at least partly mediated by a transcortical pathway (Petersen et al., 1998).
The MSR has been linked to rigidity in PD as the clinical assessment of rigidity by passive joint movements is similar to the MSR procedure and the stretch response from the stretched muscle could be responsible for the resistance observed in rigidity. Moreover, rigidity can be relieved by dorsal root resection (Rushworth, 1960) or local anesthetic block (Pollock and Davis, 1930), indicating that sensory input contributes to rigidity. In PD patients, increased M2 response has been reported in the different muscles including the triceps (Rothwell et al., 1983), flexor carpi radialis (Meara and Cody, 1993), soleus (Scholz et al., 1987, Scholz et al., 1987) and quadricep femoris (Bergui et al., 1992), but was normal in the flexor pollicis longus muscle (Rothwell et al., 1983). Some studies showed that the degree of M2 enhancement correlated with the severity of rigidity (Berardelli et al., 1983, Mortimer and Webster, 1979), but other studies reported that the M2 reflex gain was not related to rigidity scores (Rothwell et al., 1983) and changes in M2 magnitude following levodopa administration did not correspond to the improvement in rigidity (Meara and Cody, 1993). Several factors may contribute to these different findings. In MSR studies, subjects usually maintain a slight muscle contraction while sudden stretches of the joints are applied, which is different from the clinical rigidity assessment in which the patient is asked to relax (although some may not be able to) and the joints are moved slowly and not abruptly by the examiner. Besides, stretch velocity and amplitude (Powell et al., 2012), the muscle tested and the testing posture such as sitting or standing, all affect the MSR results. Therefore, increased MSR likely contributes to but is not solely responsible for rigidity in PD. For example, some studies suggested that changes in muscle properties such as increased stiffness of the muscle belly and tendon may also contribute to rigidity (Dietz et al., 1981, Marusiak et al., 2011).
6.3. Mixed nerve electrical stimulation evoked long latency reflexes
The LLR can also be assessed with electrical stimulation to mixed nerves (eLLR). The eLLR are mediated by both type Ia and type II afferent fibers. For testing of eLLR in distal hand muscle, surface EMG recordings from the abductor pollicis brevis muscle under mild background contraction is frequently used. Some studies used ulnar nerve stimulation and some used flexor carpi radialis as the target muscle. After rectification and averaging of the EMG signal, there could be four components of the response. The first component is a SLR, which is believed to be a peripheral reflex that involves the same pathway as the H-reflex. The next three components include eLLR I, eLLR II likely mediated by a transcortical route involving the motor cortex (Tataroglu et al., 2011, Deuschl et al., 1989, Tsuji and Rothwell, 2002, Chen et al., 1998b), and eLLR III which may involve the cerebellum (Claus et al., 1986).
In PD, eLLR I was found to be enhanced. It did not correlate with rigidity but was associated with the occurrence of action tremor (Deuschl and Eisen, 1999). Although eLLR II usually is not elicited with the target muscle at rest in normal subjects, a study showed that eLLR II was elicited at rest or with less than 20% maximal contraction in PD patients. This could be due to difficulty relaxing the target muscle (flexor pollicis longus/brevis) in PD patients. However, when the eLLR was measured in a small hand muscle such as the first dorsal interosseus muscle, the SLR, eLLR II and eLLR III showed no difference in PD compared to normal subjects (Noth et al., 1988). In a study in PD patients with STN-DBS, an abnormally enhanced eLLR II elicited by ulnar nerve stimulation obtained from the flexor carpi radialis muscle was restored with STN-DBS or levodopa treatment. (Marchand-Pauvert et al., 2011).
For the other forms of parkinsonism, patients with corticobasal syndrome (CBS) may have enhanced eLLR I due to reflex myoclonus (Thompson et al., 1994), but can be normal in the early stage of the disease (Deuschl and Eisen, 1999). A case report showed enlargement of both eLLR I and eLLR II in MSA patients with striatonigral degeneration (currently termed MSA-Parkinson type) (Kofler et al., 1998). PSP patients may present with relatively enlarged SEP but normal eLLR II and eLLR III (Kofler et al., 2000).
6.4. Cutaneomuscular reflex
CMR is often studied using pure sensory stimulation of the index or middle finger, with the abductor pollicis brevis or first dorsal interosseous as the target muscle. There are three excitatory components in CMR, with an early excitatory component (E1, onset at 35 ∼ 43 ms), a second excitatory component (E2, onset at 43 ∼ 60 ms) which is found in all normal adult subjects, and occasionally a third excitatory component (E3, onset at 70–82 ms). There is an inhibitory phase (I1) between E1 and E2 (Deuschl, 2003). CMR relies on pure sensory type II fibers and the I1 and E2 components depend on intact supraspinal sensory input and motor output pathways (Chen et al., 1998a). Some studies showed transient increase in motor cortical excitability that corresponds with the timing of E2 (Maertens de Noordhout et al., 1992), but other studies showed the sensory afferent faciliatory effect on the motor cortex did not correlate with the E2 peak latency, which implies that E2 was probably not generated directly from the motor cortex but may be generated by related motor circuits (Kojima et al., 2014).
In PD patients, CMR studies showed normal E1 and E2 onset latencies and amplitudes. However, the I1 component was less pronounced in PD patients (Fig. 3) and could be partially restored by dopaminergic medications (Chen et al., 1992, Fuhr et al., 1992). Patients with stimulus sensitive myoclonus associated with PD or MSA has exaggerated E2, whereas patient with stimulus sensitive myoclonus associated with CBS had synchronous activation of upper limb muscles at latencies shorter than E2 (Chen et al., 1992), suggesting stimulus sensitive myoclonus in different forms of parkinsonism may involve distinct sensorimotor circuits.
Fig. 3.

Reduced I1 response of cutaneomuscular reflex in Parkinson’s disease. The cutaneomuscular reflex obtained from abductor pollicis brevis muscle with superficial radial nerve stimulation showed reduced I1 response in two Parkinson’s disease patients (gray) compared to two normal subjects (red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
6.5. Cutaneous silent period
Cutaneous silent period (CuSP) is a spinal reflex elicited by cutaneous stimulation which leads to a short duration of EMG suppression. It typically lasts longer than 10 ms, usually about 100 ms (Leis et al., 2000). The cutaneous stimulation reaches pain threshold and involves transmission by small diameter A-delta fibers to inhibitory interneurons in spinal cord, leading to transient inhibition of alpha motor neurons (Kofler et al., 2019). Although CuSP is due to spinal inhibition, it can be affected by supraspinal inputs including the corticospinal tract. (See Kofler et al. for a review (Kofler et al., 2019)).
CuSP might be a tool to differentiate PD and atypical parkinsonism since prolonged CuSP duration was restored in PD patients after levodopa administration but not in MSA, PSP, or vascular parkinsonism (Pullman et al., 1996, Serrao et al., 2002). However, the results should be interpreted cautiously because of the low number of subjects in these studies. In contrast, others studies showed delayed and prolonged CuSP in MSA (Stetkarova et al., 2015), probably due to defects in reticulospinal and corticospinal inputs that change spinal excitability (Eusebio et al., 2007).
7. Transcranial magnetic stimulation studies
TMS has been used extensively to study the pathophysiology of parkinsonian disorders. We review the main findings here. The findings are summarized in Table 2.
Table 2.
Single and paired pulse transcranial magnetic stimulation measures in various Parkinsonian disorders.
| Single pulse TMS measures | PD | MSA | PSP | CBS |
|---|---|---|---|---|
| Motor threshold | ↓/↔ |
↔ |
↔ |
↑ |
| MEP amplitude/Recruitment curve | ↑(rest)/↓(active) | ↑ | ↑ | ↓/↔ |
| CMCT | ↔ (↑ in Parkin mutation) | ↑ | ↑ | ↔ |
| cSP | ↓ | ↑ | ↑ | ↓ |
| iSP | ↓ | ↑ | ↑ | ↓ |
| Paired pulse TMS measures | ||||
| SICI | ↓ | ↓ | ↓ | ↓ |
| LICI | ? | – | – | – |
| ICF | ↔/↓ | ↔ | ↔ | ↔ |
| SICF | ↑ | ↔ | – | – |
| SAI | ↔/↓ (ON) | ↓ | ↔ | – |
| LAI | ↓ | – | – | – |
| IHI | ↓/↔ | – | – | ↓ |
| CBI | ↓ | – | ↓ | – |
↓, decreased; ↑, increased; ↔, no change;?, uncertain/conflicting results; -, not studied; CBS, corticobasal syndrome; CBI, cerebellar inhibition; CMCT, central motor conduction time; cSP, contralateral silent period; ICF, intracortical facilitation; IHI, long interhemispheric inhibition; iSP, ipsilateral silent period; LAI, long latency afferent inhibition. LICI, long interval intracortical inhibition; MEP, motor evoked potential; MSA, multisystem atrophy, PD, Parkinson’s disease; PSP, progressive supranuclear palsy; SAI, short latency afferent inhibition; SICF, short interval intracortical facilitation; SICI, short interval intracortical inhibition.
7.1. Single pulse TMS studies
7.1.1. Motor threshold
Motor threshold (MT) is a measure of the corticospinal excitability and refers to the lowest TMS intensity needed to elicit a motor evoked potential (MEP) of certain amplitude [usually defined as 50 µV for rest MT (RMT) and 200 µV for active MT (AMT)] (Chen et al., 2008, Groppa et al., 2012). It reflects the excitability of the most sensitive group of neurons in the M1. Although most studies have reported normal RMT in PD (Ridding et al., 1995, MacKinnon et al., 2005, Ni et al., 2013), some studies have found reduced MT in PD (Valls-Sole et al., 1994, Tremblay and Tremblay, 2002). This may relate to factors such as involuntary contractions due to tremors or rigidity, disease severity and use of dopaminergic medications. AMT has been reported to be normal in PD but a study found a correlation between the severity of bradykinesia and AMT (Ellaway et al., 1995). Patients with MSA and PSP were found to have normal RMT (Morita et al., 2008, Conte et al., 2012). In patients with CBS, RMT is usually high (Kuhn et al., 2004, Pal et al., 2008).
7.1.2. MEP amplitude and recruitment curve
Recruitment curve (RC) or input–output (I/O) curve assesses the increase in MEP amplitude with higher TMS intensities (Chen et al., 2008). It assesses the strength of corticospinal projections. A study showed increased MEP amplitude and slope of RC in PD patients while at rest but these measures were decreased during voluntary muscle contraction (Valls-Sole et al., 1994). RC was normal in symptomatic PD patients with Parkin mutation compared to symptomatic gene carriers and healthy controls (Schneider et al., 2008). Patients with MSA had increased MEP amplitudes at higher stimulation intensities while patients with PSP had increased MEP amplitudes for all the intensities studied and a steeper recruitment curve while at rest (Kuhn et al., 2004). In patients of CBS, reduced MEP amplitude and a flattened I/O curve was observed at rest (Kuhn et al., 2004) but another study found no difference in RC between CBS and controls (Pal et al., 2008).
7.1.3. Central motor conduction time (CMCT)
The CMCT includes the times for excitation of motor cortical neurons, conduction through the corticospinal tract and the time for exciting spinal motor neurons to their firing threshold. It can be estimated as the conduction time between motor cortex to spinal cord by subtracting the latency from the spinal motor neuron to the muscle, known as the peripheral motor conduction time (PMCT), from the MEP latency. The formula is CMCT = MEP latency – PMCT. The F-wave is often used to estimate PMCT. The PMCT is calculated as (F + M−1)/2, where F represents the shortest F-wave latency from 10 to 20 trials and M is the M wave latency from peripheral nerve stimulation (Guérit, 2001). The 1 ms subtraction in numerator represents the estimated turnaround time of spinal motor neurons when activated antidromically. CMCT can also be measured by subtracting the latency induced by magnetic or electrical stimulation over the vertebral column from the MEP latency (Mills and Murray, 1986). However, this method may overestimate CMCT because the magnetic field activates proximal nerve roots at the intervertebral foramen exit rather than the spinal motor neurons themselves. Therefore, the duration of conduction between spinal cord and intervertebral foramen will be included in CMCT results.
To obtain the shortest latency from motor cortex to muscle, CMCT is usually measured with target muscle activated. In this situation, the MEP latency will be 1.5 to 3 ms shorter than when the target muscle is at rest (Mano et al., 1992), and has been termed “latency jump” (Caramia et al., 1993). During muscle contraction, the earliest descending corticospinal volley is more likely to cause a discharge in spinal motor neurons compared to rest since the motor neurons are often close to their firing thresholds due to ongoing descending input (Chen et al., 2008). Around 10% to 20% of maximum background force is sufficient to maintain stable CMCT latency (Guérit, 2001). It is recommended to superimpose at least five responses and then measure the shortest latency. Contraction of homologous contralateral muscles is an option for patients who are unable to produce adequate target muscle contraction (Mariorenzi et al., 1991). To induce upper extremity MEP, it is advisable to keep the induced current direction perpendicular to precentral gyrus in the posterior-anterior direction (about 45 degrees to midline and coil handle directed posteriorly when using a figure-of-8 coil). To induce lower extremity MEP, current direction should be kept perpendicular to the longitudinal fissure. Of noted, CMCT results are affected not only by the integrity of the corticospinal tract, but also the extent of recruitment of cortical motor neurons with the fastest propagation.
In PD, the CMCT results depended on the disease stage. Most studies indicated that the CMCT was shortened in PD patients, especially in the late stages, and was normal in drug naïve or early stage PD patients (Soysal et al., 2008, Fisicaro et al., 2020, Perretti et al., 2011). Levodopa could prolong the CMCT latency in PD patients (Soysal et al., 2008, Mochizuki et al., 1999, Diószeghy et al., 1999). Other studies indicated that the CMCT latency was similar to age-matched healthy subjects (Derejko et al., 2013, Diószeghy et al., 1999, Mochizuki et al., 1999). However, prolonged CMCT was consistently found in early onset PD patients with Parkin mutation (PARK2) but not in those without the mutation (Perretti et al., 2011, Schneider et al., 2008, De Rosa et al., 2006). In atypical parkinsonism, CMCT was prolonged in PSP patients, especially in those with longer disease duration (Bologna et al., 2017), but in the early stages CMCT may be normal (Fisicaro et al., 2020). CMCT was prolonged in MSA (Morita et al., 2008, Eusebio et al., 2007) and CBS (Burrell et al., 2014) patients. Besides, prolonged CMCT in lower limbs was found in patients with normal pressure hydrocephalus and could be restored after lumbar drainage (Agrawal et al., 2021). Therefore, CMCT may help to differentiate PD, PD with Parkin mutation and other atypical parkinsonisms.
7.1.4. Contralateral silent period
The contralateral silent period (cSP) refers to the pause in ongoing voluntary EMG activity following the MEP. The first part of cSP is due to decreased spinal excitability while the latter part of cSP involves cortical inhibition. cSP is reduced in patients with PD (Cantello et al., 2002) and is normalized with dopaminergic mediations (Priori et al., 1994). However, high doses of dopaminergic medications can lengthen the SP in PD which became normalized with internal globus pallidus deep brain stimulation (Chen et al., 2001). Patients with MSA and PSP have prolonged cSP but patients with CBS have reduced cSP (Kuhn et al., 2004).
7.1.5. Ipsilateral silent period (iSP)
The ipsilateral inhibitory effects induced by motor cortical TMS can be measured by the interruption of ongoing voluntary EMG activity, referred to as the ipsilateral silent period (iSP)(Chen et al., 2008). The iSP is mainly due to transcallosal inhibition (Meyer et al., 1995) but non-callosal pathways caudal to the corpus callosum (Compta et al., 2006) may also contribute. ISP onset latency is usually around 35 ms and lasts for approximately 20 ms (Fig. 4A). There are many factors that affect iSP measurement. For example, the iSP onset, end latency and transcallosal time increases with age (Petitjean and Ko, 2013), but is not significantly affected by the degree of muscle contraction or direction of TMS induced current (Kuo et al., 2017, Chen et al., 2003). The iSP duration increases with high stimulation intensity up to about 80% of stimulator output (Chen et al., 2003). Besides, the abductor pollicis brevis (APB) may be a better target muscle than the first dorsal interosseous muscle (FDI) as a second phase of inhibition due to the ipsilateral corticospinal pathway may occur in the FDI muscle (Jung and Ziemann, 2006). It has been suggested that a constant 15 ∼ 25% contraction or 100% muscle contraction with intermittent muscle relaxation between trials may be used (Hupfeld et al., 2020). The normalized iSP is calculated as the reduction in EMG area divided by the pre-stimulus mean baseline EMG level for the iSP. The optimal stimulation intensity for iSP has not determined. However, at least 60% of maximal stimulation output may be needed to reach the plateau response (Meyer et al., 1995), with measurement of iSP duration and normalized iSP (Kuo et al., 2017) recommended.
Fig. 4.

Examples of ipsilateral silent period (iSP) from a normal subject and a corticobasal syndrome patient. (A) iSP from a healthy subject. Rectified and averaged surface EMG recorded from the abductor pollicis brevis muscle with 10 trials. The vertical dashed lines indicate iSP onset and offset. The iSP onset is 31 ms and the offset time is 63 ms after TMS. The horizontal dashed line represented the mean baseline EMG level −50 to −10 ms before TMS. (B) iSP from a patient with corticobasal syndrome. Rectified, averaged surface EMG recording from the left (more affected side) APB muscle with 10 trials. There was no iSP. TMS: transcranial magnetic stimulation; EMG: electromyography.
In PD patients, iSP was shorter and smaller with stimulation of the more affected hemisphere but could be restored by levodopa (Spagnolo et al., 2013). In PD patients with mirror movement (PD-MM), the increase of iSP depth with higher stimulation intensities or degree of muscle contraction was significantly lower compared to healthy subjects, but the iSP duration in PD-MM patients was comparable to healthy subjects (Li et al., 2007). PD-MM was probably due to collateral influence of contralateral motor cortex with reduced transcallosal inhibition and intracortical inhibitory circuits (Cincotta et al., 2006, Li et al., 2007).
In atypical parkinsonism, iSP duration was significantly increased in MSA and PSP, especially in PSP (Kuhn et al., 2004). However, another study showed that iSP onset latency but not iSP duration was prolonged in PSP patients (Wolters et al., 2004). A study reported that patients with PSP Richardson type had loss of iSP whereas patients with PSP-Parkinson type showed similar iSP duration to PD patients (Wittstock et al., 2013). iSP could not be detected in CBD patients who did not present with myoclonus (Wittstock et al., 2013) (Fig. 4B).
7.2. Paired pulse TMS studies in Parkinson’s disease (Table 2)
7.2.1. Short interval intracortical inhibition (SICI)
SICI is considered a measure of cortical inhibition mediated by GABAA receptors (Ziemann, 2004, Chen et al., 2008). It is elicited by a sub-threshold conditioning stimulus (CS) followed by a test pulse at an interstimulus interval (ISI) of 1–5 ms. SICI has been found to be reduced in PD (Ammann et al., 2020, Bologna et al., 2018, Guerra et al., 2022, Ni et al., 2013, MacKinnon et al., 2005, Ridding et al., 1995) which is normalized by dopaminergic medications as well as with STN DBS (Cunic et al., 2002). In addition, SICI was reported to be normal on the less affected side and was reduced on the more affected side in newly diagnosed PD patients (Kojovic et al., 2012). The asymmetry in SICI was observed up to one year after diagnosis (Kojovic et al., 2015). SICI was found to be reduced in patients with MSA (Suppa et al., 2014), PSP (Conte et al., 2012) and CBS (Kuhn et al., 2004).
7.2.2. Long interval cortical inhibition (LICI)
LICI is elicited by a suprathreshold CS followed by a test pulse at ISI of 100–200 ms and involves GABAB receptor mediated inhibition. Some studies reported reduced (Pierantozzi et al., 2001, Chu et al., 2009) but other found no change (Sailer et al., 2003) or increased (Berardelli et al., 1996a) LICI in PD.
7.2.3. Intracortical facilitation (ICF)
ICF is elicited by a subthreshold CS followed by a test pulse at an ISI of 10–20 ms. The findings in PD have been variable with some studies showing reduced ICF (Strafella et al., 2000, Bares et al., 2003) and others finding normal ICF (Ridding et al., 1995, Berardelli et al., 1996b). ICF was normal in patients with MSA (Suppa et al., 2014), PSP (Conte et al., 2012) and CBS (Kuhn et al., 2004).
7.2.4. Short interval intracortical facilitation (SICF)
SICF is elicited by a suprathreshold pulse followed by a threshold pulse at ISIs of 1–5 ms, resulting in three peaks (around 1.5, 2.8, and 4.5 ms) and two troughs (around 2 and 3 ms). It was found to be increased in the off medication state which was normalized with dopaminergic medications (Ni et al., 2013). The changes in SICF correlated with improvement in UPDRS scores. The increased SICF may be partly responsible for reduced SICI. In a study, SICF was assessed in PD patients with levodopa induced dyskinesias (LIDs) and without LIDs. Patients were assessed at baseline and after 2 weeks of intake of 50 or 100 mg/day of safinamide (a reversible monoamine oxidase-B inhibitor). PD patients with LIDs had abnormally increased SICF, which correlated with severity of dyskinesia and was unaffected by levodopa administration. Safinamide at 50 mg per day reduced SICF while 100 mg per day normalized SICF, with both short (Guerra et al., 2019) and long-term drug administration (Guerra et al., 2021b). Another study found enhanced SICF in drug naive PD patients (Shirota et al., 2019). A recent study also found increased SICF in PD off and on dopaminergic medications (Saravanamuttu et al., 2021). Patients with MSA have been found to have normal SICF (Suppa et al., 2014).
7.2.5. Short and long latency afferent inhibition (SAI & LAI)
SAI and LAI are usually tested with median nerve stimulation followed by TMS over the M1 at 20–25 ms for SAI and at 100–200 ms for LAI. SAI is mediated by cholinergic and GABAergic cortical circuits. SAI was found to be normal in PD patients in the off medication state and was reduced on dopaminergic medications (Sailer et al., 2003). SAI is also decreased in PD patients with cognitive impairment (Nardone et al., 2017, Martin-Rodriguez and Mir, 2020). On the contrary, LAI in PD is reduced in patients with PD and is unaffected by dopaminergic medications (Sailer et al., 2003). While SAI was found to be reduced in patients with MSA (Celebi et al., 2014), it was normal in patients with PSP (Nardone et al., 2005).
7.2.6. Interhemispheric inhibition (IHI)
IHI is measured by delivering a conditioning stimulus to M1 followed by a test stimulus to the contralateral M1 (Ferbert et al., 1992). It is likely mediated by inter-hemispheric inputs mediated through the corpus callosum (Ni et al., 2009). IHI is reduced in PD patients specifically in patients with mirror movements at longer inter-stimulus intervals of 20–50 ms. Therefore, reduction in transcallosal inhibitory action may play a role in mirror movements in PD (Li et al., 2007). Previous studies have reported bilaterally reduced IHI in patients with CBS in both distal and proximal upper limb muscles (Pal et al., 2008).
7.2.7. Cerebellar inhibition (CBI)
CBI is mediated by the cerebellothalamocortical pathway and can be assessed by TMS of the cerebellum followed by M1 TMS at ISI of 5–8 ms (Ugawa et al., 1995, Pinto and Chen, 2001). CBI is decreased in patients with PD (Ni et al., 2010, Carrillo et al., 2013) and with PSP (Shirota et al., 2010).
7.3. Interactions between cortical circuits in PD
A triple-stimulus TMS paradigm can be used to investigate the interactions between cortical circuits (Ni et al., 2011). It has been found that SICI is suppressed in the presence of LICI in normal subjects in a manner consistent with presynaptic GABA-B receptor mediated inhibition of GABA release (Sanger et al., 2001), and this inhibition is decreased in PD patients in both off and on medication states (Chu et al., 2009). Triple pulse protocols investigating LAI and LICI have found that LAI reduced LICI in normal subjects but not in PD patients on or off medications (Sailer et al., 2003). The interaction between IHI and SICI has been investigated in PD. IHI reduced SICI in normal subjects and PD patients without mirror movements, but not in PD patients with mirror movements (Li et al., 2007), suggesting that reduced transcallosal inhibitory effect on intracortical inhibitory circuits may contribute to mirror movements in PD. In normal subjects, SICI facilitates SICF but this effect was absent in PD patient off medications, which was restored by dopaminergic medications (Saravanamuttu et al., 2021). These finding suggests that impaired interactions between motor cortical circuits is a pathophysiological feature of PD.
In summary, studies on cortical circuits and their interactions in PD with paired and triple pulse TMS studies have shown abnormalities in various measures of cortical excitability and intracortical circuits in PD (Table 2). Furthermore, these changes are related to stages of the disease, effects of dopaminergic medications and clinical heterogeneity of PD phenotypes. The clinical utility of these measures requires further studies.
8. Plasticity studies
8.1. Plasticity studies in PD
In TMS studies, M1 plasticity is commonly measured by post-intervention changes in MEP amplitudes (Suppa et al., 2016a, Suppa et al., 2017c, Huang et al., 2017, Suppa et al., 2022a). Current non-invasive plasticity-inducing protocols in humans such as theta burst stimulation (TBS) and paired associative stimulation (PAS) reproduce the findings of in vitro and in vivo long-term potentiation (LTP) and long-term depression (LTD) protocols used in animal models (Bliss and Lomo, 1973, Stefan et al., 2000, Huang et al., 2005, Huang et al., 2017, Mansvelder et al., 2019, Suppa et al., 2016b, Suppa et al., 2017c, Suppa et al., 2022a). Accordingly, TBS and PAS are commonly used to assess LTP/LTD-like plasticity in the human M1 in healthy subjects and in patients with movement disorders.
Early PAS studies in PD found reduced plastic change in patients off medication (Morgante et al., 2006, Ueki et al., 2006, Kacar et al., 2013). However, another study found exaggerated response to PAS in patients off therapy compared to controls (Bagnato et al., 2006). Concerning TBS studies, early studies demonstrated reduced LTP/LTD-like plasticity following TBS in PD patients (Eggers et al., 2010, Suppa et al., 2011) although another study did not report these abnormalities (Zamir et al., 2012). The reason for these inconsistencies may arise from differences in patients’ clinical features including disease duration, total daily doses of levodopa and presence of dyskinesia.
The possible effect of disease duration has been controlled for in studies designed to investigate LTP/LTD-like plasticity in de novo PD patients. These reports confirmed reduced responses to PAS and TBS in the more affected as well as in the less affected arm (Kishore et al., 2012a, Kacar et al., 2013). Another study, however, reported decreased responses to PAS only on the more affected side, whereas the less affected side was characterized by exaggerated responses interpreted as compensation (Kojovic et al., 2012). Such asymmetry in PAS-induced plasticity progressively decreased over a 12-month follow-up (Kojovic et al., 2015). A study has also suggested a correlation between MEP changes elicited by PAS and the likelihood of developing early motor complications (Kishore et al., 2017).
The impact of levodopa therapy on LTP/LTD-like plasticity has been evaluated in detail in chronically treated as well as in de novo patients with PD. In chronically treated patients, levodopa improved the abnormal PAS-induced LTP-like plasticity (Ueki et al., 2006, Bagnato et al., 2006), but not in patients with levodopa-induced dyskinesias (LIDs) (Morgante et al., 2006). By contrast, using TBS, a study (Suppa et al., 2011) demonstrated comparable responses to iTBS in PD patients on and off medications, and with or without LIDs, suggesting no beneficial effect of levodopa on TBS-induced LTP-like plasticity. In agreement with this, another study (Kishore et al., 2012b) also found no effect of acute levodopa challenge on TBS induced plasticity in de novo PD patients. The dose of levodopa also affects LTP/LTD-like plasticity in PD. In patients without LIDs and taking half their normal levodopa dose, a study (Huang et al., 2011) found no response to iTBS, while both the response to iTBS and depotentiation elicited by a specially designed TBS protocol were restored when patients took their full levodopa dose. Moreover, in PD patients with LIDs, the study found normal response to a modified facilitatory protocol only when patients received a half dose of levodopa (not eliciting LIDs), but it did not show depotentiation. Depotentiation deficit severe enough to cause a paradoxical facilitation pattern has also been reported in PD patients without LID but who may be prone to develop LID (Lago-Rodriguez et al., 2016). Different plasticity responses to levodopa according to the patients’ clinical features (stable responders to levodopa, motor fluctuations without LID and motor-fluctuations with LID) have been demonstrated by several studies (Kishore et al., 2012b, Kishore et al., 2014a, Kishore et al., 2014b).
A study (Belvisi et al., 2021) compared a large cohort of PD patients with the “mild motor-predominant” subtype (mild motor and non-motor symptoms) to those with the “diffuse malignant” subtype (combination of severe motor and non-motor manifestations). Although both subtypes had reduced responses to iTBS, patients with the “diffuse malignant” subtype had lower responses to iTBS than those with the mild motor-predominant subtype. Hence, the study confirmed earlier observations (Suppa et al., 2011) and suggested that neurophysiological parameters may represent promising biomarkers to evaluate PD subtypes and their progression (Belvisi et al., 2021).
TBS-induced LTP/LTD-like plasticity can be shaped by concurrent transcranial alternating current stimulation (tACS), a non-invasive stimulation that entrains brain oscillations, delivered at the γ frequency (Suppa et al., 2022a). Indeed, iTBS-induced LTP-like plasticity can be boosted and prolonged by concurrent γ-tACS, the effect depends on changes in GABAA receptor mediated interneuronal activity (Guerra et al., 2018, Guerra et al., 2021a, Guerra et al., 2020b). Using a combined iTBS-γ tACS, a study demonstrated that driving γ oscillations restore the LTP-like plasticity in PD patients (Guerra et al., 2020a). This finding suggested that cortical γ oscillations may play a beneficial role in modulating the LTP-like plasticity in M1 in PD (Guerra et al., 2020a). This hypothesis fits in well with a PAS study in dyskinetic PD patients treated with STN DBS (Kim et al., 2015). In the DBS-off condition, the response to PAS was reduced in patients on and off medications. By contrast, in the DBS-on condition, there was a restoration of PAS-induced responses in patients on but not off levodopa (Kim et al., 2015). These findings overall would point to a physiological link between LTP/LTD-like plasticity and oscillations in the cortico-basal ganglia motor loop (Guerra et al., 2020a, Suppa et al., 2022a, Guerra et al., 2022). In conclusion, the heterogenous findings of the above-mentioned plasticity studies are likely related to the different clinical variables of the cohorts investigated including the stage of PD, phenotype, presence or absence of motor fluctuations and dyskinesias and the patients’ therapeutic state related to the timing of levodopa doses due to the fluctuating nature of PD. Overall, most studies have demonstrated reduced LTP/LTD-like plasticity in patients with PD with possible beneficial effect of levodopa in modulating the response to TBS and PAS protocols (Suppa et al., 2016a, Suppa et al., 2017a, Bologna et al., 2016c, Huang et al., 2017).
8.2. Plasticity studies in atypical parkinsonisms
Several studies have examined LTP/LTD-like plasticity using PAS and TBS in patients with various types of atypical parkinsonisms (Bologna et al., 2017). A study (Conte et al., 2012) investigated LTP/LTD-like plasticity in patients with probable PSP. iTBS elicited abnormally enhanced responses compared with controls. Conversely, the after-effects induced by cTBS paradoxically turned from LTD-like to LTP-like plasticity (Conte et al., 2012). These changes are thought to reflect prominent neurodegeneration of inhibitory GABAergic interneurons in PSP, in addition to altered basal ganglia inputs to M1 (Conte et al., 2012, Bologna et al., 2017).
Two studies assessed LTP/LTD-like plasticity in patients with probable MSA (Kawashima et al., 2013, Suppa et al., 2014). A study (Kawashima et al., 2013) demonstrated reduced responses to PAS in patients with the parkinsonian variant of MSA (MSA-P). Another study studied both MSA-P and the cerebellar variant of MSA (MSA-C) patients found reduced responses to both iTBS and cTBS in both subgroups (Suppa et al., 2014). The abnormal LTP/LTD-like plasticity in MSA was unresponsive to levodopa (Suppa et al., 2014, Kawashima et al., 2013) and was comparable in MSA-P and MSA-C, suggesting that mechanisms responsible for the abnormal LTP/LTD-like plasticity in MSA are independent of the severity of the cerebellar involvement (Suppa et al., 2014).
A single study examined responses to TBS in patients with CBS (Suppa et al., 2016a, Di Stasio et al., 2019). When TBS was applied to the less affected M1 (contralateral to the limb manifesting only parkinsonism), both LTP and LTD-like plasticity were reduced. By contrast, the findings were more complex for the more affected M1 (contralateral to the limb manifesting parkinsonism plus other motor and non-motor symptoms). Specific clusters of motor and non-motor clinical features such as the alien limb phenomenon were associated with different neurophysiological findings such as no recordable MEPs, reduced LTP/LTD-like plasticity or even paradoxically increased LTP-like plasticity (Suppa et al., 2016a, Di Stasio et al., 2019).
Two studies investigated LTP/LTD-like plasticity in patients with frontotemporal dementia (FTD). A PAS study (Benussi et al., 2016) demonstrated reduced responses to PAS in both presymptomatic and symptomatic patients with specific genetic mutations. Another study (Di Stasio et al., 2018) examined TBS-induced plasticity and observed reduced LTP/LTD-like plasticity in FTD patients with parkinsonism but not in those without parkinsonism (Di Stasio et al., 2018).
Overall, these findings suggest that TBS and PAS may help to understand the pathophysiological basis of the clinical and neurophysiological heterogeneity in patients with atypical parkinsonism. TBS and PAS studies could potentially support the clinical diagnosis of patients with atypical parkinsonisms by disclosing specific neurophysiological patterns such as reduced plasticity responses in PD, MSA and FTD with parkinsonism, increased responses in PSP and asymmetric responses in CBS, but the utility in clinical context requires further study.
9. Myoclonus in parkinsonism
Myoclonus is defined as sudden, brief, shock-like, involuntary movements caused either by muscular contractions (positive myoclonus) or sudden cessation of muscle activities (negative myoclonus) (Fahn et al., 1986, Caviness and Brown, 2004, Shibasaki and Hallett, 2005). Myoclonus has been described in many neurodegenerative disorders and its presence has implications for the diagnosis, prognosis and management (Truong and Bhidayasiri, 2007). Myoclonus has been described in various parkinsonian disorders such as PD, MSA, CBS, DLB and FTD with parkinsonism. Early reports described myoclonus in PD patients on levodopa therapy (Klawans et al., 1975), acute epidemic encephalitis (von Economo’s encephalitis) (Ellis, 1920, Boveri, 1920) and acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication (Klawans et al., 1986). The nature and origin of myoclonus in various parkinsonian disorders and their electrophysiological characteristics will be described.
9.1. Myoclonus in idiopathic PD
Myoclonus is uncommon in PD and was initially described following levodopa therapy (Klawans et al., 1975, Klawans et al., 1975). However, myoclonus unrelated to dopaminergic therapy has been reported and can occur in the off-medication state. In a study of 17 PD patients with end-of-dose myoclonus, multifocal positive myoclonus was present in all and negative myoclonus in 70.6%. Positive myoclonus was present at rest, during posture and action (Sifoglu et al., 2017). Rest tremors were less frequent in patients with myoclonus (21.4%) than in those without myoclonus (62.5%). Repetitive myoclonus, especially during maintenance of posture, can mimic tremors (Caviness et al., 2002). Patients with PD and myoclonus have 36% increase in total α-synuclein in the primary motor cortex compared to those without myoclonus measured in post-mortem tissue (Caviness et al., 2011, Uversky, 2007, Tofaris and Spillantini, 2007, Soto and Estrada, 2008). Electrophysiologic recording showed irregular and variable EMG burst intervals, variable burst amplitudes and the absence of sinusoidal appearance (Sifoglu et al., 2017). The myoclonus was very brief (less than 50 ms), had low amplitude with a frequency ranging from 1 to 10 Hz, suggesting cortical origin (Caviness et al., 2002) and may be due to sensorimotor dysfunction (Caviness et al., 2011). Back-averaging demonstrated the presence of EEG transients prior to EMG bursts (Caviness et al., 2002, Sifoglu et al., 2017). However, giant SEP was not observed. Long loop reflex showed increased amplitude in PD patients with myoclonus during active muscle contraction (Sifoglu et al., 2017). The presence of myoclonus has been correlated with cognitive impairment in PD (Caviness et al., 2011) but was not observed in another study (Sifoglu et al., 2017). In addition to levodopa associated myoclonus, bromocriptine and amantadine can also cause myoclonus in PD (Vardi et al., 1978, Pfeiffer, 1996, Matsunaga et al., 2001).
9.2. Myoclonus in MSA
Myoclonus was observed in 31% of MSA patients and was mainly stimulus sensitive and distal predominant (Wenning et al., 1994). Reflex myoclonus was found to be more common in MSA-C than in MSA-P and PD in some studies (Gouider-Khouja et al., 1995, Rodriguez et al., 1994), but other studies reported that postural and reflex myoclonus was more frequent in MSA-P than MSA-C (Salazar et al., 2000, Okuma et al., 2005). Myoclonus in MSA has been described as “minipolymyoclonus” as they are irregular, jerky, low-amplitude hand and finger movements (Salazar et al., 2000, Okuma et al., 2005). Action induced facial myoclonus and facial myokymia has been described in MSA. These are precipitated by talking or jaw opening (Blunt et al., 1997, Lou et al., 1994). Surface EMG recordings have shown irregular synchronous bursts of 10 to 50 ms duration occurring at frequency of 5 to 7 Hz in the forearm and hand muscles in the outstretched position and not present at rest (Okuma et al., 2005). In addition, C-reflex, exaggerated E2 component of the cutaneous reflex (Chen et al., 1992), giant SEP and cortical potentials on back averaging have been demonstrated in some MSA patients, suggesting a cortical origin of the myoclonus (Okuma et al., 2005), although enlarged SEP was absent. It has been suggested that cerebellar and basal ganglia dysfunction cause increased excitability of cortical neurons (Salazar et al., 2000, Okuma et al., 2005). Some authors suggested that myoclonus could also due to brainstem hyperexcitability (Kofler et al., 1998). An association between myoclonus in MSA-C and α-synuclein pathology in the motor regions of the spinal cord has been reported, but how this related to the pathophysiology of myoclonus in MSA-C is unclear (Hwang et al., 2019).
9.3. Myoclonus in dementia with Lewy body dementias (DLB)
Myoclonus occurs in about 18% of DLB patients (Louis et al., 1997). Compared to myoclonus in PD, DLB patients had larger amplitude myoclonus occurring predominantly at rest (Caviness et al., 2003). The EMG bursts durations are less than 50 ms, and usually no giant SEP or abnormal C-reflex is observed. However, a preceding triphasic cortical potential has been observed in back-averaged EEG. The myoclonus is likely cortical in origin, but subcortical contribution cannot be ruled out. Compared to PD, the DLB myoclonus is more severe due to widespread cerebral cortical pathology that may be primarily responsible for the myoclonus (Caviness et al., 2003).
9.4. Myoclonus in corticobasal syndrome
Myoclonus is a characteristic feature of CBS. Myoclonus is predominantly distal, stimulus sensitive and more prominent on action or posture (Chen et al., 1992, Thompson et al., 1994, Rinne et al., 1994). It is observed in about two-thirds of cases at presentation and evolves over the next 2–4 years (Thompson et al., 1994). Tendon jerks, pinprick or tapping over the fingers or toes elicits the myoclonus (Thompson et al., 1994). The myoclonus is very brief (20–50 msec), and on action or following cutaneous stimulation shows clusters of 2–4 bursts with inter-burst interval of 60–80 msec (Thompson et al., 1994, Chen et al., 1992). Abnormal morphology of the SEP have been reported in CBS (Thompson et al., 1994). Distal distribution, short duration EMG bursts and stimulus sensitivity suggests cortical origin of the myoclonus (Thompson et al., 1994, Chen et al., 1992). The cortical myoclonus is probably due to the direct sensory input to primary motor cortex, abnormal hyperexcitability of the motor cortex and lack of inhibition from sensory cortex or thalamus (Thompson et al., 1994, Chen et al., 1992, Shafiq and Lang, 2002). The cortical origin is also demonstrated by presence of jerk locked cortical potentials on EEG back averaging and presence of giant SEP in some cases (Carella et al., 1997). The absence of giant SEP patients with more advanced disease has been attributed to profound parietal atrophy (Notturno et al., 2011).
9.5. Myoclonus in PSP
Myoclonus is rare in PSP and its presence favours the diagnosis of CBS. Palatal myoclonus has been described in PSP associated with hypertrophy of the inferior olivary nucleus (Suyama et al., 1997). Cortical myoclonus was demonstrated neurophysiologically in a patient with CBS but had pathologically proven PSP (Kemp et al., 2013). Short duration EMG bursts was found in arm muscles with absence of giant SEP and no cortical potential preceding the myoclonus. However, C-reflex was observed at latencies of 40 to 45 ms, consistent with cortical origin (Kemp et al., 2013). Drugs including amantadine and levodopa are also known to cause myoclonus in PSP patients.
9.6. Myoclonus in FTD
Myoclonus in FTD may present before, during, or after the development of behavioral or language disturbances and the parkinsonian symptoms are variable (Siuda et al., 2014). Myoclonus has been documented in some autosomal dominant FTD disorders such as mutations in MAPT, PGRN, CHMP2B and VCP genes, predominantly in the behavioural variant of FTD. Myoclonus is usually seen in the upper extremities, and is action induced with low amplitude (Caviness and Wszolek, 2002). The presence of myoclonus may suggest clinical progression (Caviness and Wszolek, 2002). Back averaged cortical potential was absent in most patients but was observed in one pre-symptomatic gene positive individual, suggesting a cortical origin of the myoclonus (Caviness et al., 2003). The pathophysiology of myoclonus in FTD is still not well understood.
9.7. Summary for myoclonus in parkinsonism
Myoclonus is observed in several neurodegenerative parkinsonian syndromes with varying frequency and severity. In most cases, it is observed in the distal upper limbs both at rest and with activities. It may be difficult to distinguish from fine tremors wherein electrophysiological tests are useful. Electrophysiological tests can also determine the origin of myoclonus and may help in prognostication.
10. Intraoperative microelectrode recordings
Intraoperative MER is a method where fine metal microelectrodes monitor the firing of single units in patients undergoing DBS procedures. DBS has been increasingly being used in medically refractory cases of movement disorders such as PD, dystonia and essential tremor. These movement disorders can be treated by inserting chronic indwelling electrodes in subcortical structures such as the STN, the GPi and the Vim nucleus of the thalamus. Specifically for PD, any of these targets may be chosen based on the symptom profile. The general indications are bilateral STN for cognitively intact patients with akinetic rigid symptoms, bilateral GPi for patients with akinetic rigid symptoms and less cognitive reserve or levodopa induced dyskinesias (Fasano and Lozano, 2015). Vim DBS may be offered to tremor dominant patients, and this may be a unilateral procedure contralateral to the dominant hand.
Although advances in MR imaging techniques have allowed the direct visualization of these targets with varying degrees of clarity, several factors can lead to discrepancies between the visualized virtual target and the actual final target. One factor is the time delay between selecting the imaging targets and the insertion of electrodes during surgery where there can be brain shift due to loss of cerebrospinal fluid (Halpern et al., 2008). There is continuing discussion whether intraoperative MRI or CT can supplant MER for real time target localization but a recent trial did not show significant difference between MER and intraoperative MRI guided surgery in terms of the clinical outcome of DBS therapy at one year follow-up (Liu et al., 2017). For this reason, most major DBS centers prefer to confirm tentative targets with MER to record characteristic neuronal activities and responses to microstimulation to define the exact target and surrounding structures to be avoided with submillimeter accuracy (Hutchison et al., 1994, Hutchison et al., 1997, Hutchison et al., 1998, Seifried et al., 2012, Schlaier et al., 2013). These mapping sessions last about 15–30 min per side with unique neurophysiological features for each target and will be described in turn.
10.1. MER for STN DBS in PD
Recordings from human STN neurons are comparable to those describe in early papers by Mahlon DeLong and others (DeLong et al., 1985) in single unit electrophysiology studies in non-human primates. In humans, details of neuronal firing rates and bursting patterns of the various cell types in STN have been previously described (Hutchison et al., 1998). In the anterodorsal to ventroposterior approach of the trajectory starting at 10 mm above the target in the center of the STN, the microelectrode may record bursting cells that are characteristic of the thin shell of the thalamic reticular nucleus in its anterodorsal aspect. This is a good sign of an on-target trajectory but depends on the anterior ring angle of approach, with higher angles more likely to start in reticular thalamus. Moving down in the trajectory there is a quiet area corresponding to white matter tracts of the thalamic and lenticular fasciculi with a thin strip of gray matter knows as the zona incerta (Ma, 1996). These are large amplitude units and have properties somewhat like STN but are only occasionally recorded. Below this region the recordings will show a characteristic increase in background noise and large amplitude units firing at the rate of 25 – 45 Hz with an irregular pattern. In the 12 h off medication state, the recordings have prominent beta oscillations in the spike trains that are audible as a tell-tale fluttering sound. The dorsal part of STN has higher beta power that decreases progressively toward the ventral border (Alavi et al, 2013). The LFP beta activity recorded with microelectrodes is coherent across the full dorsoventral extent of the nucleus. However, beta activity carried in spike trains was much more localized, being significantly coherent only over 1–1.5 mm distance in the track. This suggests than small focal assemblies of neurons in STN are functioning together to organize separate motor synergies. Several studies have suggested that the spatial extent of beta coherence within the STN is more correlated to the clinical scores of motor signs than the amplitude or power of the beta oscillation (Alavi et al., 2013, Zaidel et al., 2010). Coherence between neuron pairs in the STN and (substantia nigra pars reticulata) SNr was rarely observed (Alavi et al, 2013), which is not surprising considering their very different firing rates and patterns (low versus high and bursty vs regular). This suggests that subthalamic beta activity is connected to areas outside the basal ganglia through structures other than the SNr, which is a basal ganglia output nucleus in the rate model. As discussed below, the projection between STN and GPe may be a pathway that links STN beta activity with other brain areas.
A well-planned trajectory is usually at 2 – 3 mm above the tentative target in the center of the nucleus. These cells can be recorded one after the other over a 4 – 5 mm span of the STN, and this can be considered an optimal trajectory passing through the center of the nucleus. Although this intuitively suggests an optimal placement, in using a star drive or “Ben-gun“ array with a central and a lateral trajectory with two micro/macroelectrodes spaced 2 mm apart, it has been observed that both electrodes may have optimal recordings of STN neurons throughout a 6 mm length of the trajectory, but macrostimulation in the lateral track at the tentative target showed an unacceptably low threshold for activation of the internal capsule. Indeed, in an analysis of 134 electrode placements, the length of the STN trajectory did not correlate with motor outcome, but these authors emphasized test microstimulation as a mandatory tool since 37 leads were revised based on this test (Malinova et al., 2020). Passive manipulation of the limbs or voluntary movements will evoke firing rate changes (usually but not always excitatory) in the recorded neurons, termed “kinesthetic” response. Eye movement responses may also be found in the middle portion of the nucleus (Fawcett et al., 2005). STN neurons have somewhat lower firing rate and density of cells as the ventral border is approached. Below the ventral border the background noise in the recordings decreases and the SNr is normally encountered. These neurons have lower spike amplitude, much higher firing rates (80 – 100 Hz) and very regular firing pattern. A specific technique can be used to identify these neurons by passing a short 0.5 s train of 200 Hz and 3 – 5 µA current pulses through the recording electrode and observe the subsequent inhibitory effect on the neuron (Lafreniere-Roula et al., 2009). Another technique uses two microelectrodes spaced 600 µm apart and gives a single pulse through one electrode and records the evoked response with the other. SNr is unique in showing a positive going focal-evoked potential at short latency to these pulses (Prescott et al., 2009). Since this inhibitory response is not seen in STN (where a rebound burst response is typically observed), these microstimulation tests can distinguish the ventral STN from the dorsal SNr (Lafreniere-Roula et al., 2009). This physiologically defined border region between STN and SNr is usually the target for positioning the lowest of a quadripolar DBS electrode in the final implant.
Several studies have investigated the properties of STN neurons in PD patients across the spectrum of motor and non-motor activities (Yin et al., 2021, Eisinger et al., 2018). In particular, there has been much interest in the relationship between beta oscillations in the STN and PD motor symptoms (Brown, 2006, Little and Brown, 2014)(discussed further in section 11.1), and the role of STN beta oscillations in various non-motor or cognitive functional domains (Al-Ozzi et al., 2021, Eisinger et al., 2018, Spitzer and Haegens, 2017, Alanazi et al., 2021). Beta oscillations in the LFP are involved in many cortical processes such as learning and memory, attention and decision making, and it is thought that cortical and subcortical beta activities are of different origins (Spitzer and Haegens, 2017). However, the oscillation model suggests that beta oscillations in the STN derives from cortical beta activities (Brown, 2003, Hutchison et al., 2004). This may explain STN beta modulation during a visual choice preference task where patients had to choose a favorite from pairs of animal pictures (Al-Ozzi et al., 2021). Thus, beta oscillations the STN can also play a role in cognition and may be related to cognitive aspects of PD such as apathy.
10.2. MER for GPi DBS in PD
The ventrolateral GPi is a target for PD patients with disabling dyskinesias and those at risk for cognitive decline. The other major indication for GPi DBS is for cervical, generalized and secondary dystonia. A currently used technique is to start the trajectories at 10 mm above the tentative target and 2 mm above the optic tract, but in earlier procedures recordings started at 15 mm which included the globus pallidus externus (GPe) at the top of the track. In GPe, there are two cell types again described initially in the non-human primate by DeLong et al. (DeLong, 1971). In normal monkey, the main population has high frequency discharge with pauses (HFD-P, ∼85%) and the low frequency discharge with bursts neurons (LFD-B, ∼15%). In humans with PD in the 12 h off medication state, a similar profile is seen with most GPe neurons fire around 60–80 Hz with pauses (HFD-P), and the LFD-B cell type that fires around 20 Hz which makes up ∼ 11% of the total GPe population (Hutchison et al., 1994). These two types of GPe neurons are thought to be the same neurons as the prototypic and arkypallidal GP cells, respectively, more recently described in the rodent (Deffains et al., 2016). These cells have been implicated in a disynaptic interaction within the GPe to profoundly affect locomotor activity in rats (Aristieta et al., 2021). Optogenetic activation of STN neurons via the hyperdirect path drives the prototypic (HFD-P) neurons, which inhibit the arkypallidal (LFD-B) neurons, leading to increased locomotion. Activation of arkypallidal neurons by the striatal indirect pathway produces the opposite effect with global inhibition. These recent studies highlight the GPe as an important locomotor hub rather than the conventional view as a mere relay in the indirect pathway from striatum to GPi.
GPe neurons can be distinguished from GPi neurons, which have a high frequency discharge (HFD). While HFD-P neurons in the GPe can be difficult to distinguish from GPi neurons, the presence of the LFD-B neurons can identify the GPe. Between the two nuclei is the medullary laminae, which is a thin strip of white matter, and it is bounded by so called border cells (DeLong, 1971, Bezard et al., 2001, Haber, 1987). These are cholinergic neurons that populate the boundaries of GPe and GPi and occasionally the central portion, which is likely due to border cells lining the internal medullary lamina. HFD neurons are recorded throughout the length of the GPi and evoked field potentials from stimulation of an adjacent microelectrode accompanied by brief cessation of spontaneous ongoing firing may be obtained at any level in the GPi (Liu et al., 2012). The ventral border of GPi is characterized by a decrease in background noise in the recordings and absence of units. At a distance roughly 1.8 mm below the last recorded cell, the optic tract can be found which is identified by patient’s report of phosphenes in the contralateral visual field to microstimulation (100 µA, 1 s, 200 Hz, 150 µs pulse width). The top of the optic tract is the site where no phosphenes are reported when retracting the microelectrode tip. If patients are anesthetized or otherwise unable to report the visual sensations, flash evoked recordings from the optic tract can be heard if the tip is near or just on top of the optic tract. Trajectories that are too posterior may be associated with motor contractions due to stimulation of posterior limb of the internal capsule (Lozano et al., 1996).
Beta oscillatory activity is less prominent in the spike trains and LFP in the GPi compared to the STN. Some studies suggest that both neuronal firing rates and beta LFP oscillations in the GPi are modulated by cognition (Alanazi et al. 2021). Both single units and LFP in the beta band had selective responses to the attended tone in an auditory oddball paradigm, and there was more beta desynchronization during the correct than the incorrect trials. These findings showed that neuronal activity at GPi, which is the output nucleus of the basal ganglia, is involved in processing of cognitive functions and may be related to selective attention deficits.
10.3. MER for DBS of Vim thalamus for PD rest tremor
Thalamus has long been a target for movement disorders surgery and predates the use of STN and GPi interventions, and these procedures were first done under the guidance of MER before the introduction of imaging in stereotactic neurosurgery (Guiot et al., 1964, Albe-Fessard, 1973). Although there is a long history of using MER for targeting the motor thalamus, some centers dispense with it entirely and target based on structural MRI tractography of the medial lemniscus and internal capsule (King et al., 2017). With white matter suppression MRI sequences combined with Magnetization Prepared, Rapid Gradient Echo (MPRAGE) sequence, the anisotropy imposed by fibers of passage are suppressed and the segmentation of thalamic subnuclei can be clearly visualized (Iglesias et al., 2018). However, this study used fixed post-mortem brains and required considerable processing time, and the method is not currently feasible for stereotactic planning.
The method for MER depends on the tentative target. One method is to target the Vim-Ventral caudal (Vc) border to clearly define the anterior-posterior reference and then implant in front of this. An alternate method is to target a slightly more anterior location of the ventral oral posterior (Vop)/Vim border with the intention of implanting in the explored trajectory. Typical trajectories may start in Vop and then transverse Vim and then pass just in front or into the sensory Vc nucleus. The Vop has neurons with preparatory activity responses and Vim is a cerebellar relay. Both these nuclei have neurons with periodic oscillations in the firing rate in synchrony with limb tremor, termed “tremor cells” (Lenz et al., 1988). Beta activity in spike trains from Vim is an effective way to identify and map this nucleus since it appears to be present in PD, ET and pain patients (Basha et al., 2014). In practice, the first occurrence of beta activity in spike trains in the trajectory may be taken as an indication of the Vop-Vim border. This observation, along with the disappearance of thalamic beta activity with rest tremor onset, suggest that beta oscillation in thalamus is not directly related to PD pathophysiology. Since Vim receives predominant input from cerebellum, the beta oscillations likely arise from the dentate nucleus. The microelectrode recording is interleaved by microstimulation at regular 1 or 2 mm intervals and the effects on tremor and sensations are assessed in a semiquantitative manner with triaxial accelerometry being displayed over time. A short stimulus train of 100 µA for 1 s is used to test for paresthesias or effects on tremor, and if the paresthesia is mild or otherwise tolerable, then a longer 2 to 3 s train is given to assess the effects on tremor. As the sensory regions of Vc is approached, the threshold for paresthesias becomes lower, and current with lower initial amplitude may be used. Induction of paresthesia with currents less than 100 µA is usually an indication of close proximity to Vc and a threshold of less than 10 µA indicates that the electrode tip is at or within the Vc tactile border. There should be recordings of tactile responses at this site. Below the Vc, the prelemniscal radiations are found as well as the medial lemniscus and this is a quiet zone electrophysiologically, with a higher threshold of 20 µA and above for evoking hemibody paresthesias.
Similar to tremor phase reset with single pulse TMS (discussed in section 2.2), microstimulation trains (100 µA, 100 or 200 Hz, 1–3 s) in the Vim can produce tremor phase reset at the start of the train (Milosevic et al., 2018). A burst of spikes is initially elicited before silencing of the thalamic Vim neuron and tremor arrest. The initial burst led to a movement in the outstretched contralateral limb, which is likely due either orthodromic or antidromic activation of the M1 by the thalamic burst.
In summary, despite advances in real time imaging modalities, MER continues to be considered an important component of DBS lead placement in major centers that have the expertise and scientific motivation. For many centers, it remains the gold standard for confirming the location of the DBS targets by mapping the characteristic neurophysiological activities and results of microstimulation, but also advances our knowledge of the motor and non-motor pathophysiology of PD and other movement disorders.
11. Local field potential and electrocorticography recordings in Parkinson’s disease
DBS is an effective treatment for patients with PD suffering from motor complications (Schuepbach et al., 2013). During and after electrode implantation, DBS provides a unique opportunity to record neural population activity as local field potentials (LFP) directly from the target area. LFP is a population-level measure of synaptic currents and local spiking activity, similar to EEG (Buzsáki et al., 2012). Intraoperative and postoperative invasive neurophysiology studies in DBS patients have contributed significantly to our understanding of PD pathophysiology at the network level (Jenkinson et al., 2013, Kühn and Volkmann, 2017, Neumann et al., 2019). Moreover, they have sparked a novel concept of stimulation called closed-loop adaptive DBS (aDBS), which aims to move from a chronic continuous stimulation setting to a demand- dependent approach (Arlotti et al., 2018, Little et al., 2013). Advances in PD research inspired by invasive neurophysiology are summarized here.
11.1. Local field potentials
11.1.1. Beta activity as hallmark of parkinsonian bradykinesia and rigidity
A major milestone in the understanding of the relationship of neural oscillations with neurological symptoms, is the identification of exaggerated pathological beta (13–35 Hz) activity in DBS targets (Brown et al., 2001, Silberstein et al., 2003), namely the STN and GPi, in patients with PD. In the PD off-medication state, after withdrawal of dopaminergic medications, oscillatory activities are synchronized in a stereotypical pattern of high-amplitude beta activity, that are suppressed by dopaminergic medications (Kühn et al., 2006, Neumann et al., 2017b). Importantly, both the amplitude of subthalamic beta activity in the off-medication state and the medication related suppression reflect the clinical presentation of the patients (Neumann et al., 2016, Kehnemouyi et al., 2021, Kuhn et al., 2009). Specifically, bradykinesia and rigidity have been associated with exaggerated beta oscillation, but not tremor (Kühn et al., 2006, Neumann and Kühn, 2017). While invasive recordings are unavailable from healthy subjects, comparison and characterization of basal ganglia oscillatory activities were possible in patients undergoing DBS for other indications such as dystonia, Tourette syndrome and epilepsy (Lofredi et al., 2019, Miocinovic et al., 2015, Neumann et al., 2012, Neumann et al., 2018, Neumann et al., 2017a, Rektor et al., 2002, Silberstein et al., 2003, Tsang et al., 2012b). These studies showed that the presence of beta activity itself is not pathological per se. However, the amplitude and extent of beta activity in the parkinsonian off-medication state were consistently higher than in the on-medication state and other disease entities, such as dystonia (Lofredi et al., 2019, Silberstein et al., 2005). Following the early studies on the effect of levodopa on beta activity, all major findings could later be replicated for DBS. Thus, DBS can suppress beta activity, and the amount of beta activity reduction correlated with the extent of PD motor symptom alleviation (Jenkinson and Brown, 2011, Feldmann et al., 2021b, Kuhn et al., 2008, Oswal et al., 2016). However, levodopa and DBS may have different effects on beta oscillations. Whereas levodopa abolished the STN beta LFP oscillations in patients with PD, DBS decreased beta oscillations for only PD patients in whom LFP recordings showed high beta activity at baseline (Giannicola et al., 2010).
More recently, the temporal dynamics of beta oscillations have been subject of research. It was found that the relative increase in mean beta power can be explained by a prolongation of transient episodes of beta synchronization, so-called beta bursts. The presence of particularly long beta bursts in the STN and the GPi was described as characteristic of the PD off-mediation state, whereas shorter-duration bursts are similar between PD on-medication and dystonia, and may reflect normal physiology (Kehnemouyi et al., 2021, Lofredi et al., 2019, Tinkhauser et al., 2017). Beta activity can be considered a neural pattern reflective of the bradykinetic motor state that is stable over time in PD (Neumann et al., 2017b). Therefore, it is considered a valuable, state-dependent chronic biomarker, primarily in PD patients presenting with a bradykinesia/rigidity dominant PD subtype (Fig. 5).
Fig. 5.
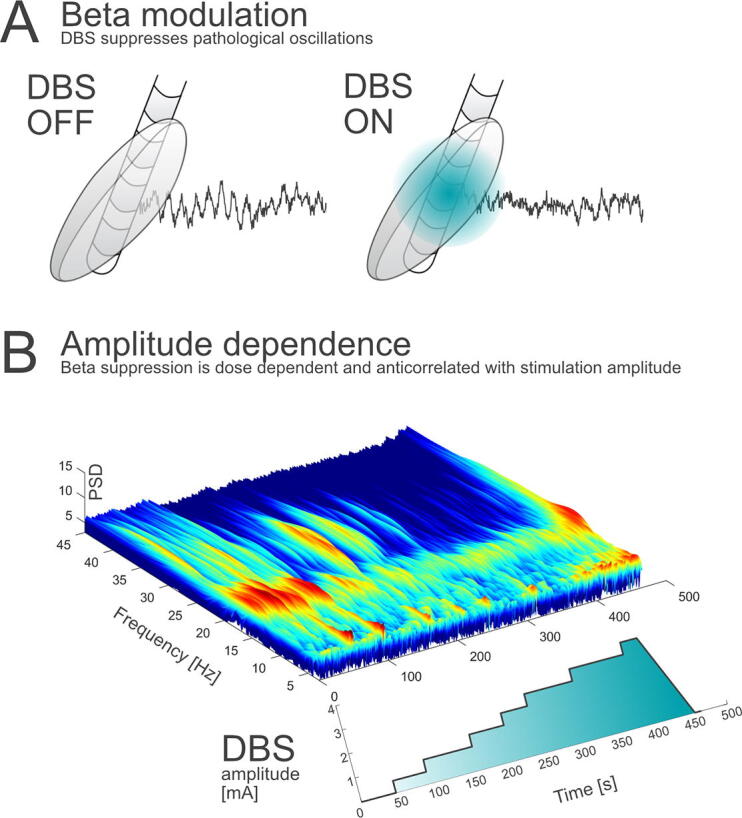
Parkinsonian beta activity in the subthalamic nucleus is suppressed with deep brain stimulation. Local field potentials (LFP) can be recorded directly through implanted DBS electrodes (A). In the medication and stimulation OFF state, the amplitude of beta oscillatory activity is increased (A left) and suppressed with deep brain stimulation (DBS) switched ON (right). Recently, the dose dependence of beta and DBS could be revealed through sensing enabled implantable devices (B; Feldmann et al., 2021a, Feldmann et al., 2021a).
11.1.2. Local field potential patterns in PD beyond beta activity
While beta activity is the most thoroughly described activity pattern in PD, other patterns of pathological activities can also be characterized through LFP recordings. Dopaminergic side effects are reflected in subthalamic LFP, where dyskinesia symptoms were associated with increased low frequency (4–8 Hz) and narrowband gamma (60 – 90 Hz) synchronization, similar to the activities seen during normal movement (Alonso-Frech et al., 2006, Jenkinson et al., 2013, Lofredi et al., 2018). Movement related gamma synchronization is reduced in the hypodopaminergic state (Lofredi et al., 2018). During freezing of gait, changes in neural entropy and disruption of low frequency synchronization have been reported (Pozzi et al., 2019, Syrkin-Nikolau et al., 2017). During sleep, there is complex modulation of beta activity that is dependent on the sleep stage (Urrestarazu et al., 2009). Moreover, activity in even higher frequency bands has been described. High frequency oscillations (HFO) at ∼ 250 Hz are not generally attenuated by dopaminergic medications in PD, but are shifted towards higher frequencies at ∼ 350 Hz (Özkurt et al., 2011, van Wijk et al., 2017). Unlike beta activity, this frequency band may also be informative regarding the presence of tremor, even in the on-medication state (Hirschmann et al., 2016).
11.2. Electrocorticography
To investigate pathological oscillations beyond the basal ganglia, intraoperative electrocorticographic recordings have further enabled characterization of PD pathophysiology. Higher broadband gamma power and higher post-movement beta synchrony were found in PD patients compared to patients with hyperkinetic movement disorders (Crowell et al., 2012). Furthermore, exaggerated beta–gamma PAC as a cortical biomarker of PD symptoms that is suppressed with DBS was described (de Hemptinne et al., 2013, de Hemptinne et al., 2015). Most importantly, it was shown that the modulation of cortical PAC with subthalamic DBS can predict PD symptom improvement (de Hemptinne et al., 2015). Some of these effects could be explained by an increased sharpness of the beta waveform in the parkinsonian state, adding waveform shape to the range of potential cortical biomarkers (Cole et al., 2017). In addition, an ECoG study demonstrated increased resting beta-gamma PAC in PD compared to epilepsy patients, whereas during motor performance PAC were similar in the two groups and showed a persistent increase for the entire duration of the motor activity. However, in the PD group, beta-gamma PAC showed greater increase in the low-beta range compared the high-beta range (Kondylis et al., 2016). The high signal-to-noise ratio, broad band neural coverage and distance to stimulation source make ECoG an important and promising technique for the investigation of advanced neurotechnology in PD research (Gilron et al., 2021).
11.3. Future outlook
11.3.1. Sensing enabled implantable devices have entered the clinic
Many important findings have been described based on intra- or perioperative experimental research settings. While both ECoG and LFP recordings have been reported to be safe (Feldmann et al., 2021a, Panov et al., 2017, Sisterson et al., 2021) and will remain a very important instrument for PD research, the experimental setting does not allow collection of information regarding the long-term stability of the described phenomena. Few sensing-enabled implantable devices have been available for research purposes only, until the first clinically United Stated Federal Food and Drug Administration and CE-approved device was released. Such devices will further impact our understanding of DBS effects and the how pathological oscillatory activities and clinical symptom are related. Examples of such findings are the first description of stable and dose-dependent effects of DBS on subthalamic beta activity (Feldmann et al., 2021b), and a combined approach to ECoG and LFP sensing that lead to the identification of cortico-subthalamic connectivity as a new and important marker for PD symptoms even months after neurostimulator implantation (Gilron et al., 2021).
11.3.2. First clinical trials on adaptive DBS
A major implication of invasive neurophysiology studies for PD patients is the development of demand-dependent adaptive DBS (Neumann et al., 2019). The first experimental studies have demonstrated that pathological beta activity can be monitored to inform an algorithm of the necessity to implement adaptive stimulation. Until recently, only few centers were able to overcome the technical barriers of such research (Arlotti et al., 2018, Little et al., 2013, Sisterson et al., 2021, Piña-Fuentes et al., 2017, Velisar et al., 2019). For example, recording and stimulating in the same target poses problems with artifact and signal-to-noise ratios that can obstruct meaningful clinical evaluations of this approach (Neumann et al., 2021). The first multicenter clinical trials of adaptive DBS are ongoing, but some practical challenges of adaptive DBS remain to be addressed. These include the choice of biomarkers, recording location (e.g., ECoG vs. STN) and control algorithms which have to be evaluated in robust clinical studies. Furthermore, the development of multivariate approaches for adaptive DBS beyond single biomarkers is an active field of research (Neumann and Rodriguez-Oroz, 2021, Merk et al., 2022).
11.4. Summary of LFP and ECoG recordings
LFP recordings from DBS and ECoG electrodes have allowed the characterization of pathological activity patterns related to PD pathophysiology. Exaggerated beta activity is a hallmark of PD bradykinesia and network synchronization phenomena can reflect specific PD motor states. With new sensing-enabled devices, further research beyond the acute intra- and perioperative stage will further enhance our understanding of brain rhythms implicated in PD. Next-generation DBS strategies will utilize oscillatory activity to adapt stimulation to concurrent demand, with potential further improvement of quality of life for patients living with PD.
12. Conclusions and future outlook
Neurophysiology studies using a variety of techniques reviewed here have advanced our knowledge of the pathophysiology of PD and parkinsonism. While some of the findings are related to dopaminergic deficits and abnormalities in the basal ganglia, many areas in the motor and non-motor networks including sensory and motor cortical areas, the cerebellum and the spinal cord are also involved. Some techniques such as tremor analysis, CMCT measurement, and myoclonus studies are useful in the diagnostic workup of parkinsonism and neurophysiology is used to accurately localize surgical targets. Future work will produce further advances in these areas but will also be instrumental in the design of new treatments such as novel applications of different non-invasive brain stimulation techniques and the development of adaptive DBS systems.
Funding
Robert Chen was supported by the Catherine Manson Chair in Movement Disorders.
Alfonso Fasano was partly supported by the University of Toronto and University Health Network Chair in Neuromodulation.
Rick Helmich was supported by the Netherlands Organization for Scientific Research (VIDI grant, # 09150172010044) and the Michael J. Fox Foundation (# MJFF-021001).
William D. Hutchison was support by the Dystonia Medical Research Foundation.
Andrea A. Kühn and Wolf-Julian Neumann were supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 424778381 – TRR 295 and Bundesministerium für Bildung und Forschung (BMBF, grant iDBS, FKZ01GQ1802).
Kaviraja Udupa was supported by Intramural research grants from NIMHANS and the Dept of Science & Technology (DST/SATYAM-19/2020/48).
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Robert Chen received honoraria from Abbvie, Merz and Ipsen, outside of the submitted work. Alfonso Fasano received honoraria for his work as consultant for Abbvie, Abbott, Boston Scientific, Ipsen, Medtronic, and Sunovion; he sits in the advisory board for Abbvie, Boston Scientific, Ceregate, and Inbrain; received speaker fees from Abbvie, Abbott, American Academy of Neurology, Boston Scientific, Brainlab, Ipsen, Medtronic, Merz, Movement Disorders Society, Sunovion, Paladin Labs, and UCB; he received royalties from Springer and has received research grants from Abbvie, Boston Scientific, Dystonia Medical Research Foundation, University of Toronto, Michael J Fox Foundation, Medtronic, and the MSA coalition. Rick Helmich has served as a consultant for Roche Pharma. William D. Hutchison has received honoraria and travel support from Medtronic Inc. Andrea A. Kuhn: Personal fees from Medtronic, personal fees from Boston Scientific, personal fees from Abbott, personal fees from Ipsen Pharma, personal fees from Teva, outside the submitted work. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abbruzzese G., Berardelli A. Sensorimotor integration in movement disorders. Mov. Disord. 2003;18:231–240. doi: 10.1002/mds.10327. [DOI] [PubMed] [Google Scholar]
- Agostino R., Currà A., Giovannelli M., Modugno N., Manfredi M., Berardelli A. Impairment of individual finger movements in Parkinson's disease. Mov. Disord. 2003;18:560–565. doi: 10.1002/mds.10313. [DOI] [PubMed] [Google Scholar]
- Agrawal A., Bhattacharya A., Kamble N., Yadav R., Pal P.K. Effect of lumbar drainage on cortical excitability in normal pressure hydrocephalus. Can. J. Neurol. Sci. 2021;48:253–258. doi: 10.1017/cjn.2020.169. [DOI] [PubMed] [Google Scholar]
- Ahmadi-Pajouh M.A., Towhidkhah F., Shadmehr R. Preparing to reach: selecting an adaptive long-latency feedback controller. J. Neurosci. 2012;32:9537–9545. doi: 10.1523/JNEUROSCI.4275-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ozzi T.M., Botero-Posada L.F., Lopez Rios A.L., Hutchison W.D. Single unit and beta oscillatory activities in subthalamic nucleus are modulated during visual choice preference. Eur. J. Neurosci. 2021;53:2220–2233. doi: 10.1111/ejn.14750. [DOI] [PubMed] [Google Scholar]
- Alanazi F.I., Al-Ozzi T.M., Kalia S.K., Hodaie M., Lozano A.M., Cohn M., et al. Neurophysiological responses of globus pallidus internus during the auditory oddball task in Parkinson's disease. Neurobiol. Dis. 2021;159 doi: 10.1016/j.nbd.2021.105490. [DOI] [PubMed] [Google Scholar]
- Alavi M., Dostrovsky J.O., Hodaie M., Lozano A.M., Hutchison W.D. Spatial extent of β oscillatory activity in and between the subthalamic nucleus and substantia nigra pars reticulata of Parkinson's disease patients. Exp. Neurol. 2013;245:60–71. doi: 10.1016/j.expneurol.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Albe-Fessard D. Electrophysiological methods for the identification of thalamic nuclei. Z Neurol. 1973;205:15–28. doi: 10.1007/BF00315957. [DOI] [PubMed] [Google Scholar]
- Alonso-Frech F., Zamarbide I., Alegre M., Rodriguez-Oroz M.C., Guridi J., Manrique M., et al. Slow oscillatory activity and levodopa-induced dyskinesias in Parkinson's disease. Brain. 2006;129:1748–1757. doi: 10.1093/brain/awl103. [DOI] [PubMed] [Google Scholar]
- Amboni M., Cozzolino A., Longo K., Picillo M., Barone P. Freezing of gait and executive functions in patients with Parkinson's disease. Mov. Disord. 2008;23:395–400. doi: 10.1002/mds.21850. [DOI] [PubMed] [Google Scholar]
- Ammann C., Dileone M., Pagge C., Catanzaro V., Mata-Marín D., Hernández-Fernández F., et al. Cortical disinhibition in Parkinson's disease. Brain. 2020;143:3408–3421. doi: 10.1093/brain/awaa274. [DOI] [PubMed] [Google Scholar]
- Antonini A., Abbruzzese G., Ferini-Strambi L., Tilley B., Huang J., Stebbins G.T., et al. Validation of the Italian version of the Movement Disorder Society-Unified Parkinson's Disease Rating Scale. Neurol Sci. 2013;34:683–687. doi: 10.1007/s10072-012-1112-z. [DOI] [PubMed] [Google Scholar]
- Aristieta A., Barresi M., Azizpour Lindi S., Barrière G., Courtand G., de la Crompe B., et al. A disynaptic circuit in the globus pallidus controls locomotion inhibition. Curr. Biol. 2021;31:707–721.e707. doi: 10.1016/j.cub.2020.11.019. [DOI] [PubMed] [Google Scholar]
- Arlotti M., Marceglia S., Foffani G., Volkmann J., Lozano A.M., Moro E., et al. Eight-hours adaptive deep brain stimulation in patients with Parkinson disease. Neurology. 2018;90:e971–e976. doi: 10.1212/WNL.0000000000005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato S., Agostino R., Modugno N., Quartarone A., Berardelli A. Plasticity of the motor cortex in Parkinson's disease patients on and off therapy. Mov. Disord. 2006;21:639–645. doi: 10.1002/mds.20778. [DOI] [PubMed] [Google Scholar]
- Bares M., Kanovsky P., Klajblova H., Rektor I. Intracortical inhibition and facilitation are impaired in patients with early Parkinson's disease: a paired TMS study. Eur. J. Neurol. 2003;10:385–389. doi: 10.1046/j.1468-1331.2003.00610.x. [DOI] [PubMed] [Google Scholar]
- Basha D., Dostrovsky J.O., Lopez Rios A.L., Hodaie M., Lozano A.M., Hutchison W.D. Beta oscillatory neurons in the motor thalamus of movement disorder and pain patients. Exp. Neurol. 2014;261:782–790. doi: 10.1016/j.expneurol.2014.08.024. [DOI] [PubMed] [Google Scholar]
- Belvisi D., Conte A., Bologna M., Bloise M.C., Suppa A., Formica A., et al. Re-emergent tremor in Parkinson's disease. Parkinsonism Relat. Disord. 2017;36:41–46. doi: 10.1016/j.parkreldis.2016.12.012. [DOI] [PubMed] [Google Scholar]
- Belvisi D., Conte A., Cutrona C., Costanzo M., Ferrazzano G., Fabbrini G., et al. Re-emergent tremor in Parkinson's disease: the effect of dopaminergic treatment. Eur. J. Neurol. 2018;25:799–804. doi: 10.1111/ene.13619. [DOI] [PubMed] [Google Scholar]
- Belvisi D., Fabbrini A., De Bartolo M.I., Costanzo M., Manzo N., Fabbrini G., et al. The pathophysiological correlates of Parkinson's disease clinical subtypes. Mov. Disord. 2021;36:370–379. doi: 10.1002/mds.28321. [DOI] [PubMed] [Google Scholar]
- Benussi A., Cosseddu M., Filareto I., Dell'Era V., Archetti S., Sofia Cotelli M., et al. Impaired long-term potentiation-like cortical plasticity in presymptomatic genetic frontotemporal dementia. Ann. Neurol. 2016;80:472–476. doi: 10.1002/ana.24731. [DOI] [PubMed] [Google Scholar]
- Berardelli A., Inghilleri M., Priori A., Marchetti P., Curra A., Rona S., et al. Inhibitory cortical phenomena studied with the technique of transcranial stimulation. Electroencephalogr. Clin. Neurophysiol. Suppl. 1996;46:343–349. [PubMed] [Google Scholar]
- Berardelli A., Rona S., Inghilleri M., Manfredi M. Cortical inhibition in Parkinson's disease. A study with paired magnetic stimulation. Brain. 1996;119:71–77. doi: 10.1093/brain/119.1.71. [DOI] [PubMed] [Google Scholar]
- Berardelli A., Rothwell J.C., Thompson P.D., Hallett M. Pathophysiology of bradykinesia in Parkinson's disease. Brain. 2001;124:2131–2146. doi: 10.1093/brain/124.11.2131. [DOI] [PubMed] [Google Scholar]
- Berardelli A., Sabra A.F., Hallett M. Physiological mechanisms of rigidity in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry. 1983;46:45–53. doi: 10.1136/jnnp.46.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardelli A., Wenning G.K., Antonini A., Berg D., Bloem B.R., Bonifati V., et al. EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson's disease. Eur. J. Neurol. 2013;20:16–34. doi: 10.1111/ene.12022. [DOI] [PubMed] [Google Scholar]
- Berendse H.W., Stam C.J. Stage-dependent patterns of disturbed neural synchrony in Parkinson's disease. Parkinsonism Relat Disord. 2007;13(Suppl 3):S440–S445. doi: 10.1016/S1353-8020(08)70046-4. [DOI] [PubMed] [Google Scholar]
- Bergui M., Lopiano L., Paglia G., Quattrocolo G., Scarzella L., Bergamasco B. Stretch reflex of quadriceps femoris and its relation to rigidity in Parkinson's disease. Acta Neurol. Scand. 1992;86:226–229. doi: 10.1111/j.1600-0404.1992.tb05075.x. [DOI] [PubMed] [Google Scholar]
- Bezard E., Boraud T., Chalon S., Brotchie J.M., Guilloteau D., Gross C.E. Pallidal border cells: an anatomical and electrophysiological study in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkey. Neuroscience. 2001;103:117–123. doi: 10.1016/s0306-4522(00)00546-7. [DOI] [PubMed] [Google Scholar]
- Bharti K., Suppa A., Tommasin S., Zampogna A., Pietracupa S., Berardelli A., et al. Neuroimaging advances in Parkinson's disease with freezing of gait: A systematic review. Neuroimage Clin. 2019;24 doi: 10.1016/j.nicl.2019.102059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore R.L., MacAskill M.R., Myall D.J., Anderson T.J. Volitional Suppression of Parkinsonian Resting Tremor. Mov Disord Clin Pract. 2019;6:470–478. doi: 10.1002/mdc3.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss T.V., Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunt S.B., Khalil N.M., Perkin G.D. Facial myokymia in multiple system atrophy. Mov. Disord. 1997;12:235–238. doi: 10.1002/mds.870120215. [DOI] [PubMed] [Google Scholar]
- Bohnen N.I., Frey K.A., Studenski S., Kotagal V., Koeppe R.A., Constantine G.M., et al. Extra-nigral pathological conditions are common in Parkinson's disease with freezing of gait: an in vivo positron emission tomography study. Mov. Disord. 2014;29:1118–1124. doi: 10.1002/mds.25929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen N.I., Muller M.L., Koeppe R.A., Studenski S.A., Kilbourn M.A., Frey K.A., et al. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology. 2009;73:1670–1676. doi: 10.1212/WNL.0b013e3181c1ded6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologna M., Guerra A., Paparella G., Giordo L., Alunni Fegatelli D., Vestri A.R., et al. Neurophysiological correlates of bradykinesia in Parkinson's disease. Brain. 2018;141:2432–2444. doi: 10.1093/brain/awy155. [DOI] [PubMed] [Google Scholar]
- Bologna M., Latorre A., Di Biasio F., Conte A., Belvisi D., Modugno N., et al. The Effect of L-Dopa/Carbidopa Intestinal Gel in Parkinson Disease Assessed Using Neurophysiologic Techniques. Clin. Neuropharmacol. 2016;39:302–305. doi: 10.1097/WNF.0000000000000184. [DOI] [PubMed] [Google Scholar]
- Bologna M., Leodori G., Stirpe P., Paparella G., Colella D., Belvisi D., et al. Bradykinesia in early and advanced Parkinson's disease. J. Neurol. Sci. 2016;369:286–291. doi: 10.1016/j.jns.2016.08.028. [DOI] [PubMed] [Google Scholar]
- Bologna M., Paparella G. Pathophysiology of rigidity in Parkinson's disease: Another step forward. Clin. Neurophysiol. 2020;131:1971–1972. doi: 10.1016/j.clinph.2020.05.013. [DOI] [PubMed] [Google Scholar]
- Bologna M., Paparella G., Fasano A., Hallett M., Berardelli A. Evolving concepts on bradykinesia. Brain. 2020;143:727–750. doi: 10.1093/brain/awz344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologna M., Suppa A., Conte A., Latorre A., Rothwell J.C., Berardelli A. Are studies of motor cortex plasticity relevant in human patients with Parkinson's disease? Clin. Neurophysiol. 2016;127:50–59. doi: 10.1016/j.clinph.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Bologna M., Suppa A., Di Stasio F., Conte A., Fabbrini G., Berardelli A. Neurophysiological studies on atypical parkinsonian syndromes. Parkinsonism Relat Disord. 2017;42:12–21. doi: 10.1016/j.parkreldis.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Borzì L., Mazzetta I., Zampogna A., Suppa A., Irrera F., Olmo G. Predicting Axial Impairment in Parkinson's Disease through a Single Inertial Sensor. Sensors (Basel) 2022;22:412. doi: 10.3390/s22020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan A.C., Strick P.L. The basal ganglia and the cerebellum: nodes in an integrated network. Nat. Rev. Neurosci. 2018;19:338–350. doi: 10.1038/s41583-018-0002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostantjopoulou S., Katsarou Z., Zafiriou D., Gerasimou G., Alevriadou A., Georgiadis G., et al. Abnormality of N30 somatosensory evoked potentials in Parkinson's disease: a multidisciplinary approach. Neurophysiol. Clin. 2000;30:368–376. doi: 10.1016/s0987-7053(00)00235-5. [DOI] [PubMed] [Google Scholar]
- Boveri P. The myoclonic form of epidemic encephalitis. BMJ. 1920;1:570. doi: 10.1136/bmj.1.3095.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozi M., Bhatia K.P. Paroxysmal exercise-induced dystonia as a presenting feature of young-onset Parkinson's disease. Mov. Disord. 2003;18:1545–1547. doi: 10.1002/mds.10597. [DOI] [PubMed] [Google Scholar]
- Braak H., Bohl J.R., Müller C.M., Rüb U., de Vos R.A., Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov. Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov. Disord. 2003;18:357–363. doi: 10.1002/mds.10358. [DOI] [PubMed] [Google Scholar]
- Brown P. Bad oscillations in Parkinson's disease. J. Neural Transm. 2006;Suppl:27–30. doi: 10.1007/978-3-211-45295-0_6. [DOI] [PubMed] [Google Scholar]
- Brown P., Oliviero A., Mazzone P., Insola A., Tonali P., Di Lazzaro V. Dopamine Dependency of Oscillations between Subthalamic Nucleus and Pallidum in Parkinson's Disease. J. Neurosci. 2001;21:1033–1038. doi: 10.1523/JNEUROSCI.21-03-01033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell J.R., Hornberger M., Vucic S., Kiernan M.C., Hodges J.R. Apraxia and motor dysfunction in corticobasal syndrome. PLoS ONE. 2014;9:e92944. doi: 10.1371/journal.pone.0092944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G., Anastassiou C.A., Koch C. The origin of extracellular fields and currents–EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci. 2012;13:407–420. doi: 10.1038/nrn3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canolty R.T., Edwards E., Dalal S.S., Soltani M., Nagarajan S.S., Kirsch H.E., et al. High gamma power is phase-locked to theta oscillations in human neocortex. Science. 2006;313:1626–1628. doi: 10.1126/science.1128115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantello R., Tarletti R., Civardi C. Transcranial magnetic stimulation and Parkinson's disease. Brain Res. Brain Res. Rev. 2002;38:309–327. doi: 10.1016/s0165-0173(01)00158-8. [DOI] [PubMed] [Google Scholar]
- Capaday C., Forget R., Fraser R., Lamarre Y. Evidence for a contribution of the motor cortex to the long-latency stretch reflex of the human thumb. J. Physiol. 1991;440:243–255. doi: 10.1113/jphysiol.1991.sp018706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramia M., Desiato M., Cicinelli P., Iani C., Rossini P. Latency jump of “relaxed” versus “contracted” motor evoked potentials as a marker of cortico-spinal maturation. Electroencephalogr. Clin. Neurophysiol. 1993;89:61–66. doi: 10.1016/0168-5597(93)90086-5. [DOI] [PubMed] [Google Scholar]
- Carella F., Ciano C., Panzica F., Scaioli V. Myoclonus in corticobasal degeneration. Mov. Disord. 1997;12:598–603. doi: 10.1002/mds.870120419. [DOI] [PubMed] [Google Scholar]
- Carrillo F., Palomar F.J., Conde V., Diaz-Corrales F.J., Porcacchia P., Fernández-Del-Olmo M., et al. Study of cerebello-thalamocortical pathway by transcranial magnetic stimulation in Parkinson's disease. Brain Stimul. 2013;6:582–589. doi: 10.1016/j.brs.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Caviness J.N., Adler C.H., Beach T.G., Wetjen K.L., Caselli R.J. Small-amplitude cortical myoclonus in Parkinson's disease: physiology and clinical observations. Mov. Disord. 2002;17:657–662. doi: 10.1002/mds.10177. [DOI] [PubMed] [Google Scholar]
- Caviness J.N., Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3:598–607. doi: 10.1016/S1474-4422(04)00880-4. [DOI] [PubMed] [Google Scholar]
- Caviness J.N., Lue L.F., Beach T.G., Hentz J.G., Adler C.H., Sue L., et al. Parkinson's disease, cortical dysfunction, and alpha-synuclein. Mov. Disord. 2011;26:1436–1442. doi: 10.1002/mds.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness J.N., Tsuboi Y., Wszolek Z.K. Clinical-electrophysiological correlation of tremor and myoclonus in a kindred with the N279K tau mutation. Parkinsonism Relat Disord. 2003;9:151–157. doi: 10.1016/s1353-8020(02)00034-2. [DOI] [PubMed] [Google Scholar]
- Caviness J.N., Wszolek Z.K. Myoclonus in pallido-ponto-nigral degeneration. Adv. Neurol. 2002;89:35–39. [PubMed] [Google Scholar]
- Celebi O., Temuçin Ç.M., Elibol B., Saka E. Cognitive profiling in relation to short latency afferent inhibition of frontal cortex in multiple system atrophy. Parkinsonism Relat Disord. 2014;20:632–636. doi: 10.1016/j.parkreldis.2014.03.012. [DOI] [PubMed] [Google Scholar]
- Chahine L.M., Kauta S.R., Daley J.T., Cantor C.R., Dahodwala N. Surface EMG activity during REM sleep in Parkinson's disease correlates with disease severity. Parkinsonism Relat Disord. 2014;20:766–771. doi: 10.1016/j.parkreldis.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.C., Chen J.T., Wu Z.A., Kao K.P., Liao K.K. Cutaneous reflexes in patients with acute lacunar infarctions. J. Neurol. Sci. 1998;159:28–37. doi: 10.1016/s0022-510x(98)00142-7. [DOI] [PubMed] [Google Scholar]
- Chen C.C., Chen J.T., Wu Z.A., Kao K.P., Liao K.K. Long latency responses in pure sensory stroke due to thalamic infarction. Acta Neurol. Scand. 1998;98:41–48. doi: 10.1111/j.1600-0404.1998.tb07376.x. [DOI] [PubMed] [Google Scholar]
- Chen C.C., Lin W.Y., Chan H.L., Hsu Y.T., Tu P.H., Lee S.T., et al. Stimulation of the subthalamic region at 20 Hz slows the development of grip force in Parkinson's disease. Exp. Neurol. 2011;231:91–96. doi: 10.1016/j.expneurol.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Chen C.C., Yeh C.H., Chan H.L., Chang Y.J., Tu P.H., Yeh C.H., et al. Subthalamic nucleus oscillations correlate with vulnerability to freezing of gait in patients with Parkinson's disease. Neurobiol. Dis. 2019;132 doi: 10.1016/j.nbd.2019.104605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Ashby P., Lang A.E. Stimulus-sensitive myoclonus in akinetic-rigid syndromes. Brain. 1992;115:1875–1888. doi: 10.1093/brain/115.6.1875. [DOI] [PubMed] [Google Scholar]
- Chen R., Cros D., Curra A., Di Lazzaro V., Lefaucheur J.P., Magistris M.R., et al. The clinical diagnostic utility of transcranial magnetic stimulation: Report of an IFCN committee. Clin. Neurophysiol. 2008;119:504–532. doi: 10.1016/j.clinph.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Chen R., Garg R.R., Lozano A.M., Lang A.E. Effects of internal globus pallidus stimulation on motor cortex excitability. Neurology. 2001;56:716–723. doi: 10.1212/wnl.56.6.716. [DOI] [PubMed] [Google Scholar]
- Chen R., Yung D., Li J.Y. Organization of ipsilateral excitatory and inhibitory pathways in the human motor cortex. J. Neurophysiol. 2003;89:1256–1264. doi: 10.1152/jn.00950.2002. [DOI] [PubMed] [Google Scholar]
- Cheron G. Is the frontal N30 component of the somatosensory evoked potentials a reliable physiological index of the dopaminergic motor pathways? Clin. Neurophysiol. 1999;110:1698–1699. doi: 10.1016/s1388-2457(99)00128-5. [DOI] [PubMed] [Google Scholar]
- Cheron G., Piette T., Thiriaux A., Jacquy J., Godaux E. Somatosensory evoked potentials at rest and during movement in Parkinson's disease: evidence for a specific apomorphine effect on the frontal N30 wave. Electroencephalogr. Clin. Neurophysiol. 1994;92:491–501. doi: 10.1016/0168-5597(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Chu J., Wagle-Shukla A., Gunraj C., Lang A.E., Chen R. Impaired presynaptic Inhibition in the motor cortex in Parkinson disease. Neurology. 2009;72:842–849. doi: 10.1212/01.wnl.0000343881.27524.e8. [DOI] [PubMed] [Google Scholar]
- Chung K.A., Lobb B.M., Nutt J.G., Horak F.B. Effects of a central cholinesterase inhibitor on reducing falls in Parkinson disease. Neurology. 2010;75:1263–1269. doi: 10.1212/WNL.0b013e3181f6128c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cincotta M., Borgheresi A., Balestrieri F., Giovannelli F., Ragazzoni A., Vanni P., et al. Mechanisms underlying mirror movements in Parkinson's disease: a transcranial magnetic stimulation study. Mov. Disord. 2006;21:1019–1025. doi: 10.1002/mds.20850. [DOI] [PubMed] [Google Scholar]
- Claus D., Schöcklmann H.O., Dietrich H.J. Long latency muscle responses in cerebellar diseases. Eur Arch Psychiatry Neurol Sci. 1986;235:355–360. doi: 10.1007/BF00381004. [DOI] [PubMed] [Google Scholar]
- Cole S.R., van der Meij R., Peterson E.J., de Hemptinne C., Starr P.A., Voytek B. Nonsinusoidal Beta Oscillations Reflect Cortical Pathophysiology in Parkinson's Disease. J. Neurosci. 2017;37:4830–4840. doi: 10.1523/JNEUROSCI.2208-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compta Y., Valls-Solé J., Valldeoriola F., Kumru H., Rumià J. The silent period of the thenar muscles to contralateral and ipsilateral deep brain stimulation. Clin. Neurophysiol. 2006;117:2512–2520. doi: 10.1016/j.clinph.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Conte A., Belvisi D., Bologna M., Ottaviani D., Fabbrini G., Colosimo C., et al. Abnormal cortical synaptic plasticity in primary motor area in progressive supranuclear palsy. Cereb. Cortex. 2012;22:693–700. doi: 10.1093/cercor/bhr149. [DOI] [PubMed] [Google Scholar]
- Crowell A.L., Ryapolova-Webb E.S., Ostrem J.L., Galifianakis N.B., Shimamoto S., Lim D.A., et al. Oscillations in sensorimotor cortex in movement disorders: an electrocorticography study. Brain. 2012;135:615–630. doi: 10.1093/brain/awr332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunic D., Roshan L., Khan F.I., Lozano A.M., Lang A.E., Chen R. Effects of subthalamic nucleus stimulation on motor cortex excitability in Parkinson's disease. Neurology. 2002;58:1665–1672. doi: 10.1212/wnl.58.11.1665. [DOI] [PubMed] [Google Scholar]
- de Hemptinne C., Ryapolova-Webb E.S., Air E.L., Garcia P.A., Miller K.J., Ojemann J.G., et al. Exaggerated phase-amplitude coupling in the primary motor cortex in Parkinson disease. Proc Natl Acad Sci U S A. 2013;110:4780–4785. doi: 10.1073/pnas.1214546110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hemptinne C., Swann N.C., Ostrem J.L., Ryapolova-Webb E.S., San Luciano M., Galifianakis N.B., et al. Therapeutic deep brain stimulation reduces cortical phase-amplitude coupling in Parkinson's disease. Nat. Neurosci. 2015;18:779–786. doi: 10.1038/nn.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa A., Volpe G., Marcantonio L., Santoro L., Brice A., Filla A., et al. Neurophysiological evidence of corticospinal tract abnormality in patients with Parkin mutations. J. Neurol. 2006;253:275–279. doi: 10.1007/s00415-006-0096-0. [DOI] [PubMed] [Google Scholar]
- Deffains M., Iskhakova L., Bergman H. Stop and Think about Basal Ganglia Functional Organization: The Pallido-Striatal “Stop” Route. Neuron. 2016;89:237–239. doi: 10.1016/j.neuron.2016.01.003. [DOI] [PubMed] [Google Scholar]
- DeLong M.R. Activity of pallidal neurons during movement. J. Neurophysiol. 1971;34:414–427. doi: 10.1152/jn.1971.34.3.414. [DOI] [PubMed] [Google Scholar]
- DeLong M.R., Crutcher M.D., Georgopoulos A.P. Primate globus pallidus and subthalamic nucleus: functional organization. J. Neurophysiol. 1985;53:530–543. doi: 10.1152/jn.1985.53.2.530. [DOI] [PubMed] [Google Scholar]
- Delong M.R., Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 2007;64:20–24. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- Delwaide P.J., Pepin J.L., Maertens de Noordhout A. Contribution of reticular nuclei to the pathophysiology of parkinsonian rigidity. Adv. Neurol. 1993;60:381–385. [PubMed] [Google Scholar]
- Delwaide P.J., Sabbatino M., Delwaide C. Some pathophysiological aspects of the parkinsonian rigidity. J. Neural Transm. Suppl. 1986;22:129–139. [PubMed] [Google Scholar]
- Derejko M., Rakowicz M., Antczak J., Inglot E., Niewiadomska M. Corticomotor excitability in drug-naive patients with Parkinson disease. Neurol. Neurochir. Pol. 2013;47:109–115. doi: 10.5114/ninp.2013.34699. [DOI] [PubMed] [Google Scholar]
- Deuschl G. In: Handbook of Clinical Neurophysiology. Hallett M., editor. Elsevier; 2003. Chapter 19 Long-latency reflexes following stretch and nerve stimulation. [Google Scholar]
- Deuschl G., Eisen A. Long-latency reflexes following electrical nerve stimulation. The International Federation of Clinical Neurophysiology. Electroencephalogr. Clin. Neurophysiol. Suppl. 1999;52:263–268. [PubMed] [Google Scholar]
- Deuschl G., Ludolph A., Schenck E., Lücking C.H. The relations between long-latency reflexes in hand muscles, somatosensory evoked potentials and transcranial stimulation of motor tracts. Electroencephalogr. Clin. Neurophysiol. 1989;74:425–430. doi: 10.1016/0168-5597(89)90031-2. [DOI] [PubMed] [Google Scholar]
- di Biase L., Brittain J.S., Shah S.A., Pedrosa D.J., Cagnan H., Mathy A., et al. Tremor stability index: a new tool for differential diagnosis in tremor syndromes. Brain. 2017;140:1977–1986. doi: 10.1093/brain/awx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasio F., Suppa A., Fabbrini A., Marsili L., Asci F., Conte A., et al. Parkinsonism is associated with altered primary motor cortex plasticity in frontotemporal dementia-primary progressive aphasia variant. Neurobiol. Aging. 2018;69:230–238. doi: 10.1016/j.neurobiolaging.2018.05.026. [DOI] [PubMed] [Google Scholar]
- Di Stasio F., Suppa A., Marsili L., Upadhyay N., Asci F., Bologna M., et al. Corticobasal syndrome: neuroimaging and neurophysiological advances. Eur. J. Neurol. 2019;26:701–e752. doi: 10.1111/ene.13928. [DOI] [PubMed] [Google Scholar]
- Dietz V., Quintern J., Berger W. Electrophysiological studies of gait in spasticity and rigidity. Evidence that altered mechanical properties of muscle contribute to hypertonia. Brain. 1981;104:431–449. doi: 10.1093/brain/104.3.431. [DOI] [PubMed] [Google Scholar]
- Dietz V., Sinkjaer T. Spastic movement disorder: impaired reflex function and altered muscle mechanics. Lancet Neurol. 2007;6:725–733. doi: 10.1016/S1474-4422(07)70193-X. [DOI] [PubMed] [Google Scholar]
- Diószeghy P., Hidasi E., Mechler F. Study of central motor functions using magnetic stimulation in Parkinson's disease. Electromyogr. Clin. Neurophysiol. 1999;39:101–105. [PubMed] [Google Scholar]
- Dirkx M.F., den Ouden H., Aarts E., Timmer M., Bloem B.R., Toni I., et al. The Cerebral Network of Parkinson's Tremor: An Effective Connectivity fMRI Study. J. Neurosci. 2016;36:5362–5372. doi: 10.1523/JNEUROSCI.3634-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx M.F., Zach H., Bloem B.R., Hallett M., Helmich R.C. The nature of postural tremor in Parkinson disease. Neurology. 2018;90:e1095–e1103. doi: 10.1212/WNL.0000000000005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx M.F., Zach H., van Nuland A., Bloem B.R., Toni I., Helmich R.C. Cerebral differences between dopamine-resistant and dopamine-responsive Parkinson's tremor. Brain. 2019;142:3144–3157. doi: 10.1093/brain/awz261. [DOI] [PubMed] [Google Scholar]
- Dirkx M.F., Zach H., van Nuland A.J., Bloem B.R., Toni I., Helmich R.C. Cognitive load amplifies Parkinson's tremor through excitatory network influences onto the thalamus. Brain. 2020;143:1498–1511. doi: 10.1093/brain/awaa083. [DOI] [PubMed] [Google Scholar]
- Drory V.E., Inzelberg R., Groozman G.B., Korczyn A.D. N30 somatosensory evoked potentials in patients with unilateral Parkinson's disease. Acta Neurol. Scand. 1998;97:73–76. doi: 10.1111/j.1600-0404.1998.tb00613.x. [DOI] [PubMed] [Google Scholar]
- Eggers C., Fink G.R., Nowak D.A. Theta burst stimulation over the primary motor cortex does not induce cortical plasticity in Parkinson's disease. J. Neurol. 2010;257:1669–1674. doi: 10.1007/s00415-010-5597-1. [DOI] [PubMed] [Google Scholar]
- Ehgoetz Martens K.A., Ellard C.G., Almeida Q.J. Does anxiety cause freezing of gait in Parkinson's disease? PLoS ONE. 2014;9:e106561. doi: 10.1371/journal.pone.0106561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehgoetz Martens K.A., Hall J.M., Georgiades M.J., Gilat M., Walton C.C., Matar E., et al. The functional network signature of heterogeneity in freezing of gait. Brain. 2018;141:1145–1160. doi: 10.1093/brain/awy019. [DOI] [PubMed] [Google Scholar]
- Ehgoetz Martens K.A., Matar E., Hall J.M., Phillips J., Szeto J.Y.Y., Gouelle A., et al. Subtle gait and balance impairments occur in idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2019;34:1374–1380. doi: 10.1002/mds.27780. [DOI] [PubMed] [Google Scholar]
- Eisinger R.S., Urdaneta M.E., Foote K.D., Okun M.S., Gunduz A. Non-motor Characterization of the Basal Ganglia: Evidence From Human and Non-human Primate Electrophysiology. Front. Neurosci. 2018;12:385. doi: 10.3389/fnins.2018.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A.E., Del Sorbo F., Romito L.M., Barzaghi C., Garavaglia B., Albanese A. Isolated limb dystonia as presenting feature of Parkin disease. J. Neurol. Neurosurg. Psychiatry. 2014;85:827–828. doi: 10.1136/jnnp-2013-307294. [DOI] [PubMed] [Google Scholar]
- Ellaway P.H., Davey N.J., Maskill D.W., Dick J.P. The relation between bradykinesia and excitability of the motor cortex assessed using transcranial magnetic stimulation in normal and parkinsonian subjects. Electroencephalogr. Clin. Neurophysiol. 1995;97:169–178. doi: 10.1016/0924-980x(94)00336-6. [DOI] [PubMed] [Google Scholar]
- Ellis A.W. The myoclonic form of acute epidemic encephalitis. Lancet. 1920;196:114–116. [Google Scholar]
- Espay A.J., Beaton D.E., Morgante F., Gunraj C.A., Lang A.E., Chen R. Impairments of speed and amplitude of movement in Parkinson's disease: a pilot study. Mov. Disord. 2009;24:1001–1008. doi: 10.1002/mds.22480. [DOI] [PubMed] [Google Scholar]
- Espay A.J., Fasano A., van Nuenen B.F., Payne M.M., Snijders A.H., Bloem B.R. “On” state freezing of gait in Parkinson disease: a paradoxical levodopa-induced complication. Neurology. 2012;78:454–457. doi: 10.1212/WNL.0b013e3182477ec0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay A.J., Giuffrida J.P., Chen R., Payne M., Mazzella F., Dunn E., et al. Differential response of speed, amplitude, and rhythm to dopaminergic medications in Parkinson's disease. Mov. Disord. 2011;26:2504–2508. doi: 10.1002/mds.23893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eusebio A., Azulay J.P., Witjas T., Rico A., Attarian S. Assessment of cortico-spinal tract impairment in multiple system atrophy using transcranial magnetic stimulation. Clin. Neurophysiol. 2007;118:815–823. doi: 10.1016/j.clinph.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Eusebio A., Chen C.C., Lu C.S., Lee S.T., Tsai C.H., Limousin P., et al. Effects of low-frequency stimulation of the subthalamic nucleus on movement in Parkinson's disease. Exp. Neurol. 2008;209:125–130. doi: 10.1016/j.expneurol.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M., Guimarães I., Cardoso R., Coelho M., Guedes L.C., Rosa M.M., et al. Speech and Voice Response to a Levodopa Challenge in Late-Stage Parkinson's Disease. Front. Neurol. 2017;8:432. doi: 10.3389/fneur.2017.00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S., Marsden C.D., Van Woert M.H. Definition and classification of myoclonus. Adv. Neurol. 1986;43:1–5. [PubMed] [Google Scholar]
- Fasano A., Aquino C.C., Krauss J.K., Honey C.R., Bloem B.R. Axial disability and deep brain stimulation in patients with Parkinson disease. Nat Rev Neurol. 2015;11:98–110. doi: 10.1038/nrneurol.2014.252. [DOI] [PubMed] [Google Scholar]
- Fasano A., Bloem B.R. Gait disorders. Continuum (Minneap Minn) 2013;19:1344–1382. doi: 10.1212/01.CON.0000436159.33447.69. [DOI] [PubMed] [Google Scholar]
- Fasano A., Canning C.G., Hausdorff J.M., Lord S., Rochester L. Falls in Parkinson's disease: A complex and evolving picture. Mov. Disord. 2017;32:1524–1536. doi: 10.1002/mds.27195. [DOI] [PubMed] [Google Scholar]
- Fasano A., Herman T., Tessitore A., Strafella A.P., Bohnen N.I. Neuroimaging of Freezing of Gait. J Parkinsons Dis. 2015;5:241–254. doi: 10.3233/JPD-150536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano A., Laganiere S.E., Lam S., Fox M.D. Lesions causing freezing of gait localize to a cerebellar functional network. Ann. Neurol. 2017;81:129–141. doi: 10.1002/ana.24845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano A., Lang A.E. Unfreezing of gait in patients with Parkinson's disease. Lancet Neurol. 2015;14:675–677. doi: 10.1016/S1474-4422(15)00053-8. [DOI] [PubMed] [Google Scholar]
- Fasano A., Lozano A.M. Deep brain stimulation for movement disorders: 2015 and beyond. Curr. Opin. Neurol. 2015;28:423–436. doi: 10.1097/WCO.0000000000000226. [DOI] [PubMed] [Google Scholar]
- Fasano A., Plotnik M., Bove F., Berardelli A. The neurobiology of falls. Neurol Sci. 2012;33:1215–1223. doi: 10.1007/s10072-012-1126-6. [DOI] [PubMed] [Google Scholar]
- Fasano A., Schlenstedt C., Herzog J., Plotnik M., Rose F.E.M., Volkmann J., et al. Split-belt locomotion in Parkinson's disease links asymmetry, dyscoordination and sequence effect. Gait Posture. 2016;48:6–12. doi: 10.1016/j.gaitpost.2016.04.020. [DOI] [PubMed] [Google Scholar]
- Fawcett A.P., Dostrovsky J.O., Lozano A.M., Hutchison W.D. Eye movement-related responses of neurons in human subthalamic nucleus. Exp. Brain Res. 2005;162:357–365. doi: 10.1007/s00221-004-2184-7. [DOI] [PubMed] [Google Scholar]
- Feldmann L.K., Neumann W.J., Faust K., Schneider G.H., Kühn A.A. Risk of Infection after Deep Brain Stimulation Surgery with Externalization and Local-Field Potential Recordings: Twelve-Year Experience from a Single Institution. Stereotact. Funct. Neurosurg. 2021;99:512–520. doi: 10.1159/000516150. [DOI] [PubMed] [Google Scholar]
- Feldmann L.K., Neumann W.J., Krause P., Lofredi R., Schneider G.H., Kühn A.A. Subthalamic beta band suppression reflects effective neuromodulation in chronic recordings. Eur. J. Neurol. 2021;28:2372–2377. doi: 10.1111/ene.14801. [DOI] [PubMed] [Google Scholar]
- Ferbert A., Priori A., Rothwell J.C., Day B.L., Colebatch J.G., Marsden C.D. Interhemispheric inhibition of the human motor cortex. J. Physiol. 1992;453:525–546. doi: 10.1113/jphysiol.1992.sp019243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisicaro F., Lanza G., Cantone M., Ferri R., Pennisi G., Nicoletti A., et al. Clinical and Electrophysiological Hints to TMS in De Novo Patients with Parkinson's Disease and Progressive Supranuclear Palsy. J Pers Med. 2020;10:274. doi: 10.3390/jpm10040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhr P., Zeffiro T., Hallett M. Cutaneous reflexes in Parkinson's disease. Muscle Nerve. 1992;15:733–739. doi: 10.1002/mus.880150618. [DOI] [PubMed] [Google Scholar]
- Garcia P.A., Aminoff M.J., Goodin D.S. The frontal N30 component of the median-derived SEP in patients with predominantly unilateral Parkinson's disease. Neurology. 1995;45:989–992. doi: 10.1212/wnl.45.5.989. [DOI] [PubMed] [Google Scholar]
- Georgiades M.J., Shine J.M., Gilat M., McMaster J., Owler B., Mahant N., et al. Hitting the brakes: pathological subthalamic nucleus activity in Parkinson's disease gait freezing. Brain. 2019;142:3906–3916. doi: 10.1093/brain/awz325. [DOI] [PubMed] [Google Scholar]
- Giannicola G., Marceglia S., Rossi L., Mrakic-Sposta S., Rampini P., Tamma F., et al. The effects of levodopa and ongoing deep brain stimulation on subthalamic beta oscillations in Parkinson's disease. Exp. Neurol. 2010;226:120–127. doi: 10.1016/j.expneurol.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Gilron R., Little S., Perrone R., Wilt R., de Hemptinne C., Yaroshinsky M.S., et al. Long-term wireless streaming of neural recordings for circuit discovery and adaptive stimulation in individuals with Parkinson's disease. Nat. Biotechnol. 2021;39:1078–1085. doi: 10.1038/s41587-021-00897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass G.A., Ahlskog J.E., Matsumoto J.Y. Orthostatic myoclonus: a contributor to gait decline in selected elderly. Neurology. 2007;68:1826–1830. doi: 10.1212/01.wnl.0000260225.46732.af. [DOI] [PubMed] [Google Scholar]
- Gouider-Khouja N., Vidailhet M., Bonnet A.M., Pichon J., Agid Y. “Pure” striatonigral degeneration and Parkinson's disease: a comparative clinical study. Mov. Disord. 1995;10:288–294. doi: 10.1002/mds.870100310. [DOI] [PubMed] [Google Scholar]
- Grey M.J., Ladouceur M., Andersen J.B., Nielsen J.B., Sinkjaer T. Group II muscle afferents probably contribute to the medium latency soleus stretch reflex during walking in humans. J. Physiol. 2001;534:925–933. doi: 10.1111/j.1469-7793.2001.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppa S., Oliviero A., Eisen A., Quartarone A., Cohen L.G., Mall V., et al. A practical guide to diagnostic transcranial magnetic stimulation: report of an IFCN committee. Clin. Neurophysiol. 2012;123:858–882. doi: 10.1016/j.clinph.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérit J-M (2001). K.R. Mills. Magnetic stimulation of the human nervous system. Oxford: Oxford university Press (1999), 333 p. 69,50£. ISBN 0-19-262986-7. Neurophysiol Clin 31: 349.
- Guerra A., Asci F., D'Onofrio V., Sveva V., Bologna M., Fabbrini G., et al. Enhancing gamma oscillations restores primary motor cortex plasticity in Parkinson's disease. J. Neurosci. 2020;40:4788–4796. doi: 10.1523/JNEUROSCI.0357-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra A., Asci F., Zampogna A., D'Onofrio V., Berardelli A., Suppa A. The effect of gamma oscillations in boosting primary motor cortex plasticity is greater in young than older adults. Clin. Neurophysiol. 2021;132:1358–1366. doi: 10.1016/j.clinph.2021.01.032. [DOI] [PubMed] [Google Scholar]
- Guerra A., Asci F., Zampogna A., D'Onofrio V., Petrucci S., Ginevrino M., et al. Gamma-transcranial alternating current stimulation and theta-burst stimulation: inter-subject variability and the role of BDNF. Clin. Neurophysiol. 2020;131:2691–2699. doi: 10.1016/j.clinph.2020.08.017. [DOI] [PubMed] [Google Scholar]
- Guerra A., Asci F., Zampogna A., D'Onofrio V., Suppa A., Fabbrini G., et al. Long-term changes in short-interval intracortical facilitation modulate motor cortex plasticity and L-dopa-induced dyskinesia in Parkinson's disease. Brain Stimul. 2021;15:99–108. doi: 10.1016/j.brs.2021.11.016. [DOI] [PubMed] [Google Scholar]
- Guerra A., Colella D., Giangrosso M., Cannavacciuolo A., Paparella G., Fabbrini G., et al. Driving motor cortex oscillations modulates bradykinesia in Parkinson's disease. Brain. 2022;145:224–236. doi: 10.1093/brain/awab257. [DOI] [PubMed] [Google Scholar]
- Guerra A., Suppa A., Bologna M., D'Onofrio V., Bianchini E., Brown P., et al. Boosting the LTP-like plasticity effect of intermittent theta-burst stimulation using gamma transcranial alternating current stimulation. Brain Stimul. 2018;11:734–742. doi: 10.1016/j.brs.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra A., Suppa A., D'Onofrio V., Di Stasio F., Asci F., Fabbrini G., et al. Abnormal cortical facilitation and L-dopa-induced dyskinesia in Parkinson's disease. Brain Stimul. 2019;12:1517–1525. doi: 10.1016/j.brs.2019.06.012. [DOI] [PubMed] [Google Scholar]
- Guiot G., Albe-Fessard D., Arfel G., Derome P. Monitoring of unit activity in the course of stereotaxic interventions. Neurochirurgie. 1964;10:427–435. [PubMed] [Google Scholar]
- Haber S. Anatomical relationship between the basal ganglia and the basal nucleus of Meynert in human and monkey forebrain. Proc. Natl. Acad. Sci. USA. 1987;84:1408–1412. doi: 10.1073/pnas.84.5.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M. Tremor: pathophysiology. Parkinsonism Relat. Disord. 2014;20(Suppl 1):S118–S122. doi: 10.1016/S1353-8020(13)70029-4. [DOI] [PubMed] [Google Scholar]
- Hallett M., DelRosso L.M., Elble R., Ferri R., Horak F.B., Lehericy S., et al. Evaluation of movement and brain activity. Clin. Neurophysiol. 2021;132:2608–2638. doi: 10.1016/j.clinph.2021.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern C.H., Danish S.F., Baltuch G.H., Jaggi J.L. Brain shift during deep brain stimulation surgery for Parkinson's disease. Stereotact. Funct. Neurosurg. 2008;86:37–43. doi: 10.1159/000108587. [DOI] [PubMed] [Google Scholar]
- Hasan H., Athauda D.S., Foltynie T., Noyce A.J. Technologies assessing limb Bradykinesia in Parkinson's disease. J Parkinsons Dis. 2017;7:65–77. doi: 10.3233/JPD-160878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmich R.C. The cerebral basis of Parkinsonian tremor: A network perspective. Mov. Disord. 2018;33:219–231. doi: 10.1002/mds.27224. [DOI] [PubMed] [Google Scholar]
- Helmich R.C., Janssen M.J., Oyen W.J., Bloem B.R., Toni I. Pallidal dysfunction drives a cerebellothalamic circuit into Parkinson tremor. Ann. Neurol. 2011;69:269–281. doi: 10.1002/ana.22361. [DOI] [PubMed] [Google Scholar]
- Helmich R.C., Van den Berg K.R.E., Panyakaew P., Cho H.J., Osterholt T., McGurrin P., et al. Cerebello-cortical control of tremor rhythm and amplitude in Parkinson's Disease. Mov. Disord. 2021;36:1727–1729. doi: 10.1002/mds.28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herter T.M., Takei T., Munoz D.P., Scott S.H. Neurons in red nucleus and primary motor cortex exhibit similar responses to mechanical perturbations applied to the upper-limb during posture. Front. Integr. Neurosci. 2015;9:29. doi: 10.3389/fnint.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschmann J., Butz M., Hartmann C.J., Hoogenboom N., Özkurt T.E., Vesper J., et al. Parkinsonian rest tremor is associated with modulations of subthalamic high-frequency oscillations. Mov. Disord. 2016;31:1551–1559. doi: 10.1002/mds.26663. [DOI] [PubMed] [Google Scholar]
- Huang Y.Z., Edwards M.J., Rounis E., Bhatia K.P., Rothwell J.C. Theta burst stimulation of the human motor cortex. Neuron. 2005;45:201–206. doi: 10.1016/j.neuron.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Huang Y.Z., Lu M.K., Antal A., Classen J., Nitsche M., Ziemann U., et al. Plasticity induced by non-invasive transcranial brain stimulation: A position paper. Clin. Neurophysiol. 2017;128:2318–2329. doi: 10.1016/j.clinph.2017.09.007. [DOI] [PubMed] [Google Scholar]
- Huang Y.Z., Rothwell J.C., Lu C.S., Chuang W.L., Chen R.S. Abnormal bidirectional plasticity-like effects in Parkinson's disease. Brain. 2011;134:2312–2320. doi: 10.1093/brain/awr158. [DOI] [PubMed] [Google Scholar]
- Hupfeld K.E., Swanson C.W., Fling B.W., Seidler R.D. TMS-induced silent periods: A review of methods and call for consistency. J. Neurosci. Methods. 2020;346 doi: 10.1016/j.jneumeth.2020.108950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado J.M., Gray C.M., Tamas L.B., Sigvardt K.A. Dynamics of tremor-related oscillations in the human globus pallidus: a single case study. Proc. Natl. Acad. Sci. USA. 1999;96:1674–1679. doi: 10.1073/pnas.96.4.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison W.D., Allan R.J., Opitz H., Levy R., Dostrovsky J.O., Lang A.E., et al. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson's disease. Ann. Neurol. 1998;44:622–628. doi: 10.1002/ana.410440407. [DOI] [PubMed] [Google Scholar]
- Hutchison W.D., Dostrovsky J.O., Walters J.R., Courtemanche R., Boraud T., Goldberg J., et al. Neuronal oscillations in the basal ganglia and movement disorders: evidence from whole animal and human recordings. J. Neurosci. 2004;24:9240–9243. doi: 10.1523/JNEUROSCI.3366-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison W.D., Lozano A.M., Davis K.D., Saint-Cyr J.A., Lang A.E., Dostrovsky J.O. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. NeuroReport. 1994;5:1533–1537. doi: 10.1097/00001756-199407000-00031. [DOI] [PubMed] [Google Scholar]
- Hutchison W.D., Lozano A.M., Tasker R.R., Lang A.E., Dostrovsky J.O. Identification and characterization of neurons with tremor-frequency activity in human globus pallidus. Exp. Brain Res. 1997;113:557–563. doi: 10.1007/pl00005606. [DOI] [PubMed] [Google Scholar]
- Hwang J., Bank A.M., Mortazavi F., Oakley D.H., Frosch M.P., Schmahmann J.D. Spinal cord α-synuclein deposition associated with myoclonus in patients with MSA-C. Neurology. 2019;93:302–309. doi: 10.1212/WNL.0000000000007949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iansek R., Huxham F., McGinley J. The sequence effect and gait festination in Parkinson disease: contributors to freezing of gait? Mov. Disord. 2006;21:1419–1424. doi: 10.1002/mds.20998. [DOI] [PubMed] [Google Scholar]
- Iglesias J.E., Insausti R., Lerma-Usabiaga G., Bocchetta M., Van Leemput K., Greve D.N., et al. A probabilistic atlas of the human thalamic nuclei combining ex vivo MRI and histology. Neuroimage. 2018;183:314–326. doi: 10.1016/j.neuroimage.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson N., Cole S.R., Voytek B., Swann N.C. Characteristics of waveform shape in Parkinson's disease detected with scalp electroencephalography. eNeuro. 2019;6 doi: 10.1523/ENEURO.0151-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.V., Nutt J.G., Carlson-Kuhta P., Stephens M., Horak F.B. Knee trembling during freezing of gait represents multiple anticipatory postural adjustments. Exp. Neurol. 2009;215:334–341. doi: 10.1016/j.expneurol.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson N., Brown P. New insights into the relationship between dopamine, beta oscillations and motor function. Trends Neurosci. 2011;34:611–618. doi: 10.1016/j.tins.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Jenkinson N., Kühn A.A., Brown P. γ oscillations in the human basal ganglia. Exp. Neurol. 2013;245:72–76. doi: 10.1016/j.expneurol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Joundi R.A., Jenkinson N., Brittain J.S., Aziz T.Z., Brown P. Driving oscillatory activity in the human cortex enhances motor performance. Curr. Biol. 2012;22:403–407. doi: 10.1016/j.cub.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung P., Ziemann U. Differences of the ipsilateral silent period in small hand muscles. Muscle Nerve. 2006;34:431–436. doi: 10.1002/mus.20604. [DOI] [PubMed] [Google Scholar]
- Kacar A., Filipovic S.R., Kresojevic N., Milanovic S.D., Ljubisavljevic M., Kostic V.S., et al. History of exposure to dopaminergic medication does not affect motor cortex plasticity and excitability in Parkinson's disease. Clin. Neurophysiol. 2013;124:697–707. doi: 10.1016/j.clinph.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Kang S.Y., Wasaka T., Shamim E.A., Auh S., Ueki Y., Lopez G.J., et al. Characteristics of the sequence effect in Parkinson's disease. Mov. Disord. 2010;25:2148–2155. doi: 10.1002/mds.23251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima S., Ueki Y., Mima T., Fukuyama H., Ojika K., Matsukawa N. Differences in dopaminergic modulation to motor cortical plasticity between Parkinson's disease and multiple system atrophy. PLoS ONE. 2013;8:e62515. doi: 10.1371/journal.pone.0062515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehnemouyi Y.M., Wilkins K.B., Anidi C.M., Anderson R.W., Afzal M.F., Bronte-Stewart H.M. Modulation of beta bursts in subthalamic sensorimotor circuits predicts improvement in bradykinesia. Brain. 2021;144:473–486. doi: 10.1093/brain/awaa394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S., Harding A.J., Halliday G.M., Mahant N., Fung V.S. Cortical limb myoclonus in pathologically proven progressive supranuclear palsy. Mov. Disord. 2013;28:1804–1806. doi: 10.1002/mds.25693. [DOI] [PubMed] [Google Scholar]
- Kim S.J., Udupa K., Ni Z., Moro E., Gunraj C., Mazzella F., et al. Effects of subthalamic nucleus stimulation on motor cortex plasticity in Parkinson disease. Neurology. 2015;85:425–432. doi: 10.1212/WNL.0000000000001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N.K.K., Krishna V., Basha D., Elias G., Sammartino F., Hodaie M., et al. Microelectrode recording findings within the tractography-defined ventral intermediate nucleus. J. Neurosurg. 2017;126:1669–1675. doi: 10.3171/2016.3.JNS151992. [DOI] [PubMed] [Google Scholar]
- Kishore A., James P., Krishnan S., Yahia-Cherif L., Meunier S., Popa T. Motor cortex plasticity can indicate vulnerability to motor fluctuation and high L-DOPA need in drug-naïve Parkinson's disease. Parkinsonism Relat Disord. 2017;35:55–62. doi: 10.1016/j.parkreldis.2016.12.005. [DOI] [PubMed] [Google Scholar]
- Kishore A., Joseph T., Velayudhan B., Popa T., Meunier S. Early, severe and bilateral loss of LTP and LTD-like plasticity in motor cortex (M1) in de novo Parkinson's disease. Clin. Neurophysiol. 2012;123:822–828. doi: 10.1016/j.clinph.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Kishore A., Popa T., Balachandran A., Chandran S., Pradeep S., Backer F., et al. Cerebellar sensory processing alterations impact motor cortical plasticity in Parkinson's disease: clues from dyskinetic patients. Cereb. Cortex. 2014;24:2055–2067. doi: 10.1093/cercor/bht058. [DOI] [PubMed] [Google Scholar]
- Kishore A., Popa T., James P., Yahia-Cherif L., Backer F., Varughese Chacko L., et al. Age-related decline in the responsiveness of motor cortex to plastic forces reverses with levodopa or cerebellar stimulation. Neurobiol. Aging. 2014;35:2541–2551. doi: 10.1016/j.neurobiolaging.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Kishore A., Popa T., Velayudhan B., Joseph T., Balachandran A., Meunier S. Acute dopamine boost has a negative effect on plasticity of the primary motor cortex in advanced Parkinson's disease. Brain. 2012;135:2074–2088. doi: 10.1093/brain/aws124. [DOI] [PubMed] [Google Scholar]
- Klawans H.L., Goetz C., Bergen D. Levodopa-induced myoclonus. Arch. Neurol. 1975;32:330–334. doi: 10.1001/archneur.1975.00490470075011. [DOI] [PubMed] [Google Scholar]
- Klawans H.L., Tanner C.M., McDermott J. Myoclonus and parkinsonism. Clin. Neuropharmacol. 1986;9:202–205. doi: 10.1097/00002826-198604000-00013. [DOI] [PubMed] [Google Scholar]
- Kofler M., Leis A.A., Valls-Solé J. Cutaneous silent periods - Part 1: Update on physiological mechanisms. Clin. Neurophysiol. 2019;130:588–603. doi: 10.1016/j.clinph.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Kofler M., Müller J., Reggiani L., Wenning G.K. Somatosensory evoked potentials in progressive supranuclear palsy. J. Neurol. Sci. 2000;179:85–91. doi: 10.1016/s0022-510x(00)00383-x. [DOI] [PubMed] [Google Scholar]
- Kofler M., Wenning G.K., Poewe W. Cortical and brain stem hyperexcitability in striatonigral degeneration. Mov. Disord. 1998;13:602–607. doi: 10.1002/mds.870130341. [DOI] [PubMed] [Google Scholar]
- Kojima S., Onishi H., Sugawara K., Miyaguchi S., Kirimoto H., Tamaki H., et al. No relation between afferent facilitation induced by digital nerve stimulation and the latency of cutaneomuscular reflexes and somatosensory evoked magnetic fields. Front. Hum. Neurosci. 2014;8:1023. doi: 10.3389/fnhum.2014.01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojovic M., Bologna M., Kassavetis P., Murase N., Palomar F.J., Berardelli A., et al. Functional reorganization of sensorimotor cortex in early Parkinson disease. Neurology. 2012;78:1441–1448. doi: 10.1212/WNL.0b013e318253d5dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojovic M., Kassavetis P., Bologna M., Parees I., Rubio-Agusti I., Beraredelli A., et al. Transcranial magnetic stimulation follow-up study in early Parkinson's disease: A decline in compensation with disease progression? Mov. Disord. 2015;30:1098–1106. doi: 10.1002/mds.26167. [DOI] [PubMed] [Google Scholar]
- Kondylis E.D., Randazzo M.J., Alhourani A., Lipski W.J., Wozny T.A., Pandya Y., et al. Movement-related dynamics of cortical oscillations in Parkinson's disease and essential tremor. Brain. 2016;139:2211–2223. doi: 10.1093/brain/aww144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A.A., Grosse P., Holtz K., Brown P., Meyer B.U., Kupsch A. Patterns of abnormal motor cortex excitability in atypical parkinsonian syndromes. Clin. Neurophysiol. 2004;115:1786–1795. doi: 10.1016/j.clinph.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Kuhn A.A., Kempf F., Brucke C., Gaynor D.L., Martinez-Torres I., Pogosyan A., et al. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory beta activity in patients with Parkinson's disease in parallel with improvement in motor performance. J. Neurosci. 2008;28:6165–6173. doi: 10.1523/JNEUROSCI.0282-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn A.A., Kupsch A., Schneider G.H., Brown P. Reduction in subthalamic 8–35 Hz oscillatory activity correlates with clinical improvement in Parkinson's disease. Eur. J. Neurosci. 2006;23:1956–1960. doi: 10.1111/j.1460-9568.2006.04717.x. [DOI] [PubMed] [Google Scholar]
- Kuhn A.A., Tsui A., Aziz T., Ray N., Brucke C., Kupsch A., et al. Pathological synchronisation in the subthalamic nucleus of patients with Parkinson's disease relates to both bradykinesia and rigidity. Exp. Neurol. 2009;215:380–387. doi: 10.1016/j.expneurol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Kühn A.A., Volkmann J. Innovations in deep brain stimulation methodology. Mov. Disord. 2017;32:11–19. doi: 10.1002/mds.26703. [DOI] [PubMed] [Google Scholar]
- Kuo Y.L., Dubuc T., Boufadel D.F., Fisher B.E. Measuring ipsilateral silent period: Effects of muscle contraction levels and quantification methods. Brain Res. 2017;1674:77–83. doi: 10.1016/j.brainres.2017.08.015. [DOI] [PubMed] [Google Scholar]
- Lafreniere-Roula M., Hutchison W.D., Lozano A.M., Hodaie M., Dostrovsky J.O. Microstimulation-induced inhibition as a tool to aid targeting the ventral border of the subthalamic nucleus. J. Neurosurg. 2009;111:724–728. doi: 10.3171/2009.3.JNS09111. [DOI] [PubMed] [Google Scholar]
- Lago-Rodriguez A., Ponzo V., Jenkinson N., Benitez-Rivero S., Del-Olmo M.F., Hu M., et al. Paradoxical facilitation after depotentiation protocol can precede dyskinesia onset in early Parkinson's disease. Exp. Brain Res. 2016;234:3659–3667. doi: 10.1007/s00221-016-4759-5. [DOI] [PubMed] [Google Scholar]
- Lauro P.M., Lee S., Akbar U., Asaad W.F. Subthalamic-Cortical Network Reorganization during Parkinson's Tremor. J. Neurosci. 2021;41:9844–9858. doi: 10.1523/JNEUROSCI.0854-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.G., Tatton W.G. Motor responses to sudden limb displacements in primates with specific CNS lesions and in human patients with motor system disorders. Can. J. Neurol. Sci. 1975;2:285–293. doi: 10.1017/s0317167100020382. [DOI] [PubMed] [Google Scholar]
- Leis A., Stokic D., Fuhr P., Kofler M., Kronenberg M., Wissel J., et al. Nociceptive fingertip stimulation inhibits synergistic motoneuron pools in the human upper limb. Neurology. 2000;55:1305–1309. doi: 10.1212/wnl.55.9.1305. [DOI] [PubMed] [Google Scholar]
- Lenz F.A., Tasker R.R., Kwan H.C., Schnider S., Kwong R., Murayama Y., et al. Single unit analysis of the human ventral thalamic nuclear group: correlation of thalamic “tremor cells” with the 3–6 Hz component of parkinsonian tremor. J. Neurosci. 1988;8:754–764. doi: 10.1523/JNEUROSCI.08-03-00754.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leodori G., Belvisi D., De Bartolo M.I., Fabbrini A., Costanzo M., Vial F., et al. Re-emergent Tremor in Parkinson's Disease: The Role of the Motor Cortex. Mov. Disord. 2020;35:1002–1011. doi: 10.1002/mds.28022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R., Hutchison W.D., Lozano A.M., Dostrovsky J.O. High-frequency synchronization of neuronal activity in the subthalamic nucleus of parkinsonian patients with limb tremor. J. Neurosci. 2000;20:7766–7775. doi: 10.1523/JNEUROSCI.20-20-07766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.Y., Espay A.J., Gunraj C.A., Pal P.K., Cunic D.I., Lang A.E., et al. Interhemispheric and ipsilateral connections in Parkinson's disease: relation to mirror movements. Mov. Disord. 2007;22:813–821. doi: 10.1002/mds.21386. [DOI] [PubMed] [Google Scholar]
- Linn-Evans M.E., Petrucci M.N., Amundsen Huffmaster S.L., Chung J.W., Tuite P.J., Howell M.J., et al. REM sleep without atonia is associated with increased rigidity in patients with mild to moderate Parkinson's disease. Clin. Neurophysiol. 2020;131:2008–2016. doi: 10.1016/j.clinph.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S., Brown P. The functional role of beta oscillations in Parkinson's disease. Parkinsonism Relat Disord. 2014;20(Suppl 1):S44–S48. doi: 10.1016/S1353-8020(13)70013-0. [DOI] [PubMed] [Google Scholar]
- Little S., Pogosyan A., Neal S., Zavala B., Zrinzo L., Hariz M., et al. Adaptive deep brain stimulation in advanced Parkinson disease. Ann. Neurol. 2013;74:449–457. doi: 10.1002/ana.23951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.D., Prescott I.A., Dostrovsky J.O., Hodaie M., Lozano A.M., Hutchison W.D. Frequency-dependent effects of electrical stimulation in the globus pallidus of dystonia patients. J. Neurophysiol. 2012;108:5–17. doi: 10.1152/jn.00527.2011. [DOI] [PubMed] [Google Scholar]
- Liu X., Zhang J., Fu K., Gong R., Chen J., Zhang J. Microelectrode Recording-Guided Versus Intraoperative Magnetic Resonance Imaging-Guided Subthalamic Nucleus Deep Brain Stimulation Surgery for Parkinson Disease: A 1-Year Follow-Up Study. World Neurosurg. 2017;107:900–905. doi: 10.1016/j.wneu.2017.08.077. [DOI] [PubMed] [Google Scholar]
- Lofredi R., Neumann W.J., Bock A., Horn A., Huebl J., Siegert S., et al. Dopamine-dependent scaling of subthalamic gamma bursts with movement velocity in patients with Parkinson's disease. Elife. 2018;7 doi: 10.7554/eLife.31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofredi R., Neumann W.J., Brücke C., Huebl J., Krauss J.K., Schneider G.H., et al. Pallidal beta bursts in Parkinson's disease and dystonia. Mov. Disord. 2019;34:420–424. doi: 10.1002/mds.27524. [DOI] [PubMed] [Google Scholar]
- Lou J.S., Valls-Solé J., Toro C., Hallett M. Facial action myoclonus in patients with olivopontocerebellar atrophy. Mov. Disord. 1994;9:223–226. doi: 10.1002/mds.870090218. [DOI] [PubMed] [Google Scholar]
- Louis E.D., Klatka L.A., Liu Y., Fahn S. Comparison of extrapyramidal features in 31 pathologically confirmed cases of diffuse Lewy body disease and 34 pathologically confirmed cases of Parkinson's disease. Neurology. 1997;48:376–380. doi: 10.1212/wnl.48.2.376. [DOI] [PubMed] [Google Scholar]
- Lourenço G., Iglesias C., Cavallari P., Pierrot-Deseilligny E., Marchand-Pauvert V. Mediation of late excitation from human hand muscles via parallel group II spinal and group I transcortical pathways. J. Physiol. 2006;572:585–603. doi: 10.1113/jphysiol.2005.102806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano A., Hutchison W., Kiss Z., Tasker R., Davis K., Dostrovsky J. Methods for microelectrode-guided posteroventral pallidotomy. J. Neurosurg. 1996;84:194–202. doi: 10.3171/jns.1996.84.2.0194. [DOI] [PubMed] [Google Scholar]
- Ma T.P. Saccade-related omnivectoral pause neurons in the primate zona incerta. NeuroReport. 1996;7:2713–2716. doi: 10.1097/00001756-199611040-00061. [DOI] [PubMed] [Google Scholar]
- Macerollo A., Chen J.C., Korlipara P., Foltynie T., Rothwell J., Edwards M.J., et al. Dopaminergic treatment modulates sensory attenuation at the onset of the movement in Parkinson's disease: A test of a new framework for bradykinesia. Mov. Disord. 2016;31:143–146. doi: 10.1002/mds.26493. [DOI] [PubMed] [Google Scholar]
- MacKinnon C.D., Gilley E.A., Weis-McNulty A., Simuni T. Pathways mediating abnormal intracortical inhibition in Parkinson's disease. Ann. Neurol. 2005;58:516–524. doi: 10.1002/ana.20599. [DOI] [PubMed] [Google Scholar]
- Maertens de Noordhout A., Rothwell J.C., Day B.L., Dressler D., Nakashima K., Thompson P.D., et al. Effect of digital nerve stimuli on responses to electrical or magnetic stimulation of the human brain. J. Physiol. 1992;447:535–548. doi: 10.1113/jphysiol.1992.sp019016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinova V., Pinter A., Dragaescu C., Rohde V., Trenkwalder C., Sixel-Döring F., et al. The role of intraoperative microelectrode recording and stimulation in subthalamic lead placement for Parkinson's disease. PLoS ONE. 2020;15:e0241752. doi: 10.1371/journal.pone.0241752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N., Pogosyan A., Marton L.F., Bolam J.P., Brown P., Magill P.J. Parkinsonian beta oscillations in the external globus pallidus and their relationship with subthalamic nucleus activity. J. Neurosci. 2008;28:14245–14258. doi: 10.1523/JNEUROSCI.4199-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning J.R., Jacobs J., Fried I., Kahana M.J. Broadband shifts in local field potential power spectra are correlated with single-neuron spiking in humans. J. Neurosci. 2009;29:13613–13620. doi: 10.1523/JNEUROSCI.2041-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano Y., Nakamuro T., Ikoma K., Sugata T., Morimoto S., Takayanagi T., et al. Central motor conductivity in aged people. Intern. Med. 1992;31:1084–1087. doi: 10.2169/internalmedicine.31.1084. [DOI] [PubMed] [Google Scholar]
- Mansvelder H.D., Verhoog M.B., Goriounova N.A. Synaptic plasticity in human cortical circuits: cellular mechanisms of learning and memory in the human brain? Curr. Opin. Neurobiol. 2019;54:186–193. doi: 10.1016/j.conb.2018.06.013. [DOI] [PubMed] [Google Scholar]
- Marchand-Pauvert V., Gerdelat-Mas A., Ory-Magne F., Calvas F., Mazevet D., Meunier S., et al. Both L-DOPA and HFS-STN restore the enhanced group II spinal reflex excitation to a normal level in patients with Parkinson's disease. Clin. Neurophysiol. 2011;122:1019–1026. doi: 10.1016/j.clinph.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Mariorenzi R., Zarola F., Caramia M., Paradiso C., Rossini P. Non-invasive evaluation of central motor tract excitability changes following peripheral nerve stimulation in healthy humans. Electroencephalogr. Clin. Neurophysiol. 1991;81:90–101. doi: 10.1016/0168-5597(91)90002-f. [DOI] [PubMed] [Google Scholar]
- Martin-Rodriguez J.F., Mir P. Short-afferent inhibition and cognitive impairment in Parkinson's disease: A quantitative review and challenges. Neurosci. Lett. 2020;719 doi: 10.1016/j.neulet.2018.06.048. [DOI] [PubMed] [Google Scholar]
- Marusiak J., Jaskólska A., Budrewicz S., Koszewicz M., Jaskólski A. Increased muscle belly and tendon stiffness in patients with Parkinson's disease, as measured by myotonometry. Mov. Disord. 2011;26:2119–2122. doi: 10.1002/mds.23841. [DOI] [PubMed] [Google Scholar]
- Matsunaga K., Uozumi T., Qingrui L., Hashimoto T., Tsuji S. Amantadine-induced cortical myoclonus. Neurology. 2001;56:279–280. doi: 10.1212/wnl.56.2.279. [DOI] [PubMed] [Google Scholar]
- Meara R.J., Cody F.W. Stretch reflexes of individual parkinsonian patients studied during changes in clinical rigidity following medication. Electroencephalogr. Clin. Neurophysiol. 1993;89:261–268. doi: 10.1016/0168-5597(93)90105-x. [DOI] [PubMed] [Google Scholar]
- Merk T., Peterson V., Köhler R., Haufe S., Richardson R.M., Neumann W.J. Machine learning based brain signal decoding for intelligent adaptive deep brain stimulation. Exp. Neurol. 2022;351 doi: 10.1016/j.expneurol.2022.113993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre T.A., Lang A.E., Ferreira J.J., Almeida V., de Carvalho M., Miyasaki J., et al. Associated movement disorders in orthostatic tremor. J. Neurol. Neurosurg. Psychiatry. 2012;83:725–729. doi: 10.1136/jnnp-2012-302436. [DOI] [PubMed] [Google Scholar]
- Meyer B.U., Roricht S., Grafin von Einsiedel H., Kruggel F., Weindl A. Inhibitory and excitatory interhemispheric transfers between motor cortical areas in normal humans and patients with abnormalities of the corpus callosum. Brain. 1995;118:429–440. doi: 10.1093/brain/118.2.429. [DOI] [PubMed] [Google Scholar]
- Miki Y., Foti S.C., Asi Y.T., Tsushima E., Quinn N., Ling H., et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain. 2019;142:2813–2827. doi: 10.1093/brain/awz189. [DOI] [PubMed] [Google Scholar]
- Mills K.R., Murray N.M. Electrical stimulation over the human vertebral column: which neural elements are excited? Electroencephalogr. Clin. Neurophysiol. 1986;63:582–589. doi: 10.1016/0013-4694(86)90145-8. [DOI] [PubMed] [Google Scholar]
- Milosevic L., Gramer R., Kim T.H., Algarni M., Fasano A., Kalia S.K., et al. Modulation of inhibitory plasticity in basal ganglia output nuclei of patients with Parkinson's disease. Neurobiol. Dis. 2019;124:46–56. doi: 10.1016/j.nbd.2018.10.020. [DOI] [PubMed] [Google Scholar]
- Milosevic L., Kalia S.K., Hodaie M., Lozano A.M., Popovic M.R., Hutchison W.D. Physiological mechanisms of thalamic ventral intermediate nucleus stimulation for tremor suppression. Brain. 2018;141:2142–2155. doi: 10.1093/brain/awy139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miocinovic S., de Hemptinne C., Qasim S., Ostrem J.L., Starr P.A. Patterns of Cortical Synchronization in Isolated Dystonia Compared With Parkinson Disease. JAMA Neurol. 2015;72:1244–1251. doi: 10.1001/jamaneurol.2015.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki Y., Oishi M., Hara M., Takasu T. Central motor conduction time in Parkinson's disease. J Clin Neurosci. 1999;6:17–19. doi: 10.1054/jocn.1996.0106. [DOI] [PubMed] [Google Scholar]
- Moore O., Peretz C., Giladi N. Freezing of gait affects quality of life of peoples with Parkinson's disease beyond its relationships with mobility and gait. Mov. Disord. 2007;22:2192–2195. doi: 10.1002/mds.21659. [DOI] [PubMed] [Google Scholar]
- Morgante F., Espay A.J., Gunraj C., Lang A.E., Chen R. Motor cortex plasticity in Parkinson's disease and levodopa-induced dyskinesias. Brain. 2006;129:1059–1069. doi: 10.1093/brain/awl031. [DOI] [PubMed] [Google Scholar]
- Morita Y., Osaki Y., Doi Y. Transcranial magnetic stimulation for differential diagnostics in patients with parkinsonism. Acta Neurol. Scand. 2008;118:159-163. doi: 10.1111/j.1600-0404.2007.00988.x. [DOI] [PubMed] [Google Scholar]
- Moro E., Hamani C., Poon Y.Y., Al-Khairallah T., Dostrovsky J.O., Hutchison W.D., et al. Unilateral pedunculopontine stimulation improves falls in Parkinson's disease. Brain. 2010;133:215–224. doi: 10.1093/brain/awp261. [DOI] [PubMed] [Google Scholar]
- Morris M.E., Iansek R., Matyas T.A., Summers J.J. Ability to modulate walking cadence remains intact in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry. 1994;57:1532–1534. doi: 10.1136/jnnp.57.12.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer J.A., Webster D.D. Evidence for a quantitative association between EMG stretch responses and Parkinsonian rigidity. Brain Res. 1979;162:169–173. doi: 10.1016/0006-8993(79)90768-6. [DOI] [PubMed] [Google Scholar]
- Muthukrishnan N., Abbas J.J., Shill H.A., Krishnamurthi N. Cueing paradigms to improve gait and posture in Parkinson's disease: A narrative review. Sensors (Basel) 2019;19 doi: 10.3390/s19245468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuraman M., Hossen A., Heute U., Deuschl G., Raethjen J. A new diagnostic test to distinguish tremulous Parkinson's disease from advanced essential tremor. Mov. Disord. 2011;26:1548–1552. doi: 10.1002/mds.23672. [DOI] [PubMed] [Google Scholar]
- Muthuraman M., Raethjen J., Koirala N., Anwar A.R., Mideksa K.G., Elble R., et al. Cerebello-cortical network fingerprints differ between essential, Parkinson's and mimicked tremors. Brain. 2018;141:1770–1781. doi: 10.1093/brain/awy098. [DOI] [PubMed] [Google Scholar]
- Nardone R., Brigo F., Versace V., Höller Y., Tezzon F., Saltuari L., et al. Cortical afferent inhibition abnormalities reveal cholinergic dysfunction in Parkinson's disease: a reappraisal. J. Neural Transm. (Vienna) 2017;124:1417–1429. doi: 10.1007/s00702-017-1775-y. [DOI] [PubMed] [Google Scholar]
- Nardone R., Florio I., Lochner P., Tezzon F. Cholinergic cortical circuits in Parkinson's disease and in progressive supranuclear palsy: a transcranial magnetic stimulation study. Exp. Brain Res. 2005;163:128–131. doi: 10.1007/s00221-005-2228-7. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Degen K., Schneider G.H., Brücke C., Huebl J., Brown P., et al. Subthalamic synchronized oscillatory activity correlates with motor impairment in patients with Parkinson's disease. Mov. Disord. 2016;31:1748–1751. doi: 10.1002/mds.26759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann W.J., Horn A., Ewert S., Huebl J., Brücke C., Slentz C., et al. A localized pallidal physiomarker in cervical dystonia. Ann. Neurol. 2017;82:912–924. doi: 10.1002/ana.25095. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Huebl J., Brücke C., Lofredi R., Horn A., Saryyeva A., et al. Pallidal and thalamic neural oscillatory patterns in tourette's syndrome. Ann. Neurol. 2018;84:505–514. doi: 10.1002/ana.25311. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Huebl J., Brücke C., Ruiz M.H., Kupsch A., Schneider G.H., et al. Enhanced low-frequency oscillatory activity of the subthalamic nucleus in a patient with dystonia. Mov. Disord. 2012;27:1063–1066. doi: 10.1002/mds.25078. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Kühn A.A. Subthalamic beta power-Unified Parkinson's disease rating scale III correlations require akinetic symptoms. Mov. Disord. 2017;32:175–176. doi: 10.1002/mds.26858. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Memarian Sorkhabi M., Benjaber M., Feldmann L.K., Saryyeva A., Krauss J.K., et al. The sensitivity of ECG contamination to surgical implantation site in brain computer interfaces. Brain Stimul. 2021;14:1301–1306. doi: 10.1016/j.brs.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann W.J., Rodriguez-Oroz M.C. Machine Learning Will Extend the Clinical Utility of Adaptive Deep Brain Stimulation. Mov. Disord. 2021;36:796–799. doi: 10.1002/mds.28567. [DOI] [PubMed] [Google Scholar]
- Neumann W.J., Staub-Bartelt F., Horn A., Schanda J., Schneider G.H., Brown P., et al. Long term correlation of subthalamic beta band activity with motor impairment in patients with Parkinson's disease. Clin. Neurophysiol. 2017;128:2286–2291. doi: 10.1016/j.clinph.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann W.J., Turner R.S., Blankertz B., Mitchell T., Kühn A.A., Richardson R.M. Toward electrophysiology-based intelligent adaptive deep brain stimulation for movement disorders. Neurotherapeutics. 2019;16:105–118. doi: 10.1007/s13311-018-00705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z., Bahl N., Gunraj C., Mazzella F., Chen R. Increased motor cortical facilitation and decreased inhibition in Parkinson's Disease. Neurology. 2013;80:1746–1753. doi: 10.1212/WNL.0b013e3182919029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z., Gunraj C., Nelson A.J., Yeh I.J., Castillo G., Hoque T., et al. Two phases of interhemispheric inhibition between motor related cortical areas and the primary motor cortex in human. Cereb. Cortex. 2009;19:1654–1665. doi: 10.1093/cercor/bhn201. [DOI] [PubMed] [Google Scholar]
- Ni Z., Muller-Dahlhaus F., Chen R., Ziemann U. Triple-pulse TMS to study interactions between neural circuits in human cortex. Brain Stimul. 2011;4:281–293. doi: 10.1016/j.brs.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Ni Z., Pinto A.D., Lang A.E., Chen R. Involvement of the cerebellothalamocortical pathway in Parkinson disease. Ann. Neurol. 2010;68:816–824. doi: 10.1002/ana.22221. [DOI] [PubMed] [Google Scholar]
- Nieuwboer A., Giladi N. Characterizing freezing of gait in Parkinson's disease: models of an episodic phenomenon. Mov. Disord. 2013;28:1509–1519. doi: 10.1002/mds.25683. [DOI] [PubMed] [Google Scholar]
- Nonnekes J., de Kam D., Geurts A.C., Weerdesteyn V., Bloem B.R. Unraveling the mechanisms underlying postural instability in Parkinson's disease using dynamic posturography. Expert Rev. Neurother. 2013;13:1303–1308. doi: 10.1586/14737175.2013.839231. [DOI] [PubMed] [Google Scholar]
- Noth J., Schürmann M., Podoll K., Schwarz M. Reconsideration of the concept of enhanced static fusimotor drive in rigidity in patients with Parkinson's disease. Neurosci. Lett. 1988;84:239–243. doi: 10.1016/0304-3940(88)90415-6. [DOI] [PubMed] [Google Scholar]
- Notturno F., Zappasodi F., Maruotti V., Marzetti L., Caulo M., Uncini A. Cortical origin of myoclonus in early stages of corticobasal degeneration. Mov. Disord. 2011;26:1567–1569. doi: 10.1002/mds.23612. [DOI] [PubMed] [Google Scholar]
- Okuma Y., Fujishima K., Miwa H., Mori H., Mizuno Y. Myoclonic tremulous movements in multiple system atrophy are a form of cortical myoclonus. Mov. Disord. 2005;20:451–456. doi: 10.1002/mds.20346. [DOI] [PubMed] [Google Scholar]
- Oswal A., Beudel M., Zrinzo L., Limousin P., Hariz M., Foltynie T., et al. Deep brain stimulation modulates synchrony within spatially and spectrally distinct resting state networks in Parkinson's disease. Brain. 2016;139:1482–1496. doi: 10.1093/brain/aww048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkurt T.E., Akram H., Zrinzo L., Limousin P., Foltynie T., Oswal A., et al. Identification of nonlinear features in cortical and subcortical signals of Parkinson's Disease patients via a novel efficient measure. Neuroimage. 2020;223 doi: 10.1016/j.neuroimage.2020.117356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkurt T.E., Butz M., Homburger M., Elben S., Vesper J., Wojtecki L., et al. High frequency oscillations in the subthalamic nucleus: a neurophysiological marker of the motor state in Parkinson's disease. Exp. Neurol. 2011;229:324–331. doi: 10.1016/j.expneurol.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Pal P.K., Gunraj C.A., Li J.Y., Lang A.E., Chen R. Reduced intracortical and interhemispheric inhibitions in corticobasal syndrome. J. Clin. Neurophysiol. 2008;25:304–312. doi: 10.1097/WNP.0b013e318182d304. [DOI] [PubMed] [Google Scholar]
- Palmer E., Ashby P. Evidence that a long latency stretch reflex in humans is transcortical. J. Physiol. 1992;449:429–440. doi: 10.1113/jphysiol.1992.sp019094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov F., Levin E., de Hemptinne C., Swann N.C., Qasim S., Miocinovic S., et al. Intraoperative electrocorticography for physiological research in movement disorders: principles and experience in 200 cases. J. Neurosurg. 2017;126:122–131. doi: 10.3171/2015.11.JNS151341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paparella G., Fasano A., Hallett M., Berardelli A., Bologna M. Emerging concepts on bradykinesia in non-parkinsonian conditions. Eur. J. Neurol. 2021;28:2403–2422. doi: 10.1111/ene.14851. [DOI] [PubMed] [Google Scholar]
- Papengut F., Raethjen J., Binder A., Deuschl G. Rest tremor suppression may separate essential from parkinsonian rest tremor. Parkinsonism Relat. Disord. 2013;19:693–697. doi: 10.1016/j.parkreldis.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Pasquereau B., DeLong M.R., Turner R.S. Primary motor cortex of the parkinsonian monkey: altered encoding of active movement. Brain. 2016;139:127–143. doi: 10.1093/brain/awv312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B., Turner R.S. Primary motor cortex of the parkinsonian monkey: differential effects on the spontaneous activity of pyramidal tract-type neurons. Cereb. Cortex. 2011;21:1362–1378. doi: 10.1093/cercor/bhq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B., Turner R.S. Primary motor cortex of the parkinsonian monkey: altered neuronal responses to muscle stretch. Front. Syst. Neurosci. 2013;7:98. doi: 10.3389/fnsys.2013.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa D.J., Reck C., Florin E., Pauls K.A., Maarouf M., Wojtecki L., et al. Essential tremor and tremor in Parkinson's disease are associated with distinct 'tremor clusters' in the ventral thalamus. Exp. Neurol. 2012;237:435–443. doi: 10.1016/j.expneurol.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Perez-Lloret S., Negre-Pages L., Damier P., Delval A., Derkinderen P., Destee A., et al. Prevalence, determinants, and effect on quality of life of freezing of gait in Parkinson disease. JAMA Neurol. 2014;71:884–890. doi: 10.1001/jamaneurol.2014.753. [DOI] [PubMed] [Google Scholar]
- Perretti A., De Rosa A., Marcantonio L., Iodice V., Estraneo A., Manganelli F., et al. Neurophysiological evaluation of motor corticospinal pathways by TMS in idiopathic early-onset Parkinson's disease. Clin. Neurophysiol. 2011;122:546–549. doi: 10.1016/j.clinph.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Petersen N., Christensen L.O., Morita H., Sinkjaer T., Nielsen J. Evidence that a transcortical pathway contributes to stretch reflexes in the tibialis anterior muscle in man. J. Physiol. 1998;512:267–276. doi: 10.1111/j.1469-7793.1998.267bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean M., Ko J.Y. An age-related change in the ipsilateral silent period of a small hand muscle. Clin. Neurophysiol. 2013;124:346–353. doi: 10.1016/j.clinph.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Pfeiffer R.F. Amantadine-induced “vocal” myoclonus. Mov. Disord. 1996;11:104–106. doi: 10.1002/mds.870110123. [DOI] [PubMed] [Google Scholar]
- Pierantozzi M., Mazzone P., Bassi A., Rossini P.M., Peppe A., Altibrandi M.G., et al. The effect of deep brain stimulation on the frontal N30 component of somatosensory evoked potentials in advanced Parkinson's disease patients. Clin. Neurophysiol. 1999;110:1700–1707. doi: 10.1016/s1388-2457(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Pierantozzi M., Palmieri M.G., Marciani M.G., Bernardi G., Giacomini P., Stanzione P. Effect of apomorphine on cortical inhibition in Parkinson's disease patients: a transcranial magnetic stimulation study. Exp. Brain Res. 2001;141:52–62. doi: 10.1007/s002210100839. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny E., Burke D. Cambridge University Press; 2012. The Circuitry Of The Human Spinal Cord: Spinal And Corticospinal Mechanisms Of Movement. [Google Scholar]
- Piña-Fuentes D., Little S., Oterdoom M., Neal S., Pogosyan A., Tijssen M.A.J., et al. Adaptive DBS in a Parkinson's patient with chronically implanted DBS: A proof of principle. Mov. Disord. 2017;32:1253–1254. doi: 10.1002/mds.26959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto A.D., Chen R. Suppression of the motor cortex by magnetic stimulation of the cerebellum. Exp. Brain Res. 2001;140:505–510. doi: 10.1007/s002210100862. [DOI] [PubMed] [Google Scholar]
- Plotnik M., Giladi N., Balash Y., Peretz C., Hausdorff J.M. Is freezing of gait in Parkinson's disease related to asymmetric motor function? Ann. Neurol. 2005;57:656–663. doi: 10.1002/ana.20452. [DOI] [PubMed] [Google Scholar]
- Pollock L.J., Davis L. Muscle tone in Parkinsonian states. Arch. Neurol. Psychiatry. 1930;23:303–319. [Google Scholar]
- Pollok B., Krause V., Martsch W., Wach C., Schnitzler A., Sudmeyer M. Motor-cortical oscillations in early stages of Parkinson's disease. J. Physiol. 2012;590:3203–3212. doi: 10.1113/jphysiol.2012.231316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma R.B., Berg D., Stern M., Poewe W., Olanow C.W., Oertel W., et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov. Disord. 2015;30:1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- Postuma R.B., Iranzo A., Hu M., Högl B., Boeve B.F., Manni R., et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019;142:744–759. doi: 10.1093/brain/awz030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D., Threlkeld A.J., Fang X., Muthumani A., Xia R. Amplitude- and velocity-dependency of rigidity measured at the wrist in Parkinson's disease. Clin. Neurophysiol. 2012;123:764–773. doi: 10.1016/j.clinph.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi N.G., Canessa A., Palmisano C., Brumberg J., Steigerwald F., Reich M.M., et al. Freezing of gait in Parkinson's disease reflects a sudden derangement of locomotor network dynamics. Brain. 2019;142:2037–2050. doi: 10.1093/brain/awz141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott I.A., Dostrovsky J.O., Moro E., Hodaie M., Lozano A.M., Hutchison W.D. Levodopa enhances synaptic plasticity in the substantia nigra pars reticulata of Parkinson's disease patients. Brain. 2009:309–318. doi: 10.1093/brain/awn322. [DOI] [PubMed] [Google Scholar]
- Priori A., Berardelli A., Inghilleri M., Accornero N., Manfredi M. Motor cortical inhibition and the dopaminergic system. Pharmacological changes in the silent period after transcranial magnetic brain stimulation in normal subjects, patients with Parkinson's disease and drug-induced parkinsonism. Brain. 1994;117:317–323. doi: 10.1093/brain/117.2.317. [DOI] [PubMed] [Google Scholar]
- Pullman S., Ford B., Elibol B., Uncini A., Su P., Fahn S. Cutaneous electromyographic silent period findings in brachial dystonia. Neurology. 1996;46:503–505. doi: 10.1212/wnl.46.2.503. [DOI] [PubMed] [Google Scholar]
- Raethjen J., Pohle S., Govindan R.B., Morsnowski A., Wenzelburger R., Deuschl G. Parkinsonian action tremor: interference with object manipulation and lacking levodopa response. Exp. Neurol. 2005;194:151–160. doi: 10.1016/j.expneurol.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Rektor I., Kuba R., Brázdil M. Interictal and ictal EEG activity in the basal ganglia: an SEEG study in patients with temporal lobe epilepsy. Epilepsia. 2002;43:253–262. doi: 10.1046/j.1528-1157.2002.28001.x. [DOI] [PubMed] [Google Scholar]
- Ridding M.C., Inzelberg R., Rothwell J.C. Changes in excitability of motor cortical circuitry in patients with Parkinson's disease. Ann. Neurol. 1995;37:181–188. doi: 10.1002/ana.410370208. [DOI] [PubMed] [Google Scholar]
- Rinne J.O., Lee M.S., Thompson P.D., Marsden C.D. Corticobasal degeneration. A clinical study of 36 cases. Brain. 1994;117:1183–1196. doi: 10.1093/brain/117.5.1183. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.E., Artieda J., Zubieta J.L., Obeso J.A. Reflex myoclonus in olivopontocerebellar atrophy. J. Neurol. Neurosurg. Psychiatry. 1994;57:316–319. doi: 10.1136/jnnp.57.3.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M.W., Mille M.L. Balance perturbations. Handb Clin Neurol. 2018;159:85–105. doi: 10.1016/B978-0-444-63916-5.00005-7. [DOI] [PubMed] [Google Scholar]
- Rossini P.M., Babiloni F., Bernardi G., Cecchi L., Johnson P.B., Malentacca A., et al. Abnormalities of short-latency somatosensory evoked potentials in parkinsonian patients. Electroencephalogr. Clin. Neurophysiol. 1989;74:277–289. doi: 10.1016/0168-5597(89)90058-0. [DOI] [PubMed] [Google Scholar]
- Rossini P.M., Paradiso C., Zarola F., Bernardi G., Caramia M.D., Margari L., et al. Brain excitability and long latency muscular arm responses: non-invasive evaluation in healthy and parkinsonian subjects. Electroencephalogr. Clin. Neurophysiol. 1991;81:454–465. doi: 10.1016/0013-4694(91)90008-r. [DOI] [PubMed] [Google Scholar]
- Rossini P.M., Traversa R., Boccasena P., Martino G., Passarelli F., Pacifici L., et al. Parkinson's disease and somatosensory evoked potentials: apomorphine- induced transient potentiation of frontal components. Neurology. 1993;43:2495–2500. doi: 10.1212/wnl.43.12.2495. [DOI] [PubMed] [Google Scholar]
- Rothwell J., Antal A., Burke D., Carlsen A., Georgiev D., Jahanshahi M., et al. Central nervous system physiology. Clin. Neurophysiol. 2021;132:3043–3083. doi: 10.1016/j.clinph.2021.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell J.C., Obeso J.A., Traub M.M., Marsden C.D. The behaviour of the long-latency stretch reflex in patients with Parkinson's disease. J. Neurol. Neurosurg. Psychiatry. 1983;46:35–44. doi: 10.1136/jnnp.46.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushworth G. Spasticity and rigidity: an experimental study and review. J. Neurol. Neurosurg. Psychiatry. 1960;23:99–118. doi: 10.1136/jnnp.23.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka E., Zarubova K., Nutt J.G., Bloem B.R. “Silly walks” in Parkinson's disease: unusual presentation of dopaminergic-induced dyskinesias. Mov. Disord. 2011;26:1782–1784. doi: 10.1002/mds.23667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer A., Molnar G.F., Paradiso G., Gunraj C.A., Lang A.E., Chen R. Short and long latency afferent inhibition in Parkinson's disease. Brain. 2003;126:1883–1894. doi: 10.1093/brain/awg183. [DOI] [PubMed] [Google Scholar]
- Salazar G., Valls-Solé J., Martí M.J., Chang H., Tolosa E.S. Postural and action myoclonus in patients with parkinsonian type multiple system atrophy. Mov. Disord. 2000;15:77–83. doi: 10.1002/1531-8257(200001)15:1<77::aid-mds1013>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Sanger T.D., Garg R.R., Chen R. Interactions between two different inhibitory systems in the human motor cortex. J. Physiol. 2001;530:307–317. doi: 10.1111/j.1469-7793.2001.0307l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanamuttu J., Radhu N., Udupa K., Baarbé J., Gunraj C., Chen R. Impaired motor cortical facilitatory-inhibitory circuit interaction in Parkinson's disease. Clin. Neurophysiol. 2021;132:2685–2692. doi: 10.1016/j.clinph.2021.05.032. [DOI] [PubMed] [Google Scholar]
- Schieppati M., Nardone A. Group II spindle afferent fibers in humans: their possible role in the reflex control of stance. Prog. Brain Res. 1999;123:461–472. doi: 10.1016/s0079-6123(08)62882-4. [DOI] [PubMed] [Google Scholar]
- Schilder J.C., Overmars S.S., Marinus J., van Hilten J.J., Koehler P.J. The terminology of akinesia, bradykinesia and hypokinesia: Past, present and future. Parkinsonism Relat Disord. 2017;37:27–35. doi: 10.1016/j.parkreldis.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Schlaier J.R., Habermeyer C., Janzen A., Fellner C., Hochreiter A., Proescholdt M., et al. The influence of intraoperative microelectrode recordings and clinical testing on the location of final stimulation sites in deep brain stimulation for Parkinson's disease. Acta Neurochir (Wien) 2013;155:357–366. doi: 10.1007/s00701-012-1592-x. [DOI] [PubMed] [Google Scholar]
- Schneider S.A., Talelli P., Cheeran B.J., Khan N.L., Wood N.W., Rothwell J.C., et al. Motor cortical physiology in patients and asymptomatic carriers of parkin gene mutations. Mov. Disord. 2008;23:1812–1819. doi: 10.1002/mds.22025. [DOI] [PubMed] [Google Scholar]
- Scholz E., Diener H.C., Noth J., Friedemann H., Dichgans J., Bacher M. Medium and long latency EMG responses in leg muscles: Parkinson's disease. J. Neurol. Neurosurg. Psychiatry. 1987;50:66–70. doi: 10.1136/jnnp.50.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuepbach W.M., Rau J., Knudsen K., Volkmann J., Krack P., Timmermann L., et al. Neurostimulation for Parkinson's disease with early motor complications. N. Engl. J. Med. 2013;368:610–622. doi: 10.1056/NEJMoa1205158. [DOI] [PubMed] [Google Scholar]
- Schwingenschuh P., Ruge D., Edwards M.J., Terranova C., Katschnig P., Carrillo F., et al. Distinguishing SWEDDs patients with asymmetric resting tremor from Parkinson's disease: a clinical and electrophysiological study. Mov. Disord. 2010;25:560–569. doi: 10.1002/mds.23019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifried C., Weise L., Hartmann R., Gasser T., Baudrexel S., Szelényi A., et al. Intraoperative microelectrode recording for the delineation of subthalamic nucleus topography in Parkinson's disease. Brain Stimul. 2012;5:378–387. doi: 10.1016/j.brs.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Serrao M., Parisi L., Valente G., Martini A., Fattapposta F., Pierelli F., et al. l-Dopa decreases cutaneous nociceptive inhibition of motor activity in Parkinson's disease. Acta Neurol. Scand. 2002;105:196–201. doi: 10.1034/j.1600-0404.2002.1o085.x. [DOI] [PubMed] [Google Scholar]
- Shafiq M., Lang A.E. Myoclonus in parkinsonian disorders. Adv. Neurol. 2002;89:77–83. [PubMed] [Google Scholar]
- Shibasaki H., Hallett M. Electrophysiological studies of myoclonus. Muscle Nerve. 2005;31:157–174. doi: 10.1002/mus.20234. [DOI] [PubMed] [Google Scholar]
- Shine J.M., Matar E., Ward P.B., Frank M.J., Moustafa A.A., Pearson M., et al. Freezing of gait in Parkinson's disease is associated with functional decoupling between the cognitive control network and the basal ganglia. Brain. 2013;136:3671–3681. doi: 10.1093/brain/awt272. [DOI] [PubMed] [Google Scholar]
- Shirota Y., Hamada M., Hanajima R., Terao Y., Matsumoto H., Ohminami S., et al. Cerebellar dysfunction in progressive supranuclear palsy: a transcranial magnetic stimulation study. Mov. Disord. 2010;25:2413–2419. doi: 10.1002/mds.23298. [DOI] [PubMed] [Google Scholar]
- Shirota Y., Ohminami S., Tsutsumi R., Terao Y., Ugawa Y., Tsuji S., et al. Increased facilitation of the primary motor cortex in de novo Parkinson's disease. Parkinsonism Relat Disord. 2019;66:125–129. doi: 10.1016/j.parkreldis.2019.07.022. [DOI] [PubMed] [Google Scholar]
- Sifoglu A., Gunduz A., Kiziltan G., Kiziltan M.E. Dopaminergic medication unrelated myoclonus is less related to tremor in idiopathic Parkinson's disease. Neurol Sci. 2017;38:679–682. doi: 10.1007/s10072-016-2793-5. [DOI] [PubMed] [Google Scholar]
- Silberstein P., Kuhn A.A., Kupsch A., Trottenberg T., Krauss J.K., Wohrle J.C., et al. Patterning of globus pallidus local field potentials differs between Parkinson's disease and dystonia. Brain. 2003;126:2597–2608. doi: 10.1093/brain/awg267. [DOI] [PubMed] [Google Scholar]
- Silberstein P., Oliviero A., Di Lazzaro V., Insola A., Mazzone P., Brown P. Oscillatory pallidal local field potential activity inversely correlates with limb dyskinesias in Parkinson's disease. Exp. Neurol. 2005;194:523–529. doi: 10.1016/j.expneurol.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Simonetta Moreau M., Meunier S., Vidailhet M., Pol S., Galitzky M., Rascol O. Transmission of group II heteronymous pathways is enhanced in rigid lower limb of de novo patients with Parkinson's disease. Brain. 2002;125:2125–2133. doi: 10.1093/brain/awf201. [DOI] [PubMed] [Google Scholar]
- Sisterson N.D., Carlson A.A., Rutishauser U., Mamelak A.N., Flagg M., Pouratian N., et al. Electrocorticography During Deep Brain Stimulation Surgery: Safety Experience From 4 Centers Within the National Institute of Neurological Disorders and Stroke Research Opportunities in Human Consortium. Neurosurgery. 2021;88:E420–E426. doi: 10.1093/neuros/nyaa592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda J., Fujioka S., Wszolek Z.K. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord. 2014;20:957–964. doi: 10.1016/j.parkreldis.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soteropoulos D.S., Baker S.N. Long-latency Responses to a Mechanical Perturbation of the Index Finger Have a Spinal Component. J. Neurosci. 2020;40:3933–3948. doi: 10.1523/JNEUROSCI.1901-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C., Estrada L.D. Protein misfolding and neurodegeneration. Arch. Neurol. 2008;65:184–189. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- Soysal A., Sobe I., Atay T., Sen A., Arpaci B. Effect of therapy on motor cortical excitability in Parkinson's disease. Can. J. Neurol. Sci. 2008;35:166–172. doi: 10.1017/s0317167100008581. [DOI] [PubMed] [Google Scholar]
- Spagnolo F., Coppi E., Chieffo R., Straffi L., Fichera M., Nuara A., et al. Interhemispheric balance in Parkinson's disease: a transcranial magnetic stimulation study. Brain Stimul. 2013;6:892–897. doi: 10.1016/j.brs.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Spitzer B., Haegens S. Beyond the status quo: A role for beta oscillations in endogenous content (Re)activation. eNeuro. 2017:4. doi: 10.1523/ENEURO.0170-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan K., Kunesch E., Cohen L.G., Benecke R., Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123:572–584. doi: 10.1093/brain/123.3.572. [DOI] [PubMed] [Google Scholar]
- Steiner L.A., Neumann W.J., Staub-Bartelt F., Herz D.M., Tan H., Pogosyan A., et al. Subthalamic beta dynamics mirror Parkinsonian bradykinesia months after neurostimulator implantation. Mov. Disord. 2017;32:1183–1190. doi: 10.1002/mds.27068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetkarova I., Kofler M., Majerova V. Cutaneous silent periods in multiple system atrophy. Biomed Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2015;159:327–332. doi: 10.5507/bp.2013.081. [DOI] [PubMed] [Google Scholar]
- Strafella A.P., Valzania F., Nassetti S.A., Tropeani A., Bisulli A., Santangelo M., et al. Effects of chronic levodopa and pergolide treatment on cortical excitability in patients with Parkinson's disease: a transcranial magnetic stimulation study. Clin. Neurophysiol. 2000;111:1198–1202. doi: 10.1016/s1388-2457(00)00316-3. [DOI] [PubMed] [Google Scholar]
- Su D., Yang S., Hu W., Wang D., Kou W., Liu Z., et al. The characteristics of tremor motion help identify Parkinson's disease and multiple system atrophy. Front. Neurol. 2020;11:540. doi: 10.3389/fneur.2020.00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppa A., Asci F., Guerra A. Transcranial magnetic stimulation as a tool to induce and explore plasticity in humans. Handb Clin Neurol. 2022;184:73–89. doi: 10.1016/B978-0-12-819410-2.00005-9. [DOI] [PubMed] [Google Scholar]
- Suppa A., Asci F., Saggio G., Di Leo P., Zarezadeh Z., Ferrazzano G., et al. Voice analysis with machine learning: one step closer to an objective diagnosis of essential tremor. Mov. Disord. 2021;36:1401–1410. doi: 10.1002/mds.28508. [DOI] [PubMed] [Google Scholar]
- Suppa A., Asci F., Saggio G., Marsili L., Casali D., Zarezadeh Z., et al. Voice analysis in adductor spasmodic dysphonia: Objective diagnosis and response to botulinum toxin. Parkinsonism Relat. Disord. 2020;73:23–30. doi: 10.1016/j.parkreldis.2020.03.012. [DOI] [PubMed] [Google Scholar]
- Suppa A., Bologna M., Conte A., Berardelli A., Fabbrini G. The effect of L-dopa in Parkinson's disease as revealed by neurophysiological studies of motor and sensory functions. Expert Rev. Neurother. 2017;17:181–192. doi: 10.1080/14737175.2016.1219251. [DOI] [PubMed] [Google Scholar]
- Suppa A., Costantini G., Asci F., Di Leo P., Al-Wardat M.S., Di Lazzaro G., et al. Voice in Parkinson's disease: a machine learning study. Front. Neurol. 2022;13 doi: 10.3389/fneur.2022.831428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppa A., Di Stasio F., Marsili L., Upadhyay N., Belvisi D., Conte A., et al. Primary motor cortex LTP/LTD-like plasticity in probable corticobasal syndrome. J. Neurophysiol. 2016;115:717–727. doi: 10.1152/jn.00755.2015. [DOI] [PubMed] [Google Scholar]
- Suppa A., Huang Y.Z., Funke K., Ridding M.C., Cheeran B., Di Lazzaro V., et al. Ten years of theta burst stimulation in humans: established knowledge, unknowns and prospects. Brain Stimul. 2016;9:323–335. doi: 10.1016/j.brs.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Suppa A., Kita A., Leodori G., Zampogna A., Nicolini E., Lorenzi P., et al. l-DOPA and freezing of gait in Parkinson's Disease: objective assessment through a wearable wireless system. Front. Neurol. 2017;8:406. doi: 10.3389/fneur.2017.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppa A., Marsili L., Belvisi D., Conte A., Iezzi E., Modugno N., et al. Lack of LTP-like plasticity in primary motor cortex in Parkinson's disease. Exp. Neurol. 2011;227:296–301. doi: 10.1016/j.expneurol.2010.11.020. [DOI] [PubMed] [Google Scholar]
- Suppa A., Marsili L., Di Stasio F., Latorre A., Parvez A.K., Colosimo C., et al. Primary motor cortex long-term plasticity in multiple system atrophy. Mov. Disord. 2014;29:97–104. doi: 10.1002/mds.25668. [DOI] [PubMed] [Google Scholar]
- Suppa A., Quartarone A., Siebner H., Chen R., Di Lazzaro V., Del Giudice P., et al. The associative brain at work: Evidence from paired associative stimulation studies in humans. Clin. Neurophysiol. 2017;128:2140–2164. doi: 10.1016/j.clinph.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Suyama N., Kobayashi S., Isino H., Iijima M., Imaoka K. Progressive supranuclear palsy with palatal myoclonus. Acta Neuropathol. 1997;94:290–293. doi: 10.1007/s004010050706. [DOI] [PubMed] [Google Scholar]
- Swann N.C., de Hemptinne C., Aron A.R., Ostrem J.L., Knight R.T., Starr P.A. Elevated synchrony in Parkinson disease detected with electroencephalography. Ann. Neurol. 2015;78:742–750. doi: 10.1002/ana.24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrkin-Nikolau J., Koop M.M., Prieto T., Anidi C., Afzal M.F., Velisar A., et al. Subthalamic neural entropy is a feature of freezing of gait in freely moving people with Parkinson's disease. Neurobiol. Dis. 2017;108:288–297. doi: 10.1016/j.nbd.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tataroglu C., Sair A., Parlaz A., Deneri E. Effects of 1-Hz repetitive transcranial magnetic stimulation on long-latency reflexes and cortical relay time. J. Clin. Neurophysiol. 2011;28:319–322. doi: 10.1097/WNP.0b013e31821c2ff0. [DOI] [PubMed] [Google Scholar]
- Thilmann A.F., Schwarz M., Töpper R., Fellows S.J., Noth J. Different mechanisms underlie the long-latency stretch reflex response of active human muscle at different joints. J. Physiol. 1991;444:631–643. doi: 10.1113/jphysiol.1991.sp018898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A., Bonanni L., Antonini A., Barone P., Onofrj M. Dopa-responsive pseudo-orthostatic tremor in parkinsonism. Mov. Disord. 2007;22:1652–1656. doi: 10.1002/mds.21621. [DOI] [PubMed] [Google Scholar]
- Thompson P.D., Day B.L., Rothwell J.C., Brown P., Britton T.C., Marsden C.D. The myoclonus in corticobasal degeneration. Evidence for two forms of cortical reflex myoclonus. Brain. 1994;117:1197–1207. doi: 10.1093/brain/117.5.1197. [DOI] [PubMed] [Google Scholar]
- Timmermann L., Florin E. Parkinson's disease and pathological oscillatory activity: is the beta band the bad guy? - New lessons learned from low-frequency deep brain stimulation. Exp. Neurol. 2012;233:123–125. doi: 10.1016/j.expneurol.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Timmermann L., Gross J., Dirks M., Volkmann J., Freund H.J., Schnitzler A. The cerebral oscillatory network of parkinsonian resting tremor. Brain. 2003;126:199–212. doi: 10.1093/brain/awg022. [DOI] [PubMed] [Google Scholar]
- Timmermann L., Wojtecki L., Gross J., Lehrke R., Voges J., Maarouf M., et al. Ten-Hertz stimulation of subthalamic nucleus deteriorates motor symptoms in Parkinson's disease. Mov. Disord. 2004;19:1328–1333. doi: 10.1002/mds.20198. [DOI] [PubMed] [Google Scholar]
- Tinkhauser G., Pogosyan A., Little S., Beudel M., Herz D.M., Tan H., et al. The modulatory effect of adaptive deep brain stimulation on beta bursts in Parkinson's disease. Brain. 2017;140:1053–1067. doi: 10.1093/brain/awx010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris G.K., Spillantini M.G. Physiological and pathological properties of alpha-synuclein. Cell. Mol. Life Sci. 2007;64:2194–2201. doi: 10.1007/s00018-007-7217-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F., Tremblay L.E. Cortico-motor excitability of the lower limb motor representation: a comparative study in Parkinson's disease and healthy controls. Clin. Neurophysiol. 2002;113:2006–2012. doi: 10.1016/s1388-2457(02)00301-2. [DOI] [PubMed] [Google Scholar]
- Truong D.D., Bhidayasiri R. Myoclonus and parkinsonism. Handb Clin Neurol. 2007;84:549–560. doi: 10.1016/S0072-9752(07)84061-9. [DOI] [PubMed] [Google Scholar]
- Tsang E.W., Hamani C., Mazzella F., Saha U., Lozano A.M., Hodaie M., et al. Subthalamic deep brain stimulation at individualized frequencies for Parkinson disease. Neurology. 2012;78:1930–1938. doi: 10.1212/WNL.0b013e318259e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang E.W., Hamani C., Moro E., Mazzella F., Lozano A.M., Hodaie M., et al. Movement related potentials and oscillatory activities in the human internal globus pallidus during voluntary movements. J. Neurol. Neurosurg. Psychiatry. 2012;83:91–97. doi: 10.1136/jnnp.2011.243857. [DOI] [PubMed] [Google Scholar]
- Tsuji T., Rothwell J.C. Long lasting effects of rTMS and associated peripheral sensory input on MEPs, SEPs and transcortical reflex excitability in humans. J. Physiol. 2002;540:367–376. doi: 10.1113/jphysiol.2001.013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y., Mima T., Kotb M.A., Sawada H., Saiki H., Ikeda A., et al. Altered plasticity of the human motor cortex in Parkinson's disease. Ann. Neurol. 2006;59:60–71. doi: 10.1002/ana.20692. [DOI] [PubMed] [Google Scholar]
- Ugawa Y., Uesaka Y., Terao Y., Hanajima R., Kanazawa I. Magnetic stimulation over the cerebellum in humans. Ann. Neurol. 1995;37:703–713. doi: 10.1002/ana.410370603. [DOI] [PubMed] [Google Scholar]
- Ulivelli M., Rossi S., Pasqualetti P., Rossini P.M., Ghiglieri O., Passero S., et al. Time course of frontal somatosensory evoked potentials. Relation to L- dopa plasma levels and motor performance in PD. Neurology. 1999;53:1451–1457. doi: 10.1212/wnl.53.7.1451. [DOI] [PubMed] [Google Scholar]
- Urrestarazu E., Iriarte J., Alegre M., Clavero P., Rodríguez-Oroz M.C., Guridi J., et al. Beta activity in the subthalamic nucleus during sleep in patients with Parkinson's disease. Mov. Disord. 2009;24:254–260. doi: 10.1002/mds.22351. [DOI] [PubMed] [Google Scholar]
- Uversky V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- Valls-Sole J., Pascual-Leone A., Brasil-Neto J.P., Cammarota A., McShane L., Hallett M. Abnormal facilitation of the response to transcranial magnetic stimulation in patients with Parkinson's disease. Neurology. 1994;44:735–741. doi: 10.1212/wnl.44.4.735. [DOI] [PubMed] [Google Scholar]
- van der Heide A., Speckens A.E.M., Meinders M.J., Rosenthal L.S., Bloem B.R., Helmich R.C. Stress and mindfulness in Parkinson's disease - a survey in 5000 patients. NPJ Parkinsons Dis. 2021;7:7. doi: 10.1038/s41531-020-00152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk B.C.M., Pogosyan A., Hariz M.I., Akram H., Foltynie T., Limousin P., et al. Localization of beta and high-frequency oscillations within the subthalamic nucleus region. Neuroimage Clin. 2017;16:175–183. doi: 10.1016/j.nicl.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi J., Glaubman H., Rabey J.M., Streifler M. Myoclonic attacks induced by L-dopa and bromocryptin in Parkinson patients: a sleep EEG study. J. Neurol. 1978;218:35–42. doi: 10.1007/BF00314716. [DOI] [PubMed] [Google Scholar]
- Velisar A., Syrkin-Nikolau J., Blumenfeld Z., Trager M.H., Afzal M.F., Prabhakar V., et al. Dual threshold neural closed loop deep brain stimulation in Parkinson disease patients. Brain Stimul. 2019;12:868–876. doi: 10.1016/j.brs.2019.02.020. [DOI] [PubMed] [Google Scholar]
- Vercruysse S., Spildooren J., Heremans E., Vandenbossche J., Levin O., Wenderoth N., et al. Freezing in Parkinson's disease: a spatiotemporal motor disorder beyond gait. Mov. Disord. 2012;27:254–263. doi: 10.1002/mds.24015. [DOI] [PubMed] [Google Scholar]
- Wenning G.K., Ben Shlomo Y., Magalhães M., Daniel S.E., Quinn N.P. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835–845. doi: 10.1093/brain/117.4.835. [DOI] [PubMed] [Google Scholar]
- Wittstock M., Pohley I., Walter U., Grossmann A., Benecke R., Wolters A. Interhemispheric inhibition in different phenotypes of progressive supranuclear palsy. J Neural Transm (Vienna) 2013;120:453–461. doi: 10.1007/s00702-012-0879-7. [DOI] [PubMed] [Google Scholar]
- Wolters A., Classen J., Kunesch E., Grossmann A., Benecke R. Measurements of transcallosally mediated cortical inhibition for differentiating parkinsonian syndromes. Mov. Disord. 2004;19:518–528. doi: 10.1002/mds.20064. [DOI] [PubMed] [Google Scholar]
- Wu T., Hallett M. The cerebellum in Parkinson's disease. Brain. 2013;136:696–709. doi: 10.1093/brain/aws360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Zhang J., Hallett M., Feng T., Hou Y., Chan P. Neural correlates underlying micrographia in Parkinson's disease. Brain. 2016;139:144–160. doi: 10.1093/brain/awv319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R., Muthumani A., Mao Z.H., Powell D.W. Quantification of neural reflex and muscular intrinsic contributions to parkinsonian rigidity. Exp. Brain Res. 2016;234:3587–3595. doi: 10.1007/s00221-016-4755-9. [DOI] [PubMed] [Google Scholar]
- Yin Z., Zhu G., Zhao B., Bai Y., Jiang Y., Neumann W.J., et al. Local field potentials in Parkinson's disease: A frequency-based review. Neurobiol. Dis. 2021;155 doi: 10.1016/j.nbd.2021.105372. [DOI] [PubMed] [Google Scholar]
- Zach H., Dirkx M.F., Roth D., Pasman J.W., Bloem B.R., Helmich R.C. Dopamine-responsive and dopamine-resistant resting tremor in Parkinson disease. Neurology. 2020;95:e1461–e1470. doi: 10.1212/WNL.0000000000010316. [DOI] [PubMed] [Google Scholar]
- Zaidel A., Spivak A., Grieb B., Bergman H., Israel Z. Subthalamic span of beta oscillations predicts deep brain stimulation efficacy for patients with Parkinson's disease. Brain. 2010;133:2007–2021. doi: 10.1093/brain/awq144. [DOI] [PubMed] [Google Scholar]
- Zamir O., Gunraj C., Ni Z., Mazzella F., Chen R. Effects of theta burst stimulation on motor cortex excitability in Parkinson's disease. Clin. Neurophysiol. 2012;123:815–821. doi: 10.1016/j.clinph.2011.07.051. [DOI] [PubMed] [Google Scholar]
- Zampogna A., Mileti I., Martelli F., Paoloni M., Del Prete Z., Palermo E., et al. Early balance impairment in Parkinson's Disease: Evidence from Robot-assisted axial rotations. Clin. Neurophysiol. 2021;132:2422–2430. doi: 10.1016/j.clinph.2021.06.023. [DOI] [PubMed] [Google Scholar]
- Zampogna A., Mileti I., Palermo E., Celletti C., Paoloni M., Manoni A., et al. Fifteen years of wireless sensors for balance assessment in neurological disorders. Sensors (Basel) 2020;20 doi: 10.3390/s20113247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann U. TMS and drugs. Clin. Neurophysiol. 2004;115:1717–1729. doi: 10.1016/j.clinph.2004.03.006. [DOI] [PubMed] [Google Scholar]