

Abstract
Motivation:
The advancement of high-throughput PPI profiling techniques results in generating a large amount of PPI data. The alignment of the PPI networks uncovers the relationship between the species that can help understand the biological systems. The comparative study reveals the conserved biological interactions of the proteins across the species. It can also help study the biological pathways and signal networks of the cells. Although several network alignment algorithms are developed to study and compare the PPI data, the development of the aligner that aligns the PPI networks with high biological similarity and coverage is still challenging.
Results:
This paper presents a novel global network alignment algorithm, BioAlign, that incorporates a significant amount of biological information. Existing studies use global sequence and/or 3D-structure similarity to align the PPI networks. In contrast, BioAlign uses the local sequence similarity, predicted secondary structure motifs, and remote homology in addition to global sequence and 3D-structure similarity. The extra sources of biological information help BioAlign to align the proteins with high biological similarity. BioAlign produces significantly better results in terms of AFS and Coverage (6-32 and 7-34 with respect to MF and BP, respectively) than the existing algorithms. BioAlign aligns a much larger number of proteins that have high biological similarities as compared to the existing aligners. BioAlign helps in studying the functionally similar protein pairs across the species.
Keywords: Network meta-analysis, computational biology, semantics
Introduction
Proteins are large biomolecules that perform important functions by interacting with other biomolecules, especially with other proteins. These interactions enable proteins to perform complex and diverse functions and to form biological pathways like metabolism. The interactions of the proteins can be represented as networks for example, signaling networks of the cells. The interactions of a set of proteins of a single species can be represented in the form of a network that is known as a PPI network. The proteins in the PPI network are represented by nodes while the interactions are represented by edges.
With the advancement of high-throughput PPI profiling techniques (such as yeast-two-hybrid, 1 filamentous fusion phage 2 ), a large amount of PPI data has been generated. This data is stored in different databases such as BioGRID, 3 HINT, 4 MINT, 5 etc. The study of the complete network of the proteins reveals more information about the biological activities of the proteins as compared to the study of the proteins in isolation. 6 The comparative study of the PPI networks helps to identify the homologous proteins and their conserved interactions across the species. This can also help in studying the healthy/disease states and in drug design.6,7
The pairwise PPI network alignment (alignment of 2 PPI networks) is the mapping of a small network over the portion of a large network. There are 2 types of alignment algorithms, (i) Local network alignment (LNA) algorithms and (ii) Global network alignment (GNA) algorithms. LNA algorithms are designed to align the communities (sub-networks) by many-many mapping between the nodes.8,9 These algorithms find the small sub-networks that are highly conserved across the species. In contrast, GNA algorithms align the nodes using one-one mapping. The goal of such aligners is to align the maximum number of nodes with high biological similarity.6-9
All the existing aligners (except SAlign 6 ) use sequence similarity and/or network topology to align the PPI networks. SAlign is the first algorithm that uses the 3D structure of the proteins along with sequence and topology. Previous studies use different types of topological measures to extract the structure of the PPI network. For example, SAlign 6 and HubAlign 10 use a Minimum-Degree-Heuristic algorithm to compute the topological scores of the nodes. ModuleAlign 11 uses Minimum-Degree-Heuristic and clustering algorithms to score the nodes. IBNAL 12 uses the similarity of the cliques (Clique-Degree-Similarity) extracted from the networks. MONACO measures the topology by iterative optimal matching of local neighborhoods around focal nodes. 13 All these algorithms combine the different scoring matrices (topology, sequence, and structure) and then use the combined score to align the PPI network using alignment algorithms. SAlign, HubAlign, and IBNAL use a Greedy algorithm for the alignment, while ModuleAlign uses the Hungarian algorithm for the alignment. Twadn 14 and SANA 15 align the nodes using a Simulated-Annealing-based optimization technique. In contrast to these studies, MAGNA++ 16 and NETAL 17 use sequence similarity and network topology, respectively. MAGNA++ uses a Genetic-Algorithm to align the nodes on the basis of sequence similarity. NETAL uses local network topological measures. The BEAMS algorithm assigns weights to the network edges based on sequence similarity and then uses a clustering-based technique for alignment. 18 The PROPER algorithm 7 is the first algorithm that does not combine the topological and biological scores. It first aligns the nodes on the basis of sequence similarity and then extends the aligned nodes using topology.
Most of the existing aligners are validated using semantic/biological similarity. The semantic similarity is a measure to compute the similarity of the proteins on the basis of their context (similarity in terms of molecular function). The semantic similarity in the PPI network domain is the average similarity between all the aligned nodes. Existing aligners do not produce alignments that have a high semantic similarity. Furthermore, some of the existing aligners (BEAMS, IBNAL, NETAL, SANA MAGNA++, and PROPER) align a fewer number of nodes (incomplete alignment). To complete the alignment, aligners align the nodes with low biological and topological scores that result in a decrease in semantic similarity. PROPER does not align low-scoring nodes and as a result, it produces alignments with comparatively high semantic similarity, but the completeness of alignment is decreased significantly. SAlign is the first algorithm that incorporates structural information in addition to sequence and topology that resultantly increasing its semantic similarity and completeness. The extra-biological information (3D structure) helps SAlign outperform all existing algorithms with a significant margin. SAlign demonstrates the impact of biological (sequence and structure) and topological information and concludes that a large contribution from topological information decreases the semantic similarity. UAlign 19 also supports this argument.
From these insights, we conclude that biological information is essential for producing high-quality alignments. To overcome the limitations of the existing studies, we develop a novel multi-stage GNA algorithm (BioAlign) that incorporates different biological sources of information. In the first stage, the close homologs (seeds) are generated on the basis of global sequence similarity, 3D structure similarity, and local sequence similarity. In the second stage, the remaining nodes are aligned using remote homology and predicted secondary structure. In the final stage, the remaining nodes are aligned using topology.
BioAlign does not compromise semantic similarity by aligning low-scoring nodes. The use of extra-biological information (local sequence similarity, remote homology-based similarity, and predicted secondary structure) and topological information help to complete the alignment with high semantic similarity and coverage. The difference between the performance of BioAlign and existing aligners is 6-32 and 7-34 in terms of semantic similarity and coverage w.r.t biological process and molecular function. Furthermore, BioAlign also aligns a larger number of nodes that have high semantic similarity as compared to existing studies.
Methods
The PPI network represents the interactions between the proteins, where nodes represent the proteins and edges represent the interactions between the proteins. is the first network that has nodes and edges. Similarly, is the second network that contains nodes and edges. The mapping of and is known as alignment. The GNA algorithms align the nodes of network with the nodes of network based on some biological and/or topological information.
BioAlign is a multi-stage approach that first generates highly similar homologs based on 3D structure similarity, global sequence similarity, and local sequence similarity. The remaining proteins are aligned using remote homology and predicted secondary structure motifs. In the final stage, BioAlign incorporates topological information (Neighborhood Expansion). The flow diagram of BioAlign is given in Supplemental Figure 1.
Stage-1
The first stage of BioAlign is based on 3D structure similarity, global sequence similarity, and local sequence similarity. The matrix of is generated that contains pairwise 3D structure similarities. Similarly, matrix and matrix contain pairwise global and local sequence similarities of the proteins, respectively. The 3D structure similarity is computed using the TMAlign tool (parameters: normalization = a (average length), transform = 1, termination = 0). 20 The global sequence similarity (in terms of bit-score) is computed using the BLAST tool (parameters: gap-open = 11, gap-extension = 1, E-value = 10, word-size = 3, matrix = blosum62). 21 The local sequence similarity is computed using the SWAlign tool (parameters: match = 2, mismatch = −1, gap-start = 1, gap-extension = 1, decay = 0, wrap = none, all the remaining parameters that include global-align, summary, user-region, and progress are set to False). To normalize the local sequence similarities, we use equation (1).
| (1) |
Where, is the local sequence similarity score of the protein pair computed by SWAlign tool. is the minimum sequence length of the protein pair. represents the percentage of identical sequence.
All the matrices are sorted on the basis of similarity scores in descending order. These sorted matrices are used to align the node pairs. BioAlign first aligns the highly similar (in terms of structure/sequence) nodes, and then aligns the comparatively low scoring nodes. All the node pairs that have 3D structure similarity , global sequence similarity (bit-score), and local sequence similarity are added to the top alignment list. These nodes are used to study the biologically similar group of proteins across the species. To increase the coverage, the remaining nodes are aligned using comparatively low structure/sequence similarity thresholds in the second phase of stage-1. All the node pairs that have 3D structure similarity , global sequence similarity (bit-score), and local sequence similarity are added to the alignment list. The average length found in local alignments of the protein pairs is , so we add an extra threshold of local sequence alignment’s length. The candidate node pairs must have a common sequence of length greater than the average length (35).
The global sequence similarity and structure similarity thresholds are tuned using a grid search technique (Supplemental Section 1). The global sequence similarity and structure similarity are directly linked with functional similarity in most cases. In contrast, the local sequence similarity can be high for functionally dissimilar protein pairs. For example, the similarity between the non-domain regions of the protein pair cannot guarantee the similarity of the function. We analyze the local sequence similarity and set the threshold so that functional similarity should not be compromised. Figure 1 shows the density plot for the local sequence similarity. From Figure 1, we can see that less than of the total protein pairs meet the set threshold. Moreover, we eliminate the protein pairs that have the aligned sequence of length less than (average length) that resultantly reduce the node pairs to less than . The strict similarity and length-based thresholds make sure that the functionally similar proteins are aligned. We set these thresholds using the grid search technique. The details of the parameter settings are provided in Supplemental Section 1.
Figure 1.
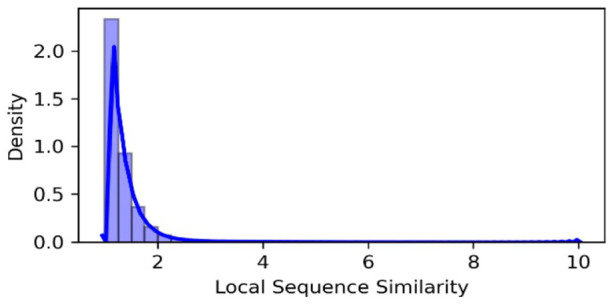
The density plot for the local sequence similarity is presented. The distribution is highly skewed. Less than 5% of the total protein pairs produce a similarity greater than 2.
| Algorithm-1: Seeds-Generation on the basis of biological scoring matrices |
|---|
| 1: Procedure: Seed Generation 2: Input: M1, M2, and M3 //Similarity Matrices 3: Input: str.t, seq.t and lseq.t //Similarity Thresholds 4: Output: Top-Nodes and Seeds 5: Seeds = [] 6: Sort M1, M2, M3 on the basis of scores 7: for all node pairs (a, b) do 8: if a ∉ Seeds and b ∉ Seeds and M1[a, b] ⩾ 0.8 then 9: Seeds.append(a, b) 10: end if 11: end for 12: for all node pairs (a, b) do 13: if a ∉ Seeds and b ∉ Seeds and M2[a, b] ⩾ 200 then 14: Seeds.append(a, b) 15: end if 16: end for 17: for all node pairs (a, b) do 18: if a ∉ Seeds and b ∉ Seeds and M3[a, b] ⩾ 4.0 then 19: if AlignmentLength(a, b) > 35 then 20: Seeds.append(a, b) 21: end if 22: end if 23: end for 24: Top-Nodes = Seeds //Top-Nodes are separated 25: for all node pairs (a, b) do 26: if a ∉ Seeds and b ∉ Seeds and M1[a, b] ⩾ str.t then 27: Seeds.append(a, b) 28: end if 29: end for 30: for all node pairs (a, b) do 31: if a ∉ Seeds and b ∉ Seeds and M2[a, b] ⩾ seq.t then 32: Seeds.append(a, b) 33: end if 34: end for 35: for all node pairs (a, b) do 36: if a ∉ Seeds and b ∉ Seeds and M3[a, b] ⩾ lseq.t then 37: if AlignmentLength(a, b) > 35 then 38: Seeds.append(a, b) 39: end if 40: end if 41: end for 42: end procedure |
Stage-2
This paper uses remote homology in the first phase of the second stage of the BioAlign algorithm. The remote homologous proteins play a vital role in the function prediction of the proteins. These proteins usually have less than 25% sequence similarity, but share similar functions and structures. The prediction of the remote homologous proteins itself is a challenging task. This paper uses a simple but novel approach to detect the remote homology between the pair of proteins.
The unaligned proteins are taken out from both networks. The homologous proteins for each unaligned protein are extracted using the PSI-BLAST tool (parameters: gap-open = 5, gap-extension = 2, E-value = 10, word-size = 11). For every unaligned pair of proteins , we count the common proteins from their homologous proteins. The unaligned pairs that have no common homologs are eliminated from the selection pool. The remaining protein pairs are sorted w.r.t the number of their common homologs. The unaligned protein pairs that have the most common homologs are aligned first. Algorithm 2 is used to align the node pairs based on remote homology.
| Algorithm-2: Alignment using Remote Homology |
|---|
| 1: Procedure: Align_Remote_Homologs 2: Input: Seeds //Output of Algorithm 1 3: Input: N1, N2 //Unique nodes of both networks 4: Input: Files contain homologous proteins for each protein 5: Output: Seeds //Extended List of Seeds 6: for all a in N1 do 7: L1a = get the list of homologous proteins from File_a 8: end for 9: for all b in N2 do 10: L2b = get the list of homologous proteins from File_b 11: end for 12: for all a in N1 do 13: for all b in N2 do 14: C[a, b] = common(L1a, L2b) //Common Homologs 15: end for 16: end for 17: sort C on the basis of Common Homologs 18: for all node pairs (a, b) do 19: if a ∉ Seeds and b ∉ Seeds and C[a, b] ⩾ 1 then 20: Seeds.append(a, b) 21: end if 22: end for 23: end procedure |
In the second phase of the stage-2, the remaining pairs are aligned using predicted secondary structure motifs. Structural biologists have found different motifs that are important in finding the functions of the proteins. Helix-Loop-Helix (HLH) and Helix-Turn-Helix (HTH) are the most important motifs that are useful in studying the protein’s function. We predict the secondary structures of the unaligned proteins using an in-house developed deep learning model. From the sequence of the predicted secondary structures, we count the number of HLH and HTH motifs for all unaligned proteins. The protein is represented in the form of a vector (of size 2) that contains the count of HLH and HTH. The differences between the vectors of all the unaligned protein pairs are calculated and sorted. The protein pairs that have small differences are aligned first. Algorithm 3 is used to align the node pairs on the basis of secondary structure motifs.
| Algorithm-3: Alignment using Secondary Structure Motifs |
|---|
| 1: Procedure: Alignment_using_SS-Motifs 2: Input: Seeds //Output of Algorithm 2 3: Input: N1, N2 // Unique Nodes of Both Networks 4: Input: SS //Predicted Secondary Structure of All Proteins 5: Output: Seeds //Extended List of Seeds 6: for all a in N1 do 7: L1a = count motifs from SSa // L1a is vector of counts of motifs (HLH, HTH) → L1a = (2,2) 8: end for 9: for all b in N2 do 10: L2b = count motifs from SSb // L1b is vector of counts of motifs (HLH, HTH) → L1a = (1,2) 11: end for 12: for all a in N1 do 13: for all b in N2 do 14: D[a, b] = Calculate Difference(L1a, L2b) 15: //Proteins of similar counts of Motifs produce small difference 16: end for 17: end for 18: sort D on the basis of vector differences 19: for all node pairs (a, b) do 20: if a ∉ Seeds and b ∉ Seeds and L1a > 0 and L2b > 0 then 21: Seeds.append(a, b) 22: end if 23: end for 24: end procedure |
Stage-3
In the final stage, the remaining nodes are aligned using network interaction information. The neighboring nodes of already aligned nodes are considered as the candidate nodes for alignment. For example, if 2 neighbors of unaligned node a are aligned previously with 2 neighbors of unaligned node b, the node pair (a, b) will be considered as the candidate node pair. All the candidate node pairs are sorted on the basis of common aligned neighbors. The node pair that has maximum aligned neighbors will be aligned first. Figure 2 further explains the working of the topological stage.
Figure 2.
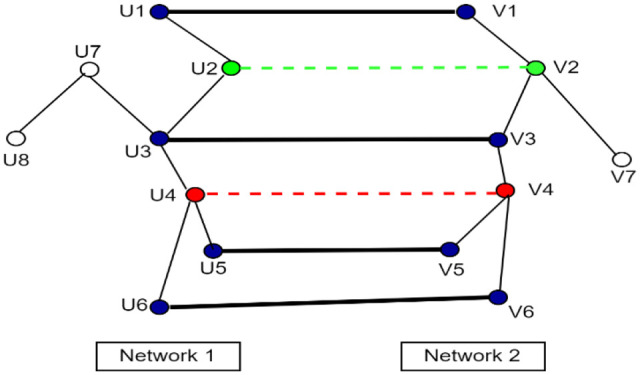
Presents the 2 networks (Network 1 on the left side and Network 2 on the right side). Blue nodes are the aligned pairs. Green and red node pairs are the candidate pairs for alignment as they have aligned neighbors. The node pair with green color (u2, v2) has a score of 2 (due to 2 aligned neighbors) while the node pair with red color has a score of 3. According to the Stage-3 algorithm, the red nodes pair will be aligned first and then the green nodes pair will be aligned. The nodes u7, u8, and v7 will not be considered as candidates for alignment as they do not have any common aligned neighbor.
Evaluation metrics
BioAlign is evaluated on the basis of Average Functional Similarity (AFS), Normalized GO-term Consistency (NGOC) and the percentage of aligned nodes. The AFS can be categorized into biological process (BP) and molecular function (MF). Two types of methods are used to calculate the functional similarity. The first type contains Lin, 22 Resnick 23 , and Schlicker et al 24 methods that are IC-based. These methods are database-dependent and result in different functional similarity for different databases. The second type is of graph-based methods that are database independent. GOGO 25 and Wang et al 26 are the most commonly used graph-based methods. A number of tools (GOSemSim, 27 NaviGO, 28 and SeSAME, 29 etc.) provide the implementation for the Wang method. GoSemSim is used by most of the recent studies so we use this tool for the implementation of Wang method. The AFS is calculated using equation (2).
| (2) |
Where, is the average functional similarity of the complete alignment ( represents all node pairs) in terms of . is the functional similarity of a node pair , calculated by Wang et al. 26 The node belongs to first network while node belongs to second network.
The ratio to the common and the total GO-terms of a pair of proteins is known as GO-term Consistency (GOC) that is calculated using equation (3).
| (3) |
Where u and v represent the protein pair. GO(u) and GO(v) represent the GO-terms of protein u and protein v, respectively.
The alignment of 2 networks is represented as the set of pairs of proteins. The GOC of all the protein pairs of the final alignment is added and divided to the network size to get the NGOC of the complete alignment (equation (4)).
| (4) |
Where a represents the protein pairs (alignment). u and v are the first and second proteins of a pair. N1 and N2 represent the number of nodes of network 1 and network 2, respectively. The maximum size of the alignment can be equal to the smaller network size.
Datasets
The networks used in this paper are presented in Table 1. The first row presents the specie name while the second and third rows present the number of nodes and edges in the networks respectively. The last row presents the percentage of proteins with available 3D structures. All the networks are collected from the HINT database 4 [Version:2019]. HINT database contains the networks of experimentally verified interactions. This paper uses all the networks that have more than 1000 interactions.
Table 1.
Data statistics that include the number of nodes, number of edges, and percentage of proteins with the 3D resolved structure are presented.
| Species Name | Mouse | Human | Yeast | Worm | Fly |
|---|---|---|---|---|---|
| Number of nodes | 744 | 10 791 | 5036 | 4486 | 7498 |
| Number of edges | 1229 | 47 427 | 19 085 | 11 496 | 25 679 |
| Nodes with 3D structure (%) | 17 | 43 | 29 | 2 | 3 |
Source: All these datasets are extracted from HINT database.
Results
This section presents a detailed comparison between the results of BioAlign and existing aligners. SAlign is a structure-aware method that incorporates biological (sequence and structure similarity) and topological information for aligning the PPI network pairs. Twadn uses 5 different node-level features and sequence similarities to align the PPI network pairs. MONACO, HubAlign, ModuleAlign, and IBNAL use sequence similarity and topological information for alignment. BEAMS adds weights to the edges on the basis of sequence similarities of the proteins and uses a module-based approach for alignment. SANA uses simulated annealing-based optimization algorithm to optimize the alignments. MAGNA++ aligns the nodes using an evolutionary algorithm (based on sequence similarity). NETAL uses only topological information for alignment. PROPER is a 2-stage approach that first generates the seeds using biological information and then extends the seeds based on their neighbors. All the algorithms use different sources of information that make the comparison interesting and give insights about the sources that are helpful in the alignment process. The comparison between all the algorithms has been made on the 5 most commonly used network pairs (Mouse-Human, Mouse-Yeast, Yeast-Human, Mouse-Worm, and Mouse-Fly). We compare the results on the basis of NGOC, AFS and the percentage of aligned nodes w.r.t MF and BP. All the aligners are used with their default parameters to produce the results.
The results of different stages of BioAlign
The BioAlign algorithm aligns the nodes in a multi-stage manner. The first stage uses global sequence similarity, 3D structure similarity, and local sequence similarity. In the first stage, BioAlign first aligns the nodes that have high sequence or structure similarities. We name these nodes as Top-Nodes. On average, BioAlign generates the alignment of Top-Nodes with AFS and w.r.t MF and BP, respectively. The percentage of aligned nodes is and w.r.t MF and BP, respectively (Table 2). The alignments of these nodes can be helpful in studying the highly similar (biologically) proteins across the species. In the second phase of stage-1, BioAlign aligns the nodes that have a relatively small sequence or structure similarity scores. This results in a decrease in AFS, but an increase in coverage. The AFS of the first stage is and w.r.t MF and BP, respectively. BioAlign aligns and nodes w.r.t MF and BP in the first stage.
Table 2.
The results of the different stages and variants of BioAlign.
| Specie pair | Measures | Top-nodes | Stage1 | Stage1 + B Stage2 (BV1) | Stage1 + T Stage3 (BV2) | Stage2 + 3 (BD) Stage1 + BT |
|---|---|---|---|---|---|---|
| Mouse-Human* | AFSMF | 0.78 | 0.78 | 0.78 | 0.78 | 0.78 |
| AFSBP | 0.68 | 0.67 | 0.67 | 0.67 | 0.67 | |
| NodesMF | 87 | 88 | 88 | 88 | 88 | |
| NodesBP | 90 | 92 | 92 | 92 | 92 | |
| Mouse-Yeast | AFSMF | 0.70 | 0.51 | 0.47 | 0.46 | 0.46 |
| AFSBP | 0.53 | 0.35 | 0.32 | 0.31 | 0.31 | |
| NodesMF | 20 | 56 | 71 | 69 | 73 | |
| NodesBP | 22 | 65 | 85 | 84 | 88 | |
| Mouse-Fly | AFSMF | 0.76 | 0.68 | 0.67 | 0.66 | 0.67 |
| AFSBP | 0.57 | 0.50 | 0.49 | 0.49 | 0.49 | |
| NodesMF | 58 | 76 | 78 | 79 | 78 | |
| NodesBP | 59 | 81 | 83 | 83 | 83 | |
| Mouse-Worm | AFSMF | 0.73 | 0.63 | 0.62 | 0.61 | 0.62 |
| AFSBP | 0.55 | 0.46 | 0.45 | 0.45 | 0.45 | |
| NodesMF | 43 | 69 | 73 | 72 | 73 | |
| NodesBP | 40 | 63 | 67 | 66 | 68 | |
| Yeast-Human | AFSMF | 0.74 | 0.60 | 0.55 | 0.55 | 0.55 |
| AFSBP | 0.57 | 0.45 | 0.42 | 0.41 | 0.41 | |
| NodesMF | 31 | 52 | 62 | 61 | 63 | |
| NodesBP | 31 | 60 | 73 | 72 | 74 | |
| Average | AFSMF | 0.74 | 0.64 | 0.62 | 0.61 | 0.62 |
| AFSBP | 0.58 | 0.48 | 0.47 | 0.47 | 0.47 | |
| NodesMF | 48 | 68 | 75 | 74 | 75 | |
| NodesBP | 49 | 72 | 80 | 79 | 81 |
Abbreviations: B, biology; T, Topology.
BV1 and BV2 use biology and topology (respectively), after stage1, to align the unaligned nodes. BD uses both biology and topology in the second stage. The bold cells present the best results of BioAlign variants.
Alignment of the mouse-human pair is completed in the first stage.
In the second stage, BioAlign can choose biological and/or topological information. The variant BV1 of BioAlign aligns the remaining nodes using biological information (remote homology and secondary structure motifs) in stage-2. In contrast, the variant BV2 aligns the remaining nodes using topological information. The percentage of unaligned nodes in Mouse-Yeast and Yeast-Human pairs is more than 20%. In both cases, variant BV1 outperforms variant BV2 in terms of AFS and coverage. In the remaining cases, the percentage of unaligned nodes is less than 5%. The difference in the results is not notable. The variant BD (default version of BioAlign) is the combination of variants BV1 and BV2. On average, the results of variant BD are comparable or better than the results of both variants in terms of AFS and coverage.
On average, the results of Stage2 are better than the results of Stage1 ( and in terms of percentage of aligned nodes w.r.t MF and BP). The AFS of Stage2 is slightly lower than Stage1. The additional information increases the coverage by 7%-9% by reducing the AFS by 1%-2%. This shows that BioAlign completes the alignment with a small decrease in AFS.
Comparison between the results of BioAlign and existing algorithms
Table 3 shows the comparison between the results of BioAlign and existing algorithms. The results of BioAlign are better or comparable to all algorithms for all network pairs on the basis of AFS w.r.t MF and BP. On average, the results of BEAMS are slightly lower than BioAlign in terms of AFS. The average performance of BioAlign is and better than Twadn in terms of AFS w.r.t MF and BP, respectively. The performance of BioAlign is 15%-19% and 16%-21% better in terms of AFS w.r.t MF and BP, respectively as compared to SAlign, MONACO, and PROPER. When we compare BioAlign with HubAlign, ModuleAlign, IBNAL, NETAL, SANA, and MAGNA++, it performs 29%-52% and 32%-51% better than these algorithms in terms of AFS w.r.t MF and BP, respectively. BEAMS, Twadn, PROPER, MONACO, and SAlign produce better results as compared to other existing aligners in terms of AFS. Comparison between the results of BioAlign and Existing Aligners. The average results of BioAlign in terms of coverage are better than all existing algorithms. BioAlign outperforms SAlign in terms of coverage by a small margin. When we compare the coverage of BioAlign with other existing algorithms, it performs 3%-11% and 4%-16% better w.r.t BP and MF, respectively. The results of SAlign, HubAlign, and Twadn are better than other existing aligners in terms of coverage.
Table 3.
Comparison between the results of BioAlign and existing techniques on 5 network pairs on the basis of AFS and percentage of aligned nodes w.r.t MF and BP. BioAlign produced high-quality alignments in terms of AFS and coverage.
| Sp. pairs | Eval. criteria | Alignment algorithms | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BA | TW | BE | MO | SA | PR | HA | MA | IB | SAN | NE | MAG | ||
| MH | AFSMF | 0.78 | 0.73 | 0.79 | 0.56 | 0.58 | 0.76 | 0.48 | 0.42 | 0.35 | 0.34 | 0.33 | 0.36 |
| AFSBP | 0.67 | 0.63 | 0.70 | 0.43 | 0.43 | 0.66 | 0.34 | 0.30 | 0.26 | 0.25 | 0.24 | 0.26 | |
| NodesMF | 88 | 86 | 85 | 82 | 82 | 87 | 78 | 74 | 72 | 75 | 73 | 76 | |
| NodesBP | 92 | 88 | 87 | 87 | 84 | 91 | 82 | 81 | 83 | 82 | 82 | 82 | |
| MY | AFSMF | 0.46 | 0.44 | 0.47 | 0.43 | 0.40 | 0.38 | 0.36 | 0.31 | 0.29 | 0.30 | 0.31 | 0.29 |
| AFSBP | 0.32 | 0.31 | 0.32 | 0.31 | 0.27 | 0.25 | 0.25 | 0.23 | 0.21 | 0.22 | 0.22 | 0.21 | |
| NodesMF | 73 | 69 | 64 | 67 | 72 | 56 | 71 | 71 | 63 | 67 | 64 | 67 | |
| NodesBP | 88 | 79 | 73 | 77 | 91 | 72 | 90 | 88 | 76 | 84 | 83 | 83 | |
| YH | AFSMF | 0.55 | 0.50 | 0.54 | 0.48 | 0.48 | 0.42 | 0.46 | 0.26 | 0.30 | 0.27 | 0.26 | 0.26 |
| AFSBP | 0.41 | 0.36 | 0.40 | 0.36 | 0.35 | 0.32 | 0.34 | 0.22 | 0.24 | 0.23 | 0.22 | 0.22 | |
| NodesMF | 63 | 60 | 55 | 59 | 64 | 57 | 63 | 60 | 58 | 61 | 60 | 59 | |
| NodesBP | 74 | 70 | 63 | 70 | 76 | 67 | 74 | 72 | 70 | 73 | 72 | 70 | |
| MF | AFSMF | 0.67 | 0.62 | 0.67 | 0.55 | 0.50 | 0.55 | 0.42 | 0.36 | 0.33 | 0.32 | 0.32 | 0.37 |
| AFSBP | 0.49 | 0.46 | 0.49 | 0.40 | 0.37 | 0.41 | 0.31 | 0.28 | 0.24 | 0.23 | 0.23 | 0.28 | |
| NodesMF | 79 | 77 | 74 | 73 | 73 | 69 | 67 | 66 | 58 | 67 | 57 | 63 | |
| NodesBP | 83 | 82 | 79 | 80 | 80 | 77 | 76 | 74 | 58 | 76 | 60 | 62 | |
| MW | AFSMF | 0.62 | 0.56 | 0.58 | 0.49 | 0.56 | 0.52 | 0.49 | 0.41 | 0.30 | 0.32 | 0.29 | 0.31 |
| AFSBP | 0.45 | 0.41 | 0.41 | 0.34 | 0.41 | 0.39 | 0.37 | 0.30 | 0.25 | 0.25 | 0.24 | 0.25 | |
| NodesMF | 73 | 68 | 67 | 66 | 75 | 61 | 73 | 71 | 62 | 59 | 62 | 64 | |
| NodesBP | 68 | 64 | 63 | 62 | 69 | 56 | 67 | 66 | 70 | 57 | 72 | 76 | |
| Avg | AFSMF | 0.62 | 0.57 | 0.61 | 0.50 | 0.50 | 0.52 | 0.44 | 0.35 | 0.31 | 0.31 | 0.30 | 0.32 |
| AFSBP | 0.47 | 0.42 | 0.46 | 0.37 | 0.37 | 0.40 | 0.32 | 0.27 | 0.24 | 0.24 | 0.23 | 0.24 | |
| NodesMF | 75 | 72 | 68 | 69 | 73 | 66 | 70 | 68 | 63 | 66 | 63 | 66 | |
| NodesBP | 81 | 77 | 72 | 75 | 80 | 73 | 77 | 76 | 71 | 74 | 74 | 75 | |
Abbreviations: BA, BioAlign; BE, BEAMS; HA, HubAlign; IB, IBNAL; MA, ModuleAlign; MAG, MAGNA++; NE, NETAL; MO, MONACO; PR, PROPER; SA, SAlign; SAN, SANA; TW, Twadn.
The percentage of aligned nodes is calculated with respect to the smaller network. AFS referred to as the average functional similarity of the complete alignment while Nodes are referred to as the percentage of nodes aligned. Bold cells represent the best results.
Although the coverage achieved by SAlign is similar to BioAlign, the AFS of SAlign is lower than BioAlign (19%-21%). In contrast, the AFS achieved by BEAMS is similar to BioAlign, the coverage of BEAMS is notably lower than BioAlign (9%-11%). The coverage achieved by HubAlign and ModuleAlign is reasonable, but the AFS produced by these algorithms is notably low as compared to BioAlign and some of the existing algorithms (Twadn, BEAMS, MONACO, PROPER, and SAlign). Although the results of PROPER in terms of AFS are reasonable, its coverage is notably low among all existing algorithms. The performance of IBNAL, NETAL, MAGNA++ is notably low in terms of both AFS and coverage. In contrast, BioAlign achieves better performance in terms of coverage as well as AFS.
A similar trend is found between the results of BioAign and existing aligners when we compare results in terms of NGOC (Table 4). Comparison between the results of BioAlign and Existing Aligners. The results of BioAlign are better than BEAMS in all cases except for Mouse-Human. The results of Twadn are inferior as compared to BioAlign and BEAMS but it outperforms all other existing algorithms. The results of SAlign, MONACO and PROPER are better than rest of the existing aligners (HubAlign, ModuleAlign, IBNAL, SANA, NETAL, and MAGNA++).
Table 4.
Comparison between the results of BioAlign and existing techniques on 5 network pairs on the basis of NGOC w.r.t MF and BP. BioAlign produced better or comparable results in terms of NGOC.
| Sp. pairs | Eval. criteria | Alignment algorithms | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BA | TW | BE | MO | SA | PR | HA | MA | IB | SAN | NE | MAG | ||
| MH | NGOCMF | 0.54 | 0.49 | 0.57 | 0.28 | 0.28 | 0.29 | 0.17 | 0.12 | 0.06 | 0.08 | 0.07 | 0.08 |
| NGOCBP | 0.48 | 0.43 | 0.52 | 0.19 | 0.17 | 0.18 | 0.08 | 0.04 | 0.01 | 0.01 | 0.01 | 0.01 | |
| MY | NGOCMF | 0.15 | 0.14 | 0.14 | 0.12 | 0.12 | 0.07 | 0.10 | 0.07 | 0.05 | 0.04 | 0.05 | 0.05 |
| NGOCBP | 0.07 | 0.06 | 0.06 | 0.05 | 0.04 | 0.02 | 0.03 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | |
| YH | NGOCMF | 0.22 | 0.18 | 0.19 | 0.17 | 0.19 | 0.15 | 0.18 | 0.07 | 0.06 | 0.07 | 0.07 | 0.07 |
| NGOCBP | 0.11 | 0.08 | 0.09 | 0.08 | 0.08 | 0.05 | 0.08 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | |
| MF | NGOCMF | 0.27 | 0.24 | 0.26 | 0.17 | 0.13 | 0.15 | 0.08 | 0.05 | 0.02 | 0.02 | 0.02 | 0.03 |
| NGOCBP | 0.14 | 0.12 | 0.13 | 0.08 | 0.07 | 0.08 | 0.04 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | |
| MW | NGOCMF | 0.21 | 0.18 | 0.18 | 0.12 | 0.16 | 0.12 | 0.13 | 0.08 | 0.03 | 0.03 | 0.03 | 0.05 |
| NGOCBP | 0.11 | 0.08 | 0.09 | 0.05 | 0.08 | 0.06 | 0.06 | 0.03 | 0.02 | 0.01 | 0.01 | 0.02 | |
| Avg | NGOCMF | 0.28 | 0.25 | 0.27 | 0.17 | 0.18 | 0.16 | 0.13 | 0.08 | 0.04 | 0.05 | 0.05 | 0.06 |
| NGOCBP | 0.18 | 0.15 | 0.18 | 0.09 | 0.09 | 0.08 | 0.06 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | |
Bold cells represent the best results.
The global PPI network alignment is a multi-objective problem that aims to align the maximum possible number of nodes with high AFS. The solutions/alignments that produce high AFS with large coverage are considered as better alignments as compared to alignments with low AFS and/or coverage. The alignments produced by BioAlign are better than the alignments produced by existing aligners as it satisfies both objectives (coverage and AFS).
The solutions of the multi-objective problems need Pareto-Front (PF) based evaluation. PF is a line that contains the best/non-dominant solutions (better in all objectives, or better in at least one objective and comparable in the other objectives). In case of maximizing both objectives, the PF will always be produced in the right-upward direction.
The results of all the aligners in terms of AFS and coverage are plotted in 2D space (Figure 3) to better understand the positions of the aligners. Some of existing aligners do not produce optimal alignments in terms of semantic similarity while some of the algorithms failed to complete alignment. To analyze the performance in terms of both objectives, we compute the difference (Euclidean) between the points (AFS, Coverage) of aligners. The results points of BioAlign, BEAMS, Twadn, and SAlign are joined by plotting lines in different colors. The light-blue line (BioAlign) contains the solutions that perform better than other solutions, so we can say that the light-blue line is the PF. When we compare the positions of BioAlign with Twadn, the difference between the points of both aligners is and in terms of MF and BP, respectively. The distance between the points of BioAlign and BEAMS is and in terms of MF and BP. The difference between the points of BioAlign and SAlign is and in terms of BP and MF, respectively. The difference between the points of BioAlign and rest of the algorithms is 12-32 and 14-34 w.r.t BP and MF, respectively. From this analysis we can say that BioAlign is rank-1 algorithm for global PPI network alignment. The ranks of Twadn, BEAMS, and SAlign are 2, 3, and 4 respectively in terms of distance.
Figure 3.
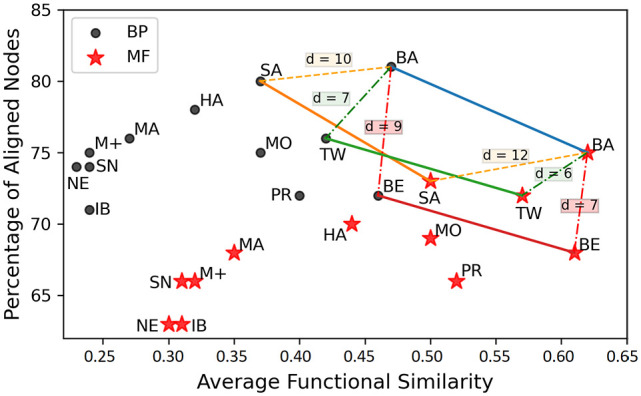
The 2D-positions of the aligners on the basis of average AFS and the percentage of aligned nodes are represented. The solutions of different aligners are represented by the lines with different colors. The best-line (light-blue) represents the Pareto-Front. The dotted lines show the difference of BioAlign with Twadn, BEAMS, and SAlign that is notably high. BioAlign outperforms all the existing aligners in terms of positions that is validated by the Pareto-Front technique.
Supplemental Table 2 shows the comparison between the results of all the existing aligners in terms of topological measures (ICS, EC, and SSS). The results of BioAlign are better than or comparable to SANA, Twadn, BEAMS, and MONACO. The remaining algorithms outperform BioAlign in terms of topological measures. Topological metrics can be misleading as the PPI network data is incomplete and noisy (high false positive rate due to inferred interactions).
Enrichment analysis
As discussed earlier, the best global aligners tend to produce alignments that have high AFS and coverage. To analyze the quality of the alignments, we show the results in terms of the percentage of aligned nodes that have high AFS (greater than and ). Figure 4 shows the number of aligned nodes that produce AFS greater than and .
Figure 4.

The number of aligned nodes that have: (A) AFS > 0.50 and (B) AFS > 0.75 are presented. BioAlign aligns a much larger number of nodes in both cases (AFS > 0.50 and AFS > 0.75). The margin between the results of BioAlign and existing aligners is notably high.
From Figure 4, we can see that BioAlign aligns more nodes that have AFS greater than and as compared to existing aligners, which indicates that BioAlign aligns the protein pairs accurately. For Mouse-Human pair, the results of the BEAMS, PROPER, and BioAlign algorithms are similar. For Mouse-Yeast pair, the results of BioAlign are slightly higher than the results of BEAMS and Twadn. For all remaining pairs, the proposed aligner outperforms all the existing aligners with a high margin. On average, BioAlign aligns more nodes that have AFS than BEAMS. BioAlign aligns 16%-31% more nodes than Twadn, SAlign, MONACO, and PROPER. When we compare BioAlign with other existing aligners, it outperforms with a margin of 53%-80% in terms of aligned nodes (Figure 4A). BioAlign aligns more nodes with AFS as compared to BEAMS. BioAlign aligns 13%-37% more nodes as compared to Twadn, SAlign, MONACO, and PROPER. When we compare BioAlign with other algorithms, it aligns 62%-91% more nodes that have AFS (Figure 4B). A similar trend is shown by all the algorithms when we compare the percentage of aligned nodes that have AFS greater than . BioAlign aligns 8%-89% more nodes that have AFS as compared to the existing aligners.
These analyses show that BioAlign aligns a much larger number of nodes that have high AFS as compared to existing techniques. Thus, BioAlign can help in studying the highly similar groups of proteins of the different networks. This study also helps in finding the functionally similar proteins among different species.
AFS of top 10% to 100% nodes
This section presents the results (AFS) of top nodes. These nodes are in the order in which they are being aligned. Figure 5 presents the AFS of all the aligners for different percentages of aligned nodes. From Figure 5, we can see that BioAlign aligns the nodes with the highest AFS among all aligners.
Figure 5.

The results in terms of AFS for different percentages of aligned nodes (10, 20 . . . 100) are presented. BioAlign outperforms all existing techniques in all cases (blue color). PROPER and BEAMS lines (green and pink colors) show incompleteness in 4 out of 5 cases. Twadn and MONACO show incompleteness in the Mouse-Yeast case. The remaining algorithms are failed to produce high AFS.
The lines of all the existing algorithms are inferior as compared to BioAlign in terms of AFS and/or completeness level. For the Mouse-Human pair, the performance of BioAlign is closed to PROPER and BEAMS. For all remaining pairs, the gap between the AFS of BioAlign and existing aligners is greater than . Although, the performance of BEAMS is closed to BioAlign for Mouse-Yeast and Mouse-Fly pairs, its completeness level is much poor. PROPER and BEAMS show incompleteness in 4 out of 5 cases. The lines of all the algorithms (except PROPER and BioAlign) are consistent (straight) w.r.t number of aligned nodes. All the aligners (except PROPER and BioAlign) use the biological and/or topological information simultaneously, so these algorithms generate consistent lines. BioAlign and PROPER align the nodes in a multi-stage manner. These aligners first align the highly similar nodes (using biological information only), and then extend the alignments using different sources of information (PROPER uses graph-percolation based method, while BioAlign uses predicted secondary structure, remote homology, and topology).
In the start (for ), the AFS of the PROPER and BEAMS aligners is similar to BioAlign, but as the number of nodes increases, the AFS decreases. Furthermore, the curves of the BEAMS and PROPER algorithm (green and pink lines in Figure 5) do not reach , which is the indication that PROPER and BEAMS generate incomplete alignments. In contrast, the proposed aligner shows relatively high consistency and its lines show completeness in all cases. In general, the short alignments (a small number of aligned nodes) produce high AFS as the mapping of a small region of a network is comparatively easier than the mapping of a large portion of a network. 6 BEAMS and PROPER generates incomplete alignments, but still achieves lower AFS than BioAlign in most of the cases. Furthermore, the decrease in the lines of BEAMS and PROPER algorithms is higher than BioAlign. This analysis shows the beneficial effect of biological sources that are used by BioAlign.
Overall, the curves of IBNAL, NETAL, MAGNA++, and ModuleAlign show the lowest results. HubAlign generates better alignments as compared to these aligners, but its AFS is lower than Twadn, MONACO, SAlign. MONACO performs better than existing aligners except for Twadn and SAlign. The results of SAlign are better than all existing algorithms except for Twadn. For Mouse-Yeast, Mouse-Worm, and Yeast-Human cases, the results of SAlign and Twadn are similar. For the remaining 2 cases, the results of Twadn are better than SAlign. Twadn is better among all existing aligners, but its difference with BioAlign is large. The completeness (coverage) and high AFS of BioAlign is validated by Figure 5.
Execution time
The average execution time of all algorithms on 5 PPI network pairs are reported in Table 5. The use of different level of information slightly increased the execution time of the proposed algorithm as compared to PROPER. Twadn and MONACO. In contrast, Bioalign takes less execution time as compared to BEAMS, HubAlign, SAlign, SANA, ModuleAlign, and MAGNA++.
Table 5.
The average execution time of the aligners on 5 datasets.
| Algorithm | Execution time |
|---|---|
| PROPER | 3 s |
| Twadn | 5 s |
| MONACO | 30 s |
| BioAlign | 48 s |
| BEAMS | 54 s |
| HubAlign | 74 s |
| SAlign | 88 s |
| SANA | 06 min |
| ModuleAlign | 26 min |
| MAGNA++ | 58 min |
Discussion
This paper presents a novel approach that incorporates different sources of biological information (global sequence similarity, 3D structure similarity, local sequence similarity, remote homology, and predicted secondary structure motifs). All the existing aligners incorporate sequence similarity and/or network topology except SAlign that includes structure similarity along with sequence similarity and network topology. From the results of the 5 network pairs, we show that BioAlign performs 8%-52% and 11%-51% better than all existing aligners (except BEAMS) in terms of AFS w.r.t MF and BP, respectively. The coverage of BioAlign is also comparable to or better than existing techniques.
The hypothesis (use of topology results in a decrease in AFS) claimed by UAlign is proved by SAlign. This paper finds a similar trend in the results of the existing aligners. SANA, IBNAL, NETAL, HubAlign, and ModuleAlign use different types of topological measures, but their AFS is not comparable to the state-of-art algorithm. MAGNA++ uses topology as a pseudo-measure to align the networks, but it generates the alignments that have low AFS. PROPER and SAlign mainly use sequence and/or structure due to which their performance is notably higher than other existing algorithms in terms of AFS. BioAlign does not incorporate topological information and achieves more accurate results than existing algorithms. All the existing aligners (except PROPER) use the scoring matrices (sequence, structure, and topology) simultaneously to align the PPI networks. The curves of these algorithms are almost consistent (Figure 5). PROPER aligns the PPI networks in 2 stages. It first aligns the nodes on the basis of sequence similarity, and then extends the aligned nodes using topology. From Figure 5, we can see that the AFS of the PROPER algorithm decreases, as the number of nodes increases (green lines of Figure 5). This indicates that the use of topological information results in a decrease in AFS. In contrast, all the biological information used by BioAlign show consistency in terms of AFS. The AFS remains high for all percentages of aligned nodes (blue lines of Figure 5). This analysis indicates that all the information sources help BioAlign in aligning the biologically similar proteins.
After stage-1, BioAlign incorporates remote homology, predicted secondary structure, and topology. In order to compare the effect of topology and biology (remote homology and predicted secondary structure), we add these metrics after stage-1 (Table 2). From the results, we conclude that the inclusion of remote homology and predicted secondary structure is beneficial as compared to the inclusion of topology. This analysis is consistent with the analysis of SAlign and UAlign. We also note that the order of the input metrics given to the BioAlign is important. Therefore, a comparison of the results of BioAlign with different orders of input metrics is given in Supplemental Table 1 (columns 4-8). From the results, we show that order of the input metrics used by BioAlign (3D structure + global sequence + local sequence + remote homology + predicted secondary structure + Topology) best maximizes the results in terms of semantic similarity as well as percentage of aligned nodes.
The AFS w.r.t MF is higher than BP in all cases. The MF of the proteins are specific and have well-defined GO-terms (annotations). In contrast, the biological processes are generic and have vague GO-terms. We also note that the percentage of aligned nodes in terms of BP is higher than MF. The generic functions of the proteins and pathways involving these proteins are commonly known, but sometimes the specific functions of the proteins are unknown.
Conclusion
This paper presents a novel multi-stage method to align the PPI networks. In contrast to existing aligners, it uses structural and sequential information (local and global) to generate the seeds. In contrast to existing aligners, it does not compromise on AFS by aligning the nodes that have low similarity (sequence or structure-wise). This results in incomplete alignment that is tackled by aligning the remaining nodes on the basis of remote homology, predicted secondary structure motifs, and topological information. All the information sources help BioAlign to align the PPI networks with high AFS and coverage. The performance of BioAlign is notably high as compared to existing aligners in terms of AFS and coverage w.r.t MF and BP. The difference between the performance of BioAlign and existing aligners is 6-32 and 7-34 w.r.t BP and MF. Moreover, BioAlign aligns a much larger number of nodes that have high AFS ( ) that is, it aligns the biologically relevant proteins.
Supplemental Material
Supplemental material, sj-pdf-1-evb-10.1177_11769343221110658 for BioAlign: An Accurate Global PPI Network Alignment Algorithm by Umair Ayub and Hammad Naveed in Evolutionary Bioinformatics
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by a grant to establish Precision Medicine Lab under the umbrella of National Center in Big Data & Cloud Computing from the Higher Education of Pakistan.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: U.A. and H.N. designed the research. U.A. implemented the research. U.A. and H.N. analyzed the results and finalized the manuscript. H.N. supervised the research.
Availability: The implementation of the BioAlign algorithm and the datasets are available at GitHub: https://github.com/cbrl-nuces/BioAlign
ORCID iD: Umair Ayub  https://orcid.org/0000-0001-8961-3978
https://orcid.org/0000-0001-8961-3978
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245-246. [DOI] [PubMed] [Google Scholar]
- 2. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315-1317. [DOI] [PubMed] [Google Scholar]
- 3. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535-D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Das J, Yu H. HINT: high-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol. 2012;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Licata L, Briganti L, Peluso D, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857-D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ayub U, Haider I, Naveed H. SAlign-a structure aware method for global PPI network alignment. BMC Bioinformatics. 2020;21:500-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kazemi E, Hassani H, Grossglauser M, Pezeshgi Modarres H. PROPER: global protein interaction network alignment through percolation matching. BMC Bioinformatics. 2016;17:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meng L, Striegel A, Milenković T. Local versus global biological network alignment. Bioinformatics. 2016;32:3155-3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guzzi PH, Milenkovic T. Survey of local and global biological network alignment: the need to reconcile the two sides of the same coin. Brief Bioinform. 2018;19:472-481. [DOI] [PubMed] [Google Scholar]
- 10. Hashemifar S, Xu J. Hubalign: an accurate and efficient method for global alignment of protein-protein interaction networks. Bioinformatics. 2014;30:i438-i444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashemifar S, Ma J, Naveed H, Canzar S, Xu J. ModuleAlign: module-based global alignment of protein-protein interaction networks. Bioinformatics. 2016;32:i658-i664. [DOI] [PubMed] [Google Scholar]
- 12. Elmsallati A, Msalati A, Kalita J. Index-based network aligner of protein-protein interaction networks. IEEE/ACM Trans Comput Biol Bioinform. 2018;15:330-336. [DOI] [PubMed] [Google Scholar]
- 13. Woo HM, Yoon BJ. MONACO: accurate biological network alignment through optimal neighborhood matching between focal nodes. Bioinformatics. 2021;37:1401-1410. [DOI] [PubMed] [Google Scholar]
- 14. Zhong Y, Li J, He J, et al. Twadn: an efficient alignment algorithm based on time warping for pairwise dynamic networks. BMC Bioinformatics. 2020;21:385-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mamano N, Hayes WB. SANA: simulated annealing far outperforms many other search algorithms for biological network alignment. Bioinformatics. 2017;33:2156-2164. [DOI] [PubMed] [Google Scholar]
- 16. Saraph V, Milenković T. MAGNA: maximizing accuracy in global network alignment. Bioinformatics. 2014;30:2931-2940. [DOI] [PubMed] [Google Scholar]
- 17. Neyshabur B, Khadem A, Hashemifar S, Arab SS. NETAL: a new graph-based method for global alignment of protein-protein interaction networks. Bioinformatics. 2013;29:1654-1662. [DOI] [PubMed] [Google Scholar]
- 18. Alkan F, Erten C. BEAMS: backbone extraction and merge strategy for the global many-to-many alignment of multiple PPI networks. Bioinformatics. 2014;30:531-539. [DOI] [PubMed] [Google Scholar]
- 19. Malod-Dognin N, Ban K, Pržulj N. Unified alignment of protein-protein interaction networks. Sci Rep. 2017;7:953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mahram A, Herbordt MC. Fast and accurate NCBI BLASTP: acceleration with multiphase FPGA-based prefiltering. In: Proceedings of the 24th ACM International Conference on Supercomputing. ACM, 2010: 73-82. [Google Scholar]
- 22. Lin D, et al. An information-theoretic definition of similarity. Citeseer. 1998;98:296-304. [Google Scholar]
- 23. Resnik P. Semantic similarity in a taxonomy: an information-based measure and its application to problems of ambiguity in natural language. J Artif Intell Res. 1999;11:95-130. [Google Scholar]
- 24. Schlicker A, Domingues FS, Rahnenführer J, Lengauer T. A new measure for functional similarity of gene products based on gene ontology. BMC Bioinformatics. 2006;7:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao C, Wang Z. GOGO: an improved algorithm to measure the semantic similarity between gene ontology terms. Sci Rep. 2018;8:15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang JZ, Du Z, Payattakool R, Yu PS, Chen CF. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23:1274-1281. [DOI] [PubMed] [Google Scholar]
- 27. Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26:976-978. [DOI] [PubMed] [Google Scholar]
- 28. Wei Q, Khan IK, Ding Z, Yerneni S, Kihara D. NaviGO: interactive tool for visualization and functional similarity and coherence analysis with gene ontology. BMC Bioinformatics. 2017;18:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Du Z, Li L, Chen CF, Yu PS, Wang JZ. G-SESAME: web tools for GO-term-based gene similarity analysis and knowledge discovery. Nucleic Acids Res. 2009;37:W345-W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-evb-10.1177_11769343221110658 for BioAlign: An Accurate Global PPI Network Alignment Algorithm by Umair Ayub and Hammad Naveed in Evolutionary Bioinformatics