

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative disease of the lower and upper motor neurons with sporadic or hereditary occurrence. Age of onset, pattern of motor neuron degeneration and disease progression vary widely among individuals with ALS. Various cellular processes may drive ALS pathomechanisms, but a monogenic direct metabolic disturbance has not been causally linked to ALS. Here we show SPTLC1 variants that result in unrestrained sphingoid base synthesis cause a monogenic form of ALS. We identified four specific, dominantly acting SPTLC1 variants in seven families manifesting as childhood-onset ALS. These variants disrupt the normal homeostatic regulation of serine palmitoyltransferase (SPT) by ORMDL proteins, resulting in unregulated SPT activity and elevated levels of canonical SPT products. Notably, this is in contrast with SPTLC1 variants that shift SPT amino acid usage from serine to alanine, result in elevated levels of deoxysphingolipids and manifest with the alternate phenotype of hereditary sensory and autonomic neuropathy. We custom designed small interfering RNAs that selectively target the SPTLC1 ALS allele for degradation, leave the normal allele intact and normalize sphingolipid levels in vitro. The role of primary metabolic disturbances in ALS has been elusive; this study defines excess sphingolipid biosynthesis as a fundamental metabolic mechanism for motor neuron disease.
ALS is a progressive neurodegenerative disease with a variable age of onset, pattern of motor neuron degeneration and disease progression1,2. In addition to the more common sporadic ALS that typically manifests between 55 and 75 years of age, pathogenic variants with monogenic inheritance underlie a growing number of familial and early-onset ALS cases. Such precise monogenic causes can provide unique mechanistic and pathophysiological insights into the disease and guide rational therapeutic designs. While a variety of cellular processes has been implicated in ALS pathogenesis3–5, a monogenic metabolic form of ALS has not been identified to date.
Serine palmitoyltransferase is a multisubunit enzyme that catalyzes the initial and rate-limiting step in sphingolipid biosynthesis by condensing l-serine and activated acyl-CoA (most commonly palmitoyl-CoA) to form long-chain bases6,7 (Extended Data Fig. 1). SPT activity requires close homeostatic regulation to ensure adequate sphingolipid synthesis and to prevent cell toxicity due to excess sphingolipids8.
Alterations in SPT activity have been linked to neurodegeneration. While complete loss of SPT function is probably not compatible with life9, overproduction of C20-sphingoid base sphingolipids in a naturally occurring mutant mouse (stellar mouse) resulted in a severe neurodegenerative phenotype10. In addition, variants in genes that encode two subunits of SPT, SPTLC1 and SPTLC2, that alter SPT amino acid substrate usage underlie hereditary sensory and autonomic neuropathy type 1 (HSAN1)11 and macular telangiectasia type 2 (ref.12). These variants increase SPT usage of l-alanine or glycine rather than l-serine and result in deoxysphingolipid synthesis. Deoxysphingolipids, which lack a critical hydroxyl moiety, cannot be efficiently degraded by the cell machinery and cause toxicity13,14. Nevertheless, no human disease has been linked to SPT overactivity.
In this study, we characterize 11 patients from seven independent families with four dominantly acting, monoallelic, pathogenic variants in SPTLC1 manifesting as childhood-onset ALS. We investigate the functional consequences of these variants on sphingolipid biosynthesis and its homeostatic regulation, as distinct from HSAN1 SPTLC1 variants. We also provide proof of concept for a precision medicine approach by developing a rational siRNA-based treatment strategy based on the identified genetic mechanism.
Results
Childhood ALS and new dominantly acting SPTLC1 variants.
We identified six individuals from six unrelated families who developed early-childhood-onset lower extremity spasticity manifesting as toe walking and gait abnormalities followed by progressive lower motor neuron-mediated weakness without sensory symptoms or signs (Supplementary Table 1 and Supplementary Video 1). The disease was universally progressive and led to loss of independent ambulation and respiratory insufficiency of variable degrees. All six individuals had upper and lower motor neuron signs and symptoms in the cranial, cervical and lumbar myotomes and thus met the revised El Escorial criteria for the clinical definition of ALS15.
Due to the early onset of the disease, a monogenic form of ALS was suspected. Next-generation or exome sequencing identified four heterozygous variants (two recurrent) in SPTLC1 (NM_006415.4) (Fig. 1a). All variants were absent in healthy individuals in the Genome Aggregation Database (gnomAD)16, were predicted to be damaging based on in silico models and impacted highly conserved amino acids (Supplementary Table 2 and Fig. 1c). Consistent with the sporadic occurrence of the disease, SPTLC1 variants occurred de novo (that is, were absent in the unaffected parents; Fig. 1b).
Fig. 1 |. SPTLC1 variants in patients with childhood-onset ALS.
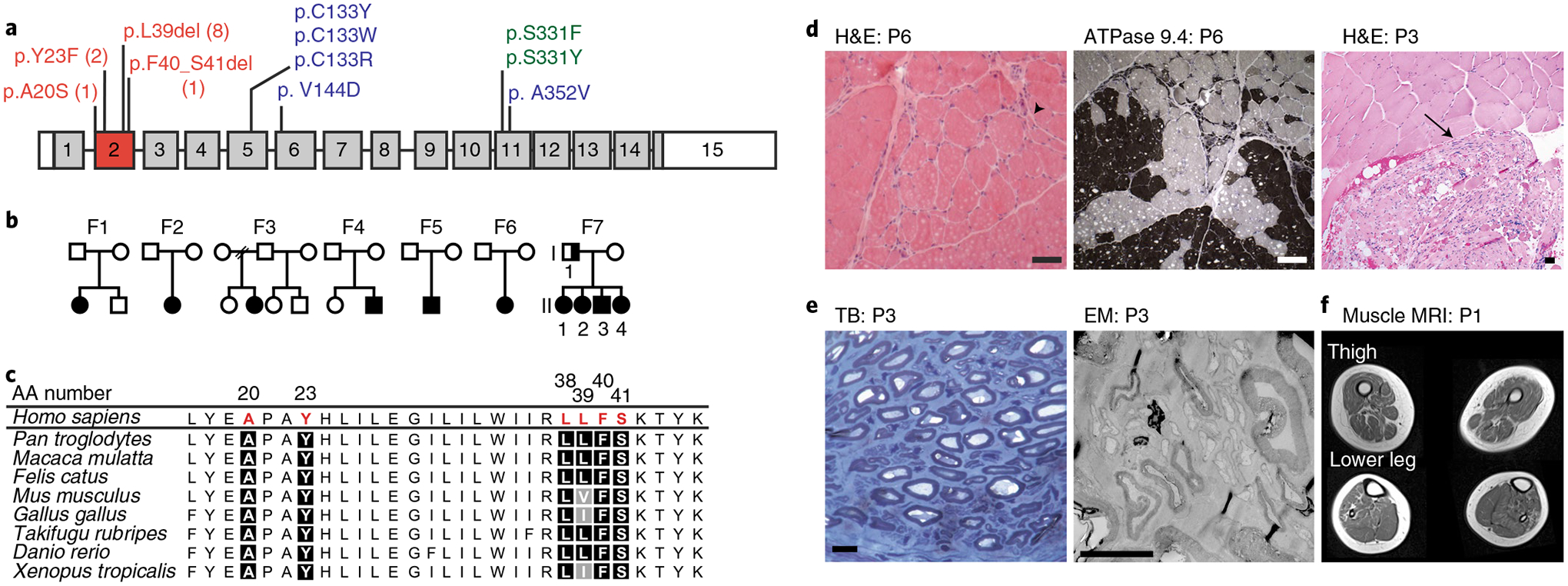
a, SPTLC1 gene with ALS variants (red) in exon 2, compared with classic (blue) and atypical (green) HSAN1 variants. The numbers for ALS variants indicate the number of individuals in this report with that variant. b, Pedigrees of families with ALS and SPTLC1 variants. Solid symbols indicate ALS/motor neuron disease; a half-solid symbol indicates sensorimotor neuropathy. Empty symbols indicate unaffected family members. F, family c, Evolutionary conservation of altered SPTLC1 amino acids associated with ALS. Due to the repeat sequence, distinction of the deletion of L38 versus L39 residues in humans is not possible. However, the presence of both residues in series is highly evolutionarily conserved. AA number, amino acid number of human SPTLC1 protein. d, Muscle biopsies showing angular atrophic fibers (arrowhead) and fiber type grouping (ATPase stain) highlighting acute and chronic neurogenic atrophy, respectively. Pyknotic nuclear clumps and grouped, fascicular atrophy (arrow) both also suggest chronic neurogenic atrophy. Scale bars, 50 μm; H&E, hematoxylin and eosin; P, patient. e, Biopsy of sural nerve (a sensory nerve). Semithin toluidine blue (TB) stain (left) and electron microscopy (EM) photograph (right) showing normal density and morphology of myelinated and unmyelinated axons. There are no degenerating axonal profiles or empty Remak Schwann cells. Scale bars, 5 μm. f, T1 axial MRI images of the lower extremities in patient 1 showing global skeletal muscle atrophy and patchy, heterogeneous T1 hyperintensity consistent with denervation-related changes in the muscle.
In an additional family (family 7) the SPTLC1 variant was inherited (Fig. 1b and Supplementary Table 1). All affected offspring initially presented with diffuse lower motor neuron disease consistent with a progressive muscular atrophy ALS phenotype2. At the time of the most recent examination, upper motor neuron findings were present in all but one affected offspring and were restricted to the lower extremities. None of the offspring had small or large fiber sensory abnormalities based on clinical examination or nerve conduction studies. In contrast, the father in the family had a mild sensorimotor axonal neuropathy. Irrespective of the variable manifestation between father and offspring, all patients in this family carried the same SPTLC1 variant (NM_006415.4:c.115_117delCTT, p.(L39del)), which was recurrent in two unrelated childhood ALS patients (patients 4 and 5; Supplementary Table 1).
Four patients had undergone a muscle biopsy, which demonstrated acute and chronic neurogenic changes (Fig. 1d) consistent with motor neuron disease. Patient 3 also had a sural nerve biopsy that failed to reveal any abnormalities in myelinated or unmyelinated sensory nerve fibers (Fig. 1e).
Imaging findings were also consistent with motor neuron disease. Skeletal muscle ultrasound in patients 1, 3 and 5 showed a patchy and striped pattern of increased echogenicity interposed with hypoechoic areas and diffuse fasciculations in the upper and lower extremities (Supplementary Video 2). Muscle magnetic resonance imaging (MRI) in patient 1 showed global muscle atrophy and areas of heterogenous infiltration with T1 hyperintensity (Fig. 1f), findings consistent with neurogenic changes in skeletal muscle.
One variant (SPTLC1 c.58 G > T) resulted in a single base-pair (bp) substitution adjacent to the splice acceptor site for exon 2 of SPTLC1, prompting us to evaluate its effect on messenger RNA splicing17. Rather than the predicted amino acid substitution p.(A20S), this variant predominantly resulted in complete skipping of exon 2, which is predicted to result in in-frame deletion of amino acids 20–56 comprising the entire first transmembrane domain of the SPTLC1 protein18. The other three ALS-associated SPTLC1 variants do not impact normal splicing (Extended Data Fig. 2) or subcellular localization of the protein (Extended Data Fig. 3a).
The ALS-associated SPTLC1 variants identified all map to the same exon and are distinct from HSAN1-associated variants (Fig. 1a), suggesting a different biochemical effect on SPT activity. Thus, we next evaluated the consequences of our newly identified SPTLC1 variants on SPT activity and substrate preference.
ALS-associated SPTLC1 variants and SPT activity.
Sera from patients with SPTLC1-related HSAN1 showed elevated levels of 1-deoxysphingolipids (and deoxymethylsphingolipids) due to SPT substrate shift from l-serine to l-alanine (and glycine) (Fig. 2a). We assayed sphingolipids in serum samples from patients with ALS-associated SPTLC1 variants and failed to detect an increase in deoxysphingolipid levels. Instead, free sphinganine and circulating ceramide levels were increased compared to controls (Fig. 2b). Similar to these findings, modeling of the variants in HEK293 SPTLC1 knockout cells showed increased de novo synthesis of 1-deoxyceramides in the classic HSAN1 variant p.(C133W), while canonical SPT product levels were increased in ALS variants (Fig. 2c).
Fig. 2 |. Elevated levels of the canonical sphingolipid products of SPT in SPTLC1-associated amyotrophic lateral sclerosis.
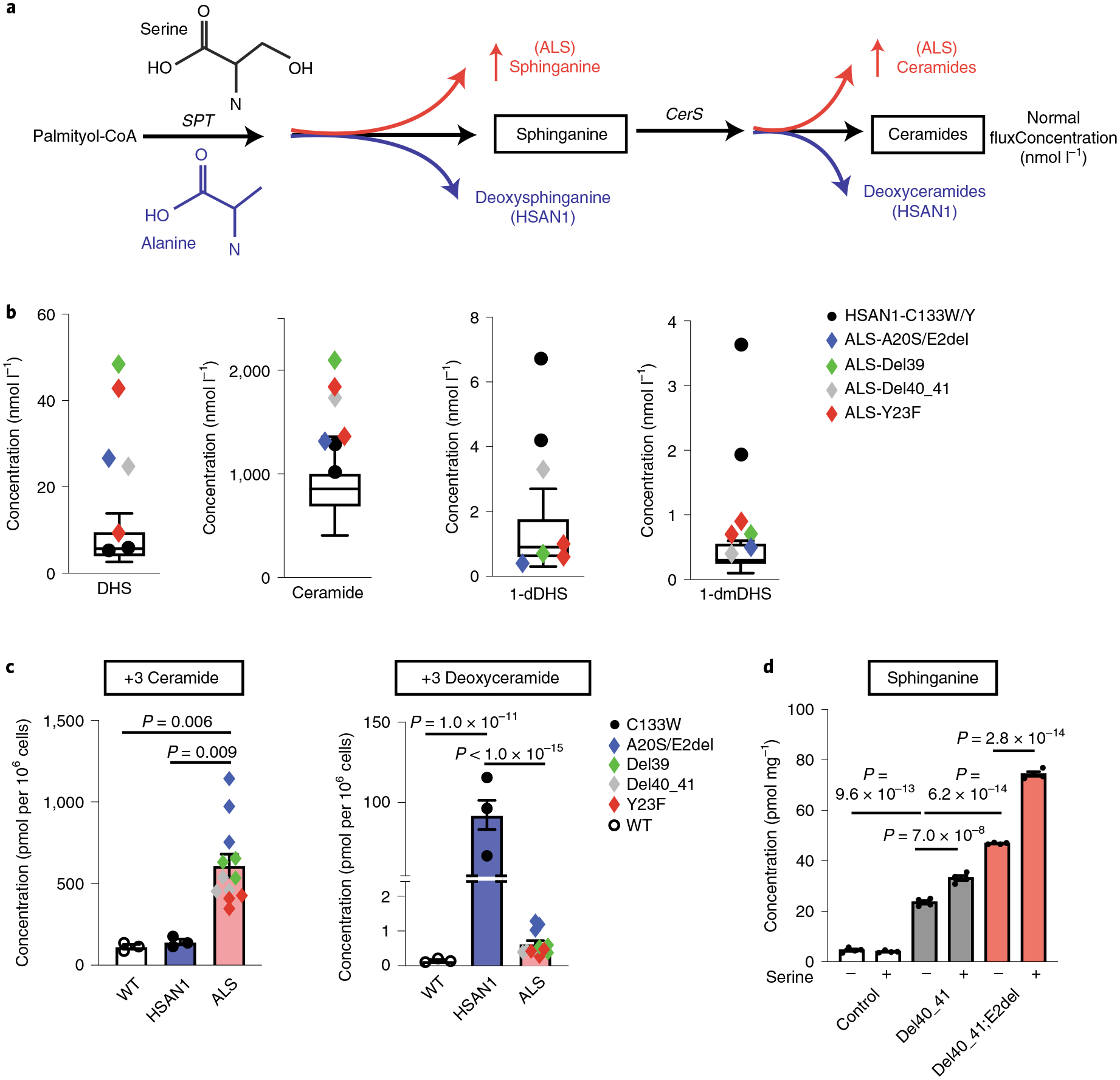
a, HSAN1-causing variants increase alanine usage by SPT leading to the formation of deoxysphingolipids (blue), while ALS-causing variants result in increased production of sphinganine and ceramides (red). Glycine (not depicted) usage by SPT similarly increases deoxymethylsphingolipids. CerS, ceramide synthases. b, Sphingolipidomic analysis of patient serum distinguishes HSAN1 (high 1-deoxydihydrosphingosine (dDHS) and 1-deoxymethyl DHS (dmDHS)) from patients with SPTLC1 ALS (high DHS, high ceramide). Thirteen control samples are represented in box-and-whisker plots as the median, first and third quartiles, and minimum and maximum. c, SPTLC1 knockout HEK293 cells were transfected with WT SPTLC1 and SPTLC1 with ALS- and HSAN1-associated variants. Sphingolipidomic analysis of de novo sphingolipid synthesis using isotope labeling (with D3, 15N-l-serine and D4-l-alanine resulting in +3 sphingolipids) shows a biochemical profile that distinguishes HSAN1 (high 1-deoxyceramides) and ALS variants (high ceramides). Three replicates for each variant were performed. d, Addition of exogenous serine (3 mM) increases canonical sphingolipid synthesis in SPTLC1 mutant iPSC-hMNs but not in controls. Error bars denote s.e.m. Four mass spectrometry measurements for each variant and condition were performed. Ordinary one-way ANOVA with Tukey’s adjustment for multiple comparisons was used for statistical comparisons.
To model the SPTLC1-associated ALS variants in a more disease-relevant cell type, we used a recently described induced pluripotent stem cell (iPSC) line that can be efficiently and uniformly differentiated into motor neuron-like cells with doxycycline supplementation19. Using CRISPR, we created one iPSC line heterozygous for p.(F40_S41del), akin to patient 3 and another line compound heterozygous for p.(F40_S41del) and a deletion of exon 2 (the consequence of the c.58 G > T variant), which would be predicted to further exacerbate the biochemical phenotype. After differentiating these into human lower motor neuron-like cells (iPSC-hMN) (Extended Data Figs. 3b,c and 4a,b), we evaluated their sphingolipid profile. ALS variant-containing iPSC-hMNs showed an increase in de novo synthesized sphingolipids compared to the wild-type (WT) isogenic control (Extended Data Fig. 4c). Sphingolipid levels were increased further in the compound heterozygous iPSC-hMN. Thus, ALS-associated SPTLC1 variants result in unrestrained SPT activity and distinctly differ from HSAN1 variants in their biochemical effects.
Serine supplementation and ALS-associated SPTLC1 variants.
Serine supplementation is a current therapeutic strategy in HSAN1 utilized to shift the equilibrium of SPT substrate use to the canonical l-serine from l-alanine and glycine20,21. This approach, however, is predicted to have no effect and possibly exacerbate the overproduction of sphingolipids in the case of an unrestrained SPT complex. Indeed, excess serine supplementation further increased the already elevated sphingolipid levels in mutant SPTLC1 iPSC-hMNs (Fig. 2d). This finding indicates that l-serine supplementation as a therapeutic strategy does not improve, and may worsen, the biochemical phenotype in patients with SPTLC1-related ALS and thus should be avoided.
Loss of inhibitory effects of ORMDLs on mutant SPT.
Similar to many enzymes, SPT activity is regulated by its downstream products. Homeostatic inhibition of SPT is readily observed by the addition of exogenous sphingosine22 or ceramides23 and requires ceramide synthase activity23 and the presence of ORMDL proteins23–25 (Fig. 3a). Recent studies have identified a specific role for the first transmembrane domain of SPTLC1 in the homeostatic regulation of SPT mediated by ORMDLs26. Thus, clustering of ALS-associated SPTLC1 variants in the first transmembrane domain of the protein (Fig. 3b) prompted closer evaluation of their effect on ORMDL-mediated regulation of SPT. Among ORMDL proteins we focused on ORMDL3, since its deficiency results in increased sphingolipid levels in the mouse central nervous system more than its analogs (Ormdl1 and Ormdl2)27. Consistently, ALS SPTLC1 variant-containing SPT did not respond to increasing levels of ORMDL3 or exogenously added ceramide as efficiently as WT SPTLC1-containing SPT (Fig. 3c,d and Supplementary Table 3). This suggests that ALS SPTLC1 variants diminish the homeostatic inhibitory effect of ceramides and ORMDLs on the SPT complex. This conclusion is strongly supported by the recently solved cryoelectron microscopy structure of the human SPT/ORMDL3 complex, which reveals that the first transmembrane domain of SPTLC1, where ALS variants cluster, makes extensive contact with the third membrane-spanning domain of ORMDL3 (refs.28,29) (Fig. 3b).
Fig. 3 |. Homeostatic regulation of SPT mediated by ORMDL proteins.
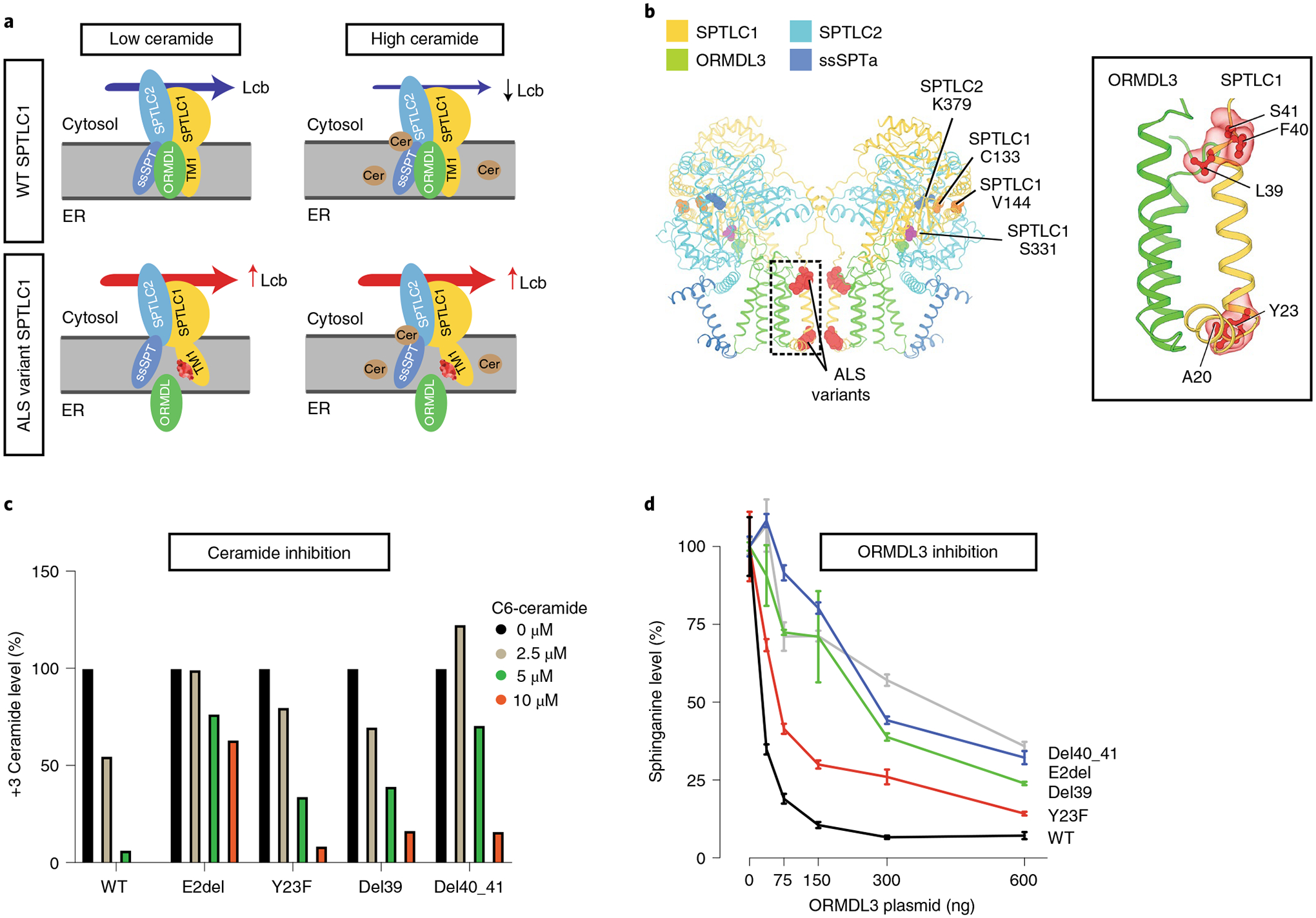
a, The SPT multisubunit complex includes SPTLC1, SPTLC2 and a small subunit (ssSPT). ORMDL proteins bind the first transmembrane domain of SPTLC1 (TM1) and inhibit SPT activity in the presence of high ceramide (Cer) levels. SPTLC1 with ALS variants in TM1 does not bind ORMDLs efficiently, and high ceramide levels are less effective in inhibition of mutant SPT activity. Lcb, long-chain base; ER, endoplasmic reticulum. b, Cryoelectron microscopy structure of the SPT/ORMDL3/ssSPTa complex. Hereditary sensory neuropathy SPTLC1 residues (for example, C133 and V144) are in the cytosolic portion of SPT, near its active site and the pyridoxal phosphate-binding residue of SPTLC2 (K379). In contrast, ALS-associated variants are near the ORMDL3-interacting transmembrane domain (inset). c, HEK293 SPTLC1 knockout cells transfected with WT or ALS-associated SPTLC1 variants. De novo synthesized ceramides (labeled with D3-15N-l-serine and D4-l-alanine) were measured in the presence of increasing, exogenously added C6-ceramide. Bar graphs show normalized +3 ceramides for each cell line. d, WT HEK293 cells were cotransfected with SPTLC2, ssSPTa, either WT or ALS variant SPTLC1 and increasing amounts of ORMDL3 plasmid. SPT activity was evaluated by measurement of sphinganine levels normalized to baseline for each cell line. Increasing ORMDL3 has a more robust inhibitory effect on sphinganine synthesis (that is, SPT activity) in WT versus ALS-associated SPTLC1 variants. Error bars are s.e.m. from five independent replicates.
Genetic and metabolic rescue by allele-specific knockdown.
In mouse models, heterozygous null Sptlc1 mutants do not have a detectable phenotype9. Human SPTLC1 is also highly tolerant of heterozygous loss-of-function variants predicted to result in haploinsufficiency (gnomAD-observed/expected number of loss-of-function variants = 0.98)16. This suggests that a 50% reduction in total SPTLC1 transcript is still sufficient to meet metabolic demands provided that SPT can be appropriately regulated. Thus we designed and validated siRNAs that specifically target two ALS-associated SPTLC1 variants without affecting the WT allele. This approach successfully reduced mutant mRNA levels (Fig. 4a) and maintained little activity against the WT allele at siRNA concentrations nearly 100-fold higher than those maximally effective against the mutant allele (~1 nM) (Extended Data Fig. 5a). In addition, allele-specific knockdown of variant SPTLC1 restored the de novo sphingolipid synthesis rate to near normal levels, rescuing the biochemical phenotype in vitro (Fig. 4b–d and Extended Data Fig. 5).
Fig. 4 |. Allele-specific knockdown of transcripts containing SPTLC1 variants associated with ALS.
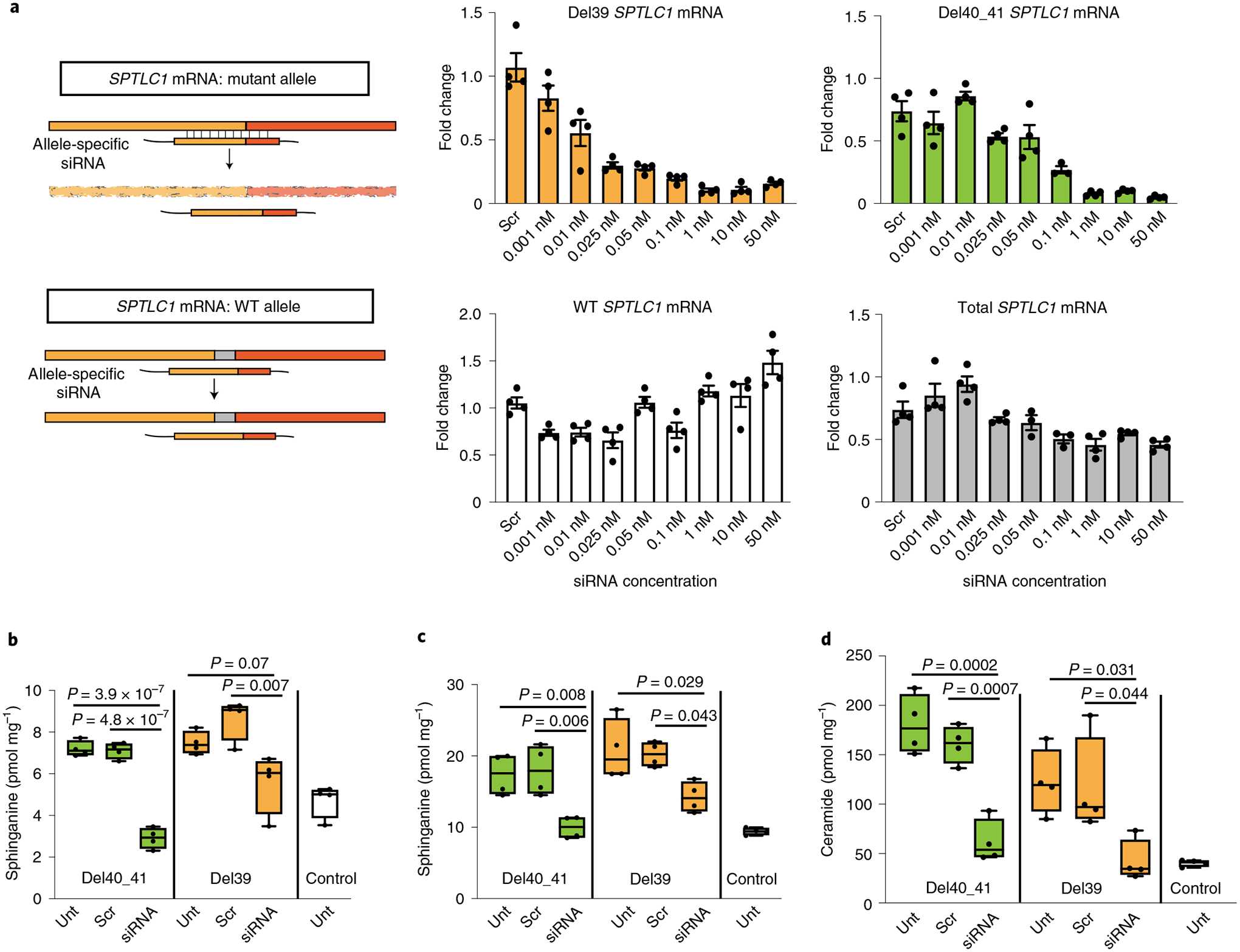
a, Allele-specific knockdown using siRNA. Precision-designed, mutation-specific siRNAs target the variant allele mRNA while not interfering with the WT transcript. Bar graphs show real-time reverse transcriptase PCR results from patient-derived fibroblasts treated with varying concentrations of allele-specific siRNAs. All values are reported as linear fold change compared to untreated cells in the same experiment. The levels of mutant allele transcripts L39del (yellow) and F40_S41del (green) show a dose-dependent decrease with increasing siRNA concentration. The levels of WT allele transcript in L39del cells (white) show no notable change, indicating allele specificity. In F40_S41del cells (gray), the level of total transcript including both mutant and WT alleles was measured and did not decrease by more than the expected 50% of total, akin to haploinsufficiency. Error bars are s.e.m. from four replicates. b–d, Corrective effect of allele-specific siRNA treatment on de novo synthesis of sphinganine (b), sphingosine (c) and ceramide (d) levels in patient-derived fibroblasts. Cells were treated with siRNA and labeled with 3,3-D2 l-serine (3 mM) 24 h before analysis. Data represented by box-and-whisker plots are the median, first and third quartiles, and minimum and maximum from four replicates. Ordinary one-way ANOVA with Tukey’s adjustment for multiple comparisons was used for statistical comparisons. Unt, untreated; scr, scrambled.
Discussion
Here we define monoallelic, dominantly acting SPTLC1 variants that cause a form of motor neuron disease that is best characterized as childhood ALS. ALS is a heterogeneous, neurodegenerative syndrome that invariably results in motor neuron degeneration and clinically manifests with upper and lower motor neuron signs and symptoms. The majority of patients in our cohort first manifested with early-childhood-onset spasticity and toe walking, a clinical feature suggestive of a related upper motor neuron disorder, hereditary spastic paraplegia (HSP), in which spasticity typically is the leading feature throughout the disease course. However, in the patients reported here, this initial manifestation was almost universally followed by diffuse and progressive lower motor neuron degeneration, including tongue fasciculations, which is highly unusual for classic forms of HSP but consistent with ALS. Complicated forms of HSP, in addition to lower extremity spasticity, may manifest with a broad range of neurodegenerative and non-neurologic phenotypic features; however, even in this group, diffuse motor neuron disease in the absence of broader neurodegeneration and extraneurologic organ system involvement is rare30. Thus, we propose classification of our SPTLC1-associated disease entity as childhood-onset ALS. Despite its childhood onset and resulting severe disability, the rate of progression of SPTLC1-associated ALS is still slower than sporadic ALS and thus mirrors other juvenile-onset hereditary ALS cases31.
The specific pathogenicity of the four ALS-associated SPTLC1 variants is strongly supported by genetic and functional data. The variants occurred de novo in six of seven families. Pathogenic de novo variants are increasingly recognized as being responsible for apparent sporadic neurodevelopmental and neurodegenerative diseases32. In addition, a distinct biochemical profile defines this class of SPTLC1 variants, which was universally confirmed in all four variants: enhanced SPT activity due to loss of ORMDL-mediated homeostatic inhibition, resulting in increased sphingolipid levels without change in their substrate specificity. Consistent with this, the variants cluster in the transmembrane domain of SPTLC1 critical for its interaction with ORMDLs. Lastly, allele-specific knockdown of two of the variants rescued the abnormal biochemical signature (Fig. 4b–d and Extended Data Fig. 5). Taken together, these data support the probable pathogenic classification of these variants (Supplementary Table 2).
We encountered a divergence from the highly consistent ALS phenotype only in the father of the one familial occurrence in our series (family 7). All of the affected offspring in family 7 carried the recurrent p.(L39del) SPTLC1 variant (also seen in families 4 and 5) and manifested with a phenotype that can formally be classified as ‘possible ALS’ or ‘suspected ALS’ based on the El Escorial criteria15. Similar phenotypic variations have also been reported in other familial forms of ALS33,34. However, the father in this family, who carries the same SPTLC1 variant, has a sensorimotor neuropathy. We do not know the underlying mechanism of this surprising phenotypic divergence in the father. Potential modifiers of the phenotype in the father in this family include variants in other enzymes and transporters in the sphingolipid synthesis pathway, serine biosynthesis and catabolic pathways and the relative abundance of SPT amino acid substrates (that is, alanine and serine), which is currently under investigation.
A striking dichotomy of biochemical consequences of SPTLC1 variants parallels the clinically distinct phenotypes of ALS and HSAN1. Classic HSAN1 typically manifests with progressive sensory loss associated with elevated serum deoxysphingolipid levels. In contrast, the patients with ALS reported here presented with childhood-onset motor neuron disease, normal sensory findings (by clinical examination, electrophysiologic studies and nerve biopsy when available) and normal deoxysphingolipid levels. Notably, distinct SPTLC1 variants (for example, p.S331F and p.S331Y) have been reported to manifest with a mixed sensorimotor neuropathy35–37. Interestingly, biochemical studies of these variants show not only elevated deoxysphingolipid production but also increased SPT activity and altered canonical sphingoid base production38–40. Thus, the biochemical consequences of SPT variants determine its associated phenotypes with an apparent, though not mutually exclusive, dichotomy: elevated deoxysphingolipids cause sensory neuropathy while SPT overactivity causes motor neuron disease (Fig. 2a). We do not yet understand why motor and sensory neurons show selective vulnerability to distinct biochemical changes in sphingolipid synthesis and homeostasis.
Overactivity of SPT and excess sphingolipid synthesis has important implications for sporadic ALS. Studies have identified altered cerebrospinal fluid lipidomic signature in patients with sporadic ALS41,42 and have correlated complex glycosphingolipids and sphingolipid metabolism with disease severity43,44. Together with our findings, these raise the possibility of a pathogenic role for disrupted sphingolipid metabolism in sporadic ALS and posit sphingolipids as potential disease biomarkers for ALS. How excess sphingolipid synthesis relates to other known cellular disturbances in ALS remains to be investigated. In addition, systematic evaluation of large genomic data repositories of sporadic and familial ALS and related motor neuron disorders, such as HSP, may be helpful in identifying disease-causing or -modifying variants in SPT complex-associated genes.
Inspired by recent advances in transcript-directed therapies, we show the feasibility of a genetic precision therapy approach using allele-specific siRNAs that selectively target the mutant SPTLC1 allele and rescue the biochemical phenotype associated with unrestrained SPT overactivity. Delivery of siRNAs to target cells—for example, motor neurons or glia, probably requires additional modifications45 but, for a severe and progressive neurodegenerative disease in children, such an approach as part of a small clinical trial may be justified46. In addition, other therapeutic approaches that aim to reduce l-serine availability by reducing its de novo synthesis (for example, inhibitors of phosphoglycerate dehydrogenase47) or direct inhibitors of SPT (for example, l-cycloserine48) may also hold promise for future interventional studies.
In summary, we report SPTLC1 as a causative gene for ALS and propose a new metabolic mechanism for motor neuron disease due to unrestrained sphingoid base synthesis. We expand the disease associations for SPTLC1 by establishing a striking dichotomy of biochemical and clinical consequences of distinct variant classes within the gene. Our allele-specific knockdown provides another example for the conceptual groundwork that spans from gene discovery to a mutation-specific therapeutic approach in small clinical trials for patients with a rare disease.
Methods
Patient recruitment, clinical evaluations and sample collection.
Patients were evaluated at different centers by neuromuscular specialists and medical geneticists as per the standards of clinical diagnosis and clinical care. A total of 11 patients (six female, five male), 8–62 years of age were evaluated. The patients were referred by their treating physicians to observational and biorepository research studies at different institutions. For the research studies (for example, skin biopsy for fibroblast cultures, research-based genetic testing, serum sample collection, muscle MRI images) we obtained written informed consent and age-appropriate assent from participants. Ethical approval for human subject research studies described in this paper was obtained from the following relevant institutional review boards: NIH/NINDS (protocol no. 12-N-0095), Washington University (protocol no. 201308083), McMaster University (protocol no. REB 14–595-T) and University of Massachusetts (protocol no. 13788_10). Although the Awaji criteria are more sensitive49, we used the more specific revised El Escorial criteria to arrive at an ALS diagnosis15.
Genetic testing.
The details of clinical and research genetic testing performed in each family with SPTLC1-associated amyotrophic lateral sclerosis are listed in Supplementary Table 4. Whole-exome sequencing or next-generation panel testing was performed in all cases. Confirmation of variants and segregation testing was performed by Sanger sequencing in all families. The SPTLC1 variants reported in this cohort are not found in the gnomAD, Decipher, Leiden or TopMed databases. An entry submitted to the GeneMatcher exchange identified a match with a patient with childhood-onset ALS-like disease but, unfortunately, a collaborative relationship did not form.
Plasmids.
Plasmids containing human SPTLC1 and SPTLC2 complementary DNAs under the control of the CMV promoter were purchased from OriGene Technologies, Inc. Variants in human SPTLC1 (corresponding to exon 2 deletion, Y23F, del39 or del40–41 amino acids) were made by QuikChange mutagenesis (Agilent Technologies) and verified by sequencing. Plasmids expressing NubG-HA-tagged ssSPTa and NubG-HA-tagged human ORMDL3 were described previously50,51.
Cell culture and transfection and heavy serine labeling.
HEK293 cells (American Type Culture Collection) were cultured in DMEM with high glucose, pyruvate and GlutaMAX (Gibco) supplemented with 10% fetal bovine serum (FBS), 100 units ml−1 penicillin and 100 μg ml−1 streptomycin. Transfections using Lipofectamine 2000 (Thermo Fisher) were performed according to the manufacturer’s instructions. Cells (6.5 × 105) were plated in six-well plates and, in all cases, transfected with an equivalent amount of SPT subunits (600 ng each of SPTLC1, SPTLC2 and ssSPTA). ORMDL3 plasmid concentration was varied from 0 to 600 ng (as indicated) and total transfected plasmid concentration was maintained using pcDNA plasmid. Cells were collected 24 h after changing the medium following transfection. In cases where deuterium-labeled serine (3,3-D2 l-serine, DLM-161, Cambridge Isotope Laboratories) was used, it was added at 3 mM for 24 h before harvesting. Cells were scraped, washed with PBS and spun. The cell pellet was suspended in PBS and divided into either glass tubes for lipid extraction or Eppendorf tubes for protein quantification.
Dermal fibroblast cultures, RNA extraction, real-time PCR and siRNA transfection.
Fibroblasts were derived and expanded from skin biopsies of patients as previously described52. Briefly, cells were expanded in high-glucose DMEM (Sigma) supplemented with 10% FBS and penicillin/streptomycin. QIAzol Lysis Reagent (Qiagen, no. 79306) was used for cell lysis and chloroform (1:5 v/v QIAzol) was added and mixed. The mixture was spun down at 12,000g for 20 min at 4 °C, the aqueous phase placed in a new tube and isopropanol (1:2 v/v QIAzole) was added to precipitate RNA. The pellet was washed with 75% EtOH solution and allowed to dry in air before resuspension in water. Quantification of RNA concentration was performed using a Nanodrop 2000 Spectrophotometer. Reverse transcription was performed using qScript XLT cDNA Supermix (Quantabio, no. 95161) according to the manufacturer’s protocol, with 500 ng of RNA.
Real-time PCR was performed using FastStart Universal Master-mix (Roche) with primers and probes specified in the section below, using 10-μl reactions in quadruplicate using a QuantStudio 6 Flex machine (Thermo Fisher). PCR parameters consisted of a primary heating stage at 50 °C for 2 min followed by 95 °C for 10 min, after which 40 cycles of 95 °C for 15 s and 60 °C for 1 min were performed. Linear fold change values were calculated using the ΔΔcycle threshold (ΔΔCt) method with PGK1 as an internal control, averaged from the replicates and normalized to untreated cells from each cell line. For the del40_41 allele, we were able to design and validate mutant-specific primers but were unable to obtain specific primers that amplified only the WT allele. Thus we compared the results using mutant-specific primers and total SPTLC1 primers to evaluate allele specificity, with a target reduction in total SPTLC1 transcript of 50%. All primers are listed in High-performance liquid chromatography and mass spectrometry analysis.
For mass spectrometry and lipidomics experiments in fibroblasts, siRNA transfection was performed using 0.5 μl cm−2 Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher, no. 13778030) in Opti-MEM (Thermo Fisher, no. 31985–070). The siRNA concentration was 10 nM for 72 h unless otherwise specified. Fibroblasts were labeled with deuterium-labeled serine (3,3-D2 l-serine, DLM-161, Cambridge Isotope Laboratories) (Fig. 4b–d) at 3 mM for 24 h before harvesting, or with isotope-labeled D3-15N-l-serine (1 mM) and D4-l-alanine (2 mM) (Cambridge Isotope Laboratories) for 16 h before harvest (Extended Data Fig. 5).
iPSC culture.
The control iPSC line WTC11 used for all experiments53 was previously engineered with an inducible motor neuron transcription factor construct19. iPSCs were cultured on Matrigel-coated (Corning, no. 354277) polystyrene tissue culture plates in E8 flex medium (Thermo Fisher, no. A2858501). Medium change was performed every 1–3 days, and cells were dissociated with Accutase (Invitrogen, no. A1110501) and passaged every 4–6 days. Passaged cells were supplemented for 1–2 days with 10 μM ROCK inhibitor (Tocris, no. 1254) to facilitate survival.
Generation of HEK293 SPTLC1 knockout cells and complementary assays.
HEK293 cells were cultured in DMEM (Sigma-Aldrich)) with 10% fetal calf serum. Cells were grown at 37 °C in a 5% CO2 atmosphere. HEK293 SPTLC1 knockout cells were generated using CRISPR–Cas9 technology. HEK293 cells were transfected with an all-in-one vector cassette, pLenti-sgRNA-Cas9-GFP (Sigma-Aldrich). Single colonies were identified and propagated for identification of deletion clones in 24 wells. Frameshift mutations introducing a premature stop were confirmed by sequencing and assessed by immunoblot.
Standard molecular biology techniques were used for the generation of V5-epitope-tagged missense ALS mutations in a pcDNA3.1-SPTLC1 backbone39. Transgenic cells expressing the vector, WT SPTLC1 or mutants were selected for growth in DMEM medium containing 400 μg ml−1 Geneticin (Thermo Fisher). Expression of WT and mutant SPTLC1-V5 proteins in knockout cells was confirmed by immunoblotting.
The l-serine labeling assay and SPT activity measurements were performed as previously described54. Cells were grown to 70–80% confluence in DMEM growth medium. The medium was changed to l-serine-free DMEM (Genaxxon Bioscience) containing 10% FBS and 1% penicillin/streptomycin supplemented with isotope-labeled D3-15N-l-serine (1 mM) and D4-l-alanine (2 mM) (Cambridge Isotope Laboratories), and cells were grown for a further 16 h (Fig. 2c). C6-ceramide supplementation was done in the labeling medium for the duration of the assay (Fig. 3c).
CRISPR–Cas9 editing of SPTLC1 in iPSCs.
To derive the c.40_41del patient mutation knock-in lines, WTC11 iPSCs were seeded in a six-well plate at 600,000 cells per well in E8 medium with RevitaCell supplement (Invitrogen). Thirty minutes later, iPSCs were transfected by a mixture of 200 μl of Opti-MEM (Invitrogen), 4 μl of Lipofectamine Stem Reagent (Invitrogen), 20 pmol of single-guide RNA (Synthego) targeting exon 2 of SPTLC1 (5’-AAAGAAGUCUGAUUAUCCAG-3’) and 1,800 ng of eSpCas9-GFP mRNA. A WY and a c.40_41del mutant ssODN (IDT) were included in the transfection as repair templates, and these contained a silent ATC-to-ATA mutation (underlined) that blocks the Cas9 PAM site to prevent recutting:
c.40_41del mutant ssODN: AGGTGTTAGAAGTGTATGTTTCATATCTTACT TGTTTTTGACTCACTCTTAGGCTCCTGCTTACCATCTTATTTTGGAAGGG ATTCTGATACTCTGGATAATCAGACTTCTTAAGACTTACAAATTACAAGAA CGATCTGATCTTACA.
GFP+ cells were individually sorted by fluorescent activated cell sorter into Matrigel-coated 96-well plates with 100 μl of E8 Flex and CloneR supplement (STEMCELL Technologies). Surviving clones were expanded after 10 days and genomic DNA was harvested. Clones were screened by PCR and restriction digest to evaluate for the AflII site created by the c.40_41del variant. We identified one clone, Del40_41;E2del, which contained the intended c.40_41del mutation on one allele and a 71-bp deletion on the other, resulting in the in-frame deletion of exon 2. This clone showed no genomic abnormalities by g-banded karyotype analysis (WiCell) and was used in experiments where mentioned. Both Del40_41 and E2del mutations were sequence confirmed in cDNA obtained from the iPSCs and after differentiation to human motor neurons. The other clones obtained were lacking the c.40_41del mutation and harbored a mixture of WT and insertion/deletion (indel) mutations. To generate a monoallelic c.del40_41 knock-in, we selected two clones that retained one WT allele and had an indel on the other allele, allowing allele targeting with new indel-specific CRISPR–Cas9 gRNAs (5’-CUUAGAGAAAAGACUUACAG-3’ or 5’-GAGAAAAGAAGUCUGAUUAG-3’) and the c.40_41del mutant ssODN. Subclones were screened with AflII digestion, sequence verified to confirm the presence of the c.40_41del mutant on one allele and analyzed for karyotypic abnormalities to yield the final Del40_41 line. Genotyping and sequencing of genomic DNA primers are listed in Supplementary Table 5.
Spinal motor neuron differentiation from iPSCs.
iPSCs were differentiated into spinal motor neurons, essentially as previously described19. The WTC11 iPSC line used here was previously modified to harbor a doxycycline-inducible construct containing the motor neuron transcription factor combination of NGN2, ISL1 and LHX3. Briefly, cells were plated in neural differentiation medium (NDM) at 20,000 cells cm−2 on Matrigel-coated dishes with 10 μM ROCK inhibitor. NDM consisted of 1:1 DMEM/F12 and Neurobasal, 1× B27 and 1× N2 (all Invitrogen) and 1× GlutaMAX (Gibco). For the first 8 days, NDM was supplemented with 1 μM retinoic acid (Sigma) and 2 μg ml−1 doxycycline (Clontech). On day 2, cells were replated onto polyethylenimine/laminin-coated dishes at 50,000 cells cm−2. From day 8, NDM was supplemented with 10 ng ml–1 BDNF and NT-3 (Fisher). Full medium changes were performed every other day until day 8, after which half the medium was changed twice weekly. For mass spectrometry experiments, differentiated iPSCs were labeled with deuterium-labeled serine (3,3-D2 l-serine, DLM-161, Cambridge Isotope Laboratories) at 3 mM concentration 24 h before harvesting (Extended Data Fig. 4c).
Immunocytochemistry and confocal microscopy.
Cells were washed in Dulbecco’s PBS (DPBS) then incubated in 4% paraformaldehyde for 10 min at room temperature. Fixation was quenched using 0.1 M glycine solution for 5 min, followed by two washes with DPBS. Cells were permeabilized in 0.25% Triton X-100 for 5 min and blocked in 10% goat serum for 1 h at room temperature. Primary antibodies in 3% goat serum were then applied overnight at 4 °C and washed three times before incubation with secondary antibody in 3% goat serum for 1 h at room temperature. Cells were washed, stained with DAPI and mounted with Fluoromount (Thermo Fisher). Images were taken using a Leica TSC SP5 II confocal microscope.
Immunoblotting.
Cell lysate was prepared by lysing cells in 1× (150 mM NaCl, 25 mM Tris-HCl pH 7.5, 1 mM EDTA, 5% glycerol and 1% NP-40) with protease inhibitor cocktail (cOmplete protease inhibitor, Sigma-Aldrich). Protein quantification was performed using the BCA assay (Thermo Fisher). Equal amounts of protein were boiled in 1× LDS Sample Buffer (Thermo Fisher) and 0.1 mM DTT. Samples were resolved by electrophoresis on a NuPAGE Novex 4–12% Bis-Tris Gel (Invitrogen) and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% milk in TBS for 1 h before primary antibody incubation overnight. The membrane was then washed and probed with secondary antibody. Bands were visualized using the Odyssey system (LI-COR) and Image Studio (LI-COR).
High-performance liquid chromatography and mass spectrometry analysis.
Pelleted cell or serum (0.1–0.2 mg of cell lysate or 20 μl of serum) was mixed with 1 ml of methanol containing 62.5 pmol of internal standards (Avanti Polar Lipids, no. LM6002), bath sonicated for several minutes and 0.5 ml of chloroform added. The mixture was incubated overnight at 48 °C. After a brief spin to pellet and remove insoluble material, the sample was dried under nitrogen. The dried sample was dissolved in 0.3 ml of mobile phase A and B at 80:20 (v/v), bath sonicated for dispersal, centrifuged to clarify and transferred to a high-performance liquid chromatography (HPLC) and mass spectrometry analysis vial for analysis. Typically, 5–20 μl of each sample was injected. The HPLC mobile-phase solvents used were (A) CH3OH:H2O:CH2O2 (74:25:1, v/v/v with 10 mM ammonium formate) and (B) CH3OH:CH2O2 (99:1, v/v with 10 mM ammonium formate). Samples were analyzed using an Agilent 1200 Series HPLC coupled to an ABSciex QTRAP 4000 MS. The column (150 mm × 4.6 mm × 5 μm Supelco Discovery BIO Wide Pore C18 HPLC Column) was pre-equilibrated with A/B (80:20 v/v) and held for 2 min after injection at 1 ml min−1. Flow rate was increased to 100% B at 7 min, held for 20 min and quickly returned to 80:20 and held for 2 min before the next injection. The mass spectrometer was set to detect compounds in multiple reaction monitoring (MRM) mode in a dual-period run, with long-chain bases detected in each run (3–9.2 min) and each class of sphingolipid (Cer, GluCer, LacCer, SM) detected in separate runs (9.2–30 min). MRM parameters are described previously55. Compounds were quantified based on the ratio of the peak to the known concentration of the representative internal standard using the ABSciex Analyst program. Each sample was normalized to protein concentration (or volume for serum). Serum samples (Fig. 2b) and cell pellets from iPSC-derived human motor neurons (Fig. 2d and Extended data Fig. 4c) and siRNA-treated fibroblasts (D2-serine supplemented; Fig. 4) were deidentified, and mass spectrometry and quantification were performed blinded (laboratory of T.M.D). For sphingolipid species at very low levels (for example, free deoxy long-chain bases) in control samples, automated peaks and quantification were inspected for accuracy and samples were remeasured if quantification was deemed unreliable.
Untargeted sphingolipidomic analysis of D3-15N-l-serine- and D4-alanine-labeled lipids was carried out essentially as previously described54 (laboratory of T.H). Frozen cell pellets were resuspended in 50 μl of PBS and extracted with 1 ml of methanol/methyl-tert-butyl ether/chloroform (MMC) (4:3:3, v/v/v) containing D7SA (d18:0), D7SO (d18:1), dhCer (d18:0/12:0), ceramide (d18:1/12:0), glucosylceramide (d18:1/8:0), SM (d18:1/18:1(D9)) and D7-S1P. Lipid extraction was performed with continuous shaking of samples in a Thermomixer (Eppendorf) at 37 °C (once, 400 r.p.m., 30 min). The single-phase supernatant was collected, dried under N2 and dissolved in 70 μl of methanol. Untargeted lipid analysis was performed on a high-resolution Q-Exactive MS analyzer (Thermo Fisher).
Antibodies, primers and siRNA sequences.
The antibodies, primers and siRNA sequences used are listed below.
Antibodies for immunocytochemistry (dilution):
Anti-HB9 antibody, Developmental Studies Hybridoma Bank, catalog no. 81.5C10 (1:10)
Anti-Choline Acetyltransferase, Aves Labs, catalog no. 6727986 (1:1,000)
Beta-III-Tubulin (Tuj1), Covance, catalog no. MMS-435P (1:5,000)
SPTLC1, BD Biosciences, catalog no. 611304 (1:1,000)
TDP-43, Proteintech, catalog no. 12892–1-AP (1:1,000)
Antibodies for immunoblotting (dilution):
GAPDH, Millipore, catalog no. MAB374 (1:10,000)
SPTLC1, BD Biosciences, catalog no. 611304 (1:5,000)
Primer, probe and siRNA sequences used in this paper are listed in Supplementary Tables 5 and 6.
Statistics and reproducibility.
Ordinary one-way analysis of variance (ANOVA) with Tukey’s adjustment for multiple comparisons was used to compare the effect of different SPTLC1 variants (Fig. 2c), sphinganine levels in iPSC-hMNs with serine supplementation (Fig. 2d) and siRNA transfection in fibroblasts on long-chain bases and sphingolipid de novo synthesis (Fig. 4b–d). Homoscedasticity in these datasets was confirmed by Bartlett and Brown–Forsythe tests. Data were not transformed before statistical analysis. The inhibitory effects of increasing ORMDL3 on SPT activity were modeled using a normalized response (that is, sphinganine concentration normalized to baseline as a percentage) versus ORMDL3 plasmid amount. Half-maximal inhibitory concentration values and corresponding 95% confidence intervals were calculated from the nonlinear least squares fit model (Table 3).
Representative micrographs.
The micrographs shown in Fig. 1d,e are representative images of a single diagnostic muscle biopsy and nerve biopsy specimen in each indicated patient. No independent replicates were performed. The PCR and agarose gel electrophoresis images shown in Extended Data Fig. 2a were independently replicated three times, with similar results. The micrographs in Extended Data Fig. 3a,b are from a single plating and immunostaining experiment for each line. The micrographs in Extended Data Fig. 4a are representative of two independent differentiation and immunostaining experiments. Representative electron microscopy images were obtained from a single plating and differentiation experiment into four replicate wells per iPSC line.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Sphingolipid biosynthesis pathway.
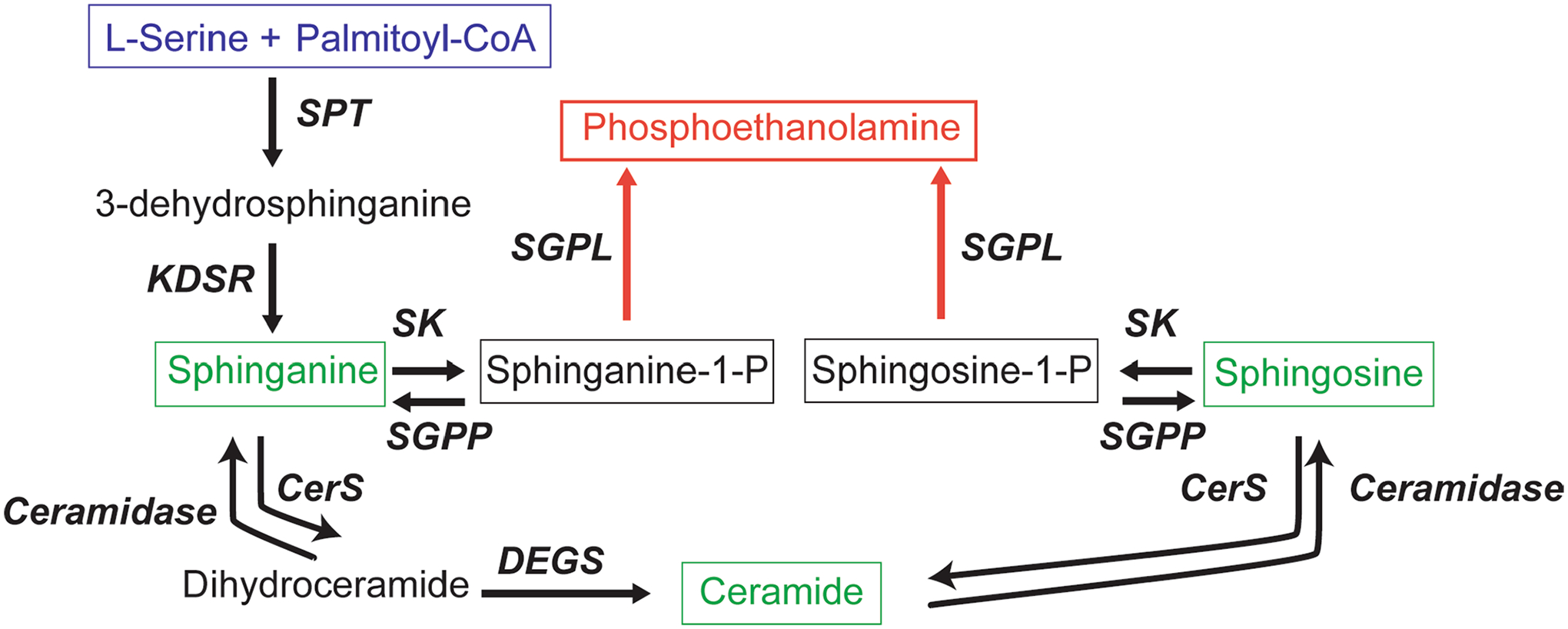
Serine palmitoyltransferase (SPT) catalyses the first and rate-limiting step in sphingolipid biosynthesis. The most abundant acyl-CoA used by SPT is palmitoyl-CoA, which after condensation with L-serine results in an 18-carbon long-chain base, 3-dehydrosphinganine. 3-dehydrosphinganine is rapidly reduced by 3-dehydrosphinganine reductase (KDSR) to form dihydrosphingosine (sphinganine). Sphinganine is acylated by a variety of ceramide synthases (CerS) to form dihydroceramides, which can be converted to ceramides by sphingolipid desaturase (DEGS). Many complex sphingolipids can be synthesized from a ceramide backbone. However, the only true exit from the pathway depends on activity of sphingosine kinase (SK) and sphingosine-1-phosphate aldolase (SGPL) to form phosphoethanolamine. SGPP = sphingosine-1-phosphate phosphatase.
Extended Data Fig. 2 |. SPTLC1 variant associated with amyotrophic lateral sclerosis and their effects on splicing.
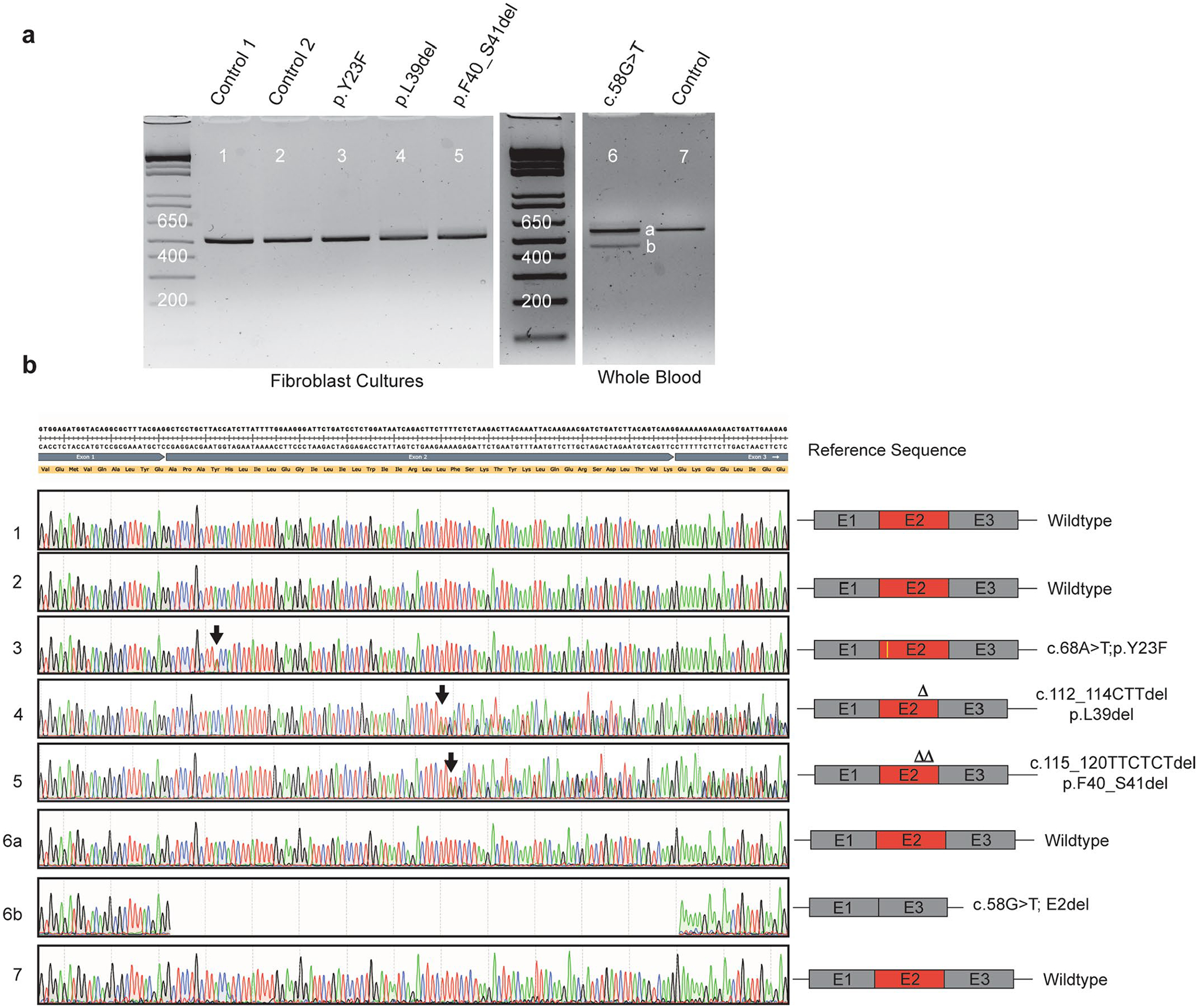
a, Gel electrophoresis of PCR amplification of cDNA obtained from patient fibroblast cultures or whole blood. Fibroblasts were not available for c.58G>T patient. PCR primers were in exon 1 and 6 of SPTLC1 (NM_006415.4). SPTLC1 c.58G>T variant resulted in two bands with an apparent difference of ~100 bp. All other variants and controls resulted in a single band. b, Sequencing of the gel purified PCR product showed that the c.58G>T variant resulted in complete skipping of exon 2 (lower band). Neither the full-length product (upper band) nor the internally deleted transcript (lower band) contained the c.58G>T variant. However, it is possible that the missense variant exists in very small amounts and escaped amplification by PCR and sequencing. All other variants were confirmed and do not affect splicing.
Extended Data Fig. 3 |. Amyotrophic lateral sclerosis associated variant SPTLC1 cellular localization.
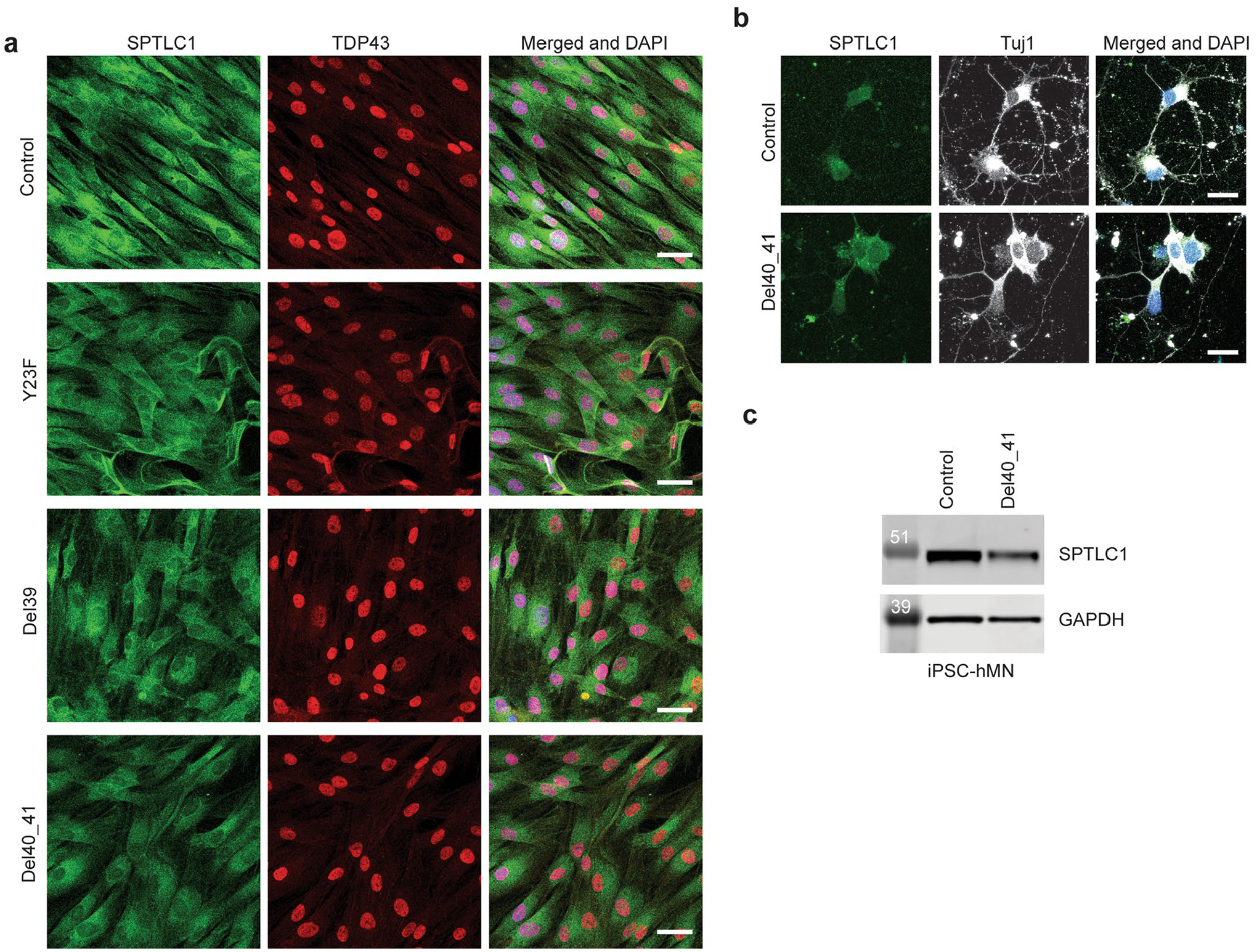
a, Immunostaining for SPTLC1 in patient derived fibroblast cultures shows normal subcellular localization in these cells, with an apparent distribution in the endoplasmic reticulum. Immunostaining for TDP43 also appears normal. Scale bar=40 μm b, Immunostaining for SPTLC1 in iPSC-derived human motor neurons with p.F40_S41 variant do not show any changes in its subcellular localization. Scale bar=20 μm c, Western blot of SPTLC1 in iPSC-derived human motor neurons confirms comparable expression levels in p.F40_S41 line and its isogenic control. Two independent differentiation experiments followed by western blotting were performed with similar results.
Extended Data Fig. 4 |. Induced pluripotent stem cell-derived human motor neurons (iPSC-hMN) with SPTLC1 ALS variants.
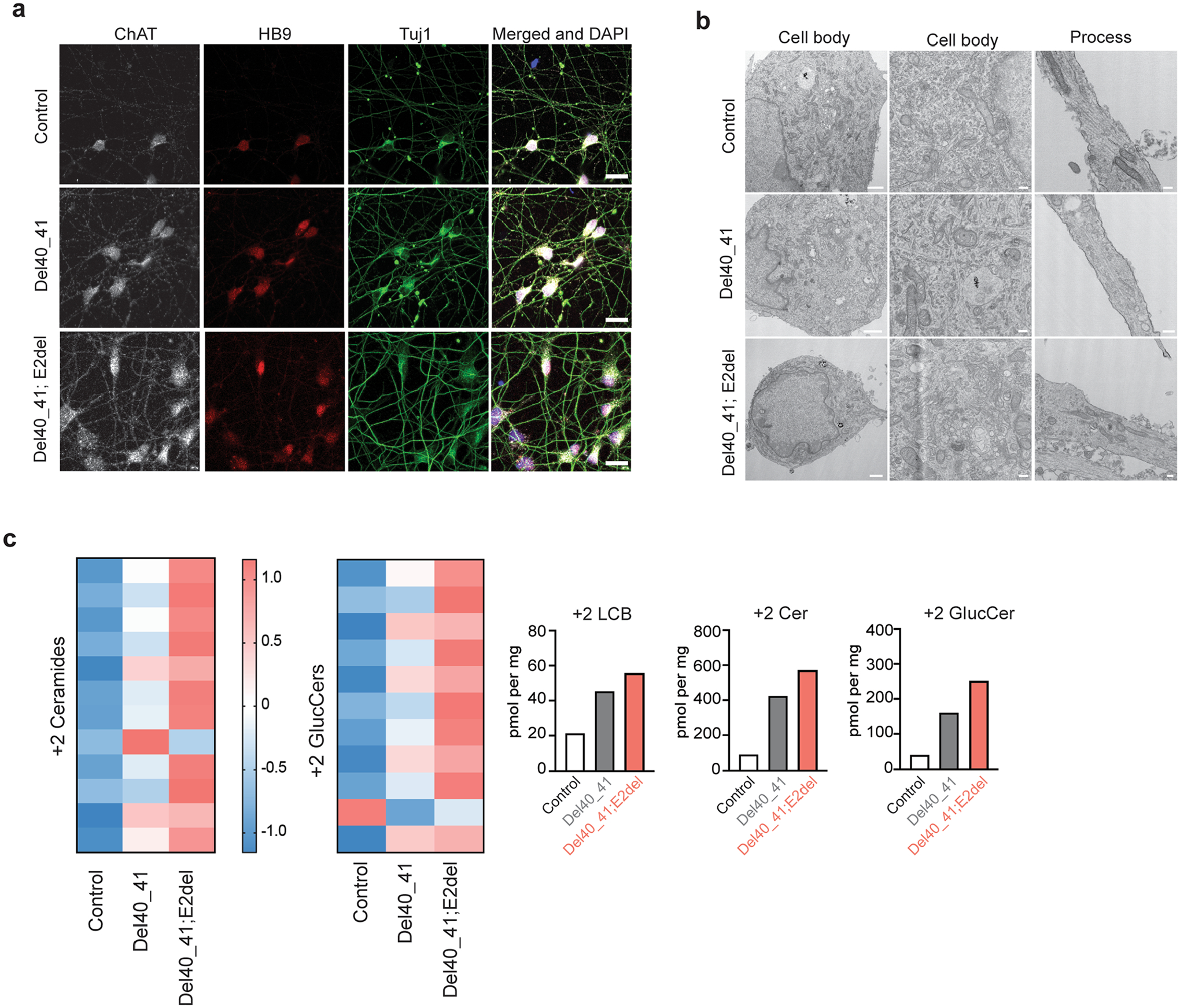
a, After differentiation by addition of doxycycline, SPTLC1 mutant iPSC-hMNs, one with heterozygous SPTLC1 p.F40_S41del variant and the other with compound heterozygous variants SPTLC1 p.F40_S41del; E2del (complete deletion of exon 2) and their isogenic control assume neuronal morphology. They express choline acetyltransferase (ChAT), Hb9 (a motor neuron specific transcription factor) as well as Tuj1 (neuronal microtubule marker). Scale bar = 20 μm b, Electron microscopy of iPSC-hMNs shows normal neuronal cell body morphology, nuclear heterochromatin, and organelles. In the processes (right), microtubules and occasional mitochondria and vesicles are noted. Scale bar left = 1 μm, middle and right = 200 nm. c, Sphingolipidomic analysis of de novo sphingolipid synthesis using isotope labelling (with 3,3-D2 L-serine) depicted in the form a heatmap with each row representing a sphingolipid species. Row Z-scores were calculated and depicted in colour. Canonical sphingolipids ceramides and glucosyl ceramides (glucCer) are increased in p.F40_S41del iPSC-hMNs compared to their isogenic control and increase further in the double mutant iPSC-hMNs. The bar graphs show the increase in total 2+ long-chain bases (LCB), ceramides (Cer), and glucosyl ceramides (GlucCer).
Extended Data Fig. 5 |. Allele specific knockdown of variant SPTLC1 mRNA.
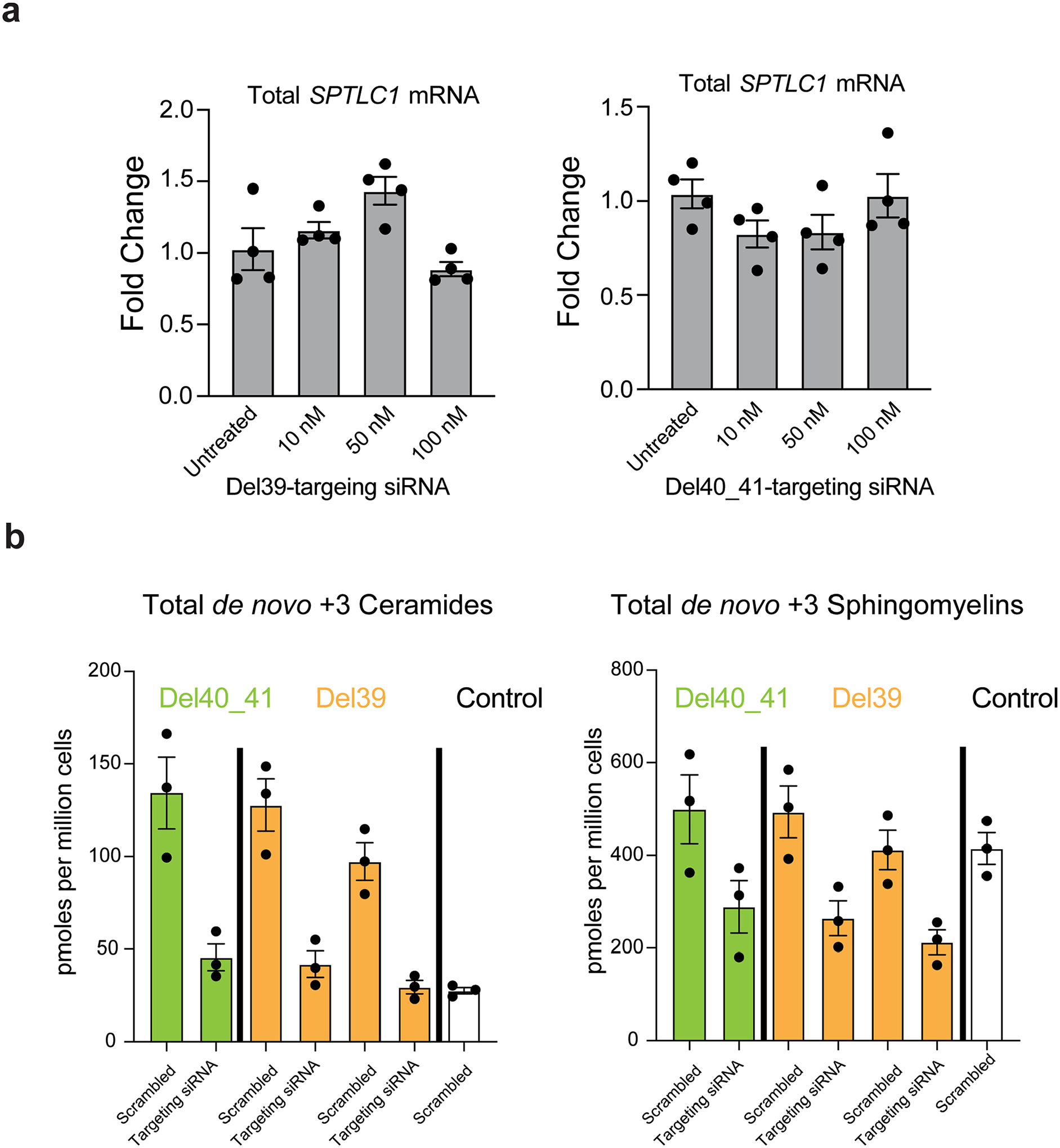
a, The two allele-specific siRNAs targeting the L39del and F40_S41del alleles do not reduce SPTLC1 mRNA levels in control cells at concentrations as high as 100nM. Error bars are SEM. Four replicates were performed. b, Sphingolipidomic analysis of patient derived fibroblasts with SPTLC1 ALS variants (F40_S41del and two patient lines with L39del) after treatment with 10 nM allele-specific siRNA targeting each corresponding variant were analysed and compared to control fibroblasts. Following the knockdown, the cells were labeled with D3,15N-L-serine and D4-L-alanine. The de novo synthesized +3 ceramide and sphingomyelin levels show a reduction in patient cells when treated with variant-specific siRNAs. Error bars are SEM. Three replicates were performed.
Supplementary Material
Acknowledgements
We thank the patients and their families for participating in our research study, C. Mendoza (NINDS/NNDCS) and G. Averion (NINDS/NNDCS) for their help in supporting the clinic, F. Eichler (Massachusetts General Hospital) for helpful discussions, and B. Smith (NINDS) and A. Nath (NINDS) for providing control serum samples. We also thank the NIH Intramural Sequencing Center for performing exome sequencing; K. Linask and J. Zhou from the NHLBI iPSC core for generating the isogenic mutant iPSC lines; and S. Lara and S. J. H. Tao Cheng in the NINDS electron microscopy facility. Work in C.G.B.’s laboratory is supported by intramural funds of NINDS/NIH. Work in T.M.D.’s laboratory was supported by CDMRP grant W81XWH-20-1-0219. C.V.L is supported by a career development grant from NIH/NINDS (K08 NS10762). T.H. is supported by Swiss National Science Foundation grant 31003A_179371. M.A.L is supported by Swiss Foundation for Research on Muscle Diseases (FSRMM). R.H.B. is funded by NIH/NINDS (R01 NS072446) and the Deater foundation. Sequencing and analysis provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) was funded by the National Human Genome Research Institute, the National Eye Institute and the National Heart, Lung and Blood Institute (grant UM1 HG008900) and, in part, by the National Human Genome Research Institute (grant R01 HG009141).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01346-1.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-021-01346-1.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01346-1.
Data availability
All data and material generated and analyzed within the main figures and supplementary material of this study are available upon request from the authors. All requests for raw and analyzed data and materials related to this article will be reviewed by the respective institution to verify whether the request is subject to any intellectual property or confidentiality obligations. Some patient-related data, including genetic sequencing data, not included in the paper or its supplements were generated as part of clinical care and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement. The following databases were searched for the presence of variants reported in this paper: the Genome aggregation database (https://gnomad.broadinstitute.org), the Decipher database (https://www.deciphergenomics.org), the Leiden database (https://www.lovd.nl) and the TopMed database (https://bravo.sph.umich.edu/freeze8/hg38/).
References
- 1.Al-Chalabi A & Hardiman O The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol 9, 617–628 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Swinnen B & Robberecht W The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol 10, 661–670 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Brown RH & Al-Chalabi A Amyotrophic lateral sclerosis. N. Engl. J. Med 377, 162–172 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Lee Y et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannwarth S et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breslow DK Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb. Perspect. Biol 5, a013326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison PJ, Dunn TM & Campopiano DJ Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep 35, 921–954 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G & Bieberich E Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv. Biol. Regul 70, 51–64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hojjati MR, Li Z & Jiang XC Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim. Biophys. Acta 1737, 44–51 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Zhao L et al. Elevation of 20-carbon long chain bases due to a mutation in serine palmitoyltransferase small subunit b results in neurodegeneration. Proc. Natl Acad. Sci. USA 112, 12962–12967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bejaoui K et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet 27, 261–262 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Gantner ML et al. Serine and lipid metabolism in macular disease and peripheral neuropathy. N. Engl. J. Med 381, 1422–1433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gable K et al. A disease-causing mutation in the active site of serine palmitoyltransferase causes catalytic promiscuity. J. Biol. Chem 285, 22846–22852 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penno A et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J. Biol. Chem 285, 11178–11187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks BR, Miller RG, Swash M Munsat TL & World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord 1, 293–299 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong HY et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han G et al. The topology of the Lcb1p subunit of yeast serine palmitoyltransferase. J. Biol. Chem 279, 53707–53716 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Fernandopulle MS et al. Transcription factor-mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol 79, e51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fridman V et al. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology 92, e359–e370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garofalo K et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J. Clin. Invest 121, 4735–4745 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandon EC, van Echten G, Birk R, Schmidt RR & Sandhoff K Sphingolipid biosynthesis in cultured neurons. Down-regulation of serine palmitoyltransferase by sphingoid bases. Eur. J. Biochem 198, 667–674 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Siow DL & Wattenberg BW Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J. Biol. Chem 287, 40198–40204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis DL, Gable K, Suemitsu J, Dunn TM & Wattenberg BW The ORMDL/Orm-serine palmitoyltransferase (SPT) complex is directly regulated by ceramide: reconstitution of SPT regulation in isolated membranes. J. Biol. Chem 294, 5146–5156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hjelmqvist L et al. ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biol 3, RESEARCH0027 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han G et al. The ORMs interact with transmembrane domain 1 of Lcb1 and regulate serine palmitoyltransferase oligomerization, activity and localization. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 245–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke BA et al. The Ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. eLife 8, e51067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Xie T, Liu P, Wang L & Gong X Structural insights into the assembly and substrate selectivity of human SPT-ORMDL3 complex. Nat. Struct. Mol. Biol 28, 249–257 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Wang Y et al. Structural insights into the regulation of human serine palmitoyltransferase complexes. Nat. Struct. Mol. Biol 28, 240–248 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blackstone C Hereditary spastic paraplegia. Handb. Clin. Neurol 148, 633–652 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Connolly O et al. A systematic review of genotype phenotype correlation across cohorts having causal mutations of different genes in ALS. J. Pers. Med 10, 58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicolas G & Veltman JA The role of de novo mutations in adult-onset neurodegenerative disorders. Acta Neuropathol 137, 183–207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cervenakova L et al. Progressive muscular atrophy variant of familial amyotrophic lateral sclerosis (PMA/ALS). J. Neurol. Sci 177, 124–130 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET & Brown RH Jr. Limited corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4V mutation in the copper/zinc superoxide dismutase gene. Ann. Neurol 43, 703–710 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Auer-Grumbach M et al. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur. J. Med. Genet 56, 266–269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suh BC et al. Early-onset severe hereditary sensory and autonomic neuropathy type 1 with S331F SPTLC1 mutation. Mol. Med. Rep 9, 481–486 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Rotthier A et al. Genes for hereditary sensory and autonomic neuropathies: a genotype–phenotype correlation. Brain 132, 2699–2711 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bode H et al. HSAN1 mutations in serine palmitoyltransferase reveal a close structure–function–phenotype relationship. Hum. Mol. Genet 25, 853–865 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Suriyanarayanan S et al. A novel variant (Asn177Asp) in SPTLC2 causing hereditary sensory autonomic neuropathy type 1C. Neuromolecular Med 21, 182–191 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Harmon JM et al. Topological and functional characterization of the ssSPTs, small activating subunits of serine palmitoyltransferase. J. Biol. Chem 288, 10144–10153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blasco H et al. Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci. Rep 7, 17652 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cutler RG, Pedersen WA, Camandola S, Rothstein JD & Mattson MP Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol 52, 448–457 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Dodge JC et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 112, 8100–8105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henriques A et al. Sphingolipid metabolism is dysregulated at transcriptomic and metabolic levels in the spinal cord of an animal model of amyotrophic lateral sclerosis. Front. Mol. Neurosci 10, 433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alterman JF et al. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat. Biotechnol 37, 884–894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med 381, 1644–1652 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacold ME et al. Corrigendum: a PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol 12, 656 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Lowther J et al. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol. Biosyst 6, 1682–1693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carvalho MD & Swash M Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph. Lateral Scler 10, 53–57 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Han G et al. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl Acad. Sci. USA 106, 8186–8191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta SD et al. Expression of the ORMDLS, modulators of serine palmitoyltransferase, is regulated by sphingolipids in mammalian cells. J. Biol. Chem 290, 90–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohassel P et al. Dominant collagen XII mutations cause a distal myopathy. Ann. Clin. Transl. Neurol 6, 1980–1988 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyaoka Y et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 11, 291–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lone MA et al. Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proc. Natl Acad. Sci. USA 117, 15591–15598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merrill AH Jr., Sullards MC, Allegood JC, Kelly S & Wang E Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 36, 207–224 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and material generated and analyzed within the main figures and supplementary material of this study are available upon request from the authors. All requests for raw and analyzed data and materials related to this article will be reviewed by the respective institution to verify whether the request is subject to any intellectual property or confidentiality obligations. Some patient-related data, including genetic sequencing data, not included in the paper or its supplements were generated as part of clinical care and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement. The following databases were searched for the presence of variants reported in this paper: the Genome aggregation database (https://gnomad.broadinstitute.org), the Decipher database (https://www.deciphergenomics.org), the Leiden database (https://www.lovd.nl) and the TopMed database (https://bravo.sph.umich.edu/freeze8/hg38/).