

Abstract
Background:
Aldosterone is a critical pathological driver for cardiac and renal disease. We recently discovered that MANP, a novel atrial natriuretic peptide (ANP) analog, possessed more potent aldosterone inhibitory action than ANP in vivo. MANP and NP-augmenting therapy sacubitril/valsartan are under investigations for human hypertension treatment. Understanding the elusive mechanism of aldosterone inhibition by natriuretic peptides (NPs) remains to be a priority. Conflicting results were reported on the roles of the particulate guanylyl cyclase A receptor (pGC-A) and NP clearance receptor (NPRC) in aldosterone inhibition. Furthermore, the function of protein kinase G (PKG) and phosphodiesterases (PDE) on aldosterone regulation are not clear.
Methods:
In the present study, we investigated the molecular mechanism of aldosterone regulation in a human adrenocortical cell line H295R and in mice.
Results:
We first provided evidence to show that pGC-A, not NPRC, mediates aldosterone inhibition. Next, we confirmed that MANP inhibits aldosterone via PDE2 not PKG, with specific agonists, antagonists, siRNA silencing, and fluorescence resonance energy transfer (FRET) experiments. Further, the inhibitory effect is mediated by a reduction of intracellular Ca2+ levels. We then illustrated that MANP directly reduces aldosterone synthase CYP11B2 expression via PDE2. Lastly, in PDE2 knockout mice, consistent with in vitro findings, embryonic adrenal CYP11B2 is markedly increased.
Conclusions:
Our results innovatively explore and expand the NP/pGC-A/cGMP/PDE2 pathway for aldosterone inhibition by MANP in vitro and in vivo. Additionally, our data also support the development of MANP as a novel ANP analog drug for aldosterone excess treatment.
Keywords: Natriuretic peptide, aldosterone, PDE2, Ca2+, H295R, cGMP, Knockout, Bay 60-7550
Graphical Abstract

Introduction
Aldosterone is a hormone produced by adrenal glomerulosa cells which plays a key role in blood pressure regulation through the regulation of sodium homeostasis 1. Apart from its classical role in sodium and water retention, excessive aldosterone also mediates pathophysiologic fibrosis, hypertrophy, cell injury, and organ remodeling in the heart, vasculature and kidney contributing to hypertension, heart failure, and chronic kidney disease 1–6. Drug discovery targeting the aldosterone receptor to date has resulted in the wide use of mineralocorticoid receptor (MR) antagonists for multiple cardiorenal disease states with improved outcomes 7,8. However, the use of MR antagonists in part is limited by a counter-regulatory increase in aldosterone which may limit efficacy 9. Thus efforts in drug development have centered on direct aldosterone inhibition by targeting aldosterone synthase CYP11B2. To date, progress in this area has been mixed although recently there has been a report of a well-tolerated and specific CYP11B2 inhibitor in healthy human subjects 10,11.
Atrial natriuretic peptide (ANP) is a cardiac hormone which plays an important role in blood pressure homeostasis through vasodilating and natriuretic actions via activation of the particulate guanylyl cyclase A receptor (pGC-A) and production of the second messenger cGMP 12–16. Importantly, ANP is also a key endogenous inhibitor of aldosterone production 17 while in a seminal study may also inhibit the MR 18. Such properties support a therapeutic role for ANP in disease states such as hypertension. Yet, the instability of ANP due to its susceptibility to degradation by neprilysin has limited its use to acute indications in which ANP is administered intravenously 16,19.
MANP (also called ZD100 or frameshift ANP) is a novel ANP analog that was bioengineered by us to overcome the pharmacodynamic limitations of ANP. Dickey et al. reported MANP to be highly resistant to neprilysin degradation compared to ANP 20. In healthy canines, MANP potently reduced circulating aldosterone levels which was more sustained than ANP 21. Aldosterone suppression by MANP was also reported in animal models of hypertension and hypertensive heart failure 22,23. Following the robust blood pressure lowering effects observed in the pilot phase 1 clinical study in patients with resistant hypertension 24, MANP is now entering phase 2 clinical trials for further evaluations. Additionally, strategies to augment endogenous NP levels exemplified by sacubitril therapy have been shown to treat hypertension more efficiently than angiotensin receptor blocker alone 25.
Thus far, the mechanisms of aldosterone suppression by NPs are not clearly defined. Previous studies report pGC-A activation suppresses aldosterone, while studies also suggest that the NP clearance receptor (NPRC) plays a suppressive role 26–28. Further, results are conflicting regarding the key cGMP/protein kinase G (PKG) signaling pathway in aldosterone regulation 29,30. Additionally, in vitro evidence also suggested that cGMP activates phosphodiesterases (PDEs), specifically cGMP-stimulated PDE2, which itself may lead to aldosterone inhibition by lowering cAMP levels 31,32. However, thus far clear evidence is lacking in delineating the detailed mechanism of NP-mediated aldosterone suppression in vitro. Further, no in vivo study has been performed to understand the function of PDE2 and CYP11B2. As MANP has now entered clinical trials, it is a high priority to define the mechanisms through which NP suppresses aldosterone.
Here we report for the first time molecular mechanisms of aldosterone inhibition by MANP in a human adrenal cortical cell line H295R. We demonstrated that MANP directly inhibits aldosterone production in adrenal cortical cells stimulated by angiotensin II (ANGII), forskolin, and KCl. We then investigated if the inhibitory effect is mediated by the pGC-A/cGMP/PDE2 pathway and the role of NPRC and PKG. We also assessed if intracellular Ca2+ in human adrenal cells activated by ANGII could be suppressed by MANP. We then determined the role of aldosterone synthase CYP11B2 in vitro. Finally, we assessed CYP11B2 expression in PDE2 knockout mice.
Methods
The authors declare that all supporting data are available within the article or in the Supplemental Material.
Cell Culture and Western Blotting
H295R cells (ATCC, Manassas, VA) were cultured and maintained with DMEM:F12 medium supplemented with ITS Premix (Corning, NY), 2.5% Nu-serum, 100 U/mL penicillin and streptomycin. For western blotting analysis, cells lysates were collected with NP-40 containing protease inhibitors. Primary antibodies recognizing pGC-A (R&D systems, Minneapolis, MN), NPRC (Abcam, Waltham, MA), PDE2 (FabGennix, Frisco, TX), PKG I (Cell Signaling, Danvers, MA), GAPDH (Cell Signaling) and anti-IgG secondary antibodies (Santa Cruz Biotechnology, Dallas, TX) were used.
Statistical Analysis
All in vitro studies were performed for at least a total of 3 independent experiments and data are expressed as means±SE. Two-tailed, unpaired t-tests were used for comparisons between two groups. 2-way ANOVA were performed in studies in which multiple time points were involved. Data normality was assessed with Shapiro-Wilk test and non-parametric Mann-Whitney test was applied for data that were not normally distributed. Significance analysis was performed with Prism 8 (Graphpad, La Jolla, LA), and statistical significance was accepted as p<0.05.
Results
MANP cGMP Activation and Aldosterone Production
As illustrated in Figure S1A, western blotting demonstrated the presence of pGC-A in H295R cells. Treatment of H295R cells with MANP for 10 min increased intracellular cGMP generation in a dose-dependent manner from 10−8 to 10−6 M compared to control group (Figure 1A). A similar trend was seen in a period of 24-hour treatment (Table S1). ANGII stimulation at concentrations of 10−8 to 10−6 M increased aldosterone levels approximately 3 to 5 fold in H295R cells in comparison with the control group (Figure 1B). Importantly, treatment with MANP from 10−8 to 10−6 M inhibited aldosterone stimulation induced by ANGII (Figure 1C, Figure S2A). Further, forskolin, a direct activator of adenylyl cyclase producing cAMP, stimulated aldosterone generation underscoring the role of cAMP as an activator of aldosterone, while MANP inhibited this stimulation (Figure 1D). MANP also suppressed aldosterone production induced by exogenous KCl (Figure S2A).
Figure 1.

cGMP production and aldosterone inhibition in H295R cells. (A) intracellular cGMP generation by MANP treatment (10−8, 10−7, or 10−6 M) for 10 min. (B) aldosterone production stimulated by ANGII (10−8, 10−7, or 10−6 M). (C) aldosterone inhibition by MANP (10−8, 10−7, or 10−6 M) in the presence of 10−8 M ANGII. (D) aldosterone inhibition by MANP (10−7 or 10−6 M) in the presence of 10−6 M forskolin (FSK, a cAMP activator). Aldosterone levels were described as fold changes from Control (Ctrl) group. n=3 each group. * p<0.05 vs. Control, # p<0.05 vs. ANGII or FSK. Data are expressed as means ± SE. Unpaired t-test or non-parametric Mann-Whitney test was performed.
NPRC, PKG and Aldosterone Inhibition
Western blotting analysis reveals NPRC and PKG expression in H295R cells (Figure S1B). To investigate if NPRC also mediates an aldosterone-inhibitory effect, we conducted experiments using NPRC specific agonist cANF(4-23) (cANF) and antagonist AP-811 (AP). The ligand cANF(4-23) binds to NPRC only and does not activate guanylyl cyclase receptors. As shown in Figure 2A, aldosterone production was suppressed by MANP, but not by cANF(4-23). Additionally, the inhibitory effect by MANP was not affected by NPRC antagonist AP-811 (Figure 2B).
Figure 2.

Aldosterone suppression by MANP is not affected by NPRC or PKG actions in H295R cells. (A) aldosterone production by 10−7 M MANP or 10−6 M NPRC agonist, cANF(4-23) in the presence of 10−8 M ANGII. (B) aldosterone production by 10−7 M MANP in the presence of 10−8 M ANGII with or without 10−6 M NPRC antagonist, AP-811 (AP). (C) aldosterone production by various concentrations of a membrane-permeable, non-specific cGMP analog, cGMP-AM (cGAM) in the presence of 10−8 M ANGII. (D) aldosterone production by a range of membrane-permeable, PKG-specific cGMP analog, 8-pCTP-cGMP (10−12, 10−11, 10−10, 10−9, 10−8, 10−7, 10−6, or 10−5 M) in the presence of 10−8 M ANGII. (E) aldosterone production by 10−7 M MANP in the presence of 10−8 M ANGII and 10−6 M PKG-specific blocker, Rp-8-pCPT-cGMPS. Aldosterone levels were described as fold changes from Control (Ctrl) group. n=3 each group. * p<0.05 vs. Control, # p<0.05 vs. ANGII. Data are expressed as means ± SE. Unpaired t-test or non-parametric Mann-Whitney test was performed.
We then validated that a non-specific cGMP analog, cGMP-AM successfully recapitulated the aldosterone-suppressing actions of MANP in H295R cells (Figure 2C). Two major downstream protein targets of cGMP are PKG and PDEs, therefore we explored which downstream molecule mediates the aldosterone suppression of MANP. We employed a wide range of concentrations from 10−12 to 10−5 M of a membrane-permeant PKG activator, 8-pCTP-cGMP, which failed to reduce aldosterone levels (Figure 2D). In parallel, we employed the PKG inhibitor Rp-8-pCPT-cGMPS which also did not affect aldosterone reduction induced by MANP (Figure 2E).
PDE2 and MANP Inhibition of Aldosterone
Figure 3A illustrates the ability of IBMX, a non-specific PDE inhibitor, to reverse the aldosterone-inhibitory effect of MANP. Of note, cilostamide, a PDE3 specific (cGMP-inhibited PDE, increases cAMP levels) inhibitor, reduced aldosterone levels stimulated by ANGII (Figure S2B and C). Figure 3B reports our investigation of PDE2 (cGMP-stimulated PDE) in MANP-mediated aldosterone inhibition. Importantly, the PDE2 inhibitor Bay 60-7550 mimicked the inhibitory effect of IBMX on MANP-induced suppression of aldosterone. Specifically, a dose-dependent response was observed with 3 concentrations of Bay 60-7550 at 10−8 to 10−6 M with a significant change achieved in 10−6 M group (Figure 3B). We further validated the role of PDE2 with siRNA interference. Consistently, knockdown of PDE2 with a PDE2 specific siRNA also significantly blocked the aldosterone inhibitory effect by MANP (Figure S1C and Figure 3B).
Figure 3.

Aldosterone suppression by MANP is mediated by phosphodiesterase 2 (PDE2) in H295R cells. (A) aldosterone production by 10−7 M MANP in the presence of 10−8 M ANGII with 5X10−4 M of the PDE non-specific inhibitor, IBMX. (B) aldosterone production by 10−7 M MANP in the presence of 10−8 M ANGII and PDE2 specific inhibitor, Bay 60-7550 (10−8, 10−7, or 10−6 M) or in cells transfected with 10−7 M PDE2 specific siRNA. Aldosterone levels were described as fold changes from Control (Ctrl) group. n=3 each group. * p<0.05 vs. Control, # p<0.05 vs. ANGII, $ p<0.05 vs. MANP. Data are expressed as means ± SE. Unpaired t-test or non-parametric Mann-Whitney test was performed.
MANP Increases PDE2 Activity
We firstly confirmed that a cAMP analog, 8-Br-cAMP increases aldosterone production in H295R cells (Ctrl: 1.00±0.04, 8-Br-cAMP 10−5 M: 1.02±0.04, 8-Br-cAMP: 10−4 M 1.49±0.01), which is also consistent with forskolin results observed in Figure 1D. To further validate the role of PDE2 as a target of MANP, we employed the state-of-art Epac2-camps cAMP biosensor based FRET imaging of H295R cells in the presence or absence of MANP so as to determine whether MANP enhanced PDE2 activity. FRET CFP/YFP ratio quantifies cAMP levels and PDE2 activity is inversely proportional to FRET ratio. By monitoring cAMP responses in real-time using FRET technology, as illustrated in Figure 4A and B, we first confirmed that forskolin (FSK), a known cAMP activator, increases FRET signal i.e. cAMP levels. MANP treatment 10 min prior to FSK stimulation reduces cAMP levels (activates PDE2). Furthermore, inhibiting PDE2 activity by Bay 60-7550 in MANP treated cells reversed MANP’s FRET-lowering effect, which suggests an increase of cAMP and a reduction of PDE2 activity by Bay 60-7550. In Figure 4C and D, we transfected cells with PDE2 specific siRNA and then treated the cells with MANP in the presence of FSK. We found that lowering PDE2 expression by siRNA, similar to Bay 60-7550 inhibition, induced an increase of FRET signal, corresponding to cAMP enhancement and PDE2 activity reduction.
Figure 4.
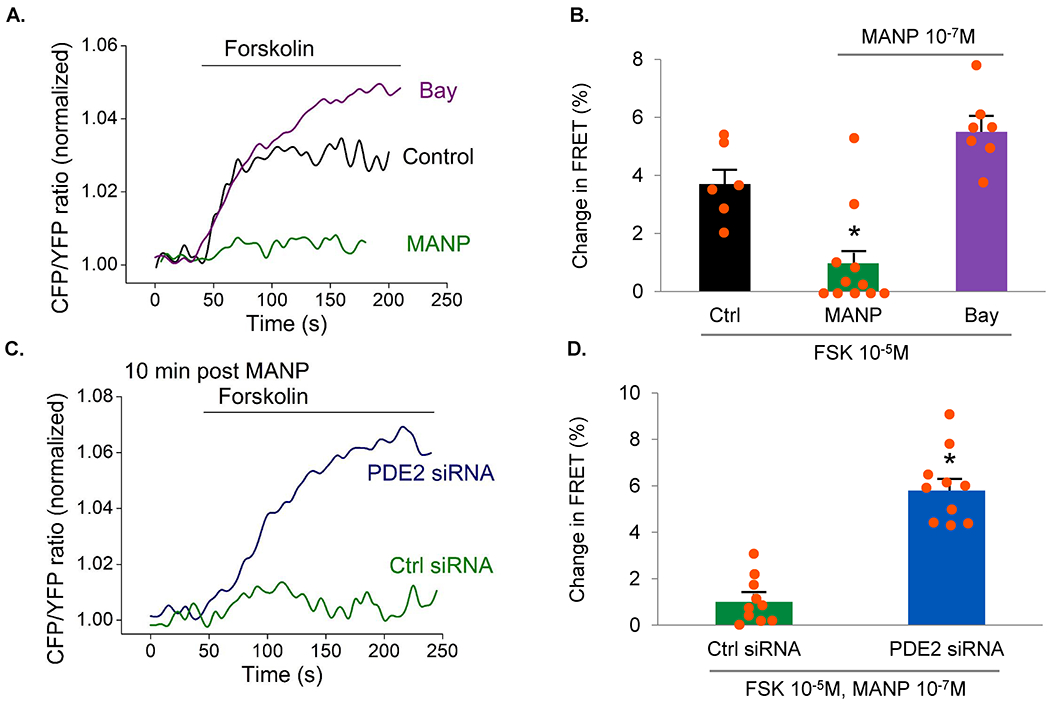
PDE2 activity measured by fluorescence resonance energy transfer (FRET) based biosensor Epac2-camps expressed in H295R cells using adenovirus. The use of FRET provides us with a tool to measure cAMP activity in which FRET ratio is inversely related to PDE2 activity. (A) Examples of live cell imaging traces. Changes in FRET signal (expressed as CFP/YFP ratio reflecting intracellular cAMP levels) by 10−7 M MANP, or by 10−6 M Bay 60-7550 plus MANP, in the presence of 10−5 M forskolin (a cAMP activator) were measured. Control group was 10−5 M forskolin stimulation only. For MANP group, cells were pretreated with 10−7 M MANP for 10 min followed by forskolin stimulation. For Bay group, cells were pretreated with 10−6 M Bay 60-7550, MANP followed by forskolin stimulation. (B) Quantification of the experiments shown in (A) as a % FRET changes. PDE2 activity is inversely related to FRET signals. Control group, n=6. MANP group, n=11, Bay group, n=7. (C) Examples of live cell imaging traces in siRNA study. Control siRNA group used negative control siRNA. In PDE2 siRNA group, cells were transfected with PDE2 siRNA. 48 hours post-transfection, both groups received 10−7 M MANP pretreatment for 10 min and 10−5 M forskolin treatment. (D) Quantification of the experiments shown in (C) as a % FRET changes. Control siRNA group, n=10. PDE2 siRNA group, n=10. * p<0.05 vs. Control. Data are expressed as means ± SE. Unpaired t-test was performed.
MANP May Regulate Intracellular Ca2+ Levels
Figure 5 illustrates intracellular Ca2+ flux traces under different conditions. Firstly, 10−8 M ANGII markedly increased intracellular Ca2+ levels (Figure 5A). Pretreatment with MANP (10−7 M) significantly delayed and reduced intracellular Ca2+ levels stimulated by ANGII. The suppression of Ca2+ levels by MANP was abolished with 10−6 M PDE2 inhibitor Bay 60-7550 treatment. Further, the Ca2+ channel blocker efonidipine at 10−6 M blocked Ca2+ elevation induced by ANGII and Bay 60-7550. Meanwhile, treatment with efonidipine at 3 concentrations (10−8 to 10−6 M) abrogated aldosterone elevation by Bay 60-7550 (Figure 5B). We then transfected cells with PDE2 siRNA and treated the cells with efonidipine in the presence of ANGII and MANP. In PDE2 siRNA treated cells, 10−6 M efonidipine suppressed aldosterone increase mediated by PDE2 siRNA (Figure 5C).
Figure 5.

Intracellular Ca2+ levels and aldosterone production regulated by Ca2+ levels in H295R cells. (A) Relative Ca2+ levels were calculated based upon recorded fluorescence signals in Fura-2 loaded H295R cells. Fold difference F/F0 was used as relative Ca2+ concentration values. The groups are as follows: ANGII group, 10−8 M ANGII perfusion; MANP group, 10−7 M MANP 5 min pretreatment plus ANGII perfusion; Bay 60-7550 group, 10−6 M Bay 60-7550 15 min pretreatment followed by MANP 5 min treatment plus ANGII perfusion; efonidipine (Efo) group, 10−6 M efonidipine 15 min pretreatment and then Bay 60-7550 15 min treatment followed by MANP 5 min treatment plus ANGII perfusion. n=7-10 for each group. (B) aldosterone production by a serial of concentrations of Ca2+ blocker, efonidipine (10−8, 10−7, or 10−6 M) in the presence of 10−8 M ANGII, 10−7 M MANP, and 10−6 M Bay 60-7550. (C) aldosterone production by 10−6 M Efo in cells transfected with 10−7 M PDE2 siRNA. Aldosterone levels were described as fold changes from Control group (in siRNA experiments, control group transfected with negative control siRNA) that received only treatment buffer (panel B and C). (D) Aldosterone synthase CYP11B2 gene expression by real-time PCR in H295R cells. CYP11B2 expression by 10−8 M ANGII, 10−7 M MANP in the presence of ANGII, or 10−6 M Bay 60-7550 plus MANP in the presence of ANGII. CYP11B2 gene expression was normalized to GAPDH. n=3 each group. * p<0.05 vs. Control, # p<0.05 vs. ANGII group, $ p<0.05 vs. Bay 60-7550 group or PDE2 siRNA group. Data are expressed as means ± SE. 2-way ANOVA was used for panel A and unpaired t-test or non-parametric Mann-Whitney test was performed was performed for panels B-D.
Aldosterone Synthase CYP11B2 Gene Expression
As illustrated in our Graphic Abstract, the final step in the regulation of aldosterone production is aldosterone synthase CYP11B2. As shown in Figure 5D, ANGII markedly upregulated aldosterone synthase CYP11B2 gene expression in H295R cells. Importantly, MANP significantly reduced CYP11B2 expression stimulated by ANGII by approximately 20%. Additionally, the PDE2 inhibitor Bay 60-7550 reversed MANP’s inhibitory effect further supporting the role for PDE2 in MANP induced suppression of aldosterone production.
Pde2 Knockout Mice
Lastly, we investigated Cyp11b2 mRNA expression by manipulating Pde2 levels in vivo. Of note, knockdown of Pde2 in heterozygotes mice showed a trend of Cyp11b2 elevation. Complete deletion of Pde2 causes embryonic lethality 33 and thus studies were performed in E14.5 pups. In Pde2 homozygous knockout mice embryos, we found the expression of Cyp11b2 was dramatically increased compared to wild type embryos (Figure 6).
Figure 6.

Cyp11b2 gene expression in Pde2 genetic mice. (A) Adult adrenal relative Cyp11b2 expression in wildtype (WT) and heterozygotes (HET) mice. (B) Embryonic adrenal relative Cyp11b2 expression in WT and knockout (KO) mice embryos. n=3-6 for different groups. * p<0.05 vs. wild type. Data are expressed as means ± SE. Unpaired t-test or non-parametric Mann-Whitney test was performed.
Discussion
In the present study, we for the first time report the comprehensive mechanisms of aldosterone suppression in vitro and in vivo by the novel pGC-A activator and ANP analog MANP. We firstly showed that MANP suppressed aldosterone production stimulated by ANGII, forskolin, or KCl in human adrenal cells. Importantly, we then demonstrated that the aldosterone inhibitory effect is mediated solely by pGC-A and not by the NPRC receptor. We also elucidated the inhibitory action of MANP was transduced via cGMP and its downstream target PDE2 not PKG. Further, we report that intracellular Ca2+ flux is involved in this process, in which MANP may lower intracellular Ca2+ concentration activated by ANGII leading to reduced aldosterone production. Lastly, PDE2 mediates the inhibition of aldosterone synthase CYP11B2 by MANP in vitro and in vivo. Our results advance insights in MANP mediated-aldosterone suppressing molecular mechanism and support the continuing clinical development of MANP in disease states of aldosterone excess.
The development of ANP as a chronic therapeutic for cardiovascular and renal disease has been limited by its need to be administered intravenously due to rapid degradation by neprilysin 16,19. MANP has emerged as a highly innovative ANP analog bioengineered to go beyond ANP. Indeed, we have recently reported the sustained blood pressure lowering action of MANP for 7 days with once daily subcutaneous injection in experimental models 34. Further, once daily and for three days subcutaneous administration of MANP has been tested in preliminary studies in humans with hypertension 24. To date, MANP has been reported to suppress aldosterone in canine models of hypertension and heart failure and in human 22–24. Previous studies have also reported that pGC-A activators such as ANP suppressed aldosterone in vitro and in vivo 26,27,35. Yet the molecular mechanisms of natriuretic peptide-mediated aldosterone inhibition remain unclear. Conflicting mechanisms include functional receptor (pGC-A or NPRC) and downstream messengers and mediators (PKG or PDE). Furthermore, there is a scarcity of data to link intracellular Ca2+ concentration and pGC-A pathway in aldosterone regulation. Lastly, in vivo mechanistic studies of PDE2 mediated aldosterone modulation are lacking.
In our current study, we firstly demonstrated that MANP inhibits aldosterone production stimulated by ANGII, forskolin, or KCl. Potential causes for seemingly lower values of aldosterone by forskolin or KCl may be due to modest cAMP pathway machinery or low potassium channel activity in H295R cells. Although the levels of aldosterone stimulation by different compounds vary, MANP consistently suppresses aldosterone production. Furthermore, we have clearly established the receptor pathway that mediates MANP suppression of aldosterone in human adrenal cells in vitro, which provides a key, novel insight for our understanding of NP biology. Although both pGC-A and NPRC were demonstrated in our study to be present in human adrenal cells, only pGC-A mediates the aldosterone inhibitory effect. Our study reports that the pGC-A/cGMP pathway leads to aldosterone inhibition, while NPRC does not play a role. Further, we showed that a non-specific cGMP analog cGMP-AM recapitulates the actions of MANP. Altogether, our findings support the pGC-A/cGMP pathway suppresses aldosterone production.
Conflicting results are especially pronounced with regards to the role of the cGMP/PKG pathway in the regulation of aldosterone production 29,30. In our study, the PKG-specific cGMP analog 8-pCPT-cGMP at concentrations ranging from 10−12 to 10−5 M did not affect aldosterone levels. A previous study that showed this analog stimulated aldosterone production, the concentrations used were 3X10−4 M and above 29, which we believe are excessively high. Concentrations higher than 10−5 M may not faithfully reflect the physiological responses given the intracellular cGMP concentrations we observed in the current study. In fact, studies have shown that 8-pCPT-cGMP also binds to other proteins such as protein kinase A and PDEs at high concentrations 36, which may then stimulate aldosterone production. Importantly, in the current study, the PKG specific inhibitor Rp-8-pCPT-cGMPS did not block the aldosterone-suppressive role of MANP. Previously, genetic mice studies reported that PKG knockout littermates presented similar levels of aldosterone compared with wild type 30, which support the absence of a major role for MANP activation of PKG in suppression of aldosterone production. Nevertheless, a direct measurement of PKG activity would be useful to confirm the effectiveness of PKG inhibition/activation experiments performed in our study.
Our finding of aldosterone inhibition by MANP via cGMP/PDE2 is in line with previous studies. MacFarland et al. reported that PDEs exist in adrenal cells and Nikolaev et al. demonstrated that ANP induces PDE2 activation 31,32. Notably, PDE2 cleaves both cGMP and cAMP, and its activity can also be enhanced by cGMP. Indeed, PDE2 is also called a cGMP-stimulated PDE. Combined with PDE2 inhibition studies and real-time PDE2 activity by FRET, we validated that MANP increases PDE2 activity in human adrenal cells. To mention, a steady-state level (i.e. 24-hour period) of cAMP was not determined, which may be required to confirm our current PDE2 hypothesis in the regulation of aldosterone produced over a 24-hour period. Nevertheless, our study provides unique, novel insights connecting MANP and PDE2 activity on aldosterone inhibition. Therapeutic strategies to activate PDE2 may thus provide important clinical relevance for aldosterone suppression and hypertension treatment. Additionally, the potent aldosterone-lowering effects with PDE3 specific inhibitor cilostamide underscores the differential regulations by PDE family enzymes e.g. PDE2, PDE3. The existence of compartmentation and different pools of cGMP/cAMP 37,38 in cardiomyocytes or smooth muscle cells suggests a closer look at the differential localization of PDE2/3 would be helpful to further advance the mechanistic work of aldosterone regulation.
Our studies went beyond the known cGMP/PDE2 signaling by also expanding our understanding of Ca2+ pathway in MANP mediated aldosterone suppression. It is well known that intracellular Ca2+ levels and related downstream pathways are one of the most critical mediators for aldosterone production in adrenal cells with one study employing rat adrenal cells with ANP stimulation 39. In the current study, we demonstrated that MANP may reduce intracellular Ca2+ levels in human adrenal cells and PDE2 inhibitor Bay 60-7550 may increase intracellular Ca2+ levels. Further, the Ca2+ blocker efonidipine reduced aldosterone production upregulated by PDE2 inhibition. Previously, we also showed that MANP, in human vascular smooth muscle cells, inhibits and delays Ca2+ elevation evoked by ANGII 34. A Ca2+ lowering effect observed in both adrenal and vascular cells may represent a common feature of Ca2+ modulation by MANP. Additionally, studies support ANP inhibition of calcium influx in rat and bovine adrenal glomerulosa cells with techniques e.g. calcium patch-clamp experiments 39,40. Our current study did not perform in-depth calcium channel patch-clamp experiments, nor record continuous Ca2+. Single trace Ca2+ recordings and aldosterone regulation by calcium blocker reflect an action of Ca2+ in MANP mediated aldosterone suppression, while the definite role of Ca2+ in MANP/cGMP pathway remains unknown. Our data suggests that PDE2 activation by MANP could result in lower intracellular Ca2+ levels. It is also plausible to speculate that the aldosterone inhibitory effect by MANP involves both PDE2 activation and intracellular Ca2+ reduction, with the two pathways inter-connected. Future efforts are required to address this unanswered question.
In addition, the molecular mechanism by which aldosterone is suppressed by cAMP and Ca2+ is not completely addressed by the current study. Understanding the contribution and interconnected relationship of cAMP and Ca2+ remains a key issue, although our findings indicate both are suppressed by PDE2 and both are involved in MANP-mediated aldosterone suppression. The specific downstream molecular targets of aldosterone regulation by Ca2+ and cAMP are not fully addressed and more studies are needed.
Lastly, our data supports MANP as a direct aldosterone synthase inhibitor, which is consistent with previous reports of the CYP11B2 expression suppressed by native NPs that activate pGC-A 41. We also demonstrated that PDE2 is involved in the CYP11B2 expression regulation. More importantly, we conducted in vivo mouse genetic studies and confirmed our in vitro mechanistic findings. Indeed in adrenal glands taken from adult Pde2 heterozygous KO mice (a similar trend) and embryonic Pde2 homozygous KO mice, Cyp11b2 mRNA expression was increased. Thus our in vivo results, combined with the comprehensive in vitro studies, support a key role of PDE2 in CYP11B2 gene regulation. Of note, we did not measure CYP11B2 protein expression, especially with the use of an antibody recognizing B2 subtype e.g. not B1, in which future endeavors are needed to confirm our mRNA findings.
CYP11B2 gene transcription by ANGII is also controlled by intracellular Ca2+ levels and further activation of downstream transcription factors such as nuclear receptor subfamily 4 group A member 1, cAMP response element-binding protein 42. Future studies are required to confirm the potential regulatory effect of MANP on these transcription factors. Nonetheless, as the efforts to develop aldosterone synthase inhibitors for states of aldosterone excess have been mixed, our studies have established an important therapeutic role for MANP as an aldosterone synthase inhibitor. Additionally, MANP-mediated aldosterone suppression is a multifactorial process, consisting of direct aldosterone synthase CYP11B2 inhibition and the suppression of aldosterone upstream regulators such as renin and ANGII. These properties complement MANP’s renal and cardiovascular enhancing properties further supporting its continuing clinical development for cardiovascular, renal, and metabolic disease.
MANP (or mutant ANP) was initially discovered from a family of atrial fibrillation 43. Although in canine studies, compared to vehicle 22, MANP intravenous infusion did not induce atrial fibrillation or heart rate abnormalities, research designated to evaluate heart rate changes and a close monitoring of heart rate in future clinical studies would be helpful.
Studies have reported suppressing PDEs activity such as PDE5 inhibitor therapy reduces blood pressure in human subjects 44. Thus targeting specific PDEs to modulate cyclic nucleotides or aldosterone levels may represent a novel, effective method for hypertension treatment. In fact, we observed that activating PDE2 with designer NPs or inhibiting PDE3 activity leads to aldosterone suppression. Our findings illustrate the possibility of suppressing PDE3 activity or enhancing PDE2 activity to lower aldosterone levels and thus may support the concept of targeting PDE2/3 in hypertension treatment.
In conclusion, we report the molecular mechanisms of aldosterone inhibition of the novel ANP analog MANP, an innovative pGC-A activator now entering clinical trials for cardiovascular disease. This is the first comprehensive study (in vitro and in vivo) elucidating the signaling pathways regulating aldosterone production by NPs, encompassing the upstream receptors (pGC-A, NPRC), second messenger (cGMP) to downstream mediators (PDE2, PKG, Ca2+, CYP11B2). In this study we establish a key role for the pGC-A/cGMP/PDE2 pathway contributing to the aldosterone-lowering properties with aldosterone synthase. Importantly, our study highlights the translational value and therapeutic potential of MANP for aldosterone-excess states such as hypertension and heart failure.
Limitations
Our study has several limitations. Firstly, our in vitro studies were performed in immortalized adrenocortical H295R cells. Although H295R is the most widely used cell model for aldosterone research, validation studies in human or bovine primary adrenocortical cells are needed. In fact, published studies report a more profound aldosterone-inhibitory effect with ANP in rat or bovine adrenal glomerulosa cells than in H295R cells 26,45–48. Possible causes may include compromised responses to aldosterone stimulators 49, impaired CREB pathway 50, and modest expression of pGC-A and PDE2 in H295R cells. Additionally, the in vivo genetic mice study used a constitutive PDE2 knockout model. Given the potential systemic actions of PDE2, organ inter-communications and adult/embryonic differences, we could not rule out the possibility of other factors in addition to PDE2 contributing to aldosterone regulation by cGMP. Thus, a future project disrupting PDE2 in the adrenal gland specifically is warranted. In addition, assessing cellular Ca2+ changes in attenuated PDE2 expression/activity (siRNA knockdown or specific inhibitor) conditions would be helpful to validate the role of PDE2 in MANP-mediated Ca2+ regulation. Limitations also include lack of mechanistic studies to delineate cAMP/calcium relationship and protein kinase G activity determination.
Perspectives
This current study uncovers the molecular pathway for aldosterone inhibition by a designer natriuretic peptide (NP) in vitro and in vivo. Our results support the unique aldosterone suppression by particulate guanylyl cyclase (pGC-A) receptor, followed by cGMP generation and phosphodiesterase 2 (PDE2) activation, which then inhibits aldosterone synthase CYP11B2 expression. Based upon these mechanistic findings, MANP, a novel NP analog, may thus represent a promising therapeutic for aldosterone excess e.g. hypertension.
Supplementary Material
Novelty and Relevance.
What Is New?
MANP, a novel natriuretic peptide analog, inhibits aldosterone via particulate guanylyl cyclase receptor and cGMP activation in vitro.
Natriuretic peptide clearance receptor and cGMP-dependent protein kinase G are not involved in aldosterone regulation in vitro.
The inhibition of aldosterone relies on phosphodiesterase 2 activation and calcium inhibition.
In mice, consistent with in vitro findings, PDE2 knockout increases aldosterone synthase CYP11B2 expression.
What is Relevant?
Natriuretic peptides, including endogenously produced and chemically synthesized, inhibit aldosterone, a critical pathological driver for cardiovascular and renal disease.
Clinical/Pathophysiological Implications?
Our study sheds light on the molecular pathway regulating aldosterone production by natriuretic peptides. Increasing cGMP and PDE2 activity may evolve as an especially useful strategy for aldosterone synthase and aldosterone inhibition. Therapeutics to augment endogenous natriuretic peptides with neprilysin inhibitors (e.g. sacubitril) or exogenous natriuretic peptides administration may represent a new therapeutic avenue for hypertension management.
Sources of Funding
The work was supported by grants from the American Heart Association Postdoctoral Fellowship 826869 (Y.C.) and National Heart, Lung and Blood Institute (NHLBI), grant number R01 HL136340 (J.C.B.) and Deutsche Forschungsgemeinschaft NI 1301/7-1 (V.O.N.).
Abbreviations and nomenclature
- ANGII
angiotensin II
- ANP
atrial natriuretic peptide
- Bay
Bay 60-7550, PDE2 specific inhibitor
- cAMP
3’,5’, cyclic adenosine monophosphate
- cGMP
3’,5’, cyclic guanosine monophosphate
- Cilostamide
PDE3 specific inhibitor
- CYP11B2
cytochrome p450 family 11 subfamily b member 2
- Efo
efonidipine, a commonly used Ca2+ channel blocker
- FRET
fluorescence resonance energy transfer
- FSK
forskolin
- IBMX
3-Isobutyl-1-methylxanthine
- KCl
potassium chloride
- MANP
mutant atrial natriuretic peptide
- NP
natriuretic peptide
- NPRC
natriuretic peptide clearance receptor
- PDE2
phosphodiesterase 2
- pGC-A
particulate guanylyl cyclase A receptor
- PKG
cGMP dependent protein kinase G
Footnotes
Disclosure
John C. Burnett, Jr is the inventor for MANP and Mayo Clinic has licensed MANP to E-STAR Bio. Other authors have nothing to disclose.
References
- 1.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nature Reviews Nephrology. 2013;9:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsumura K, Fujii K, Oniki H, Oka M, Iida M. Role of aldosterone in left ventricular hypertrophy in hypertension. American journal of hypertension. 2006;19:13–18. doi: 10.1016/j.amjhyper.2005.05.013 [DOI] [PubMed] [Google Scholar]
- 3.Freel EM, Mark PB, Weir RA, McQuarrie EP, Allan K, Dargie HJ, McClure JD, Jardine AG, Davies E, Connell JM. Demonstration of blood pressure-independent noninfarct myocardial fibrosis in primary aldosteronism: a cardiac magnetic resonance imaging study. Circulation Cardiovascular imaging. 2012;5:740–747. doi: 10.1161/circimaging.112.974576 [DOI] [PubMed] [Google Scholar]
- 4.Bai M, Chen Y, Zhao M, Zhang Y, He JC, Huang S, Jia Z, Zhang A. NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am J Physiol Renal Physiol. 2017;312:F556–f564. doi: 10.1152/ajprenal.00332.2016 [DOI] [PubMed] [Google Scholar]
- 5.Schiffrin EL. Effects of aldosterone on the vasculature. Hypertension. 2006;47:312–318. doi: 10.1161/01.HYP.0000201443.63240.a7 [DOI] [PubMed] [Google Scholar]
- 6.Buglioni A, Cannone V, Cataliotti A, Sangaralingham SJ, Heublein DM, Scott CG, Bailey KR, Rodeheffer RJ, Dessì-Fulgheri P, Sarzani R. Circulating aldosterone and natriuretic peptides in the general community: relationship to cardiorenal and metabolic disease. Hypertension. 2015;65:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. New England Journal of Medicine. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 8.Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, Ford I, Cruickshank JK, Caulfield MJ, Salsbury J. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. The Lancet. 2015;386:2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AJ. Hypertension: renin–angiotensin–aldosterone system alterations. Circ Res. 2015;116:960–975. [DOI] [PubMed] [Google Scholar]
- 10.Calhoun DA, White WB, Krum H, Guo W, Bermann G, Trapani A, Lefkowitz MP, Ménard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo-and active-controlled phase 2 trial. Circulation. 2011;124:1945–1955. [DOI] [PubMed] [Google Scholar]
- 11.Bogman K, Schwab D, Delporte M-L, Palermo G, Amrein K, Mohr S, De Vera Mudry MC, Brown MJ, Ferber P. Preclinical and early clinical profile of a highly selective and potent oral inhibitor of aldosterone synthase (CYP11B2). Hypertension. 2017;69:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol Rev. 2016;96:751–804. doi: 10.1152/physrev.00022.2015 [DOI] [PubMed] [Google Scholar]
- 13.Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH, Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nat Rev Cardiol. 2014;11:403–412. doi: 10.1038/nrcardio.2014.64 [DOI] [PubMed] [Google Scholar]
- 14.Blanton RM. cGMP Signaling and Modulation in Heart Failure. Journal of cardiovascular pharmacology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Preedy MEJ, Baliga RS, Hobbs AJ. Multiplicity of Nitric Oxide and Natriuretic Peptide Signaling in Heart Failure. 2020;75:370–384. doi: 10.1097/fjc.0000000000000724 [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Burnett JC Jr. Biochemistry, Therapeutics, and Biomarker Implications of Neprilysin in Cardiorenal Disease. Clin Chem. 2017;63:108–115. doi: 10.1373/clinchem.2016.262907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atarashi K, Mulrow P, Franco-Saenz R. Effect of atrial peptides on aldosterone production. J Clin Invest. 1985;76:1807–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakagawa H, Oberwinkler H, Nikolaev VO, Gaßner B, Umbenhauer S, Wagner H, Saito Y, Baba HA, Frantz S, Kuhn M. Atrial natriuretic peptide locally counteracts the deleterious effects of cardiomyocyte mineralocorticoid receptor activation. Circulation: Heart Failure. 2014;7:814–821. [DOI] [PubMed] [Google Scholar]
- 19.Saito H, Ogihara T, Nakamaru M, Hara H, Higaki J, Rakugi H, Tateyama H, Minamino T, Iinuma K, Kumahara Y. Hemodynamic, renal, and hormonal responses to alpha-human atrial natriuretic peptide in patients with congestive heart failure. Clin Pharmacol Ther. 1987;42:142–147. [DOI] [PubMed] [Google Scholar]
- 20.Dickey DM, Yoder AR, Potter LR. A familial mutation renders atrial natriuretic Peptide resistant to proteolytic degradation. The Journal of Biological Chemistry. 2009;284:19196–19202. doi: 10.1074/jbc.M109.010777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKie PM, Cataliotti A, Huntley BK, Martin FL, Olson TM, Burnett JC Jr. A human atrial natriuretic peptide gene mutation reveals a novel peptide with enhanced blood pressure-lowering, renal-enhancing, and aldosterone-suppressing actions. Journal of the American College of Cardiology. 2009;54:1024–1032. doi: 10.1016/j.jacc.2009.04.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKie PM, Cataliotti A, Boerrigter G, Chen HH, Sangaralingham SJ, Martin FL, Ichiki T, Burnett JC Jr. A novel atrial natriuretic peptide based therapeutic in experimental angiotensin II mediated acute hypertension. Hypertension. 2010;56:1152–1159. doi: 10.1161/HYPERTENSIONAHA.110.159210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKie PM, Cataliotti A, Ichiki T, Sangaralingham SJ, Chen HH, Burnett JC Jr. M - atrial natriuretic peptide and nitroglycerin in a canine model of experimental acute hypertensive heart failure: differential actions of 2 cGMP activating therapeutics. Journal of the American Heart Association. 2014;3:e000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen HHW S Iyer SR Sangaralingham J Cannone V Burnett JC A First-In-Human Study Of MANP: A Novel Atrial Natriuretic Peptide Analogue In Human Hypertension. Hypertension. 2021;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams B, Cockcroft JR, Kario K, Zappe DH, Brunel PC, Wang Q, Guo W. Effects of Sacubitril/Valsartan Versus Olmesartan on Central Hemodynamics in the Elderly With Systolic Hypertension: The PARAMETER Study. Hypertension. 2017;69:411–420. doi: 10.1161/hypertensionaha.116.08556 [DOI] [PubMed] [Google Scholar]
- 26.Oda S, Sano T, Morishita Y, Matsuda Y. Pharmacological profile of HS-142-1, a novel nonpeptide atrial natriuretic peptide (ANP) antagonist of microbial origin. II. Restoration by HS-142-1 of ANP-induced inhibition of aldosterone production in adrenal glomerulosa cells. J Pharmacol Exp Ther. 1992;263:241–245. [PubMed] [Google Scholar]
- 27.Olson LJ, Lowe DG, Drewett JG. Novel natriuretic peptide receptor/guanylyl cyclase A-selective agonist inhibits angiotensin II- and forskolin-evoked aldosterone synthesis in a human zona glomerulosa cell line. Molecular pharmacology. 1996;50:430–435. [PubMed] [Google Scholar]
- 28.Anand-Srivastava M, Sairam M, Cantin M. Ring-deleted analogs of atrial natriuretic factor inhibit adenylate cyclase/cAMP system. Possible coupling of clearance atrial natriuretic factor receptors to adenylate cyclase/cAMP signal transduction system. Journal of Biological Chemistry. 1990;265:8566–8572. [PubMed] [Google Scholar]
- 29.Gambaryan S, Butt E, Marcus K, Glazova M, Palmetshofer A, Guillon G, Smolenski A. cGMP-dependent protein kinase type II regulates basal level of aldosterone production by zona glomerulosa cells without increasing expression of the steroidogenic acute regulatory protein gene. J Biol Chem. 2003;278:29640–29648. doi: 10.1074/jbc.M302143200 [DOI] [PubMed] [Google Scholar]
- 30.Spiessberger B, Bernhard D, Herrmann S, Feil S, Werner C, Luppa PB, Hofmann F. cGMP-dependent protein kinase II and aldosterone secretion. Febs J. 2009;276:1007–1013. doi: 10.1111/j.1742-4658.2008.06839.x [DOI] [PubMed] [Google Scholar]
- 31.MacFarland RT, Zelus BD, Beavo JA. High concentrations of a cGMP-stimulated phosphodiesterase mediate ANP-induced decreases in cAMP and steroidogenesis in adrenal glomerulosa cells. The Journal of biological chemistry. 1991;266:136–142. [PubMed] [Google Scholar]
- 32.Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time monitoring of the PDE2 activity of live cells: hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. The Journal of biological chemistry. 2005;280:1716–1719. doi: 10.1074/jbc.C400505200 [DOI] [PubMed] [Google Scholar]
- 33.Assenza MR, Barbagallo F, Barrios F, Cornacchione M, Campolo F, Vivarelli E, Gianfrilli D, Auletta L, Soricelli A, Isidori AM, et al. Critical role of phosphodiesterase 2A in mouse congenital heart defects. Cardiovasc Res. 2018;114:830–845. doi: 10.1093/cvr/cvy030 [DOI] [PubMed] [Google Scholar]
- 34.Chen Y, Schaefer JJ, Iyer SR, Harders GE, Pan S, Sangaralingham SJ, Chen HH, Redfield MM, Burnett JC Jr. Long-term blood pressure lowering and cGMP-activating actions of the novel ANP analog MANP. Am J Physiol Regul Integr Comp Physiol. 2020;318:R669–r676. doi: 10.1152/ajpregu.00354.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards AM, Nicholls MG, Espiner EA, Ikram H, Yandle TG, Joyce SL, Cullens MM. Effects of alpha-human atrial natriuretic peptide in essential hypertension. Hypertension. 1985;7:812–817. doi: 10.1161/01.hyp.7.5.812 [DOI] [PubMed] [Google Scholar]
- 36.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, et al. Cyclic nucleotide analogs as probes of signaling pathways. Nature methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277 [DOI] [PubMed] [Google Scholar]
- 37.Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ Res. 2015;117:707–719. doi: 10.1161/circresaha.114.305892 [DOI] [PubMed] [Google Scholar]
- 38.Feiteiro J, Verde I, Cairrão E. Cyclic guanosine monophosphate compartmentation in human vascular smooth muscle cells. Cellular signalling. 2016;28:109–116. doi: 10.1016/j.cellsig.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 39.Chartier L, Schiffrin EL. Role of calcium in effects of atrial natriuretic peptide on aldosterone production in adrenal glomerulosa cells. Am J Physiol. 1987;252:E485–491. doi: 10.1152/ajpendo.1987.252.4.E485 [DOI] [PubMed] [Google Scholar]
- 40.Barrett PQ, Isales CM, Bollag WB, McCarthy RT. Modulation of Ca2+ channels by atrial natriuretic peptide in the bovine adrenal glomerulosa cell. Canadian journal of physiology and pharmacology. 1991;69:1553–1560. doi: 10.1139/y91-231 [DOI] [PubMed] [Google Scholar]
- 41.Liang F, Kapoun AM, Lam A, Damm DL, Quan D, O’Connell M, Protter AA. B-Type natriuretic peptide inhibited angiotensin II-stimulated cholesterol biosynthesis, cholesterol transfer, and steroidogenesis in primary human adrenocortical cells. Endocrinology. 2007;148:3722–3729. doi: 10.1210/en.2006-1599 [DOI] [PubMed] [Google Scholar]
- 42.Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. Mol Cell Endocrinol. 2004;217:67–74. doi: 10.1016/j.mce.2003.10.011 [DOI] [PubMed] [Google Scholar]
- 43.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC Jr., Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–165. doi: 10.1056/NEJMoa0706300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kloner RA. Cardiovascular effects of the 3 phosphodiesterase-5 inhibitors approved for the treatment of erectile dysfunction. Circulation. 2004;110:3149–3155. doi: 10.1161/01.Cir.0000146906.42375.D3 [DOI] [PubMed] [Google Scholar]
- 45.Kudo T, Baird A. Inhibition of aldosterone production in the adrenal glomerulosa by atrial natriuretic factor. Nature. 1984;312:756–757. doi: 10.1038/312756a0 [DOI] [PubMed] [Google Scholar]
- 46.Schiebinger RJ, Kem DC, Brown RD. Effect of atrial natriuretic peptide on ACTH, dibutyryl cAMP, angiotensin II and potassium-stimulated aldosterone secretion by rat adrenal glomerulosa cells. Life Sci. 1988;42:919–926. doi: 10.1016/0024-3205(88)90391-8 [DOI] [PubMed] [Google Scholar]
- 47.Goodfriend TL, Elliott ME, Atlas SA. Actions of synthetic atrial natriuretic factor on bovine adrenal glomerulosa. Life Sci. 1984;35:1675–1682. doi: 10.1016/0024-3205(84)90179-6 [DOI] [PubMed] [Google Scholar]
- 48.Elliott ME, Goodfriend TL. Effects of atrial natriuretic peptide, angiotensin, cyclic AMP, and potassium on protein phosphorylation in adrenal glomerulosa cells. Life Sci. 1987;41:2517–2524. doi: 10.1016/0024-3205(87)90436-x [DOI] [PubMed] [Google Scholar]
- 49.Xing Y, Rainey WE, Apolzan JW, Francone OL, Harris RB, Bollag WB. Adrenal cell aldosterone production is stimulated by very-low-density lipoprotein (VLDL). Endocrinology. 2012;153:721–731. doi: 10.1210/en.2011-1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nogueira EF, Rainey WE. Regulation of aldosterone synthase by activator transcription factor/cAMP response element-binding protein family members. Endocrinology. 2010;151:1060–1070. doi: 10.1210/en.2009-0977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.