

Abstract
Recent findings:
The continuing discoveries of genetic causes for dystonia syndromes are transforming our view of these disorders. They share unexpectedly common underlying mechanisms, including dysregulation in neurotransmitter signaling, gene transcription, and quality control machinery. The field has further expanded to include forms recently associated with endolysosomal dysfunction.
Purpose of review:
We describe here how such mechanisms shared by different genetic forms can give rise to motor performance dysfunctions with a clinical aspect of dystonia.
Summary:
The discovery of biological pathways shared between different monogenic dystonias is an important conceptual advance in the understanding of the underlying mechanisms, with a significant impact on the pathophysiological understanding of clinical phenomenology. The functional relationship between dystonia genes could revolutionize current dystonia classification systems, classifying patients with different monogenic forms based on common pathways. The most promising effect of these advances is on future mechanism-based therapeutic approaches.
Keywords: dystonia, genetics, environment, molecular mechanisms, movement disorders
Introduction
Dystonia is a collection of physical signs that are variably aggregated in different syndromes (1). The current definition and classification of dystonia provide a common language that is used to describe and classify these diverse syndromes (2,3). Dystonia is characterized by hyperkinetic features, such as involuntary postures and movements, including tremor; but at the same time displays hypokinetic features, particularly a slowing of voluntary movement (4,5).
The complexity of clinical presentations have been ordered by the current classification scheme (2). This two-axis system has been well received and widely adopted (6). According to axis 1, dystonia can occur in isolation (isolated dystonia), or in combination with another movement disorder, or with additional neurological and medical problems. Etiology of dystonia (axis 2) can be idiopathic (unknown), inherited (of genetic origin) or acquired (environmental) (2). Many genes are known to cause dystonia; a gene list aggregated by phenotype has been recently published (1), but the listing is far from complete. Whole-genome sequencing of an unselected patient population with dystonia showed that approximately 12% of non-acquired dystonia patients carried a pathogenic gene mutation (7). This suggests that dystonia is idiopathic in the vast majority of patients, with a minority of cases being inherited. There is a high degree of phenotypic overlap in patients with inherited dystonia and their phenotypic differences are of limited use for guessing any specific genetic etiology (8*). Performing wide gene panels or whole exome/genome sequencing is warranted for cases where an inherited etiology is considered (9).
The complex derangement of voluntary movement observed in dystonia is currently interpreted as due to the disorganization of an anatomical network including widespread motor and sensory brain regions (10). Network derangement can occur in patients who suffer from discrete brain lesions and have a relatively homogeneous dystonia phenotype (11) as well as in patients with inherited disorders (12).
As the number of genes associated with dystonia and the phenotypic heterogeneity of cases sharing the same genetic cause increase, the correlation between clinical and molecular findings seems to lead to apparent paradoxes. We here try to shed light on these paradoxes, unraveling how shared molecular mechanisms explain similar phenotypes in different monogenic forms, and on the other hand how heterogeneity in dystonia can be attributable to a common underlying mechanism.
How can so many different genes cause the same disorder?
The continuous progress in identifying genetic causes of dystonias represents a huge opportunity to understand the biological complexity underlying pathogenesis. The more dystonia genes emerge in different forms, the more we are beginning to discern the existence of previously unexplored biological convergences among the underlying molecular mechanisms. Many pathways of convergence have been described. The main ones include dopamine transmission, gene transcription machinery, endoplasmic reticulum (ER) -mediated cellular stress response, and more recently also vesicular/endolysosomal trafficking. It is therefore possible that the pathology underlying the spectrum of individual monogenic dystonias may ultimately be attributed to abnormalities in a few convergent pathways. There are several prior reviews of some of these pathways (13–16), so only the most important and recent ones are highlighted here.
Dopamine Transmission
Defects in nigrostriatal dopamine transmission have been firmly implicated in dystonia pathogenesis. Based on the nature of enzymatic defects they can be grouped in: a) enzymatic defects in BH4 metabolism: GTP cyclohydrolase 1 (GCH1), sepiapterin reductase (SPR), dihydropteridine reductase (DHPR), 6-pyruvoyl tetrahydropterin synthase (PTS), pterin-4a-carbinolamine dehydratase (PCD) deficiencies b) Primary neurotransmitter synthesis defects: tyrosine hydroxylase (TH), aromatic amino acid decarboxylase deficiency (DDC) c) Monoamine transportopathies (SLC6A3 and SLC18A2) (17,18*)
Genetic variants in GCH1, SPR, and PTS lead to dystonias by altering the biosynthetic pathway of tetrahydrobiopterin (BH4). BH4 deficiency is characterized by insufficient synthesis of the monoamine neurotransmitters dopamine and serotonin due to a disturbance of BH4 biosynthesis or recycling. Recently, guidelines according to the SIGN (Scottish Intercollegiate Guidelines Network) methodology were proposed by evaluating all available evidence for the diagnosis and treatment of BH4 deficiencies (19). Beyond the dysfunction linked to dopamine transport and signaling in the striatum, other potential mechanisms are emerging. A proteomic analysis on TH knock in mice revealed that nearly 20% of the differentially regulated striatal proteins are encoded by genes reported to be involved in inherited disorders characterized by dystonic features, suggesting shared mechanisms across many different forms of dystonia (20*).
More hints on the mechanisms underlying these disorders come from the discovery of new genes. DNAJC12, a gene encoding a heat shock protein that likely increases TH stability in nigrostriatal dopaminergic neurons, was found to be causative in cases with intellectual disability, developmental delay, often in combination with prominent dystonia and parkinsonism (21,22). More recently, independent reports associated mutations of the NR4A2 gene, previously implicated in a neurodevelopmental disorder with autism, intellectual disability, and epilepsy (23), to cases presenting with early-onset dystonia and dopa-responsive dystonia-parkinsonism (24–26). Finally, new achievements from experimental models suggest interesting links between other monogenic forms of isolated dystonia and impaired transmission and dopaminergic signaling (27,28*). Altogether, these observations provide strong support for abnormal dopamine transmission as a shared mechanism for multiple different types of dystonia.
Gene transcription/regulation
Abnormal regulation of gene transcription for dystonia is supported by the identification of dystonia-causing mutations in THAP1 and KMT2B (29,30). KMT2B, encoding a histone H3 methyltransferase, is involved in chromatin plasticity and epigenetic control during early development (31). A recent study demonstrated that KMT2B loss of function leads to a non-random DNA hypermethylation selectively involving promoters and other regulatory regions that control gene expression (32**). Importantly, this study proved the feasibility of validating childhood-onset associated pathogenic KMT2B variants by methylation analysis on peripheral blood DNA.
Recent observations associated pathogenic variants within the YY1 gene with dystonia (33–35). YY1 encodes yin and yang 1, a zinc-finger transcription factor involved in neurodevelopment, which is known to have an established role in oligodendrocyte lineage progression. An interplay between THAP1 and YY1 has been suggested from several lines of evidence. THAP1 loss may alter brain myelination by significantly reducing DNA binding of YY1 (36*). Also, alterations in various neurotransmitter release cycle pathways, extracellular matrix organization, deoxyribonucleic acid methylation, and differential expression of transcription factors, including YY1 and THAP1 itself emerged from a transcriptome analysis in induced pluripotent stem cells (iPSCs)-derived neurons of THAP1 manifesting versus non-manifesting patients (37*).
We are only starting to understand the DNA regulatory mechanism complex linked to transcription factors genes associated with dystonia. Recently, THAP1, YY1, and HCF1 were found to bind directly to the promoter of SHLD1, a component of the Shieldin complex, which shields double-strand DNA breaks from nucleolytic resection (38*). Altogether, these observations suggest that another shared mechanism for many types of dystonia may involve abnormal neural development. A critical role for developmental processes has been recognized before, including specific time windows in which the abnormality may occur (39).
Quality Control Machinery and Trafficking
A recent area of mechanistic convergence among different dystonia genes is the quality control machinery. ER-associated degradation (ERAD) is the major pathway by which folding of proteins is continuously monitored and misfolded polypeptides are transferred from the ER to the cytosol to be degraded by the proteasome (40). The ER undergoes continuous dynamic remodeling, especially in response to specific stimuli, as in the unfolded protein response (UPR). Downstream UPR, the cytosolic signaling cascades mediate restoration of ER status by arresting protein translation, via PERK‐mediated phosphorylation of the translation factor eIF2α (eukaryotic initiation factor 2 alpha), and by transcriptional upregulation of lumenal oxidoreductases and chaperones, and ERAD (41). Substrates of ERAD are limited to proteins. On the other hand, the autophagy-lysosome system is able to degrade parts of the ER, including membranes. Proteins, membranes and organelles are delivered to the lysosomes via different routes including ER-phagy, ERES-microautophagy, or vesicular transport (Fig. 1). Interestingly, newly discovered monogenic forms of dystonia are linked at different level to the quality control machinery (42).
Figure 1.
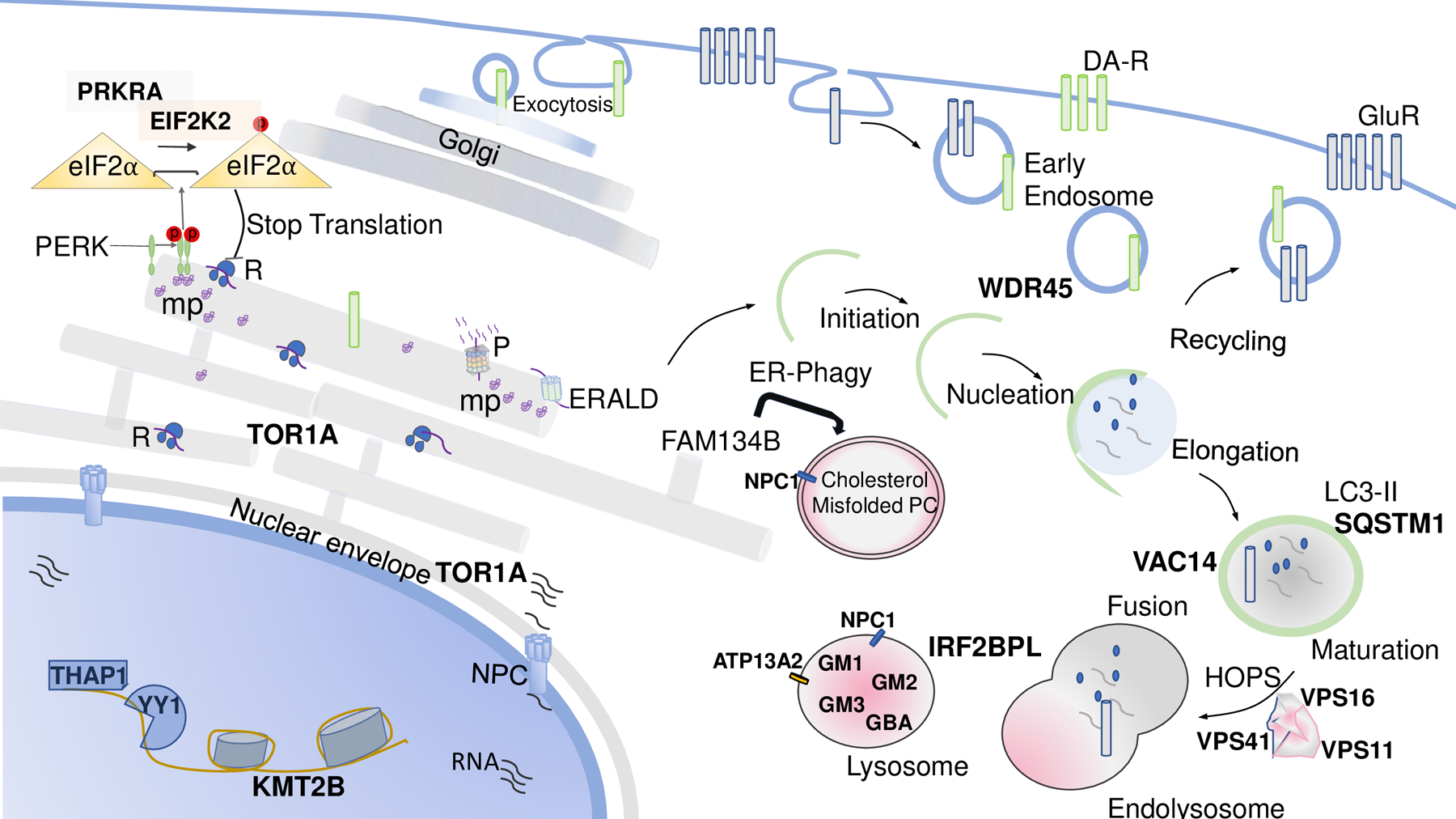
Schematic representation of converging biological processes associated to dystonia syndromes. Abbreviations: DA-R, Dopamine Transporters; GluR, Glutamate Transportes; HOPS, homotypic fusion and vacuole protein sorting; mp, misfolded proteins; NPC, nuclear pore complex; R, ribosome; P, proteasome; PERK, PKR-like ER kinase; PC, procollagen.
The TOR1A gene encodes for torsinA, a protein embedded in the lumen of the endoplasmic reticulum and the endomembrane space of nuclear envelope (43,44). Different functional roles have been attributed to torsinA, including being a component of the cytoskeleton and the nuclear envelope, and involvement in the secretory pathway and synaptic vesicle machinery. Experimental models using transgenic rodents, and more recently iPSCs-derived neurons from patients carrying the common TOR1A ΔGAG deletion support the impairment of nuclear envelope and impaired nucleocytoplasmic transport eventually leading to mislocalization of mRNAs and proteins (45). In addition, torsinA associates with proteins implicated in ERAD, where fibroblasts from TOR1A-associated dystonia patients are more sensitive to ER stress and less able to degrade proteins that have to undergo ERAD-degradation (46). Several reports linked expression of this gene with dysregulation of eIF2α (47).
A new actor recently implicated the pathogenesis of dystonia is the eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) (48**). EIF2AK2 phosphorylates eIF2α in the presence of ER stress. Of note, EIF2AK2 activity is regulated by the interferon-inducible double-stranded RNA-dependent Protein Kinase Activator A (PRKRA), whose mutations cause a rare form of young onset parkinsonism-dystonia (49,50). To further support the molecular link with stress response, fever-induced decompensation has recently been described in a patient carrying an EIF2AK2 novel variant (51,52).
The autophagy-lysosomal system has been implicated in the pathogenesis of dystonia at different levels. The homotypic fusion and protein sorting (HOPS) complex physiologically facilitates the fusion of autophagosomes with lysosomes, which is a critical step in the process of cellular clearing and protein homeostasis. Recent publications reported genetic variants in genes encoding central parts of the HOPS complex to cause dystonia of various degrees, which were grouped under the term HOPS-associated neurological disorders (53–56**). Three of the six vacuolar protein sorting-C proteins, forming the HOPS complex, have been identified to cause autosomal-dominant (VPS16) or autosomal-recessive (VPS11, VPS16, and VPS41) dystonia. In most cases, dystonic features first presented in infancy (VPS41) or adolescence (VPS11), whereas onset in VPS11-mutated patients was in adulthood with progressive generalization. Additional symptoms, including epilepsy, polyneuropathy, optic neuropathy and spasticity, have been described for manifestations in infancy. Functional studies on patient’s cells documented impairment of the autophagolysosome for all three gene mutations (57**).
Furthermore, lysosomal storage disorders have been reported to cause combined dystonia, which has been particularly recognized in cases of Niemann-Pick type C, fucosidosis, GM1, and GM2 gangliosidosis, and Gaucher’s disease type 3. Recently, GM3 deficiency has been linked with dystonic phenotypes (58). The I1061T NPC1 mutant accumulates in the ER lumen and is degraded by two independent pathways functioning in a complementary fashion, in part by ER-phagy in a FAM134B- and autophagy-dependent process; in part, it is triaged through ERAD by the proteasome (59). Dystonic phenotypes have been associated with other endo-lysosomal and autophagy related genes — WDR45, VAC14 and ATP13A2 — further establishes the growing link between lysosomal dysfunction and dystonia (57). In addition, biallelic loss-of-function SQSTM1 variants were associated with progressive dystonia, chorea, ataxia, and vertical gaze palsy in several patients (60), and pathogenic IRF2BPL variants were identified in cases featuring dystonia as part of a complex neurodevelopmental syndrome (61,62). Intriguingly, enlarged lysosomes filled with an osmiophilic material were reported in a skin biopsy from a patient with a pathogenic IRF2BPL variant, suggesting a possible crucial role in lysosomal function (63).
How much the dysfunction of the autophagy-lysosomal system will contribute to the pathogenesis of isolated dystonias, rather than more complex forms, remains to be understood. Of course, the discovery of these forms opens a window to future possible treatments targeting the lysosome in different forms of dystonia.
How can a single gene cause multiple different phenotypes?
Most dystonia genes are associated with significant heterogeneity of clinical phenotypes. For example, a common GAG deletion in TOR1A is typically associated with dominantly inherited childhood-onset dystonia that begins in one leg and then spreads to a generalized pattern (8). However, the same gene deletion can also begin elsewhere before spreading to generalized dystonia, or cause focal dystonia that does not spread, and remains limited to the neck, the larynx, or to another region. Sometimes, onset is delayed until adulthood. The same deletion has also been reported to cause a tremor without overt dystonia. Finally, the deletion is only 30% penetrant, which means it causes no apparent clinical phenotype in 70% of carriers.
The same type of clinical heterogeneity can be seen with genes responsible for other isolated dystonias, such as THAP1 (8). Genetic variants in THAP1 are typically associated with segmental craniocervical dystonia with onset in adolescence or early adulthood. However, genetic variants may also be associated with onset in other age groups, focal dystonia affecting different body parts, generalized dystonia, and tremor without overt dystonia. About half of the carriers of a pathogenic variant in THAP1 are non-penetrant. Significant clinical heterogeneity also occurs for other genes associated with isolated dystonia such as ANO3 or GNAL. These observations for the isolated dystonias raise questions regarding the mechanisms responsible for so much phenotypic heterogeneity from a single gene. Phenotypic heterogeneity can express as quantitative or qualitative differences, whereby genetic variations influence the clinical severity or the clinical expression.
Different genetic variants may have different consequences. The mechanisms responsible for this remarkable clinical heterogeneity among the isolated dystonias are not known. However, multiple clues have been obtained from other disorders where dystonia is combined with additional neurological or medical problems. The first potential mechanism involves how severely the genetic variant impacts the function of the associated protein. Some variants cause complete loss of protein function, while others produce only partial loss of function. Variants causing complete loss of function often produce a severe or earlier onset phenotype, while those causing partial loss of function often produce less severe or later-onset phenotypes. One well known example involves the HPRT1 gene, which encodes for the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGprt) (64). Genetic variants in HPRT1 that result in complete loss of HGprt function result in Lesch-Nyhan disease, characterized by infantile-onset severe generalized dystonia, along with cognitive and behavioral problems. Genetic variants in HPRT1 that permit a very small amount of residual enzyme function may result in less severe generalized dystonia, with little or no cognitive or behavioral problems. Genetic variants in HPRT1 that permit even more residual enzyme function may produce very mild dystonia or even focal dystonia (65).
This mechanism of variable loss of protein function has been established for numerous other genes as well, and could explain clinical heterogeneity for genes where there are numerous different genetic variants that could have different effects on their protein products, such as THAP1, ANO3 or GNAL.
Dystonia genes interact with other genes, a mechanism that could account for clinical heterogeneity associated with a single dystonia gene. An interaction with other genes could exaggerate the pathological effects of the dystonia gene, or could compensate for its defects. There is no evidence supporting this potential mechanism for any of the isolated dystonia genes; but there are data supporting this mechanism for combined dystonia syndromes. One example involves the GCH1 gene, which causes dopa-responsive dystonia that is often combined with parkinsonism (18). This disease is also associated with considerable variation in the clinical phenotype, with onset in children or adults, focal and generalized forms, and varying requirements for levodopa. Pathogenic variants in GCH1 are only partly penetrant, with symptomatic females outnumbering males approximately 4:1, even for the same genetic variant in the same family (66). These observations provide evidence that genes on the sex chromosomes have a sizable influence on the expression of dystonia. What genes are responsible for sex differences associated with GCH1 variants is unknown. There could be genes on the X-chromosome that make females more vulnerable, or there could be genes on the Y-chromosome that protect males. It is interesting to note that there is a significant female predominance for most other types isolated dystonia as well (67*).
Dystonia genes interact with environmental influences, providing a third mechanism that might account for clinical heterogeneity associated with a single dystonia gene. There are numerous epidemiological studies that have disclosed these potential influences on the incidence and severity of different types of isolated dystonias, although the mechanisms are not well understood. Some of the best illustrations of the gene-environment interaction come from other types of dystonia that are combined with additional problems. One particularly well-studied example is glutaric aciduria type 1, associated with the GCDH gene (68). Individuals with pathogenic defects in GCDH may remain virtually symptom-free for their entire lives. However, if they experience a high fever during a specific developmental window (usually less than 2 years of age), they may suffer an acute encephalopathic crisis with striatal necrosis and permanently disabling severe generalized dystonia with parkinsonism. This fever is typically caused by a common infectious disease, such as a respiratory virus or a gastrointestinal bacterium. Fortunately, the mechanisms responsible for fever-induced striatal necrosis have been sufficiently well characterized and effective treatments have been developed (68).
Another well-established gene-environment relationship involves GLUT1, where some genetic variants cause paroxysmal dystonia (69). Symptoms occur in children or young adults, and they are reliably triggered by fasting. This gene encodes a transporter that is essential for moving glucose from the blood into the brain. Genetic variants that produce a weak transporter are adequate for maintaining brain glucose when serum glucose levels are high, but they are unable to maintain brain glucose when blood glucose levels drop during fasting (70). Useful treatments involve avoidance of fasting, administration of triheptanoin as an alternative energy source that does not require the glucose transporter, or a ketogenic diet, where ketones replace glucose as energy source.
Another interesting gene-environment interaction involves the ATP1A3 gene, which is associated with rapid-onset dystonia-parkinsonism (71). Pathogenic variants in this gene may again remain silent for life, but symptoms can be triggered by psychological or physical stress. Similar to variants associated with GCDH, symptoms associated with ATP1A3 begin rapidly, over a period of hours or days, and they often become permanent. However, symptoms associated with ATP1A3 typically first emerge in teenagers or young adults, not in children less than 2 years old. Another difference between GCDH and ATP1A3 is that there is no evidence for striatal necrosis associated with ATP1A3. The biological mechanisms responsible for stress-induced dystonia associated with ATP1A3 are not known. However, it is interesting to note that stress transiently exaggerates symptoms in many different types of dystonia, both inherited and idiopathic.
Although these example disorders of gene-environment interaction are all quite rare, they show how disease associated with a single gene can be markedly influenced by common environmental factors such as infectious diseases, diet, brain stimulation, or even psychological stress.
Conclusion
Dystonia is a complex and intriguing movement disorder both for the clinician and the neuroscientist. Inherited dystonias provide the opportunity to understand the complexity of mechanisms that cause these clinical features.
Key Points.
New genetic discoveries reveal common mechanisms underlying the pathogenesis of dystonia
Endolysosomal trafficking defect is emerging as a new pathogenetic mechanism in dystonia
Dystonia genes interaction with environmental factors account for clinical heterogeneity
Financial Support
HAJ is supported by grants to The Dystonia Coalition (NS065701, TR001456, NS116025) which is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported by the Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), and the National Institute of Neurological Diseases and Stroke (NINDS). He is also supported by NIH R01 grants (NS109242, NS119758, NS119831).
Conflicts of Interest
AA has received honoraria from Merz Pharma and Ipsen Pharma and unrestricted research support from Boston Scientific.
H. A. Jinnah has grant support (recent, active, or pending) from the US government (National Institutes of Health), private philanthropic organizations (Cure Dystonia Now), and industry (Addex, Aeon, Revance, Jazz). Dr. Jinnah has also served on advisory boards or as a consultant for Addex, Allergan, Apello, CoA Therapeutics, Cavion, Daiichi Sankyo, Ipsen, PureTech, Retrophin, Revance and Takaha/Ene. He has received honoraria or stipends for lectures or administrative work from the International Parkinson’s Disease and Movement Disorders Society. Dr. Jinnah has also served on the Scientific Advisory Boards for several private foundations including the Benign Essential Blepharospasm Research Foundation, Cure Dystonia Now, the Dystonia Medical Research Foundation, the Tourette Association of America, and Tyler’s Hope for a Cure. He also is principle investigator for the Dystonia Coalition, which has received the majority of its support through the NIH (grants NS116025, NS065701 from the National Institutes of Neurological Disorders and Stroke TR001456 from the Office of Rare Diseases Research at the National Center for Advancing Translational Sciences). The Dystonia Coalition has received additional material or administrative support from industry sponsors (Allergan Inc. and Merz Pharmaceuticals) as well as private foundations (The Benign Essential Blepharospasm Foundation, Cure Dystonia Now, The Dystonia Medical Research Foundation, and The National Spasmodic Dysphonia Association).
ADF has received honoraria from Sanofi Genzyme and Zambon for Scientific Advisory Board and invited lecturers.
References
- 1.Albanese A, Di Giovanni M, Lalli S. Dystonia: diagnosis and management. Eur J Neurol. 2019. Jan;26(1):5–17. [DOI] [PubMed] [Google Scholar]
- 2.Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VSC, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013. Jun;28(7):863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinnah HA, Albanese A. The New Classification System for the Dystonias: Why Was it Needed and How was it Developed? Mov Disord Clin Pract. 2014. Dec;1(4):280–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, Marsden CD. The pathophysiology of primary dystonia. Brain. 1998. Jul;121 (Pt 7:1195–212. [DOI] [PubMed] [Google Scholar]
- 5.Shaikh AG, Wong A, Zee DS, Jinnah HA. Why are voluntary head movements in cervical dystonia slow? Parkinsonism Relat Disord. 2015. Jun;21(6):561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasikumar S, Albanese A, Krauss JK, Fasano A. Implementation of the Current Dystonia Classification from 2013 to 2018. Mov Disord Clin Pract. 2019. Mar;6(3):250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar KR, Davis RL, Tchan MC, Wali GM, Mahant N, Ng K, et al. Whole genome sequencing for the genetic diagnosis of heterogenous dystonia phenotypes. Parkinsonism Relat Disord. 2019. Dec;69:111–8. [DOI] [PubMed] [Google Scholar]
- 8.*.Lange LM, Junker J, Loens S, Baumann H, Olschewski L, Schaake S, et al. Genotype-Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov Disord. 2021. May;36(5):1086–103. [DOI] [PubMed] [Google Scholar]; This review provides a comprehensive overview of the current knowledge of hereditary isolated dystonia.
- 9.Pozojevic J, Beetz C, Westenberger A. The importance of genetic testing for dystonia patients and translational research. J Neural Transm. 2021. Apr;128(4):473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schirinzi T, Sciamanna G, Mercuri NB, Pisani A. Dystonia as a network disorder: a concept in evolution. Curr Opin Neurol. 2018. Aug;31(4):498–503. [DOI] [PubMed] [Google Scholar]
- 11.Corp DT, Joutsa J, Darby RR, Delnooz CCS, van de Warrenburg BPC, Cooke D, et al. Network localization of cervical dystonia based on causal brain lesions. Brain. 2019. Jun;142(6):1660–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebrahimi-Fakhari D, Van Karnebeek C, Münchau A. Movement Disorders in Treatable Inborn Errors of Metabolism. Mov Disord. 2019. May;34(5):598–613. [DOI] [PubMed] [Google Scholar]
- 13.Keller Sarmiento IJ, Mencacci NE. Genetic Dystonias: Update on Classification and New Genetic Discoveries. Curr Neurol Neurosci Rep. 2021. Feb;21(3):8. [DOI] [PubMed] [Google Scholar]
- 14.Jinnah HA, Sun YV. Dystonia genes and their biological pathways. Neurobiol Dis. 2019. Sep;129:159–68. [DOI] [PubMed] [Google Scholar]
- 15.Balint B, Mencacci NE, Valente EM, Pisani A, Rothwell J, Jankovic J, et al. Dystonia. Nat Rev Dis Prim. 2018. Sep;4(1):25. [DOI] [PubMed] [Google Scholar]
- 16.Weisheit CE, Pappas SS, Dauer WT. Inherited dystonias: clinical features and molecular pathways. Handb Clin Neurol. 2018;147:241–54. [DOI] [PubMed] [Google Scholar]
- 17.Cherian A, Paramasivan NK, Divya KP. Dopa-responsive dystonia, DRD-plus and DRD look-alike: a pragmatic review. Acta Neurol Belg. 2021. Jun;121(3):613–23. [DOI] [PubMed] [Google Scholar]
- 18.*.Weissbach A, Pauly MG, Herzog R, Hahn L, Halmans S, Hamami F, et al. Relationship of Genotype, Phenotype, and Treatment in Dopa-Responsive Dystonia: MDSGene Review. Mov Disord. 2022. Feb;37(2):237–52. [DOI] [PubMed] [Google Scholar]; In this systematic review provides a comprehensive overview and analysis of genetic, clinical, treatment, and biochemical data in the six most frequent monogenic forms of DRD
- 19.Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson TS, Sivri HS, Yildiz Y, et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH(4)) deficiencies. Orphanet J Rare Dis. 2020. May;15(1):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.*.Briscione MA, Dinasarapu AR, Bagchi P, Donsante Y, Roman KM, Downs AM, et al. Differential expression of striatal proteins in a mouse model of DOPA-responsive dystonia reveals shared mechanisms among dystonic disorders. Mol Genet Metab. 2021. Aug;133(4):352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study discovered differentially regulated proteins through a proteomic approach in a mouse model of DOPA-responsive dystonia. Interestingly, nearly 20% of the differentially regulated striatal proteins are associated with pathogenic variants that cause inherited disorders with dystonia
- 21.Anikster Y, Haack TB, Vilboux T, Pode-Shakked B, Thöny B, Shen N, et al. Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability. Am J Hum Genet. 2017. Feb;100(2):257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straniero L, Guella I, Cilia R, Parkkinen L, Rimoldi V, Young A, et al. DNAJC12 and dopa-responsive nonprogressive parkinsonism. Ann Neurol. 2017. Oct;82(4):640–6. [DOI] [PubMed] [Google Scholar]
- 23.Reuter MS, Krumbiegel M, Schlüter G, Ekici AB, Reis A, Zweier C. Haploinsufficiency of NR4A2 is associated with a neurodevelopmental phenotype with prominent language impairment. Am J Med Genet A. 2017. Aug;173(8):2231–4. [DOI] [PubMed] [Google Scholar]
- 24.Wirth T, Mariani LL, Bergant G, Baulac M, Habert M-O, Drouot N, et al. Loss-of-Function Mutations in NR4A2 Cause Dopa-Responsive Dystonia Parkinsonism. Mov Disord. 2020. May;35(5):880–5. [DOI] [PubMed] [Google Scholar]
- 25.Winter B, Krämer J, Meinhardt T, Berner D, Alt K, Wenzel M, et al. NR4A2 and Dystonia with Dopa Responsiveness. Vol. 36, Movement disorders : official journal of the Movement Disorder Society. United States; 2021. p. 2203–4. [DOI] [PubMed] [Google Scholar]
- 26.Jesús S, Hinarejos I, Carrillo F, Martínez-Rubio D, Macías-García D, Sánchez-Monteagudo A, et al. NR4A2 Mutations Can Cause Intellectual Disability and Language Impairment With Persistent Dystonia-Parkinsonism. Neurol Genet. 2021. Feb;7(1):e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu-Taeger L, Ott T, Bonsi P, Tomczak C, Wassouf Z, Martella G, et al. Impaired dopamine- and adenosine-mediated signaling and plasticity in a novel rodent model for DYT25 dystonia. Neurobiol Dis. 2020. Feb;134:104634. [DOI] [PubMed] [Google Scholar]
- 28.*.Downs AM, Fan X, Kadakia RF, Donsante Y, Jinnah HA, Hess EJ. Cell-intrinsic effects of TorsinA(ΔE) disrupt dopamine release in a mouse model of TOR1A dystonia. Neurobiol Dis. 2021. Jul;155:105369. [DOI] [PMC free article] [PubMed] [Google Scholar]; By conditionally expressing Tor1a(ΔE) in either dopamine neurons or cholinergic interneurons in mice, the authors demonstrated that the defect in striatal dopamine release is caused by the action of the Tor1a(ΔE) mutation within dopamine neurons.
- 29.Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009. Mar;41(3):286–8. [DOI] [PubMed] [Google Scholar]
- 30.Zech M, Boesch S, Maier EM, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am J Hum Genet. 2016. Dec;99(6):1377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Zhao L, Tian X, Peng C, Gong F, Chen Y. Crystal Structure of MLL2 Complex Guides the Identification of a Methylation Site on P53 Catalyzed by KMT2 Family Methyltransferases. Structure. 2020. Oct;28(10):1141–1148.e4. [DOI] [PubMed] [Google Scholar]
- 32.**.Ciolfi A, Foroutan A, Capuano A, Pedace L, Travaglini L, Pizzi S, et al. Childhood-onset dystonia-causing KMT2B variants result in a distinctive genomic hypermethylation profile. Clin Epigenetics. 2021. Aug;13(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that a distinctive DNA hypermethylation pattern is associated with DYT28, providing a novel epigenetic approach to increase diagnostic accuracy in case of ambiguous genetic findings
- 33.Carminho-Rodrigues MT, Steel D, Sousa SB, Brandt G, Guipponi M, Laurent S, et al. Complex movement disorder in a patient with heterozygous YY1 mutation (Gabriele-de Vries syndrome). Vol. 182, American journal of medical genetics. Part A. United States; 2020. p. 2129–32. [DOI] [PubMed] [Google Scholar]
- 34.Zorzi G, Keller Sarmiento IJ, Danti FR, Bustos BI, Invernizzi F, Panteghini C, et al. YY1-Related Dystonia: Clinical Aspects and Long-Term Response to Deep Brain Stimulation. Vol. 36, Movement disorders : official journal of the Movement Disorder Society. United States; 2021. p. 1461–2. [DOI] [PubMed] [Google Scholar]
- 35.Malaquias MJ, Damásio J, Mendes A, Freixo JP, Magalhães M. A Case of YY1-Related Isolated Dystonia with Severe Oromandibular Involvement. Vol. 36, Movement disorders : official journal of the Movement Disorder Society. United States; 2021. p. 2705–6. [DOI] [PubMed] [Google Scholar]
- 36.*.Yellajoshyula D, Rogers AE, Kim AJ, Kim S, Pappas SS, Dauer WT. A pathogenic DYT-THAP1 dystonia mutation causes hypomyelination and loss of YY1 binding. Hum Mol Genet. 2021. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrated that the YY1 mutation that do not occur on residues critical for DNA binding, the F81L mutation, impairs THAP1 transcriptional activity and disrupts CNS myelination.
- 37.*.Baumann H, Ott F, Weber J, Trilck-Winkler M, Münchau A, Zittel S, et al. Linking Penetrance and Transcription in DYT-THAP1: Insights From a Human iPSC-Derived Cortical Model. Mov Disord. 2021. Jun;36(6):1381–91. [DOI] [PubMed] [Google Scholar]; Using a trascriptome approach in iPSCs from manifesting and nonmanifesting THAP1 mutation carriers and controls, the authors show that transcriptional alterations during cortical development influence DYT-THAP1 pathogenesis and penetrance
- 38.*.Shinoda K, Zong D, Callen E, Wu W, Dumitrache LC, Belinky F, et al. The dystonia gene THAP1 controls DNA double-strand break repair choice. Mol Cell. 2021. Jun;81(12):2611–2624.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Here, the authors report that the transcription factors THAP1, YY1, and HCF1 act as a a transcriptional network that directly controls DSB repair choice by binding directly to the SHLD1 promoter. This finding suggests a potential link between DNA damage and pathogenic mutations in these dystonia genes.
- 39.Li J, Kim S, Pappas SS, Dauer WT. CNS critical periods: implications for dystonia and other neurodevelopmental disorders. JCI insight. 2021. Feb;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fregno I, Molinari M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit Rev Biochem Mol Biol. 2019. Apr;54(2):153–63. [DOI] [PubMed] [Google Scholar]
- 41.Wilkinson S ER-phagy: shaping up and destressing the endoplasmic reticulum. FEBS J. 2019. Jul;286(14):2645–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Leonibus C, Cinque L, Settembre C. Emerging lysosomal pathways for quality control at the endoplasmic reticulum. FEBS Lett. 2019. Sep;593(17):2319–29. [DOI] [PubMed] [Google Scholar]
- 43.Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, et al. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000. May;9(9):1403–13. [DOI] [PubMed] [Google Scholar]
- 44.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci U S A. 2004. Jan;101(3):847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding B, Tang Y, Ma S, Akter M, Liu M-L, Zang T, et al. Disease Modeling with Human Neurons Reveals LMNB1 Dysregulation Underlying DYT1 Dystonia. J Neurosci. 2021. Mar;41(9):2024–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nery FC, Armata IA, Farley JE, Cho JA, Yaqub U, Chen P, et al. TorsinA participates in endoplasmic reticulum-associated degradation. Nat Commun. 2011. Jul;2:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beauvais G, Watson JL, Aguirre JA, Tecedor L, Ehrlich ME, Gonzalez-Alegre P. Efficient RNA interference-based knockdown of mutant torsinA reveals reversibility of PERK-eIF2α pathway dysregulation in DYT1 transgenic rats in vivo. Brain Res. 2019. Mar;1706:24–31. [DOI] [PubMed] [Google Scholar]
- 48.**.Kuipers DJS, Mandemakers W, Lu C-S, Olgiati S, Breedveld GJ, Fevga C, et al. EIF2AK2 Missense Variants Associated with Early Onset Generalized Dystonia. Ann Neurol. 2021. Mar;89(3):485–97. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors identified EIF2AK2 variants implicated in early onset generalized dystonia. These findings provide direct evidence for a key role of a dysfunctional eIF2α pathway, which orchestrates the cellular stress response, in the pathogenesis of dystonia.
- 49.Camargos S, Scholz S, Simón-Sánchez J, Paisán-Ruiz C, Lewis P, Hernandez D, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008. Mar;7(3):207–15. [DOI] [PubMed] [Google Scholar]
- 50.Zech M, Castrop F, Schormair B, Jochim A, Wieland T, Gross N, et al. DYT16 revisited: exome sequencing identifies PRKRA mutations in a European dystonia family. Mov Disord. 2014. Oct;29(12):1504–10. [DOI] [PubMed] [Google Scholar]
- 51.Magrinelli F, Moualek D, Tazir M, Pacha LA, Verghese A, Bhatia KP, et al. Heterozygous EIF2AK2 Variant Causes Adolescence-Onset Generalized Dystonia Partially Responsive to DBS. Mov Disord Clin Pract. 2022. Feb;9(2):268–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waller SE, Morales-Briceño H, Williams L, Mohammad SS, Fellner A, Kumar KR, et al. Possible EIF2AK2-Associated Stress-Related Neurological Decompensation with Combined Dystonia and Striatal Lesions. Mov Disord Clin Pract. 2022. Feb;9(2):240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai X, Chen X, Wu S, Liu W, Zhang X, Zhang D, et al. Homozygous mutation of VPS16 gene is responsible for an autosomal recessive adolescent-onset primary dystonia. Sci Rep. 2016. May;6:25834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.**.Monfrini E, Cogiamanian F, Salani S, Straniero L, Fagiolari G, Garbellini M, et al. A Novel Homozygous VPS11 Variant May Cause Generalized Dystonia. Ann Neurol. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]; This study associated for the first time pathogenic variants of VPS11, a gene encoding a subunit of the HOPS complex, with adult-onset dystonia.
- 55.**.Sanderson LE, Lanko K, Alsagob M, Almass R, Al-Ahmadi N, Najafi M, et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain. 2021. Apr;144(3):769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; Here, the authors describe five unrelated families all carrying homozygous variants in VPS41 leading to protein dysfunction and provided evidence of VPS41 role in neurodevelopment in zebrafish model.
- 56.**.Steel D, Zech M, Zhao C, Barwick KES, Burke D, Demailly D, et al. Loss-of-Function Variants in HOPS Complex Genes VPS16 and VPS41 Cause Early Onset Dystonia Associated with Lysosomal Abnormalities. Ann Neurol. 2020. Nov;88(5):867–77. [DOI] [PubMed] [Google Scholar]; This study associate for the first time dystonia syndromes to pathogenic variants in VPS41 and VPS16, and revealed the previously unrecognised role of HOPS complex dysfunction in the pathogenesis of dystonia
- 57.**.Monfrini E, Zech M, Steel D, Kurian MA, Winkelmann J, Di Fonzo A. HOPS-associated neurological disorders (HOPSANDs): linking endolysosomal dysfunction to the pathogenesis of dystonia. Brain. 2021. Oct;144(9):2610–5. [DOI] [PubMed] [Google Scholar]; Here the forms of dystonia associated with genes that encode the HOPS complex are described under the hat of HOPSANDs with a description of the common features and pathogenetic mechanisms
- 58.Wang AS, Kilbane C. Dystonia Due to GM3 Synthase Deficiency. Vol. 9, Movement disorders clinical practice. 2022. p. 236–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, et al. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun. 2018. Sep;9(1):3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zúñiga-Ramírez C, de Oliveira LM, Kramis-Hollands M, Algarni M, Soto-Escageda A, Sáenz-Farret M, et al. Beyond dystonia and ataxia: Expanding the phenotype of SQSTM1 mutations. Parkinsonism Relat Disord. 2019. May;62:192–5. [DOI] [PubMed] [Google Scholar]
- 61.Marcogliese PC, Shashi V, Spillmann RC, Stong N, Rosenfeld JA, Koenig MK, et al. IRF2BPL Is Associated with Neurological Phenotypes. Am J Hum Genet. 2018. Aug;103(2):245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prilop L, Buchert R, Woerz S, Gerloff C, Haack TB, Zittel S. IRF2BPL mutation causes nigrostriatal degeneration presenting with dystonia, spasticity and keratoconus. Vol. 79, Parkinsonism & related disorders. England; 2020. p. 141–3. [DOI] [PubMed] [Google Scholar]
- 63.Ginevrino M, Battini R, Nuovo S, Simonati A, Micalizzi A, Contaldo I, et al. A novel IRF2BPL truncating variant is associated with endolysosomal storage. Mol Biol Rep. 2020. Jan;47(1):711–4. [DOI] [PubMed] [Google Scholar]
- 64.Fu R, Ceballos-Picot I, Torres RJ, Larovere LE, Yamada Y, Nguyen KV, et al. Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain. 2014. May;137(Pt 5):1282–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jinnah HA, Ceballos-Picot I, Torres RJ, Visser JE, Schretlen DJ, Verdu A, et al. Attenuated variants of Lesch-Nyhan disease. Brain. 2010. Mar;133(Pt 3):671–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furukawa Y, Lang AE, Trugman JM, Bird TD, Hunter A, Sadeh M, et al. Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology. 1998. Apr;50(4):1015–20. [DOI] [PubMed] [Google Scholar]
- 67.*.Rafee S, O’Riordan S, Reilly R, Hutchinson M. We Must Talk about Sex and Focal Dystonia. Mov Disord. 2021. Mar;36(3):604–8. [DOI] [PubMed] [Google Scholar]; In this viewpoint paper, the authors argue that the most distinctive aspects of adult onset isolated focal dystonia are the marked sex-related differences demonstrated by epidemiological, clinical, and laboratory studies in patients with adult onset dystonia
- 68.Strauss KA, Williams KB, Carson VJ, Poskitt L, Bowser LE, Young M, et al. Glutaric acidemia type 1: Treatment and outcome of 168 patients over three decades. Mol Genet Metab. 2020. Nov;131(3):325–40. [DOI] [PubMed] [Google Scholar]
- 69.Gras D, Roze E, Caillet S, Méneret A, Doummar D, Billette de Villemeur T, et al. GLUT1 deficiency syndrome: an update. Rev Neurol (Paris). 2014. Feb;170(2):91–9. [DOI] [PubMed] [Google Scholar]
- 70.Valente EM, Albanese A. “Gluing” phenotypes together: the case of GLUT1. Vol. 77, Neurology. United States; 2011. p. 934–5. [DOI] [PubMed] [Google Scholar]
- 71.Heinzen EL, Arzimanoglou A, Brashear A, Clapcote SJ, Gurrieri F, Goldstein DB, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 2014. May;13(5):503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]