

Abstract
Background:
Individuals with late-onset Pompe disease (LOPD) and the common c.-32–13 T > G variant are widely thought to have milder, adult-onset disease. This belief, and the consequent low suspicion of clinical involvement in children, has led to delays in diagnosis and treatment initiation in patients with early onset of symptoms. Previous reports of LOPD in children do not include description of the early-onset phenotype. This description of signs and symptoms, some of which are subtle and less known, is important to facilitate prompt identification and appropriate treatment in symptomatic children.
Methods:
Retrospective chart review of a cohort of 84 LOPD patients with the c.-32–13 T > G variant was conducted to identify patients diagnosed clinically (as opposed to through newborn screening) who had clinically documented symptom-onset within the first two years of life.
Results:
Four patients had early onset of symptoms, with age at onset ranging from 10 days to 20 months. Initial symptoms included delay in achievement of gross motor milestones, signs of proximal muscle weakness, swallow and feeding difficulties, and sleep apnea. Early and characteristic alterations in posture and movement were identified in all patients. Age at diagnosis ranged from 10 months to 26 months. Median age at enzyme replacement therapy (ERT) initiation was 23.5 months. Despite ERT, progression of musculoskeletal involvement and residual muscle weakness was evident in all patients, as evidenced by ptosis, myopathic facies, scoliosis, lumbar lordosis, scapular winging, and trunk and lower extremity weakness. Standardized functional assessments showed gross motor function below age level as measured by the Alberta Infant Motor Scales, the Peabody Developmental Motor Scales-2, the Bruininks-Oseretsky Test of Motor Proficiency, Second Edition, and the six-minute walk test.
Conclusions:
Onset of symptoms including delay in achievement of gross motor milestones, signs of proximal muscle weakness, swallow and feeding difficulties, and sleep apnea in the first two years of life is not uncommon in individuals with LOPD and the c.-32–13 T > G variant. Patients with early-onset disease appear to have a more, rapid and severe progression of disease with persistent residual muscle deficits which partially improve with higher doses of ERT. Careful evaluation for specific and characteristic patterns of posture and movement in patients with this variant is necessary to identify those who have early onset of disease. Increased awareness of the early-onset signs and symptoms may also enable early identification of disease onset in children who are diagnosed through newborn screening.
Keywords: Pompe disease, Glycogen storage disease type II, Enzyme replacement therapy, IVS variant, Six minute walk test, Late-onset Pompe disease, C.-32–13 T > G
1. Introduction
Pompe disease is caused by deficiency of the enzyme acid α-glucosidase (GAA). Classic infantile onset Pompe disease (IOPD) presents in the first year of life with profound hypotonia, generalized muscle weakness and hypertrophic cardiomyopathy. Late-onset Pompe disease (LOPD) is characterized by weakness- primarily in the trunk, proximal lower limb, and respiratory muscles- and can present from the first year of life to as late as the sixth decade [1,2]. The most common LOPD pathogenic variant in Caucasians is the splice variant c.-32–13 T > G in intron 1 of the GAA gene, also known as the common IVS1 variant, found on at least one allele in 68% to 90% of Caucasian patients with LOPD [3–5]. This splice-site variant allows production of low levels of normal GAA and was traditionally believed to present with a mild, adult onset phenotype due to the presence of residual enzyme activity [3,6]. However, there is increasing evidence that the clinical presentation associated with the c.-32–13 T > G variant has a broader spectrum than previously recognized [3,7].
Patients with early childhood and juvenile onset Pompe disease associated with the common c.-32–13 T > G variant have been previously reported. However, these reports are retrospective in nature and lack detailed phenotypic characterizations of the nature and extent of musculoskeletal involvement in the first one to two years of life [8–10]. At this time, there is limited awareness within the pediatric medical community including physical therapists (PTs) regarding clinical characteristics of LOPD that may present early in life. As a result, many patients remain undiagnosed for several years or are diagnosed incorrectly with non-specific developmental delay and/or hypotonia when young, and with other genetic and muscle disorders when older, such as limb-girdle muscular dystrophy, or polymyositis [31]. The diagnostic delay for this group of patients can be up to 12 years [11].
The addition of Pompe disease to the recommended uniform screening panel (RUSP) for newborns in March 2015 has resulted in the identification of patients with IOPD and LOPD in infancy. However, only eight states currently include Pompe disease in their newborn screening (NBS) panels, which indicates that the majority of children with LOPD are still diagnosed in a clinical setting. To avoid diagnostic delays and misdiagnoses, it is important that physicians are aware of the early and subtle signs of musculoskeletal involvement in children with LOPD. A better understanding of the early-onset phenotype of LOPD in children with the common IVS variant will guide timely treatment initiation and management plans for the increasing numbers of patients identified via NBS. We present four patients heterozygous for the c.-32–13 T > G variant and a second pathogenic variant with symptom onset in the first two years of life. These patients represent the first published cohort with a detailed description of early-onset LOPD signs and symptoms and well documented evolution of clinical course before and after ERT.
2. Methods
Medical records of 84 LOPD patients followed at Duke University Medical Center (DUMC) with the c.-32–13 T > G variant were reviewed to identify patients who were clinically diagnosed with Pompe disease. Patients with symptom onset in the first two years of life and receiving ERT were included in the study. Patients who met the above criteria but did not have detailed description of their early symptom evolution and follow up were excluded from this report in an attempt to be precise about the early course and evolution of LOPD in infancy. Children diagnosed through NBS or those with a positive family history in an older sibling were excluded so as to accurately capture an unbiased early-onset phenotype. Four patients, who were compound heterozygous for the c.-32–13 T > G variant and a second pathogenic variant, with reliably documented disease onset in the first two years of life, and were diagnosed clinically were included in this study. Diagnosis of Pompe disease was made by GAA enzyme activity assay and confirmed by pathogenic variant analysis. Medical records were reviewed to determine age at symptom onset, initial symptoms, age at diagnosis, clinical and physical examination findings and progression of clinical course. Physical therapy assessments included qualitative assessment of posture, movement, and musculoskeletal status, as well as standardized assessments including the Alberta Infant Motor Scale (AIMS), Gross Motor Function Measure-88 (GMFM-88), six minute walk test (6MWT), the Peabody Developmental Motor Scales-2nd Edition (PDMS-2), and the Bruininks-Oseretsky Test of Motor Proficiency, 2nd Edition (BOT-2). All standardized assessments were administered and scored in accordance with standardized test procedures specific for each assessment. 6MWT was administered in accordance with American Thoracic Society (ATS) guidelines and was reported as distance walked, with calculation of percent predicted, amount below the mean, and whether within or below the range for age [12–16]. Occupational therapy and speech therapy evaluations assessed oropharyngeal muscle weakness and included barium swallow study. Laboratory assessments including AST, ALT, creatinine kinase (CK), urinary glucose tetra-saccharide (Glc4) and complete metabolic profile, cardiac evaluation including electrocardiography (ECG) and echocardiogram (ECHO), were collected systematically from chart review. Written informed consent was obtained from a parent or guardian for all individuals as part of Duke Institutional Review Board approved protocols.
3. Results
Four patients out of 84 (4.8%) with LOPD and the c.-32–13 T > G variant, two males and two females, were diagnosed clinically and identified with early-onset symptoms. All were compound heterozygous for the c.-32–13 T > G variant and a second pathogenic variant (Table 1). Age at symptom onset ranged between 10 days and 20 months. Age at diagnosis ranged from 10 months to 26 months. Diagnostic delay of 6 months to 18 months was seen in this cohort (median 9.75 months). Median age at ERT initiation was 23.5 months.
Table 1.
Description of patients with the compound heterozygous C.-32–13 T > G variant and early-symptom onset.
| Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Current age (in years) | 13.3 | 7.53 | 9.11 | 2.52 |
| Sex | Male | Female | Male | Female |
| Genotype | c.-32–13 T > G/ c.525delT | c.-32–13 T > G/ c.1827delC | c.-32–13 T > G/ c.2501_2502delCA | c.-32–13 T > G/ c.118C > T |
| Age at symptom onset | 5 months | 10 days | 6 months | 20 months |
| Age at diagnosis | 10 months | 15 months | 24 months | 26 months |
| Age at ERT initiation | 20 months | 19 months | 27 months | 28 months |
| Method of diagnosis | Muscle biopsy | Muscle biopsy | Muscle biopsy | Enzyme activity assay in blood |
| Antibody titers peak (last) | 3200 (3200) | Undetectable | 800 (〈100) | Not available |
| ACE genotype | I/I | Not available | Not available | I/D |
4. Case reports
4.1. Patient 1
A 5-month-old Caucasian male presented with delayed motor development and feeding difficulties. Workup revealed elevated CK, ALT and AST levels. Muscle biopsy at age 9 months showed muscle fibers containing intensely PAS positive vacuoles, consistent with Pompe disease. The diagnosis was confirmed by deficient GAA activity on skin fibroblasts (residual enzyme activity of 2.2 nmol/min/mg protein; 4.35% of normal) at age 10 months. Sequencing of the GAA gene revealed c.-32–13 T > G- and c.525delT (p.Glu176Argfs*45) variants. ECG and ECHO were normal. A clinical opinion was sought at DUMC at age 15 months due to parental concerns. History revealed progression of symptoms such as delayed motor skills, easy fatigability, swallowing difficulty especially with solids, and snoring with likely apneic episodes from age 5 months. At age 15 months, he was able to sit independently, crawl, pull to stand, take a few steps while cruising, and had a pincer grasp. He had generalized hypotonia, head lag when pulled to sit, myopathic facies, and hypertrophied calf muscles (Table 2). EMG showed profuse myotonic discharges consistent with Pompe disease. A barium swallow study demonstrated numerous episodes of penetration with thickened liquids and an episode of aspiration with thin liquids. PFT showed a normal crying vital capacity of 91%. Pulmonary somnography revealed obstructive apneas and hypopneas, central apneas with accompanying oxygen desaturations, oxygenation and ventilation were essentially normal. He was not started on ERT as he did not qualify for the “Myozyme expanded access program” (ClinicalTrials.gov # NCT00074932) at that time.
Table 2.
Comparison of clinical symptoms at disease onset and current status.
| Patient ID | Clinical symptoms | Gross motor/Ambulatory status | Musculoskeletal | Pulmonary | Gastrointestinal | Genitourinary |
|---|---|---|---|---|---|---|
| #1 | At onset | Delayed walking* | Easy fatigability | Sleep apnea | Aspiration during feeding | None |
| Current | Walks unassisted; wide-based gait with frequent tripping and falls; uses motorized scooter for long distances | Ptosis; weakness of neck flexors, shoulder abductors, hip abductors; paraspinal muscles & ankle dorsiflexors; spinal extension with lumbar lordosis, scoliosis, scapular winging, wide based, foot-slapping gait | Unassisted ventilation | None | None | |
| #2 | At onset | Delayed sitting, pull-to-stand, and walking | Lethargy, hypotonia | Apneic episodes | Feeding difficulty, vomiting, diarrhea | None |
| Current | Walks unassisted; wide-based gait | Myopathic facies; scapular winging; weakness of neck flexors, hip abductors, ankle dorsiflexors; genu recurvatum, and medial pronation at ankle, elbow hyperextension; stepto gait pattern on stairs | Unassisted ventilation; New onset snoring and sleep disturbances/apnea; suspected aspiration pneumonia | Choking on saliva, persistent gastroesophageal reflux, | Urinary retention and overflow incontinence, chronic constipation | |
| #3 | At onset | Poor head control till age 6 months | Needing assistance to pull to stand from a prone position, wide-based gait with lumbar lordosis, | Weak cry | None | None |
| Current | Walks unassisted; wide-based gait with frequent tripping and falls | Ptosis; lingual muscle weakness, lumbar lordosis, scapular winging; weakness of neck flexors and abdominals; core muscle weakness, decreased hip extensor and hip abductor strength; scoliosis | Unassisted ventilation | Cramping, vomiting, diarrhea | None | |
| #4 | At onset | Delayed walking | Unsteady gait with frequent falls | Apnea and bradycardia as newborn | None | None |
| Current | Walks unassisted; unsteady and abnormal gait with frequent falls; scoots in sitting for mobility | Delayed motor skills; poor pincer grasp; lumbar lordosis, scapular winging; weakness of abdominals, hip extensors, abductors and adductors, ankle plantar flexors, and neck flexors | Unassisted ventilation; Snoring, apneic pauses |
Lost to follow-up at this age.
At age 19 months, he demonstrated further regression in his motor skills precipitated by a viral illness. Regression of milestones, progressive neuromuscular weakness and deteriorating pulmonary function qualified him for ERT through the Myozyme clinical trial and was initiated at 20 mg/kg every other week (EOW) at age 20 months (Table 1). At age 3.3 years, 20 months post-initiation of ERT, he was an active toddler, walking independently. Parents reported motor regression in the previous year following two episodes of viral illnesses. He had coxa valga deformity, clinical plateau, and rising urine glucose tetrasaccharide (glc4) levels. He had residual muscle deficits on PT assessments. These led to increase in his ERT dose to 30 mg/kg EOW by the treating physician at age 3.4 years, to reach a goal of 40 mg/kg EOW at age 3.6 years. Progressive scoliosis was noted from age 6.5 years. ERT dose was increased to 40 mg/kg/week at age 9.8 years. At follow-up at age 10 years, he continued to walk independently without assistive devices, used a motorized scooter for long distance mobility, participated in sports in school and enjoyed recreational swimming. However, he had persisting core muscle and proximal muscle weakness as evidenced by neck flexor weakness, an anterior pelvic tilt, lumbar lordosis, scapular winging, posterior and lateral trunk lean during stance, wide based gait, and frequent tripping due to decreased foot clearance. Gross motor function on standardized assessments of functional strength that offer calculation of age equivalencies, showed a decline in age equivalency prior to initiation of ERT, from 11 to 12 months to 10–11 months, at 15 and 19 months respectively, as measured by the AIMS (Table 4). Gradual improvements in age equivalency, though still below age level, are apparent following initiation of ERT, from 11 months at 2.58 years to 3.58 years at 5.3 years on the stationary subset on the PDMS-2; with function in walking showing improvement in age equivalencies from 14 months at 2.58 years to 2.3 years at 5.3 years on the PDMS-2. On the BOT-2, there is stabilization at < 4 years on the Running Speed and Agility at 8 and 9 years of age. The GMFM, which does not offer age equivalencies but assesses functional strength over time (Table 3), showed improvement from 45.7% at 19 months to 94.65% at 10.2 years of age. Functional measure of strength peaked at age equivalency of 4:4 to 4:5 years of age as measured by the Strength Subtest of the BOT-2, and has been maintained, although age equivalency on other the Subtests, Running and Agility, which requires use of strength in higher level measures of agility show stabilization at younger age (< 4) and lower level, as skill demands increase (Table 4). Physical examination revealed mild ptosis of the left eye and increasing scoliosis. Progressive thoracolumbar scoliosis required surgery at age 12 years. He continues to receive ERT locally at 40 mg/kg/week since age 9.8 years. Throughout the clinical course patient maintained low antibody titers 〈3200) to ERT. 6MWT distance has been consistently below the mean for age, initially over 2 standard deviations below the mean and below the range for age and gender, improving to 1 standard deviation below the mean, and within the range for age. Percent-predicted on the 6MWT, has remained below 80% over time but has improved steadily on ERT from 67.84% to 77.79% (Table 3).
Table 4.
Performance on standardized functional assessments offering age equivalencies / percentile function for age.
| Patient ID | AIMS (age range: birth to 18 months of age) | PDMS-2 (age range: birth to 6 years of age) |
BOT-2 Strength and Agility (age range: 4 ½ to 21 years of age) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stationary Subtest | Locomotion Subtest | |||||||||||
| #1 | Age at testing: | 15 mo. | 19 mo. | 2.58 yrs. | 3.3 yrs. | 5.3 yrs. | 2.58 yrs. | 3.3 yrs. | 5.3 yrs. | 8 yrs. | 9.6 yrs. | 10.2 yrs. |
| Percentile: | < 5th | < 5th | 9th | 5th | 9th | 1st | 1st | 2nd | < 1% | < 1% | ||
| Age equivalency | 11–12 mo.** | 10–11 mo.** | 11 mo. | 11 mo. | 3.58 yrs. | 14 mo. | 14 mo. | 2.3 yrs. | Running speed and agility: | |||
| < 4 yrs. | < 4 yrs. | * | ||||||||||
| Strength: | ||||||||||||
| < 4 yrs. | 4:4 to 4:5 yrs. | 4:4 to 4:5 yrs. | ||||||||||
| Scaled Scores | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Running speed & agility: | |||
| 4 | 3 | * | ||||||||||
| Strength: | ||||||||||||
| 3 | 5 | 5 | ||||||||||
| Standard Score | N/A | 6 | 5 | 6 | 3 | 3 | 4 | 20 | 21 | |||
| ***Descriptive category | Abnormal | Below average | Poor | Below average | Very Poor | Very poor | Poor | Well Below Average | Well Below Average | Well Below Average | ||
| #2 | Age at testing | 1.7 yrs. | 2.2 yrs. | 2.2 yrs. | Below age range for BOT-2 administration | |||||||
| Percentile | < 5th | 25th | 5th | |||||||||
| Age equivalency | ~ 13 mo. ** | 18 mo. | 17 mo. | |||||||||
| Scaled score | N/A | N/A | ||||||||||
| Standard Score | N/A | 8 | 5 | |||||||||
| ***Descriptive Category | Abnormal | Average | Poor | |||||||||
| #3 | Age at testing | Above age range for AIMS administration | Above age range for PDMS-2 administration | 9 yrs. | ||||||||
| Percentile | 7th | |||||||||||
| Age equivalency | Running speed and agility: | |||||||||||
| 6:6 to 6:8 | ||||||||||||
| Strength: | ||||||||||||
| 5:4 to 5:5 | ||||||||||||
| Scaled Score | Running speed and agility: | |||||||||||
| 9 | ||||||||||||
| Strength: | ||||||||||||
| 7 | ||||||||||||
| Standard score | 35 | |||||||||||
|
***Descriptive Category |
Below average | |||||||||||
| #4 | Age at testing | Above age range for AIMS administration | 2.52 yrs. | 2.52 yrs. | Below age range for BOT-2 administration | |||||||
| Percentile | 2nd | 1st | ||||||||||
| Scaled Score | N/A | N/A | ||||||||||
| Standard Score | 4 | 3 | ||||||||||
| ***Descriptive Category | Poor | Very poor | ||||||||||
AIMS: Alberta infant motor scales; BOT-2: Bruininks-Oseretsky Test of Motor Proficiency, Second Edition; PDMS-2: Peabody Developmental Motor Scales-2nd Edition; yrs.: years;
Descriptive categories (low to high):
AIMS: abnormal → suspicious → normal.
PDMS-2: very poor → poor → below average → average → above average → superior → very superior.
BOT-2: well below average → below average → average → above average →Well above average.
Based on 50th percentile norms.
Running speed and agility not measured at this visit.
Table 3.
Change in muscle function and biomarker levels over time.
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Patient 4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age in years | 1.6* | 2.6 | 8 | 9.6 | 10.2 | 1.7* | 2.2 | 3.5 | 2.62* | 6.9 | 7.8 | 9 | 2.52 |
| 6MWT | too young for 6MWT | too young for 6MWT | too young for 6MWT | too young for 6MWT | |||||||||
| Meters; | 368.5; | 401.1; | 458; | 373; | 386; | 464; | |||||||
| Mean for age†† | 483 ± 40 | 496 ± 53 | 506 ± 45 | 463 ± 40 | 488 ± 35 | 496 ± 53 | |||||||
| Range for age | 400–600 | 395–600 | 400–601 | 380–600 | 438–583 | 395–600 | |||||||
| In / Below Range | Below | Within | Within | Below | Below | Within | |||||||
| SD below mean | 2.86 | 1.79 | 1.06 | 2.25 | 2.91 | 0.60 | |||||||
| % predicted*** | 62.42% | 63.98% | 70.90% | 70.09% | 69.64% | 76.67% | |||||||
| % predicted**** | 67.84% | 70.86% | 77.79% | 72.38% | 75.02% | 79.41% | |||||||
| GMFM (total percent score) | 45.7 | 67.63 | 93.70 | 92.66 | 94.65 | 67.32 | 93.69 | 96.8 | 97.91 | 65.52 | |||
| Dimension A (%) | 43 | 96.08 | 98.04 | 98.04 | 98.04 | 52.94 | 98.04 | 100 | 100 | 94.12 | |||
| Dimension B (%) | 78.33 | 90 | 98.33 | 98.33 | 98.33 | 86.67 | 96.67 | 100 | 100 | 70 | |||
| Dimension C (%) | 64.29 | 78.57 | 100 | 95.24 | 95.24 | 73.8 | 100 | 100 | 100 | 59.52 | |||
| Dimension D (%) | 33.33 | 51.28 | 84.62 | 89.74 | 87 | 71.79 | 82.05 | 92.31 | 92.31 | 69.23 | |||
| Dimension E (%) | 6.9 | 22.22 | 87.5 | 81.94 | 94.44 | 51.39 | 69.4† | 91.67 | 91.67 | 97.22 | 34.72 | ||
| GSGC** | 12 | 10 | 10 | 10 | 11 | 9 | |||||||
| CK (U/L) | 2736 | 2642 | 1038 | 1139 | 687 | 239 | 143 | 128 | 1063 | 961 | 1694 | ||
| Urinary Glc4 (mmol/MCN) | 55 | 21.1 | 18.8 | 17.8 | 6.6 | 2.1 | 2.6 | 34.9 | 9.4 | 13.3 | 9.3 | 14.9 | |
Assessment prior to ERT initiation;
Range: 4 to 27 lower scores better;
other dimensions not evaluated;
mo.: months; 6MW: 6 min walk test; AIMS: Alberta infant motor scales, BOT-2: Bruininks-Oseretsky Test of Motor Proficiency, Second Edition; GMFM: Gross Motor Function Measure; GSGC: Gait, Stairs, Gowers, Chair score; PDMS-2: Peabody Developmental Motor Scales-2nd Edition.
: according to [12]
: according to [15](regression equation includes age, gender, height, procedure not completed in accordance with ATS guidelines - child pushed wheel),
: according to [16] (regression equation includes age, gender, height, weight), done in accordance with ATS guidelines. MCN = moles of creatinine.
4.2. Patient 2
A Caucasian female, one of fraternal twins, was born at term and presented at day 10 of life with concerns of poor feeding, lethargy and vomiting. At age 2 months, she developed breathing difficulty, especially in the supine position with apneic episodes during sleep. She had hypotonia and developmental delay with easy fatigability while her twin was developing normally. Due to concerns of developmental delay and elevated AST, ALT, and CK, a muscle biopsy was done at age 14 months by a pediatric neurologist, which showed vacuolation with PAS positive glycogen stores suggestive of Pompe disease. Diagnosis was confirmed by low GAA enzyme activity of 0.7 pmol/punch/h (normal 0.59 to 3.88 pmol/punch/h) on blood assay. Molecular testing revealed c.-32–13 T > G and c.1827delC pathogenic variants in the GAA gene. At initial clinical evaluation at DUMC at age 18 months, history revealed delayed gross motor milestones, including sitting with support at 9–10 months, pulling-to-stand at 14 months and walking at 17 months (Table 1). On clinical examination, there was failure to thrive with weight less than 5th percentile for age, height between 10th and 25th percentile and head circumference closer to 50th percentile. She had myopathic facies, scapular winging and neck flexor weakness. PT assessment revealed wide-based gait with anterior pelvic tilt, weakness of neck flexors and abdominals, decreased strength in shoulder girdle muscles with scapular winging, a tendency towards genu recurvatum and plantar flexion (Table 2). She was started on alglucosidase alfa 20 mg/kg EOW at age 19 months. Her gross motor function improved, initially below the 5th percentile for age on the AIMS at 1.7 years of age prior to ERT, and increasing after initiation of ERT to the 25th percentile on the Stationary Subtest and the 5th percentile on the Locomotion Subtest on the PDMS-2 at 2.2 years of age, though signs of residual weakness persisted (Table 4). At age 3.5 years, she could walk and run independently. On the GMFM, which does not offer age equivalencies but assesses functional strength over time (Table 3), she showed improvement from 51.39% at 2.2 years to 69.4% at 3.5 years on Dimension E (Walking Running and Jumping) and her myopathic facies had improved, but she showed decreased functional use of ankle dorsiflexion during ambulation; with reports of frequent falls and complaints of leg pain. She had difficulty swallowing saliva, persistent gastroesophageal reflux, and feeding problems. A gastrostomy tube was placed to help with her feeding. She also complained of urinary retention and chronic constipation. Parents reported increased hypernasality of voice, new onset snoring and sleep disturbances/apnea. She had a poor cough and was unable to lie flat due to choking on her saliva which led to repeated hospital admissions for upper respiratory infections and suspected aspiration pneumonia. ECHO and ECG had been normal. A swallow study, pulmonary function tests, polysomnography and gastroenterology consult were recommended, in addition to continuing physical therapy and ERT. She did not mount an antibody response to ERT during the course of treatment. This patient has since been lost to follow up.
4.3. Patient 3
This Caucasian male patient was diagnosed at age 2 years with Pompe disease. He had difficulty sitting unsupported and holding head upright at 6 months, unsteady wide-based gait and frequent falls at 12 months, and tired easily compared to his peers (Table 1). Around age 18–20 months, he was noticed to have difficulty in rolling from supine to prone position, required upper body assistance or had to push on his thighs to stand. At age 21 months, he could not get up unassisted after a fall, needed increasing assistance to stand from sitting or kneeling positions and had difficulty climbing stairs which brought him to medical attention. On initial evaluation by a neurologist at age 23 months, he was noted to have a soft voice with a weak cry, truncal hypotonia, proximal muscle weakness, prominent lordosis, scapular winging, and scoliosis. Positive Gower’s sign and elevated CK, AST and ALT levels led to the work up for neuromuscular disorders including Duchenne and Becker’s muscular dystrophy among others, with normal results. Muscle biopsy showed increased lysosomal vacuoles and PAS positive glycogen storage indicative of Pompe disease, confirmed by molecular testing. He was heterozygous for the c.-32–13 T > G and c.2501_2502delCA (p.T834 fs) variants in the GAA gene. He was initiated on ERT 20 mg/kg EOW at age 2.2 years. At age 2.6 years, 5 months post ERT, there was improvement in muscle strength, decreased lordosis and improved hypernasality. At age 3.8 years, he continued to be fully ambulatory but with an abnormal gait and persistent weakness in his extremities. At age 6.7 years, he had hyporeflexia, atrophy of proximal lower limb muscles and difficulty in consuming large meals, which triggered parental referral to DUMC. At evaluation at DUMC at age 6.9 years, he had a wide-based gait and needed support while standing up and climbing stairs. Additionally, he had mild ptosis, lingual muscle weakness and terminal neck flexor weakness. There was scapular winging and scoliosis with a Cobb angle of 40° for which a back brace was prescribed. At age 7.8 years, he continued to be fully ambulatory without assistance but reported easy fatigability compared to peers. PT assessment showed postural deviations including posterior trunk lean and lateral shift during stance, scapular winging, pectus excavatum, lower rib flaring, decreased trunk rotation and genu recurvatum (Table 3). At age 9 years, he was active in school, roller-skating and swimming. He reported frequent tripping and falls due to weak ankle dorsiflexion. He also reported cramping, vomiting and diarrhea unrelated to the time of ERT (Table 2). He had no reported speech or swallowing difficulties. His PFTs, ECG and ECHO have been normal over time. Distance walked on 6MWT has always been below the mean for age, but has improved over time on ERT, initially > 2 standard deviations below the mean but eventually < 1 standard deviation below the mean, initially below, but now within, the range for age. Percent predicted for age, gender, height, and weight has remained below 80% over time, but has improved from 72.38 to 79.41. He remains below average overall, at the 7th percentile in gross motor function, as measured by the BOT-2 (Table 4). On the GMFM, which does not offer age equivalencies but assesses functional strength over time (Table 3), he has improved steadily from 93.69% at age 6.0 to 97.91% at 9 years of age.
4.4. Patient 4
This 2.5 year old Caucasian girl was born preterm at 36 weeks and needed NICU admission and CPAP briefly in the newborn period for apnea and bradycardia. The first year of her life was otherwise uneventful, but with delay in achieving motor milestones of crawling, standing, and walking. She sat unsupported at 6 months, crawled at 12 months, stood unsupported at 20 months, and began walking without support at 22 months. At 24 months, her local physical therapist noted decreased strength in core and bilateral lower extremities, impaired muscle tone, delay in motor skill acquisition, impaired gait pattern, and impaired standing posture. Her PT regimen was increased from weekly 45 min to twice weekly for 30 min. However, her gait became more unsteady, she was falling more, and tended to turn her legs inward. She also reported pain and soreness in her legs. She was referred by her PCP to a neuromuscular specialist where she was noted to have proximal muscle weakness evident by positive Gowers sign and waddling gait. Concern was raised for spinal muscular atrophy for which genetic testing was ordered. When the test results turned out to be negative, she was tested for Pompe disease. At age 26 months, she was diagnosed with Pompe disease by blood GAA enzyme assay which showed a level of 1.80 pmol/punch/h (normal > 3.88). Mutation analysis identified her GAA variants as c.-32–13 T > G and C.118C > T (Table 1). She was initiated on ERT 20 mg/kg/infusion EOW at age 28 months. At her evaluation at DUMC at age 30 months she was able to walk, but due to frequent tripping and falling, she was scooting in sitting on the floor for mobility. She had lumbar lordosis and bilateral winged scapulae. She had pronation of the feet bilaterally. She had a pincer grasp, but with weakness in thumb abduction which affects her grasp efficiency and endurance. PT assessments noted decreased strength in her abdominals, hip extensors, abductors and adductors, ankle plantar flexors, and neck flexors. She had delayed gross motor skills including inability to negotiate stairs in standing (with or without assistance), inability to run, inability to initiate jumping, and use of compensatory patterns of transitioning from supine to sitting, reflecting decreased strength and decreased use of abdominal obliques. Gowers sign was present during transition to standing, and was at the 2nd and 1st percentile for age on the PDMS-2 on the stationary and locomotion scales respectively. On the GMFM, which does not offer age equivalencies but assesses functional strength over time (Table 3), she scored 65.52%. She was noted to have gait abnormalities such as increased pelvic rotation bilaterally during ambulation, and decreased hip extension and push off bilaterally. Her cardiology evaluations have been normal. She exhibits asthma, snoring, apneic pauses, mouth breathing, and nasal congestion (Table 2). Pulmonary function tests revealed mild reversible obstructive airways disease. Her vital capacity was 69% of predicted which improved to 105% post-bronchodilator.
5. Discussion
The four patients reported here with LOPD and the c.-32–13-T > G variant had several identifiable common features. The most common symptoms of early disease onset were delayed achievement of motor milestones with characteristic patterns of posture and movement reflecting weakness typical of Pompe disease (4/4), feeding difficulty (2/4) and sleep apnea (2/4). Despite ERT, all had residual muscle weakness in the form of ptosis, lumbar lordosis, scoliosis, scapular winging, trunk and limb weakness, gait deviations, decreased function for age on standardized gross motor assessments, swallowing difficulty, oropharyngeal dysphagia, and gastrointestinal and genitourinary involvement in childhood. On standardized assessments of gross motor function that offer calculation of age equivalencies (AIMS, PDMS-2, and BOT-2), all patients were functioning below age level before initiation of ERT and remained below age level on ERT, but all showed improvement after starting on ERT. The two who were old enough to complete the 6MWT (patients 1 and 3) have consistently been below 80% of percent predicted, but both improved over time on ERT, one increasing 89.5 m distance and 10.13% of predicted, and the other increasing 91 m distance and 7.03% of predicted, both moving from below the range for age to within the age for range.
A closer look at the evolution of muscle function as measured by GMFM in Table 3 is enlightening and shows that in most patients, Dimensions D (standing) and E (walking, running, and jumping) show the greatest residual deficits over time, especially when considering the age at which 100% should be achieved by typically developing children. Typically developing children should be able to complete all items on the GMFM by age 5 years, achieving a score of 100% on all dimensions, which was not achieved by any of the children in this study, even up to 10.2 years of age, although steady increases in scores on the GMFM were observed over time in all the children. While > 95% was achieved in Dimensions A, B, and C at all testing points when over age 5 years, scores on Dimensions D and E ranged from 81.94 to 97.22, with all scores but one below 95%, even at ages 10.2 and 9, essentially twice the age at which 100% should be achieved in typically developing children, with even the highest Dimension D and E score of 97.22 showing a difference from typical function that is over the minimal clinically important difference (MCID), showing a large effect size of having a diagnosis of Pompe disease. This suggests that analyzing Dimensional scores rather than only total GMFM scores may offer greater insight into motor function and change over time when monitoring treatment response at follow-up. Individual item analysis may be enlightening over time: for example in those functioning at higher levels on Dimension D and E, but without 100% function on Dimension A, weakness in neck flexors, classic in Pompe disease, may be a uniform finding in many that prohibits achievement of 100% score by limiting achievement of antigravity head lifting in supine.
This cohort had clinical features which are seen in adult-onset LOPD, but with earlier manifestation than typically reported. This suggests that patients whose symptoms begin earlier are likely to have a more rapidly progressive course with more severe musculoskeletal involvement. Presence of progressive scoliosis in 2/4 patients with need for bracing or corrective surgery is a case in point. A study from the Pompe Registry showed that there is an increased risk of scoliosis in the subset of LOPD patients with early symptom onset [17]. The possibility that these more subtle motor manifestations may be present more widely and earlier than previously realized in individuals with the c.-32–13-T > G variant is important to consider. Many adults with LOPD report a history of subtle motor impairments that were dismissed early on, or considered variations of normal, recognized only retrospectively as being potential early signs of motor impairment which may have reflected early muscle involvement (i.e., fatiguing on long walks, never being able to do a sit up, having difficulty keeping up with peers in higher level gross motor activities). The importance of early recognition of muscle involvement is supported by muscle MRI studies which show early evidence of muscle deterioration on MRI prior to more overt clinical manifestation or measurement of weakness, suggesting an opportunity for intervention to preserve muscle integrity preventatively in order to avoid muscle damage that may later be irrevocable [18–20]. We have identified some characteristic early gross-motor features which may be predictive of early musculoskeletal involvement (Table 5, Fig. 1). Detection of these features requires evaluation of patients, especially those diagnosed through NBS, by an experienced pediatric physical therapist.
Table 5.
Characteristic early gross motor features of LOPD- Posture and movement*
| - “developmental delay” – specifically, delay in achievement of gross motor skills reflecting strength (e.g.: sitting, achievement of sitting, creeping on hands and knees, standing, walking, jumping, running) |
| - head lag when pulled to sit (reflecting neck flexor weakness) |
| - hypotonia |
| - sitting posture characterized by rounded back, head tipped back into extension, myopathic faces with a tendency towards slight open mouth position |
| - scapular winging (reflecting decreased strength in shoulder girdle and scapular stabilizing musculature) |
| - decreased use of abdominals, especially abdominal obliques (may flip to prone to push back up into sitting rather than rotating up to sitting using abdominals |
| - lower rib flaring (reflecting decreased use of abdominal obliques) |
| - lower extremity positional tendency towards: flexion, abduction, external rotation |
| - tendency to scoot in sitting for floor mobility (rather than creeping on hands and knees) |
| - increased anterior pelvic tilt and lumbar lordosis in upright for age (reflecting decreased use of force couple between hip extensors and abdominals) |
| - difficulty with “activities of elevation against gravity” e.g.: pulling to stand, walking, climbing (including stairs), jumping, skipping, hopping, running |
| - possible hypermobility with elbow hyperextension and genu recurvatum |
| - possible hypoextensibility in plantar flexors, especially across 2 joints (knee and ankle), tendency towards varus foot positioning with plantar fascia tightness or excessive pronation |
| - delayed independent ambulation with characteristic atypical posture and gait (wide base, increased anterior pelvic tilt, lumbar lordosis, spinal extension, posterior and/or lateral trunk lean during stance phase of gait, decreased development of medial longitudinal arch (i.e. tendency towards pronation or “flat feet”), decreased use of ankle dorsiflexion during swing phase of gait (steppage or foot-slap gait); may include compensatory lower extremity internal rotation during gait, especially when older |
| - muscle pain, fatigue |
May be mild.
Fig. 1.
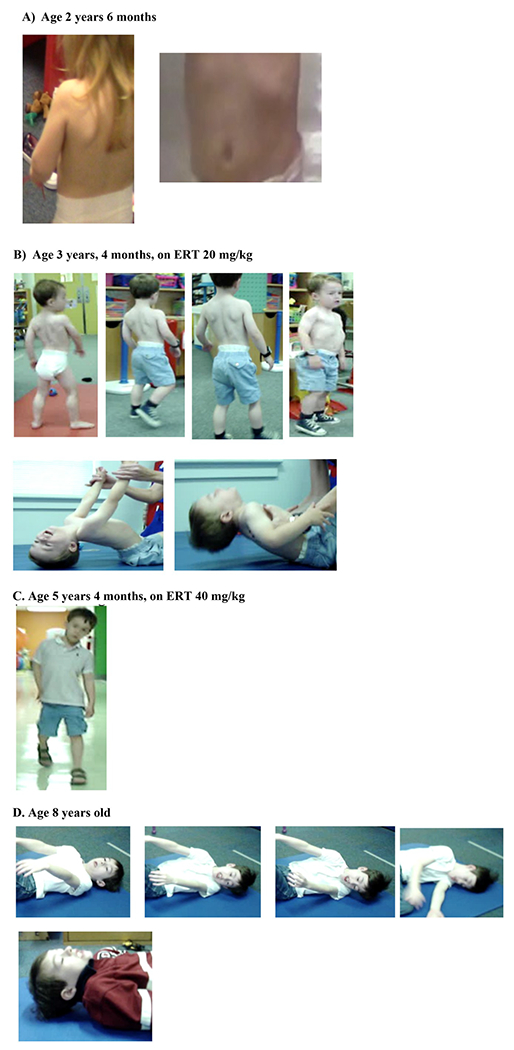
Early-onset musculoskeletal phenotype in children LOPD.
A) Age 2 years, 6 months.
Scapular winging and Lower rib flaring.
B) Age 3 years, 4 months, on ERT 20 mg/kg.
Anterior pelvic tilt with excessive hip flexion.
Spinal hyperextension and asymmetry.
Scapular winging.
Increased width of base of support.
Lateral trunk lean during stance phase of gait.
Decreased ankle dorsiflexion during swing phase of gait.
Enlarged calves.
Lower rib flaring.
Neck flexor weakness.
C) Age 5 years 4 months (on ERT 40 mg/kg):
Continued lateral & posterior trunk lean during stance phase of gait.
D) Age 8 years old.
Gowers maneuver required to achieve standing: cannot achieve standing without 1 hand on knee and 1 hand on floor.
Attempting to lift head to do sit-up but cannot lift head.
Attempting to lift head but cannot use antigravity neck flexion to lift head.
Persistent neck flexor weakness.
Another adult-onset Pompe disease symptom seen earlier in this cohort is oropharyngeal muscle weakness which manifested as feeding and swallowing difficulty. While bulbar muscle weakness has been documented in classic IOPD and in adults with LOPD, this is the first report in early-onset LOPD [21,22]. Sleep apnea was also noted in two patients here; which is reported in adults but for the first time in children with early-onset LOPD here [23]. Similarly, this is the first report of adult-onset symptoms of gastrointestinal and genitourinary such as cramping, constipation and incontinence in early-onset patients with the c.-32–13-T > G variant [24,25].
Despite their early presentation (10 days-22 months), there was a diagnostic delay of 4 months to 18 months in these patients, which is longer than that in patients with classic IOPD who are usually diagnosed within a few weeks to months of birth. Diagnostic delays as long as twelve years have been reported in the Pompe Registry study in children presenting with age at onset ≤12 months of age and no cardiomyopathy [11]. These four patients had a relatively short diagnostic delay as they were aggressively evaluated due to their more severe phenotype. Interestingly, diagnosis was confirmed in all four by neurologists, which further illuminates the diagnostic odyssey in these children with developmental delay due to Pompe disease, primarily a musculoskeletal disorder. In all four, diagnosis was done by muscle biopsy and/or GAA enzyme assay. With the availability of blood based assays, one can now diagnose Pompe disease without invasive procedures. A greater awareness of early symptoms and diagnostic modalities of Pompe disease among physicians and PTs could prevent unwarranted invasive tests such as muscle biopsy and facilitate early diagnosis.
The lack of cardiac involvement in these patients was a likely significant contributor to the diagnostic delay. There is the perception that IOPD with classic cardiomyopathy presents in childhood while LOPD does not present until adulthood, especially in those with the common c.-32–13-T > G variant. This “leaky” splice-site variant allows production of low levels of normal GAA which likely prevents cardiomyopathy, yet there can be early presentation, as early as the first days to months of life. There could be several factors contributing to this early-onset, rapidly progressive phenotype such as other genetic and epigenetic factors such as environment, diet and exercise [3,26]. The non-IVS1 GAA variants seen in the four patients with early-onset reported in our cohort were nonsense or frameshift variants. Two of these variants, namely the c.525delT and c.1827delC, were present in 12 patients each (totally 24/83; 28.9%) in our LOPD cohort, all of who had disease-onset in adulthood, suggesting that the presence of these particular non-IVS/IVS1 genotype combinations are insufficient to cause the early-onset phenotype. It is widely known that patients with other non-IVS1 nonsense/missense variants can present with LOPD at a young age, thereby ruling out nonsense/missense mutation type solely as the possible explanation for this phenotype (Table 6). Patients 1 and 4, whose ACE genotypes were available, did not have the unfavorable D/D alleles eliminating this as a likely reason for early onset. Further exploration of genetic modifiers responsible for this phenotype may be facilitated by whole-exome sequencing or RNA-seq analysis, which is beyond the scope of this paper.
Table 6.
Selected published studies on early-onset LOPD.
| Study | Patients | c.-32–13 T > G variant an d another pathogenic variant | Mean age at symptom onset | First symptoms | Age at diagnosis (years) | Follow-up period | Selected motor, respiratory outcomes on ERT |
|---|---|---|---|---|---|---|---|
| Bembi et al., 2010 | N=24 (7 juveniles, 17 adults) | No genotype data reported on any patient | 2.5 (±1.3) years | not described | 2.8 (±1.4) | 36 months | Improved 6MWT and stabilization of FEV1 |
| Deroma et al., 2014 | N=8 | Present in 7 patients | 6 months – 9 years | ↑CK, walking abnormalities, difficulty in climbing stairs (presented as grouped data) | 1 year to 9 years, 7 months | At least 72 months | Improved 6MWT and FVC |
| Van Capelle et al., 2016 | N=31 | Present in 21 patients | 2.6 years (0.5–13 years) | Motor delay, limb-girdle muscle weakness, diarrhea, fatigue | 4.0 | —(Cross-sectional study) | Over 50% had weakness of neck flexors, hip extensor and flexors and shoulder abductors, scoliosis, myopathic facies, low-absent reflexes. |
These four patients show that patients with LOPD and the c.-32–13 T > G variant can present earlier than previously known and adds important clinical description to previous reports in Table 6 [7,8,10]. The few reports describing the early symptoms in juvenile LOPD patients with the c.-32–13 T > G variant described grouped data of patient cohorts (Table 6). Unlike the detailed description of each individual patient as described in this report, these studies do not provide an adequate descriptive account of the early-onset phenotype and clinical course [8–10]. Moreover, these studies do not report on many of the early-onset features presented in our study.
Special attention is given to this particular GAA variant to dispel the prevailing notion that this is a mild phenotype and therefore unlikely to present at an early age. This notion can delay recognition of early musculoskeletal symptoms and thereby, treatment initiation. The implementation of NBS for Pompe disease in eight states in the USA makes it possible to diagnose these patients early. Current treatment guidelines for asymptomatic LOPD patients diagnosed through NBS recommend ERT initiation at symptom-onset [27]. Hence it is imperative that physicians and other healthcare providers are aware of early signs and symptoms of disease onset. We have recently reported the presence of subtle but specific signs of musculoskeletal involvement in LOPD patients diagnosed through NBS [28]. The current study makes a distinction from the cohort reported in our previous study in that here we report the presence of early-onset phenotype in patients who were clinically diagnosed and therefore represents a true evolution of the phenotype as opposed to one merely detected early through NBS, prenatal diagnosis or positive family history. Early treatment initiation is important because ERT can stabilize and partially improve muscle function, but cannot reverse previously sustained muscle damage [20,29]. Studies from Taiwan where NBS for Pompe disease is implemented have shown that the delay of even a few days in ERT initiation in IOPD can change motor function outcomes [30]. The persistent muscle weakness in all four patients here, despite an ERT dose of 40 mg/kg/week in patient 1, underscores the importance of timely treatment.
It is important to note that antibody titers remained low in the three out of four patients in whom titers were tested, which underscores the fact that the clinical decline and residual muscle deficits observed in these patients is not due to the presence of high antibody titers.
While this study reports on the early-onset phenotype in only 4/84 patients in our cohort with LOPD and the C-32–13-T > G variant, it has to be pointed out that we included only patients with a clinically documented history of early symptoms. Many more patients reported a retrospective history of subtle motor impairments but where not included in the manuscript due to the lack of clear documentation of their symptoms. It is therefore possible that the true magnitude of early-onset phenotype associated with this GAA variant is likely under-recognized and under-reported and in actuality much greater than currently reported in literature.
6. Conclusions
To summarize, the description of the clinical course presented here highlights that LOPD patients with heterozygous c.-32–13 T > G variant can present early in the first year of life and can have rapid disease progression and severe phenotype. The clinical presentation of LOPD with onset in infancy, has certain specific clinical features such as delayed achievement of motor milestones and characteristic alterations of posture and movement during motor development that are typical of Pompe disease in childhood. These characteristic features reflect the emergence of specific patterns of muscle weakness early in development, alters musculoskeletal development and creates risk factors for compromise of musculoskeletal integrity and further development. Other early symptoms include feeding difficulties; failure to thrive; and sleep apnea. It is especially important to raise awareness of these early features of Pompe disease associated with the c.-32–13 T > G variant at this time when NBS for Pompe disease has been added to the RUSP in United States.
Acknowledgements
The authors are thankful to Sophia’s Pompe Strong Team, The Emerson and Barbara Kampen Foundation, and Mr. and Mrs. Allen Boger for their generous support towards research in Pompe disease. The authors are thankful to the Lysosomal Storage Disease Network for their support allowing for continued research into Pompe disease. This research was supported in part by the Lysosomal Disease Network, a part of the National Institutes of Health Rare Diseases Clinical Research Network (RDCRN) - Grant number (5U54NS065768-05) (Subaward number N004689101). The Lysosomal Disease Network is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NCATS. This consortium is funded through a collaboration between NCATS, NINDS, and NIDDK. The content is solely thae responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. the funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding
No external funding sources were used for this study.
Conflict of interest disclosures
The authors, MH, MR and HC declare that they have no competing interests. LB, SLA, and LEC have received honoraria from Genzyme Corporation. PSK has received research/grant support from Sanofi Genzyme, Amicus, Valerion, and AskBio. PSK has recevied honoraria from Sanofi Genzyme, Amicus, and AskBio. LEC has participated in research funded by Genzyme Corporation, the Leal Foundation, Valerion, and Roivant Sciences. PSK serves as an Advisory Board member for Sanofi Genzyme, AMicus, Baebies, and AskBio. LEC is a member of the Pompe Registry North American Board of Advisors for Genzyme Corporation of Sanofi. PSK has equity in Actus Theraputics, which is developing gene therapy for Pompe disease.
List of abbreviations
- 6MWT
six minute walk test
- AIMS
Alberta infant motor scales
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATS
American Thoracic society
- BOT-2
Bruininks-Oseretsky Test of Motor Proficiency, 2nd Edition
- CK
creatine kinase
- ECG
electrocardiography
- ECHO
echocardiography
- EOW
every other week
- ERT
enzyme replacement therapy
- GMFM
Gross Motor Function Measure
- GSGC
Gait, Stairs, Gowers, Chair score
- GAA
acid α–glucosidase
- IOPD
infantile Pompe disease
- IVS
intervening sequence
- LOPD
late-onset Pompe disease
- NBS
newborn screening
- PDMS-2
Peabody Developmental Motor Scales-2nd Edition
- PFT
pulmonary function test Primary care provider
- RUSP
recommended uniform screening panel
Footnotes
Ethics approval and consent to participate
All patients and or parents/guardians gave written informed consent under Duke Institutional Review Board reviewed Protocol, # Pro00010830.
Consent for publication
Written consent for publication was obtained from the concerned patients’ parent or legal guardian mentioned in this study.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Byrne BJ, Kishnani PS, Case LE, Merlini L, Muller-Felber W, Prasad S, van der Ploeg A, Pompe disease: design, methodology, and early findings from the Pompe Registry, Mol. Genet. Metab 103 (1) (2011) 1–11. [DOI] [PubMed] [Google Scholar]
- [2].Kishnani PS, Howell RR, Pompe disease in infants and children, J. Pediatr 144 (5 Suppl) (2004) S35–S43. [DOI] [PubMed] [Google Scholar]
- [3].Kroos MA, Pomponio RJ, Hagemans ML, Keulemans JL, Phipps M, Deriso M, Palmer RE, Ausems MG, Van der Beek NA, Van Diggelen OP, et al. , Broad spectrum of Pompe disease in patients with the same c.-32-13T- > G haplotype, Neurology 68 (2) (2007) 110–115. [DOI] [PubMed] [Google Scholar]
- [4].Laforet P, Laloui K, Granger B, Hamroun D, Taouagh N, Hogrel JY, Orlikowski D, Bouhour F, Lacour A, Salort-Campana E, et al. , The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease, Rev. Neurol. (Paris) 169 (8-9) (2013) 595–602. [DOI] [PubMed] [Google Scholar]
- [5].Huie ML, Chen AS, Tsujino S, Shanske S, Dimauro S, Engel AG, Hirschhorn R, Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (−13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT->CT) mutation, Hum. Mol. Genet 3 (12) (1994) 2231–2236. [DOI] [PubMed] [Google Scholar]
- [6].Laforet P, Nicolino M, Eymard PB, Puech JP, Caillaud C, Poenaru L, Fardeau M, Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation, Neurology 55 (2000). [DOI] [PubMed] [Google Scholar]
- [7].van Capelle CI, van der Meijden JC, van den Hout JMP, Jaeken J, Baethmann M, Voit T, Kroos MA, Derks TGJ, Rubio-Gozalbo ME, Willemsen MA, et al. , Childhood Pompe disease: clinical spectrum and genotype in 31 patients, Orphanet Journal of Rare Diseases 11 (2016) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bembi B, Pisa FE, Confalonieri M, Ciana G, Fiumara A, Parini R, Rigoldi M, Moglia A, Costa A, Carlucci A, et al. , Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II, J. Inherit. Metab. Dis 33 (6) (2010) 727–735. [DOI] [PubMed] [Google Scholar]
- [9].van Capelle CI, van der Meijden JC, van den Hout JM, Jaeken J, Baethmann M, Voit T, Kroos MA, Derks TG, Rubio-Gozalbo ME, Willemsen MA, et al. , Childhood Pompe disease: clinical spectrum and genotype in 31 patients, Orphanet J Rare Dis 11 (1) (2016) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deroma L, Guerra M, Sechi A, Ciana G, Cisilino G, Dardis A, Bembi B, Enzyme replacement therapy in juvenile glycogenosis type II: a longitudinal study, Eur. J. Pediatr 173 (6) (2014) 805–813. [DOI] [PubMed] [Google Scholar]
- [11].Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J, Pompe Registry Boards of a: timing of diagnosis of patients with Pompe disease: data from the Pompe registry, Am. J. Med. Genet. A 161A (10) (2013) 2431–2443. [DOI] [PubMed] [Google Scholar]
- [12].Lammers AE, Hislop AA, Flynn Y, Haworth SG, The 6-minute walk test: normal values for children of 4-11 years of age, Arch. Dis. Child 93 (6) (2008) 464–468. [DOI] [PubMed] [Google Scholar]
- [13].Laboratories ATSCoPSfCPF, ATS statement: guidelines for the six-minute walk test, Am. J. Respir. Crit. Care Med 166 (1) (2002) 111–117. [DOI] [PubMed] [Google Scholar]
- [14].Henricson E, Abresch R, Han JJ, Nicorici A, Goude Keller E, Elfring G, Reha A, Barth J, McDonald CM, Percent-predicted 6-minute walk distance in duchenne muscular dystrophy to account for maturational influences, PLoS Curr 4 (2012) RRN1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Geiger R, Strasak A, Treml B, Gasser K, Kleinsasser A, Fischer V, Geiger H, Loeckinger A, Stein JI, Six-minute walk test in children and adolescents, J. Pediatr 150 (4) (2007) 395–399 399 e391–392. [DOI] [PubMed] [Google Scholar]
- [16].Ulrich S, Hildenbrand FF, Treder U, Fischler M, Keusch S, Speich R, Fasnacht M, Reference values for the 6-minute walk test in healthy children and adolescents in Switzerland, BMC Pulm Med 13 (2013) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roberts M, Kishnani PS, van der Ploeg AT, Müller-Felber W, Merlini L, Prasad S, Case LE, The prevalence and impact of scoliosis in Pompe disease: Lessons learned from the Pompe Registry, Mol. Genet. Metab 104 (4) (2011) 574–582. [DOI] [PubMed] [Google Scholar]
- [18].Carlier RY, Laforet P, Wary C, Mompoint D, Laloui K, Pellegrini N, Annane D, Carlier PG, Orlikowski D, Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: Involvement patterns, Neuromuscul. Disord 21 (11) (2011) 791–799. [DOI] [PubMed] [Google Scholar]
- [19].Gaeta M, Musumeci O, Mondello S, Ruggeri P, Montagnese F, Cucinotta M, Vinci S, Milardi D, Toscano A, Clinical and pathophysiological clues of respiratory dysfunction in late-onset Pompe disease: New insights from a comparative study by MRI and respiratory function assessment, Neuromuscul. Disord 25 (11) (2015) 852–858. [DOI] [PubMed] [Google Scholar]
- [20].Carlier PG, Azzabou N, de Sousa PL, Hicks A, Boisserie JM, Amadon A, Carlier RY, Wary C, Orlikowski D, Laforet P, Skeletal muscle quantitative nuclear magnetic resonance imaging follow-up of adult Pompe patients, J. Inherit. Metab. Dis 38 (3) (2015) 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hobson-Webb LD, Jones HN, Kishnani PS, Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement, Neuromuscul. Disord 23 (4) (2013) 319–323. [DOI] [PubMed] [Google Scholar]
- [22].Jones HN, Muller CW, Lin M, Banugaria SG, Case LE, Li JS, O’Grady G, Heller JH, Kishnani PS, Oropharyngeal dysphagia in infants and children with infantile Pompe disease, Dysphagia 25 (4) (2010) 277–283. [DOI] [PubMed] [Google Scholar]
- [23].Boentert M, Karabul N, Wenninger S, Stubbe-Drager B, Mengel E, Schoser B, Young P, Sleep-related symptoms and sleep-disordered breathing in adult Pompe disease, Eur. J. Neurol 22 (2) (2015) 369–376 (e327). [DOI] [PubMed] [Google Scholar]
- [24].McNamara ER, Austin S, Case L, Wiener JS, Peterson AC, Kishnani PS, Expanding our understanding of lower urinary tract symptoms and incontinence in adults with pompe disease, JIMD Rep 20 (2015) 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Karabul N, Skudlarek A, Berndt J, Kornblum C, Kley RA, Wenninger S, Tiling N, Mengel E, Plockinger U, Vorgerd M, et al. , Urge incontinence and gastrointestinal symptoms in adult patients with pompe disease: a cross-sectional survey, JIMD Rep 17 (2014) 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Slonim AE, Bulone L, Goldberg T, Minikes J, Slonim E, Galanko J, Martiniuk F, Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy, Muscle Nerve 35 (1) (2007) 70–77. [DOI] [PubMed] [Google Scholar]
- [27].Kronn DFD-S D, Hwu W, Jones SA, Nakamura K, Okuyama T, Swoboda KJ, Kishnani PS, on behalf of the Pompe Disease Newborn Screening Working Group: Management of Confirmed Newborn-Screened Patients with Pompe Disease across the Disease Spectrum, Pediatrics 140 (Supplement 1) (2017) S24–S45. [DOI] [PubMed] [Google Scholar]
- [28].Rairikar MV, Case LE, Bailey LA, Kazi ZB, Desai AK, Berrier KL, Coats J, Gandy R, Quinones R, Kishnani PS, Insight into the phenotype of infants with Pompe disease identified by newborn screening with the common c.-32–13T > G “late-onset” GAA variant, Mol. Genet. Metab 122 (2017) 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chien YH, Lee NC, Chen CA, Tsai FJ, Tsai WH, Shieh JY, Huang HJ, Hsu WC, Tsai TH, Hwu WL: Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J. Pediatr 2015, 166(4):985–991 e981–982. [DOI] [PubMed] [Google Scholar]
- [30].Yang CF, Yang CC, Liao HC, Huang LY, Chiang CC, Ho HC, Lai CJ, Chu TH, Yang TF, Hsu TR, et al. , Very early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes, J. Pediatr 169 (2016) 174–180 (e171). [DOI] [PubMed] [Google Scholar]
- [31].Kishnani PS, et al. , Pompe disease diagnosis and management guideline, Genet. Med 8 (5) (May 2006) 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.