

Abstract
BACKGROUND:
Several definitions have attempted to stratify metastatic castrate-sensitive prostate cancer (mCSPC) into low and high-volume states. However, at this time, comparison of these definitions is limited. Here we aim to compare definitions of metastatic volume in mCSPC with respect to clinical outcomes and mutational profiles.
METHODS:
We performed a retrospective review of patients with biochemically recurrent or mCSPC whose tumors underwent somatic targeted sequencing. 294 patients were included with median follow-up of 58.3 months. Patients were classified into low and high-volume disease per CHAARTED, STAMPEDE, and two numeric (≤3 and ≤5) definitions. Endpoints including radiographic progression-free survival (rPFS), time to development of castration resistance (tdCRPC), and overall survival (OS) were evaluated with Kaplan–Meier survival curves and log-rank test. The incidence of driver mutations between definitions were compared.
RESULTS:
Median OS and tdCRPC were shorter for high-volume than low-volume disease for all four definitions. In the majority of patients (84.7%) metastatic volume classification did not change across all four definitions. High volume disease was significantly associated with worse OS for all four definitions (CHAARTED: HR 2.89; p < 0.01, STAMPEDE: HR 3.82; p < 0.01, numeric ≤3: HR 4.67; p < 0.01, numeric ≤5: HR 3.76; p < 0.01) however, were similar for high (p = 0.95) and low volume (p = 0.79) disease across all four definitions. Those with discordant classification tended to have more aggressive clinical behavior and mutational profiles. Patients with low-volume disease and TP53 mutation experienced a more aggressive course with rPFS more closely mirroring high-volume disease.
CONCLUSIONS:
The spectrum of mCSPC was confirmed across four different metastatic definitions for clinical endpoints and genetics. All definitions were generally similar in classification of patients, outcomes, and genetic makeup. Given these findings, the simplicity of numerical definitions might be preferred, especially when integrating metastasis directed therapy. Incorporation of tumor genetics may allow further refinement of current metastatic definitions.
INTRODUCTION
Metastasis has classically been considered a binary state, which has heavily influenced treatment paradigms. However, a new theory of metastasis was proposed in the 1990s, one that hypothesized metastasis as spectrum rather than a simple binary entity resulting in the postulation of a low-volume metastatic state perhaps amenable to cure with definitive treatment, termed oligometastasis [1]. Subsequent work has aimed to categorize patients with metastatic prostate cancer by splitting disease into high- and low-volume states for treatment and risk stratification purposes [2–4]. Two such definitions, from the CHAARTED and STAMPEDE trials which aimed to investigate systemic therapies in metastatic castration sensitive prostate cancer (mCSPC), utilized a combination of metastasis location (bone and visceral metastases) and number when risk stratifying patients. Other definitions, emerging from investigations of metastasis directed therapy (MDT), typically used a cut-off of 3–5 lesions when incorporating stereotactic ablative radiation (SABR) into the treatment paradigm of oligometastatic patients [5–9].
While all these definitions of metastatic disease volume appear to be effective in risk stratifying patients, those from the CHAARTED and STAMPEDE trials represent a more nuanced classification that incorporate both lesion location and number as compared to the simplified numerical definitions of simple lesion enumeration used for MDT [5, 6, 8]. Despite their individual efficacy there remain questions regarding these definitions. For example, neither take into account timing of metastasis development, which includes synchronous (de novo) and metachronous (recurrent) disease. Additionally, there is no consensus surrounding the use of these definitions. Thus, here we aim to compare four commonly utilized definitions of metastatic volume to investigate their differences in clinical outcomes as well as genetic profiles in order to better understand their ability to risk stratify patients with mCSPC.
MATERIALS AND METHODS
Following institutional review board approval, we retrospectively reviewed patients from a single academic institution with mCSPC (synchronous and metachronous) or biochemical recurrence (BCR) who underwent nextgeneration somatic sequencing (NGS) from 12/2013 to 4/2020 of either their primary tumor or castration-sensitive metastatic site. NGS was performed either through Foundation One CDx (324-gene panel) or Personal Genome Diagnostics CancerSELECT 125 (125-gene panel) platforms. Additional inclusion criteria included prior definitive treatment to the prostate in patients with either BCR (micrometastatic disease only) or metachronous mCSPC. Follow-up data and clinical endpoints were collected through serial physical examination, conventional imaging, and prostate-specific antigen (PSA) measurements.
Patients were classified into BCR, “low-volume”, and “high-volume” metastatic disease utilizing four definitions. Patients classified as BCR had a rising PSA following definitive therapy to the primary tumor without any radiographic evidence of gross metastatic disease at last follow-up using conventional imaging. Patients were classified as BCR independent of the following four definitions. The four definitions in our analysis included those from CHAARTED, modified STAMPEDE, and two commonly utilized numerical definitions of low- (sometimes also referred to as oligometastatic) vs high (sometimes also referred to as polymetastatic) volume disease. High-volume disease per CHAARTED criteria was defined as the presence of visceral metastasis or ≥4 bone lesions with ≥1 beyond the vertebral bodies and pelvis; others were defined as low-volume [2]. High-volume disease per modified STAMPEDE was defined as ≥4 bone metastases regardless of location or any visceral metastases, with others classified as low-burden disease [10]. The two commonly utilized numerical definitions of low-volume were ≤3 and ≤5 lesions, regardless of location, with >3 and >5 metastases classified as high-volume, defined in agreement between two physicians.
Analysis aimed to compare these definitions in their ability to stratify patients by predetermined clinical endpoints including radiographic progression-free survival (rPFS), time to development of castrate-resistant prostate cancer (tdCRPC), and overall survival (OS) from time of initial diagnosis. rPFS was defined as time from first development of metastatic disease to new or radiographically enlarging known metastatic lesions. The endpoint tdCRPC was defined as time from diagnosis to castration-resistance according to Prostate Cancer Working Group 3 criteria [11]. OS was defined from time of initial diagnosis to death. Patients without events were censored at the last imaging date for rPFS, last PSA for tdCRPC, and last patient contact for OS. Kaplan-Meier (KM) survival curves were generated for BCR, low- and high-volume disease across all four definitions for the abovementioned clinical outcomes and compared utilizing the log-rank test. Among patients with metastatic disease, clinical outcomes were similarly evaluated between mutational status of driver mutations and pathways of interest including TP53, WNT, TMPRSS2-ERG, cell cycle, SPOP, DNA double strand break repair (DDSB), and PI3K/AKT/mTOR. Mutations which demonstrated significant differences in clinical outcomes were further stratified by low- and high-volume disease. Clinical outcomes of this volumetric-genetic classification were further analyzed utilizing KM survival curves.
Frequency of driver mutations and pathways of interest were reported for BCR, low- and high-volume disease groups across all four definitions. Differences in the frequency of driver mutations within each metastatic category were compared using a Pearson’s χ2. For all analyses, a p value <0.05 was considered statistically significant. All statistical analyses were performed with IBM SPSS Statistics, version 25.
RESULTS
A total of 294 patients with BCR or mCSPC underwent somatic NGS. Baseline patient and tumor characteristics can be found within Supplemental Table 1. Median follow-up was 58.3, and 77.7 months for the entire cohort and biochemically recurrent groups, respectively. Within the entire cohort, 15.3% (n = 45) of patients were biochemically recurrent at last follow-up. Patients with metastatic disease were classified utilizing four distinct definitions for volume of disease with 31.3% (n = 92/294), 35.4% (n = 104/294), 37.4% (n = 110/294), and 30.6% (n = 90/294) classified as high-volume disease by CHAARTED, STAMPEDE, numerical >3, and numerical >5, respectively.
Fig. 1A–C demonstrates Kaplan–Meier survival curves for rPFS, tdCRPC, and OS for BCR, low- and high-volume disease across all four definitions. Within the entire cohort, five-year OS and tdCRPC was significantly different between BCR, low- and high-volume disease across all four definitions (p < 0.01). The five-year rPFS was significantly different among low- and high-volume disease when using the STAMPEDE (p = 0.04), numerical ≤3 (p < 0.01) and numerical ≤5 (p = 0.03) and trended towards significance with the CHAARTED (p = 0.09) definition (Table 1). When comparing disease volume across definitions, Cox regression demonstrated similar hazard ratios for rPFS (CHAARTED: HR 1.38; p = 0.09, STAMPEDE: HR 1.45; p = 0.04, numerical >3: HR 1.73; p < 0.01, numerical >5: HR 1.49; p = 0.04), tdCRPC (CHARTED: HR 2.44; p < 0.01, STAMPEDE: HR 3.03; p < 0.01, numerical >3: HR 3.35; p < 0.01, numerical >5: HR 3.51; p < 0.01), and OS (CHARTED: HR 2.89; p < 0.01, STAMPEDE: HR 3.82; p < 0.01, numerical >3: HR 4.67; p < 0.01, numerical >5: HR 3.76; p < 0.01), for high and low-volume disease across all four definitions (Table 1). Finally, cox regression demonstrated no significant differences between any of the high-or low- volume definitions with regard to rPFS, tdCRPC, or OS (Table 2).
Fig. 1. Clinical outcomes for metastatic castration sensitive prostate cancer stratified by disease volume.
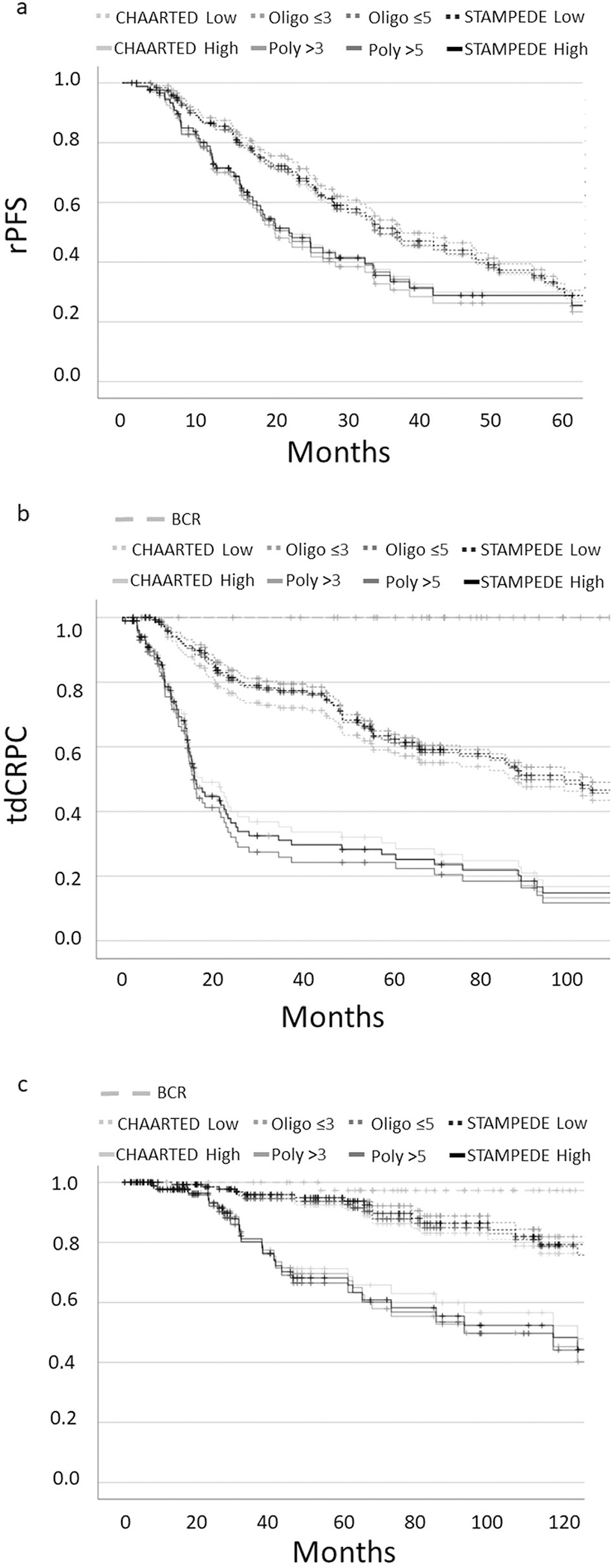
Kaplan Meier Survival Curves for rPFS (1a), tdCRPC (1b), and OS (1c).
Table 1.
5-year radiographic progression-free survival, time to castration resistance, and overall survival and cox regression comparing high vs low volume disease across all four definitions.
| BCR | Low volume | High volume | p value | HR (95%CI) | p value | |
|---|---|---|---|---|---|---|
| 5-Year rPFS | ||||||
| CHAARTED | NA | 30% | 30% | 0.09 | 1.38 (0.96–1.98) | 0.09 |
| STAMPEDE | NA | 31% | 29% | 0.04 | 1.45 (1.01–2.07) | 0.04 |
| Numeric ≤3 | NA | 33% | 26% | <0.01 | 1.73 (1.21–2.48) | <0.01 |
| Numeric ≤5 | NA | 31% | 29% | 0.03 | 1.49 (1.04–2.14) | 0.04 |
| 5-year tdCRPC | ||||||
| CHAARTED | 100% | 57% | 28% | <0.01 | 2.44 (1.74–3.43) | <0.01 |
| STAMPEDE | 100% | 61% | 25% | <0.01 | 3.03 (2.16–4.27) | <0.01 |
| Numeric ≤3 | 100% | 63% | 25% | <0.01 | 3.35 (2.38–4.73) | <0.01 |
| Numeric ≤5 | 100% | 60% | 22% | <0.01 | 3.51 (2.49–4.95) | <0.01 |
| 5-Year OS | ||||||
| CHAARTED | 97% | 92% | 71% | <0.01 | 2.89 (1.60–5.24) | <0.001 |
| STAMPEDE | 97% | 95% | 68% | <0.01 | 3.82 (2.08–7.02) | <0.001 |
| Numeric ≤3 | 97% | 95% | 70% | <0.01 | 4.67 (2.50–8.74) | <0.001 |
| Numeric ≤5 | 97% | 94% | 66% | <0.01 | 3.76 (2.07–6.83) | <0.001 |
BCR biochemical recurrence, rPFS radiographic progression free survival, tdCRPC time to castration resistance, OS Overall Survival
Table 2.
Cox regression comparing between high- and low-volume disease definitions for rPFS, tdCRPC, and OS.
| Definitions | HR (95%CI) | p value |
|---|---|---|
| High volume | ||
| rPFS (vs Numeric >3) | ||
| Numeric >5 | 0.95 (0.65–1.38) | 0.78 |
| CHAARTED | 0.91(0.62–1.33) | 0.62 |
| STAMPEDE | 0.92 (0.64–1.32) | 0.64 |
| tdCRPC (vs Numeric >3) | ||
| Numeric >5 | 1.11 (0.79–1.54) | 0.55 |
| CHAARTED | 0.91 (0.65–1.27) | 0.59 |
| STAMPEDE | 0.98 (0.71–1.35) | 0.89 |
| OS (vs Numeric >3) | ||
| Numeric >5 | 1.02 (0.59–1.75) | 0.96 |
| CHAARTED | 0.87 (0.51–1.51) | 0.63 |
| STAMPEDE | 0.95 (0.56–1.60) | 0.84 |
| Low Volume | ||
| rPFS (vs Numeric ≤3) | ||
| Numeric ≤5 | 1.10 (0.78–1.54) | 0.59 |
| CHAARTED | 1.09 (0.77–1.53) | 0.63 |
| STAMPEDE | 1.13 (0.81–1.58) | 0.48 |
| tdCRPC (vs Numeric ≤3) | ||
| Numeric ≤5 | 1.09 (0.77–1.53) | 0.64 |
| CHAARTED | 1.07 (0.75–1.51) | 0.72 |
| STAMPEDE | 1.21 (0.87–1.70) | 0.26 |
| OS (vs Numeric ≤3) | ||
| Numeric ≤5 | 1.28 (0.66–2.48) | 0.47 |
| CHAARTED | 1.14 (0.57–2.29) | 0.71 |
| STAMPEDE | 1.38 (0.71–2.68) | 0.34 |
rPFS radiographic progression free survival, tdCRPC time to castration resistance, OS Overall Survival
Given the overall similarity in outcomes, we assessed the congruence in classification between these definitions. Compared to the numerical ≤3 definition, 92%, 88%, and 93% of patients were similarly classified by numerical ≤5, CHAARTED, and STAMPEDE, respectively with 95.2% concordance between CHAARTED and STAMPEDE definitions (Fig. 2). Further, of patients with metastatic disease, 133 (53.4%) patients were classified as low-volume by all four definitions, and 78 (31.3%) classified as high-volume by all four definitions. Only 38 patients (15.3%) were re-classified into a different metastatic group when categorizing into the four metastatic definitions, the majority of which (63.2%) were CHAARTED low volume, but numerical >3 high volume disease. This discordant group had outcomes similar to high volume disease with 5-year PFS, tdCRPC, and OS of 27%, 33% and 73%, respectively.
Fig. 2. Patient population similarities between disease volumes.
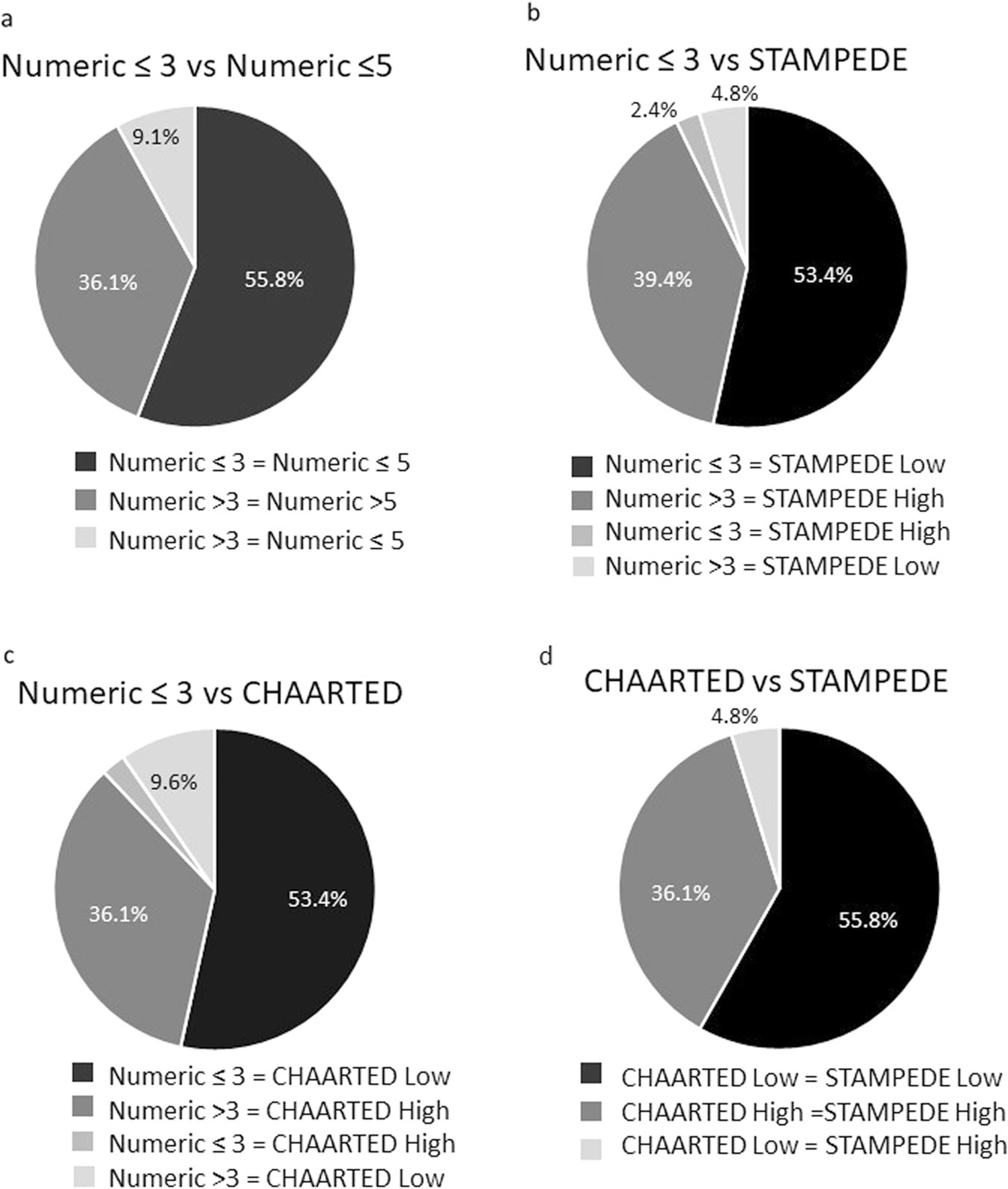
Similarities in classification for Oligometastatic ≤3 and Oligometastatic ≤5 (2a), CHAARTED (2b), and STAMPEDE definitions (2c).
We next aimed to assess the mutational landscape of driver mutations of interest across all four definitions. Oncoprint profiles for each definition are shown in Supplemental Fig. 1. Across all four definitions, the frequency of driver mutations in WNT, cell cycle, TP53, and PI3K/AKT/mTOR were significantly different between BCR, low, and high-volume disease (Fig. 3, supplemental Fig. 2) with higher incidence of mutations in patients with higher volumes of metastatic disease. However, incidence of mutations was similar for high and low-volume groups, respectively, when comparing across the four definitions for all genes and pathways of interest (Fig. 3). Patients with discordant metastatic volume classification as mentioned above (i.e. CHAARTED low volume and numerical >3) tended to have incidences of mutations in driver genes similar to high volume patients, in line with their poorer clinical outcomes (TP53: 50%, WNT: 16.6%, DDSB: 25%, PI3K/Akt/mTOR: 37.5%, cell cycle: 16.6%, SPOP: 8.3%).
Fig. 3. Frequency of driver mutations across the spectrum of mCSPC.
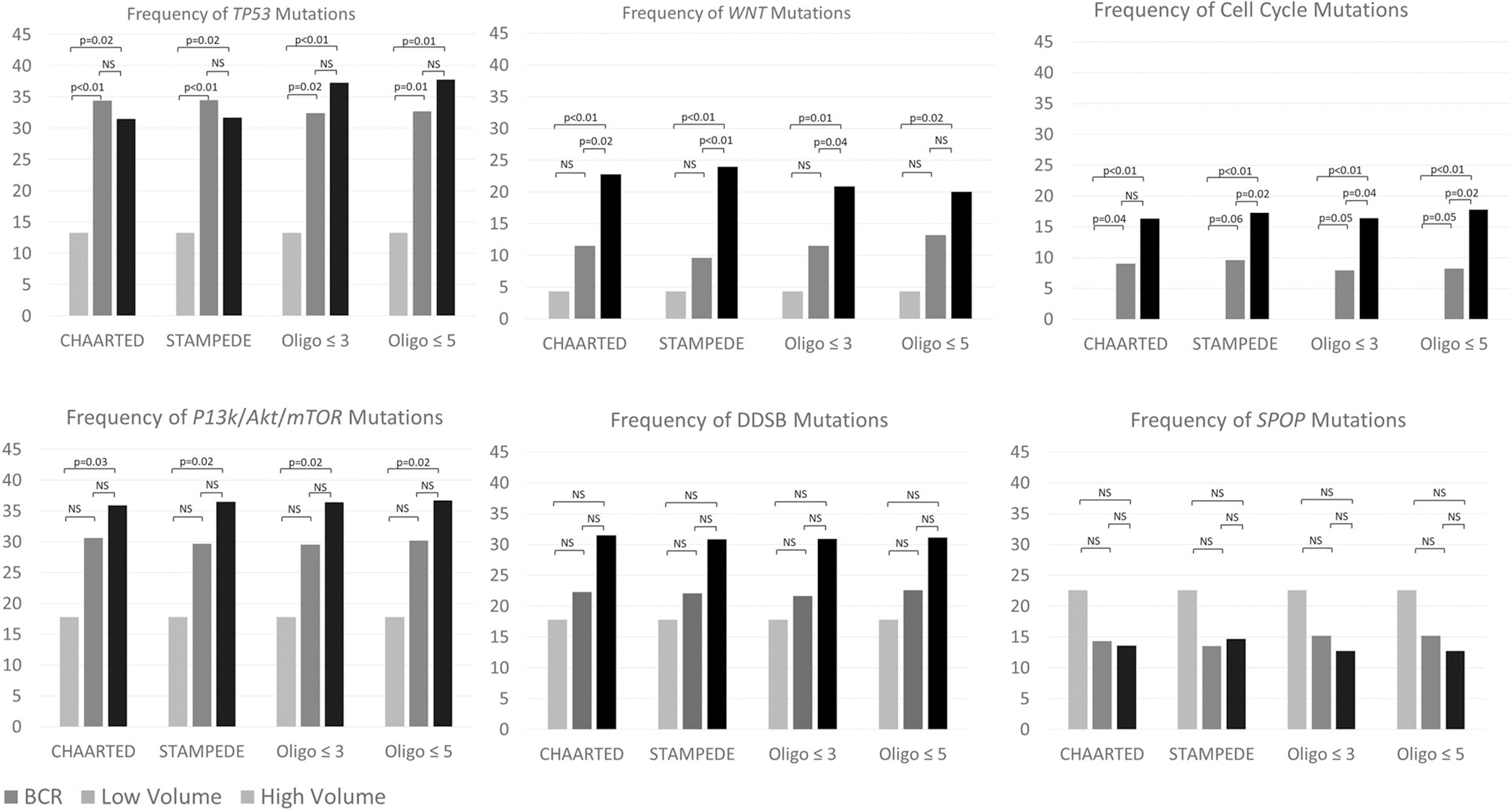
Frequency of driver mutations increases with higher disease volume.
We further evaluated the prognostic significance of driver mutations on rPFS, tdCRPC, and OS in patients with metastatic disease. Among the six genes and pathways of interest detailed above, only TP53 mutations were found to have significantly worse 5-yr rPFS (21% vs 35%, p < 0.01) and tdCRPC (37% vs 53%, p < 0.01) [Supplemental Table 2]. Given the prognostic role of TP53 mutations in patients with metastatic disease, we then evaluated outcomes of patients with high and low-volume disease stratified by TP53 mutational status (Fig. 4, Supplemental Fig. 3–5). Although patients with high-volume disease had similar outcomes regardless of mutation status, patient with low-volume, TP53 mutated disease behaved more aggressively. Specifically, this low-volume mutated cohort had rPFS more congruent with high-volume disease and demonstrated an intermediate risk within tdCRPC and OS.
Fig. 4. Clinical outcomes stratified by TP53 mutational status.
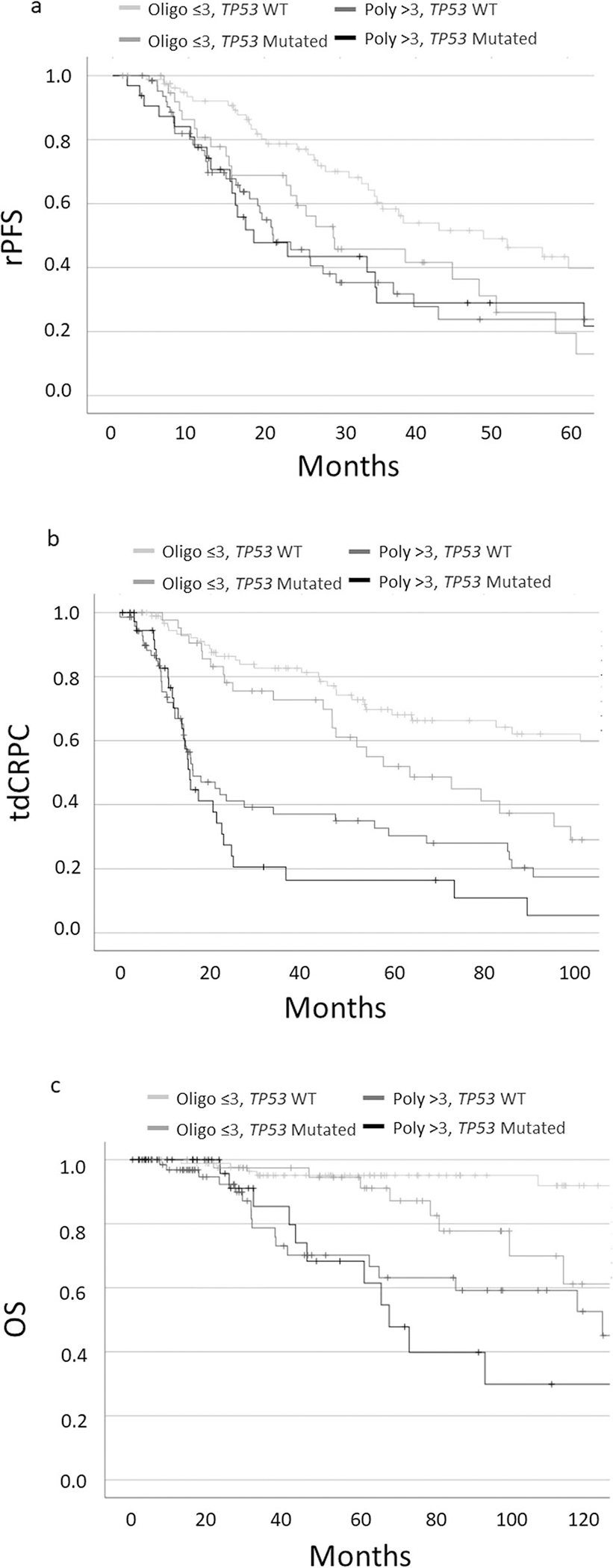
Kaplan Meier Survival Curves of Oligo- and Polymetastatic (≤3) patients stratified by TP53 mutational status for radiographic progression-free survival (4a), time to castration resistant prostate cancer (4b), and overall survival (4c).
DISCUSSION
Here we report clinical outcomes and the mutational landscape for current widely used definitions of mCSPC and demonstrate the remarkable similarity of outcomes across all four definitions. Specifically, we have shown congruence in (1) how these definitions classify patients, (2) clinical outcomes in each metastatic group, and (3) the underlying mutational spectrum across each metastatic volume definition. Additionally, we have demonstrated the incidence of driver mutations increases consistently across metastatic volume in all four definitions. Finally, we show that TP53 mutational status can further risk stratify patients with low-volume disease who may have a more aggressive clinical course.
Interestingly, although these four definitions are distinct and, in some cases, seemingly quite different, there was minimal variability in how they classify patients in practice, with only 13% of patients receiving a discrepant classification when comparing across all definitions. These similarities may provide some insight into patterns of spread and underlying metastatic biology in mCSPC. Most notably, lymph node only high-volume (numerical definition) disease and visceral low volume (numerical definition) disease are uncommon clinical situations in mCSPC. This is also consistent with work by Pasoglou et al. in identifying patterns of metastatic deposits in recurrent prostate cancer which identified lymph node only polymetastatic disease accounts for only 17% of patients with metachronous polymetastatic disease [12]. Additionally, Gupta et al. assessed the distribution of disease with prostate-specific membrane antigen/positron emission tomography (PSMA-PET) among 179 patients with newly diagnosed or recurrent high-risk (Gleason 9/10 or PSA > 20 ng/mL) prostate cancer with 0–3 metastases on conventional imaging. Within this cohort, only 5 patients (2.8%) were found to have visceral metastases, further demonstrating the rarity of visceral metastases in oligometastatic CSPC [13]. Given the rarity of visceral metastasis in mCSPC we were unable to assess the significance of these patients in CHAARTED or STAMPEDE definitions.
In addition to the above noted advances in systemic therapy, MDT represents a new paradigm for the treatment of oligometastatic prostate cancer. Within metachronous mCSPC, the STOMP and ORIOLE trials randomized patients with three or fewer metastases to observation or MDT. STOMP and ORIOLE trials demonstrated MDT conferred an improvement in five-year ADTfree survival (34% vs 8%; p = 0.006) and 6-month progression-free survival (81% vs 39%; p = 0.005), respectively [5, 14]. However, there is no universally accepted definition of oligometastasis at this time, and most commonly a simple enumeration of three or five lesions is used. The similar outcomes of the four groups compared in this study raises the question of the best definition to use and the implications of their differences. From a practical standpoint, simple enumeration might lend itself best for selection of MDT, as a pertinent limiting factor in treatment with MDT is the number of lesions. Future trials will have to factor in these differences in definitions to definitively address these questions. Additionally, future work will need to understand how advanced imaging with PSMA-PET can improve detection of occult metastatic disease potentially leading to category migration.
Here we have demonstrated the mutational landscape of mCSPC is generally in line with prior reports with frequency of mutations in driver mutations lying between localized and castration resistance disease. Hamid et al previously reported an incidence of 37% and 28% for TP53 and PTEN mutations, similar to what we demonstrate here [15–20]. Notably, the cohort reported here appears to have a lower than expected mutation frequency in cell cycle genes including Rb1. Additionally, while rates of DDSB mutations were in line with other studies such as PROREPAIR-B, rates of BRCA mutations, which are likely prognostic, were lower in our cohort. This may reflect our cohort consisted of mCSPC compared to castration resistant disease in PROREPAIR-B, or that our population was heterogeneous [21]. Both of these findings may limit the generalizability of our findings. In the future, genetic factors might be able to resolve differences between these disease volume categorizations and provide a more precise definition of oligometastasis. For example, here we show that patients with low-volume metastatic disease with TP53 mutations appear to have a more aggressive clinical course with time to rPFS in line with high-volume disease. Further analysis of tumor genetics may be able to better identify aggressive biology disease masquerading as low-volume disease and conversely allow more accurate identification of indolent biology disease presenting with high-volume metastasis. Interestingly, rates of TP53 mutations only increased across the spectrum of disease when using a numerical only volume definition. Taken together, these data provide evidence that TP53 mutations might play an important role in providing additional information beyond lesion number and location when defining oligometastasis. Furthermore, TP53 mutations have previously been shown to be associated with more aggressive disease and higher burden of metastatic disease, however our group has also demonstrated that individuals with numerical definitions of high volume disease and TP53 wildtype tumors have times to progression and development of castrate-resistance similar to individuals with the numerical definition of low volume (oligometastatic) disease [17, 22–25]. Thus, genetics hold the potential to refine definitions of oligometastasis and aid in treatment selection in the future, however more investigation is needed.
This study has several limitations, most prominently that this cohort was a retrospectively identified cohort and therefore outcomes were not prospectively defined, and even-though all patients were managed in a similar fashion by a small group of oncologists at our institution, heterogeneity of treatment may have impacted outcomes in ways we did not mitigate for in our analysis. Additionally, although we did not detect a difference between these various definitions, it is possible our sample size was inadequate to detect small differences in outcomes. Further, the inclusion of both metachronous and synchronous disease in one cohort might limit interpretation as these are likely two distinct biologic entities that may require different definitions in the future. Other limitations include that the majority of samples genomically profiled were from the primary tumor rather than metastatic castration-sensitive sites which may miss mutations acquired during disease progression. However, there does appear to be some evidence that driver mutations, especially DNA damage repair related, are early truncal events and thus a large proportion of mutations were likely captured through sequencing of primary tissue [26]. Further limitations related to primary prostate cancer multifocality and sampling of tissue via biopsies may have led to missing of relevant clonal alterations. Finally, two different sequencing assays were utilized and genomic alterations were limited to mutations without any information regarding mutation zygosity.
CONCLUSIONS
Here we demonstrate similarities of four commonly utilized definitions of mCSPC in terms of patient classifications, clinical outcomes, and incidence of pathogenic driver mutations across the spectrum of disease. Given the concordance between these definitions, numerical definitions of disease volume might be preferred given its simplicity, especially when utilizing MDT due to the practical limitations of treating multiple lesions. Future work is needed to further refine risk stratification by incorporating timing, location, and genomics of disease.
Supplementary Material
Footnotes
COMPETING INTERESTS
Consultant for Pfizer, Exelexis. ESA: Patent holder/licenser for Qiagen; Consultant for Amgen, Astellas, AstraZeneca, Bayer, Clovis, Dendreon, Eli Lilly and Co. ESSA, GlaxoSmithKline, Jannsen, Medivation, Merck, and Sanofi; Research grant recipient from AstraZeneca, Bristol Myers-Squibb, Celgene, Clovis, Dendreon, Genentech, Janssen, Johnson & Johnson, Merck, Novartis, Sanofi, Tokai. PO: Consultant for Bayer, Janssen, Curium; Research grant recipient from Varian, Bayer. AG: Relationship with Janssen and Astellas; Royalties from ICR. AR: Speaker’s Bureau for Blue Earth and Janssen; Stock Options from Decipher Biosciences; Consultant to Astellas. PT: Research funding from Astellas Pharm, Bayer Healthcare and RefleXion Medial Inc; Consultant for RefleXion, Grants from RefleXion; Personal fees from Noxopharm, Janssen-Taris Biomedical, Myovant and AstraZeneca; Holds a patent 9114158-Compounds and Methods of Use in Ablative Radiotherapy licensed to Natsar Pharm.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41391-021-00484-4.
Reprints and permission information is available at http://www.nature.com/reprints
REFERENCES
- 1.Hellman S, Weichselbaum RR. Oligometastases. J Clin Oncol. 1995;13:8–10. [DOI] [PubMed] [Google Scholar]
- 2.Kyriakopoulos CE, Chen YH, Carducci MA, Liu G, Jarrard DF, Hahn NM, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: long-term survival analysis of the randomized phase III E3805 CHAARTED Trial. J Clin Oncol. 2018;36:1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker CC, James ND, Brawley CD, Clarke NW, Hoyle AP, Ali A, et al. Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): a randomised controlled phase 3 trial. Lancet 2018;392:2353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips R, Shi WY, Deek M, Radwan N, Lim SJ, Antonarakis ES, et al. Outcomes of observation vs stereotactic ablative radiation for oligometastatic prostate cancer: The ORIOLE phase 2 randomized clinical trial. JAMA Oncol. 2020;6:650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez DR, Tang C, Zhang J, Blumenschein GR Jr, Hernandez M, Lee JJ, et al. Local consolidative therapy vs. maintenance therapy or observation for patients with oligometastatic non-small-cell lung cancer: long-term results of a multi-institutional, phase II, randomized study. J Clin Oncol. 2019;37:1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutera P, Clump DA, Kalash R, D’Ambrosio D, Mihai A, Wang H, et al. Initial results of a multicenter phase 2 trial of stereotactic ablative radiation therapy for oligometastatic cancer. Int J Radiat Oncol Biol Phys. 2019;103:116–22. [DOI] [PubMed] [Google Scholar]
- 8.Palma DA, Olson R, Harrow S, Gaede S, Louie AV, Haasbeek C, et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. Lancet 2019;393:2051–8. [DOI] [PubMed] [Google Scholar]
- 9.Ost P, Reynders D, Decaestecker K, Fonteyne V, Lumen N, De Bruycker A, et al. Surveillance or metastasis-directed therapy for oligometastatic prostate cancer recurrence: a prospective, randomized, multicenter phase II trial. J Clin Oncol. 2018;36:446–53. [DOI] [PubMed] [Google Scholar]
- 10.Ali A, Hoyle A, Haran ÁM, Brawley CD, Cook A, Amos C, et al. Association of bone metastatic burden with survival benefit from prostate radiotherapy in patients with newly diagnosed metastatic prostate cancer: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2021;7:555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol. 2016;34:1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasoglou V, Michoux N, Van Damme J, Van Nieuwenhove S, Halut M, Triqueneaux P, et al. Pattern of metastatic deposit in recurrent prostate cancer: a whole-body MRI-based assessment of lesion distribution and effect of primary treatment. World J Urol. 2019;37:2585–95. [DOI] [PubMed] [Google Scholar]
- 13.Gupta SK, Watson T, Denham J, Shakespeare TP, Rutherford N, McLeod N, et al. Prostate-specific membrane antigen positron emission tomography-computed tomography for prostate cancer: distribution of disease and implications for radiation therapy planning. Int J Radiat Oncol Biol Phys. 2017;99:701–9. [DOI] [PubMed] [Google Scholar]
- 14.Ost P, Reynders D, Decaestecker K, Fonteyne V, Lumen N, Bruycker AD, et al. Surveillance or metastasis-directed therapy for oligometastatic prostate cancer recurrence (STOMP): Five-year results of a randomized phase II trial. J Clin Oncol. 2020;38:10. [Google Scholar]
- 15.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;116:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B, et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol. 2019;76:89–97. [DOI] [PubMed] [Google Scholar]
- 18.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell 2013;153:666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–45. [DOI] [PubMed] [Google Scholar]
- 20.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64. [DOI] [PubMed] [Google Scholar]
- 21.Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-B: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37:490–503. [DOI] [PubMed] [Google Scholar]
- 22.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22:1520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam PJ, Van Oyen P, et al. TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2019;25:1766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deek MP, Van der Eecken K, Phillips R, Parikh NR, Isaacsson Velho P, Lotan TL, et al. The mutational landscape of metastatic castration-sensitive prostate cancer: The Spectrum Theory Revisited. Eur Urol. 2021;80:632–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schweizer MT, Sivakumar S, Tukachinsky H, Coleman I, De Sarkar N, Yu EY, et al. Concordance of DNA repair gene mutations in paired primary prostate cancer samples and metastatic tissue or cell-free DNA. JAMA Oncol. 2021;7:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.