

Abstract
MAPT encodes the microtubule-associated protein tau, which is the main component of neurofibrillary tangles (NFTs) and found in other protein aggregates. These aggregates are among the pathological hallmarks of primary tauopathies such as frontotemporal dementia (FTD). Abnormal tau can also be observed in secondary tauopathies such as Alzheimer’s disease (AD) and synucleinopathies such as Parkinson’s disease (PD). On top of pathological findings, genetic data also links MAPT to these disorders. MAPT variations are a cause or risk factors for many tauopathies and synucleinopathies and are associated with certain clinical and pathological features in affected individuals. In addition to clinical, pathological, and genetic overlap, evidence also suggests that tau and alpha-synuclein may interact on the molecular level, and thus might collaborate in the neurodegenerative process. Understanding the role of MAPT variations in tauopathies and synucleinopathies is therefore essential to elucidate the role of tau in the pathogenesis and phenotype of those disorders, and ultimately to develop targeted therapies. In this review, we describe the role of MAPT genetic variations in tauopathies and synucleinopathies, several genotype-phenotype and pathological features, and discuss their implications for the classification and treatment of those disorders.
Keywords: MAPT, neurodegenerative disorders, tauopathies, synucleinopathies, genetic variation
A pathological hallmark of several neurodegenerative disorders is accumulation of misfolded proteins aggregates. For instance, accumulation of misfolded microtubule-associated protein tau (MAPT) is the hallmark of tauopathies, which may be further classified as primary and secondary. In primary tauopathies, tau is the main aggregated protein, as it is the case in frontotemporal dementia (FTD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), Pick’s disease (PiD) chronic traumatic encephalopathy (CTE), and primary age-related tauopathy (PART). In secondary tauopathies, other misfolded proteins are present alongside tau, such as amyloid beta (Aβ) in Alzheimer’s disease (AD)1. Similarly, synucleinopathies are characterized by the accumulation of misfolded alpha-synuclein and include Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA)2–4. Interestingly, evidence suggests that tau and alpha-synuclein might collaborate in neurodegeneration by co-localizing and promoting misfolding and accumulation of one another5–7. Tau pathology is also frequently observed in synucleinopathies and vice-versa5. However, a clear cause-effect between misfolded protein aggregates and neurodegeneration has not been demonstrated in most tauopathies and synucleinopathies, and it is possible that these aggregates are a byproduct of neurodegeneration, or that misfolded protein aggregation is a protective mechanism that becomes overwhelmed in individuals with disease8. Supporting this hypothesis, tau and alpha-synuclein pathology can also be seen in healthy individuals and are positively correlated with age8. For these reasons, elucidating the role of misfolded protein aggregation and understanding the role of tau in neurodegeneration is important for both tauopathies an synucleinopathies. While genetic variations in MAPT, the gene encoding tau, is a cause or risk factor for many tauopathies and synucleinopathies9, their role in the pathogenesis and phenotype of most of those disorders needs to be clarified. Herein, we describe the roles of MAPT variations in the phenotype and pathological features of tauopathies and synucleinopathies, and discuss the implications of these findings for disease classification and therapy.
MAPT and Tau: structure, function and interaction with alpha-synuclein
MAPT, located on chromosome 17q21.31, is composed of 15 exons and encodes the microtubule associated protein tau. The gene has two main haplotypes due to a 900 kb inversion on chromosome 17q2110. The H1 haplotype (direct orientation) is observed in all populations and has normal patterns of genetic variation, resulting in multiple different subhaplotypes11. The H2 haplotype (inverse orientation) is almost exclusive to individuals of European ancestry and has a prevalence of about 20% in this population12. Although the H2 haplotype sequence differs significantly from the H1 haplotype, sequence diversity and variability within the haplotype itself is limited10. Tau is particularly present in neurons of the central nervous system, especially in axons. Six known isoforms are expressed in the adult human brain (figure 1).
Figure 1.
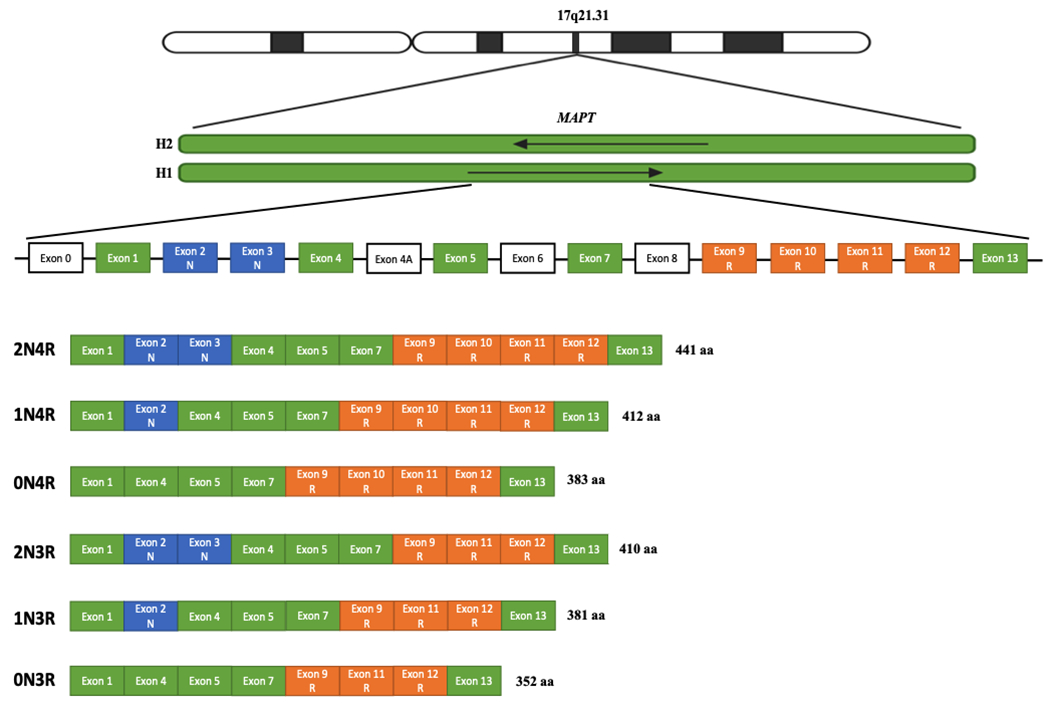
Structure of the six tau isoforms expressed in the adult human brain
Human MAPT is located on chromosome 17q21.31 and has two main haplotypes: H1 (direct orientation) and H2 (inverse orientation). The gene contains 15 exons, including exon 0, which encodes the 5’ untranslated region (UTR). Tau isoforms can have zero, one, or two amino-terminal inserts based on alternative splicing of exons 2 and 3 (0N, 1N, 2N; shown in blue), The insertion of exon 3 is dependent on the presence of exon 2. Tau isoforms can also include three or four microtubule-binding repeats, depending on splicing of exon 10 (3R or 4R; shown in orange). Exon 4A corresponds to the alternative exon located between exons 4 and 5, and is expressed outside of the brain. Exon 6 is also expressed outside the brain, while exon 8 insertion has not been reported in humans.
The protein has four functional domains: an N-terminal projection domain, a proline-rich domain, a microtubule-binding domain (R), and a C-terminus13. The major role of tau in the central nervous system is to promote microtubules assembly, involved in neurite polarity, axon elongation, and axonal transport13. These functions are influenced by the exonic composition of MAPT isoforms: compared to their counterparts with three microtubule-binding domains (3R) (figure 1), 4R tau isoforms have greater affinity for microtubules and lead to increased microtubule assembly. Tau has other roles in addition to its association with microtubules, including regulation of neuronal plasticity, nucleolar organization, and DNA protection against oxidative stress1. Tau can also undergo multiple posttranslational modifications (PTMs) that regulate its activity. The most notable of these is phosphorylation, which decreases the affinity of tau for tubulin and facilitates its dissociation from microtubules. Less is known about the influence of other PTMs and epigenetic mechanisms on MAPT, but they are also likely involved in tauopathies. For instance, tau acetylation prevents degradation of phosphorylated tau, and autopsy studies have shown increased levels of tau acetylation in different tauopathies14–16.
Soluble tau can accumulate in the cytoplasm in the form of nonfibrillar aggregates known as pre-tangles, which eventually undergo conformational changes and form intracellular, insoluble, densely packed neurofibrillary tangles (NFTs)17. Accumulation of NFTs, and possibly to a greater extent their precursors, might lead to neuronal dysfunction and death by disrupting microtubule assembly, axonal transport and mitochondrial trafficking18.
Alpha-synuclein, encoded by SNCA, is the main component of proteins aggregates that are the pathological hallmark of synucleinopathies: Lewy bodies (LBs) in Parkinson’s disease (PD) and dementia with LBs (DLB), and glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA). In addition to being involved in these three disorders, alpha-synuclein may interact with tau and might contribute to tauopathy via several mechanisms, including inhibition of tau-tubulin binding, tau hyperphosphorylation, initiation of tau polymerization, and promotion of tau aggregation5. Conversely, dysfunction of tau-mediated axonal transport may promote accumulation of tau and alpha-synuclein in neurons19. Alpha-synuclein and tau also co-localize in axons and Lewy bodies, and they synergistically promote the fibrillation and accumulation of each other in vitro and in vivo6,7,20. There is some pathological overlap between tauopathies and synucleinopathies. LBs are observed in more than half of AD autopsies, and the majority of PD patients also have some AD pathology21,22.
Tauopathies
Frontotemporal Dementia
FTD is a clinically and neuropathologically heterogeneous group of neurodegenerative disorders characterized by frontotemporal lobar degeneration (FTLD), executive dysfunction, behavioral abnormalities, personality changes, and progressive speech and language difficulties. Affected individuals might also develop motor symptoms such as parkinsonism with or without atypical features23–26. The three main clinical subtypes of FTD are behavioral variant FTD (bvFTD), progressive non-fluent aphasia (PNFA), and semantic dementia (SD). Pathologically, FTLD is categorized based on the type of inclusion observed, the two most common being tau (FTDL-tau) and TAR DNA-binding protein 43 (TDP-43, FTDL-TDP)23,27. The clinical and pathological features of FTD and its subtypes are described in table 1. Although clinicopathological correlations have been established, clinical presentation cannot reliably predict autopsy findings. Most cases of SD are associated with FTLD-TDP, while tau inclusions are seen in most cases of PNFA. Individuals with bvFTD can exhibit FTLD-tau, FTLD-TDP, or other types of pathology28–30. Brain atrophy patterns seem to correlate more closely with clinical phenotype (Table 1)31.
Table 1.
Clinical and pathological features of main tauopathies and synucleinopathies
| Disease | Proteinopathy type | Motor features | Cognitive features | Protein accumulation pattern | Brain atrophy and neuronal degeneration pattern | References |
|---|---|---|---|---|---|---|
| FTD (FTLD-tau) | Primary tauopathy | Parkinsonism with symmetrical bradykinesia, postural rigidity, absence of resting tremor, and minimal or absent response to levodopa. Features can overlap with CBS or PSP. | bvFTD: personality changes, disinhibition, apathy, impulsivity, loss of empathy, hyperorality, compulsive behavior, repetitive movements, ritualistic behaviors, and stereotypy of speech. Deficits in executive tasks with relative sparing of episodic memory and visuospatial skills. PNFA: impaired speech production and non-fluent speech, impaired comprehension of complex sentences. Spared single word comprehension and object knowledge. SD: impaired word comprehension, loss of semantic memory, surface dyslexia or dysgraphia. Spared repetition and speech production. |
FTLD-tau pathologic subtypes: AGD - accumulation of 4R tau-containing argyrophilic grains in the medial temporal lobe. GGT - 4R-tau containing globular glial inclusions and neuronal globular or tangle-like inclusions. Pathology can overlap with PSP and CBD. |
bvFTD - Bilateral atrophy of the frontal lobes, insula, anterior cingulate, and anterior temporal lobes. PNFA - atrophy of the anterior perisylvian cortex, mostly in the dominant hemisphere. SD - asymmetrical atrophy of the dominant anterior inferior temporal lobe. |
23,26,31–33,36,211–214 |
| PSP | Primary tauopathy | Heterogeneous phenotype with many possible variants. Ocular motor dysfunction, dysarthria, postural instability, and bradykinesia / akinesia are common. Can also present as CBS. | Behavioral and/or language impairment similar to subtypes of FTD | Accumulation of 4R-tau NFTs in subcortical structures, especially the subthalamic nucleus, basal ganglia, and brainstem. The NFTs may be associated with 4R-tau positive tufted astrocytes, oligodendroglial coiled bodies, and neuropil threads. | Atrophy of the subthalamic nucleus and brainstem tegmentum, depigmentation of the substantia nigra. | 33,79 |
| CBD | Primary tauopathy | CBS: asymmetric levodopa-resistant parkinsonism, dystonia, myoclonus, orobuccal or limb apraxia, alien limb phenomena, cortical sensory deficit. Can present similarly to PD or PSP. | Can present similarly to FTD or AD-like dementia. | Accumulation of 4R-tau positive neuronal inclusions, white and grey matter threads, coiled bodies, and astrocytic plaques. NFTs and corticobasal bodies are seen. | Asymmetric focal cortical atrophy and depigmentation of the substantia nigra. | 85,96 |
| PiD | Primary tauopathy | Most commonly presents as bvFTD, but can also present as other types of FTD and CBS. | 3R-tau Pick bodies, predominantly in the dentate gyrus and temporal lobes. | Cerebral atrophy predominant in frontal and temporal lobes. | ||
| CTE | Primary tauopathy | Usually late in disease: parkinsonism and, in some cases, motor neuron disease. | Irritability, aggressively, impulsivity, depressive symptoms, and memory impairment. Speech abnormalities and dementia can develop later. | Focal deposition of tau-positive NFTs, tangles, threads, and astrocytes that is most pronounced around cortical sulci and penetrating vessels. The medial temporal lobe, basal ganglia, diencephalon, and brainstem can also be involved, especially later in the disease. TDP-43, Aβ, and alpha-synuclein deposition can also be observed. | Generalized atrophy of the cerebral cortex, notably medial temporal lobes, diencephalon, and mamillary bodies. | 104,218 |
| PART | Primary tauopathy | Usually not observed. | Can be associated with memory loss in aging. Can also present with progressive decline in episodic and semantic memory, processing speed, and attention. Symptoms are usually milder and progress more slowly than in AD. | Accumulation of 3R and 4R-tau NFTs mostly in the medial temporal lobe, basal forebrain, brainstem, and olfactory bulb and cortex. The NFTs are identical to those of AD, but Aβ deposits are not seen. Symptom severity correlates with NFT burden. | Neocortical and medial temporal lobe atrophy might be observed in some individuals. | 106,219,220 |
| AD | Secondary tauopathy | Extrapyramidal symptoms or parkinsonism can be present late in disease but are not common nor predominant. | Short-term memory loss, visuospatial impairment, and executive dysfunction. Language and behavior impairment typically occur later. Can also develop personality changes, loss of empathy, and obsessive / compulsive behaviors. | NFTs and Aβ extracellular neuritic plaques. NFTs accumulate in a stereotypical manner: 1) entorhinal cortex and hippocampus 2) other limbic structures such as the amygdala and thalamus, 3) in neocortex. LBs are also seen in most cases, predominantly in the amygdala. |
Predominant atrophy of medial, basal, lateral temporal, and medial parietal lobes. | 208–210 |
| PD | Synucleinopathy | Asymmetric and levodopa-responsive parkinsonism. Shuffling, festinating gait. Postural instability and autonomic dysfunction can occur later in disease. | Sleep dysfunction, depression, anxiety, apathy. Dementia occurs late in disease. | Accumulation of misfolded alpha-synuclein into LBs, predominantly in the substancia nigra. NFTs and Aβ plaques can also be identified. | Loss of dopaminergic neurons, predominantly in the ventrolateral area of the substantia nigra pars compacta. | 2.135,215 |
| DLB | Synucleinopathy | Parkinsonism (often less levodopa-responsive than PD), dysautonomia. | Dementia, fluctuating cognition, visual hallucinations, RBD, apathy, depression. | LBs predominantly in the brainstem. LBs can also accumulate in limbic structures and in the cerebral cortex, particularly the frontal and temporal lobes. Aβ plaques with or without NFTs can be seen. | Pallor of the substantia nigra, atrophy of the midbrain, hypothalamus, substantia innominate, lateral prefrontal cortex, and left premotor cortex. | 3,170,216 |
| MSA | Synucleinopathy | Levodopa-unresponsive parkinsonism (can be responsive to levodopa, especially early in disease), pyramidal signs, cerebellar abnormalities, and autonomic dysfunction. Predominant symptoms depend on subtype: MSA-P (parkinsonism) and MSA-C (cerebellar dysfunction). | Sleep dysfunction, depression, anxiety, attention deficit. Presence of dementia should prompt consideration of alternative diagnosis, such as DLB. | Presence of argyrophilic GCIs that are alpha-synuclein and tau-positive. | Neuronal loss and gliosis in the striatonigral and olivopontocerebellar systems. | 4,179,217 |
Abbreviations: FTD: frontotemporal dementia; FTLD: frontotemporal lobar degeneration; CBS: corticobasal syndrome; PSP: progressive supranuclear palsy; bvFTD: behavioral variant frontotemporal dementia; PNFA: progressive non-fluent aphasia; SD: semantic dementia; AGD: argyrophilic grain disease; GGT: globular glial tauopathy; CBD: corticobasal degeneration; NFTs: neurofibrillary tangles; PD: Parkinson’s disease; AD: Alzheimer’s disease; PiD: Pick’s disease; CTE: chronic traumatic encephalopathy; TDP-43: TAR DNA-binding protein-43; Aβ: amyloid beta; PART: primary age-related tauopathy; LBs: Lewy bodies; DLB: dementia with Lewy bodies; RBD: rapid eye movement sleep behavior disorder; MSA: multiple system atrophy; GCIs: glial cytoplasmic inclusions
FTLD-tau can be further divided based on the tau isoform contained in the inclusions. Pick’s disease (PiD) is associated with aggregation of 3R-tau, while 4R-tau is seen in argyrophilic grain disease (AGD) and globular glial tauopathy (GGT). Pathological patterns that can be seen in FTLD-tau can also include CBD and PSP (table 1)32. The pathology of the different subtypes of FTLD-tau and other tauopathies has been described in detail in a recent review33.
More than 50 pathogenic MAPT mutations have been reported in FTLD, comprising 5-20% of familial cases depending on the population34,35. The phenotype of affected individuals is highly variable, even in patients with the same variant from the same family. The different phenotypes described in FTLD-tau and corresponding MAPT mutations are shown in figure 29,25,36–63. Pathological features seen in MAPT-associated FTLD-tau are similar to the different FTLD-tau subtypes seen in sporadic cases and include PiD, AGD, GGT, CBD, and PSP32,37,64–66. The vast majority of pathogenic mutations are either coding mutations affecting microtubule-binding repeats and their flanking region (exons 9-13, figure 2), reducing their ability to bind to microtubules, or variants in exon 10 and intron 10 that lead to differential splicing and a relative increase in 4R tau. Accordingly, mutations in exon 10 and intron 10 are mostly associated with predominantly 4R FTLD-tau (AGD, GGT, CBD, PSP), while most mutations in other exons are associated with predominant 3R pathology (PiD)37. The p.V337M and p.R406W mutations, in exon 12 and 13 respectively, have been associated with mixed 3R and 4R isoforms and paired, straight, helical tau filaments36. The molecular pathophysiology of specific MAPT mutations and the associated pathological and neuroimaging features has been comprehensively reviewed36. Unlike other mutations, the p.A152T MAPT variant is not a disease-causing mutation but rather a risk factor for the development of FTD67,68. The role of MAPT haplotypes in FTD is unclear. While some studies have found an association between FTD and the H169−71 or H272,73 haplotype, other studies, including a meta-analysis of the H2 haplotype, have found no such association74–77. There are contradicting results regarding the effect of the H2 haplotype on AAO of FTD70,71,73,77,78. Overall, it seems that these haplotypes do not have a major role in FTD, whereas rare MAPT mutations are important in FTD.
Figure 2.
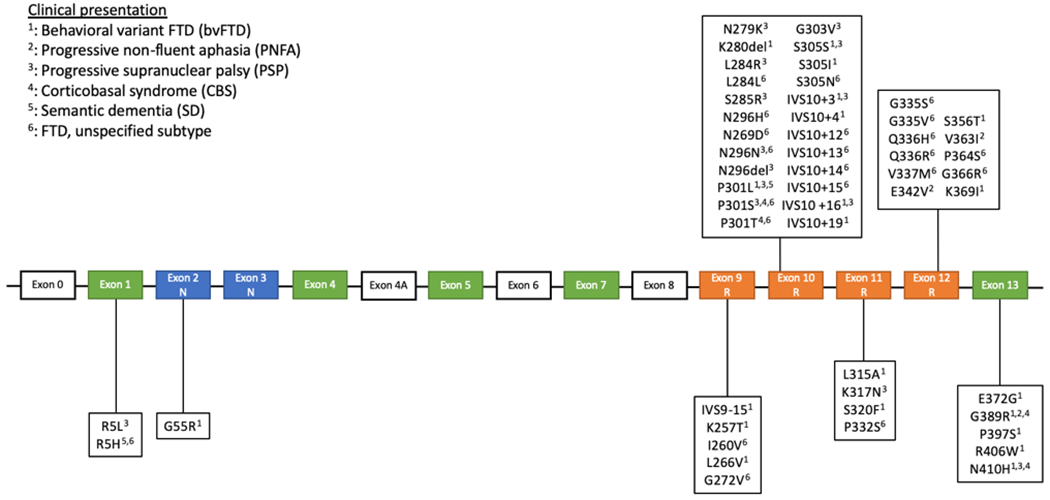
MAPT mutations and associated clinical presentations in frontotemporal lobar degeneration
The vast majority of reported mutations are in exons 9-13 and in the intronic region following exon 10. The most commonly reported presentations are bvFTD, PSP, and unspecified types of FTD. The mutations associated with a bvFTD and unspecified FTD presentation are well distributed among exons 9-13, while all but 3 mutations presenting with a PSP phenotype are within exon 10 and the intronic region following it. The four mutations associated with a CBS presentation are located in exons 10 and 13. The two mutations associated with PNFA are in exon 12 and 13 and the two SD-associated mutation are located in exons 1 and 10. All the intronic mutations are associated with either bvFTD or an unspecified FTD subtype. PSP was the other clinical presentation observed in intronic mutations. The three most commonly reported mutations are N279K, P301L, and IVS10+16. Exons in orange encode the microtubule-binding repeats, while exons in blue encode the amino-terminal inserts.
Progressive supranuclear palsy
PSP is traditionally considered an atypical parkinsonian syndrome with a heterogeneous phenotype79. The main clinical and pathological features of PSP are detailed in table 1. Most identified pathogenic MAPT mutations (summarized in figure 2) are located in exon 10 and intron 10, which is consistent with the 4R-tau pathology seen in PSP. However, families with autosomal dominant PSP with MAPT mutations are rare42,80,81, and the main role of MAPT in PSP is as the most important genetic risk factor rather than a monogenic cause. Numerous studies have shown that the MAPT H1c subhaplotype and the H2 haplotype are respectively associated with an increased and decreased PSP risk75,82–85. The increased PSP risk associated with the H1c subhaplotype may be explained by increased levels of MAPT gene expression and proportion of 4R isoform86–88. An increase in N-terminal exon-containing MAPT transcripts may explain the reduced risk seen with the H2 haplotype89.
Despite the clear association between the MAPT H1c subhaplotype and risk of PSP, only few studies have investigated correlations between MAPT variations and clinicopathological phenotype. No difference in age at onset (AAO), motor and cognitive symptoms, and survival between heterozygous and homozygous H1 PSP patients have been reported90. In addition, similar rates of cognitive impairment and no significant difference in tau burden and Braak NFT stage were found when comparing H1/H1 and H1/H2 carriers with PSP91. This lack of significant impact of tau haplotypes on PSP pathology was also previously noted92,93. Interestingly, another study analyzed cognitive performance in 305 PSP patients, and found that carriers of the H1c subhaplotype had better performance in terms of executive function, attention, and general cognitive function94. The H1c subhaplotype has also been associated with a more severe pathological phenotype95. Compared to FTD, in PSP the H1 subhaplotypes and the H2 haplotypes seem to have a more prominent role.
Corticobasal degeneration
CBD is a pathological entity that most commonly presents clinically as corticobasal syndrome (CBS), a form of atypical parkinsonism. The pathological features and possible clinical phenotypes of CBD are described in Table 196. CBD and PSP show some MAPT-related genetic overlap: MAPT mutations are a rare cause of CBD, and the H1 MAPT haplotype and H1c subhaplotype are well-established risk factors for CBD; the H2 haplotype is also associated with reduced risk11,75,97–100. Genotype-phenotype correlations studies of MAPT in CBD are even more limited than they are for PSP. The only such study found no correlation between tau pathology burden and MAPT haplotype in 36 H1/H1 and 9 non-H1/H1 CBD autopsy cases93. There are major limiting factors for determining the influence of MAPT on the clinical and pathological characteristics of CBD, including the significant clinicopathological overlap with other tauopathies, the rarity of CBD (and even lower prevalence of non-H1/H1 patients), and necessity of autopsy for a definitive diagnosis.
Pick’s disease
PiD is a unique pathological entity, as it is the only pure 3R-tau tauopathy101. Its main pathological features and the associated clinical presentations are described in table 1. Factors that contribute to the 3R-tau nature of Pick bodies include the disease-specific patterns of tau filament phosphorylation and folding seen in PiD41,102. MAPT mutations associated with PiD tend to be localized outside of exon and intron 10, as these mutations typically lead to a relative increase and 4R-tau and are associated with corresponding 4R-tau pathologies37. Some pathological features seen in PiD are associated with specific mutations. For instance, the MAPT p.K257T and p.S320F mutations are associated with Pick bodies and axonal inclusions, while the p.L266V and p.L315R mutations are also associated with glial inclusions. In contrast to 4R-tauopathies, the MAPT H1 haplotype does not seem to be associated with PiD103. This lack of association might explain some of the conflicting results seen in association studies of MAPT haplotypes in FTD, as the different studies may have different proportions of patients with PiD pathology. This emphasizes the importance of pathological confirmation of disease, or preferably, identification of reliable biomarkers that can distinguish between the pathologies. This may also indicates that PiD pathology might be associated with distinct diseases mechanisms and will have different responses to treatment. This distinction is therefore also important for future therapy development and participation in clinical trials.
Chronic traumatic encephalopathy
CTE is a pathologically defined neurodegenerative disease that usually begins to develop about 10 years after repetitive traumatic brain injury104. Its clinical and pathological features are described in table 1. Although it is considered a tauopathy, TDP-43, Aβ, and alpha-synuclein deposition can also be observed104. Genetic studies in CTE are limited, and no association with MAPT haplotypes or variants have been reported so far104,105. Further studies are needed to elucidate the genetics of CTE.
Primary age-related tauopathy
PART is a neuropathological pattern commonly observed in older individuals106. Its pathology and associated clinical features are described in table 1. Despite the fact that research on PART has increased in the last years, it remains an understudied entity. The H1 MAPT haplotype was overrepresented in two small studies that included a total of 30 patients107,108, which are too underpowered to determine whether this association is true. Next steps in the study of PART involve a better characterization of the clinical spectrum associated with the pathological features, and genetic association studies that will evaluate the impact of MAPT on its incidence, pathology, and symptomatology.
Alzheimer’s disease
AD is the most prevalent neurodegenerative disorder and the most common cause of dementia109. The pathology and symptomatology of the disease is described in table 1. It is considered a secondary tauopathy: in addition to intracellular NFTs, AD is also characterized by aggregation of Aβ in the form of extracellular neuritic plaques. The two proteins may act synergistically in a positive feedback loop to promote their aggregation: Aβ leads to tau hyperphosphorylation, while extracellular secreted tau leads to increased level of Aβ110–112. A recent study has shown that the biochemical characteristics of tau vary among individuals with AD, and that features that promote tau seeding, such as high levels of high-molecular weight, hyperphosphorylated, oligomeric tau are associated with increased tau burden and a more severe clinical phenotype113. Early case-control genetic studies of MAPT in AD have been inconsistent in the identification of risk variants87,114–120. More recent studies, including meta-analyses, have identified an association between the H1 haplotype (particularly the H1c subhaplotype) and increased risk of AD and an association with reduced risk with the H2 haplotype75,88,89,121–123. In addition, the A allele of the rs393152 single nucleotide polymorphism (SNP), which is located within the extended MAPT locus, was found to be significantly associated with increased risk of AD in a meta-analysis88.
There is currently no definitive evidence that monogenic forms of AD are caused by MAPT mutations 124–127. Table 2 summarizes the genetic findings related to MAPT in AD. Genotype-phenotype studies of MAPT in AD are limited: non-replicated studies have shown an association between the H1 haplotype and a higher prevalence of symptoms in individuals who met AD pathological criteria at autopsy128, a decreased NFT and Aβ plaque burden129, and a faster rate of cognitive decline in affected individuals130. Studies correlating cerebrospinal fluid levels of tau and MAPT mutations have shown conflicting results131–134.
Table 2.
Reported MAPT genetic findings in tauopathies and synucleinopathies
| Disease | Causative variants | Risk variants and haplotypes | Phenotype-genotype correlations | References |
|---|---|---|---|---|
| FTD | Numerous causative variants reported; see figure 1. | p.A152T is a risk variant. Role of MAPT haplotype is unclear. | Earlier age of onset, higher prevalence of movement disorder, and lower prevalence of language disorder compared to other genetic causes of FTD. Earlier AAO compared to sporadic cases of FTD. H2: possibly earlier AAO. See figure 1 for phenotypes associated with reported causative variants. |
66–68,70–73,75–78,194,221 |
| PSP | Numerous causative variants reported; see figure 1. | H1c: increased risk. H2: decreased risk. H1d, H1g, and H1o: increased risk (one large study). H1b and H1q: increased risk (one small study each). |
H1c: better cognitive performance (one study), more severe pathological phenotype (one study). H1: earlier AAO (one study). |
82–84,93–95,99,193,222 |
| CBD | Numerous causative variants reported; see figure 1. | H1c: increased risk. H2: decreased risk. |
No correlation between tau pathology burden and MAPT haplotype (one study). | 99 |
| PiD | Numerous causative variants reported; see figure 1. | MAPT haplotype are not associated with PiD risk. | See FTD. | 41,43,50 |
| CTE | None. | No significant association found (one study) | None reported. | 105 |
| PART | None. | H1 overrepresented in two small studies. | None reported. | 107,108 |
| AD | p.R406W mutation and 17q21.31 microduplication have shown AD-like phenotype but lack Aβ pathology. | H1 and H1c: increased risk. H2: decreased risk. Unclear association with pA152T variant rs393152: increased risk with A allele |
H1: faster cognitive decline, decreased NFTs and Aβ burden, lower prevalence of symptoms among those that met AD pathological criteria (one study each). Conflicting results for genotype associated with CSF tau levels. |
68,69,88,123,125–134,190,192 |
| PD | None. | H1: increased risk. H2: decreased risk. |
H1: decreased cognitive performance, increased risk of PD dementia (mixed results), non-tremor dominant phenotype, hallucinations. H1p: PD dementia (one study). H1h: non-tremor dominant PD (one study). Intronic MAPT SNPs (rs2435207 and rs11079727): later onset of motor symptoms in LRRK2-associated PD. Mixed genotype-pathology findings. |
88,138.148–162,168,169,195,224–226 |
| DLB | None. | p.A152T, H1/H1 diplotype, and H1g subhaplotype are potential risk factors (mixed results). | H1: Increased alpha-synuclein deposition, particularly in brainstem regions (one small cohort). | 171–173,177,178 |
| MSA | None. | H1, H1j and H1x: increased risk. H1e and H2: reduced risk. (one study each) |
No association between genotype and AAO (one study). | 180,181,186 |
Abbreviations: FTD: frontotemporal dementia; AAO: age at onset; FTLD: frontotemporal lobar degeneration; PSP: progressive supranuclear palsy; CBD: corticobasal degeneration; PiD: Pick’s disease; CTE: chronic traumatic encephalopathy; PART: primary age-related tauopathy; AD: Alzheimer’s disease; NFTs: neurofibrillary tangles; Aβ: amyloid beta; CSF: cerebrospinal fluid; PD: Parkinson’s disease; DLB: dementia with Lewy bodies; MSA: multiple system atrophy
Despite the fact that NFTs are a pathological hallmark of the disease, the influence of MAPT variations seems to be minor in AD. An issue that can complicate genetic studies is the significant clinical overlap between AD and other tauopathies and the lack of neuropathological confirmation in many cases. Further research is required to clarify the impact of MAPT variation on the clinicopathological phenotype of AD.
Synucleinopathies
Although this group of diseases is defined by the presence of alpha-synuclein accumulation, tau pathology is quite common, and a role for MAPT genetic variants have been demonstrated with different degrees of certainty.
Parkinson Disease
PD is the most common synucleinopathy135. The main clinical manifestations and pathological findings of PD are described in table 1. Despite being considered a synucleinopathy, NFTs (and Aβ plaques) can also be found in PD, with a higher tau pathology burden in those who also developed dementia136. The MAPT H1 haplotype is consistently identified as one of the strongest genetic risk factors for PD, while the H2 haplotype is associated with reduced risk75,137–141. Subhaplotype analysis have yielded inconsistent results between different populations142–145 or no association with PD137,146,147. Some studies have suggested genotype-phenotype correlations, yet many of those were not replicated, and therefore need to be considered cautiously. For example, several studies suggested that MAPT variants affect cognition in PD, demonstrating an association of the H1 haplotype with decreased cognitive performance148,149 and increased risk of dementia150–153. However, these results were not replicated in other studies154–160. Other features that have been reported to be more common in H1/H1 PD patients include a non-tremor dominant phenotype161,162 and hallucinations155. Although one study reported an association between the H2 haplotype-tagging SNP rs8070723 and an older PD AAO163, other studies found no such association164–167. Reports of correlations between MAPT variations and PD pathological findings have shown inconsistent results including increased LB burden associated with the H1168 or H2 haplotype169, decreased NFTs in patients with the H1 haplotype129, or no association between MAPT genotype and pathology159.
The role of the MAPT H1 haplotype as a risk factor for PD is well-established. Larger, and preferably longitudinal studies, are required to properly examine whether MAPT variants have true effects on the clinical course of PD.
Dementia with Lewy bodies
DLB is one of the most common causes of dementia170. Table 1 describes the main clinical and pathological findings associated with the disease. Genetic association studies investigating MAPT as a potential risk factor for DLB have found overrepresentation of the p.A152T171 variant, H1/H1 diplotype172, and H1g subhaplotype173 in affected individuals. However, these results have not been reported in three large studies174–176. In an autopsy cohort of 22 DLB patients, an increased alpha-synuclein deposition the brains of H1/H1 patient was noted, particularly in brainstem regions177,178. These results have yet to be replicated in other cohorts. Despite the fact that tau pathology is not uncommon in DLB, current evidence on a possible influence of MAPT variation on disease risk or phenotype is limited and suggest a minor effect at best. Further studies are needed to clarify these potential associations and the role of MAPT in DLB.
Multiple system atrophy
MSA, the third major synucleinopathy, is a rare neurodegenerative disorder that can have a broad range of manifestations179. The clinical and pathological findings of the disease are described in table 1. Few studies on the role of MAPT variation in MSA have been published. A positive association between the H1 haplotype and MSA has been reported180 , while another study181 suggested that the H1j and H1x subhaplotype are risk factors for MSA. The H1e and H2 haplotypes were associated with reduced risk. However, these studies included only 59 and 213 MSA cases, respectively. Other studies did not identify associations between MAPT and MSA, although they only genotyped a limited number of variants182–185. In another study, no difference in haplotype distribution when comparing 22 early-onset (defined by the authors as ≤ 55 years) and 18 late-onset MSA cases was found186. Similar to DLB, current evidence, although limited, suggest that MAPT variations are either not significant or have a minor role in MSA.
Overlap between tauopathies and synucleinopathies
Tauopathies and synucleinopathies share various clinical, pathological, and genetic characteristics. Cognitive impairment is a feature of all the reviewed disorders (except MSA), and parkinsonism is also common in PD, DLB, MSA, PSP, and CBD. In fact, the four latter diseases are commonly considered as “Parkinson-plus” syndromes187. Parkinsonism can also be seen in FTD, CTE and late-stage AD (table 1). Tau pathology may be present in all of these disorders, either as a primary or secondary finding in tauopathies, and also as a component of other inclusions such as Lewy bodies and glial cytoplasmic inclusions in synucleinopathies (table 1). In addition, the MAPT H1 haplotype is associated with increased risk of PSP, CBD, AD, PD, and potentially FTD, PART, DLB, and MSA (table 2). This overlap may suggest possible interactions between alpha-synuclein and tau, and/or shared mechanisms of neurodegeneration. Another hypothesis is that different MAPT variants, and/or tau or alpha-synuclein accumulation make neurons more vulnerable in general, and neurodegeneration occurs due to additional factors. Indeed, different alterations in MAPT could contribute to the variation in observed phenotypes. For instance, the A allele of the H1-tagging rs242557 SNP is associated with increased risk of PSP and CBD but not PD139,188. If tau and alpha-synuclein are themselves pathogenic, it is possible that their co-occurrence in tauopathies and synucleinopathies is the result of a positive feedback mechanism, where the predominant protein promotes its own accumulation by enhancing misfolding and aggregation of its counterpart. Further studies are required to determine the causes and implications of the potential interactions between tauopathies and synucleinopathies, notably because current evidence is insufficient to determine whether misfolded alpha-synuclein and tau aggregates are pathogenic, protective, both (at different disease phases) or by-products of neurodegeneration8. Better understanding of these interactions could have important implications for future research and therapeutics.
Discussion – The role of MAPT in the spectrum of neurodegeneration
The current review highlights the potential roles of MAPT variation and the tau protein in tauopathies and synucleinopathies. These diseases vary in terms of age of onset, clinical manifestations, pathology, and pattern of inheritance (table 1). They also show some overlap. This overlap is even more prominent in FTD, PSP, and CBD, where the symptomatology of one disease can be associated with the pathology of another. In a recent study, 310 patients with a syndrome likely to be caused by frontotemporal lobar degeneration (bvFTD, PNFA, SD, PSP, CBD) were evaluated, and it was found that 62% of patients met the diagnostic criteria for more than one disorder. The same study also showed increased syndromic overlap in 46 patients at follow-up and a continuous spectrum of association between brain atrophy and clinical manifestations189. Genetic evidence also reinforces the concept of a spectrum, as the H1 haplotype is associated with PSP, CBD, and potentially FTD, and the fact that the same MAPT mutations might cause different clinical syndromes (figure 2). PSP and CBD also share several genetic risk factors outside of MAPT85,100.
The role of MAPT in neurodegenerative disorders extends beyond genetics and involves epigenetics and PTMs of tau. Tau hypomethylation was reported in AD190 (although not seen in two prior studies191,192), PSP95,193,194, and in the putamen of PD patients195. Tau hyperphosphorylation leads to its aggregation and is the most studied PTM, but other mechanisms are also likely involved in the pathogenesis of tauopathies and related disorders. Tau acetylation prevents degradation of phosphorylated tau, and post-mortem studies have shown increased levels of tau acetylation in AD, CBD, PSP, and other tauopathies14–16. Using cryo-electron microscopy, two recent studies have shown that tau filaments have disease-specific structures and folding patterns that are influenced by distinct PTM signatures that involve a combination of acetylation, ubiquitination, trimethylation, and phosphorylation196,197. Another potential factor that explains the clinical spectrum seen in tauopathies is that the proportion of different tau isoforms varies between different brain regions and between healthy and diseased individuals198.
The role of tau in neurodegeneration, in addition to its microtubule-related functions, may also involve other pathophysiologic pathways such as oxidative stress and mitochondrial dysfunction1,18. This may partially explain the failure of tau-targeting medications in clinical trials of AD199 and other tauopathies200. This would also provide insight on why the risk of developing PD, and potentially other synucleinopathies, is influenced by MAPT variations, beyond the co-localization and synergistic fibrillation of tau and alpha-synuclein. Another factor to consider is that the 17q21.31 region, where MAPT is located, contains other candidate genes for neurodegeneration, such as NSF and KANSL1, which could potentially contribute to the overrepresentation of the H1 haplotype in several neurodegenerative disorders201–203. NSF is involved in synaptic vesicle exocytosis and is a substrate of the PD-associated gene LRRK2204. Recently, de novo heterozygous NSF variants were reported in two unrelated children with early infantile epileptic encephalopathy205. KANSL1 is involved in chromatin modification and neuronal process, and KANSL1 haploinsufficiency has been shown to be sufficient to cause 17q21.31 syndrome, a rare multi-system disorder that is notably associated with intellectual disability, hypotonia, and epilepsy206.
Beyond its role as a risk factor, MAPT seems to be a disease modifier, as numerous correlations between genotype and motor, non-motor, and pathological features of different diseases have been reported (table 2). However, many of these associations need to be more clearly defined, as they were either observed in a small number of studies or not replicated. Better insight into these genotype-phenotype associations may lead to a better understanding of mechanisms of different symptoms, and to patient stratification for prognosis, treatment, and inclusion in clinical trials.
Concluding remarks
Despite the unclear mechanisms by which tau accumulation is associated with neurodegeneration, further understanding of the genetics MAPT is essential to improve disease classification and genotype-phenotype correlations of tauopathies and synucleinopathies. Furthermore, classification systems incorporating genetic information might better represent distinct pathogenic mechanisms and therefore better guide the development of new therapies and inclusion criteria in future clinical trials. In addition to genetics, should these criteria be based on specific symptoms, clinical diagnosis, genotype, brain atrophy patterns on imaging, potential biomarkers, or a combination of multiple factors? Currently, there is no definitive answer to this question, although it is clear that the current classification system, which is mainly based on clinical symptoms, has several limitations that might impede the implementation of targeted therapies. For instance, patients with similar clinical presentations of frontotemporal dementia may have 3R and 4R tauopathies and require different treatments, as these pathologies might be associated with different underlying mechanisms. This distinction might also expand to 4R tauopathy subtypes, as the different protein inclusions observed in AGD, GGT, PSP, and CBD could reflect distinct pathogenic mechanisms responding differently to therapy. In that case, more reliable pathological markers would have to be identified, since clinical manifestations and genotypes can only hint at a specific type of pathology but not reliably predict it. Whether tau-targeting drugs will prove to be beneficial in neurodegenerative diseases is still to be determined, since thus far no tau-targeting therapy has been successful in clinical trials199,200. However, this does not exclude MAPT and tau as therapeutic targets of interest. For instance, MAPT mutations and haplotype leading to increased tau expression could be targeted with antisense nucleotides (ASO), which could decrease tau expression via several mechanisms including splicing modulation, translational arrest, and targeted degradation207. It is also possible that only certain genetic subtypes of tauopathies (and potentially synucleinopathies) could respond to tau therapy. In any case, further understanding of the role of MAPT in neurodegenerative disorders will be crucial, and might lead to a better understanding of neurodegeneration, the mechanisms of specific diseases, the development of therapies that either target tau or biologically related mechanisms, and the selection of specific individuals who may benefit from such treatments.
Acknowledgements
ZGO is supported by the Fonds de recherche du Québec–Santé Chercheur-Boursier award and is a Parkinson Canada New Investigator awardee. OAR is supported by the National Institutes of Health (NIH; R01 NS78086; U54 NS100693 and U54 NS110435), the US Department of Defense (W81XWH-17-1-0249), The Michael J. Fox Foundation, American Parkinson Disease Association (APDA) Center for Advanced Research, a Lewy Body Dementia Association Research Center of Excellence, the Mayo Clinic LBD Functional Genomics Program and The Little Family Foundation. Figures were created with Biorender.com
Conflicts of interest
ZGO Received consultancy fees from Lysosomal Therapeutics Inc. (LTI), Idorsia, Prevail Therapeutics, Inceptions Sciences (now Ventus), Ono Therapeutics, Denali, Handl Therapeutics, Neuron23 and Deerfield.
References
- 1.Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016;126(Pt 3):238–292. [DOI] [PubMed] [Google Scholar]
- 2.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–1157. [DOI] [PubMed] [Google Scholar]
- 3.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. [DOI] [PubMed] [Google Scholar]
- 4.Stefanova N, Bucke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol. 2009;8(12):1172–1178. [DOI] [PubMed] [Google Scholar]
- 5.Moussaud S, Jones DR, Moussaud-Lamodiere EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62(4):389–397. [DOI] [PubMed] [Google Scholar]
- 7.Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. [DOI] [PubMed] [Google Scholar]
- 8.Espay AJ, Vizcarra JA, Marsili L, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92(7):329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang CC, Xing A, Tan MS, Tan L, Yu JT. The Role of MAPT in Neurodegenerative Diseases: Genetics, Mechanisms and Therapy. Mol Neurobiol. 2016;53(7):4893–4904. [DOI] [PubMed] [Google Scholar]
- 10.Zody MC, Jiang Z, Fung HC, et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet. 2008;40(9):1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pittman AM, Myers AJ, Abou-Sleiman P, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42(11):837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37(2):129–137. [DOI] [PubMed] [Google Scholar]
- 13.Strang KH, Golde TE, Giasson BI. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. 2019;99(7):912–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67(6):953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin DJ, Cohen TJ, Grossman M, et al. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain. 2012;135(Pt 3):807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irwin DJ, Cohen TJ, Grossman M, et al. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol. 2013;183(2):344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran G, Udgaonkar JB. Mechanistic studies unravel the complexity inherent in tau aggregation leading to Alzheimer’s disease and the tauopathies. Biochemistry. 2013;52(24):4107–4126. [DOI] [PubMed] [Google Scholar]
- 18.Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT. Are tangles as toxic as they look? J Mol Neurosci. 2011;45(3):438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, Bush AI. Tau protein: relevance to Parkinson’s disease. Int J Biochem Cell Biol. 2010;42(11):1775–1778. [DOI] [PubMed] [Google Scholar]
- 20.Guo JL, Covell DJ, Daniels JP, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154(1):103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olney NT, Spina S, Miller BL. Frontotemporal Dementia. Neurol Clin. 2017;35(2):339–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devenney EM, Ahmed RM, Hodges JR. Frontotemporal dementia. Handb Clin Neurol. 2019;167:279–299. [DOI] [PubMed] [Google Scholar]
- 25.Baizabal-Carvallo JF, Jankovic J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat Rev Neurol. 2016;12(3):175–185. [DOI] [PubMed] [Google Scholar]
- 26.Karageorgiou E, Miller BL. Frontotemporal lobar degeneration: a clinical approach. Semin Neurol. 2014;34(2):189–201. [DOI] [PubMed] [Google Scholar]
- 27.Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry. 2011;82(5):476–486. [DOI] [PubMed] [Google Scholar]
- 28.Mann DMA, Snowden JS. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017;27(6):723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain. 2014;137(Pt 4):1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spinelli EG, Mandelli ML, Miller ZA, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol. 2017;81(3):430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sieben A, Van Langenhove T, Engelborghs S, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124(3):353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci. 2011;45(3):384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs GG. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41(1):3–23. [DOI] [PubMed] [Google Scholar]
- 34.Rainero I, Rubino E, Michelerio A, D’Agata F, Gentile S, Pinessi L. Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct Neurol. 2017;32(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forrest SL, Kril JJ, Stevens CH, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2018;141(2):521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Zee J, Rademakers R, Engelborghs S, et al. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006;129(Pt 4):841–852. [DOI] [PubMed] [Google Scholar]
- 39.Spillantini MG, Yoshida H, Rizzini C, et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol. 2000;48(6):939–943. [DOI] [PubMed] [Google Scholar]
- 40.Grover A, England E, Baker M, et al. A novel tau mutation in exon 9 (1260V) causes a four-repeat tauopathy. Exp Neurol. 2003;184(1):131–140. [DOI] [PubMed] [Google Scholar]
- 41.Tacik P, DeTure M, Hinkle KM, et al. A Novel Tau Mutation in Exon 12, p.Q336H, Causes Hereditary Pick Disease. J Neuropathol Exp Neurol. 2015;74(11):1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogaki K, Li Y, Takanashi M, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord. 2013;19(1):15–20. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi K, Kidani T, Ujike H, et al. Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17) with a missense mutation of S305N closely resembling Pick’s disease. J Neurol. 2003;250(8):990–992. [DOI] [PubMed] [Google Scholar]
- 44.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. [DOI] [PubMed] [Google Scholar]
- 45.Baba Y, Baker MC, Le Ber I, et al. Clinical and genetic features of families with frontotemporal dementia and parkinsonism linked to chromosome 17 with a P301S tau mutation. J Neural Transm (Vienna). 2007;114(7):947–950. [DOI] [PubMed] [Google Scholar]
- 46.Deramecourt V, Lebert F, Maurage CA, et al. Clinical, neuropathological, and biochemical characterization of the novel tau mutation P332S. J Alzheimers Dis. 2012;31(4):741–749. [DOI] [PubMed] [Google Scholar]
- 47.McCarthy A, Lonergan R, Olszewska DA, et al. Closing the tau loop: the missing tau mutation. Brain. 2015;138(Pt 10):3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Momeni P, Wickremaratchi MM, Bell J, et al. Familial early onset frontotemporal dementia caused by a novel S356T MAPT mutation, initially diagnosed as schizophrenia. Clin Neurol Neurosurg. 2010;112(10):917–920. [DOI] [PubMed] [Google Scholar]
- 49.Iseki E, Matsumura T, Marui W, et al. Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 2001;102(3):285–292. [DOI] [PubMed] [Google Scholar]
- 50.Pickering-Brown SM, Baker M, Nonaka T, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127(Pt 6):1415–1426. [DOI] [PubMed] [Google Scholar]
- 51.Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology. 2017;88(8):758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erro ME, Zelaya MV, Mendioroz M, et al. Globular glial tauopathy caused by MAPT P301T mutation: clinical and neuropathological findings. J Neurol. 2019;266(10):2396–2405. [DOI] [PubMed] [Google Scholar]
- 53.Lin HC, Lin CH, Chen PL, Cheng SJ, Chen PH. Intrafamilial phenotypic heterogeneity in a Taiwanese family with a MAPT p.R5H mutation: a case report and literature review. BMC Neurol. 2017;17(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohn-Hokke PE, Wong TH, Rizzu P, et al. Mutation frequency of PRKAR1B and the major familial dementia genes in a Dutch early onset dementia cohort. J Neurol. 2014;261(11):2085–2092. [DOI] [PubMed] [Google Scholar]
- 55.Stanford PM, Shepherd CE, Halliday GM, et al. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain. 2003;126(Pt 4):814–826. [DOI] [PubMed] [Google Scholar]
- 56.Rossi G, Bastone A, Piccoli E, et al. New mutations in MAPT gene causing frontotemporal lobar degeneration: biochemical and structural characterization. Neurobiol Aging. 2012;33(4):834 e831–836. [DOI] [PubMed] [Google Scholar]
- 57.Neumann M, Diekmann S, Bertsch U, Vanmassenhove B, Bogerts B, Kretzschmar HA. Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics. 2005;6(2):91–95. [DOI] [PubMed] [Google Scholar]
- 58.Borrego-Ecija S, Antonell A, Puig-Butille JA, et al. Novel P397S MAPT variant associated with late onset and slow progressive frontotemporal dementia. Ann Clin Transl Neurol. 2019;6(8):1559–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–825. [DOI] [PubMed] [Google Scholar]
- 60.Spina S, Murrell JR, Yoshida H, et al. The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol. 2007;113(4):461–470. [DOI] [PubMed] [Google Scholar]
- 61.Llado A, Ezquerra M, Sanchez-Valle R, Rami L, Tolosa E, Molinuevo JL. A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. Eur J Neurol. 2007;14(8):e9–10. [DOI] [PubMed] [Google Scholar]
- 62.Yasuda M, Nakamura Y, Kawamata T, Kaneyuki H, Maeda K, Komure O. Phenotypic heterogeneity within a new family with the MAPT p301s mutation. Ann Neurol. 2005;58(6):920–928. [DOI] [PubMed] [Google Scholar]
- 63.Hayashi S, Toyoshima Y, Hasegawa M, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. 2002;51(4):525–530. [DOI] [PubMed] [Google Scholar]
- 64.Finger EC. Frontotemporal Dementias. Continuum (Minneap Minn). 2016;22(2 Dementia):464–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lashley T, Rohrer JD, Mead S, Revesz T. Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol. 2015;41(7):858–881. [DOI] [PubMed] [Google Scholar]
- 66.van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coppola G, Chinnathambi S, Lee JJ, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21(15):3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee SE, Tartaglia MC, Yener G, et al. Neurodegenerative disease phenotypes in carriers of MAPT p.A152T, a risk factor for frontotemporal dementia spectrum disorders and Alzheimer disease. Alzheimer Dis Assoc Disord. 2013;27(4):302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferrari R, Wang Y, Vandrovcova J, et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. 2017;88(2):152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verpillat P, Camuzat A, Hannequin D, et al. Association between the extended tau haplotype and frontotemporal dementia. Arch Neurol. 2002;59(6):935–939. [DOI] [PubMed] [Google Scholar]
- 71.Hughes A, Mann D, Pickering-Brown S. Tau haplotype frequency in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Exp Neurol. 2003;181(1):12–16. [DOI] [PubMed] [Google Scholar]
- 72.Ghidoni R, Signorini S, Barbiero L, et al. The H2 MAPT haplotype is associated with familial frontotemporal dementia. Neurobiol Dis. 2006;22(2):357–362. [DOI] [PubMed] [Google Scholar]
- 73.Kaivorinne AL, Kruger J, Kuivaniemi K, et al. Role of MAPT mutations and haplotype in frontotemporal lobar degeneration in Northern Finland. BMC Neurol. 2008;8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laws SM, Friedrich P, Diehl-Schmid J, et al. Genetic analysis of MAPT haplotype diversity in frontotemporal dementia. Neurobiol Aging. 2008;29(8):1276–1278. [DOI] [PubMed] [Google Scholar]
- 75.Zhang CC, Zhu JX, Wan Y, et al. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. 2017;8(27):44994–45007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dilliott AA, Evans EC, Farhan SMK, et al. Genetic Variation in the Ontario Neurodegenerative Disease Research Initiative. Can J Neurol Sci. 2019;46(5):491–498. [DOI] [PubMed] [Google Scholar]
- 77.Borroni B, Yancopoulou D, Tsutsui M, et al. Association between tau H2 haplotype and age at onset in frontotemporal dementia. Arch Neurol. 2005;62(9):1419–1422. [DOI] [PubMed] [Google Scholar]
- 78.Laws SM, Perneczky R, Drzezga A, et al. Association of the tau haplotype H2 with age at onset and functional alterations of glucose utilization in frontotemporal dementia. Am J Psychiatry. 2007;164(10):1577–1584. [DOI] [PubMed] [Google Scholar]
- 79.Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim HJ, Jeon BS, Yun JY, Seong MW, Park SS, Lee JY. Screening for MAPT and PGRN mutations in Korean patients with PSP/CBS/FTD. Parkinsonism Relat Disord. 2010;16(4):305–306. [DOI] [PubMed] [Google Scholar]
- 81.Donker Kaat L, Boon AJ, Azmani A, et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology. 2009;73(2):98–105. [DOI] [PubMed] [Google Scholar]
- 82.Im SY, Kim YE, Kim YJ. Genetics of Progressive Supranuclear Palsy. J Mov Disord. 2015;8(3):122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heckman MG, Brennan RR, Labbe C, et al. Association of MAPT Subhaplotypes With Risk of Progressive Supranuclear Palsy and Severity of Tau Pathology. JAMA Neurol. 2019;76(6):710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yokoyama JS, Karch CM, Fan CC, et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017;133(5):825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alonso Adel C, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279(33):34873–34881. [DOI] [PubMed] [Google Scholar]
- 87.Myers AJ, Pittman AM, Zhao AS, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25(3):561–570. [DOI] [PubMed] [Google Scholar]
- 88.Desikan RS, Schork AJ, Wang Y, et al. Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol Psychiatry. 2015;20(12):1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allen M, Kachadoorian M, Quicksall Z, et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. 2014;6(4):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Litvan I, Baker M, Hutton M. Tau genotype: no effect on onset, symptom severity, or survival in progressive supranuclear palsy. Neurology. 2001;57(1):138–140. [DOI] [PubMed] [Google Scholar]
- 91.Koga S, Parks A, Kasanuki K, et al. Cognitive impairment in progressive supranuclear palsy is associated with tau burden. Mov Disord. 2017;32(12):1772–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu WK, Le TV, Adamson J, et al. Relationship of the extended tau haplotype to tau biochemistry and neuropathology in progressive supranuclear palsy. Ann Neurol. 2001;50(4):494–502. [DOI] [PubMed] [Google Scholar]
- 93.Robinson JL, Yan N, Caswell C, et al. Primary Tau Pathology, Not Copathology, Correlates With Clinical Symptoms in PSP and CBD. J Neuropathol Exp Neurol. 2020;79(3):296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gerstenecker A, Roberson ED, Schellenberg GD, et al. Genetic influences on cognition in progressive supranuclear palsy. Mov Disord. 2017;32(12):1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Allen M, Burgess JD, Ballard T, et al. Gene expression, methylation and neuropathology correlations at progressive supranuclear palsy risk loci. Acta Neuropathol. 2016;132(2):197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Houlden H, Baker M, Morris HR, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–1706. [DOI] [PubMed] [Google Scholar]
- 98.Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47(3):374–377. [DOI] [PubMed] [Google Scholar]
- 99.Cruchaga C, Vidal-Taboada JM, Ezquerra M, et al. 5’-Upstream variants of CRHR1 and MAPT genes associated with age at onset in progressive supranuclear palsy and cortical basal degeneration. Neurobiol Dis. 2009;33(2):164–170. [DOI] [PubMed] [Google Scholar]
- 100.Kouri N, Ross OA, Dombroski B, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6:7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kertesz A Pick Complex: an integrative approach to frontotemporal dementia: primary progressive aphasia, corticobasal degeneration, and progressive supranuclear palsy. Neurologist. 2003;9(6):311–317. [DOI] [PubMed] [Google Scholar]
- 102.Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561(7721):137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goedert M Tau filaments in neurodegenerative diseases. FEBS Lett. 2018;592(14):2383–2391. [DOI] [PubMed] [Google Scholar]
- 104.Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130(6):877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abdolmohammadi B, Dupre A, Evers L, Mez J. Genetics of Chronic Traumatic Encephalopathy. Semin Neurol. 2020. [DOI] [PubMed] [Google Scholar]
- 106.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Janocko NJ, Brodersen KA, Soto-Ortolaza AI, et al. Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol. 2012;124(5):681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Santa-Maria I, Haggiagi A, Liu X, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol. 2012;124(5):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108(14):5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71(4):505–508. [DOI] [PubMed] [Google Scholar]
- 112.Bright J, Hussain S, Dang V, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging. 2015;36(2):693–709. [DOI] [PubMed] [Google Scholar]
- 113.Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Abraham R, Sims R, Carroll L, et al. An association study of common variation at the MAPT locus with late-onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(8):1152–1155. [DOI] [PubMed] [Google Scholar]
- 115.Russ C, Powell JF, Zhao J, et al. The microtubule associated protein Tau gene and Alzheimer’s disease--an association study and meta-analysis. Neurosci Lett. 2001;314(1–2):92–96. [DOI] [PubMed] [Google Scholar]
- 116.Mukherjee O, Kauwe JS, Mayo K, Morris JC, Goate AM. Haplotype-based association analysis of the MAPT locus in late onset Alzheimer’s disease. BMC Genet. 2007;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Myers AJ, Kaleem M, Marlowe L, et al. The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum Mol Genet. 2005;14(16):2399–2404. [DOI] [PubMed] [Google Scholar]
- 118.Laws SM, Friedrich P, Diehl-Schmid J, et al. Fine mapping of the MAPT locus using quantitative trait analysis identifies possible causal variants in Alzheimer’s disease. Mol Psychiatry. 2007;12(5):510–517. [DOI] [PubMed] [Google Scholar]
- 119.Cousin E, Mace S, Rocher C, et al. No replication of genetic association between candidate polymorphisms and Alzheimer’s disease. Neurobiol Aging. 2011;32(8):1443–1451. [DOI] [PubMed] [Google Scholar]
- 120.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23. [DOI] [PubMed] [Google Scholar]
- 121.Gerrish A, Russo G, Richards A, et al. The role of variation at AbetaPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer’s disease. J Alzheimers Dis. 2012;28(2):377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu QY, Yu JT, Miao D, et al. An exploratory study on STX6, MOBP, MAPT, and EIF2AK3 and late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34(5):1519 e1513–1517. [DOI] [PubMed] [Google Scholar]
- 123.Zhou F, Wang D. The associations between the MAPT polymorphisms and Alzheimer’s disease risk: a meta-analysis. Oncotarget. 2017;8(26):43506–43520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sassi C, Guerreiro R, Gibbs R, et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(12):2881 e2881–2881 e2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ygland E, van Westen D, Englund E, et al. Slowly progressive dementia caused by MAPT R406W mutations: longitudinal report on a new kindred and systematic review. Alzheimers Res Ther. 2018;10(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rademakers R, Dermaut B, Peeters K, et al. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum Mutat. 2003;22(5):409–411. [DOI] [PubMed] [Google Scholar]
- 127.Le Guennec K, Quenez O, Nicolas G, et al. 17q21.31 duplication causes prominent tau-related dementia with increased MAPT expression. Mol Psychiatry. 2017;22(8):1119–1125. [DOI] [PubMed] [Google Scholar]
- 128.Monsell SE, Mock C, Fardo DW, et al. Genetic Comparison of Symptomatic and Asymptomatic Persons With Alzheimer Disease Neuropathology. Alzheimer Dis Assoc Disord. 2017;31(3):232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wider C, Ross OA, Nishioka K, et al. An evaluation of the impact of MAPT, SNCA and APOE on the burden of Alzheimer’s and Lewy body pathology. J Neurol Neurosurg Psychiatry. 2012;83(4):424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peterson D, Munger C, Crowley J, et al. Variants in PPP3R1 and MAPT are associated with more rapid functional decline in Alzheimer’s disease: the Cache County Dementia Progression Study. Alzheimers Dement. 2014;10(3):366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kauwe JS, Cruchaga C, Mayo K, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci U S A. 2008;105(23):8050–8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Babic Leko M, Willumsen N, Nikolac Perkovic M, et al. Association of MAPT haplotype-tagging polymorphisms with cerebrospinal fluid biomarkers of Alzheimer’s disease: A preliminary study in a Croatian cohort. Brain Behav. 2018;8(11):e01128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cruchaga C, Kauwe JS, Harari O, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78(2):256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Elias-Sonnenschein LS, Helisalmi S, Natunen T, et al. Genetic loci associated with Alzheimer’s disease and cerebrospinal fluid biomarkers in a Finnish case-control cohort. PLoS One. 2013;8(4):e59676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 136.Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14(9):626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li J, Ruskey JA, Arnulf I, et al. Full sequencing and haplotype analysis of MAPT in Parkinson’s disease and rapid eye movement sleep behavior disorder. Mov Disord. 2018;33(6):1016–1020. [DOI] [PubMed] [Google Scholar]
- 138.Chang D, Nalls MA, Hallgrimsdottir IB, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wider C, Vilarino-Guell C, Jasinska-Myga B, et al. Association of the MAPT locus with Parkinson’s disease. Eur J Neurol. 2010;17(3):483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8(3):e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Healy DG, Abou-Sleiman PM, Lees AJ, et al. Tau gene and Parkinson’s disease: a case-control study and meta-analysis. J Neurol Neurosurg Psychiatry. 2004;75(7):962–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fidani L, Kalinderi K, Bostantjopoulou S, et al. Association of the Tau haplotype with Parkinson’s disease in the Greek population. Mov Disord. 2006;21(7):1036–1039. [DOI] [PubMed] [Google Scholar]
- 143.Yu L, Huang J, Zhai D, et al. MAPT rs242562 and GSK3B rs334558 are associated with Parkinson’s Disease in central China. BMC Neurosci. 2014;15:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Skipper L, Wilkes K, Toft M, et al. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am J Hum Genet. 2004;75(4):669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vandrovcova J, Pittman AM, Malzer E, et al. Association of MAPT haplotype-tagging SNPs with sporadic Parkinson’s disease. Neurobiol Aging. 2009;30(9):1477–1482. [DOI] [PubMed] [Google Scholar]
- 146.Winkler S, Konig IR, Lohmann-Hedrich K, Vieregge P, Kostic V, Klein C. Role of ethnicity on the association of MAPT H1 haplotypes and subhaplotypes in Parkinson’s disease. Eur J Hum Genet. 2007;15(11):1163–1168. [DOI] [PubMed] [Google Scholar]
- 147.Zabetian CP, Hutter CM, Factor SA, et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson’s disease. Ann Neurol. 2007;62(2):137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Winder-Rhodes SE, Hampshire A, Rowe JB, et al. Association between MAPT haplotype and memory function in patients with Parkinson’s disease and healthy aging individuals. Neurobiol Aging. 2015;36(3):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morley JF, Xie SX, Hurtig HI, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov Disord. 2012;27(4):512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Goris A, Williams-Gray CH, Clark GR, et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann Neurol. 2007;62(2):145–153. [DOI] [PubMed] [Google Scholar]
- 151.Seto-Salvia N, Clarimon J, Pagonabarraga J, et al. Dementia risk in Parkinson disease: disentangling the role of MAPT haplotypes. Arch Neurol. 2011;68(3):359–364. [DOI] [PubMed] [Google Scholar]
- 152.Williams-Gray CH, Mason SL, Evans JR, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry. 2013;84(11):1258–1264. [DOI] [PubMed] [Google Scholar]
- 153.Evans JR, Mason SL, Williams-Gray CH, et al. The natural history of treated Parkinson’s disease in an incident, community based cohort. J Neurol Neurosurg Psychiatry. 2011;82(10):1112–1118. [DOI] [PubMed] [Google Scholar]
- 154.Mata IF, Leverenz JB, Weintraub D, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014;71(11):1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Papapetropoulos S, Farrer MJ, Stone JT, et al. Phenotypic associations of tau and ApoE in Parkinson’s disease. Neurosci Lett. 2007;414(2):141–144. [DOI] [PubMed] [Google Scholar]
- 156.Ezquerra M, Campdelacreu J, Gaig C, et al. Lack of association of APOE and tau polymorphisms with dementia in Parkinson’s disease. Neurosci Lett. 2008;448(1):20–23. [DOI] [PubMed] [Google Scholar]
- 157.Factor SA, Steenland NK, Higgins DS, et al. Disease-related and genetic correlates of psychotic symptoms in Parkinson’s disease. Mov Disord. 2011;26(12):2190–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yarnall AJ, Breen DP, Duncan GW, et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology. 2014;82(4):308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Compta Y, Parkkinen L, O’Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain. 2011;134(Pt 5):1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Paul KC, Rausch R, Creek MM, et al. APOE, MAPT, and COMT and Parkinson’s Disease Susceptibility and Cognitive Symptom Progression. J Parkinsons Dis. 2016;6(2):349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Di Battista ME, Pascale E, Purcaro C, et al. Clinical subtypes in Parkinson’s disease: the impact of MAPT haplotypes. J Neural Transm (Vienna). 2014;121(4):353–356. [DOI] [PubMed] [Google Scholar]
- 162.Pascale E, Di Battista ME, Rubino A, et al. Genetic Architecture of MAPT Gene Region in Parkinson Disease Subtypes. Front Cell Neurosci. 2016;10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging. 2016;37:209 e201–209 e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Blauwendraat C, Heilbron K, Vallerga CL, et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord. 2019;34(6):866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Elbaz A, Ross OA, Ioannidis JP, et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann Neurol. 2011;69(5):778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang Y, Wang G, Rowe D, et al. SNCA Gene, but Not MAPT, Influences Onset Age of Parkinson’s Disease in Chinese and Australians. Biomed Res Int. 2015;2015:135674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bandres-Ciga S, Ahmed S, Sabir MS, et al. The Genetic Architecture of Parkinson Disease in Spain: Characterizing Population-Specific Risk, Differential Haplotype Structures, and Providing Etiologic Insight. Mov Disord. 2019;34(12):1851–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Robakis D, Cortes E, Clark LN, et al. The effect of MAPT haplotype on neocortical Lewy body pathology in Parkinson disease. J Neural Transm (Vienna). 2016;123(6):583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heckman MG, Kasanuki K, Diehl NN, et al. Parkinson’s disease susceptibility variants and severity of Lewy body pathology. Parkinsonism Relat Disord. 2017;44:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Labbe C, Ogaki K, Lorenzo-Betancor O, et al. Role for the microtubule-associated protein tau variant p.A152T in risk of alpha-synucleinopathies. Neurology. 2015;85(19):1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cervera-Carles L, Pagonabarraga J, Pascual-Sedano B, et al. Copy number variation analysis of the 17q21.31 region and its role in neurodegenerative diseases. Am J Med Genet B Neuropsychiatr Genet. 2016;171B(2):175–180. [DOI] [PubMed] [Google Scholar]
- 173.Labbe C, Heckman MG, Lorenzo-Betancor O, et al. MAPT haplotype H1G is associated with increased risk of dementia with Lewy bodies. Alzheimers Dement. 2016;12(12):1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Guerreiro R, Ross OA, Kun-Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 2018;17(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bras J, Guerreiro R, Darwent L, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet. 2014;23(23):6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chia R, Sabir MS, Bandres-Ciga S, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into the complex genetic architecture. bioRxiv. 2020:2020.2007.2006.185066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Colom-Cadena M, Gelpi E, Marti MJ, et al. MAPT H1 haplotype is associated with enhanced alpha-synuclein deposition in dementia with Lewy bodies. Neurobiol Aging. 2013;34(3):936–942. [DOI] [PubMed] [Google Scholar]
- 178.Colom-Cadena M, Gelpi E, Charif S, et al. Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2013;72(12):1203–1212. [DOI] [PubMed] [Google Scholar]
- 179.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372(3):249–263. [DOI] [PubMed] [Google Scholar]
- 180.Vilarino-Guell C, Soto-Ortolaza AI, Rajput A, et al. MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology. 2011;76(7):670–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Labbe C, Heckman MG, Lorenzo-Betancor O, et al. MAPT haplotype diversity in multiple system atrophy. Parkinsonism Relat Disord. 2016;30:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Morris HR, Vaughan JR, Datta SR, et al. Multiple system atrophy/progressive supranuclear palsy: alpha-Synuclein, synphilin, tau, and APOE. Neurology. 2000;55(12):1918–1920. [DOI] [PubMed] [Google Scholar]
- 183.Chen Y, Cao B, Ou R, et al. Association analysis of the GRN rs5848 and MAPT rs242557 polymorphisms in Parkinson’s disease and multiple system atrophy: a large-scale population-based study and meta-analysis. Int J Neurosci. 2016;126(10):947–954. [DOI] [PubMed] [Google Scholar]
- 184.Sailer A, Scholz SW, Nalls MA, et al. A genome-wide association study in multiple system atrophy. Neurology. 2016;87(15):1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gu X, Chen Y, Zhou Q, et al. Analysis of GWAS-linked variants in multiple system atrophy. Neurobiol Aging. 2018;67:201 e201–201 e204. [DOI] [PubMed] [Google Scholar]
- 186.Morris HR, Schrag A, Nath U, et al. Effect of ApoE and tau on age of onset of progressive supranuclear palsy and multiple system atrophy. Neurosci Lett. 2001;312(2):118–120. [DOI] [PubMed] [Google Scholar]
- 187.Sjostrom AC, Holmberg B, Strang P. Parkinson-plus patients--an unknown group with severe symptoms. J Neurosci Nurs. 2002;34(6):314–319. [DOI] [PubMed] [Google Scholar]
- 188.Ezquerra M, Pastor P, Gaig C, et al. Different MAPT haplotypes are associated with Parkinson’s disease and progressive supranuclear palsy. Neurobiol Aging. 2011;32(3):547 e511–546. [DOI] [PubMed] [Google Scholar]
- 189.Murley AG, Coyle-Gilchrist I, Rouse MA, et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain. 2020;143(5):1555–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Iwata A, Nagata K, Hatsuta H, et al. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum Mol Genet. 2014;23(3):648–656. [DOI] [PubMed] [Google Scholar]
- 191.Bakulski KM, Dolinoy DC, Sartor MA, et al. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29(3):571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. 2009;68(8):880–891. [DOI] [PubMed] [Google Scholar]
- 193.Huin V, Deramecourt V, Caparros-Lefebvre D, et al. The MAPT gene is differentially methylated in the progressive supranuclear palsy brain. Mov Disord. 2016;31(12):1883–1890. [DOI] [PubMed] [Google Scholar]
- 194.Li Y, Chen JA, Sears RL, et al. An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy. PLoS Genet. 2014;10(3):e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Coupland KG, Mellick GD, Silburn PA, et al. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro. Mov Disord. 2014;29(13):1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arakhamia T, Lee CE, Carlomagno Y, et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell. 2020;180(4):633–644 e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580(7802):283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Trabzuni D, Wray S, Vandrovcova J, et al. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21(18):4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6(1):e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tsai RM, Miller Z, Koestler M, et al. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients With Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Steinberg KM, Antonacci F, Sudmant PH, et al. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet. 2012;44(8):872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Soutar MPM, Melandri D, Annuario E, et al. Regulation of mitophagy by the NSL complex underlies genetic risk for Parkinson’s disease at Chr16q11.2 and on the MAPT H1 allele. bioRxiv. 2020:2020.2001.2006.896241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Soto-Beasley AI, Walton RL, Valentino RR, et al. Screening non-MAPT genes of the Chr17q21 H1 haplotype in Parkinson’s disease. Parkinsonism & Related Disorders. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Belluzzi E, Gonnelli A, Cirnaru MD, et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol Neurodegener. 2016;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Suzuki H, Yoshida T, Morisada N, et al. De novo NSF mutations cause early infantile epileptic encephalopathy. Ann Clin Transl Neurol. 2019;6(11):2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zollino M, Orteschi D, Murdolo M, et al. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44(6):636–638. [DOI] [PubMed] [Google Scholar]
- 207.Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9–21. [DOI] [PubMed] [Google Scholar]
- 208.Scarmeas N, Hadjigeorgiou GM, Papadimitriou A, et al. Motor signs during the course of Alzheimer disease. Neurology. 2004;63(6):975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Portet F, Scarmeas N, Cosentino S, Helzner EP, Stern Y. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Warren JD, Rohrer JD, Rossor MN. Clinical review. Frontotemporal dementia. BMJ. 2013;347:f4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Benussi A, Padovani A, Borroni B. Phenotypic Heterogeneity of Monogenic Frontotemporal Dementia. Front Aging Neurosci. 2015;7:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Snowden JS, Adams J, Harris J, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7–8):497–505. [DOI] [PubMed] [Google Scholar]
- 214.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011;24(6):542–549. [DOI] [PubMed] [Google Scholar]
- 215.Dickson DW. Neuropathology of Parkinson disease. Parkinsonism Relat Disord. 2018;46 Suppl 1:S30–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lowe J Neuropathology of dementia with Lewy bodies. Handb Clin Neurol. 2008;89:321–330. [DOI] [PubMed] [Google Scholar]
- 217.Nagaishi M, Yokoo H, Nakazato Y. Tau-positive glial cytoplasmic granules in multiple system atrophy. Neuropathology. 2011;31(3):299–305. [DOI] [PubMed] [Google Scholar]
- 218.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jefferson-George KS, Wolk DA, Lee EB, McMillan CT. Cognitive decline associated with pathological burden in primary age-related tauopathy. Alzheimers Dement. 2017;13(9):1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Teylan M, Mock C, Gauthreaux K, et al. Cognitive trajectory in mild cognitive impairment due to primary age-related tauopathy. Brain. 2020;143(2):611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gasca-Salas C, Masellis M, Khoo E, et al. Characterization of Movement Disorder Phenomenology in Genetically Proven, Familial Frontotemporal Lobar Degeneration: A Systematic Review and Meta-Analysis. PLoS One. 2016;11(4):e0153852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rademakers R, Melquist S, Cruts M, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet. 2005;14(21):3281–3292. [DOI] [PubMed] [Google Scholar]
- 223.Baba Y, Putzke JD, Tsuboi Y, et al. Effect of MAPT and APOE on prognosis of progressive supranuclear palsy. Neurosci Lett. 2006;405(1–2):116–119. [DOI] [PubMed] [Google Scholar]
- 224.Gan-Or Z, Bar-Shira A, Mirelman A, Gurevich T, Giladi N, Orr-Urtreger A. The age at motor symptoms onset in LRRK2-associated Parkinson’s disease is affected by a variation in the MAPT locus: a possible interaction. J Mol Neurosci. 2012;46(3):541–544. [DOI] [PubMed] [Google Scholar]
- 225.Golub Y, Berg D, Calne DB, et al. Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkinsonism Relat Disord. 2009;15(7):539–541. [DOI] [PubMed] [Google Scholar]
- 226.Labbe C, Lorenzo-Betancor O, Ross OA. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol. 2016;132(4):515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Caillet-Boudin ML, Buee L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]