

Abstract
Apoptosis is a common cell death program that is important in human health and disease. Signaling in apoptosis is largely driven through protein–protein interactions. The BCL-2 family proteins function in protein–protein interactions as key regulators of mitochondrial poration, the process that initiates apoptosis through the release of cytochrome c, which activates the apoptotic caspase cascade leading to cellular demolition. The BCL-2 pore-forming proteins BAK and BAX are the key executors of mitochondrial poration. We review the state of knowledge of protein–protein and protein–lipid interactions governing the apoptotic function of BAK and BAX, as determined through X-ray crystallography and NMR spectroscopy studies. BAK and BAX are dormant, globular α-helical proteins that participate in protein–protein interactions with other pro-death BCL-2 family proteins, transforming them into active, partially unfolded proteins that dimerize and associate with and permeabilize mitochondrial membranes. We compare the protein–protein interactions observed in high-resolution structures with those derived in silico by AlphaFold, making predictions based on combining experimental and in silico approaches to delineate the structural basis for novel protein–protein interaction complexes of BCL-2 family proteins.
Protein–protein interactions in apoptotic signaling
Protein–protein interactions are essential in biology. To name a few, they provide the rigid, yet dynamic, infrastructure of the cytoskeleton needed for shaping and strengthening cells, mediate transport across membranes, define molecular machines such as the ribosome, help package DNA into nucleosomes, organize biomolecular condensates, and govern cellular pathway signaling throughout the lifespan of an organism (1, 2). Protein–protein interactions define the apoptotic pathway, a form of programmed cell death implicated in human health and disease (3). In this review we briefly describe key signaling hubs in the extrinsic and intrinsic apoptotic pathways (Figure 1) (4) and delve into the details of apoptosis initiation by BCL-2 family proteins, emphasizing the intricate protein–protein interactions underlying mitochondrial poration from a structural biology perspective.
Figure 1. Apoptosis regulation through protein–protein interactions.

The apoptotic pathway is engaged extrinsically through death receptor signaling and intrinsically through mitochondrial poration by the effector BCL-2 family proteins BAK and BAX. Protein–protein interactions mediate many of the steps in apoptosis, culminating in the activation of initiator and executor caspases, the latter of which perform widespread cellular proteolysis, resulting in cell death. In this review we detail our latest understanding of the regulation of effector BCL-2 proteins by protein–protein interactions. tBID was recently shown to mediate apoptosis in cells lacking BAK, BAX, and BOK [42], and it is reclassified here as a pro-death initiator and effector (*).
The cumulative endpoint in apoptotic signaling is the activation of apoptotic caspases, the cysteine–aspartic proteases that execute widespread intracellular proteolysis and cellular destruction (Figure 1) (5). Extrinsic activation of apoptosis involves the activation of death receptors by extracellular transmembrane receptor–ligand interactions (e.g., Fas ligand [FasL] binding to the Fas receptor). Death receptor clustering promotes the assembly of the receptor-associated intracellular scaffold FADD (FAS-associated death domain protein), which in turn recruits initiator caspases 8 and 10 and activates them via proximity-induced oligomerization (Figure 1) (6–10). Active initiator caspases cleave and activate executioner caspases 3 and 7 in mitochondrial-independent type I cells and the BCL-2 protein BID in mitochondrial-dependent type II cells (Figure 1) (11, 12). The main protein–protein interactions governing extrinsic apoptosis signaling are mediated by the death domains present in death receptors, FADD, and initiator caspases (13, 14).
Apoptosis is also activated intrinsically through the engagement of BCL-2 family proteins in response to pathophysiological stimuli such as DNA damage induced by radiation or chemotherapy. The BCL-2 family proteins regulate the integrity of the outer mitochondrial membrane by defining a dynamic protein–protein interaction network whose balance or imbalance ultimately dictates whether a cell survives or dies by apoptosis (Figure 1) (3, 4, 15). During apoptosis, pro-death BCL-2 family proteins are activated to induce mitochondrial outer membrane permeabilization (MOMP) or mitochondrial poration, releasing cytochrome c and other proapoptotic factors that activate the caspase cascade downstream. Cytochrome c interacts with the β-propeller domains (containing WD40 repeats) of apoptotic protease-activating factor 1 (APAF-1), inducing conformational changes in and oligomerization of APAF-1 to form a seven-spoked apoptosome platform for activating initiator caspase 9 (16). Recruitment of caspase 9 to the apoptosome is mediated by protein–protein interactions between the CARD domain of APAF-1 (another death domain) and that of caspase 9, which oligomerizes the latter, promoting its proximity-induced activation (17, 18). Active caspase 9 then cleaves and activates executioner caspases 3 and 7 (Figure 1).
BCL-2 family proteins function through protein–protein interactions
The BCL-2 family proteins include pro-death BCL-2 homology 3 (BH3)-only initiators and effectors, as well as pro-survival guardians (Figure 1) (15). The BCL-2 fold, shared by effectors and guardians, forms an α-helical globular domain with the central hydrophobic helix ⍺5 surrounded by seven amphipathic helices (Figure 2) (19–21). Helices α2–α5 define the canonical hydrophobic groove, which binds BH3 ligands (22) and the C-terminal transmembrane (TM) targeting domain (helix ⍺9) in BAX (21). Because of this configuration, BAX is cytosolic, whereas BAK and the guardians are mainly targeted to the outer mitochondrial membrane (OMM). Localization is dynamically established by the shuttling of effectors (and guardians) between the cytosol and the OMM at different rates in a process known as retrotranslocation (23, 24).
Figure 2. Asymmetric BH3-in-groove protein–protein interactions activate BAK and BAX.

The transient hit-and-run activation mechanism of BAK and BAX involves their binding to BH3-only activators followed by autoactivation in trans, both of which processes share a BH3-in-groove structural basis while cooperating in activation signal amplification. BAX is cytosolic, undergoing additional triggering by BH3-only activating ligands to displace its transmembrane-targeting C-terminal helix from the activation groove and facilitate its targeting to the outer mitochondrial membrane (OMM). Representative structures of BAK and BAX in the apo and BH3 ligand–bound states are shown. Activation destabilizes effectors, promoting their unfolding and association with membranes. Hydrophobic residues in the BH3 ligand engage hydrophobic pockets P(0)–P(5), and they have been identified above the sequence alignment and in the cartoon-surface representation of the BAK BH3–BAK complex. The conserved Asp in the BH3, D(s), forms a salt bridge with the conserved Arg in the groove, R(s).
The BCL-2 family protein–protein interactions are categorized into those mediated by the pore-forming effectors and those mediated by the pro-survival guardians, both of which protein types act as receptors for BH3-only initiators (15). The guardians are also the receptors of active effectors (25). The effectors self-associate and oligomerize into apoptotic pores that are thought to be proteolipidic in nature (reviewed in (26, 27)), although the basis of poration is incompletely defined structurally. Initiation of the mitochondrial apoptotic pathway upregulates or activates BH3-only initiators, including BAD, BID, BIK, BIM, BMF, HRK, NOXA, and PUMA, which have low affinities for the effectors and high affinities for the guardians, although some initiators, such as BAD, are inert towards the effectors (28, 29). Based on their ability to activate the effectors, some BH3-only initiators have been further categorized as direct effector activators; these include BID, BIM, PUMA, NOXA and potentially others (25, 30–40). The BH3-only initiators are intrinsically disordered except for BID, which has a globular architecture resembling that of guardians and effectors (41). We note that tBID, the active form of BID produced through proteolysis, was recently shown to act as a pore-forming effector in cells that are genetically devoid of the entire BCL-2 family repertoire including BAK, BAX, and BOK (42) (Figure 1). The role of tBID as effector of mitochondrial poration is intriguing and merits additional investigations to identify patho-physiological relevance beyond those governing the immune response against Shigella infection which appear to be tBID-dependent and BAK- and BAX-independent (42).
The BH3 domains of BH3-only initiators adopt amphipathic helical conformations when bound to the hydrophobic grooves of effectors and guardians (15, 43). Although the various BH3 domains of BCL-2 family proteins share sequence similarities, the mechanisms and specificities of the various family members are driven by subtleties in their protein–protein interactions. Focusing on the effectors, we will highlight our latest structural understanding of these interactions, which have been resolved experimentally by X-ray crystallography and NMR spectroscopy. We will focus on BH3 domain ligand–induced conformational changes in effectors, which we compare with models derived in silico by using AlphaFold, and on effector dimerization and interactions with membrane lipids and bilayers.
Asymmetric BH3-in-groove interactions trigger effectors to initiate apoptosis
To initiate apoptosis, the BCL-2 effectors BAK and BAX undergo transitions from soluble dormant monomeric proteins to membrane-embedded proteins exhibiting heterogeneous oligomerization to form apoptotic pores. We define dormant BAK as being folded into the BCL-2 globular domain as observed in the structure of apo BAK (PDB 2imt) (44). Dormant BAK is not associate with pro-survival guardians but is known to associate with VDAC2 as recently reviewed (45). For brevity we exclude BAK interactions with VDAC2 from our discussion and our figures. Effector conformational transitions are enabled through direct activation by BH3-only initiators or through autoactivation in trans, wherein the BH3 domain of an active effector engages a dormant effector (46). The BH3 domains of BH3-only initiators and effectors, but not those of guardians, can activate BAK or BAX to various extents in liposomal or mitochondrial permeabilization assays as peptides or as chimeras in which they replace the BH3 domain of BID (25, 47, 48). The BH3 domains most potent in activating effectors are from BID, BIM, BAK, and BAX (15). Activator BH3 ligands have been shown to bind the noncanonical trigger site in BAX (helices α1 and α6 and the α1–α2 loop), promoting its allosteric transformation, dissociation of its α9 helix from the canonical groove, and its targeting to the OMM (Figure 2) (31, 49, 50). OMM-targeted BAK and BAX engage activating BH3 ligands at the canonical groove (helices α2–α5) to activate and further change their conformations to dynamic open states exhibiting partially unfolded N-termini (approximately 60–65 residues containing the BH4) and transiently exposed BH3 domains (26, 51–53).
Chemically stapled BIM BH3, termed stabilized ⍺-helix of BCL-2 domain (SAHB), has been shown by paramagnetic resonance enhancement NMR to bind as a helix at the noncanonical trigger site in BAX (31). Binding at helices α1 and α6 displaces the α1–α2 loop to induce allosteric ripples through the protein core that are felt in the canonical groove, destabilizing the binding of TM (α9) (Figure 2) (49). BAX BH3 SAHB and PUMA BH3 SAHB have also been shown to bind the noncanonical trigger site (50, 54). Binding of tBID at a similar site in BAK is supported biochemically (55) and, more recently, binding of BMF BH3 and HRK BH3 to BAK was mapped to a site resembling the noncanonical trigger site in BAX (40), although how this binding induces BAK opening is unclear in the absence of high-resolution structural investigations.
The activation of effectors by BH3 ligand engagement at the canonical groove is understood as a result of high-resolution structural analyses of BAK and BAX bound to the BH3 domain of BID, BIM, BAK, or BAX (Table S1, Figures 2, 3) (28, 29, 46, 56, 57). These structures have revealed a recognition mechanism that is shared by different BH3 peptides at the canonical activation groove. Hydrophobic pockets P(0)–P(5), which are occluded in the apo form, open to accommodate hydrophobic residues of the BH3 ligand. A salt bridge between an Arg residue (R127 in BAK, R109 in BAX) in the groove helix α5 and a conserved Asp residue in the BH3 domain locks the ligand in the P(0)–P(5) configuration [salt bridge R(s)–D(s) in Figure 2]. The structures of BAK bound to BID BH3, BIM BH3, and BAK BH3 have revealed how these BH3 ligands destabilize the fold of BAK by altering the electrostatic network that stabilizes helix α1 at the bottom of the activation groove (29, 46, 56). Specifically in BAK, four electrostatic contacts between helices α1 (R42, E46), α3 (D90), and α5 (R137) that are observed in the apo form are disrupted and replaced by one or two electrostatic contacts between helices α1 (R42, E46) and α2 (N86) in the BH3-bound form, promoting the unfolding of helices α1 and α2 at the membranes. Missense mutagenesis in the electrostatic network that stabilizes helix α1, including E46A, R42A, and R137A, promoted spontaneous (auto)activation of BAK at lower protein levels compared to WT BAK in liposome permeabilization assays, providing biochemically support for the role of this network in BAK stabilization and activation. Together, these structures have affirmed the longstanding hit-and-run model which hypothesizes that transient binding of activators induces conformational changes to activate BAK (58). Direct activation and autoactivation share a similar mechanism and cooperate in the amplification of BAK activation signaling (Figures 2 and 3).
Figure 3. Asymmetric BH3-in-groove effector activation destabilizes contacts with inhibitory helix ⍺1.
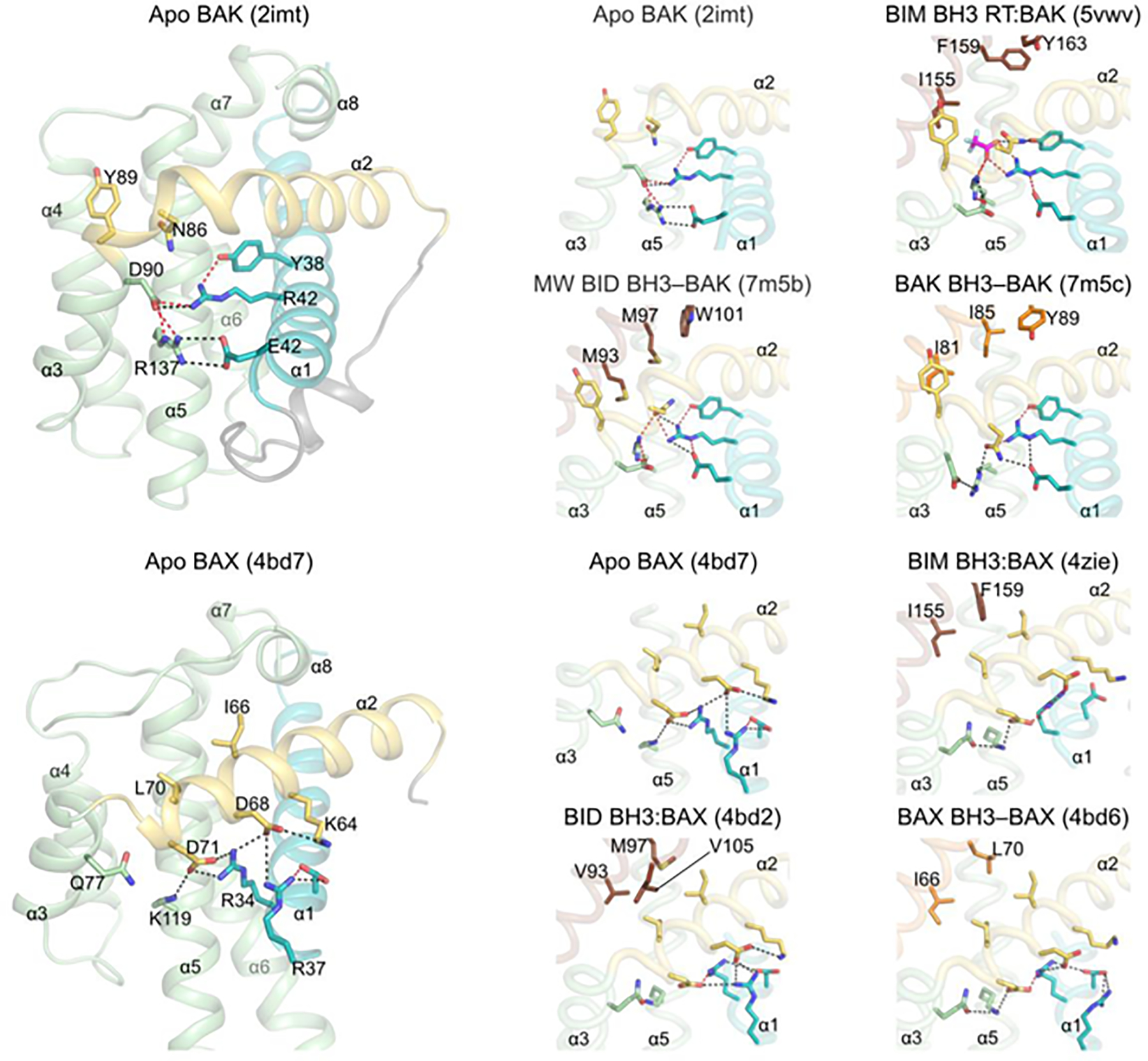
Apo effectors exhibit an electrostatic network at the bottom of the activation groove that is destabilized in directly activated and autoactivated complexes. This was recently elucidated for structures of activator BH3 ligands bound to BAK in which electrostatic contacts with helix α1 are shifted from helices α3 and α5 (apo BAK) to helix α2 (reference 44). Helices α1 and α2 become dynamic and are exposed after activation. We present a retrospective analysis of BH3-activated BAX complexes, which suggests that electrostatic contacts divergent from those seen in BAK rearrange upon activation. Electrostatic contacts between helices α2 and α5 in apo BAX are destabilized in favor of those between helices α1 and α2. We note that BH3 ligand induced a conformational change involving a major shift in helix α3, which may also contribute to BAX destabilization.
It is currently unclear how the binding of BH3 ligand to BAX-ΔTM (mimicking OMM-targeted BAX) destabilizes its fold, although the formation of ligand-induced cavities has been proposed. Here we have examined the electrostatic network at the bottom of the activation groove, which in apo BAX stabilizes helices α1 and α2 through two Arg residues (R34 and R37) in helix α1 forming hydrogen bonds to two Asp residues (D68 and D71) in helix α2. D71 also forms hydrogen bonds to K119 in helix α5. This network is rearranged in the BH3-bound complexes, with helix α3 moving out of the groove, which brings Q77 into proximity to K119 for hydrogen bonding (Figure 3). These changes possibly weaken the K119–D71 hydrogen bond, which in the BID BH3:BAX complex is lost altogether. Remarkably, early mutagenesis studies of this electrostatic network have suggested that D71A mutagenesis promotes spontaneous BAX activation, causing full targeting of the mutant to mitochondria and robust apoptosis initiation. However, R34A, R34E, K119A, and R34A/K119A mutants behaved similarly to WT BAX, exhibiting cytosolic localization and no spontaneous activation suggesting that they may adopt a different ground (dormant) state than D71A BAX (59). Our hypothesis that the BH3 ligand-induced rearrangement of the electrostatic network stabilizing helix α1 in BAX is important in BAX activation and helix α1 release, merits additional investigation based on contemporary knowledge of this system. Additional support for the rearrangement of this network in regulating BAX activation comes from studies using antibodies that bind to epitopes located in the α1–α2 loop immediately after the helix α1 C-terminus; one of the antibodies activated BAX by binding the epitope and destabilizing the C-terminus of helix α1 (60). The structural and mutagenesis data support the electrostatic network playing a critical role in stabilizing helix α1 in BAX activation by BH3 ligands.
Comparison of protein–protein interactions of BH3-bound effectors derived in silico and by high-resolution structural analysis
The landscape of high-resolution structures of BH3-bound effectors is incomplete because of the technical limitations of structural elucidation by X-ray crystallography and NMR spectroscopy in resolving the remaining complexes (Table S1). We tested the performance of AlphaFold (DeepMind), a revolutionary artificial intelligence–based structure elucidation tool that predicts protein structures with remarkable accuracy based purely on the protein sequence (61–63). Using AlphaFold-multimer (63) running on the St. Jude Children’s Research Hospital cluster, we modeled the WT BID BH3–activated, the mutant peptide MW [with substitutions at the hydrophobic residue positions M(3) and W(5)] BID BH3–activated and BAK BH3–activated complexes of BAK and BIM BH3–activated and BAX BH3–activated complexes of BAX and compared the models with the corresponding crystal structures (Figure 4). We used the full-length BAK and BAX sequences, whose structures are modeled in AlphaFold with helix α9 bound to the canonical activation groove (61, 62). We note that the confidence level for the helix α9 modeling in the groove is high for BAX and low for BAK.
Figure 4. Crystal and alphafold2 structures of BH3 ligand–activated BAK and BAX.

A. Overlays of structures of BAK and BAX bound to BH3 activating ligands as determined by crystallography (gray) or AlphaFold-multimer (rainbow colors) reveal excellent overall similarity as judged by the low root mean square deviation (using the align function in PyMOL). Although many of the side chains exhibit similar rotamers throughout the complex, side-chain rotamers within the helix α1 electrostatic network differ between the crystal and AlphaFold models. We note that the AlphaFold model of full-length BAK places the C-terminal TM helix α9 in the activation groove (not shown). However, unlike apo BAX, which exhibits high confidence values in α9, the confidence values of ⍺9 are low in apo BAK. The BH3 ligands displace the TM region in BAK and BAX in AlphaFold-multimer models.
B. In silico mutagenesis of the TM helix α9 in the BAK BH3:BAK AlphaFold complexes showing the confidence level colored on the structure.
C. Overlay of AlphaFold structures for WT and MW BID BH3:BAK complexes.
With the BH3 domain sequences of WT or MW BID, WT BIM, WT BAK, and WT BAX included (Table S2), AlphaFold-multimer generated BAK and BAX complexes with the BH3 ligand bound to the activation groove, and interestingly with the TM α9 displaced yet maintaining helicity (Figure 4, Table S2). Displacement of the TM helix α9 by the BH3 activators of BAK and BAX is subject to ongoing debate with two opposing viewpoints: 1) the TM is allosterically displaced by the BH3 ligands binding at the non-canonical trigger site of BAX (described above, Figure 1) or 2) the BH3 ligands directly compete with the TM at the canonical activation groove; our AlphaFold results support the latter model (Figure 4A). Intriguingly, the TM region is still predicted as an α-helix after being displaced from the binding groove, albeit with lower confidence values compared to the rest of the protein (Figure 4B). While on one hand recent work has validated the ability of AlphaFold to accurately predict the fold of transmembrane proteins, on the other hand a significant correlation has been reported between low confidence AlphaFold predictions and intrinsic disorder (64, 65). To address the question whether this helical prediction is a result of overfitting by the program, we introduced helix breaking mutations in silico. A gradual diminution of the AlphaFold confidence level was observed within the TM helix upon L199P and V197G/V198G substitutions and only a severe in the BAK BH3–BAK complexes and observed within the displaced TM helix upon the substitutions, and only a severe V197G/V198G/L199G/L200G substitution results in a nearly complete unfolding of the helix (Figure 4B). Thus, the prediction result appears to reflect the intrinsic helicity in of the transmembrane domain of BAK. Pertinently, AlphaFold predicts helicity in the BH3 region of the BH3-only proteins BAD, BIM, BIK, BMF, HRK, NOXA, but not that of PUMA (https://alphafold.ebi.ac.uk/). We previously showed that these BH3 peptides adopt random coil conformations in solution by circular dichroism (47), and others have concluded based on Φ value analysis (a method based on mutagenesis for probing folding pathways) that the BH3-only proteins encode folding pathways within their intrinsically disordered sequence (66). We note the difficulty of studying isolated helices in solution including the amphipathic BH3 regions and the highly hydrophobic TM regions which easily aggregate in biological buffers. AlphaFold provides a complementary platform to investigate to make educated hypotheses regarding these structural motifs including the effect of mutations.
Examination of side-chain rotamers for the top five ranked AlphaFold models of BH3-bound effectors overlaid on the corresponding crystal structures indicates excellent similarity throughout the structure, including most of the side chains (rmsd < 1.2Å, Figure 4A). We note an exception at the bottom of the activation groove within the helix α1 electrostatic network, which is divergent (Figure 4A). The NMR structure of BAK bound to BID BH3 SAHB (PDB ID 2m5b) also fails to define accurately the helix α1 electrostatic network as observed in the crystal structures (29). While the ability of AlphaFold to accurately represent the outcome of point mutations is under question, AlphaFold-multimer model of the WT BID BH3–BAK complex is highly similar to the MW BID BH3–BAK complex, underscoring the utility and incredible robustness of AlphaFold-multimer for mutagenesis studies of BCL-2 family protein complexes (Figure 4C). Based on this analysis, we conclude that AlphaFold-multimer can be used reliably to predict the overall structure of complexes of BH3 ligand–bound effectors. A caveat to this approach is that important mechanistic details may not be captured accurately with AlphaFold-multimer (e.g., side-chain rotamers and hydrogen bonds in critical regulatory regions such as the helix α1 electrostatic network). Therefore, high-resolution structure determination, preferably by X-ray crystallography, remains essential for elucidating mechanistic details of BH3 ligand–bound effectors.
Symmetric BH3-in-groove effector dimers as building blocks of apoptotic pores
Effector activation promotes conformational changes that transform dormant globular monomers to poorly understood open-state molecules that oligomerize [(15)]. These conformational changes were deduced initially from crystal structures of domain-swapped dimers induced by activation using octylmaltoside detergent or BH3 activators + CHAPS detergent (28, 67). Those structures have revealed that helices α2–α5 (the core) dissociate from helices α6–α8 (the latch). The fold of swapped dimers is identical to that of monomers apart from the swap point at the α5–α6 junction where the α-helix is extended. The swapped dimers are off pathway but have been used extensively as surrogates for structural investigations of effector complexes with BH3 ligands (reviewed in (15)).
More recently, the conformational changes have been characterized for truncated α2–α8 BAK activated with C12E8 detergent (68). This construct exhibits symmetric BH3-in-groove dimerization of the α2–α5 core and an extended conformation of the α6–α8 latch (Figure 5A, left). The α2–α8 dimer has a hydrophobic face that presumably interacts with acyl chains of membrane phospholipids, possibly enabling its intercalation into the outer leaflet of the OMM (Figure 5B–5D). Importantly, the α2–α8 dimer in micellar C12E8 detergent is active in mitochondrial and liposomal permeabilization assays, in which the detergent is diluted to submicellar concentrations, suggesting that it is the building block of apoptotic pores (68). Compared to monomeric BAK, the α2–α8 dimer has similar overall geometry for α3–α5, resembling that in the closed state, but differs in the orientation of α2 and α6–α8, which extend away from the core (Figure 5A, right). Activation of BAK by binding of BH3 ligands to the activation groove uses a BH3-in-groove geometry that is similar, albeit asymmetric, to that of symmetric α2–α5 core dimers (Figure 5A, right). Biochemical probing of the BH3-in-groove interactions showed that mutations of the activation groove of the BH3 domain blocked BAK autoactivation in trans but not direct BAK activation, as BAK-mediated poration still ensued, although it was diminished in extent with the mutants as compared to WT BH3 (46). This suggests there is heterogeneity in the mechanism of membrane poration by activated, opened BAK, because these mutations are predicted to block symmetric α2–α5 core dimerization.
Figure 5. Symmetric BH3-in-groove effector dimers as building blocks of apoptotic pores.

Upon activation, effectors undergo major changes in conformation involving the separation of helix α1, the core (helices α2–α5), and the latch (helices α6–α8).
A. A truncated BAK construct (helices α2–α8) forms symmetric BH3-in-groove dimers in activating detergent, capturing the latch in an extended helix conformation relative to that in monomeric BAK (left). Asymmetric and symmetric BH3-in-groove dimers exhibit similarity in the BH3-binding groove, although they differ considerably in the orientation of the BH3 and latch (right).
B, C. Structures of symmetric BH3-in-groove BAK dimers (B) and BAX dimers (C) have been determined for the truncated α2–α5 apo core, for the core bound to phospholipids or fatty acids, and for bicelle-bound conformations, all of which are similar overall. The orientation of structural representations has been achieved by aligning helix α5 of the gray monomers.
D. Symmetric BH3-in-groove effector dimers interact with the lipid bilayer in multiple ways to destabilize and permeabilize the OMM through heterogeneous proteolipidic or toroidal apoptotic pores. The TM regions target the effectors to the OMM and have been shown to mediate oligomerization possible through parallel or antiparallel interactions.
Mechanistic support for the association of α2–α5 core dimers with membranes derives from structural studies of BAK core dimers bound to phospholipids and fatty acids, including PE, PC, PS, PG, and lysoPC (Figure 5B, Table S3) (69). The phosphate in the polar head group of phospholipids is coordinated by the W125 backbone amide in helix α5 of one protomer and the side chains of Y136 and R137 of the second protomer, although the latter is within hydrogen bonding range in only a few of the phospholipid-bound structures (Figures 5A, 5B). The phospholipid sn2 acyl chains are not well resolved but localize within the vast hydrophobic surface generated by helices α4 and α5 of the α2–α5 core dimers. The sn1 acyl chains are also poorly defined, protruding towards other dimers in the crystal structures that captured trimeric and dimeric assemblies of the α2–α5 dimers (Table S3). Biochemical probing through mutagenesis and lipid–protein crosslinking supports a mechanism whereby lipids bridge adjacent dimers (Figure 5D) (69). Bridging of active BAK and BAX through protein–protein crosslinking has also been shown at helix α6 within the latch, suggesting there is heterogeneous proximity of the latch within effector oligomers (70, 71). Support for the role of α2–α5 core dimers in the pore lumen lining stems from structural studies of BAX α2–α5 core dimer in bicelles (72) as proposed originally based on investigations with double electron–electron resonance electron paramagnetic resonance (73). Rather than intercalating within the leaflet of the bilayer, the BAX α2–α5 core dimer binds to and caps the DMPC bilayer replacing DHPC at the rim of bicelles (Figure 5C, 5D) (72). Of note, the α2–α5 core dimers of BAK and BAX are quite dynamic, adopting concave or flat configurations relative to the hydrophobic core face, and they may regulate membrane curvature and bilayer capping, respectively, at different stages of the poration process (Figures 5B–5D). Effector dimers are predicted to form arcs and rings on the OMM that initially release cytochrome c followed by mitochondrial DNA as the pores enlarge (74–77).
Effector oligomerization is enabled also by the TM regions, which function not only in membrane targeting, but in bridging adjacent dimers (Figure 5D). Although structures of intact effectors in lipid bilayers are lacking, biochemical evidence supports the role of TM in BAK and BAX oligomerization (78–80). Additionally, DEER EPR analysis has suggested that parallel TM regions may enable the lining of the pore lumen by the core dimers of BAK (81). On the other hand, the isolated TM of BAK does not oligomerize in nanodiscs suggesting the possible involvement of other factors in native membranes enabling TM-mediated oligomerization (52). Presumably, once the lipid bilayer is permeabilized the TM could flip from the outer leaflet to the inner leaflet of the OMM bilayer at the pore lumen as proposed in the clamp model (73) (Figure 5D). Because of their heterogeneity brought about by disorder of dimeric building blocks (82), the structure of the apoptotic pores has not yet been elucidated at high resolution. Overall, however, effector dimerization is a key intermediate stage promoting efficient mitochondrial poration and apoptotic response.
Outlook for effector BCL-2 family protein–protein and protein–lipid interactions
High-resolution structural investigations have resulted in remarkable recent advances in our understanding of effector-mediated protein–protein interactions and their interactions with membranes. The development of AlphaFold and AlphaFold-multimer as powerful in silico algorithms for structural elucidation provides a great opportunity to model the effector protein–protein interactome. Elucidating the structures of the apoptotic pores formed by full-length BAK, BAX, BOK, and tBID represents a challenging new frontier in the field of effector structural biology. Structural characterization of apoptotic pores produced by BID and BOK is not yet available. tBID does not form arcs or rings in super-resolution microscopy supporting the possibility of monomer-mediated membrane rupture (42). The opened conformation of tBID in detergent micelles was investigated by NMR indicating the absence of a folded hydrophobic core, yet it exhibits extensive interactions via its helices with the micelle as a monomer (83). A similar interaction of tBID with the OMM may mediate its rupture. Mutagenesis has suggested that helix 6 but not the BH3 is critical in tBID-mediated poration. In contrast, BOK is predicted to spontaneously oligomerize in the presence of membranes, although it is unclear if BOK forms the core dimers similar to BAK and BAX. Hybrid approaches combining high-resolution and medium-resolution techniques, in silico modeling, and molecular dynamics will be essential in deciphering the dynamic conformations of apoptotic pores, which may differ mechanistically among the growing list of effectors.
Supplementary Material
Perspectives.
Mitochondrial poration by the pore-forming, effector BCL-2 family proteins is the most critical regulatory step in apoptosis initiation upon which a cell commits to dying.
For more than two decades the activation of effector BCL-2 family proteins in apoptosis has been highly controversial. Structures of activated effectors triggered by BH3 domain ligands have clarified the importance of cooperativity of effector direct activation and autoactivation. Effector dimerization is essential for efficient mitochondrial poration. Effector dimers are the building blocks of apoptotic pores.
Elucidating the structures of full-length apoptotic pores in the presence of membranes remains a major goal that will reveal exactly how the effectors oligomerize within proteolipidic pores into higher order structures that have been visualized as arcs and rings in situ by super-resolution microscopy.
Acknowledgements
The authors thank Keith A. Laycock for scientific editing of the manuscript. Funding for this study was supported by ALSAC and NIGMS (R01GM129470).
Footnotes
Conflict of Interest
The authors declare no conflict of interests.
References
- 1.Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019;176(3):419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alberts BJ A; Lewis J; Raff M; Roberst K; Walter P Molecular Biology of the Cell. sixth edition ed. Alberts BJ,A; Lewis J; Raff M; Roberst K; Walter P, editor. New York: Garland Science; 2002 November 19, 2014. [Google Scholar]
- 3.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20(3):175–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2019. [DOI] [PubMed] [Google Scholar]
- 5.Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol. 2020;38:567–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Driscoll PC. Structural studies of death receptors. Methods Enzymol. 2014;545:201–42. [DOI] [PubMed] [Google Scholar]
- 7.Fox JL, Hughes MA, Meng X, Sarnowska NA, Powley IR, Jukes-Jones R, et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate. Nat Commun. 2021;12(1):819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Q, Fu TM, Cruz AC, Sengupta P, Thomas SK, Wang S, et al. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol Cell. 2016;61(4):602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell. 2016;64(2):236–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan L, Fu TM, Zhao W, Zhao L, Chen W, Qiu C, et al. Higher-Order Clustering of the Transmembrane Anchor of DR5 Drives Signaling. Cell. 2019;176(6):1477–89 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao Z, Mak TW. Type I and type II pathways of Fas-mediated apoptosis are differentially controlled by XIAP. J Mol Cell Biol. 2010;2(2):63–4. [DOI] [PubMed] [Google Scholar]
- 12.Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813(4):558–63. [DOI] [PubMed] [Google Scholar]
- 13.Ferrao R, Wu H. Helical assembly in the death domain (DD) superfamily. Curr Opin Struct Biol. 2012;22(2):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moldoveanu T, Czabotar PE. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb Perspect Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou M, Li Y, Hu Q, Bai XC, Huang W, Yan C, et al. Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015;29(22):2349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su TW, Yang CY, Kao WP, Kuo BJ, Lin SM, Lin JY, et al. Structural Insights into DD-Fold Assembly and Caspase-9 Activation by the Apaf-1 Apoptosome. Structure. 2017;25(3):407–20. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Zhou M, Hu Q, Bai XC, Huang W, Scheres SH, et al. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A. 2017;114(7):1542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24(5):677–88. [DOI] [PubMed] [Google Scholar]
- 20.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381(6580):335–41. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103(4):645–54. [DOI] [PubMed] [Google Scholar]
- 22.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275(5302):983–6. [DOI] [PubMed] [Google Scholar]
- 23.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145(1):104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015;34(1):67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44(4):517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bleicken S, Assafa TE, Stegmueller C, Wittig A, Garcia-Saez AJ, Bordignon E. Topology of active, membrane-embedded Bax in the context of a toroidal pore. Cell Death Differ. 2018;25(10):1717–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flores-Romero H, Ros U, Garcia-Saez AJ. Pore formation in regulated cell death. EMBO J. 2020;39(23):e105753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152(3):519–31. [DOI] [PubMed] [Google Scholar]
- 29.Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20(5):589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17(10):1270–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455(7216):1076–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–58. [DOI] [PubMed] [Google Scholar]
- 33.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36(3):487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525–35. [DOI] [PubMed] [Google Scholar]
- 35.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–42. [DOI] [PubMed] [Google Scholar]
- 36.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110(11):E986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–92. [DOI] [PubMed] [Google Scholar]
- 38.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330(6009):1390–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, et al. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24(2):199–210. [DOI] [PubMed] [Google Scholar]
- 40.Ye K, Meng WX, Sun H, Wu B, Chen M, Pang YP, et al. Characterization of an alternative BAK-binding site for BH3 peptides. Nat Commun. 2020;11(1):3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96(5):615–24. [DOI] [PubMed] [Google Scholar]
- 42.Flores-Romero H, Hohorst L, John M, Albert MC, King LE, Beckmann L, et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 2022;41(2):e108690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644(2–3):83–94. [DOI] [PubMed] [Google Scholar]
- 44.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24(5):677–88. [DOI] [PubMed] [Google Scholar]
- 45.Birkinshaw RW, Iyer S, Lio D, Luo CS, Brouwer JM, Miller MS, et al. Structure of detergent-activated BAK dimers derived from the inert monomer. Mol Cell. 2021;81:2123–34.e5. [DOI] [PubMed] [Google Scholar]
- 46.Singh G, Guibao CD, Seetharaman J, Aggarwal A, Grace CR, McNamara DE, et al. Structural basis of BAK activation in mitochondrial apoptosis initiation. Nat Commun. 2022;13(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286(1):491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hockings C, Anwari K, Ninnis RL, Brouwer J, O’Hely M, Evangelista M, et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015;6:e1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dengler MA, Robin AY, Gibson L, Li MX, Sandow JJ, Iyer S, et al. BAX Activation: Mutations Near Its Proposed Non-canonical BH3 Binding Site Reveal Allosteric Changes Controlling Mitochondrial Association. Cell Rep. 2019;27(2):359–73 e6. [DOI] [PubMed] [Google Scholar]
- 50.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40(3):481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandow JJ, Tan IK, Huang AS, Masaldan S, Bernardini JP, Wardak AZ, et al. Dynamic reconfiguration of pro-apoptotic BAK on membranes. EMBO J. 2021;40(20):e107237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sperl LE, Ruhrnossl F, Schiller A, Haslbeck M, Hagn F. High-resolution analysis of the conformational transition of pro-apoptotic Bak at the lipid membrane. EMBO J. 2021;40(20):e107159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell. 2008;30(3):369–80. [DOI] [PubMed] [Google Scholar]
- 54.Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, et al. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chemistry & biology. 2013;20(7):888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li MX, Tan IKL, Ma SB, Hockings C, Kratina T, Dengler MA, et al. BAK alpha6 permits activation by BH3-only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci U S A. 2017;114(29):7629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF, et al. Conversion of Bim-BH3 from Activator to Inhibitor of Bak through Structure-Based Design. Mol Cell. 2017;68(4):659–72 e9. [DOI] [PubMed] [Google Scholar]
- 57.Robin AY, Krishna Kumar K, Westphal D, Wardak AZ, Thompson GV, Dewson G, et al. Crystal structure of Bax bound to the BH3 peptide of Bim identifies important contacts for interaction. Cell Death Dis. 2015;6:e1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14(16):2060–71. [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou H, Hou Q, Hansen JL, Hsu YT. Complete activation of Bax by a single site mutation. Oncogene. 2007;26(50):7092–102. [DOI] [PubMed] [Google Scholar]
- 60.Robin AY, Iyer S, Birkinshaw RW, Sandow J, Wardak A, Luo CS, et al. Ensemble Properties of Bax Determine Its Function. Structure. 2018;26(10):1346–59 e5. [DOI] [PubMed] [Google Scholar]
- 61.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Zidek A, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596(7873):590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans RONMP A; Antropova N; Senior A; Green T; Zidek A; Bates R; Blackwell S; Yim J; Ronnberger O; Bodenstein S; Zielinski M; Bidgland A; Potapenko A; Cowie A; Tunyasuvunakool K; Jain R; Clancy E; Kohli P; Jumper J; Hassabis D Protein complex prediction with AlphaFold-Multimer. BioRxiv. 2022. [Google Scholar]
- 64.Ruff KM, Pappu RV. AlphaFold and Implications for Intrinsically Disordered Proteins. J Mol Biol. 2021;433(20):167208. [DOI] [PubMed] [Google Scholar]
- 65.Hegedus T, Geisler M, Lukacs GL, Farkas B. Ins and outs of AlphaFold2 transmembrane protein structure predictions. Cell Mol Life Sci. 2022;79(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crabtree MD, Mendonca C, Bubb QR, Clarke J. Folding and binding pathways of BH3-only proteins are encoded within their intrinsically disordered sequence, not templated by partner proteins. J Biol Chem. 2018;293(25):9718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55(6):938–46. [DOI] [PubMed] [Google Scholar]
- 68.Birkinshaw RW, Iyer S, Lio D, Luo CS, Brouwer JM, Miller MS, et al. Structure of detergent-activated BAK dimers derived from the inert monomer. Mol Cell. 2021. [DOI] [PubMed] [Google Scholar]
- 69.Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, et al. BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol. 2020;27(11):1024–31. [DOI] [PubMed] [Google Scholar]
- 70.Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36(4):696–703. [DOI] [PubMed] [Google Scholar]
- 71.Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012;19(4):661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lv F, Qi F, Zhang Z, Wen M, Kale J, Piai A, et al. An amphipathic Bax core dimer forms part of the apoptotic pore wall in the mitochondrial membrane. EMBO J. 2021;40(14):e106438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, Garcia-Saez AJ, Bordignon E. Structural model of active Bax at the membrane. Mol Cell. 2014;56(4):496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cosentino K, Hertlein V, Jenner A, Dellmann T, Gojkovic M, Pena-Blanco A, et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol Cell. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378). [DOI] [PubMed] [Google Scholar]
- 76.Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35(4):389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hauseman ZJ, Harvey EP, Newman CE, Wales TE, Bucci JC, Mintseris J, et al. Homogeneous Oligomers of Pro-apoptotic BAX Reveal Structural Determinants of Mitochondrial Membrane Permeabilization. Mol Cell. 2020;79(1):68–83 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, et al. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015;22(10):1665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, et al. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35(2):208–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mandal T, Shin S, Aluvila S, Chen HC, Grieve C, Choe JY, et al. Assembly of Bak homodimers into higher order homooligomers in the mitochondrial apoptotic pore. Sci Rep. 2016;6:30763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uren RT, O’Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, et al. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Tjandra N. Structural insights of tBid, the caspase-8-activated Bid, and its BH3 domain. J Biol Chem. 2013;288(50):35840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.