

Abstract
Background
A multistep pathogenesis of myeloid leukemia including mutations in epigenetic, spliceosome, and signaling genes has been recently demonstrated in a preclinical model but is poorly validated in patients.
Methods
Clinical, phenotypic, and biologic features were compared between three distinct molecularly defined CMML cohorts including TET2 monomutated patients (T, n = 10), TET2/SRSF2 bimutated patients (TS, n = 19), and patients who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN, n = 14).
Results
Median survival was 90, 45, and 9 months, respectively (p = .001). Whereas no patient in the T and TS group transformed into acute myeloid leukemia (AML), 6/14 patients in the TSN group had AML at study entry or transformed during follow‐up. Leukocyte counts, blast cell counts, and LDH levels were significantly higher in TSN vs. TS and T, respectively, whereas hemoglobin and platelet values were not significantly different. Increased growth factor‐independent myeloid colony formation was restricted to TSN but not found in T and TS, respectively. The proportion of patients showing in vitro myelomonocytic skewing in T, TS, and TSN was 0%, 56%, and 100%, respectively (p = .010).
Conclusion
Our results demonstrate that the model of multistep pathogenesis in CMML can be recapitulated in patients regarding clinical, phenotypic, and biologic features.
Keywords: CMML, NRAS, pathogenesis, SRSF2, TET2
1. What is the NEW aspect of your work?
A multistep pathogenesis of myeloid leukemia, including mutations in epigenetic, spliceosome, and signaling genes has been recently demonstrated in a preclinical model but is poorly validated in patients.
2. What is the CENTRAL finding of your work?
Based on survival, risk of transformation, laboratory parameters, growth factor‐independent myeloid colony formation, and myelomonocytic skewing, three distinct molecularly defined CMML cohorts, including TET2 monomutated patients, TET2/SRSF2 bimutated patients, and patients who had NRAS mutations in addition to TET2/SRSF2 comutations can be clearly discriminated.
3. What is (or could be) the SPECIFIC clinical relevance of your work?
The demonstration of multistep pathogenesis in CMML patients may be relevant for clinical management.
1. INTRODUCTION
Recently, a multistep pathogenesis of myeloid leukemia has been elegantly demonstrated in a preclinical model reported by Wang et al. 1 By sequentially introducing one mutated gene of the epigenetic machinery such as ASXL1, one mutated gene of the spliceosome such as SRSF2, and one mutated gene of cell signaling such as NRAS by CRISPR‐mediated gene editing of induced pluripotent stem cells (iPSC), the authors generated cell lines that exhibited progressive malignant features capturing clonal hematopoiesis, myelodysplastic syndrome, and transplantable acute leukemia, respectively. Most importantly, they identified dysregulation of inflammatory signaling as an early and persistent event in leukemogenesis and a promising early therapeutic target.
The validation of pathogenetic concepts which have been developed in preclinical models by clinical data is important because it improves the translational significance and increases the probability that treatment strategies based on these models are working in patients. The "Austrian Biodatabase of Chronic Myelomonocytic Leukemia“ (ABCMML) has been reported to be a representative and useful real‐life data source for further biomedical research. 2 In this database, we collected epidemiologic, hematologic, biochemical, clinical, immunophenotypic, cytogenetic, molecular, and biologic data of patients with CMML from different Austrian centers over 30 years. Having these data, we can correlate molecular subgroups of CMML with clinical outcome, phenotypic features, and biologic characteristics. Using molecular data from the ABCMML, we selected patients who had TET2 mutations alone (T), those who had only TET2/SRSF2 comutations (TS) and those who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN) for further analysis in order to validate findings based on preclinical models in patients.
2. PATIENTS AND METHODS
2.1. Patients
Recently, we have shown that the ABCMML may be used as a representative and useful real‐life data source for biomedical research. 2 In this database, we retrospectively collected epidemiologic, hematologic, biochemical, clinical, immunophenotypic, cytogenetic, molecular, and biologic data of patients with CMML from different centers. The diagnosis of CMML and leukemic transformation was according to the WHO criteria. 3 , 4 , 5 Clinical and laboratory routine parameters were obtained from patient records. A detailed central manual retrospective chart review was carried out to ensure data quality before analysis of data from institutions.
43 CMML patients collected between 1.1.1990 and 30.11.2021 qualified for our study regarding their molecular profile. This research has been approved by the ethic committee of the City of Vienna on 10 June 2015 (ethic code: 15–059‐VK).
2.2. Molecular studies
Genomic DNA was isolated from mononuclear cell (MNC) fractions of these blood samples according to standard procedures. The mutational status of CMML‐related protein coding genes was determined by targeted amplicon sequencing using the MiSeq platform (Illumina). Details regarding gene panel, library preparation, and data processing have been reported previously 2 and are given in the Supporting Information. Only variants with an allelic frequency (VAF) ≥5%, a described population frequency (MAF) <1%, and an annotated pathogenic effect (or probability >90% of being pathogenic) were included, with pathogenicity determined according to databases as shown in Table S1 and published studies.
2.3. Colony assay
In one of our centers (Medical University of Vienna), the assessment of hematopoietic colony formation in vitro has been an integral part of the diagnostic work up in patients with suspected myeloid malignancies for many years. 6 Colony‐forming unit‐granulocyte/macrophage (CFU‐GM) and burst‐forming unit‐erythroid (BFU‐E) growth was assessed in semisolid cultures using a modification of the clonal assay described by Fauser & Messner 7 with and without growth factors as previously described by us. 8 , 9 Mononuclear cells (MNC) were isolated from peripheral blood (PB) of patients by Ficoll‐Hypaque density gradient centrifugation (density 1.077 g/ml, 400 g for 40 min). The low‐density cells were collected from the interface between density solution and plasma, washed twice, and resuspended in Iscove‘s modified Dulbecco‘s medium (GIBCO). PBMNCs were cultured in 0.9% methylcellulose, 30% fetal calf serum (FCS; Biomedica), 10% bovine serum albumin (Sigma), α‐thioglycerol (10−4 mol/L), and Iscove‘s modified Dulbecco‘s medium. For the stimulation of progenitor cells, cultures were supplemented with recombinant human granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) (10 ng/ml; R&D Systems), rh‐interleukin‐3 (10 ng/ml; R&D Systems), and erythropoietin (EPO, 2 U/ml; Roche). Stimulated cultures were plated in duplicates at 100 × 103 PBMNC/ml. Unstimulated cultures were plated in duplicates or triplicates, respectively, at 25–100 × 103 PBMNC/ml. Plates were incubated at 37°C, 5% CO2, and full humidity. After a culture period of 14 days, cultures were examined under an inverted microscope. Aggregates with more than 40 translucent, dispersed cells were counted as CFU‐GM. Bursts containing more than 100 red colored cells were scored as BFU‐E. Progenitor cell data are expressed as mean values from cultures.
2.4. Statistical analysis
The log‐rank test was used to determine whether individual parameters were associated with the overall survival (OS). OS was defined as the time from sampling to death (uncensored) or last follow‐up (censored). Time to AML transformation was defined as the time of sampling to the time of transformation to secondary AML (uncensored) or death/last contact (censored). Univariate Cox regression analyses were performed to determine hazard ratios and confidence intervals of potential prognostic factors and multivariate Cox regression analyses to investigate the relation of their prognostic impact to each other. Dichotomous variables were compared between different groups with the use of the chi‐square test. The Mann–Whitney U‐test was used to compare two and the Kruskal‐Wallis test to compare three unmatched groups when continuous variables were not normally distributed. Results were considered significant at p < .05. Statistical analyses were performed with the SPSS v. 27 (SPSS Inc); the reported p‐values were two‐sided.
3. RESULTS
3.1. Patients
The characteristics of patients from our ABCMML have been reported previously. 2 In this study, we included 43 patients including 4 patients in transformation at study entry for three distinct molecularly defined CMML cohorts representing TET2 monomutated patients (T, n = 10), TET2/SRSF2 bimutated patients (TS, n = 19), and patients who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN, n = 14). A comprehensive list of individual patient characteristics is given in Table S2. The median age was 76 years (50–92), the percentage of males 63% (27/43). There was no statistically significant difference between the three cohorts regarding age, but the percentage of males was higher in the TSN group (12/14, 86%) as compared with the other two groups (15/29, 52%).
3.2. Clinical outcome
Figure 1 shows the Kaplan‐Meier plots for overall survival (OS) in CMML patients with TET2 mutations alone (T), those with only a TET2/SRSF2 comutations (TS) and those who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN). Kaplan‐Meier plots clearly discriminated the three cohorts with a median survival of 90, 45, and 9 months, in the T, TS, and TSN group, respectively (p = .001). As shown in Table S3, other parameters with potential impact on prognosis were also studied in univariate Cox regression analyses, including CMML category, increased white blood cell (WBC) count, decreased hemoglobin (Hb) level, decreased platelet (PLT) count, the presence of adverse cytogenetics, and increased spontaneous myeloid colony formation. According to these analyses, increased WBC counts, CMML subcategories and growth factor‐independent CFU‐GM formation were significantly associated with poor survival. It has to be mentioned that the sample size in this study was limited, and, considering the hazard ratios exceeding 1, some parameters may have reached statistical significance with higher patient numbers. To determine the relation of the prognostic impact of the molecular subtypes with other prognostic parameters, multivariate Cox regression analyses were performed adjusting for these factors. As shown in Table S4, molecular subtypes retained their significance in the presence of WBC and CMML subcategories but lost their prognostic significance if adjusted for spontaneous myeloid colony formation.
FIGURE 1.
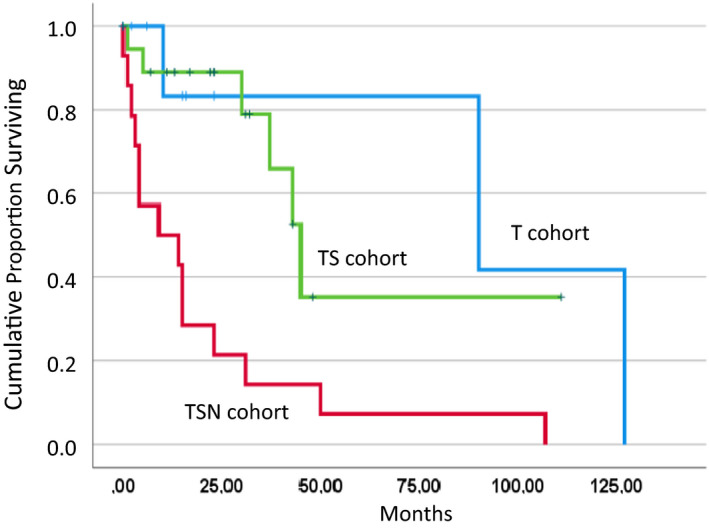
Overall survival indicated by Kaplan‐Meier plots in three distinct molecularly defined CMML cohorts including TET2 monomutated patients (T, n = 10), TET2/SRSF2 bimutated patients (TS, n = 19), and patients who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN, n = 14)
Also, the risk of transformation was different between the three cohorts as shown in Figure 2. Whereas there was no transformation seen in the T and TS group, respectively, four patients in the TNS group had already CMML‐derived AML at study entry (n = 4) and two patients transformed into AML during follow‐up resulting in a transformation incidence of 43% (6/14) (p < .001).
FIGURE 2.
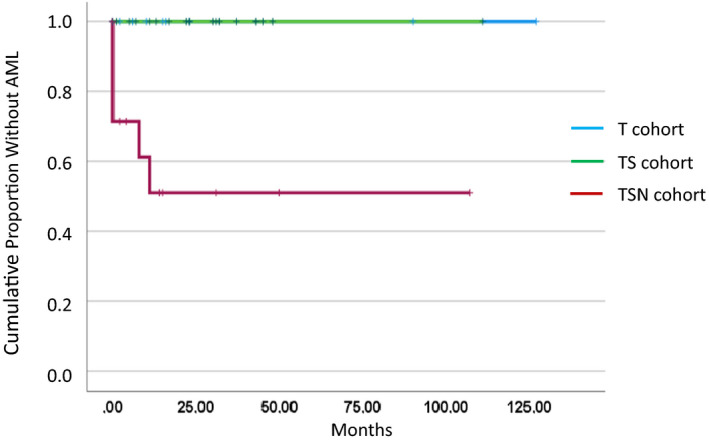
Time to AML transformation in three distinct molecularly defined CMML cohorts including TET2 monomutated patients (T, n = 10), TET2/SRSF2 bimutated patients (TS, n = 19), and patients who had NRAS mutations in addition to TET2/SRSF2 comutations (TSN, n = 14)
3.3. Phenotypic features
The phenotypic features including white blood cell counts (WBC), hemoglobin values, platelet counts, blast cell counts, and LDH levels of the three patient cohorts are given in Table 1. WBC counts and blast cell counts were significantly higher in TSN vs. TS and T, respectively. There was a significant and continuous increment of LDH levels from T to TS to TSN. No significant differences were observed among the three cohorts regarding hemoglobin and platelet values. The proportion of patients with splenomegaly was 29% (2/7), 25% (3/12), and 44% (4/9) in cohorts T, TS, and TSN, respectively, which was not statistically significant (p = .474).
TABLE 1.
Phenotypic features stratified by the 3 patient cohorts
|
T (n = 10) |
TS (n = 19) |
TSN (n = 14) |
p‐value | |
|---|---|---|---|---|
|
Leukocytes G/L; median (range) Evaluable = 43 |
5.9 (3.1–98) | 8.4 (2.9–55.1) | 34.0 (7.8–107.4) | .002 |
|
Hemoglobin g/dl; median (range) Evaluable = 43 |
11.8 (6.5–14.4) | 11.7 (8.4–14.3) | 11.0 (7.0–13.8) | .547 |
|
Platelets G/L; median (range) Evaluable = 43 |
108 (29–507) | 102 (7–345) | 92 (16–288) | .652 |
|
PB blasts %; median (range) Evaluable = 39 |
0 (0–3) | 0 (0–0) | 0 (0–57) | .006 |
|
LDH U/L; median (range) Evaluable = 27 |
181 (167–351) | 238 (68–672) | 348 (204–644) | .008 |
Abbreviations: T, CMML patients who had TET2 mutations alone; TS, CMML patients who had only TET2/SRSF2 comutations; TSN, CMML patients who had NRAS mutations in addition to TET2/SRSF2 comutations.
3.4. Biologic characteristics
We have shown that spontaneous formation of CFU‐GM is a functional surrogate parameter of RAS‐pathway activation. 10 , 11 , 12 The spontaneous formation of CFU‐GM in normal individuals (median 4.8/105 PBMNC, range 3.5–8.5) has been reported by us previously. 13 As shown in Figure 3, growth factor‐independent myeloid colony formation was significantly increased in cohort TSN vs. TS and T, respectively (0.018), but there was no difference between TS and T, respectively (p = .218). Median values and ranges of CFU‐GM/105 MNC were 0.5 (0–3), 3 (0–35), and 59 (3–202), in T, TS, and TSN, respectively (Table S5). Myelomonocytic skewing is another important in vitro characteristic in CMML and is indicated by a higher number of myeloid over erythroid colonies from PBMNC. 14 , 15 As we have reported previously, there was a high prevalence of myelomonocytic skewing in CMML patients (120/146, 82%), whereas this phenomenon was rare in normal individuals (1/98, 1%). 15 Stimulated myeloid and erythroid colony formation in CMML patients in this study is shown in Table 2. The proportion of patients with in vitro myelomonocytic skewing in T, TS, and TSN was 0% (0/4), 56% (5/9), and 100% (5/5), respectively (p = .010).
FIGURE 3.
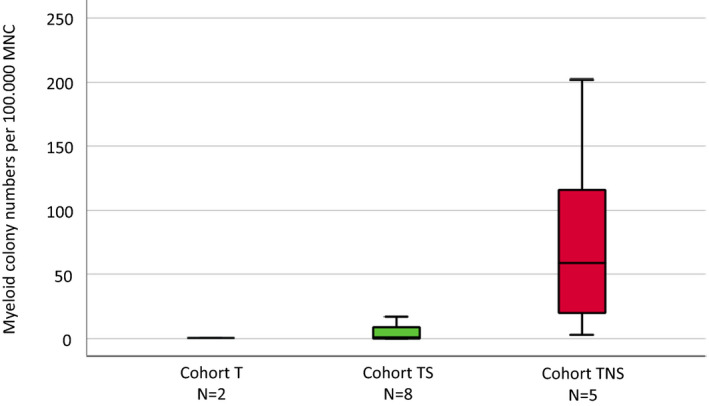
Box plots showing the distribution of spontaneous colony numbers in the three patient cohorts including median values, minimum values, and maximum values, respectively. Cultures were plated in duplicates at 25–100 × 103 PBMNC/ml. Aggregates with more than 40 translucent, dispersed cells were counted as CFU‐GM. CFU‐GM data from patients are expressed as mean values from cultures
TABLE 2.
Stimulated in vitro formation of myeloid and erythroid colonies in CMML patients
| CFU‐GM/100.000 PBMNC | BFU‐E/100.000 PBMNC | CFU‐GM>BFU‐E | |
|---|---|---|---|
| Cohort T | |||
| Pat 1 | 0 | 1 | ‐ |
| Pat 6 | 13 | 19 | ‐ |
| Pat 8 | 10 | 12 | ‐ |
| Pat 10 | 0 | 2 | ‐ |
| Cohort TS | |||
| Pat 12 | 6 | 0 | + |
| Pat 15 | 37 | 10 | + |
| Pat 17 | 75 | 65 | + |
| Pat 20 | 14 | 5 | + |
| Pat 21 | 2 | 10 | ‐ |
| Pat 24 | 12 | 19 | ‐ |
| Pat 26 | 6 | 10 | ‐ |
| Pat 27 | 5 | 5 | ‐ |
| Pat 29 | 21 | 1 | + |
| Cohort TSN | |||
| Pat 30 | 46 | 0 | + |
| Pat 36 | 7 | 5 | + |
| Pat 41 | 311 | 101 | + |
| Pat 42 | 267 | 0 | + |
| Pat 43 | 175 | 0 | + |
| Controls, median (range), n = 80 | 9 (1–44) | 33 (5–91) | |
4. DISCUSSION
In our study, we provide evidence that the multistep pathogenesis of myeloid leukemia that has been demonstrated in a preclinical model by sequential introduction of mutated genes of the epigenetic machinery, the spliceosome, and cell signaling 1 can be recapitulated in patients with CMML. We show that the differences in clinical outcome, phenotype, and biologic characteristics capture distinct stages of this disease.
There is already some evidence in the literature that the evolution of CMML is a stepwise process. TET2 mutated clones can be detected in a small fraction of older subjects with clonal, but non‐leukemic hematopoiesis. 16 Results of single‐cell clonal tracking experiments indicate that a TET2 mutation, when present, is often the earliest recurrent genetic event in CMML. 14 A paper by Ricci et al. strongly suggested for the first time that RAS mutations function as secondary events that contribute to the development of MP‐CMML. 17 In this paper, in two patients who progressed from MDS‐CMML to MP‐CMML, RAS mutations which were predominant in the myeloproliferative phase were by allele‐specific PCR, already detectable at low levels at the time of myelodysplastic phase, documenting the expansion of a RAS‐mutated clone in concomitance with CMML evolution. In preclinical models recapitulating CMML, it has been shown that somatic loss‐of‐function mutations in Tet2 and activating Nras mutations exert cooperating effects and accelerate disease progression. 18 , 19 Moreover, also Asxl1 loss cooperates with oncogenic Nras in mice to reprogram immune microenvironment and drive leukemic transformation. 20 In a retrospective analysis in 337 patients, we have demonstrated a correlation of RAS‐pathway mutations and spontaneous myeloid colony growth with progression and transformation in CMML. 11
The genotypic and phenotypic evolution in a single patient with chronic myelomonocytic leukemia was reported by us recently. 21 The combination of TET2 and SRSF2 mutations is very frequently observed in CMML and highly specific for myeloid neoplasm with monocytosis. 22 Therefore, we decided to use TET2 as the prototype gene of the epigenetic machinery and not the ASXL1 gene that has been used by Wang et al. in their preclinical model. 1 Whereas molecular aberrations of the T and TS cohort were restricted to TET2 alone or the TET2/SRSF2 comutation, patients in the TSN group had often more additional mutations including NRAS. We did this to get a sufficient patient number for the third cohort. In fact, the median number of mutations was 5 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 in this group. This represents the real‐life situation since some reports have indicated that more advanced CMML shows a higher number of mutations than less advanced disease. 23
Regarding clinical outcome, there is a clear discrimination between the three patient cohorts. Patients in the TSN cohort had a median survival of only 9 months which is compatible with other reports of advanced CMML. 24 In our study, we observed transformation into AML only in this subgroup but not in the cohorts with TET2 mutation alone or patients with only a TET2/SRSF2 comutation. This is not unexpected, since NRAS mutations have been shown to be an adverse prognostic factor regarding survival and AML evolution, and therefore, have been integrated in the clinical/molecular CMML‐specific prognostic scoring system (CPSS‐Mol). 25
Also, the higher number of WBC and blast cells is in agreement with previous reports since NRAS mutations in CMML have been associated with myeloproliferation. 2 , 20 Although there was a numerical decline of hemoglobin and platelet values in the TSN group, this difference was not statistically significant. The relatively low patient numbers in the three cohorts may explain this finding since anemia and thrombocytopenia are established prognostic parameters in larger CMML cohorts. 2
We recently were able to show that growth factor‐independent CFU‐GM formation may be a functional surrogate of RAS‐pathway activation. 10 , 11 , 12 The biologic basis for this observation was provided by Padron et al. when he reported hypersensitivity of CMML progenitors using phospho‐STAT5 flow cytometry. 26 Moreover, our group could show in a small retrospective study that CMML patients with high spontaneous CFU‐GM growth (>100/105 PBMNC) have an inferior prognosis as compared with patients with low myeloid colony formation suggesting a clinical significance of the original observation. 27 These results have been recently extended in a much larger CMML patient cohort indicating that spontaneous myeloid colony formation was compared with other established single prognostic factors, the strongest predictor regarding OS. 28 This may indicate that in vitro cultures using unmanipulated MNC may be a more global test that covers different aspects of malignancy better than any of the single parameters. In this study, we show that significantly increased spontaneous myeloid colony formation is restricted to the patient group containing NRAS mutations. This is in agreement with our previous findings which show a high correlation between high spontaneous CFU‐GM formation and the presence of mutations in the RASopathy genes and indicate that hyperactivation of the RAS‐pathway plays a critical pathogenetic role in the advanced phase of CMML which is characterized by high leukocyte counts, an increase of blast cells and a high risk of transformation to AML.
Myelomonocytic skewing has been proposed as a key phenomenon in the pathophysiology of CMML. In a seminal paper using mutation‐specific discrimination analysis of single‐cell‐derived colonies in 28 patients with CMML, Itzykson et al. could show that the main features of this disease are early clonal dominance, arising at the CD34+/CD34‐ stage of hematopoiesis, and granulomonocytic differentiation skewing of multipotent and common myeloid progenitors. 14 Furthermore, we could demonstrate that myelomonocytic skewing as determined by semisolid cultures can separate subgroups of CMML patients with a different phenotype, a different genotype, and a different prognosis. 15 In this study, we found that myelomonocytic skewing was not observed in the CMML patients with a TET2 mutation alone, but was seen in more than half of the patients who had only a TET2/SRSF2 mutation and in all patients who had additional NRAS mutations. This indicates that the preferential expansion of myelomonocytopoiesis over erythropoiesis in CMML is a biologic phenomenon starting at the CMML stage characterized by the comutation of genes of the epigenetic and the spliceosome regulation and further parallels the progression of CMML into more advanced disease.
We have not systematically studied patients with reactive monocytosis. However, we found in our dataset single patients who could be categorized as reactive monocytosis due to the fact that the monocytosis was transient and patients never developed a myeloid malignancy. In three of them, in vitro cultures have been performed, and in two of these patients, NGS analysis has been done. In none of these patients, myelomonocytic skewing was seen in semisolid cultures and none had molecular aberrations which were covered by our NGS gene panel. In the literature, hematopoietic progenitor cell colony growth has been already demonstrated to differentiate CMML from reactive monocytosis. Whereas either leukemic or pseudonormal growth was seen in CMML patients, this pattern was not observed in patients with reactive monocytosis. 29 Regarding the role of molecular aberrations in patients with monocytosis, it has been shown that somatic mutations were detected in 79% of patients with confirmed diagnosis of CMML but also in 57% of patients with nondiagnostic bone marrow features. 30 The OS in nondiagnostic mutated patients, however, was indistinguishable from those with CMML and significantly worse than in unmutated patients. These data indicate that NGS may be a very sensitive tool to detect patients with a myeloid clone earlier than the features of CMML found in BM.
There are several limitations that must be considered in our study. Most of the information used in this study was derived from retrospective real‐world data that were not collected systematically or prospectively. Thus, not every parameter was available in all patients. In addition, data from patient records were obtained over many years and from many different centers. However, real‐world data have recently been recognized as an important way to get insights into the routine management and the natural history of rare diseases. 31 CMML is a rare disease and adequate patient numbers for a systematic and prospective study are not easy to collect within a limited time frame. Moreover, the ABCMML provides information derived from molecular as well as from functional studies, and therefore allows a more comprehensive view and deeper insight into the complex pathophysiology of this hematologic malignancy. 2
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
KG designed the project, collected, analyzed and interpreted the data, and wrote the manuscript. EJ, AB, MG, and RM performed the experiments. TG performed the administration of data. TN, MP, SM‐S, RS, AZ, HS, LÖ, RK, GH, and PV collected the data. All authors reviewed the draft manuscript and approved the final version for submission. All authors have read and agreed to the published version of the manuscript.
Supporting information
Tab S1‐S5
ACKNOWLEDGMENT
None.
Geissler K, Jäger E, Barna A, et al. Multistep pathogenesis of chronic myelomonocytic leukemia in patients. Eur J Haematol. 2022;109:50–57. 10.1111/ejh.13768
Funding information
This study was supported by the “Gesellschaft zur Erforschung der Biologie und Therapie von Tumorkrankheiten ‐ ABCMML‐112015”
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, (KG), upon reasonable request.
REFERENCES
- 1. Wang T, Pine AR, Kotini AG, et al. Sequential CRISPR gene editing in human IPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell Stem Cell. 2021;28(6):1074‐1089.e7. doi: 10.1016/j.stem.2021.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geissler K, Jäger E, Barna A, et al. The Austrian biodatabase for chronic myelomonocytic leukemia (ABCMML): a representative and useful real‐life data source for further biomedical research. Wien Klin Wochenschr. 2019;131(17–18):410‐418. doi: 10.1007/s00508-019-1526-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292‐2302. doi: 10.1182/blood-2002-04-1199 [DOI] [PubMed] [Google Scholar]
- 4. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937‐951. doi: 10.1182/blood-2009-03-209262 [DOI] [PubMed] [Google Scholar]
- 5. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. doi: 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 6. Ohler L, Geissler K, Hinterberger W. Diagnostic and prognostic value of colony formation of hematopoietic progenitor cells in myeloid malignancies. Wien Klin Wochenschr. 2003;115(13–14):537‐546. doi: 10.1007/bf03041036 [DOI] [PubMed] [Google Scholar]
- 7. Fauser AA, Messner HA. Identification of megakaryocytes, macrophages, and eosinophils in colonies of human bone marrow containing neurtophilic granulocytes and erythroblasts. Blood. 1979;53(5):1023‐1027. [PubMed] [Google Scholar]
- 8. Geissler K, Ohler L, Födinger M, et al. Interleukin 10 inhibits growth and granulocyte/macrophage colony‐stimulating factor production in chronic myelomonocytic leukemia cells. J Exp Med. 1996;184(4):1377‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geissler K, Peschel C, Niederwieser D, et al. Potentiation of granulocyte colony‐stimulating factor‐induced mobilization of circulating progenitor cells by seven‐day pretreatment with interleukin‐3. Blood. 1996;87(7):2732‐2739. [PubMed] [Google Scholar]
- 10. Geissler K, Jäger E, Barna A, et al. Chronic myelomonocytic leukemia patients with RAS pathway mutations show high in vitro myeloid colony formation in the absence of exogenous growth factors. Leukemia. 2016;30(11):2280‐2281. doi: 10.1038/leu.2016.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geissler K, Jäger E, Barna A, et al. Correlation of RAS‐pathway mutations and spontaneous myeloid colony growth with progression and transformation in chronic myelomonocytic leukemia—A retrospective analysis in 337 patients. IJMS. 2020;21(8):3025. doi: 10.3390/ijms21083025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geissler K. Molecular pathogenesis of chronic myelomonocytic leukemia and potential molecular targets for treatment approaches. Front Oncol. 2021;11, 751668. doi: 10.3389/fonc.2021.751668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oehler L, Foedinger M, Koeller M, et al. Interleukin‐10 inhibits spontaneous colony‐forming unit‐granulocyte‐macrophage growth from human peripheral blood mononuclear cells by suppression of endogenous granulocyte‐macrophage colony‐stimulating factor release. Blood. 1997;89(4):1147‐1153. [PubMed] [Google Scholar]
- 14. Itzykson R, Kosmider O, Renneville A, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186‐2198. doi: 10.1182/blood-2012-06-440347 [DOI] [PubMed] [Google Scholar]
- 15. Geissler K, Jäger E, Barna A, et al. Myelomonocytic skewing in chronic myelomonocytic leukemia: phenotypic, molecular and biologic features and impact on survival. Eur J Haematol. 2021;106(5):627‐633. doi: 10.1111/ejh.13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179‐1181. doi: 10.1038/ng.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ricci C, Fermo E, Corti S, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16(8):2246‐2256. doi: 10.1158/1078-0432.CCR-09-2112 [DOI] [PubMed] [Google Scholar]
- 18. Kunimoto H, Meydan C, Nazir A, et al. Cooperative epigenetic remodeling by TET2 loss and NRAS mutation drives myeloid transformation and MEK inhibitor sensitivity. Cancer Cell. 2018;33(1):44‐59.e8. doi: 10.1016/j.ccell.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin X, Qin T, Zhao M, et al. Oncogenic N‐Ras and Tet2 haploinsufficiency collaborate to dysregulate hematopoietic stem and progenitor cells. Blood Adv. 2018;2(11):1259‐1271. doi: 10.1182/bloodadvances.2018017400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. You X, Liu F, Binder M, et al. Asxl1loss cooperates with oncogenic nras in mice to reprogram immune microenvironment and drive leukemic transformation. Blood. 2021;139(7):1066‐1079. doi: 10.1182/blood.2021012519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geissler K, Jäger E, Gurbisz M. Genotypic and phenotypic evolution in a patient with chronic myelomonocytic leukemia. Leuk Res Rep. 2019;12:100185. doi: 10.1016/j.lrr.2019.100185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itzykson R, Fenaux P, Bowen D, et al. Diagnosis and treatment of chronic myelomonocytic leukemias in adults: recommendations from the European hematology association and the European LeukemiaNet. HemaSphere. 2018;2(6):e150. doi: 10.1097/HS9.0000000000000150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mason CC, Khorashad JS, Tantravahi SK, et al. Age‐related mutations and chronic myelomonocytic leukemia. Leukemia. 2016;30(4):906‐913. doi: 10.1038/leu.2015.337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patnaik MM, Pierola AA, Vallapureddy R, et al. Blast phase chronic myelomonocytic leukemia: Mayo‐MDACC collaborative study of 171 cases. Leukemia. 2018;32(11):2512‐2518. doi: 10.1038/s41375-018-0143-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elena C, Gallì A, Such E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128(10):1408‐1417. doi: 10.1182/blood-2016-05-714030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Padron E, Painter JS, Kunigal S, et al. GM‐CSF‐dependent PSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068‐5077. doi: 10.1182/blood-2012-10-460170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sagaster V, Ohler L, Berer A, et al. High spontaneous colony growth in chronic myelomonocytic leukemia correlates with increased disease activity and is a novel prognostic factor for predicting short survival. Ann Hematol. 2004;83(1):9‐13. doi: 10.1007/s00277-003-0743-9 [DOI] [PubMed] [Google Scholar]
- 28. Geissler K, Jäger E, Barna A, et al. Molecular basis and clinical application of growth‐factor‐independent in vitro myeloid colony formation in chronic myelomonocytic leukemia. IJMS. 2020;21(17):6057. doi: 10.3390/ijms21176057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Birrer A, Jusufi F, Bernimoulin M, et al. Hematopoietic progenitor cell colony growth differentiates chronic myelomonocytic leukemia from reactive monocytosis. Eur J Haematol. 2008;81(4):267‐272. doi: 10.1111/j.1600-0609.2008.01115.x [DOI] [PubMed] [Google Scholar]
- 30. Cargo C, Cullen M, Taylor J, et al. The use of targeted sequencing and flow cytometry to identify patients with a clinically significant monocytosis. Blood. 2019;133(12):1325‐1334. doi: 10.1182/blood-2018-08-867333 [DOI] [PubMed] [Google Scholar]
- 31. Khozin S, Blumenthal GM, Pazdur R. Real‐world data for clinical evidence generation in oncology. JNCI. 2017;109(11):1‐5. doi: 10.1093/jnci/djx187 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tab S1‐S5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, (KG), upon reasonable request.