

SUMMARY
The non‐essential supernumerary maize (Zea mays) B chromosome (B) has recently been shown to contain active genes and to be capable of impacting gene expression of the A chromosomes. However, the effect of the B chromosome on gene expression is still unclear. In addition, it is unknown whether the accumulation of the B chromosome has a cumulative effect on gene expression. To examine these questions, the global expression of genes, microRNAs (miRNAs), and transposable elements (TEs) of leaf tissue of maize W22 plants with 0–7 copies of the B chromosome was studied. All experimental genotypes with B chromosomes displayed a trend of upregulated gene expression for a subset of A‐located genes compared to the control. Over 3000 A‐located genes are significantly differentially expressed in all experimental genotypes with the B chromosome relative to the control. Modulations of these genes are largely determined by the presence rather than the copy number of the B chromosome. By contrast, the expression of most B‐located genes is positively correlated with B copy number, showing a proportional gene dosage effect. The B chromosome also causes increased expression of A‐located miRNAs. Differentially expressed miRNAs potentially regulate their targets in a cascade of effects. Furthermore, the varied copy number of the B chromosome leads to the differential expression of A‐located and B‐located TEs. The findings provide novel insights into the function and properties of the B chromosome.
Keywords: B chromosome, gene expression, maize, dosage‐sensitive modulation, microRNA
Significance Statement
The non‐essential maize (Zea mays) B chromosome has wide applications in various genetic studies and genetic engineering of minichromosomes, yet little is known about how it impacts global gene expression. In the transcriptome analysis of maize plants with 0–7 copies, the B chromosome was found to impact the expression of thousands of A‐chromosome‐located genes in a predominant dosage‐insensitive manner while the B‐located genes generally show a proportional gene dosage effect. This property is in contrast to A chromosome aneuploidy, which modulates genes in a dosage‐sensitive fashion.
INTRODUCTION
Supernumerary B chromosomes (Bs) are non‐essential chromosomes that can be present or absent from some individuals within a population. They are found in a variety of species including flowering plants, gymnosperms, fungi, and animals (Jones and Houben 2003; Jones and Rees 1982). The maize (Zea mays) B chromosome was first identified in the 1920s (Kuwada 1925; Longley 1927; Randolph 1928). The B chromosome does not pair or recombine with the A chromosomes at meiosis. The inheritance of the B chromosome is non‐Mendelian and follows its own evolutionary pathway (Beukeboom 1994). Maize B chromosomes have a special accumulation mechanism in that they non‐disjoin at the second pollen mitosis followed by preferential fertilization of the egg by the sperm containing the B chromosomes (Carlson 1988; Roman 1948). This non‐disjunction property makes the maize B chromosome a powerful tool when it translocates with the A chromosomes. Maize with stable B‐A translocations has wide applications in various genetic studies, including experimental mapping, chromosomal dosage studies, chromosomal behavior, and genetic engineering of minichromosomes (Beckett 1991; Birchler and Veitia 2012; Cody et al. 2015; Liu et al. 2015).
B chromosomes are usually highly heterochromatic. The maize B chromosome contains a minute short arm, a centromeric knob, a proximal euchromatin region, four blocks of distal heterochromatin, and a distal euchromatin region (Cheng et al. 2016; McClintock 1933). It is widely accepted that B chromosomes are derived from the A chromosomes (Camacho et al. 2000; Jones and Rees 1982). Maize A and B chromosomes share a collection of highly repetitive sequences including CentC, CRM, CentA, and knob repeats (Alfenito and Birchler 1993; Lamb et al. 2005; Stark et al. 1996; Viotti et al. 1985). Retrotransposons were found on the maize B chromosome by analyzing DNA sequencing data and fluorescence in situ hybridization (FISH) images. Some of these retrotransposons or repetitive sequences derived from the B chromosome are B‐specific while others are shared with the A chromosomes (Blavet et al. 2021; Huang et al. 2016; Lamb et al. 2005; Stark et al. 1996; Theuri et al. 2005). The maize B centromere repeat sequence bears a similarity to that of heterochromatic knobs and the maize Cent4, a sequence associated with the centromere of chromosome 4 (Alfenito and Birchler 1993; Page et al. 2001).
Although the B chromosome of maize is dispensable for the normal growth and reproduction of the plant and has yet to be proven to have any beneficial effects (Randolph 1941), it is not totally inert. The maize B chromosome is capable of impacting the A chromosomes by increasing their recombination frequencies (Hanson 1969; Rhoades 1968). When the B chromosome is present at high copy numbers (usually over 10), it causes various morphological effects including a reduction in fertility, decreased vigor, and increased nuclear and cell size (Randolph 1941). In addition, in a recent study, the B chromosome has been shown to impact the expression of over 100 genes transcribed from the A chromosomes (Huang et al. 2016). A few genes located on the B chromosome were identified with high sequence similarity but sometimes acquired multiple SNPs, insertions, or deletions compared to their A homologs (Huang et al. 2016). Some of these B‐located genes are transcribed, indicating that the B chromosome is not inactive (Huang et al. 2016). The sequence of the maize B chromosome has been assembled and published (Blavet et al. 2021). Recently, 758 predicted protein‐coding genes in 125.9 Mb of B chromosome sequence were identified, at least 88 of which are expressed (Blavet et al. 2021). Analysis of the gene and transposable element (TE) content of the B chromosome against the A chromosomes suggests the current content of the B chromosome is a result of a continuous transfer from the A chromosomal complement for millions of years (Blavet et al. 2021).
Despite all the advances in the study of the B chromosome, little is known about how the B chromosome impacts the expression of the A chromosomes or the factors that may participate in the regulation of the A chromosome expression. It is also unclear whether the increase of B copy number would have a cumulative effect on gene expression of the A chromosomes. Given that the B chromosome is a powerful tool in various genetics studies, an understanding of how it impacts the expression of the A chromosomes is crucial. Furthermore, there is a lack of knowledge about how genes on the B chromosome respond to changes in B copy number. It is interesting to examine if genes on the B chromosome as a non‐essential chromosome would show gene dosage effects, dosage compensation, and trans‐acting inverse dosage effects, which are normally observed in aneuploidy of essential chromosomes (Birchler et al. 2001; Shi et al. 2021; Yang et al. 2021). Therefore, we performed a comprehensive study analyzing the expression of genes, microRNAs (miRNAs), and TEs of maize W22 plants with 0–7 Bs. This comparison would allow us to better understand how gene expression of the A and B chromosomes is impacted by the B chromosome. This study would also determine if changes in expression levels of miRNAs, if any, would impact the expression of their mRNA targets. Having a dosage series of 0–7 Bs will further help us to define how genes, miRNAs, and TEs respond to changes in B copy number. Such analyses are also an interesting topic to be investigated in terms of how a non‐essential chromosome with an accumulation mechanism evolves. Additionally, changes in dosage of part of the genome (aneuploidy) that contains essential genes/chromosomes have been known to produce severe phenotypic consequences and genome‐wide changes in gene expression (Birchler et al. 2001; Blakeslee 1934; Blakeslee et al. 1920; Hou et al. 2018; Shi et al. 2021; Veitia et al. 2008; Yang et al. 2021). It is intriguing to compare how the varied copy number of a non‐essential chromosome affects gene expression.
RESULTS
The B chromosome causes changes in the expression of A‐located and B‐located genes
To examine the effect of the B chromosome on gene expression, we performed mRNA sequencing (mRNA‐seq) on a collection of maize W22 plants with 0 through 7 copies of Bs (Table S1). We refer to the control genotype as 0B and to the experimental genotypes as 1B through 7B. W22 plants with Bs did not exhibit any morphological defect compared to the control (Figure S1a). Results of cell area measurement suggest the epidermal cell area is unchanged in plants with 0B compared to 5B (Figure S1b; Student's t‐test, P > 0.05). Sequence reads were processed and mapped to the W22 reference genome, the B chromosome sequence, and organellar genomes (Blavet et al. 2021; Bosacchi et al. 2015; Clifton et al. 2004; Springer et al. 2018). Of uniquely mapped reads, 98.9% were mapped to the A chromosomes, whereas only 0.8% were mapped to the B chromosome on average (Table S1). To better illustrate the effect of the B chromosome on the A chromosome and on itself, we further divided genes into A‐located genes and B‐located genes. This mRNA‐seq experiment detected the expression of 20 910 A‐located and 273 B‐located genes (mean of normalized counts across all genotypes over 0.5).
Plotting the distribution of ratios of experimental to control read counts provides a broad view of trends of gene expression, especially for those that are subtly modulated with experimental and control values close to each other. Normalized counts of biological replicates were averaged and ratios of each gene of each experimental condition to the control were plotted as a histogram. Ratio distributions of A‐located genes display a spread of modulations with a trend of slight downregulation with the median of ratios slightly below 1.0 (Figure 1a), whereas ratio distributions of B‐located genes show a trend of upregulation (Figure 1b; Figure S2). Scatter plots were used to demonstrate the fold change values and significance of differential gene expression (DGE) as cross‐validation to the ratio distributions (Figure S3). The log2(fold change [FC]) value of gene expression for each comparison and the false discovery rate (FDR)‐adjusted P‐value of significance were generated by Cuffdiff (Trapnell et al. 2012; Trapnell et al. 2013). The expression of thousands of genes was affected by the B chromosome. Interestingly, 1984 A‐located differentially expressed genes (DEGs) were upregulated and 1595 A‐located DEGs were downregulated in all experimental genotypes compared to the control (Figure 2a). Moreover, 47 B‐located DEGs were upregulated and six B‐located DEGs were downregulated in all seven comparisons (Figure 2b). Overall, for both A‐located and B‐located DEGs, a greater number of DEGs is upregulated than downregulated (Figure 2). Ratio distributions of A‐located genes show a slight downregulation because ratios of a greater proportion of genes are below 1.0 (approximately 51.6% on average for seven comparisons) than being above 1.0 (approximately 48.4%). However, such variation is trivial and is not significant in the DGE analysis, as there is a greater number of upregulated A‐located DEGs than downregulated A‐located DEGs. Therefore, the A‐located genes show a predominant effect of a slight upregulation. As for the B‐located genes, both ratio distribution and DGE analysis display upregulation of gene expression. Although a peak is observed below 1.0 in the ratio distributions of B‐located genes, there is a greater proportion of genes with ratios above 1.0 (approximately 60.0%) than those below 1.0 (approximately 40.0%). This phenomenon is likely caused by the clustering of a subset of genes below 1.0 rather than an overall trend of downregulation in comparison to A‐located genes. Further validation of the mRNA‐seq experiment came from the examination of gene expression differences using an external spike‐in by quantitative PCR (qPCR) (Figure S4). Five genes with distinct expression patterns were selected. Ratios of DGE and trends of induction or repression revealed in the qPCR assay generally match with those in the mRNA‐seq experiment (Pearson correlation: R = 0.97, P = 3.6e−26).
Figure 1.
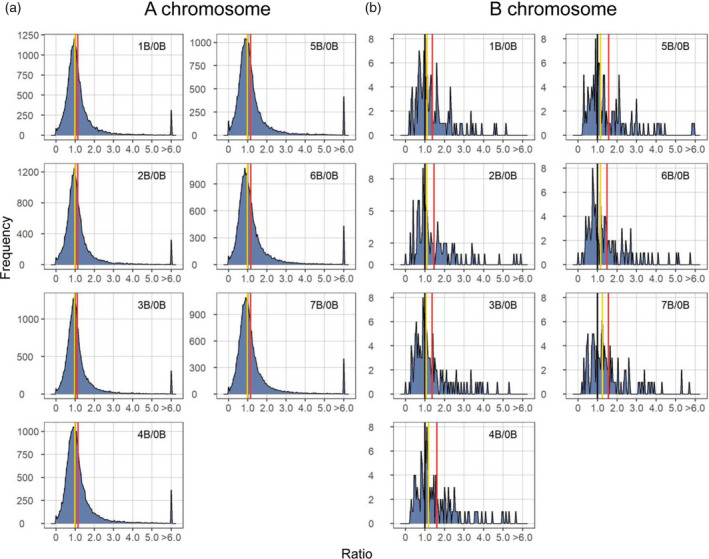
Ratio distributions of gene expression in each experimental genotype compared with the control. (a) Ratio distributions of A‐located genes. (b) Ratio distributions of B‐located genes with outliers (ratio > 6 or < 1/6) removed. Normalized read counts for each gene were averaged across biological replicates and were then used for the generation of ratios comparing each experimental group to the control. Gene ratios for each comparison were plotted on the x‐axis with a bin width of 0.05. The y‐axis denotes the number of genes per bin (frequency). A ratio of 1.00 represents no change in the experimental genotype versus the control and is labeled with vertical lines in black. The mean and median of ratios were computed and demarcated with labeled vertical lines in red and yellow, respectively.
Figure 2.
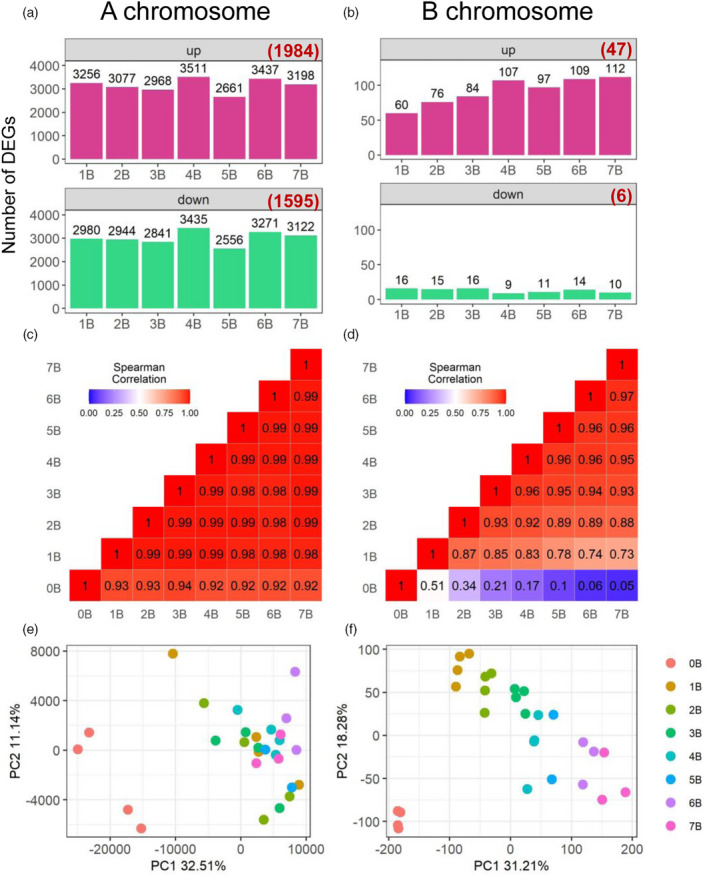
Expression patterns of A‐located genes and B‐located genes. (a, b) Number of A‐located (a) and B‐located (b) differentially expressed genes (DEGs). DEGs were determined as described in the ‘Experimental Procedures’ section. Numbers in brackets represent DEGs in all seven comparisons. (c, d) Heatmap of pairwise correlation (Spearman) matrix between the gene expression level of plants from one experimental or control condition to another. The Spearman correlation coefficient (SCC) was produced by comparing normalized read counts of A‐located genes (c) or B‐located genes (d) averaged across all biological replicates in one genotype to those in another genotype (e.g., 0B versus 1B). The number in each box represents the SCC in the pairwise comparison between two genotypes on the x‐ and y‐axes. The color scale level of each box is reflected by the value of SCC as shown in the color scale bar in the top left corner. (e, f) PCA plots of the expression levels of A‐located genes (e) and B‐located genes (f).
Expression of the majority of A‐located genes is not sensitive to B dosage
As shown in Figure 1a, the trend of slight upregulation of A‐located genes in general does not shift with increased B copy number in the experimental genotype. In addition, the number of A‐located DEGs does not vary much from 1B to 7B (Figure 2a). Therefore, we hypothesized that the overall change in expression of A‐located genes is not correlated with B copy number. To test this hypothesis, Spearman correlation coefficients (SCCs) were computed to examine the correlation of gene expression in different genotypes, and Pearson correlation coefficients (PCCs) were calculated to investigate if the expression of a gene responds to altered B copy number in a dosage‐sensitive manner. The Pearson correlation evaluates the linear relationship, while the Spearman correlation evaluates the monotonic relationship between two variables. Therefore, the Spearman correlation was implemented in producing a pairwise correlation matrix to evaluate the correlation between the gene expression level of plants of different genotypes. SCCs of the expression level of A‐located genes between each experimental genotype with different number of Bs is approaching 1.0 (e.g., 1B versus 7B), whereas SCCs between any experimental genotype (1B to 7B) and the control (0B) are between 0.92 and 0.94 (Figure 2c). This result suggests that changes in the global expression of A‐located genes are more likely to be caused by the presence of the B chromosome, rather than the B copy number.
Further, to examine if there is a linear relationship between gene expression and the B copy number in the B dosage series, a PCC between the expression level of each gene and the number of Bs (0–7) in its corresponding genotype was generated (Figure 3). Here we define B‐dosage‐sensitive genes as those whose expression levels have a strong positive or negative correlation with B copy number (PCC > 0.8 or < −0.8, Figure 3b,f). Out of 20 910 A‐located expressed genes, 1876 (approximately 9%) are sensitive to B dosage based on this criterion (Figure 3a). All other genes are defined as B‐dosage‐insensitive genes (−0.8 ≤ PCC ≤ 0.8), exhibiting a trend of induction (0.2 < PCC ≤ 0.8, Figure 3e) or repression (−0.8 ≤ PCC < −0.2, Figure 3c) regardless of the B copy number, or unchanged regardless of the presence of the B chromosome (−0.2 ≤ PCC ≤ 0.2, Figure 3d). Further validation of this observation came from a principal component analysis (PCA) of expression levels of A‐located genes (Figure 2e). Biological replicates from the 0B group clustered differently from the 1–7B group. All the above‐mentioned results suggest the majority of A‐located genes are not sensitive to changes in B dosage; in other words, the effect of the maize B chromosome on A‐located genes depends on the presence or absence of the B chromosome rather than the B dosage.
Figure 3.
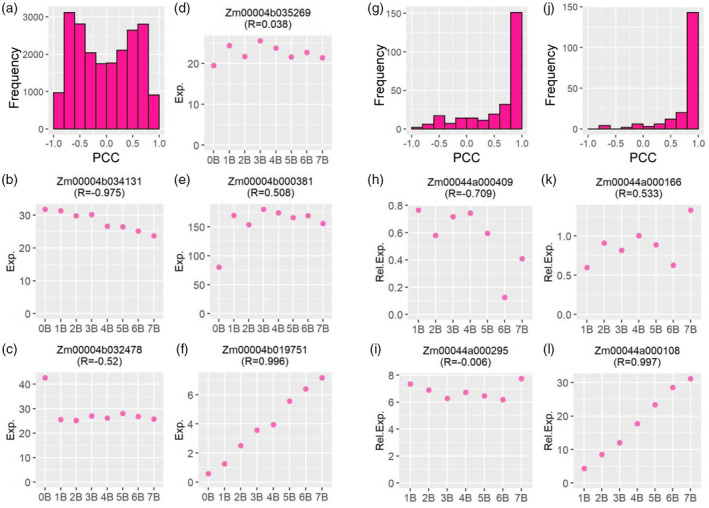
Gene expression level and its Pearson correlation coefficient (PCC) representing B‐dosage sensitivity. (a, g, j) Histogram of PCCs between the expression level of A‐located (a) or B‐located (g, j) genes and the B copy number. (a, g) The PCC for each gene was computed by comparing normalized read counts averaged across all biological replicates for each genotype (gene expression level of 0B to 7B) to the B copy number (0–7). Then the PCC for each gene was plotted into a histogram on the x‐axis with a bin width of 0.2. The y‐axis denotes the number of genes (frequency) per bin. (j) Histogram of PCC between the expression level of B‐located genes and the B copy number (1B–7B), with counts of 0B deducted from 1B–7B. (b–f) Example genes from (a) with distinct expression patterns and different values of PCC. The y‐axis shows the normalized read counts (Exp.) averaged across all biological replicates for each genotype on the x‐axis. (h, i, k, l) Example genes from (j) with distinct expression patterns and different values of PCC. The y‐axis denotes the expression level relative to 0B (Rel. Exp.) averaged across all biological replicates for each genotype on the x‐axis. The label indicates the gene locus and its PCC value (R).
Expression of the majority of B‐located genes exhibits a proportional gene dosage effect
In contrast to A‐located genes, B‐located genes in the B dosage series show an overall distinct expression pattern. Ratio distributions of B‐located genes demonstrate a trend of upregulation correlated with the B copy number, with the median of ratios rising from 1.10 (1B/0B) to 1.25 (7B/0B) (Figures 1a and 4; Table S2). There is an increase in the number of upregulated DEGs with ascending B copy number (Figure 2a). SCCs of the expression levels of B‐located genes between each genotype vary from 0.05 to 0.97 (Figure 2d). Any two groups with less variation in the B copy number display a higher degree of correlation. In addition, a PCA plot of expression levels of B‐located genes shows distinct grouping by their B copy number (Figure 2f). Furthermore, PCCs representing the B‐dosage sensitivity of B‐located genes demonstrate that expression of most B‐located genes is sensitive to B dosage, as PCCs of 153 out of 273 (approximately 56%) expressed B‐located genes fall in the range of >0.8 or <−0.8 (Figure 3g). Unexpectedly, the expression of many B‐located genes in the 0B group is not zero (Table S3), likely due to the high sequence similarity between A‐located and B‐located genes (see ‘Discussion’ section for more details). Therefore, we used expression levels of the 0B group as the baseline and subtracted counts of the 0B group from 1B–7B groups. Afterward, we excluded B‐located genes with negative values for further analysis and redid the histogram of PCCs demonstrating the correlation between B copy number (1–7) and gene expression levels (Figure 3j). In this case, 143 out of the 196 (approximately 73%) B‐located genes being expressed are B‐dosage‐sensitive (PCC > 0.8; e.g., Figure 3l), which exhibit a proportional gene dosage effect in response to changes in B copy number. Expression of 6 (3%) genes is negatively correlated with B copy number (e.g., Figure 3h); whereas 9 (5%) of them are dosage‐compensated (−0.2 ≤ PCC ≤ 0.2; e.g., Figure 3i). Thirty‐eight (19%) genes exhibit a trend of induction but that is not proportional to the B copy number (0.2 < PCC ≤ 0.8; e.g., Figure 3k). In sum, all evidence supports a strong linear correlation between the expression of B‐located genes and the B copy number. In other words, most B‐located genes show a proportional gene dosage effect to changes in B copy number.
Figure 4.
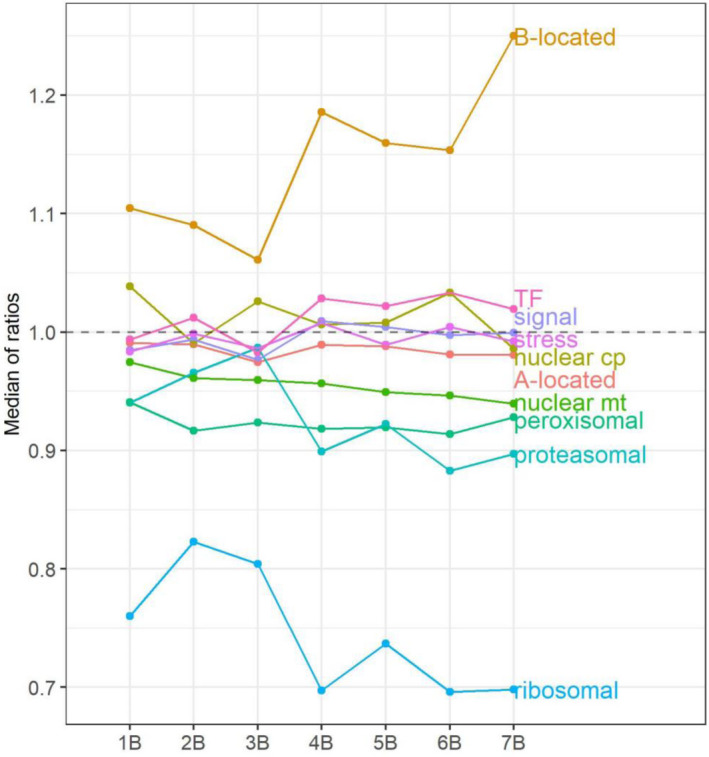
Median of ratios of A‐ and B‐located genes, and each functional class of A‐located genes. The x‐axis refers to the B copy number of the experimental group, whereas the y‐axis denotes the median of ratios in each distribution (Figures 1 and 5; Figures S7–S13), computed as in Table S2.
A‐located genes with different responses to B dosage may participate in distinct biological processes
To evaluate if those A‐located genes that display various dosage responses to the B chromosome are involved in related biological processes, we performed Gene Ontology (GO) enrichment analysis on B‐dosage‐sensitive genes and B‐dosage‐insensitive genes whose expression is induced, repressed, silenced, or unchanged by the presence of the B chromosome. No term was found to be over‐ or underrepresented in B‐dosage‐sensitive A‐located genes that positively correlate with B copy number, whereas terms related to ribonucleoprotein complex assembly (GO:0022618), positive regulation of cellular protein metabolic process (GO:0032270), and cytoplasmic translation (GO:0002181) are enriched in B‐dosage‐sensitive genes with a negative correlation with B copy number (Table S4). This result suggests that these two groups of genes are likely to have different functions.
Genes with weak or no correlation with B copy number show underrepresentation of term DNA duplex unwinding (GO:0032508) and overrepresentation of term regulation of cellular process (GO:0050794) (Table S4). We observed overrepresentation of terms related to photosynthesis and light reaction (GO:0019684), circadian rhythm (GO:0007623), lipid biosynthetic process (GO:0008610), a few metabolic processes, etc., and underrepresentation of DNA repair (GO:0006281), ribonucleoprotein complex biogenesis (GO:0022613), and telomere maintenance (GO:0000723) in genes with induced expression. In addition, overrepresentation of many terms was found in genes with repressed expression, including maturation of LSU‐rRNA from tricistronic rRNA transcript (SSU‐rRNA, 5.8S rRNA, LSU‐rRNA) (GO:0000463), cellular response to oxidative stress (GO:0034599), protein folding (GO:0006457), translation (GO:0006412), etc. Sixty‐two genes are lowly expressed or not expressed at our sampling stage with the presence of the B chromosome in contrast to the control, including a few putative transcription factors (TFs) (Table S5). However, we did not observe an overrepresentation of any terms in this group (Table S4).
Ratios of extreme values form peaks of outliers (ratio > 6) in the ratio distribution of A‐located genes (Figure 1a). Examination of the function and the expression pattern of those genes would reveal the cause of these extreme ratios. GO enrichment analysis was performed on genes with ratios over 6 that appear in all seven comparisons. We did not observe over‐ or underrepresentation of any term (Table S4). Therefore, these extreme values were not caused by defects in plant growth or development, nor induced by stress.
Meanwhile, we examined the degree of dosage sensitivity to the B copy number of these A‐located outliers. This group is heavily populated with genes whose expression is positively correlated with B copy number, with 65 out of 254 (approximately 26%) outliers being B‐dosage‐sensitive (PCC > 0.8) (Figure S5). It is likely that the expression of these genes is extremely low or approaching zero in the absence of the B chromosome. When their expression is induced by the B chromosome, computing the ratio of expression levels of the experimental genotype to the control (approximately 0) would generate an extreme value. B‐dosage‐sensitive genes compose most of the B‐located outliers, with 107 out of 122 (approximately 88%) outliers having PCCs over 0.8 (Figures S2 and S5). The extreme values are caused by the fact that B‐located genes are not present in the control group.
A‐located genes in different functional groups exhibit diverse responses to the B chromosome
Given that we observed overrepresentation or underrepresentation of various biological processes in A‐located genes that exhibit different dosage responses to the varied B copy number, the B chromosome may impact the expression of A‐located genes through different cellular processes and pathways. Therefore, we examined the expression of A‐located genes coming from several functional groups to evaluate if diverse groups of genes respond to aneuploidy of the B chromosome in different manners. Ratio distributions were plotted for each functional group, along with mean, median, and standard deviation (SD) for each distribution (Table S2), for statistical determinations of deviation of the distributions from normal (Table S2), to compare two distributions for differences with Kolmogorov–Smirnov (K–S) tests of significance (Table S6), or to compare variances across groups using Bartlett's test (Table S7). Scatter plots for the significance of DGE were performed as a complement, with the number of upregulated and downregulated A‐located DEGs summarized in Table S8. Medians of each ratio distribution (with outliers excluded) and histograms of PCCs to illustrate the B‐dosage sensitivity of each functional group were plotted (Figure 4; Figure S6). Genes of different functional groups were selected as described in a previous study (Johnson et al. 2020).
TFs distribute differently compared to all genes, indicated by K–S tests (Figure S7; P < 0.05, Table S6). Ratio distribution of TFs shows higher expression levels in contrast to all A‐located genes, with an increase of mean and median of ratios (Table S2; Figure 4). A greater fraction of genes in this group is positively correlated with B copy number than those in all genes (Figure 3a; Figure S6). By contrast, components of signal transduction genes have similar distributions and trends of expression compared to all genes (Figure 4; Figures S6 and S8). Distributions of ratios for structural components of the ribosome and subunits of the proteasome are significantly different from all genes (Figure 5; Figure S9), statistically testified by K–S and Bartlett's tests (P ≈ 0, Tables S6 and S7). Structural components of the ribosome show elevated degrees and a narrower spread of downmodulation than all genes (Figures 4 and 5). In contrast, proteasomal genes exhibited a narrower spread of normal distribution with a slight trend of downregulation compared to all genes (Figure 4; Figure S9; P > 0.05, Table S2). A larger fraction of genes in these two categories is negatively correlated with B copy number than all A‐located genes (Figure S6). Similar distributions between stress‐related genes and all A‐located genes indicate that the B chromosome does not induce any stress‐related responses in normal growth conditions (Figure S10). This result matches our observation that the B dosage series did not display any stress‐related morphological defects and that stress‐related terms are not enriched in the outliers (Figure S1 and Table S4).
Figure 5.
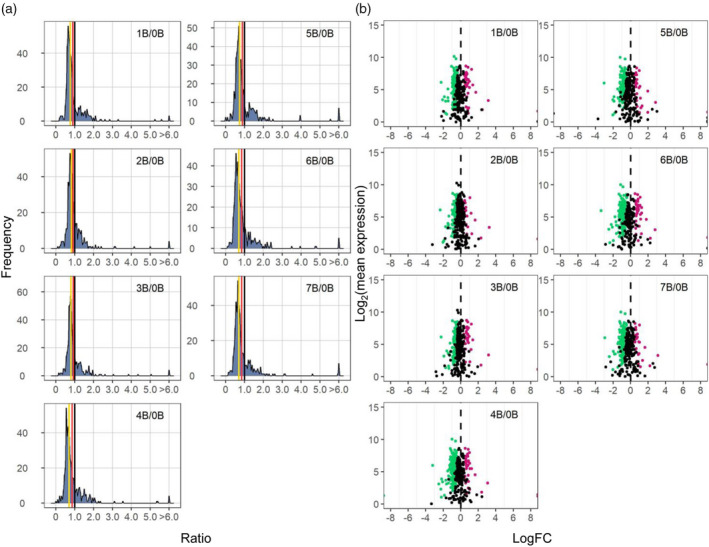
Ratio distributions and scatter plots for the functional class of structural components of the ribosome. (a) Ratio distributions of structural components of the ribosome, generated as described in Figure 1. (b) Scatter plots of structural components of the ribosome. The x‐axis represents the log2(fold change) values of each experimental genotype to the control, whereas the y‐axis denotes the mean of normalized counts of each experimental genotype and the control. Data points with q < 0.05 and a corresponding log(FC) >0 are depicted in magenta, while points with q < 0.05 and a corresponding log(FC) <0 are depicted in green. Otherwise, they are shown in black. A ratio of 0 represents no change (black vertical dashed line).
Plant cells contain two semi‐autonomous organelles, the chloroplast and the mitochondrion. The chloroplast and mitochondrion each contain a small genome that relies largely on nuclear factors for maintenance and expression (Newton et al. 2009). Distributions of nuclear genes that are subsequently transported to the chloroplast (nuclear chloroplast) or mitochondrion (nuclear mitochondrial) are different compared to all genes (Figures S11 and S12), confirmed by K–S and Bartlett's tests (P < 0.05, Tables S6 and S7). More nuclear chloroplast DEGs are upregulated with a slight increase of mean and median of ratios (Figure 4; Tables S2 and S8). The upregulation of nuclear chloroplast genes is cross‐verified by the overrepresentation of photosynthesis‐related genes in the group of genes whose expression was induced by the B chromosome (Table S4). As for nuclear mitochondrial genes, there is a trend of downregulation compared to all genes, considering a decrease of mean and median of ratios and a larger proportion of DEGs being downregulated (Figure 4; Tables S2 and S8). In addition, more genes in this group show a negative correlation with B copy number than those in all A‐located genes (Figure 3a; Figure S6). Peroxisomes are highly dynamic organelles and are involved in a wide range of plant processes (Hu et al. 2012). Peroxisomes do not contain DNA or ribosomes and therefore are devoid of organelle‐synthesized components. Distributions of peroxisomal‐targeted genes are very similar to all A‐located genes (Figure S13).
Differential expression of many miRNAs correlates with their predicted targets
miRNAs are approximately 21‐nt regulatory RNAs processed from short hairpin precursors encoded by MIR genes. Many functions as essential post‐transcriptional regulators of endogenous genes in affecting plant development and stress responses (Cui et al. 2017). Differential expression of miRNAs could contribute to changes in gene expression observed in this study. Therefore, we performed small RNA sequencing (sRNA‐seq) to evaluate this possibility. In brief, filtered counts were mapped to the reference genome using the ShortStack tool, which allows comprehensive annotation and quantification of sRNA genes (Axtell 2013; Axtell and Meyers 2018; Shahid and Axtell 2014). Read counts were normalized to reads per million mapped reads (RPM) values. Ninety‐eight miRNA genes (or clusters defined by ShortStack) were identified, all of which are located on the A chromosomes. Forty‐nine of those clusters were previously known loci annotated in miRbase version 22 (Table S9) (Kozomara et al. 2019). Log(FC) values and the FDR‐adjusted P‐values of significance in the scatter plots were generated by Empirical Analysis of Digital Gene Expression Data in R (edgeR) (McCarthy et al. 2012; Robinson et al. 2010). Ratio distributions, computation of PCC, and statistical tests were performed using the same method for analyzing mRNA‐seq. Ratio distributions of miRNAs generally display a trend of upregulation in 1–7B in contrast to 0B with mean and median both above 1.00, with more differentially expressed miRNAs (DEMs) being upregulated than downregulated (Figure 6; Figure S14; Table S2). Only 10 out of 98 clusters (approximately 10%) encoding miRNAs are sensitive to B dosage (PCC > 0.8 or < −0.8; Figure 6). Expression of six dosage‐sensitive miRNAs is positively correlated with the B copy number (PCC > 0.8) (Figure S15). Four of them belong to the miR166 family, which is involved in the regulation of shoot apical meristem development (Zhu et al. 2011). Two of the four miRNAs that show a negative correlation with B dosage are members of the miR393 family, which controls root system architecture in response to nitrate availability (Vidal et al. 2010), while one is from the miR171 family, which participates in the regulation of gibberellin signaling, auxin homeostasis, and chlorophyll biosynthesis (Figure S15) (Huang et al. 2017b; Ma et al. 2014).
Figure 6.
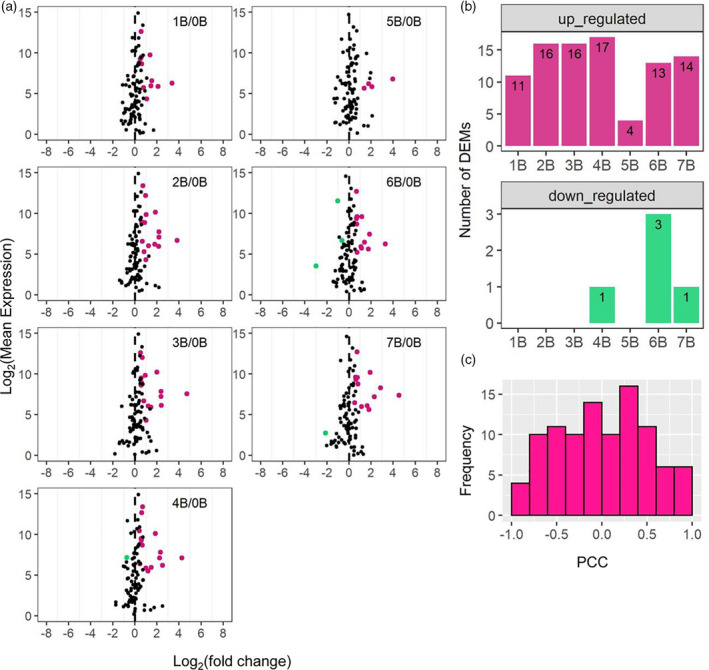
Scatter plots of A‐located miRNAs, the number of differentially expressed miRNAs (DEMs), and histograms of Pearson correlation coefficients (PCCs). (a) Scatter plots of A‐located miRNAs. Significance (false discovery rate [FDR] < 0.05) was determined by edgeR. Otherwise, plots were generated as in Figure 5b. (b) The number of A‐located DEMs determined as in (a). (c) Histogram of PCCs of A‐located miRNAs plotted as in Figure 3.
Twenty miRNAs were differentially expressed in at least two experimental genotypes compared to the control. To examine if these DEMs are associated with changes in mRNA expression, targets of DEMs were predicted by psRNAtarget (Table S10) (Dai et al. 2018). After data treatment as described in the ‘Experimental Procedures’ section, 146 predicted interactions between DEMs were characterized and their corresponding mRNA targets were identified (Table S11). The relationship between an miRNA and its mRNA target was determined by the Pearson correlation between the expression levels of the two. Twenty‐seven interactions show a strong linear relationship with P‐values of Pearson correlation of <0.05. Nineteen of these interactions display a negative correlation (Table S11). As a complement, interactions of DEMs and their targets were identified by summarizing data generated from degradome sequencing from former studies in maize (see ‘Experimental Procedures’ section). In total, 104 interactions were identified, 33 of which show significant correlation (P < 0.05; Table S12). Eight of these significant interactions were identified by both psRNATarget and degradome sequencing (Tables S11 and S12). Also, we computed the Pearson correlation between the expression levels of the miRNA (or the target) and the B copy number, which represents their degree of sensitivity to B dosage (Tables S11 and S12). For the B‐dosage‐sensitive DEMs, none of their targets with significant correlation fall in the range of being B‐dosage‐sensitive (Tables S11 and S12). Therefore, it is not likely that the differential expression of B‐dosage‐sensitive genes is regulated by miRNAs.
Even so, miRNA could impact gene expression in a B‐dosage‐insensitive manner. While examining these interactions, we found one of the predicted targets is a TF targeted by miR171 (Figure S16). It belongs to the family of GAI, RGA, and SCR (GRAS) TFs, which play critical roles in plant growth and development such as gibberellic acid (GA) signaling and phytochrome A signal transduction (Cenci and Rouard 2017; Hirsch and Oldroyd 2009; Pysh et al. 1999). In addition, a selenium‐binding protein (Zm00004b018026) is targeted by members of the miR398 family (Figure S16). Members of the miR166 family target a few genes with different functions (Figure S17). They negatively regulate the expression of the mitochondrial RNA editing factor RNA editing‐interacting protein 3/multiple organellar RNA editing factor 3 (RIP3/MORF3) (Bentolila et al. 2013; Takenaka et al. 2012). Furthermore, miR167c is involved in the regulation of several genes, many of which are auxin response factors (Figure S18). In sum, the expression of many DEMs correlates with the expression of their mRNA targets possibly involved in transcription, organellar RNA processing, auxin response, etc. Differential expression of some genes in the B dosage series is likely to be impacted by miRNAs in a B‐dosage‐insensitive manner.
The B chromosome triggers the differential expression of transposable elements
The maize genome is largely composed of TEs. The highly heterochromatic B chromosome contains numerous repetitive sequences such as retrotransposons (Blavet et al. 2021). Therefore, an increase of the B copy number may result in changes in the suppressive state of TEs on both A and B chromosomes. To address this hypothesis, we examined the expression levels of TEs in the B dosage series. TE exemplar sequences were generated from TEs that were identified by a structural annotation of transposons in the W22v2 genome and the B chromosome sequence (Blavet et al. 2021; Springer et al. 2018). Reads from mRNA‐seq were mapped to TE exemplar sequences that only contain non‐overlapping TEs. Ratio distributions of A‐located TEs mapped to the TE exemplar sequences did not show a trend of up‐ or downregulation (Figure S19a), with medians of ratios approximating 1.0 (Table S2; Figure S20). However, differential expression analysis of TEs showed that there are more upregulated differentially expressed TEs (DETs) than downregulated DETs (Figure S19c; Table S13). The majority of A‐located TEs did not show a high degree of B‐dosage sensitivity when assayed globally, as mean and medians of ratios did not change much from 1B/0B to 7B/0B (Table S2; Figure S20). When analyzed on a per TE basis, 7% of A‐located expressed TEs and 21% of DETs are B‐dosage‐sensitive (Figure S21a). In addition, there is an increase of upregulated DETs from 1B/0B to 7B/0B while there is little variation between numbers of downregulated DETs from 1B/0B to 7B/0B (Table S13). In sum, only a small fraction of A‐located TEs is B‐dosage‐sensitive.
Ratio distributions of B‐located TEs did not change with an increase in the B dosage (Figure S19b). However, there is an increase of upregulated DETs and a decrease of downregulated DETs from 1B/0B to 7B/0B (Figure S19d; Table S13). Similar to the expression patterns of B‐located genes, the expression of many B‐located TEs is not eliminated in the control group (0B). Therefore, the expression level of B‐located TEs in the 0B group was deducted from those of all experimental groups (1–7B), and TEs with negative values were excluded for further analysis when examining the expression pattern of individual B‐located TEs. By applying this procedure, the proportion of B‐dosage‐sensitive B‐located TEs increases from 36% to 69%, and that of B‐dosage‐sensitive B‐located DETs increases from 80% to 82% (Figure S21b).
TEs are classified into two major classes distinguished by their different modes of transposition. Class I TEs are retrotransposons that use reverse transcriptase for transposition via an RNA intermediate, whereas Class II TEs are DNA transposons that transpose directly from DNA to DNA (Kidwell and Lisch 1997). For both A‐located and B‐located TEs, Class I and Class II TEs have distinct ratio distributions, confirmed by K–S tests (P < 0.05, Table S6; Figures S22 and S23). Compared to those of Class II TEs, distributions of Class I TEs contain a second peak around ratio 0.50, suggesting more TEs being downregulated in this class compared with the control (Figures S22 and S23). This observation is cross‐validated by clusters of green data points in the scatter plots of Class I TEs but not in those of Class II TEs (Figures S22 and S23). Furthermore, the proportion of upregulated Class II DETs out of all Class II DETs is much greater than the proportion of upregulated Class I DETs out of all Class I DETs (Table S13). All the above observations apply to both A‐located and B‐located TEs. Although A‐located and B‐located TEs from the same class display similar features, they show different degrees of B‐dosage sensitivity. The overall trend of the expression of A‐located Class I and Class II TEs is not correlated with B copy number, with the mean and median of each distribution being relatively constant with the increase of B copy number (Figures S22 and S23; Table S2). By contrast, ratio distributions of B‐located Class II TEs show an overall trend of upregulation in response to increased B copy number, with medians of ratios increasing from 0.87 (1B/0B) to 1.12 (7B/0B); whereas those of B‐located Class I TEs show an overall trend of downregulation, with medians decreasing from 1.32 (1B/0B) to 1.12 (7B/0B) (Figures S20, S22, and S23; Table S2). Thus, B‐located TEs exhibit a greater degree of B‐dosage sensitivity compared with A‐located TEs.
In sum, all the above observations indicate an impact of the B chromosome on the expression of A‐located and B‐located TEs. Although ratio distributions of A‐located and B‐located TEs did not show much variation in the experimental groups compared with the control, results from DGE analysis revealed a slight upregulation of detectable TE expression. Approximately 6% of A‐located TEs and 29% of B‐located TEs from the exemplar sequence are significantly upregulated, whereas 3% of A‐located and 3% of B‐located TEs are significantly downregulated. The effect of B copy number on the expression of A‐located TEs is not as significant as that on B‐located TEs, with more B‐located TEs being sensitive to changes in B copy number. Class I and Class II TEs on both A and B chromosomes show distinct expression patterns under the impact of the B chromosome.
DISCUSSION
Although the maize B chromosome was discovered about a century ago (Kuwada 1925; Longley 1927; Randolph 1928), it has been thought to be an essentially inert chromosome. The maize B chromosome has recently been shown to affect gene expression of the A chromosomes (Huang et al. 2016). However, little was known about how it impacts the A chromosomes transcriptionally and/or post‐transcriptionally. In this study, we performed mRNA‐seq and sRNA‐seq on samples from maize plants with 0 to 7 B chromosomes to assay the expression of A‐located and B‐located genes, miRNAs, and TEs in response to changes in B copy number.
Comparison with another study related to B chromosome dosage
In a previous study performed by Huang et al. (2016), 130 A‐located DEGs were detected in comparison between B73 lines with and without the maize B chromosome. Here, we were able to identify more genes under the influence of the B chromosome, with 7962 A‐located DEGs (38% of all expressed A‐located genes) in at least two experimental conditions and 3579 A‐located DEGs (17% of all expressed A‐located genes) in all experimental conditions with Bs compared to the control. Thus, we further confirmed that the maize B chromosome is not inert, and it impacts the expression of the A chromosomes. The difference between the number of DEGs identified in Huang et al. and this study could also be caused by the following reasons. (i) Fourteen‐day leaf tissue from B73 inbred with 0, 1, and 6 Bs was used in Huang et al., whereas 45‐day leaf tissue from W22 inbred with 0–7 Bs was used in this study. (ii) Genes with log(FC) ≥ 3 and FDR < 0.05 computed by edgeR were counted as DEGs in Huang et al., while genes with q < 0.05 computed by Cuffdiff were counted as DEGs in this study. Results from Huang et al. suggested that the maize B influences the A chromosome transcription with a stronger effect when comparing B73 with 6 Bs to B73 with 1 B. The analysis here indicates that a stronger effect only occurs for a small fraction of genes. While analysis of a few B‐located genes and long terminal repeats in Huang et al. is performed through de novo assembly of B‐specific reads unmapped to the maize B73 reference genome together with PCR validation, the recently published B chromosome sequence allowed a more extensive analysis. Our results reveal that 53 B‐located genes (19% of all expressed B‐located genes) are differentially expressed in all seven experimental genotypes when Bs are present compared to those when the B is absent.
Various responses of A‐ and B‐located genes to changes in B dosage
Responses of A‐located genes to varied B dosage are different from B‐located genes assayed both globally and on a per gene basis. While examining the expression of A‐located genes globally, distributions of these genes did not show a significant variation with the increase of B copy number in the experimental group, nor was there an increase in the number of A‐located DEGs being observed from 1B to 7B. In contrast to the A‐located genes, a trend of increased expression of B‐located genes is observed in the ratio distributions of experimental groups with 1–7 Bs compared with the control, correlated with the increase of B copy number. An increase in the number of upregulated B‐located DEGs is also observed. The difference in the trend of global expression of A‐located and B‐located genes is cross‐verified by the PCA plot and Spearman pairwise correlation matrix. While investigating the impact of B dosage on gene expression on a per gene basis, increasing the number of Bs only has a cumulative impact on 9% of expressed A‐located genes. Thus, the expression of most A‐located genes is not sensitive to changes in B copy number. Only 0.5% of B‐dosage‐sensitive A‐located genes have B‐located homologs, as determined by a previous study (Blavet et al. 2021). Therefore, the fact that these A‐located genes are sensitive to B dosage is more likely to be caused by direct regulation from the B chromosome, rather than a result of mismapped reads due to sequence similarity between A and B chromosomes. By contrast, 73% of expressed B‐located genes are B‐dosage‐sensitive. In other words, most B‐located genes show a proportional dosage effect with the varied B dosage. In sum, varied B dosage affects the expression of the A chromosomes in a B‐dependent but B‐dosage‐insensitive manner. The main differential gene expression is caused by the presence or the absence of the B chromosome. By contrast, changing the dosage of the B chromosome influences expression of the B‐located genes in a B‐dosage‐sensitive manner.
Genes located on the A chromosomes respond to changes in B dosage differently. Various GO terms are over‐ or under‐represented in groups of genes with disparate responses to B dosage, indicating A‐located genes with diverse dosage response to change of B copy number may participate in different biological processes and pathways. In addition, A‐located genes from functional classes of TFs, genes encoding structural components of the ribosome and proteasome, and nuclear chloroplast and mitochondrial genes are distributed differently compared with all A‐located genes. Components of the ribosome exhibit the most significantly different distribution in comparison to all A‐located genes, with a narrow range spread of downregulation. When examining expression patterns of individual genes of different functional classes, expression of a larger fraction of ribosomal, proteasomal, and nuclear mitochondrial genes, respectively, shows a negative correlation with an increase of B copy number, while that of TFs exhibits a positive correlation. Therefore, A‐located genes from different functional classes display disparate responses to changes in B dosage.
The apparent expression of many B‐located genes in the 0B group is not zero, likely due to the high sequence similarity between the genes on the B chromosomes and their A paralogs, as the gene content of the B chromosome appears to result from continuous transfer from the A chromosomes (Blavet et al. 2021). To test this hypothesis, two types of reads from the four biological replicates of the 0B group were gathered: (i) all reads uniquely mapped to the B chromosome and (ii) reads uniquely mapped to 125 B‐located genes that are expressed (averaged expression level > 0.5) in the condition of 0B. Eight pools of reads were then mapped to the A chromosomes as the reference genome. Approximately 68% of all reads uniquely mapped to the B chromosome could be remapped to the A chromosomes, whereas 96% of reads uniquely mapped to B‐located genes that are expressed in the absence of the B chromosome are remapped to the A chromosomes on average (Table S14). A Student's t‐test demonstrates a significant difference between the mapping rate of the two groups (P ≈ 0). Thus, the unexpected counts of B‐located genes in the condition of 0B are caused by mismapped reads due to sequence similarity, which could not be sorted by the current mapping tool and/or sequencing technique. Further, reads uniquely mapped to the B chromosome only compose 0.2–1.7% of uniquely mapped reads, with a strong positive correlation between B copy number and the percentage of the B chromosome mapped reads (P ≈ 0; R = 0.95). The low contribution of the B chromosome mapped reads indicates the sparse gene density and degeneration of most genes on the B chromosome.
Haploid and diploid maize with a stable B–A translocation was used to study the effect of aneuploidy on global gene expression (Shi et al. 2021; Yang et al. 2021). Plants in the experimental group of these studies have part or the equivalent of a whole B chromosome or more. Results of ratio distributions and DGE analysis indicate that global gene modulation observed in A chromosome disomies, trisomies, and tetrasomies are trivially impacted by the B chromosome portion (Shi et al. 2021; Yang et al. 2021). Therefore, compared to observed global gene modulation in two genomic imbalance studies, the 141‐Mb B chromosome has limited influence on the expression of A chromosomes. For example, the varied copy number of a region in the long arm of chromosome 1 (TB‐1La; the size of region 145.7 Mb) in haploid or diploid conditions resulted in much greater changes in global gene expression compared to haploids or diploids with Bs, respectively (Shi et al. 2021; Yang et al. 2021). This observation suggests that dosage‐sensitive regulators that transpose to the B chromosome are mainly selected against and basically only genes that help maintain the B would be retained.
The function of miRNAs in gene regulation under the impact of the B chromosome
Ninety‐eight miRNA genes were detected in this study, all of which are located on the A chromosomes. Huang et al. reported the identification of three novel B‐derived miRNAs using MiRDeep2 in a previous study (Huang et al. 2020). Two of these sequences were identified to be mapped to the B chromosome in this study; however, they were not characterized as miRNAs by the criteria in ShortStack. About 10% of A‐located miRNAs display sensitivity to changes in B dosage. Although miRNAs in the experimental groups are upregulated compared with the control, ratios of miRNAs in all experimental genotypes show a similar degree of upregulation. Furthermore, the number of DEMs did not vary much from the 1B/0B comparison to the 7B/0B comparison. All these observations indicate that most miRNAs are regulated in a B‐dependent, but B‐dosage‐insensitive manner assayed both globally and on a per gene basis.
Expression levels of DEMs and many of their targets predicted by psRNATarget or collected from degradome sequencing data show a significant correlation. The association between these components is supported by other studies. For instance, the interaction between miR171 and GRAS genes has been characterized in a few plant species, including Arabidopsis, maize, tomato (Solanum lycopersicum), and Larix kaempferi (Ge et al. 2016; Huang et al. 2017b; Li et al. 2014; Ma et al. 2014; Wang et al. 2010). In Arabidopsis, miR171 acts to negatively regulate GRAS (or Scarecrow‐Like) genes in mediating gibberellin‐regulated chlorophyll biosynthesis under light conditions (Ma et al. 2014; Wang et al. 2010). In addition, one of the targets of miR166, Zm00004b039240, encodes an ethylene receptor potentially involved in the ethylene‐activated signaling pathway. The miR166 family has been found to be negatively regulated by exogenous ethylene treatment (Pei et al. 2013; Zhao et al. 2017). Furthermore, four targets associated with miR167c are auxin response factors. miR167 is reported to regulate the expression of auxin response factors in various species including Arabidopsis, rice (Oryza sativa), and soybean (Glycine max) (Liu et al. 2012; Pashkovskiy et al. 2016; Wang et al. 2015).
Expression patterns of miRNAs and their targets suggest that miRNAs are likely to affect mRNA target expression in a B‐dosage‐insensitive manner. If the mRNA target functions as a major player in gene regulation, it may lead to a gene regulatory cascade. For instance, GRAS33, whose expression is repressed by the B chromosome putatively due to the rising level of miR171, co‐expresses with 26 genes in a gene co‐expression network using maize mRNA‐seq data (Huang et al. 2017a). Of these genes co‐expressed with GRAS33, 54% are differentially expressed in the B dosage series. Expression levels of six of them are significantly correlated with that of GRAS33 (Pearson correlation, P < 0.05), four showing a positive correlation and two showing a negative correlation (Table S15). Most of these genes co‐expressing with GRAS33 fall in the range of being B‐dosage‐insensitive. Two of them also encode TFs (Zm00004b002945, Homeobox‐transcription factor 84; Zm00004b021538, G2‐like‐transcription factor 49). In sum, miRNAs likely play a role in regulating changes in gene expression caused by the B chromosome.
The impact of the B chromosome on transcriptome size
It is possible that the change in expression caused by the B chromosome is a result of the varied transcriptome size. However, the B dosage does not have a cumulative effect on the transcriptome size of the B dosage series based on the following observations. (i) Only a small fraction of A‐located genes and TEs are B‐dosage‐sensitive. (ii) Although most B‐located genes and TEs are B‐dosage‐sensitive, reads mapped to B‐located genes only compose approximately 0.25% of reads uniquely mapped to A and B chromosomes on average, while reads mapped to B‐located TEs cover approximately 7% of reads uniquely mapped to all TE exemplar sequences. (iii) It is generally accepted that there is a linear relationship between cell size, transcriptome size, and ploidy in Arabidopsis and maize (Miller et al. 2012; Robinson et al. 2018; Shi et al. 2021; Tsukaya 2013). Another study reported that a cell cycle regulator, KIP‐related protein 4, uses DNA content as an internal scale to regulate cell size in the Arabidopsis shoot stem cell niche (D'Ario et al. 2021). That the epidermal cell size is unchanged when comparing 5B to 0B suggests the transcriptome size of mRNA does not increase with more Bs. Therefore, a lack of changes in global gene and TE expression and in cell size correlated with B copy number indicates a lack of correlation between transcriptome size and B copy number.
The B chromosome causes a slight upregulation of global gene expression, and this effect is dependent on the presence of the B chromosome regardless of the B copy number. Therefore, it is possible that the presence of the B chromosome would lead to varied transcriptome size, but the variation is not correlated with the B dosage. However, this hypothesis is rejected based on the following evidence. First, cell size is not significantly different in 0B compared with 5B. Second, the total RNA was spiked with the External RNA Controls Consortium (ERCC) before rRNA removal and library preparation. If the transcriptome size of mRNAs/TEs changes because of increasing B copy number, the same amount of total RNA would contain the same amount of ERCC but a varied amount of mRNA/TE transcripts. Thus, we would expect variations in ratios of reads uniquely mapped to the reference genome (genome for mRNA‐seq or TE exemplar sequences, respectively) to reads uniquely mapped to ERCC sequences. Our result demonstrates that ratios regarding mRNAs and TEs are not statistically different, respectively, in the experimental genotypes compared with the control (Student's t‐test, P > 0.05) (Figure S24). In addition, considering that the expression of genes and TEs is not regulated towards the opposite direction, it is unlikely that the effect on the mRNA transcriptome size is canceled by the impact on the TE transcriptome size. In sum, the size of the transcriptome of mRNA or TE remains constant in the B dosage series and is unlikely to be the cause of changes in global gene expression.
Concluding remarks
Compared to the aneuploidy of the A chromosomes, the varied copy number of the B chromosome is clearly less detrimental (Hou et al. 2018; Shi et al. 2021; Yang et al. 2021). The highly heterochromatic B chromosome contains a limited number of actively transcribed genes. However, our results demonstrated that the B chromosome is not totally inert, as the analysis of mRNA‐seq data reveals that 273 out of 758 predicted B‐located genes are actively transcribed in leaf tissue. Other B‐encoded genes may be expressed in other tissues. In addition, the B chromosome causes differential expression of genes, miRNAs, and TEs (summarized in Figure 7). Varied B dosage mainly causes an overall B‐dosage‐insensitive regulation of A‐located genes, miRNAs, and TEs, with a small fraction being B‐dosage‐sensitive, while changes in B copy number lead to a predominant B‐dosage‐sensitive modulation of B‐located genes and TEs. B‐located genes are likely to be involved in the regulation of A chromosome expression. Furthermore, A‐located miRNAs participate in the regulation of A chromosome gene expression under the impact of the B chromosome and could potentially modulate its targets in a cascade of effects. In summary, the presence of the B chromosome results in changes in gene expression both transcriptionally and post‐transcriptionally. An increase of B copy number causes both cumulative effects and non‐cumulative effects on the expression of genes, miRNAs, and TEs.
Figure 7.
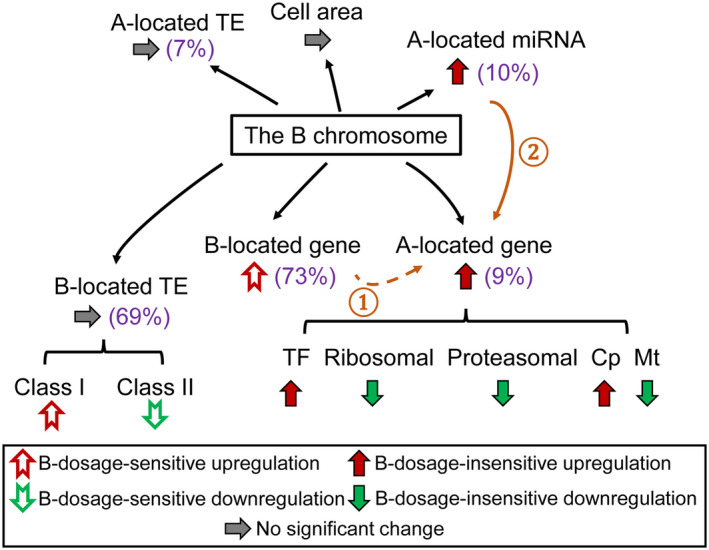
Summary of the impact of the B chromosome. Values in brackets represent the percentage of B‐dosage‐sensitive genes, miRNAs, and TEs, respectively. Varied B dosage leads to an overall B‐dosage‐insensitive upregulation of A‐located genes and miRNAs, with genes from different functional classes responding differently. Ascending B copy number causes a trend of B‐dosage‐sensitive upregulation of B‐located genes. Although no global change is observed for A‐ and B‐located TEs, B‐located TEs from different classes show trends of B‐dosage‐sensitive effects. Cell area does not vary significantly with an increase of B dosage. ①: B‐dosage‐sensitive A‐located genes are likely to be regulated by B‐dosage‐sensitive B‐located genes. ②: A‐located miRNAs modulate the expression of A‐located targets in a B‐dosage‐insensitive manner.
EXPERIMENTAL PROCEDURES
Plant material and morphological analysis
To screen for maize plants with B chromosomes, we performed FISH on a collection of segregating maize W22 seeds with different numbers of B chromosomes (Kato et al. 2004; Kato et al. 2011). A probe targeting telomere sequences (TTTAGGG)(n) homologous to portions of the B‐specific element ZmBs was used in the hybridization to visualize the B chromosome (Lamb et al. 2005). Maize plants with 1–7 copies of Bs were saved and grown in the Sears greenhouse at the University of Missouri‐Columbia under the following conditions: 16 h light, 25°C day/20°C night. W22 wild‐type plants with no B were used as the control for the experiment. Experimental groups with 5, 6, and 7 Bs contain 2, 3, and 3 biological replicates, respectively. All other experimental groups and the control group each contain 4 biological replicates (Table S1). The central 12‐inch (30.48‐cm) portion of leaf tissue without the mid‐rib was collected from the fifth and sixth leaves from the bottom of the plant 45 days after germination. The tissue was frozen in liquid nitrogen and then stored at −80°C for further analysis.
Forty‐five‐day‐old plants were used to obtain the epidermal cell imprints for cell area measurement as described previously (Ferris et al. 2002; Ferris and Taylor 1994; Shi et al. 2021; Yao et al. 2011). Imprints were taken from the abaxial surface of the second leaf from the bottom of each selected leaf at similar positions and were examined by a brightfield microscope (Olympus). For each plant, four imprints were taken. We measured the area of 25 epidermal cells from each imprint using Fiji (Schindelin et al. 2012). We calculated the mean value for the area of 100 cells for each plant and performed Student's t‐tests of means for statistical significance.
Nucleic acid isolation and RNA‐seq library construction
Total RNA and sRNA were co‐extracted from maize leaves using the mirVana™ miRNA Isolation Kit (Thermo Fisher Scientific). For mRNA‐seq, total RNA was treated and libraries were prepared as described in previous studies (Shi et al. 2021; Yang et al. 2021). Libraries were sequenced on the NextSeq500 platform using 75 bp single‐end sequencing. sRNA libraries were generated directly from total RNA with the TruSeq Small RNA Library Preparation Kit (Illumina) and were sequenced on the NextSeq500 platform. High‐throughput sequencing was performed at the University of Missouri DNA Core Facility.
mRNA‐seq data processing
mRNA‐seq data were processed as described in a previous study (Shi et al. 2021). In short, adaptors were trimmed, and low‐quality reads were removed. Subsequently, reads mapped to ERCC and maize chloroplast and mitochondrial genomes were excluded for mapping. The remaining reads were mapped to the maize reference genome W22v2, the maize chloroplast and mitochondrial genomes, and the B chromosome sequence using TopHat2 with default parameters (Blavet et al. 2021; Bosacchi et al. 2015; Clifton et al. 2004; Kim et al. 2013; Springer et al. 2018). In total, 11–19 million uniquely mapped non‐organellar read counts were obtained for each plant (Table S1). Normalized read counts were generated by Cuffdiff and lowly expressed genes (≤3 out of 28 samples with normalized count ≥ 1) were excluded (Shi et al. 2021; Trapnell et al. 2013). PCA plots were generated in R using normalized read counts.
To examine the expression of TEs, we generated TE exemplar sequences based on TEs identified and annotated in maize genome W22v2 and the maize B chromosome reference sequence (Blavet et al. 2021; Springer et al. 2018). In total, 57 955 A‐located and 2292 B‐located TEs that are most likely to be intact and do not overlap with other TEs were selected according to the GFF3 file, converted to a FASTA format file, and used as the reference for alignment. Adaptor‐trimmed and quality‐filtered mRNA reads were mapped to the organellar genome as described above and then mapped to the reference using bowtie2 (Langmead and Salzberg 2012). Uniquely mapped reads were selected for further analysis. RPKM normalization was performed and lowly expressed TEs (≤3 out of 28 samples with RPKM ≥ 1) were removed.
sRNA‐seq data processing
sRNA reads were trimmed for adaptors and filtered for a low‐quality score as described above. Reads matching known structural RNAs (rRNAs, tRNAs, sn‐RNAs, and sno‐RNAs) from Rfam 14.0 (Daub et al. 2015) and reads with poly(A) tails were removed before further analysis. Reads between 18 and 24 nt in length were aligned to the maize reference genome W22v2, the maize chloroplast and mitochondrial genomes, and the B chromosome sequence (Blavet et al. 2021; Bosacchi et al. 2015; Clifton et al. 2004; Springer et al. 2018) using ShortStack version 3.8.3 (−mincov 20) (Axtell 2013; Axtell and Meyers 2018; Shahid and Axtell 2014). ShortStack identifies islands with significant alignment coverage of sRNA reads, reports the predominant sRNA size of each cluster as Dicercall, analyzes putative RNA hairpins, and annotates MIRNA loci (Axtell 2013; Shahid and Axtell 2014). ShortStack did not detect any B‐located miRNA in the B dosage series. MIRNA loci annotated by ShortStack were further confirmed by aligning the sequence of each cluster against miRNAs reported in miRBase 22 (Kozomara et al. 2019) and the transcript sequences in the RefSeq database at NCBI (O'Leary et al. 2016) restricted to organism Z. mays with BLASTN (Altschul et al. 1990). In parallel, miRNAs were annotated by their locations on the A chromosomes using W22v2 genome annotation as a reference (Table S9). Then, RPM normalization was performed and lowly expressed miRNAs (≤3 out of 28 samples with RPM ≥ 1) were filtered out.
Statistical analysis
All statistical tests were performed with extreme values (ratio > 6 or < 1/6) excluded. The mean, median, and SD of each comparison were computed (Table S2). P‐values of normality check were generated by Lilliefors (K–S) tests (Table S2). P‐values used for similarity determinations between two ratio distributions were calculated by the K–S test (Table S6). Bartlett's test was performed to test if variances are equal across different groups (Table S7).
Ratio distribution and scatter plots
Ratio distribution plots were generated as described in previous studies (Hou et al. 2018; Shi et al. 2020; Shi et al. 2021; Yang et al. 2021). In brief, means of normalized counts of biological replicates of each experimental genotype (1–7 Bs, respectively) and the control group (0B) were computed. The ratio was generated by dividing the mean of treatment counts by the mean of control counts and was plotted into a histogram. Mean and median of ratios with outliers excluded (ratio > 6 or < 1/6) were computed and demarcated with labeled vertical lines in red and yellow, respectively.
We performed DGE analysis with Cuffdiff and differential miRNA/TE expression analysis with edgeR to test for the significance of fold change values between each treatment group to the control group (McCarthy et al. 2012; Robinson et al. 2010; Trapnell et al. 2012). The FDR‐adjusted P‐value or q value and log(FC) were used for scatter plots. The log(FC) value between treatment and control was plotted on the x‐axis, while the sum of normalized counts of the treatment and control group was plotted on the y‐axis. Data points with FDR (q value) < 0.05 and a corresponding log(FC) > 0 were depicted in magenta, while points with FDR (q value) < 0.05 and a corresponding log(FC) < 0 were depicted in green. Otherwise, they were designated in black.
Gene Ontology enrichment analysis
All gene model names from the W22 genome were translated to B73 (AGPv4) using the Translate Gene Model IDs tool from MaizeGDB for GO enrichment analysis (Andorf et al. 2016). Analysis was performed using the PANTHER Overrepresentation Test tool via Fisher's exact test with Bonferroni‐corrected P‐value (Thomas et al. 2003). Categories with adjusted P < 0.05 were defined as statistically significant and are displayed in Table S4. Criteria for categorizing genes into groups are described below.
We computed the PCCs between the expression level of each gene to the B copy number in the corresponding genotypes (0 to 7). Genes positively correlated with B copy number (PCC ≥ 0.8), negatively correlated with B copy number (PCC ≤ −0.8), and unchanged regardless of B copy number (−0.2 ≤ PCC ≤ 0.2) were determined by values of their PCC. Genes whose expression is induced (or repressed) by B copy number are based on the following criteria: genes are significantly differentially expressed with FDR < 0.05 and log(FC) > 0 (log(FC) < 0 for repressed genes) in all experimental genotypes (1B to 7B); genes in 0B have the minimum (maximum for repressed genes) number of read counts in comparing to experimental conditions (1–7 Bs); genes with PCC between 0.2 and 0.8 (−0.8 to −0.2 for repressed genes). Silenced genes are a subgroup of the repressed genes with an additional condition that the gene ratio of the experiment to control is less than 1/6. Outliers are genes with gene ratio > 6 in all experimental conditions compared to the control.
microRNA target prediction
Differential gene expression analysis was performed to clusters defined as MIRNA loci by edgeR as described above. DEMs with significance (FDR < 0.05) in only one experimental condition (e.g., only in the 1B/0B comparison but not the others) are more likely to be caused by experimental variation. Therefore, clusters significantly differentially expressed in at least two experimental conditions compared to the control were selected for further analysis. The ‘major RNA’ sequence generated by ShortStack is hypothesized to be the sequence of mature miRNA for each cluster. Sequences of these major RNAs were extracted and used for target prediction. The prediction was done using psRNATarget with an expected value of 3 or less (Table S10) (Dai et al. 2018). psRNATarget could predict if an miRNA functions through target cleavage or translation repression. Considering that we can only detect changes in the transcription level, targets with type ‘translation repression’ were filtered out. Degradome sequencing data were collected from various studies regarding miRNA regulation (Fu et al. 2017; Gong et al. 2015; Li et al. 2017; Liu et al. 2014; Wu et al. 2016; Zhang et al. 2019; Zhao et al. 2013; Zhao et al. 2016; Zhou et al. 2016). Afterward, we translated gene model names of these targets from B73 to W22. Targets with significant differential expression in at least two experimental conditions compared to the control were retained. To examine the correlation between the expression of an miRNA and its targets in each experimental and control condition (0B to 7B), normalized read counts of miRNAs and their targets were averaged across biological replicates and were then used for the computation of PCCs (Tables S11 and S12).
Quantitative PCR
One microgram of total RNA was spiked in 2 μl 1:100 diluted ERCC RNA Spike‐In Mix (Thermo Fisher Scientific). cDNA was synthesized using the SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) with gene‐specific primers. Quantitative real‐time PCR (qRT‐PCR) was performed with a StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific) and the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). Two nanograms of cDNA was used as the template for qRT‐PCR. Three technical replicates were carried out for each sample. RNA accumulation levels of selected genes were measured in relation to levels of ERCC‐00111 as a control. Primers are listed in Table S16.
CONFLICT OF INTEREST
None of the authors have a competing interest.
Supporting information
Figure S1. The phenotype of experimental genotypes (1B‐7B) and the control (0B) and the cell size comparison.
Figure S2. Ratio distributions of expression of B‐located genes in each experimental genotype compared with the control with outliers (ratio > 6 or < 1/6) included.
Figure S3. Scatter plots of DGE in each experimental genotype compared with the control.
Figure S4. Gene ratios measured by qPCR compared with those computed by RNA‐seq.
Figure S5. Gene expression level of outliers and PCC with B copy number (ratio > 6 or < 1/6).
Figure S6. Histogram of PCC between the expression level of A‐located genes from each functional class and B copy number.
Figure S7. Ratio distributions and scatter plots of DGE for the functional class of TFs.
Figure S8. Ratio distributions and scatter plots of DGE for the functional class of signal transduction.
Figure S9. Ratio distributions and scatter plots of DGE for the functional class of structural proteins of the proteasome.
Figure S10. Ratio distributions and scatter plots of DGE for the functional class of stress‐related genes.
Figure S11. Ratio distributions and scatter plots of DGE for the functional class of nuclear chloroplast genes.
Figure S12. Ratio distributions and scatter plots of DGE for the functional class of nuclear mitochondrial genes.
Figure S13. Ratio distributions and scatter plots of DGE for the functional class of peroxisomal‐targeted genes.
Figure S14. Ratio distributions of A‐located miRNAs and B copy numbers.
Figure S15. Expression levels of miRNAs that are sensitive to B dosage.
Figure S16. Expression levels of miRNAs and their targets in the B dosage series.
Figure S17. Expression levels of the family members of zma‐MIR166 and their targets in the B dosage series, indicating different members of the same family have the same targets.
Figure S18. Expression levels of zma‐MIR167c and its targets in the B dosage series.
Figure S19. Ratio distributions and scatter plots of differential expression for all TEs.
Figure S20. Median of ratios of A‐located and B‐located TEs in different superfamilies.
Figure S21. Histogram of PCC between the expression level of each TE and B copy number representing their B‐dosage sensitivity.
Figure S22. Ratio distributions and scatter plots of differential expression for Class I TEs.
Figure S23. Ratio distributions and scatter plots of differential expression for Class II TEs.
Figure S24. Ratios of reads uniquely mapped to the reference genome (a) and the TE exemplar (b) to those uniquely mapped to ERCC sequences.
Table S1. Read mapping statistics and group information of mRNA‐seq and sRNA‐seq experiments.
Table S2. The mean, median, SD, and normality tests for each distribution listed in the order of mention in the text.
Table S3. Expression levels and information of predicted B‐located genes.
Table S4. GO enrichment analysis on genes from different subgroups.
Table S5. Normalized counts and ratios of genes silenced in the B dosage series.
Table S6. Distribution comparisons with K–S tests of significance listed in the order of mention in the text.
Table S7. Comparisons of distribution variances with Bartlett's test listed in the order of mention in the text.
Table S8. Number of DEGs for each functional class.
Table S9. Clusters of MIRNA loci identified by ShortStack and their annotations.
Table S10. Target prediction of DEMs in at least two experimental conditions compared to the control.
Table S11. Correlation between the expression levels of DEMs and their targets predicted by psRNATarget.
Table S12. Correlation between the expression levels of DEMs and their targets found in degradome sequencing data.
Table S13. Number of A‐located (a) and B‐located (b) DETs in each TE superfamily.
Table S14. Number and percentage of reads of B‐located genes mapped to the A chromosome.
Table S15. Correlation between the expression level of GRAS33 and the genes that co‐express with it. ‘R’ refers to the Pearson correlation between a gene and the corresponding B copy number, representing its degree of B‐dosage sensitivity computed as described in Figures 3a and 6c.
Table S16. Primer sequences used in qPCR.
ACKNOWLEDGMENTS
This research was supported by National Science Foundation grant IOS‐1545780.
DATA AVAILABILITY STATEMENT
All sequencing data were deposited at the Gene Expression Omnibus (GEO) repository under accession number GSE127500 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127500). The following secure token has been created to allow review of record GSE127500 while it remains in private status: alcjoomqbfktzwp.
REFERENCES
- Alfenito, M.R. & Birchler, J.A. (1993) Molecular characterization of a maize B chromosome centric sequence. Genetics, 135, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. & Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andorf, C.M. , Cannon, E.K. , Portwood, J.L. , Gardiner, J.M. , Harper, L.C. , Schaeffer, M.L. et al. (2016) MaizeGDB update: new tools, data and interface for the maize model organism database. Nucleic Acids Research, 44, D1195–D1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell, M.J. (2013) ShortStack: comprehensive annotation and quantification of small RNA genes. RNA, 19, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell, M.J. & Meyers, B.C. (2018) Revisiting criteria for plant microRNA annotation in the era of big data. The Plant Cell, 30, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett, J.B. (1991) Cytogenetic, genetic and plant breeding applications of B–A translocations in maize. In: Gupta, P.K. , Tsuchiya, T. (Eds.), Developments in plant genetics and breeding. Amsterdam: Elsevier, pp. 493–529. [Google Scholar]
- Bentolila, S. , Oh, J. , Hanson, M.R. & Bukowski, R. (2013) Comprehensive high‐resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genetics, 9, e1003584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beukeboom, L.W. (1994) Bewildering Bs: an impression of the 1st B‐chromosome conference. Heredity, 73, 328–336. [Google Scholar]
- Birchler, J.A. , Bhadra, U. , Bhadra, M.P. & Auger, D.L. (2001) Dosage‐dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Developmental Biology, 234, 275–288. [DOI] [PubMed] [Google Scholar]
- Birchler, J.A. & Veitia, R.A. (2012) Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proceedings of the National Academy of Sciences of the United States of America, 109, 14746–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeslee, A.F. (1934) New Jimson weeds from old chromosomes. The Journal of Heredity, 25, 81–108. [Google Scholar]
- Blakeslee, A.F. , Belling, J. & Farnham, M.E. (1920) Chromosomal duplication and Mendelian phenomena in Datura mutants. Science, 52, 388–390. [DOI] [PubMed] [Google Scholar]
- Blavet, N. , Yang, H. , Su, H. , Solanský, P. , Douglas, R.N. , Karafiátová, M. et al. (2021) Sequence of the supernumerary B chromosome of maize provides insight into its drive mechanism and evolution. Proceedings of the National Academy of Sciences of the United States of America, 118, e2104254118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosacchi, M. , Gurdon, C. & Maliga, P. (2015) Plastid genotyping reveals the uniformity of cytoplasmic male sterile‐T maize cytoplasms. Plant Physiology, 169, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, J.P.M. , Sharbel, T.F. & Beukeboom, L.W. (2000) B‐chromosome evolution. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 355, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, W.R. (1988) B chromosomes as a model system for nondisjunction. In: Vig, B.K. and Sandberg, A.A. (Eds.) Aneuploidy: induction and test systems (part B). New York: Alan R. Liss Inc., pp. 199–207. [Google Scholar]
- Cenci, A. & Rouard, M. (2017) Evolutionary analyses of GRAS transcription factors in angiosperms. Frontiers in Plant Science, 8, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y.‐M. , Feng, Y.‐R. , Lin, Y.‐P. & Peng, S.‐F. (2016) Cytomolecular characterization and origin of de novo formed maize B chromosome variants. Chromosome Research, 24, 183–195. [DOI] [PubMed] [Google Scholar]
- Clifton, S.W. , Minx, P. , Fauron, C.‐R. , Gibson, M. , Allen, J.O. , Sun, H. et al. (2004) Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiology, 136, 3486–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cody, J.P. , Swyers, N.C. , McCaw, M.E. , Graham, N.D. , Zhao, C. & Birchler, J.A. (2015) Minichromosomes: vectors for crop improvement. Agronomy, 5, 309–321. [Google Scholar]
- Cui, J. , You, C. & Chen, X. (2017) The evolution of microRNAs in plants. Current Opinion in Plant Biology, 35, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X. , Zhuang, Z. & Zhao, P.X. (2018) psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Research, 46, W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ario, M. , Tavares, R. , Schiessl, K. , Desvoyes, B. , Gutierrez, C. , Howard, M. et al. (2021) Cell size controlled in plants using DNA content as an internal scale. Science, 372, 1176–1181. [DOI] [PubMed] [Google Scholar]
- Daub, J. , Eberhardt, R.Y. , Tate, J.G. & Burge, S.W. (2015) Rfam: annotating families of non‐coding RNA sequences. Methods in Molecular Biology, 1269, 349–363. [DOI] [PubMed] [Google Scholar]
- Ferris, R. , Long, L. , Bunn, S.M. , Robinson, K.M. , Bradshaw, H.D. , Rae, A.M. et al. (2002) Leaf stomatal and epidermal cell development: identification of putative quantitative trait loci in relation to elevated carbon dioxide concentration in poplar. Tree Physiology, 22, 633–640. [DOI] [PubMed] [Google Scholar]
- Ferris, R. & Taylor, G. (1994) Stomatal characteristics of four native herbs following exposure to elevated CO2 . Annals of Botany, 73, 447–453. [Google Scholar]
- Fu, R. , Zhang, M.I. , Zhao, Y. , He, X. , Ding, C. , Wang, S. et al. (2017) Identification of salt tolerance‐related microRNAs and their targets in maize (Zea mays L.) using high‐throughput sequencing and degradome analysis. Frontiers in Plant Science, 8, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, F. , Luo, X.U. , Huang, X. , Zhang, Y. , He, X. , Liu, M. et al. (2016) Genome‐wide analysis of transcription factors involved in maize embryonic callus formation. Physiologia Plantarum, 158, 452–462. [DOI] [PubMed] [Google Scholar]
- Gong, S. , Ding, Y. , Huang, S. & Zhu, C. (2015) Identification of miRNAs and their target genes associated with sweet corn seed vigor by combined small RNA and degradome sequencing. Journal of Agricultural and Food Chemistry, 63, 5485–5491. [DOI] [PubMed] [Google Scholar]
- Hanson, G.P. (1969) B‐chromosome‐stimulated crossing over in maize. Genetics, 63, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, S. & Oldroyd, G.E.D. (2009) GRAS‐domain transcription factors that regulate plant development. Plant Signaling & Behavior, 4, 698–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, J. , Shi, X. , Chen, C. , Islam, M.S. , Johnson, A.F. , Kanno, T. et al. (2018) Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proceedings of the National Academy of Sciences of the United States of America, 115, E11321–E11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Baker, A. , Bartel, B. , Linka, N. , Mullen, R.T. , Reumann, S. et al. (2012) Plant peroxisomes: biogenesis and function. The Plant Cell, 24, 2279–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Vendramin, S. , Shi, L. & McGinnis, K.M. (2017a) Construction and optimization of a large gene coexpression network in maize using RNA‐seq data. Plant Physiology, 175, 568–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W. , Du, Y. , Zhao, X. & Jin, W. (2016) B chromosome contains active genes and impacts the transcription of A chromosomes in maize (Zea mays L.). BMC Plant Biology, 16, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W. , Peng, S. , Xian, Z. , Lin, D. , Hu, G. , Yang, L. et al.(2017b) Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnology Journal, 15, 472–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y.‐H. , Peng, S.‐F. , Lin, Y.‐P. & Cheng, Y.‐M. (2020) The maize B chromosome is capable of expressing microRNAs and altering the expression of microRNAs derived from A chromosomes. Chromosome Research, 28, 129–138. [DOI] [PubMed] [Google Scholar]
- Johnson, A.F. , Hou, J. , Yang, H. , Shi, X. , Chen, C. , Islam, M.S. et al. (2020) Magnitude of modulation of gene expression in aneuploid maize depends on the extent of genomic imbalance. Journal of Genetics and Genomics, 47, 93–103. [DOI] [PubMed] [Google Scholar]
- Jones, N. & Houben, A. (2003) B chromosomes in plants: escapees from the A chromosome genome? Trends in Plant Science, 8, 417–423. [DOI] [PubMed] [Google Scholar]
- Jones, R.N. & Rees, H. (1982) B chromosomes, 1st edition. New York: Academic Press. [Google Scholar]
- Kato, A. , Lamb, J.C. , Albert, P.S. , Danilova, T. , Han, F. , Gao, Z. et al. (2011) Chromosome painting for plant biotechnology. Methods in Molecular Biology, 701, 67–96. [DOI] [PubMed] [Google Scholar]
- Kato, A. , Lamb, J.C. & Birchler, J.A. (2004) Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proceedings of the National Academy of Sciences of the United States of America, 101(37), 13554–13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell, M.G. & Lisch, D. (1997) Transposable elements as sources of variation in animals and plants. Proceedings of the National Academy of Sciences of the United States of America, 94, 7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. & Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara, A. , Birgaoanu, M. & Griffiths‐Jones, S. (2019) miRBase: from microRNA sequences to function. Nucleic Acids Research, 47, D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwada, Y. (1925) On the number of chromosomes in maize. Shokubutsugaku Zasshi, 39, 227–234. [Google Scholar]
- Lamb, J.C. , Kato, A. & Birchler, J.A. (2005) Sequences associated with A chromosome centromeres are present throughout the maize B chromosome. Chromosoma, 113, 337–349. [DOI] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Peng, T. , Wang, Q. , Wu, Y. , Chang, J. , Zhang, M. et al. (2017) Development of incompletely fused carpels in maize ovary revealed by miRNA, target gene and phytohormone analysis. Frontiers in Plant Science, 8, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W.‐F. , Zhang, S.‐G. , Han, S.‐Y. , Wu, T. , Zhang, J.‐H. & Qi, L.‐W. (2014) The post‐transcriptional regulation of LaSCL6 by miR171 during maintenance of embryogenic potential in Larix kaempferi (Lamb.) Carr. Tree Genetics & Genomes, 10, 223–229. [Google Scholar]
- Liu, H. , Jia, S. , Shen, D. , Liu, J. , Li, J. , Zhao, H. et al. (2012) Four AUXIN RESPONSE FACTOR genes downregulated by microRNA167 are associated with growth and development in Oryza sativa . Functional Plant Biology, 39, 736. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Qin, C. , Chen, Z. , Zuo, T. , Yang, X. , Zhou, H. et al. (2014) Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics, 15, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Su, H. , Pang, J. , Gao, Z. , Wang, X.‐J. , Birchler, J.A. et al. (2015) Sequential de novo centromere formation and inactivation on a chromosomal fragment in maize. Proceedings of the National Academy of Sciences of the United States of America, 112, E1263–E1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley, A.E. (1927) Supernumerary chromosomes in Zea mays . Journal of Agricultural Research, 35, 769–784. [Google Scholar]
- Ma, Z. , Hu, X. , Cai, W. , Huang, W. , Zhou, X. , Luo, Q. et al. (2014) Arabidopsis miR171‐targeted scarecrow‐like proteins bind to GT cis‐elements and mediate gibberellin‐regulated chlorophyll biosynthesis under light conditions. PLoS Genetics, 10, e1004519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, D.J. , Chen, Y. & Smyth, G.K. (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Research, 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B. (1933) The association of non‐homologous parts of chromosomes in the mid‐prophase of meiosis in Zea mays . Cell and Tissue Research, 19, 191–237. [Google Scholar]
- Miller, M. , Zhang, C. & Chen, Z.J. (2012) Ploidy and hybridity effects on growth vigor and gene expression in Arabidopsis thaliana hybrids and their parents. G3 (Bethesda), 2, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, K.J. , Stern, D.B. & Gabay‐Laughnan, S. (2009) Mitochondria and chloroplasts. In: Bennetzen, J.L. and Hake, S. (Eds.) Handbook of maize: genetics and genomics. New York: Springer, pp. 481–503. Available from: 10.1007/978-0-387-77863-1_24 [DOI] [Google Scholar]
- O'Leary, N.A. , Wright, M.W. , Brister, J.R. , Ciufo, S. , Haddad, D. , McVeigh, R. et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research, 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, B.T. , Wanous, M.K. & Birchler, J.A. (2001) Characterization of a maize chromosome 4 centromeric sequence: evidence for an evolutionary relationship with the B chromosome centromere. Genetics, 159, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashkovskiy, P.P. , Kartashov, A.V. , Zlobin, I.E. , Pogosyan, S.I. & Kuznetsov, V.V. (2016) Blue light alters miR167 expression and microRNA‐targeted auxin response factor genes in Arabidopsis thaliana plants. Plant Physiology and Biochemistry, 104, 146–154. [DOI] [PubMed] [Google Scholar]
- Pei, H. , Ma, N. , Chen, J. , Zheng, Y.I. , Tian, J.I. , Li, J. et al. (2013) Integrative analysis of miRNA and mRNA profiles in response to ethylene in rose petals during flower opening. PLoS One, 8, e64290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysh, L.D. , Wysocka‐Diller, J.W. , Camilleri, C. , Bouchez, D. & Benfey, P.N. (1999) The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW‐LIKE genes. The Plant Journal, 18, 111–119. [DOI] [PubMed] [Google Scholar]
- Randolph, L.A. (1928) Types of supernumerary chromosomes in maize. The Anatomical Record, 41, 102. [Google Scholar]
- Randolph, L.F. (1941) Genetic characteristics of the B chromosomes in maize. Genetics, 26, 608–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades, M.M. (1968) Studies on the cytological basis of crossing over. In: Peacock, W.J. and Brock, R.D. (Eds.), Replication and recombination of genetic material. Canberra, Australia: Australian Academy of Sciences, pp. 229–241. [Google Scholar]
- Robinson, D.O. , Coate, J.E. , Singh, A. , Hong, L. , Bush, M. , Doyle, J.J. et al. (2018) Ploidy and size at multiple scales in the Arabidopsis sepal. The Plant Cell, 30, 2308–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. & Smyth, G.K. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman, H. (1948) Directed fertilization in maize. Proceedings of the National Academy of Sciences of the United States of America, 34(2), 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. et al. (2012) Fiji: an open‐source platform for biological‐image analysis. Nature Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahid, S. & Axtell, M.J. (2014) Identification and annotation of small RNA genes using ShortStack. Methods, 67, 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, X. , Chen, C. , Yang, H. , Hou, J. , Ji, T. , Cheng, J. et al. (2020) The gene balance hypothesis: epigenetics and dosage effects in plants. In: Spillane, C. and McKeown, P. (Eds.) Plant epigenetics and epigenomics. methods in molecular biology. New York: Springer US, pp. 161–171. Available from: 10.1007/978-1-0716-0179-2_12 [DOI] [PubMed] [Google Scholar]
- Shi, X. , Yang, H. , Chen, C. , Hou, J. , Hanson, K.M. , Albert, P.S. et al. (2021) Genomic imbalance determines positive and negative modulation of gene expression in diploid maize. The Plant Cell, 33, 917–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, N.M. , Anderson, S.N. , Andorf, C.M. , Ahern, K.R. , Bai, F. , Barad, O. et al. (2018) The maize W22 genome provides a foundation for functional genomics and transposon biology. Nature Genetics, 50, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Stark, E.A. , Connerton, I. , Bennett, S.T. , Barnes, S.R. , Parker, J.S. & Forster, J.W. (1996) Molecular analysis of the structure of the maize B‐chromosome. Chromosome Research, 4, 15–23. [DOI] [PubMed] [Google Scholar]
- Takenaka, M. , Zehrmann, A. , Verbitskiy, D. , Kugelmann, M. , Hartel, B. & Brennicke, A. (2012) Multiple organellar RNA editing factor (MORF) family proteins are required for RNA editing in mitochondria and plastids of plants. Proceedings of the National Academy of Sciences of the United States of America, 109, 5104–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theuri, J. , Phelps‐Durr, T. , Mathews, S. & Birchler, J. (2005) A comparative study of retrotransposons in the centromeric regions of A and B chromosomes of maize. Cytogenetic and Genome Research, 110, 203–208. [DOI] [PubMed] [Google Scholar]
- Thomas, P.D. , Campbell, M.J. , Kejariwal, A. , Mi, H. , Karlak, B. , Daverman, R. et al. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Research, 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Hendrickson, D.G. , Sauvageau, M. , Goff, L. , Rinn, J.L. & Pachter, L. (2013) Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nature Biotechnology, 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. et al. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nature Protocols, 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaya, H. (2013) Does ploidy level directly control cell Size? Counterevidence from Arabidopsis genetics. PLoS One, 8, e83729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia, R.A. , Bottani, S. & Birchler, J.A. (2008) Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends in Genetics, 24, 390–397. [DOI] [PubMed] [Google Scholar]
- Vidal, E.A. , Araus, V. , Lu, C. , Parry, G. , Green, P.J. , Coruzzi, G.M. et al. (2010) Nitrate‐responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana . Proceedings of the National Academy of Sciences of the United States of America, 107, 4477–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viotti, A. , Privitera, E. , Sala, E. & Pogna, N. (1985) Distribution and clustering of two highly repeated sequences in the A and B chromosomes of maize. Theoretical and Applied Genetics, 70, 234–239. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Mai, Y.‐X. , Zhang, Y.‐C. , Luo, Q. & Yang, H.‐Q. (2010) MicroRNA171c‐targeted SCL6‐II, SCL6‐III, and SCL6‐IV genes regulate shoot branching in Arabidopsis. Molecular Plant, 3, 794–806. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Li, K. , Chen, L. , Zou, Y. , Liu, H. , Tian, Y. et al. (2015) MicroRNA167‐directed regulation of the auxin response factors GmARF8a and GmARF8b Is required for soybean nodulation and lateral root development. Plant Physiology, 168, 984–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F.‐Y. , Tang, C.‐Y. , Guo, Y.‐M. , Yang, M.‐K. , Yang, R.‐W. , Lu, G.‐H. et al. (2016) Comparison of miRNAs and their targets in seed development between two maize inbred lines by high‐throughput sequencing and degradome analysis. PLoS One, 11, e0159810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Shi, X. , Chen, C. , Hou, J. , Ji, T. , Cheng, J. et al. (2021) Predominantly inverse modulation of gene expression in genomically unbalanced disomic haploid maize. The Plant Cell, 33, 901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, H. , Kato, A. , Mooney, B. & Birchler, J.A. (2011) Phenotypic and gene expression analyses of a ploidy series of maize inbred Oh43. Plant Molecular Biology, 75, 237–251. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Xie, S. , Han, J. , Zhou, Y.U. , Liu, C. , Zhou, Z. et al. (2019) Integrated transcriptome, small RNA, and degradome analysis reveals the complex network regulating starch biosynthesis in maize. BMC Genomics, 20, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, F. , Wang, C. , Han, J. , Zhu, X. , Li, X. , Wang, X. et al. (2017) Characterization of miRNAs responsive to exogenous ethylene in grapevine berries at whole genome level. Functional & Integrative Genomics, 17, 213–235. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Xu, Z. , Mo, Q. , Zou, C. , Li, W. , Xu, Y. et al. (2013) Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Annals of Botany, 112, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Xue, Y. , Yang, H. , Li, H. , Sun, G. , Zhao, X. et al. (2016) Genome‐Wide identification of miRNAs and their targets involved in the developing internodes under maize ears by responding to hormone signaling. PLoS One, 11, e0164026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Xu, Z. , Duan, C. , Chen, Y. , Meng, Q. , Wu, J. et al. (2016) Dual transcriptome analysis reveals insights into the response to Rice black‐streaked dwarf virus in maize. Journal of Experimental Botany, 67, 4593–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , Hu, F. , Wang, R. , Zhou, X. , Sze, S.‐H. , Liou, L. et al. (2011) Arabidopsis argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell, 145, 242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The phenotype of experimental genotypes (1B‐7B) and the control (0B) and the cell size comparison.
Figure S2. Ratio distributions of expression of B‐located genes in each experimental genotype compared with the control with outliers (ratio > 6 or < 1/6) included.
Figure S3. Scatter plots of DGE in each experimental genotype compared with the control.
Figure S4. Gene ratios measured by qPCR compared with those computed by RNA‐seq.
Figure S5. Gene expression level of outliers and PCC with B copy number (ratio > 6 or < 1/6).
Figure S6. Histogram of PCC between the expression level of A‐located genes from each functional class and B copy number.
Figure S7. Ratio distributions and scatter plots of DGE for the functional class of TFs.
Figure S8. Ratio distributions and scatter plots of DGE for the functional class of signal transduction.
Figure S9. Ratio distributions and scatter plots of DGE for the functional class of structural proteins of the proteasome.
Figure S10. Ratio distributions and scatter plots of DGE for the functional class of stress‐related genes.
Figure S11. Ratio distributions and scatter plots of DGE for the functional class of nuclear chloroplast genes.
Figure S12. Ratio distributions and scatter plots of DGE for the functional class of nuclear mitochondrial genes.
Figure S13. Ratio distributions and scatter plots of DGE for the functional class of peroxisomal‐targeted genes.
Figure S14. Ratio distributions of A‐located miRNAs and B copy numbers.
Figure S15. Expression levels of miRNAs that are sensitive to B dosage.
Figure S16. Expression levels of miRNAs and their targets in the B dosage series.
Figure S17. Expression levels of the family members of zma‐MIR166 and their targets in the B dosage series, indicating different members of the same family have the same targets.
Figure S18. Expression levels of zma‐MIR167c and its targets in the B dosage series.
Figure S19. Ratio distributions and scatter plots of differential expression for all TEs.
Figure S20. Median of ratios of A‐located and B‐located TEs in different superfamilies.
Figure S21. Histogram of PCC between the expression level of each TE and B copy number representing their B‐dosage sensitivity.
Figure S22. Ratio distributions and scatter plots of differential expression for Class I TEs.
Figure S23. Ratio distributions and scatter plots of differential expression for Class II TEs.
Figure S24. Ratios of reads uniquely mapped to the reference genome (a) and the TE exemplar (b) to those uniquely mapped to ERCC sequences.
Table S1. Read mapping statistics and group information of mRNA‐seq and sRNA‐seq experiments.
Table S2. The mean, median, SD, and normality tests for each distribution listed in the order of mention in the text.
Table S3. Expression levels and information of predicted B‐located genes.
Table S4. GO enrichment analysis on genes from different subgroups.
Table S5. Normalized counts and ratios of genes silenced in the B dosage series.
Table S6. Distribution comparisons with K–S tests of significance listed in the order of mention in the text.
Table S7. Comparisons of distribution variances with Bartlett's test listed in the order of mention in the text.
Table S8. Number of DEGs for each functional class.
Table S9. Clusters of MIRNA loci identified by ShortStack and their annotations.
Table S10. Target prediction of DEMs in at least two experimental conditions compared to the control.
Table S11. Correlation between the expression levels of DEMs and their targets predicted by psRNATarget.
Table S12. Correlation between the expression levels of DEMs and their targets found in degradome sequencing data.
Table S13. Number of A‐located (a) and B‐located (b) DETs in each TE superfamily.
Table S14. Number and percentage of reads of B‐located genes mapped to the A chromosome.
Table S15. Correlation between the expression level of GRAS33 and the genes that co‐express with it. ‘R’ refers to the Pearson correlation between a gene and the corresponding B copy number, representing its degree of B‐dosage sensitivity computed as described in Figures 3a and 6c.
Table S16. Primer sequences used in qPCR.
Data Availability Statement
All sequencing data were deposited at the Gene Expression Omnibus (GEO) repository under accession number GSE127500 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127500). The following secure token has been created to allow review of record GSE127500 while it remains in private status: alcjoomqbfktzwp.