

Summary
Sphagnum peatmosses are fundamental members of peatland ecosystems, where they contribute to the uptake and long‐term storage of atmospheric carbon. Warming threatens Sphagnum mosses and is known to alter the composition of their associated microbiome. Here, we use a microbiome transfer approach to test if microbiome thermal origin influences host plant thermotolerance.
We leveraged an experimental whole‐ecosystem warming study to collect field‐grown Sphagnum, mechanically separate the associated microbiome and then transfer onto germ‐free laboratory Sphagnum for temperature experiments. Host and microbiome dynamics were assessed with growth analysis, Chla fluorescence imaging, metagenomics, metatranscriptomics and 16S rDNA profiling.
Microbiomes originating from warming field conditions imparted enhanced thermotolerance and growth recovery at elevated temperatures. Metagenome and metatranscriptome analyses revealed that warming altered microbial community structure in a manner that induced the plant heat shock response, especially the HSP70 family and jasmonic acid production. The heat shock response was induced even without warming treatment in the laboratory, suggesting that the warm‐microbiome isolated from the field provided the host plant with thermal preconditioning.
Our results demonstrate that microbes, which respond rapidly to temperature alterations, can play key roles in host plant growth response to rapidly changing environments.
Keywords: climate change, heat tolerance, microbiome transfer, moss, peatland, Sphagnum, symbiosis, synthetic communities
Introduction
Sphagnum peat mosses are fundamental ecosystem engineers (Clymo & Hayward, 1982; van Breemen et al., 1995), contributing to the construction of bog and peatland systems that occupy just 3% of the global land surface yet store c. 30% of all soil carbon (Gorham, 1991; Yu et al., 2010). In boreal regions, Sphagnum production can increase with modest warming (Robroek et al., 2007b; Hupperts et al., 2021), but these positive effects are not entirely generalizable (Gunnarsson et al., 2004) and are expected to be offset by water stress from surface drying (Robroek et al., 2007a) and more extreme warming events in the future (Bragazza, 2008; Bragazza et al., 2016; Norby et al., 2019). The competitive success and productivity of this keystone genus is largely dependent on symbiotic interactions with microbial associates (Lindo et al., 2013; Weston et al., 2014; Kostka et al., 2016), through which c. 35% of atmospheric nitrogen fixed by diazotrophic bacteria in the microbiome is transferred to the Sphagnum host (Berg et al., 2013). Currently, however, we lack a basic understanding of how warming influences Sphagnum–microbiome interactions and how these interactions influence host acclimation and adaptation to elevated temperature.
Sphagnum symbiosis is characterized by an intimate association with dinitrogen (N2)‐fixing cyanobacteria on the host cell surface and within water‐filled hyaline cells (Granhall & Hofsten, 1976; Basilier et al., 1978; Basilier, 1979, 1980). Hyaline cells help nonvascular mosses retain water and also provide a buffered environment for the microbiome that is less harsh than the external pore water, which is characterized by fluctuating temperature spikes and low pH (Clymo & Hayward, 1982). Phylogenetic evidence suggests that bacterial methanotrophs are also important N2‐fixing members of the Sphagnum microbiome in boreal peat bogs (Kip et al., 2010; Liebner & Svenning, 2013; Larmoia et al., 2014; Vile et al., 2014). These methanotrophs not only fix N2 but also supply 5–20% of the CO2 necessary for host photosynthesis as a byproduct of methane oxidation (Raghoebarsing et al., 2005). In addition to the prominent N2‐fixing bacteria, Sphagnum spp. host a diverse array of heterotrophic bacteria, archaea (Kostka et al., 2016), fungi (Kostka et al., 2016), protists (Lamentowicz & Mitchell, 2005; Jassey et al., 2015) and viral symbionts (Stough et al., 2018) within a complex food web structure. Results from a whole ecosystem peatland warming experiment indicate that elevated temperatures are associated with changes in the Sphagnum microbial biomass (Basińska et al., 2020), microbial community (Carrell et al., 2019; Reczuga et al., 2020), reduced N2 fixation (Carrell et al., 2019) and reduced Sphagnum biomass production (Norby et al., 2019). It remains unknown whether the warming‐altered microbiome influences host acclimation, growth and production, and if so, in what manner.
Disentangling the effects of Sphagnum symbiotic interactions in the context of climate change is complicated by our inability to predict whether and how mutually beneficial interactions will persist under variable environments. In N2‐fixing legumes (Heath et al., 2010) and coral systems (Cunning et al., 2015; Bay et al., 2016; Howells et al., 2016; Baker et al., 2018), for example, altered environmental conditions can increase the cost of the interaction relative to the benefits (i.e. the cost : benefit ratio), resulting in breakdown of mutualism and can even lead to antagonistic interactions. One strategy for maintaining a favorable cost : benefit ratio is partner switching, that is the substitution of one symbiont for another. In corals, for example, the negative effect of elevated temperatures on host performance can be tempered by replacing symbiont partners with more thermotolerant species (Bay et al., 2016; Howells et al., 2016). Another strategy is the habitat‐adapted symbiosis paradigm that does not emphasize partner choice, but instead proposes that endophytes adapt to stress in a habitat‐specific manner and can confer the same functional stress tolerance to their plant hosts (Rodriguez et al., 2008). Because it is not known whether endophytes are locally adapted or differentiated by environmental sorting, the term ‘adaptation’ is applied loosely (Giauque et al., 2019). Nonetheless, habitat‐associated benefits from endophytes originating from extreme temperatures and salinities can benefit host plants subjected to the same environmental extremes (Redman et al., 2011). By contrast, the habitat origin of fungal endophytes along a rainfall gradient had little effect on the drought responses of Panicum virgatum (Giauque & Hawkes, 2013). A more explicit test of habitat‐associated effects relative to evolutionary history and physiological traits was carried out by Giauque et al. (2019). They found little support for the idea that fungal endophyte phylogenetic relatedness predicts host benefits, but did find some evidence that microbes that had experienced similarly stressful environments could benefit their hosts. However, the host benefit was not as strong as in previous studies (Rodriguez et al., 2008), in which fungal endophytes were isolated from more extreme environments. Further complicating the habitat‐adaptation paradigm is our lack of understanding of the underlying mechanism in conferring host benefits.
Given the importance of bacteria for Sphagnum performance and ecosystem biogeochemistry (Raghoebarsing et al., 2005; Kip et al., 2010; Lindo et al., 2013; Kostka et al., 2016), we sought to determine the influence of habitat origin on host acclimation to thermal stress. To investigate this experimentally, we mechanically separated the microbiome from field‐grown Sphagnum plants collected under an ambient and a warm condition, transferred the constituent microbes to axenic plants, and then exposed the new host plants to short‐term heat stress. To assess host and bacterial dynamics, we performed growth analysis, Chla fluorescence imaging, metagenomics, metatranscriptomics and 16S rDNA profiling. The transfer of environmentally conditioned microbiomes to axenic plants, which is analogous to microbiome transplant studies in medical research, allowed us to test the following hypotheses: the isolation and transfer of a microbiome adapted to a warming origin can transmit thermotolerance to the plant host, the warming environment selects for microbial symbionts that can maintain nitrogen transfer with the plant at elevated temperatures, and the warming‐adapted microbiome elicits a host gene response enriched in nitrogen metabolism and abiotic stress response.
Materials and Methods
Study site and field sampling
The Spruce and Peatland Responses Under Changing Environments (SPRUCE) experiment, located in the S1 bog of the Marcell Experimental Forest (47°30.4760′N, 93°27.1620′W; 418 m above mean sea level), MN, USA, uses a regression‐based design at the whole‐ecosystem scale to produce nominal warming of ambient +0, +2.25, +4.5, +6.75 and +9°C in a Picea mariana–Sphagnum spp. raised bog ecosystem with open‐top chamber systems (Hanson et al., 2016). Heating of the soil was initiated in June 2014 and aboveground air heating began in June 2015. A full discussion of the experimental details and ecosystem description is available (Hanson et al., 2016). To obtain field‐conditioned microbiomes, 100 g of living green stems of fully hydrated, hollow dwelling Sphagnum angustifolium (Russow) C.E.O. Jensen was collected from ambient +0°C (ambient) and ambient +9°C (elevated) plots in August 2016 and August 2017. The collected stem portion typically included capitula and 2–3 cm of living stem. The Sphagnum material of each microbiome was placed in an individual sterile bag and shipped to Oak Ridge National Laboratory on blue ice.
Microbiome transfer to axenic Sphagnum and warming treatment
To isolate the microbiomes from each field‐conditioned microbiome, a sample of 100 g of tissue was diced with a sterile razor blade and pulverized in PBS with a mortar and pestle. The resulting suspension was filtered through Mira Cloth, centrifuged to pellet the microbes, and then resuspended in 500 ml BG11 ‐N medium (pH 5.5). Glycerol stocks of 100 ml of inoculum were frozen and stored at −80°C. Axenic tissue‐culture Sphagnum was propagated via clonal shoot propagation of Sphagnum generated from ethanol sterilized Sphagnum spores collected from the S1 bog. A single capitulum of axenic S. angustifolium was added to each well of a 12‐well plate and inoculated with 2 ml of ambient‐microbiome, warm‐microbiome or sterile media. The repeated 2017 experiment used the same experimental design, except that the number of replicate plants was increased from n = 6 to n = 12. The Sphagnum angustifolium genotype was the same as that sequenced by the DOE JGI (https://phytozome‐next.jgi.doe.gov/info/Sfallax_v1_1). The sealed plates were placed into growth chambers with a 350 μmol m– 2 s–1 of photosynthetically active radiation (PAR), 12 h : 12 h, light : dark cycle, programmed to either ambient or elevated field plot temperatures. August 2016 field plot temperatures from 6 h blocks were averaged from each day, resulting in a cycle of four temperatures. August 2017 temperatures did not differ from those in August 2016, so the same temperature profile was used for incubations for both years (Supporting Information Table S1). The plates were randomly distributed throughout the reach‐in growth chamber and positions were randomly shuffled weekly to account for possible edge effects or light variation across the chamber. Sufficient RNA could not be isolated from all treatments in 2016 so the incubation duration of the 2016 experiment was reduced from 4 to 3 wk in the 2017 experiment.
To determine if the growth response of inoculated Sphagnum resulted from residual plant molecules or the microbiome obtained during the isolation of thermal‐origin microbiomes, we repeated the experiment with a heat‐killed microbial control from glycerol stocks of the thermal‐origin microbiomes. Glycerol stocks were thawed, microbes were pelleted via centrifugation, rinsed and resuspended in BG11 ‐N, and an aliquot of glycerol stock was heat‐killed. Each microbiome, heat‐killed control or no microbial control were applied to axenic S. angustifolium and placed in growth chambers for temperature treatments (Table S1).
Measurement of growth and photosynthesis
To measure growth, images from the top of each plate were collected weekly, and surface area was measured using the ImageJ software (Schneider et al., 2012). The change in surface area was determined as a proxy for growth (Heck et al., 2021). To estimate maximal photosystem II (PSII) quantum yield, Chl fluorescence parameters were measured weekly with a FluorCam FC800 (PSI, Bruno, Czech Republic) after 20 min of dark adaptation. Maximum quantum yield (QY_max) was determined using the FluorCam 7 software.
Normality of the data was checked using the Shapiro–Wilk test before checking homoskedasticity of variances using Levene’s test in the R package car (Fox & Weisberg, 2018). Growth rate (mm d−1) and total growth over the duration of the experiment were rank‐transformed before two‐way ANOVA to assess the influence of experimental temperature and donor microbiome. Fluorescence (F v/F m) was measured in moss as a proxy for photosynthetic activity throughout the experiment. However, to highlight the greatest differences in donor microbiome and experimental temperature combinations, only fluorescence data from the last week were used. Fluorescence data were also rank‐transformed before using a two‐way ANOVA. ⍺ = 0.05 was used to denote statistical significance in both two‐way ANOVA and Tukey’s honest significant difference (HSD) post hoc analyses. Growth and fluorescence statistics were analyzed using R v.3.5.1 (R Core Team, 2021).
16S rDNA and ITS sequencing of community profiles
To characterize bacterial/archaeal and fungal components of the microbiomes of inocula and final microbiomes of the laboratory experiments, each sample (n = 3 for inocula and n = 5 for laboratory experiments) was pulverized in liquid N2, and DNA was extracted from 50 mg of material using the DNeasy PowerPlant Pro Kit (Qiagen). Extracted DNA was taken to the Genomics Core at the University of Tennessee, Knoxville, TN, USA, for library preparation and sequencing on an Illumina MiSeq (San Diego, CA, USA). Libraries were prepped for the 16S rRNA gene by means of a two‐step PCR approach with a mixture of custom 515F and 806R primers (Cregger et al., 2018) to characterize archaeal/bacterial communities, and for the ITS2 gene region with a custom mixture of primers to characterize the fungal community. Samples were pooled in equal concentrations and sequenced on the MiSeq with negative control samples.
Microbial sequences were processed with the Qiime 2 v.2018.11 platform (Bolyen et al., 2019). Paired sequences were demultiplexed with the plugin demux and quality‐filtered (denoised, dereplicated, chimera‐filtered and pair‐end merged) and processed into sequence variants (SVs) with the Dada2 plugin (Callahan et al., 2016). Taxonomy was assigned using a pretrained Naive Bayes classifier based on the Greengenes v.13_8 database (99% operational taxonomic units (OTUs)) that are trimmed to the 515F/806R primer pair for 16S rDNA and based on the UNITE databse (99% OTUs) for ITS2. Sequences assigned as chloroplast or mitochondria were removed. Microbial diversity was calculated based on a subsample of 19 000 sequences to fit the size of the smallest library. SV‐based alpha diversity (Shannon diversity) and beta diversity (Bray–Curtis) were calculated using the phyloseq 1.30.0 (McMurdie & Holmes, 2013) package in R (R Core Team, 2021). Beta diversity was visualized using nonmetric multidimensional scaling ordination (NMDS) based on Bray–Curtis similarity distances. A permutational multivariate analysis of variance (PERMANOVA) with 999 permutations was used to calculate the significance of clustering of samples by microbial and chamber treatment. The correlation between microbial diversity and Sphagnum growth was assessed using Pearson correlation.
Metatranscriptomics profiling
Only 2017 experimental samples were profiled for metatranscriptomics as sufficient RNA could not be isolated from 2016 experimental samples. Cryogenically stored samples from the end of the experiment were ground in liquid nitrogen, and total RNA was extracted using a method combining CTAB lysis buffer and the Spectrum Total Plant RNA extraction kit (Sigma) as described previously (Timm et al., 2016). RNA quality and quantity were determined using a NanoDrop Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Total RNA (3 µg) of three biological replicates was sent to Macrogen (Seoul, South Korea), where libraries were prepared with TruSEq Stranded RNA with Ribo‐Zero‐Plant and sequenced on an Illumina HiSeq 2500 in Rapid Run mode (paired‐end, 2 × 150 nt).
Metatranscriptome reads were partitioned into S. angustifolium and microbial transcripts by mapping reads to the S. angustifolium v.1.0 genome using Bbmap v.38.22. Microbial transcripts were processed using the Samsa v.2.2.0 pipeline (Westreich et al., 2016), except that differentially expressed SEED functional gene ontologies (Overbeek et al., 2014) were identified using Limma‐voom v.3.11 (Ritchie et al., 2015) with multiple testing correction using false discovery rate (FDR). Taxonomic classification of microbial transcripts was performed by mapping reads to the metagenome assembly using BamM v.1.7.3 and transferring the taxonomic classification of metagenomic gene models and metagenome‐assembled genome (MAG) assignments to mapped transcripts.
To identify differentially expressed (DE) S. angustifolium genes, S. angustifolium read‐pairs were mapped to the S. angustifolium v1.0 reference genome using RSubread v.2.3.0 (Liao et al., 2019) and analyzed using Limma‐voom v.3.11. Enrichment of Mapman4 ontology bins (Schwacke et al., 2019) in the set of DE genes was determined using the Mapman desktop application v.3.6.0RC1 (Thimm et al., 2014). The statistical significance of Mapman ontology bins was determined using a Kruskal–Wallis test with multiple testing correction using FDR in R v.3.6.1. The log2(fold change) (LFC) of Mapman4 ontology bins was determined by averaging LFC across DE genes within each bin. For all analyses we used a FDR‐corrected P‐value < 0.05 to determine statistically significant results.
Metagenomics of the starting inoculum and phylogenetic analysis of cyanobacterial MAGs
A composite ambient‐microbiome and a composite warming‐microbiome sample were sequenced as an Illumina TruSeq PCR‐Free library on an Illumina 2500 in Rapid Run mode (paired‐end, 2 × 150 nt). Full details are provided in Methods S1.
Results
Plant host performance in response to experimental temperature is dependent on the thermal origin of the microbiome
In both years, a donor microbiome that matched the experimental thermal conditions conferred the greatest increase in host growth (Fig. 1b). Host benefits from the microbiome were especially apparent under experimental warming conditions: in 2016 and 2017, moss receiving a warm‐microbiome exhibited an increase in growth of 89 and 87%, respectively, relative to plants receiving the mock control (Fig. 1b; Tables S2–S4). By contrast, under experimental warming conditions moss inoculated with a temperature‐mismatched microbiome (i.e. receiving ambient‐microbiome and warming treatment) did not perform statistically significantly better than moss without a microbiome (Table S4), indicating that a microbiome preadapted to warming conditions specifically is conferring a growth advantage in the warming chamber. Host benefits from temperature‐mismatched microbiomes were least prominent under ambient experimental treatment, in which growth increases ranged from 3% to 17% following inclusion of a warm‐microbiome (F temperature : microbiome = 32.01; P < 0.05 for 2017; Table S3). Throughout the experiment, moss photosynthetic activity and response to temperature and microbiome origin were evaluated by monitoring Chla fluorescence (F v/F m). The results mirrored the growth analysis: F v/F m values were 11–45% higher when microbiome thermal origin matched experimental temperature (Tables S5, S6; Figs S1, S2).
Fig. 1.
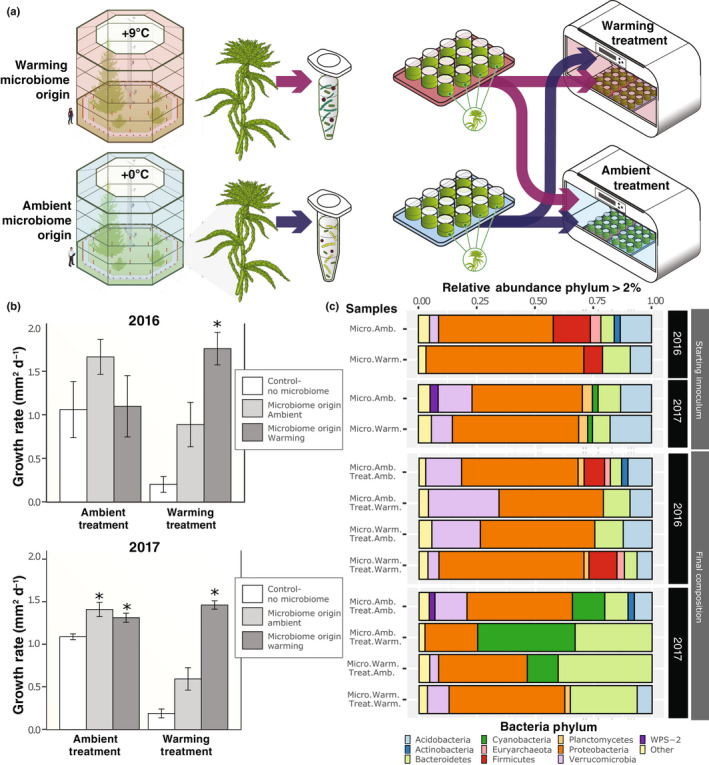
(a) Experimental approach and design: field‐collected donor moss microbiomes collected from ambient or warming conditions were transferred to germ‐free recipient moss (Sphangum angustifolium), and the resulting communities were then placed in an ambient or warm growth chamber. (b) Average moss growth rate under ambient or warming treatments, as a function of the thermal origin of the microbiome. Error bars represent standard error of the mean of n = 6 for 2016, n = 12 for 2017. (c) Relative abundance of microbiome phyla, determined by 16S rDNA amplicon sequencing of the starting field‐collected inoculum (n = 3 of each composite sample) from ambient or warming experimental plots, and the final compositions of experimental samples (n = 6 for each condition). An asterisk indicates statistical significance (P < 0.05) based on a Tukey’s HSD post hoc test of the percentage change of total growth between moss with a microbiome and moss without a microbiome within the same chamber.
To test the viability of the contribution of the conditioned microbiome and molecules collected during inoculum isolation, we repeated the conditioned microbiome experiment with glycerol stocks of +0°C and +9°C microbiomes collected from the SPRUCE field site. Moss phenotypes of heat‐killed controls were consistent with no microbe controls (P > 0.99) while growth rates were highest in temperature‐matched microbiome treatments and lower in discordant microbiome treatments (Fig. S3). Hence, the thermal tolerance conferred by the microbial inoculums is from direct contact with living organisms, and not an indirect consequence or nutrients, signaling compounds or other molecules acquired during isolation.
Habitat origin and thermal treatment conditions structure the starting microbiome and resultant microbial community
Amplicon sequencing produced an average of 83 847 ± 53 040 reads after quality filtering (Table S7). In the 2016 inoculum, we retained 82% of the ambient‐microbiome SVs and 85% of the warm‐microbiome SVs compared to the unprocessed field‐microbiome. In 2017, the ambient‐microbiome and warm‐microbiome represented 76 and 89% respectively of the SVs identified in the field‐microbiomes. Across the treatments, we recovered an average of 482 SVs with an average of 350 bacterial/archaeal SVs and an average of 107 fungal SVs. In 2017, the initial community structure of the S. angustifolium field‐collected inoculum differed between thermal origins (Adonis, R 2 = 0.92133, P = 0.009) and 2016 (Adonis, R 2 = 0.53, P = 0.1) (Fig. S4). The bacterial phylum Proteobacteria dominated all reads (21–68%) with cyanobacterira increasing in abundance in 2017 (15%) compared to 2016 (3%) (Fig. 1c). At the class level, the ambient‐microbiome consisted largely of Alphaproteobacteria (32%) and Clostridia (16%) in 2016, whereas Alphaproteobacteria (30%) and Acidobacteria (11%) were most abundant in 2017 (Table S8). Within the warm‐microbiome, Gammaproteobacteria were highly abundant in 2016 (43%), but only constituted 15% of the community abundance in 2017, with the difference compensated for by an additional increase in Alphaproteobacteria abundance (30%). Despite between‐year differences in community composition at the class level within thermal regimes, the growth benefits provided to the plant were strikingly consistent (Fig. 1b).
Sphagnum‐associated microbial communities responded to 4 wk of thermal treatment conditions regardless of year, thermal origin or growth temperature. NMDS ordinations of the microbiome Bray–Curtis distance matrix revealed that the community composition of the warm‐microbiomes responded similarly across thermal treatments, whereas ambient‐microbiome structure varied to a greater extent both across and within thermal treatments (Fig. S5). To determine whether changing community structure influenced microbial diversity, we estimated the Shannon diversity index for each treatment condition at the conclusion of the study in both years (Fig. 2a,b). Microbial Shannon diversity was highest when microbiome thermal origin matched chamber treatment temperatures; conversely, discordant combinations resulted in substantially lower microbial Shannon diversity (Fig. 2a,b). The ambient‐microbiome had the highest diversity under matched (i.e. ambient) treatment conditions in both years (ANOVA, P < 0.01). Similarly, the warm‐origin microbiome had the highest diversity under the warming treatment in both years (ANOVA, P < 0.01). Detailed class‐level community composition assignments are provided in Table S9. Given that greater phylogenetic diversity is likely to be accompanied by greater metabolic and functional diversity, we expected that microbial diversity would be associated with enhancements in plant acclimation to stressful warming conditions, reflected by improved growth. As expected, bacterial and archaea Shannon diversity (as inferred from 16S rDNA) at the end of the experiment was correlated with Sphagnum growth (Pearson correlation, r = 0.744, P = 0.003; Fig. 2b). By contrast, ITS‐derived fungal Shannon diversity estimates did not correlate with moss growth (Pearson correlation, r = −0.204, P = 0.403; Fig. S6). The fungal communities did not vary greatly across treatments with the identification of just one phylum level (Table S10), three class levels (Table S11), and RNA sequencing (RNA‐seq) reads that only constituted 0.6% of the total (Tables S12), so we largely focus on the bacterial component of the microbiome in the following sections.
Fig. 2.
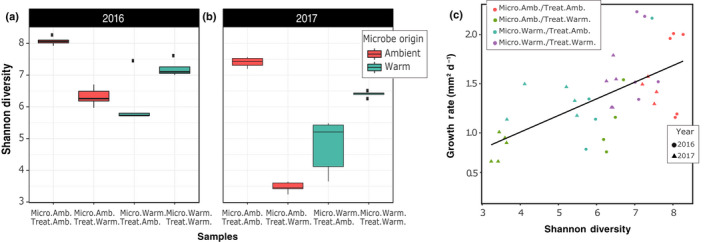
Microbial diversity change in response to habitat origin and experimental temperature. Shannon diversity index of the microbiome at the conclusion of the experiments in 2016 (a) and 2017 (b), based on 16S rDNA amplicon data for five replicates of each condition in each year. Microbiomes had lower Shannon diversity when the thermal origin and experimental treatment were mismatched (i.e. ambient origin in warming treatment or warming origin in ambient treatment) (ANOVA, P < 0.01). Lines within the boxplots represent median, 25th and 75th percentile values, while whiskers are defined by the largest value not greater than 1.5× the interquartile range (IQR) and the smallest value not less than 1.5× the IQR. (c) Sphagnum angustifolium growth rate was linearly correlated with microbiome Shannon diversity (Pearson correlation, r = 0.744, P = 0.003) at the conclusion of the experiment.
Metagenome and metatranscriptome analyses reveal changes in symbiotic microbe abundance and composition in response to thermal origin and temperature treatment
Host thermal acclimation and productivity varied with microbiome origin. To further explore how community dynamics influence host thermal acclimation, we used metagenomics and metatranscriptomics to identify both plant and microbial gene sets responsive to thermal and microbiome conditions (Fig. 3). For metagenome assemblies, DNA sequencing reads mapping to the S. angustifolium genome (https://phytozome‐next.jgi.doe.gov/info/Sfallax_v1_1) were removed, and the remaining reads were coassembled into 4 762 069 contigs with an N50 of 1261 bp. Binning of metagenome contigs yielded 45 MAGs with a quality score ≥ 70 with ≤ 5% contamination (Table S12). The high‐quality MAG standard of > 90% complete and < 5% contamination (Bowers et al., 2017) was met for 28 of our genomes, whereas 13 and nine MAGs are > 95% and > 97% complete, respectively. Taxonomic assignments and Blast hits from annotated proteins were resolved to the lowest taxonomic level using Checkm (Parks et al., 2015) and Diamond (Buchfink et al., 2014) (Table S12). For metatranscriptomes, we generated 429.6 GB of RNA‐seq data across three replicates for each treatment. On average, 40.93 ± 7.39 million reads passed quality filtering per sample across all thermal treatments and microbiome conditions (Table S13). In samples derived from plants receiving a microbiome transfer, c. 65% of reads aligned to the Sphagnum genome, except in the discordant case when plants received ambient‐microbiome followed by warming treatment. Under that condition, the plants were severely stressed, and only 12.4% of the reads aligned to the Sphagnum genome (Fig. S7).
Fig. 3.
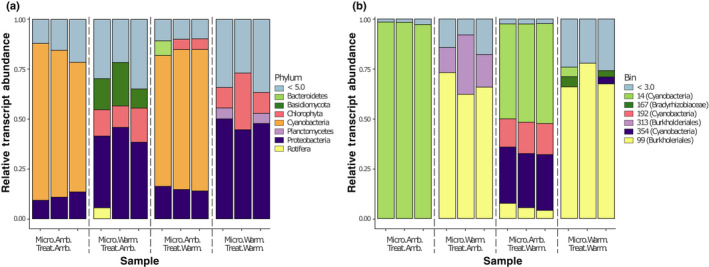
Microbial community transcriptional profile response to temperature treatment. Relative abundance of microbial transcripts mapping to metagenome contigs for (a) major phyla and (b) metagenome‐assembled genomes (MAGs). Each bar represents a metatranscriptome sample for the ambient‐microbiome (Micro. Amb.) or warming‐microbiome (Micro. Warm) under either the ambient (Treat. Amb.) or warming (Treat. Warm) treatment. Colors indicate (a) phyla or (b) MAGs; light blue represents (a) phyla with < 5% of mapped transcripts or (b) MAGs with < 3% of mapped transcripts.
To expand on the amplicon‐based community composition results (Fig. 1c; Table S8) and determine which microbial members are transcriptionally active, we categorized transcriptional profiles based on taxonomic composition. Under matched ambient origin and experimental temperature, microbial transcripts were mostly from Cyanobacteria symbionts (72.5 ± 6.9%), followed by Proteobacteria (11.2 ± 2.2%) (Fig. 3a). Under matched warm‐microbiome and warm‐temperature treatment, Cyanobacteria transcript reads were largely absent, and the metatranscriptome was mainly derived from Proteobacteria (47.5 ± 2.7%), Chlorophyta (16.39 ± 10.4%) and Planctomycetes (4.9 ± 0.57%). Results from mismatched origin and experimental conditions more closely reflected their microbiome origin communities (Table S8). This finding was also reflected in a multidimensional scaling analysis using level 3 SEED functional annotation, in which cluster variation was explained more based on microbial origin rather than experimental temperature (Fig. S8).
Experimental warming increases transcript abundance from alternative cyanobacteria members, signaling possible symbiont exchange
Differences in the composition and abundance of Sphagnum‐associated cyanobacteria in response to warming are important because these organisms are some of the key symbionts with Sphagnum mosses (Granhall & Hofsten, 1976), and the exchange of symbionts with more thermotolerant forms have been implicated in host thermotolerance in other systems (Rodriguez et al., 2008). To explore this further, we taxonomically refined three of our high‐quality cyanobacteria MAGs with a phylogenetic tree reconstruction using an additional 109 cyanobacterial genomes (Fig. 4). We found that all three cyanobacterial MAGs belong to the heterocystous B1 clade of Cyanobacteria, which also contains known plant associates (Shih et al., 2013). To determine which of the cyanobacteria are most responsive to thermal conditions, we aligned the microbial RNA‐seq reads from the end of the experiment onto the MAGs. Of all non‐Sphagnum RNA‐seq reads, 31.6 ± 8.7% mapped onto cyanobacteria MAGs for matched ambient‐microbiome and ambient temperature conditions. This percentage decreased to 26.1 ± 1.1% when plants receiving ambient‐microbiomes were subjected to discordant warming treatment. Further, microbiomes originating from warming field conditions contained negligible levels of cyanobacterial RNA‐seq reads (0.1–0.08%). Cyanobacterial reads predominantly aligned to MAG bin 14 (98 ± 0.7%), but to a considerably lesser extent (49 ± 0.6%) when placed under warming experimental conditions. The decrease in bin 14 RNA‐seq reads was accompanied by an increase in reads from Cyanobacteria bin 354 (28 ± 0.6%) and bin 192 (15 ± 0.9%) (Fig. 3b). Due to sampling constraints, we did not normalize the results of RNA‐seq analysis to community abundance changes.
Fig. 4.
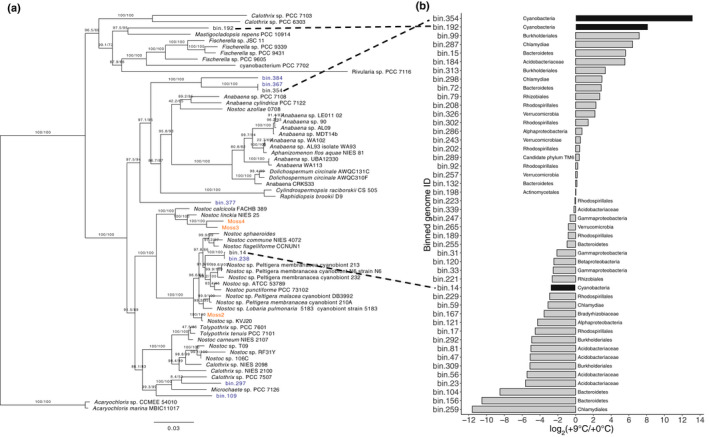
Bar chart representing Cyanobacteria log2(fold change) of counts per million reads mapping to metagenomic bins between ambient‐microbiome and warm‐microbiome metagenome samples. Phylogeny of selected Cyanobacteria and log2(fold change) of metagenomic bins. (a) Maximum‐likelihood phylogram where the numbers at nodes indicate UFBoot2 and Shimodaira–Hasegawa‐like approximate likelihood ratio support. Branch lengths indicate estimated substitutions per site. Metagenomic bin taxa labels are colored by source from either within peatland responses under changing environments (SPRUCE) (blue) or from Warshan et al. (2017) (orange). (b) Bar chart representing log2(fold change) of metagenomic bins between ambient‐microbiome and warm‐microbiome metagenomes. Black bars indicate cyanobacterial metagenome‐assembled genomes (MAGs) recovered in this work.
Host plant transcriptional response to warming
Given that the warming environment alters community composition in a way that benefits host plant acclimation to warming, we hypothesized that the warming environment selects for more thermotolerant symbionts that are able to maintain nitrogen exchange with the plant at elevated temperatures. If this is the case, we would expect that plant and microbial transcriptional patterns relating to N transport and metabolism would be similar between matched origin and temperature conditions (i.e. warm‐microbiome + warming treatment or ambient‐microbiome + ambient treatment). Functional ontology enrichment analysis across all conditions revealed that in plants receiving an ambient‐microbiome and ambient treatment, gene expression was enriched for pathways involved in N metabolism, including ammonium transporters, ammonium assimilation and glutamine synthetase, as well as growth‐related ontologies including photosynthesis, cytoskeletal elongation and hormonal regulation (Fig. 5a). This was also apparent on an LFC basis when comparing plants with an ambient‐microbiome between temperature conditions (Dataset S1). In this case, ambient treatment plants corroborated enrichment analysis with induced ammonium transport (LFC 4.0, P = 2.0 × 10–2) and glutamine synthetase (LFC 2.4, P = 1.1 × 10–2). In addition, 153 of 178 genes within the photosynthesis ontology were induced, with PSII light‐harvesting complex II most strongly affected (LFC 2.1, P = 2.48 × 10–6). In addition, we noted differences in fatty acid synthesis, especially in desaturation and elongation (LFC 2.74, P = 2.97 × 10–6), phenolic secondary metabolite production (LFC 2.86, P = 1.77 × 10–5), cell wall expansion (LFC 6.5, P = 2.7 × 10–6), phytohormone signaling with noncysteine‐rich peptides (LFC 4.63, P = 8.1 × 10–4), jasmonic acid synthesis (LFC 2.36, P = 4 × 10–2) and response to external stimuli (LFC 2.53, P = 3.2 × 10–2). As expected, heat shock proteins (HSPs) were responsive to warming, especially the HSP70 family, of which 15 members were induced (LFC 1.2, P = 6.5 × 10–3).
Fig. 5.
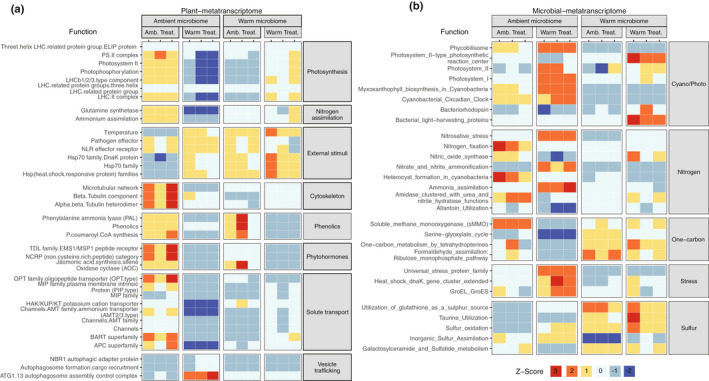
Plant and microbial ontology enrichment analysis. Heatmap of z‐scores for (a) Mapman4 ontology categories enriched in differentially expressed Sphagnum angustifolium genes and (b) differentially expressed SEED level 3 categories related to nitrogen, one‐carbon and sulfur metabolism; cyanobacterial/photosynthesis; and stress. Differential expression was defined as |log2(fold change) > 1| and corrected P < 0.05.
Plant ontology enrichment analysis did not support the hypothesis that the warm‐microbiome would provision the plant with N at warming treatment conditions (Fig. 5a). Likewise, there was no support for this hypothesis on an LFC basis when comparing RNA‐seq profiles from plants with warm‐microbiomes across temperature treatments (Dataset S1). Despite the apparent lack of microbially provided fixed N, the warm‐microbiome still provided growth benefits to warming‐treated plants (Fig. 1b), and this was also apparent in RNA‐seq enrichment analysis of growth‐related ontologies. Specifically, plants exposed to warming that received warm‐microbiomes exhibited enrichment for photosynthesis – PSII light harvesting complex II (LFC 1.4, P = 1.9 × 10–3), cell wall expansion (LFC 2.5, P = 1.3 × 10–5) and phenolic secondary metabolite production (LFC 2.9, P = 2.0 × 10–8).
Microbial RNA‐seq DE analysis of functional ontologies supported the notion that the warming‐altered cyanobacteria community is not fixing N, and is therefore not provisioning the plant with N. DE enrichment analysis revealed that microbial N metabolism differed dramatically between both treatments and origins (Fig. 5b; Table S14). Indeed, exposure to warming decreased N‐fixation ontology gene expression by 53.4‐fold (Fig. 5b; Table S13). Moreover, there was no enrichment evidence for N‐fixation among the warm‐microbiomes, regardless of temperature treatment. This was also apparent with LFC when comparing microbes from a warm‐microbiome to those from an ambient‐microbiome under ambient treatment conditions where genes enriched for N‐fixation substantially decreased (LFC −8.0, P = 1.6 × 10–9) (Dataset S2). The same conditions also showed an increase in sulfur‐related metabolism via taurine utilization (LFC 1.5, P = 2.0 × 10–4) and the utilization of glutathione as a sulfur source (LFC 2.7, P = 1.8 × 10–7). Although we could not obtain direct evidence for N‐fixation in this study due to sample size restrictions, these observations corroborate previous 15N2‐based fixation rates reported at the same field site where our warm‐ and ambient‐inocula were obtained (Carrell et al., 2019).
How the warm‐microbiome influences host plant photosynthesis and growth temperature acclimation remains to be elucidated, but we can glean clues from communities composed of discordant warm‐microbes at ambient experimental temperatures. In that case, enrichment for the HSP70 family is induced without heat (Fig. 5a). This trend was also observed on an LFC basis where 33 out of 35 detected HSPs were induced (Dataset S1).
Discussion
The establishment of constructed communities derived from microbiome transfers, coupled with comparative metatranscriptomics, revealed several novel aspects of microbial contributions to plant temperature response. First, plants receiving a microbiome from a high‐temperature environment exhibited enhanced photosynthetic and growth acclimation responses to similarly warm environments. Second, the warm‐microbiome was less diverse than the ambient‐microbiome, but contained transcripts from a more diverse set of cyanobacteria, suggesting symbiont swapping or replacement. Finally, the warm‐microbiome transferred a thermotolerant phenotype to the plant through host transcriptional reprogramming involving heat shock response and hormonal regulation genes.
Our results demonstrate that the originating thermal habitat of the microbiome has a dramatic effect on Sphagnum host acclimation to elevated temperatures. These results were consistent across two years of field‐collected donor inocula and two independent laboratory experiments. Although the fact that plants benefit from microbial relationships is well known, the transfer of microbially acquired habitat‐specific abiotic stress tolerance to recipient plants was reported much more recently, and to date has largely been limited to endophytic fungi (e.g. Giauque & Hawkes, 2013; Giauque et al., 2019). In an early example of this, Redman et al. (2002) collected a native North American grass, Dichanthelium lanuginosum, endemic to geothermal sites with soil temperatures reaching up to 50°C. After isolation of a Curvularia sp. fungus and reinoculation onto endophyte‐free plants, thermotolerance was conferred to the recipient plant host. This approach of isolating endophytic fungi from plants endemic to extreme habitats in an attempt to confer habitat‐associated benefits has been tested a number of times with both successful (Rodriguez et al., 2008; Redman et al., 2011) and mixed results (Giauque & Hawkes, 2013; Giauque et al., 2019). In all cases, the microbial component focused on fungi and was constrained to single‐member strain‐based studies.
The microbiome transfer approach used in this study allowed us to test habitat‐associated benefits from a broader set of organisms that is more representative of the dynamic coevolving community. However, this strategy made it difficult to relate specific taxa to recipient host benefits. For example, the warm‐inocula differed substantially between years, even at the phylum level, yet both showed trends in providing host thermal benefits. This is consistent with the idea that microbial community taxonomic composition is not necessarily a clear indication of community function. Indeed, functional similarity independent of taxonomic group has been reported in other systems, including human gut (Moya & Ferrer, 2016) and microalgae (Burke et al., 2011) microbiomes. Hence, the challenge is to look beyond taxonomic association and determine what components of the microbiome are responsible for conferring thermotolerance on the host plant.
One possible mechanism for enhanced host temperature tolerance is the replacement of primary symbionts with more thermotolerant symbionts. Sphagnum mosses have long been known to host N2‐fixing Cyanobacteria as symbionts (Granhall & Hofsten, 1976; Basilier et al., 1978; Basilier, 1979, 1980). More recently, they have been shown to associate with a suite of bacteria, including those that oxidize methane into CO2, as well as a number of viral, archaea and protists (reviewed by Kostka et al., 2016). The influence of warming on these symbionts, especially the Cyanobacteria, would directly affect host nutrient status and productivity. Our metagenome analysis assembled three cyanobacterial MAGs. DNA and RNA‐seq reads mapping to the binned MAG genomes indicated that Sphagnum plants were primarily colonized by a single cyanobacterial member from the genus Nostoc. With increasing temperature, this Nostoc MAG decreased in abundance, while two additional cyanobacterial MAGs increased in abundance, indicating a possible exchange for more thermotolerant members of the clade. Precedent for symbiont shuffling has been provided in coral systems. Corals host algal symbiont communities that are genetically diverse and susceptible to symbiont loss due to environmental stress and ensuing coral bleaching events. However, the stress events leading to the bleaching, as well as the bleaching itself, provide an opportunity for replacement of symbionts with organisms that are more suitable to the new environmental condition, such as those with higher stress tolerance (reviewed by Apprill, 2020). The coral system also demonstrates the potential role of the surrounding bacterial community in coral thermotolerance. This was elegantly demonstrated by Ziegler et al. (2017), who showed that long‐term temperature elevation modified the composition of the bacterial community, and that particular bacterial taxa could predict coral thermotolerance. However, the coral system is not amenable to germ‐free host strains or microbiome transfers, making it difficult to quantify the contribution of the microbiome to host thermotolerance.
From the results of this study, it is difficult to discern whether host thermal benefits from the donor microbiome are driven by community change from primary Nostoc cyanobacterial symbionts, or instead by the surrounding microbial community. Our hypothesis that the key cyanobacterial symbiont was augmented by additional thermotolerant cyanobacteria to maintain N2 fixation at elevated temperatures was not entirely supported. Although we observed an increase in cyanobacteria diversity and ontology enrichment for photosynthesis, thereby providing support for exchange with thermotolerant symbionts, the metatranscriptome analysis yielded no evidence for N2‐fixation under warming. Caution must be observed as evidence for N2‐fixation in the current study is based on gene transcriptional analysis and not direct measures for 15N2 incorporation. However, this finding is consistent with a previous field study (Carrell et al., 2019) at the same SPRUCE site, where 16S rDNA amplicon profiling, nifH quantitative reverse transcriptase PCR and 15N2 incubation assays revealed a decrease in nifH‐containing N‐fixing bacteria and a reduction in 15N2 incorporation in response to warming.
In addition to N metabolism, recent studies have identified a role for sulfur exchange within feathermoss – and Sphagnum – cyanobacterial symbioses (Warshan et al., 2017; Stuart et al., 2020; Carrell et al., 2021). For example, Stuart et al. (2020) found that targeted mutatgenesis of the cyanobacterial alkane sulfonate monooxygenase resulted in an inability to colonize feathermoss. In addition, Carrell et al. (2021) used metabolic cross‐feeding and spatial metabolite profiling to discover that Sphagnum provided sulfur‐rich choline‐O‐sulfate, taurine and sulfoacetate, which were subsequently depleted by the Nostoc symbiont. Within the current study, the warm‐microbiome showed enrichment for bacterially related sulfur metabolism regardless of treatment temperature. Whether bacterial sulfur metabolism is specific to the cyanbacterial component of the microbiome and the role sulfur metabolism plays in Sphagnum–microbiome functioning remains to be elucidated.
Despite the lack of evidence for microbial N2‐fixation in contributing to plant thermotolerance, the metatranscriptome analysis did reveal a role for heat shock and hormonal reprogramming as potential host pathways underlying the microbial transfer of thermotolerance. Multiple studies have shown that insertional mutants or antisense transgenics for HSP70 fail to acquire thermotolerance, while the overexpression of HSP70 seems to enhance thermotolerance (Larkindale et al., 2007). Within the model plant Arabidopsis thaliana, the HSP70s represent a multigene family whose proteins are found within all subcellular compartments of the cell where they can refold stress‐denatured proteins and prevent aggregation of denatured proteins (Sung et al., 2001). Within the current study, plants grown at ambient temperatures were enriched for Hsp70 gene family transcripts when they received a warm‐microbiome, but not when they received an ambient‐microbiome. Shekhawat et al. (2021) also observed an increase in HSP70 gene expression in Arabidopsis and wheat plants when colonized by the Enterobacter sp. root endophyte, yet this was only observed under heat stress and not ambient temperature conditions. Furthermore, our metatranscriptome analysis did not show microbial‐mediated repression of the Hsp90 family or induction of the Hsp101 family, both of which have been implicated in the heat shock response. While microbially mediated repression of Arabidopsis Hsp90, leading to elevated thermotolerance, was previously demonstrated in the desert‐dwelling fungus Paraphaeosphaeria quadriseptata (Mclellan et al., 2007), results from the current study only show a role for HSP70s.
In addition to HSP70 reprogramming, microbial thermotolerance may have been transferred via hormonal pthways. The warm‐microbiome elicited host plant expression of genes contributing to jasmonic acid synthesis (via allene oxide cyclase (AOC)). Jasmonic acid is a key phytohormone contributing to both abiotic and biotic stress responses, and has been implicated in flowering plant thermotolerance (Clarke et al., 2009). AOC synthesizes 12‐oxo‐phytodienoic acid (OPDA), which is a signaling compound and intermediate in the jasmonic acid biosynthesis pathway. In the liverwort Marchantia polymorpha, overexpression of AOC increases OPDA, suggesting that its function is similar to that of its homologs in flowering plants (Yamamoto et al., 2015). However, AOC overexpression in M. polymorpha decreases growth. Likewise, the warm‐microbiome induced expression of enzymes involved in the production of phenolic compounds, including phenylalanine ammonia lyase (PAL), which has been implicated in both temperature response and disease resistance (Huang et al., 2010). In contrast to the heat shock response, the jasmonic acid and phenolic ontology enrichments disappeared after the plants were exposed to warming. Furthermore, the ambient‐microbiome elicted some aspects of jasmonic acid synthesis as well indicating that more research is needed to determine if cross‐talk with the heat shock response occurs. Thus, it remains to be determined whether these compounds are contributing to a beneficial thermal preconditioning or instead reflect a defensive response.
One unexpected observation was that the warm‐microbiome elicited the induction of the heat shock response in plants that were never exposed to elevated temperatures. Thermotolerance can be acquired by previous exposure to a sublethal temperature stress (Kotak et al., 2007). Similarly, plants associated with beneficial rhizosphere microbes can more rapidly mount a defense response to biotic and abiotic stressors (Conrath et al., 2006). Although there is a considerable body of literature on the biotic aspects of microbially induced plant priming, increasing evidence suggests that plants can also be primed against abiotic stressors. For example, Ali et al. (2009) found that a Pseudomonas sp. strain isolated from pigeon pea endemic to an arid region conferred enhanced survival and growth on sorghum seedlings exposed to elevated temperatures. This early example has been corroborated with multiple plant hosts and microbial strains, yet the underlying genetic mechanisms remain to be identified (Yamamoto et al., 2015). In most cases studied thus far, microbially induced resistance to abiotic stress has been studied in individual strains or small community consortia. By contrast, in this study we examined how microbial dynamics within more complex communities interact to influence host physiology and growth.
Conclusions
Our findings provide a starting point for future studies that systematically decouple inherent host acclimation responses to challenging environmental conditions from those of the associated microbiome. A key benefit of the microbiome transfer and constructed community approaches described here is that they allow the coevolved host–microbiome consortia collected from extreme environmental conditions to be separated and tested across a range of thermal experimental conditions. Our observation that the microbiome can transmit thermotolerant phenotypes has a number of implications. It sets the stage for moving beyond the current notion that plants are restricted to ‘adapt or migrate’ strategies for survival to rapidly changing environmental conditions (Lau & Lennon, 2012). The current study provides an alternative perspective on these outcomes by showing that thermotolerant phenotypes can be rapidly transmitted to plant hosts. We anticipate multiple challenges as the findings of our studies are transferred beyond the laboratory into ecological systems. First, additional research is needed to determine the extent to which inter‐ and intraspecific genetic variation influences the plant’s ability to receive microbial benefits, and if so, to identify the causal alleles. Bringing this goal closer to reality, a genome sequencing campaign representing some 78 species within the c. 300‐member genus Sphagnum, as well as the development of high‐density genetic maps from sequencing of a 200‐member pedigree cross, are currently underway (Weston et al., 2018). Second, the identification of responsible microbial taxa is challenged by large community diversity, complex community interactions and strain isolation limitations. These experimental tests could take multiple forms, including the dilution and sequencing of donor microbiomes or strain isolation and testing in our demonstrated plate‐based experimental system or repeat transfers of field conditioned microbiomes paired with isotope tracing and metatranscriptomics. Within the context of this study, such an approach could determine whether microbial benefits are mainly a function of swapping primary cyanobacteria symbionts for more thermotolerant members, or whether additional microbial members are driving the host phenotype. In closing, the current study revealed that warming altered microbial community structure in a manner that induced the plant heat shock response, especially the HSP70 family and jasmonic acid production. Hsp70 induction occurred even without a warming treatment, suggesting that the warm‐microbiome itself can induce plant thermal acclimation.
Author contributions
AAC and DJW designed research; AAC, TJL, DJW, DAP, SSJ and JG performed research; AAC, TJL, KGMC and DLC analyzed data; JS, PJH, AJS, AAC, TJL, DJW, DAP, DLC, KGMC and JHL wrote the manuscript.
Supporting information
Dataset S1 Log fold change of RNA‐seq profiles from plants with ambient‐ and warm‐microbiomes across temperature treatments and ambient and warming microbiome treatments.
Dataset S2 Log fold change of RNA‐seq profiles from plant microbiomes with ambient and warming microbiomes treatments.
Fig. S1 Average moss fluorescence in 2016 at the end of the experiment.
Fig. S2 Average moss fluorescence in 2017 at the end of the experiment.
Fig. S3 Average moss growth rate under ambient or warming treatments.
Fig. S4 Bacterial/archaeal amplicon nonmetric multidimensional scaling (NMDS) ordination of the Bray–Curtis distance matrix of inoculum and field.
Fig. S5 Bacterial/archaeal amplicon nonmetric multidimensional scaling (NMDS) ordination of the Bray–Curtis distance matrix of samples at the end of the experiment.
Fig. S6 Linear correlation of plant growth rate and fungal (ITS) Shannon diversity at the conclusion of the experiment.
Fig. S7 Boxplot of percentage of metatranscriptomic reads mapping to Sphagnum, metagenome assembly and metagenome assembled genomes.
Fig. S8 MDS of the top 500 most variable microbial SEED level 3 categories.
Methods S1 Metagenome sequencing and analysis.
Table S1 Incubation temperature and light cycle for 2016 and 2017 laboratory experiments.
Table S2 Summary growth rate and total moss growth over 4 wk.
Table S3 Two‐way ANOVA tables of total moss growth.
Table S4 Percentage change of total moss growth between microbiome transfers.
Table S5 Two‐way ANOVA tables of moss fluorescence (F v/F m).
Table S6 Percentage change of fluorescence at harvest.
Table S7 Number of paired‐end bacterial/archaeal amplicon reads.
Table S8 Bacterial/archaeal amplicon‐based community abundance of starting inoculum.
Table S9 Bacterial/archaeal amplicon‐based community abundance at the end of laboratory incubations.
Table S10 Fungal amplicon (ITS) phylum‐level taxonomy of microbiomes at the end of the laboratory experiment.
Table S11 Fungal (ITS) class level taxonomy of microbiomes at the end of the laboratory experiment.
Table S12 Descriptive statistics and log2 fold change of metagenome assembled genomes (MAG).
Table S13 Number of paired‐end metatranscriptomic reads passing quality control.
Table S14 Log2 fold change of SEED subsystem level 3 gene ontologies for the microbial fraction of metatranscriptomic reads.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We are grateful for the editor and reviewers for detailed and constructive comments, presubmission comments from Dr Gustaf Granath and field site maintenance from Robert Nettles III. Collection of starting microbial inocula was made possible through the SPRUCE project, which is supported by the Office of Science; Biological and Environmental Research (BER); US Department of Energy (DOE), grant/award no. DE‐AC05–00OR22725. Experimentation, sample collection and analyses were supported by the DOE BER Early Career Research Program. Oak Ridge National Laboratory is managed by UT‐Battelle, LLC, for the US DOE under contract no. DE‐AC05‐00OR22725. AJS was supported by NSF DEB‐1737899, 1928514. The work conducted by the US DOE Joint Genome Institute (JGI) is supported by the Office of Science of the US Department of Energy under contract no. DE‐AC02‐05CH11231. We thank the DOE JGI and collaborators for prepublication access to the S. angustifolium (formerly S. fallax) genome sequence.
Data availability
Raw 16S and ITS sequence files can be found on NCBI using the BioProject ID PRJNA644113. Raw metagenome and metatranscriptome sequence files can be found on NCBI using the BioProject ID PRJNA644538.
References
- Ali SZ, Sandhya V, Grover M, Kishore N, Rao LV, Venkateswarlu B. 2009. Pseudomonas sp. strain AKM‐P6 enhances tolerance of sorghum seedlings to elevated temperatures. Biology and Fertility of Soils 46: 45–55. [Google Scholar]
- Apprill A. 2020. The role of symbioses in the adaptation and stress responses of marine organisms. Annual Review of Marine Science 12: 291–314. [DOI] [PubMed] [Google Scholar]
- Baker DM, Freeman CJ, Wong JCY, Fogel ML, Knowlton N. 2018. Climate change promotes parasitism in a coral symbiosis. ISME Journal 12: 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilier K. 1979. Moss‐associated nitrogen fixation in some mire and coniferous forest environments around Uppsala, Sweden. Lindbergia 5: 84–88. [Google Scholar]
- Basilier K. 1980. Fixation and uptake of nitrogen in sphagnum blue‐green algal associations. Oikos 34: 239–242. [Google Scholar]
- Basilier K, Granhall U, Stenström T‐A, Stenstrom T‐A. 1978. Nitrogen fixation in wet minerotrophic moss communities of a subarctic mire. Oikos 31: 236–246. [Google Scholar]
- Basińska AM, Reczuga MK, Gąbka M, Stróżecki M, Łuców D, Samson M, Urbaniak M, Leśny J, Chojnicki BH, Gilbert D et al. 2020. Experimental warming and precipitation reduction affect the biomass of microbial communities in a Sphagnum peatland. Ecological Indicators 112: 106059. [Google Scholar]
- Bay LK, Doyle J, Logan M, Berkelmans R, Bay LK. 2016. Recovery from bleaching is mediated by threshold densities of background thermo‐tolerant symbiont types in a reef‐building coral. Royal Society Open Science 3: 160322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg A, Danielsson Å, Svensson BH. 2013. Transfer of fixed‐N from N2‐fixing cyanobacteria associated with the moss Sphagnum riparium results in enhanced growth of the moss. Plant and Soil 362: 271–278. [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al‐Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon‐Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe‐Fadrosh EA et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome‐assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology 35: 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragazza L. 2008. A climatic threshold triggers the die‐off of peat mosses during an extreme heat wave. Global Change Biology 14: 2688–2695. [Google Scholar]
- Bragazza L, Buttler A, Robroek BJM, Albrecht R, Zaccone C, Jassey VEJ, Signarbieux C. 2016. Persistent high temperature and low precipitation reduce peat carbon accumulation. Global Change Biology 22: 4114–4123. [DOI] [PubMed] [Google Scholar]
- van Breemen N. 1995. How Sphagnum bogs down other plants. Trends in Ecology & Evolution 10: 270–275. [DOI] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2014. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. 2011. Bacterial community assembly based on functional genes rather than species. Proceedings of the National Academy of Sciences, USA 108: 14288–14293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. 2016. DADA2: high‐resolution sample inference from Illumina amplicon data. Nature Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell AA, Kolton M, Glass JB, Pelletier DA, Warren MJ, Kostka JE, Iversen CM, Hanson PJ, Weston DJ. 2019. Experimental warming alters the community composition, diversity, and N2 fixation activity of peat moss (Sphagnum fallax) microbiomes. Global Change Biology 25: 2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell AA, Veličković D, Lawrence TJ, Bowen BP, Louie KB, Carper DL, Chu RK, Mitchell HD, Orr G, Markillie LM et al. 2021. Novel metabolic interactions and environmental conditions mediate the boreal peatmoss‐cyanobacteria mutualism. ISME Journal 29: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SM, Cristescu SM, Miersch O, Harren FJM, Wasternack C, Mur LAJ. 2009. Jasmonates act with salicylic acid to confer basal thermotolerance in Arabidopsis thaliana . New Phytologist 182: 175–187. [DOI] [PubMed] [Google Scholar]
- Clymo RS, Hayward PM. 1982. The ecology of Sphagnum . In: Smithn AJE, ed. Bryophyte ecology. Dordrecht, the Netherlands: Springer, 229–289. [Google Scholar]
- Conrath U, Beckers GJM, Flors V, García‐Agustín P, Jakab G, Mauch F, Newman M‐A, Pieterse CMJ, Poinssot B, Pozo MJ et al. 2006. Priming: getting ready for battle. Molecular Plant–Microbe Interactions 19: 1062–1071. [DOI] [PubMed] [Google Scholar]
- Cregger MA, Veach AM, Yang ZK, Crouch MJ, Vilgalys R, Tuskan GA, Schadt CW. 2018. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunning R, Silverstein RN, Baker AC. 2015. Investigating the causes and consequences of symbiont shuffling in a multi‐partner reef coral symbiosis under environmental change. Proceedings of the Royal Society B: Biological Sciences 282: 20141725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, Weisberg S. 2018. An R companion to applied regression. New York, NY, USA: Sage Publications. [Google Scholar]
- Giauque H, Connor EW, Hawkes CV. 2019. Endophyte traits relevant to stress tolerance, resource use and habitat of origin predict effects on host plants. New Phytologist 221: 2239–2249. [DOI] [PubMed] [Google Scholar]
- Giauque H, Hawkes CV. 2013. Climate affects symbiotic fungal endophyte diversity and performance. American Journal of Botany 100: 1435–1444. [DOI] [PubMed] [Google Scholar]
- Gorham E. 1991. Northern peatlands: role in the carbon cycle and probable responses to climatic warming. Ecological Applications 1: 182–195. [DOI] [PubMed] [Google Scholar]
- Granhall ULF, Hofsten AV. 1976. Nitrogenase activity in relation to intracellular organisms in Sphagnum mosses. Physiologia Plantarum 36: 88–94. [Google Scholar]
- Gunnarsson U, Granberg G, Nilsson M. 2004. Growth, production and interspecific competition in Sphagnum: effects of temperature, nitrogen and sulphur treatments on a boreal mire. New Phytologist 163: 349–359. [DOI] [PubMed] [Google Scholar]
- Hanson PJ, Gill AL, Xu X, Phillips JR, Weston DJ, Kolka RK, Riggs JS, Hook LA. 2016. Intermediate‐scale community‐level flux of CO2 and CH4 in a Minnesota peatland: putting the SPRUCE project in a global context. Biogeochemistry 129: 255–272. [Google Scholar]
- Heath KD, Stock AJ, Stinchcombe JR. 2010. Mutualism variation in the nodulation response to nitrate. Journal of Evolutionary Biology 23: 2494–2500. [DOI] [PubMed] [Google Scholar]
- Heck MA, Lüth VM, van Gessel N, Krebs M, Kohl M, Prager A, Joosten H, Decker EL, Reski R. 2021. Axenic in vitro cultivation of 19 peat moss (Sphagnum L.) species as a resource for basic biology, biotechnology, and paludiculture. New Phytologist 229: 861–876. [DOI] [PubMed] [Google Scholar]
- Howells EJ, Abrego D, Meyer E, Kirk NL, Burt JA. 2016. Host adaptation and unexpected symbiont partners enable reef‐building corals to tolerate extreme temperatures. Global Change Biology 22: 2702–2714. [DOI] [PubMed] [Google Scholar]
- Huang J, Gu M, Lai Z, Fan B, Shi K, Zhou YH, Yu JQ, Chen Z. 2010. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiology 153: 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupperts SF, Gerber S, Nilsson MC, Gundale MJ. 2021. Empirical and Earth system model estimates of boreal nitrogen fixation often differ: a pathway toward reconciliation. Global Change Biology 27: 5711–5725. [DOI] [PubMed] [Google Scholar]
- Jassey VEJ, Signarbieux C, Hättenschwiler S, Bragazza L, Buttler A, Delarue F, Fournier B, Gilbert D, Laggoun‐Défarge F, Lara E et al. 2015. An unexpected role for mixotrophs in the response of peatland carbon cycling to climate warming. Scientific Reports 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kip N, van Winden JF, Pan Y, Bodrossy L, Reichart G‐J, Smolders AJP, Jetten MSM, Damsté JSS, Op den Camp HJM. 2010. Global prevalence of methane oxidation by symbiotic bacteria in peat‐moss ecosystems. Nature Geoscience 3: 617–621. [Google Scholar]
- Kostka JE, Weston DJ, Glass JB, Lilleskov EA, Shaw AJ, Turetsky MR. 2016. The Sphagnum microbiome: new insights from an ancient plant lineage. New Phytologist 211: 57–64. [DOI] [PubMed] [Google Scholar]
- Kotak S, Larkindale J, Lee U, von Koskull‐Döring P, Vierling E, Scharf KD. 2007. Complexity of the heat stress response in plants. Current Opinion in Plant Biology 10: 310–316. [DOI] [PubMed] [Google Scholar]
- Lamentowicz M, Mitchell EAD. 2005. The ecology of testate amoebae (protists) in Sphagnum in north‐western Poland in relation to peatland ecology. Microbial Ecology 50: 48–63. [DOI] [PubMed] [Google Scholar]
- Larkindale J, Mishkind M, Vierling E. 2007. Plant responses to high temperature. Plant Abiotic Stress 100: 144. [Google Scholar]
- Larmoia T, Leppanen SM, Tuittila ES, Aarva M, Merila P, Fritze H, Tiirola M. 2014. Methanotrophy induces nitrogen fixation during peatland development. Proceedings of the National Academy of Sciences, USA 111: 734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JA, Lennon JT. 2012. Microbe‐mediated adaptation to novel environments. Proceedings of the National Academy of Sciences, USA 109: 14058–14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Research 47: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Svenning MM. 2013. Environmental transcription of mmoX by methane‐oxidizing Proteobacteria in a subarctic palsa peatland. Applied and Environmental Microbiology 79: 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindo Z, Nilsson MC, Gundale MJ. 2013. Bryophyte‐cyanobacteria associations as regulators of the northern latitude carbon balance in response to global change. Global Change Biology 19: 2022–2035. [DOI] [PubMed] [Google Scholar]
- Mclellan CA, Turbyville TJ, Kithsiri Wijeratne EM, Kerschen A, Vierling E, Queitsch C, Whitesell L, Gunatilaka AAL. 2007. A rhizosphere fungus enhances Arabidopsis thermotolerance through production of an HSP90 inhibitor. Plant Physiology 145: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A, Ferrer M. 2016. Functional redundancy‐induced stability of gut microbiota subjected to disturbance. Trends in Microbiology 24: 402–413. [DOI] [PubMed] [Google Scholar]
- Norby RJ, Childs J, Hanson PJ, Warren JM. 2019. Rapid loss of an ecosystem engineer: S phagnum decline in an experimentally warmed bog. Ecology and Evolution 9: 12571–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M et al. 2014. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Research 42: D206–D214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2021. R: a language and environment for statistical computing, v.3.6.1, R 4.1. Vienna, Austria: R Foundation for Statistical Computing. [WWW document] URL https://www.R‐project.org/ [accessed 1 August 2021]. [Google Scholar]
- Raghoebarsing AA, Smolders AJP, Schmid MC, Rijpstra WIC, Wolters‐Arts M, Derksen J, Jetten MSM, Schouten S, Sinninghe Damsté JS, Lamers LPM et al. 2005. Methanotrophic symbionts provide carbon for photosynthesis in peat bogs. Nature 436: 1153–1156. [DOI] [PubMed] [Google Scholar]
- Reczuga MK, Seppey CVW, Mulot M, Jassey VEJ, Buttler A, Słowińska S, Słowiński M, Lara E, Lamentowicz M, Mitchell EAD. 2020. Assessing the responses of Sphagnum micro‐eukaryotes to climate changes using high throughput sequencing. PeerJ 8: e9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman RS, Kim YO, Woodward CJDA, Greer C, Espino L, Doty SL, Rodriguez RJ. 2011. Increased fitness of rice plants to abiotic stress via habitat adapted symbiosis: a strategy for mitigating impacts of climate change. PLoS ONE 6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman RS, Sheehan KB, Stout RG, Rodriguez RJ, Henson JM. 2002. Thermotolerance generated by plant/fungal symbiosis. Science 298: 1581. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu DI, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Research 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robroek BJM, Limpens J, Breeuwer A, Crushell PH, Schouten MGC. 2007a. Interspecific competition between Sphagnum mosses at different water tables. Functional Ecology 21: 805–812. [Google Scholar]
- Robroek BJM, Limpens J, Breeuwer A, Schouten MGC. 2007b. Effects of water level and temperature on performance of four Sphagnum mosses. Plant Ecology 190: 97–107. [Google Scholar]
- Rodriguez RJ, Henson J, Van Volkenburgh E, Hoy M, Wright L, Beckwith F, Kim Y‐O, Redman RS. 2008. Stress tolerance in plants via habitat‐adapted symbiosis. ISME Journal 2: 404–416. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nature Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacke R, Ponce‐Soto GY, Krause K, Bolger AM, Arsova B, Hallab A, Gruden K, Stitt M, Bolger ME, Usadel B. 2019. MapMan4: a refined protein classification and annotation framework applicable to multi‐omics data analysis. Molecular Plant 12: 879–892. [DOI] [PubMed] [Google Scholar]
- Shekhawat K, Saad MM, Sheikh A, Mariappan K, Al‐Mahmoudi H, Abdulhakim F, Eida AA, Jalal R, Masmoudi K, Hirt H. 2021. Root endophyte induced plant thermotolerance by constitutive chromatin modification at heat stress memory gene loci. EMBO Reports 22: e51049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PM, Wu D, Latifi A, Axen SD, Fewer DP, Talla E, Calteau A, Cai F, Tandeau de Marsac N, Rippka R et al. 2013. Improving the coverage of the cyanobacterial phylum using diversity‐driven genome sequencing. Proceedings of the National Academy of Sciences, USA 110: 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stough JMA, Kolton M, Kostka JE, Weston DJ, Pelletier DA, Wilhelm SW. 2018. Diversity of active viral infections within the Sphagnum microbiome. Applied and Environmental Microbiology 84: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart RK, Pederson ER, Weyman PD, Weber PK, Rassmussen U, Dupont CL . 2020. Bidirectional C and N transfer and a potential role for sulfur in an epiphytic diazotrophic mutualism. ISME Journal 14: 3068–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung DY, Vierling E, Guy CL. 2001. Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiology 126: 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. 2014. MAPMAN: a user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Timm CM, Pelletier DA, Jawdy SS, Gunter LE, Henning JA, Engle N, Aufrecht J, Gee E, Nookaew I, Yang Z et al. 2016. Two poplar‐associated bacterial isolates induce additive favorable responses in a constructed plant‐microbiome system. Frontiers in Plant Science 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vile MA, Kelman Wieder R, Živković T, Scott KD, Vitt DH, Hartsock JA, Iosue CL, Quinn JC, Petix M, Fillingim HM et al. 2014. N2‐fixation by methanotrophs sustains carbon and nitrogen accumulation in pristine peatlands. Biogeochemistry 121: 317–328. [Google Scholar]
- Warshan D, Espinoza JL, Stuart RK, Richter RA, Kim S‐Y, Shapiro N, Woyke T, C Kyrpides N, Barry K, Singan V et al. 2017. Feathermoss and epiphytic Nostoc cooperate differently: expanding the spectrum of plant–cyanobacteria symbiosis. ISME Journal 11: 2821–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston DJ, Timm CM, Walker AP, Gu L, Muchero W, Schmutz J, Shaw AJ, Tuskan GA, Warren JM, Wullschleger SD. 2014. Sphagnum physiology in the context of changing climate: emergent influences of genomics, modelling and host‐microbiome interactions on understanding ecosystem function. Plant, Cell & Environment 38: 1737–1751. [DOI] [PubMed] [Google Scholar]
- Weston DJ, Turetsky MR, Johnson MG, Granath G, Lindo Z, Belyea LR, Rice SK, Hanson DT, Engelhardt KA, Schmutz J et al. 2018. The sphagnome project: enabling ecological and evolutionary insights through a genus‐level sequencing project. New Phytologist 217: 16–25. [DOI] [PubMed] [Google Scholar]
- Westreich ST, Korf I, Mills DA, Lemay DG. 2016. SAMSA: a comprehensive metatranscriptome analysis pipeline. BMC Bioinformatics 17: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Ohshika J, Takahashi T, Ishizaki K, Kohchi T, Matusuura H, Takahashi K. 2015. Functional analysis of allene oxide cyclase, MpAOC, in the liverwort Marchantia polymorpha . Phytochemistry 116: 48–56. [DOI] [PubMed] [Google Scholar]
- Yu Z, Loisel J, Brosseau DP, Beilman DW, Hunt SJ. 2010. Global peatland dynamics since the Last Glacial Maximum. Geophysical Research Letters 37: 1–5. [Google Scholar]
- Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR. 2017. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nature Communications 8: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1 Log fold change of RNA‐seq profiles from plants with ambient‐ and warm‐microbiomes across temperature treatments and ambient and warming microbiome treatments.
Dataset S2 Log fold change of RNA‐seq profiles from plant microbiomes with ambient and warming microbiomes treatments.
Fig. S1 Average moss fluorescence in 2016 at the end of the experiment.
Fig. S2 Average moss fluorescence in 2017 at the end of the experiment.
Fig. S3 Average moss growth rate under ambient or warming treatments.
Fig. S4 Bacterial/archaeal amplicon nonmetric multidimensional scaling (NMDS) ordination of the Bray–Curtis distance matrix of inoculum and field.
Fig. S5 Bacterial/archaeal amplicon nonmetric multidimensional scaling (NMDS) ordination of the Bray–Curtis distance matrix of samples at the end of the experiment.
Fig. S6 Linear correlation of plant growth rate and fungal (ITS) Shannon diversity at the conclusion of the experiment.
Fig. S7 Boxplot of percentage of metatranscriptomic reads mapping to Sphagnum, metagenome assembly and metagenome assembled genomes.
Fig. S8 MDS of the top 500 most variable microbial SEED level 3 categories.
Methods S1 Metagenome sequencing and analysis.
Table S1 Incubation temperature and light cycle for 2016 and 2017 laboratory experiments.
Table S2 Summary growth rate and total moss growth over 4 wk.
Table S3 Two‐way ANOVA tables of total moss growth.
Table S4 Percentage change of total moss growth between microbiome transfers.
Table S5 Two‐way ANOVA tables of moss fluorescence (F v/F m).
Table S6 Percentage change of fluorescence at harvest.
Table S7 Number of paired‐end bacterial/archaeal amplicon reads.
Table S8 Bacterial/archaeal amplicon‐based community abundance of starting inoculum.
Table S9 Bacterial/archaeal amplicon‐based community abundance at the end of laboratory incubations.
Table S10 Fungal amplicon (ITS) phylum‐level taxonomy of microbiomes at the end of the laboratory experiment.
Table S11 Fungal (ITS) class level taxonomy of microbiomes at the end of the laboratory experiment.
Table S12 Descriptive statistics and log2 fold change of metagenome assembled genomes (MAG).
Table S13 Number of paired‐end metatranscriptomic reads passing quality control.
Table S14 Log2 fold change of SEED subsystem level 3 gene ontologies for the microbial fraction of metatranscriptomic reads.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Raw 16S and ITS sequence files can be found on NCBI using the BioProject ID PRJNA644113. Raw metagenome and metatranscriptome sequence files can be found on NCBI using the BioProject ID PRJNA644538.