

Summary
Precision oncology has made significant advances, mainly by targeting actionable mutations in cancer driver genes. Aiming to expand treatment opportunities, recent studies have begun to explore the utility of tumor transcriptome to guide patient treatment. Here we introduce SELECT (SynthEtic LEthality and rescue-mediated precision onCology via the Transcriptome), a precision oncology framework harnessing genetic interactions to predict patient response to cancer therapy from the tumor transcriptome. SELECT is tested on a broad collection of 35 published targeted and immunotherapy clinical trials from 10 different cancer types. It is predictive of patients’ response in 80% of these clinical trials and in the recent multi-arm WINTHER trial. The predictive signatures and the code are made publicly available for academic use, laying a basis for future prospective clinical studies.
eTOC blurb
SELECT is a precision oncology framework to analyze the tumor transcriptome. The synthetic lethal partners of drug targets are predictive of patients’ response to targeted and immunotherapies across a large number of clinical trials.
Introduction
There have been significant advances in precision oncology, with an increasing adoption of sequencing tests that identify targetable mutations in cancer driver genes. Aiming to complement these efforts by considering genome-wide tumor alterations at additional “-omics” layers, recent studies have begun to explore the utilization of transcriptomics data to guide cancer patients’ treatment (Beaubier et al., 2019; Hayashi et al., 2020; Rodon et al., 2019; Tanioka et al., 2018; Vaske et al., 2019; Wong et al., 2020). These studies have reported encouraging results, testifying to the potential of such approaches to complement mutation panels and increase the likelihood that patients will benefit from genomics-guided, precision treatments. However, current approaches for utilizing tumor transcriptomics data to guide patient treatments are still of heuristic exploratory nature, raising the need for developing and testing new systematic approaches.
Here we present a precision oncology framework, SELECT, aimed at selecting the best drugs for a given patient based on the tumor transcriptome. Unlike recent transcriptome-based approaches that are focused on matching drugs based on the expression of their targets (Beaubier et al., 2019; Rodon et al., 2019), our approach is based on identifying and utilizing the broader scope of genetic interactions (GI) of drug targets, which provide biologically testable biomarkers for therapy response prediction. We focus on two major types of GIs that are highly relevant to predicting the response to cancer therapies: (1) Synthetic lethal (SL) interactions, which describe the relationship between two genes whose concomitant inactivation, but not their individual inactivation, reduces cell viability (e.g., an SL interaction that is widely used in the clinic is PARP inhibitors on the background of disrupted DNA repair) (Lord and Ashworth, 2017). (2) Synthetic rescue (SR) interactions, which denote a type of genetic interactions where a change in the activity of one gene reduces the cell’s fitness but an alteration of another gene’s activity (termed its SR partner) rescues cell viability (e.g., the rescue of Myc alterations by BCL2 activation in lymphomas (Eischen et al., 2001)). These SR interactions are of interest because when a gene is targeted by a small molecule inhibitor or an antibody, the tumor may respond by up or down regulating its rescuer gene(s), conferring resistance to therapies.
To identify the SL and SR partners of cancer drugs, we leverage on our two recently published computational pipelines (Lee et al., 2018; Sahu et al., 2019), which identify genetic dependencies that are supported by multiple layers of omics data, including in vitro functional screens, patient tumor DNA and RNA sequencing data, and phylogenetic profile similarity across multiple species. Applying these pipelines, we have previously successfully identified a Gq-driver mutation as marker for FAK inhibitor SL treatment in uveal melanoma (Feng et al., 2019) and a synergistic SL combination for treating melanoma and pancreatic tumors with Asparaginase and MAPK inhibitors (Pathria et al., 2019). We also identified SR interactions that mediate resistance to checkpoint therapies in melanoma (Sahu et al., 2019). However, the fundamental question whether genetic dependencies inferred from multi-omics tumor data can be used to determine efficacious therapeutics for individual cancer patients has not been addressed so far. Here we present and study a computational framework, termed SELECT, to address this challenge. The end result is a systematic approach for robustly predicting patients’ response to targeted and immune therapies across tens of different treatments and cancer types, offering an alternative way to complement existing mutation-based approaches.
Results
Overview of the SELECT framework and the analysis
We collected cancer patient pre-treatment transcriptomics profiles together with therapy response information from numerous publicly available databases, surveying Gene Expression Omnibus (GEO), ArrayExpress and the literature, and a new unpublished cohort of anti-PD1 treatment in lung adenocarcinoma. Overall, we could find 48 such datasets that include both transcriptomics and clinical response data, spanning 13 chemotherapy, 14 targeted therapy and 21 immunotherapy datasets across 13 different cancer types. This test collection is at a scale surpassing all previous efforts to predict patients ‘ response to anti-cancer treatments, at least to our knowledge (Supplemental Table 1; the transcriptomics profiles and the treatment outcome information are publicly available for the 23 out of 48 datasets and those are made accessible; see Data and Code Availability).
The SELECT framework consists of two basic steps: (A) For each drug whose response we aim to predict, we first identify the clinically relevant pan-cancer GIs (the interactions found to be shared across many cancer types) of the drug’s target genes (Law et al., 2014). (B) The identified GI partners of the drug emerging from step (A) are then used to predict a given patient’s response to a given treatment based on her/his tumor’s gene expression (see Figure 1A,B and the STAR Methods for a complete description of this process both SL and SR interactions, the latter used to predict response to immune checkpoint therapy). The prediction of response to targeted and chemotherapy drugs is based on the SL partners of the drug targets, while the prediction of response to checkpoint therapy is based on the SR partners of these drugs’ targets.
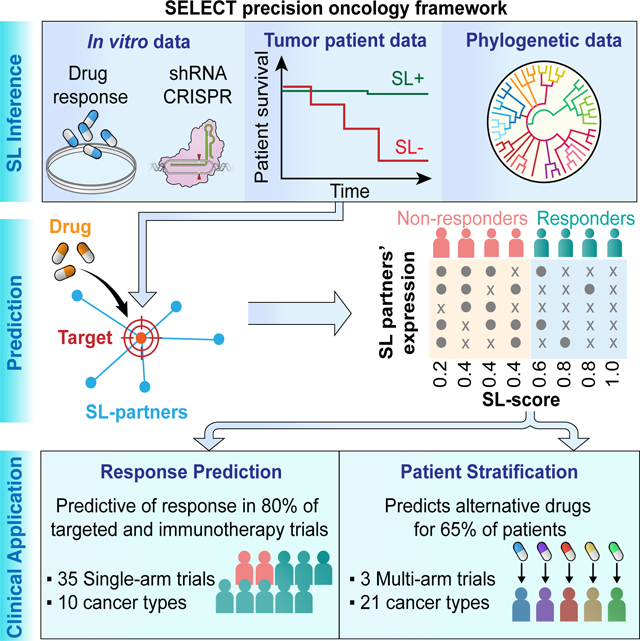
Figure 1. SELECT precision oncology framework.

(A-B) The SELECT precision oncology framework is composed of two steps: (i) identifying SL interaction partners of drug targets and (ii) predicting drug response in patients using SL partners. (A) The SL partners (gene P) of the drug target genes (gene T) are supported by genetic dependencies in cell lines, patient tumor data, and phylogenetic profiles. (B) The identified SL partners of the drug target genes are used to compute an SL-score to predict the response to the given therapy. See also Supplemental Figure 1,2, Supplemental Table 2.
Here we provide a short overview of the pertaining SL and SR pipelines, as follows:
(A). Identifying SL interaction partners of drug targets (Figure 1A).
(A.1) Following a modified version of ISLE (Lee et al., 2018), we begin by generating an initial pool of SL drug target interactions for targeted therapy: For each drug we compile a list of initial candidate SL pairs of its targets by analyzing cancer cell line dependencies with RNAi, CRISPR/Cas9, or pharmacological inhibition (Aguirre et al., 2016; Barretina et al., 2012; Basu et al., 2013; Cheung et al., 2011; Cowley et al., 2014; Iorio et al., 2016; Marcotte et al., 2012; Marcotte et al., 2016; Tsherniak et al., 2017). Among these candidate SL pairs, we select those that are more likely to be clinically relevant by analyzing the TCGA data, identifying pairs that are significantly associated with better patient survival. Finally, among the candidate pairs that remain after these two steps we select those pairs that are supported by a phylogenetic profiling analysis (Lee et al., 2018). The top significant SL partners that pass all these filters form the candidate pool of SL partners of the specific drug in hand. However, this typically results in hundreds of significant candidate GI partners for each drug, a number which needs to be markedly reduced to obtain generalizable and biologically meaningful biomarker stratification signatures. (A.2) In the second step, SELECT generates a reduced set of interaction partners to make therapy response predictions, by choosing the top 25 SL pairs found as the compact SL signature of each drug. This number of optimal SL set size is based on a minimal amount of supervised learning performed analyzing just one single targeted dataset, as described in detail in STAR Methods. The following step describes how these SL pairs are then used to predict the response of a given patient to targeted therapies.
(B). Predicting drug response in patients using the drug’s SL partners identified in step (A) (Figure 1B).
The SL partners of the drug identified in step (A) are then used to predict a given patient’s response to a given treatment based on her/his tumor’s gene expression. This is based on the notion that a drug will be more effective against the tumor when its SL partners are down-regulated, because when the drug inhibits its targets, more SL interactions will become jointly down-regulated and hence ‘activated’. To quantify the extent of such predicted lethality, we assign an SL-score denoting the fraction of down-regulated SL partners of a drug in a given tumor (STAR Methods). The larger the fraction of SL partners that are down-regulated, the higher the SL-score and the more likely the patient is predicted to respond to the given therapy. Predictions of patient response to checkpoint therapy are based on SR pairs of the drug targets, which yield a stronger signal than their SL partners in this case. The process to infer the SR pairs of drugs and then their SR scores in each patient is conceptually analogous to the process described above for targeted therapies (focusing on the top 10 pairs). The SR score of a drug in a given patient quantifies the fraction of its down-regulated SR partner genes based on the patient’s tumor transcriptomics, and hence the likelihood of resistance to the given therapy (STAR Methods). In defining responders vs non-responders, in all predictions made we followed the criteria used in each of the clinical trials as explicitly listed in Supplemental Table 1. In most cases, we take CR and PR as responders and SD and PD as non-responders, but there are some exceptions in cohorts where the number of samples are unevenly distributed or the response was not evaluated based on RECIST criteria, as detailed in STAR Methods and Supplemental Table 1.
We emphasize that the SL/SR partners were inferred only once analyzing cancer cell line and TCGA data and their set size was optimized by training on only one single clinical trial dataset, prior to their application to a large collection of other test clinical trial datasets (STAR Methods). In other words, the transcriptomic profiles and treatment outcome data available in all the independent test clinical trial cohorts were never used in the SL and SR inference and hence the latter serve as independent test sets (Figure 1B). Importantly, throughout the study we used the same, fixed, sets of parameters in making the predictions for targeted and immunotherapies. Taken together, these procedures markedly reduce the well-known risk of obtaining over-fitted predictors that would fail to predict on datasets other than those on which they were originally built.
SELECT prediction of response to targeted cancer therapies
We start by analyzing four melanoma cohorts treated with BRAF inhibitors, for which pretreatment transcriptomics data and response information are available (Hugo et al., 2015; Kakavand et al., 2017; Rizos et al., 2014). Applying SELECT, we identified the 25 most significant SL partners of BRAF (Supplemental Table 2), where the number 25 was determined from training on one single dataset and kept fixed thereafter in all targeted therapies predictions (see STAR Methods for the details of parameter choices; Supplemental Figures 1,2). As expected, we find that responders have higher SL-scores than non-responders in the three melanoma-BRAF cohorts for which therapy response data is available (Figure 2A). Quantifying the predictive power via the use of the standard area under the receiver operating characteristics curve (AUC of the ROC curve) measure, we find AUCs greater than 0.7 in all three datasets, and an aggregate performance of AUC=0.71 when the three cohorts are merged (Figure 2B). As some datasets do not have a balanced number of responders and non-responders, we additionally quantified the resulting performance via precision-recall curves (often used as supplement to the routinely used ROC curves, Supplemental Figure 3A). As evident from the latter, one can choose a single classification threshold that successfully captures most true responders while misclassifying less than half of the non-responders. Even though all patients in these three cohorts have either a BRAF V600E or V600K mutation, there is still a large variability in their response. SELECT successfully captures some of this variability to predict the patients who will respond better to BRAF inhibition.
Figure 2. SELECT stratifies melanoma patients for BRAF inhibitors based on the expression of BRAF SL partners.

(A) SL-scores are significantly higher in responders (green) vs non-responders (red) based on Wilcoxon rank-sum test after multiple hypothesis correction. For false discovery rates: * denotes 10% and ** denotes 5%. (B) ROC curves depicting the prediction accuracy of the response to BRAF inhibition using SL-scores in the three melanoma cohorts (red, yellow, blue) and their aggregation (green). The stars denote the point of the maximal Fl-score. (C) Bar graphs show the predictive accuracy in terms of Area Under the Curve (AUC) of ROC curve (Y-axis) of SL-based predictors (red) and controls including several known transcriptomics-deduced metrics (IFNg signature, proliferation index, cytolytic score, and the drug target expression levels) and several interaction-based scores (based on randomly chosen partners, randomly chosen PPI partners of the drug target gene(s), the identified SL partners of other cancer drugs, and experimentally identified SL partners) in the three BRAF inhibitor cohorts (X-axis). (D) Bar graphs showing the fraction of responders in the patients with high SL-scores (top tertile; green) and low SL-scores (bottom tertile; purple). The grey line denotes the overall response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SL group and depletion of responders in the low-SL group (compared to their baseline frequency in the cohort). (E,F) Kaplan-Meier curves depicting the survival of patients with low (yellow) vs high (blue) BRAF SL-scores (top vs. bottom tertile of SL-scores) of (E) GSE50509 (Rizos et al., 2014) and (F) independent (unseen) BRAF inhibitor clinical trials (Wongchenko et al., 2017). Patients with high SL-scores show better prognosis, as expected. The logrank P-value and median survival difference are denoted. See also Supplemental Figure 2,3, Supplemental Table 2,5.
Reassuringly, the SL based prediction accuracy levels are overall higher compared to those obtained by several published transcriptomic based predictors, including the proliferation index (Whitfield et al., 2006), IFNg signature (Ayers et al., 2017), cytolytic score (Rooney et al., 2015), or the expression of the drug target gene itself (BRAF in this case). SL based prediction accuracy levels are also better than other interaction-based scores, including the fraction of down-regulated randomly selected genes, the fraction of in vitro experimentally determined SL partners (i.e. genes that pass the first step of Figure 1A), the fraction of the inferred SL partners of other drugs, or the fraction of down-regulated protein-protein interaction partners of drug targets (all of sizes similar to the SL set; empirical P<0.001) (Figure 2C). Although studies have reported that V600E is associated with better response to vemurafenib than V600K patients (Pires da Silva et al., 2019), this has not been observed in the three melanoma cohorts that we analyzed, where the BRAF 600E mutant patients are not significantly enriched in responders. The BRAF SL-score and the BRAF mutation status are not significantly correlated, demonstrating that SL-score is not a mere readout of BRAF oncogenic mutation states (Supplemental Figure 3B). The patients with high SL-score (defined as those in the top tertile) show significantly higher rate of response than the overall response rate, and the patients with low SL-score (in the bottom tertile) show the opposite trend (Figure 2D). We observe that patients with higher SL-scores showed significantly better treatment outcome in terms of progression-free survival in the dataset analyzed above where this data was available to us (Figure 2E); the integrated analysis of large-scale BRAF inhibitor clinical trials (Wongchenko et al., 2017) (N=621) also shows that the SL-score is associated with significantly improved progression-free survival (Figure 2F). As expected, the SL partners of BRAF were found to be enriched with the functional annotation ‘regulation of GTPase mediated signal transduction’ (Fisher exact test P<0.002).
We next test the prediction accuracy of the SL-based approach on a variety of targeted therapies and cancer types. We collected 23 additional publicly available datasets from clinical trials of cytotoxic agents and targeted cancer therapies, each one containing both pre-treatment transcriptomics data and therapy response information. This compendium includes breast cancer patients treated with lapatinib (Guarneri et al., 2015), tamoxifen (Desmedt et al., 2009), or gemcitabine (Julka et al., 2008); colorectal cancer patients treated with irinotecan (Graudens et al., 2006), multiple myeloma patients treated with bortezomib (Terragna et al., 2016) or dexamethasone (Manojlovic et al., 2017), acute myeloid leukemia treated with gemtuzumab (Bolouri et al., 2018), and hepatocellular carcinoma patients treated with sorafenib (Pinyol et al., 2019). In each dataset, we first identified the SL interaction partners of the targets of the drug given in that trial (STAR Methods; Supplemental Table 2) and based on those, we computed the SL-score of each sample from its transcriptomics.
We first note that SELECT mostly fails in predicting the response to cytotoxic agents, obtaining AUC>0.7 in only 2 out of 11 of these datasets, where RECIST information was available. This is not surprising given that the SL approach relies on the specificity and correct identification of the targets of each drug, and cytotoxic agents typically have a multitude of targets which are often poorly defined. This is in difference from the more recently developed targeted and checkpoint therapies which have better defined targets. Indeed, we find that higher SL-scores are associated with better response in 4 out of 7 of targeted therapy datasets we could test. The results for the therapies that are successfully predicted (AUC’s all greater than 0.7) are presented in Figures 3A,B, and the pertaining precision-recall curves are shown in Supplemental Figure 3C. The predictive performance of a variety of expression-based control predictors is random (Figure 3C). Patients with high SL-scores (within top tertile) have significantly higher response rates than the overall response rates, and the patients with low SL-scores (within bottom tertile) show the opposite trend (Supplemental Figure 3D). Importantly, in the four datasets where we have survival information, we observe that patients with higher SL-scores also have improved overall survival (Figures 3D-G).
Figure 3. SELECT stratifies patients for targeted therapies across different cancer types.

(A) SL-scores are significantly higher in responders (green) vs non-responders (red) based on Wilcoxon ranksum test after multiple hypothesis correction. For false discovery rates: * denotes 10%, ** denotes 5%, and *** denotes 1%. Cancer types are noted on the top of each dataset. (B) ROC curves for breast cancer patients treated with lapatinib (GSE66399) (Guarneri et al., 2015), tamoxifen (GSE16391) (Desmedt et al., 2009), gemcitabine (GSE8465) (Julka et al., 2008), colorectal cancer patients treated with irinotecan (GSE3964) (Graudens et al., 2006), multiple myeloma patients treated with bortezomib (GSE68871) (Terragna et al., 2016), and hepatocellular carcinoma patients treated with sorafenib (GSE109211) (Pinyol et al., 2019). The circles denote the point of maximal F1-score. (C) Bar graphs show the predictive accuracy in terms of AUCs (Y-axis) of SL-based predictors and a variety of controls specified earlier in Figure 2C (X-axis). (D-G) Kaplan-Meier curves depicting the survival of patients with low vs high SL-scores of (D) multiple myeloma patients treated with dexamethasone (Manojlovic et al., 2017), and (E) acute myeloid leukemia patients treated with gemtuzumab (Bolouri et al., 2018), (F) breast cancer treated with tamoxifen (GSE16391) (Desmedt et al., 2009), and (G) breast cancer cohorts treated with taxane-anthracycline (GSE32603) (Magbanua et al., 2015), where X-axis denotes survival time and Y-axis denotes the probability of survival. Patients with high SL-scores (top-tertile, blue) show better prognosis than the patients with low SL-scores (bottom tertile, yellow), as expected. The logrank P-values and median survival differences (or 80-percentile survival differences if survival exceeds 50% at the longest time point) are denoted in the figure. Tumor type abbreviations: MM, multiple myeloma; CRC, colorectal cancer; BRCA, breast invasive carcinoma; AML, acute myeloid leukemia; and LIHC, liver hepatocellular carcinoma. See also Supplemental Figure 3, Supplemental Table 2,5.
SELECT prediction of response to immune checkpoint blockade
We next turn to study the ability of the SELECT to predict clinical response to immune checkpoint inhibitors. To identify the SR interaction partners that are predictive of the response to anti-PD1/ PDL1 and anti-CTLA4 therapy, we introduced a few modifications in the published pipelines (Lee et al., 2018; Sahu et al., 2019), considering the characteristics of immune checkpoint therapy (STAR Methods). For anti-PD1/PDL1 therapy, where the antibody blocks the physical interaction between PD1 and PDL1, we considered the interaction term (i.e. the product of PD1 and PDL1 gene expression values) in identifying the SR partners of the treatment (STAR Methods). For anti-CTLA4 therapy, where the precise mechanism of action involves several ligand/receptor interactions (Wei et al., 2017), we focused on the CTLA4 itself, using its protein expression levels as they are likely to better reflect the activity than the mRNA levels. Using this immune-tailored version of SELECT, we analyzed the TCGA data to identify the SL and SR partners of PD1/PDL1 and of CTLA4. We could not identify statistically significant SL interaction partners of these checkpoint targets but did identify significant pan-cancer SR interactions. In the latter, the inactivation of the target gene of a drug is compensated by the downregulation of another gene (termed the partner rescuer gene). Given an immune checkpoint drug and pre-treatment tumor transcriptomics data from a patient, we quantify the fraction f of SR partners that are downregulated in the tumor. We define 1-f as its SR-score, where tumors with higher SR scores have less ‘active’ rescuers and are hence expected to respond better to the therapy (STAR Methods).
To evaluate the accuracy of SR-based predictions, we collected a set of 21 immune checkpoint therapy datasets that included both pre-treatment transcriptomics data and therapy response information (either by RECIST or PFS), overall comprising of 1,021 patients (Supplemental Table 1). These datasets include melanoma (Chen et al., 2016; Gide et al., 2018; Huang et al., 2019; Liu et al., 2019; Nathanson et al., 2017; Prat et al., 2017; Van Allen et al., 2015), non-small cell lung cancer (Cho et al., 2020; Damotte et al., 2019; Hwang et al., 2020; Thompson et al., 2020), renal cell carcinoma (Braun et al., 2020; Miao et al., 2018), metastatic gastric cancer (Kim et al., 2018) and urothelial carcinoma (Snyder et al., 2017) cohorts treated with anti-PD1/PDL1, anti-CTLA4 or their combination. Indeed, we find that overall higher SR-scores are associated with better response to immune checkpoint blockade (Figure 4A), with AUCs greater than 0.7 in 15 out of 18 datasets where RECIST information is available (Figure 4B, for corresponding precision-recall curves see Supplemental Figure 3E). Notably, SELECT remains predictive when multiple datasets of the same cancer types are combined for melanoma, non-small cell lung cancer or kidney cancer (Figure 4C).
Figure 4. SELECT stratifies patients for immune checkpoint therapy across different cancer types.

(A) SR-scores are significantly higher in responders (green) vs non-responders (red) based on Wilcoxon ranksum test after multiple hypothesis correction. For false discovery rates: * denotes 20%, ** denotes 10%, *** denotes 5%, and **** denotes 1%. Cancer types are noted on the top of each dataset. Results are shown for melanoma (Chen et al., 2016; Gide et al., 2018; Liu et al., 2019; Nathanson et al., 2017; Prat et al., 2017; Van Allen et al., 2015), non-small cell lung cancer (Cho et al., 2020; Damotte et al., 2019; Hwang et al., 2020; Thompson et al., 2020), renal cell carcinoma (Braun et al., 2020; Miao et al., 2018), and metastatic gastric cancer (Kim et al., 2018) treated with anti-PD1/PDL1, anti-CTLA4 or their combination, and our new lung adenocarcinoma cohort treated with anti-PD1 (GSE166449). (B-C) ROC curves showing the prediction accuracy obtained with the SELECT framework (B) in the 15 different datasets and (C) across their cancer type-specific aggregation in melanoma, non-small cell lung cancer and kidney cancer. The circles denote the point of maximal F1-score. (D) Bar graphs show the predictive accuracy in terms of AUC (Y-axis) of SR-based predictors and controls across the 15 different cohorts (X-axis) (control predictors are similar to those described in Figure 2C, with the addition of T-cell exhaustion and CD8+ T-cell abundance). (E-H) Kaplan-Meier curves depicting the survival of patients with low vs high SR-scores in (E) anti-PD1/CTLA4 combination-treated melanoma (Gide et al., 2018), (F) nivolumab/pembrolizumab-treated melanoma (Liu et al., 2019), (G) atezolizumab-treated urothelial cancer (Snyder et al., 2017), and (H) nivolumab-treated melanoma (Riaz et al., 2017) cohorts. Patients with high SR- scores (blue; over top tertile) show better prognosis than the patients with low SR-scores (yellow; below bottom tertile), and the logrank P-values and median survival differences are denoted. Tumor type abbreviations: STAD, stomach adenocarcinoma, SKCM, skin cutaneous melanoma; NSCLC, non-small cell lung cancer; and KIRC, kidney renal clear cell carcinoma. See also Supplemental Figure 3, Supplemental Table 4.
The prediction accuracy of SELECT is overall superior to a variety of expression-based controls (Figure 4D) including T-cell exhaustion markers (Wherry et al., 2007) and the estimated CD8+ T-cell abundance (Newman et al., 2015). Notably, SELECT is also predictive for a new unpublished dataset of lung adenocarcinoma patients treated with pembrolizumab, an anti-PD1 checkpoint inhibitor, at Samsung Medical Center (STAR Methods, Figures 4A,B,D (denoted as ‘new SMC dataset’)). As expected, patients with high SR-scores (in the top tertile) are enriched with responders, while patients with low SR-scores (in the bottom tertile) are enriched with non-responders (Supplemental Figure 3F). We also analyzed three recently published anti-PD1 glioblastoma trials (Cloughesy et al., 2019; Schalper et al., 2019; Zhao et al., 2019, where even though their overall efficacy was moderate, the SR-score shows a considerable predictive signal (Supplemental Figure 3G-H). Finally, the SR-scores are also predictive of progression-free or overall patient survival in four different checkpoint inhibition cohorts where this data was available (Figures 4E – H).
The predicted SR partners of PD1/PDL1 and CTLA4 (Figure 5A) are enriched for T-cell apoptosis and response to IL15 pathways (Supplemental Figure 3I). They include key immune genes such as CD4, CD8A, and CD274, and PPI interaction partners of PD1/PDL1 and CTLA4 such as CD44, CD27 and TNFRSF13B. The contribution of individual SR partners to the response prediction is different across different datasets from different cancer types (Figure 5B), where CD4, CD27, and CD8A play an important role in many samples. Taken together, these results testify that the SR partners of PD1/PDL1 and CTLA4 serve as effective biomarkers for checkpoint response across a wide range of cancer types.
Figure 5. Meta-analysis of SELECT SR partners for immune checkpoint therapy.

(A) The SR partners of PD1-PDL1 interaction (left) and CTLA4 (right), where red circles denote SR partners, yellow circles denote checkpoint targets, purple circles denote genes that belong to immune pathways, and cyan circles denote a protein-protein interaction (based on STRING database (Szklarczyk et al., 2015)) with PD1/PDL1 or CTLA4, respectively. (B) A heatmap showing the association of individual SR partners’ gene expression (Y-axis) with anti-PD1 response in the 12 clinical trial cohorts (X-axis). The significant point-biserial correlation coefficients are color-coded (P<0.1), and the cancer types of each cohort are denoted on the top of the heatmap. (C) The SR-based predicted response rates for different TCGA cancer types (Y-axis) correlate with the objective response rates observed in independent clinical trials across these cancer types (X-axis) (Spearman R=0.45, P<0.08), with a regression line (blue). Tumor type abbreviations: UCEC, uterine corpus endometrial carcinoma; STAD, stomach adenocarcinoma, SKCM, skin cutaneous melanoma; SARC, sarcoma; PRAD, prostate adenocarcinoma; PAAD, pancreatic adenocarcinoma; OV, ovarian serous cystadenocarcinoma; NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; LIHC, liver hepatocellular carcinoma; KIRC, kidney renal clear cell carcinoma; HNSC, head-neck squamous cell carcinoma; GBM, glioblastoma multiforme; ESCA, esophageal carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; BRCA, breast invasive carcinoma; and BLCA, bladder carcinoma. See also Supplemental Figure 3.
To study if tumor-specific SR scores can explain the variability observed in the objective response rates (ORR) of different tumor types to immune checkpoint therapy, we computed the SR-scores for anti-PD1 therapy for each tumor sample in the TCGA (STAR Methods). Comparing these scores to the threshold SR score for determining responders, we computed the fraction of predicted responders to anti-PD1 therapy in each cancer type in the TCGA cohort. We then compared these predicted cancer-specific fractions to the actual ORR observed in anti-PD1 clinical trials of 16 cancer types (Lee and Ruppin, 2019; Yarchoan et al., 2017). Notably, we find that these two measures significantly correlate (Figure 5C), demonstrating that SR-scores are effective predictors of ORR to checkpoint therapy across different cancer types.
Summing up over the three classes of the drugs that we studied, SELECT achieves greater than 0.7 AUC predictive performance levels in 24 out of 39 datasets containing RECIST response information, spanning 2 out of 11 non-targeted cytotoxic agents, 7 out of 10 targeted therapies and 15 out of 18 immunotherapy cohorts (including our new SMC dataset) (Figure 6). Adding the 7 (out of 9) additional datasets where SL/SR-score is predictive of progression-free or overall survival (1 chemo-, 3 targeted- and 3 immuno-therapy), it is predictive in 31 out of 48 cohorts overall (65%), and in 28 out of 35 (80%) among the targeted and checkpoint therapies. Notably, these accuracies are markedly better than those obtained using a range of control predictors.
Figure 6. Overall prediction accuracy of SELECT precision oncology framework.

The bar graphs show the overall predictive accuracy of SELECT for chemotherapy (red), targeted therapy (green) and immunotherapy (purple) in 24 different cohorts encompassing 8 different cancer types and 9 treatment options (for which discrete response information like RECIST was given). Tumor type abbreviations: STAD, stomach adenocarcinoma; NSCLC, non-small cell lung cancer; MM, multiple myeloma; LIHC, liver hepatocellular carcinoma; KIRC, kidney renal clear cell carcinoma; CRC, colorectal cancer; BRCA, breast invasive carcinoma; and BLCA, bladder carcinoma.
A retrospective analysis of the WINTHER trial
To evaluate SELECT in a multi-arm basket clinical trial setting, we performed a retrospective analysis of the recent WINTHER trial data, the first large-scale basket prospective clinical trial that has incorporated transcriptomics data for cancer therapy in adult patients with advanced solid tumors (Rodon et al., 2019). This multi-center study had two arms, one recommending treatment based on actionable mutations in a panel of cancer driver genes and another, based on the patients’ transcriptomics data. We considered the gene expression data of 71 patients with 50 different targeted treatments (single or combinations) for which significant SL partners were identified. One patient had a complete response, 7 had a partial response and 11 were reported to have stable disease (labeled as responders), while 52 had progressive disease (labeled as non-responders).
We first applied SELECT to identify the SL partners for each of the drugs prescribed in the study (STAR Methods). The resulting SL-scores of the therapies used in the trial are significantly higher in responders than non-responders (Wilcoxon rank-sum P<0.05, Figure 7A). Notably, the SL-scores of the drugs given to each patient are predictive of the actual responses observed in the trial (AUC=0.72, Figure 7B, an SL-score of 0.44 chosen as optimal threshold with maximal F1-score (Supplemental Figure 4A-B)). As shown in Figure 7C, the prediction accuracy of SL-score is superior to that of control expression-based predictors. This reassuring predictive signal led us to evaluate how many patients are predicted to benefit from the set of drugs employed in the trial, if the treatment choices would have been guided by SELECT?
Figure 7. SELECT analysis of the WINTHER trial.

(A) Responders (CR, PR, and SD; red) show significantly higher SL-scores compared to non-responders (PD; green) (Wilcoxon rank- sum P<0.05). (B) SL-scores are predictive of response to the different treatments prescribed at the trial (AUC of R0C=0.72). The black circle denotes the point of maximal F1-score (corresponding to an SL-score threshold of 0.44). (C) Bar graphs show the predictive accuracy in terms of AUC (X-axis) of SL-based predictors and different controls (Y-axis) (control types are similar to those described in Figure 2C). (D) (top panel) Comparison of the SL-scores (Y-axis) of the treatments actually prescribed in the WINTHER trial (blue) and the SL-scores of the best therapy identified by our approach (red) across all 71 patients; samples are ordered by the difference in the two SL-scores. (bottom panel) A more detailed display of the SL-scores of the treatment given in the trial (bottom row) and of all candidate therapies (all other rows), for all 71 patients (the treatments considered are denoted in every column). Blue boxes denote the highest SL-scoring treatments predicted for each patient. Cancer types of each sample are color-coded at the bottom of the figure. (E-F) SELECT recommendations for two individual patients in the WINTHER trial. The X-axis denotes the SL-score and the Y-axis lists the different cohort treatments. The drugs given in WINTHER trial are colored in blue and the top prediction by SELECT are in red. (G) A bar graph showing the frequency (X-axis) of the drugs (Y-axis) predicted to be most effective across the WINTHER cohort. (H) The correlation between the estimated coverage of top-predicted drugs in the WINTHER cohort (Y-axis) and in a TEMPUS cohort of corresponding cancer types (n=98). See also Supplemental Figure 4-7, Supplemental Table 5.
To answer this question in a systematic manner, we identified the top predicted drugs in every patient individually by computing the SL-scores of all cohort drugs based on the patients’ tumor transcriptomics (STAR Methods). For approximately 94% (67/71) of the patients we could identify therapies that have higher SL-scores than those of the drugs prescribed to them in the WINTHER trial (Figure 7D). Based on the 0.44 optimal classification threshold identified earlier, we estimate that 65% (46/71) of the patients would respond to the new treatments (with 10% false positive rate), compared to 27% that responded (based on either targeted DNA sequencing or transcriptomics) in the original trial. Of the 52 non-responders reported in the original trial, we find that 62% (32/52) can be matched with predicted effective therapies (with 10% false positive rate) (Figure 7D and Supplemental Figure 4C). We note that while this analysis focuses on SL based treatment recommendations, obviously, other non-SL mechanisms may underlie patients’ response to different drugs.
Notably, Figure 7D quite clearly shows a trend where patients in the WINTHER trial that were predicted to respond well to one of the drugs used in the trial, were often predicted to respond to quite a few of these drugs. Following this observation, we considered the mean value of the SL-scores across all the drugs that were used in the WINTHER trial, which we term mean SL-score of a given sample. This score represents the tendency of a tumor to respond to any targeted therapy given in the cohort, a surrogate for its overall vulnerability. Notably, we find that SELECT is more predictive in the patient cohorts where the mean SL-score is high (computed over all the samples in a given cohort) than in those where it is low (Supplemental Figure 5). This analysis indicates that SELECT works better in cohorts where the tumors are overall more vulnerable (as quantified by the mean SL-score). This is quite notable as it suggests that SELECT may actually offer more targeted treatment opportunities in more advanced tumors (with more genomic and transcriptional alterations).
To illustrate the potential future application of SELECT for patient stratification, we briefly describe here two individual cases arising in the WINTHER data analysis. The first involves a 75-year-old male colon cancer patient who was treated with cabozantinib because of p53 and APC mutations, and the patient indeed responded to the therapy. SELECT also recommends the treatment of cabozantinib, bringing additional support to the treatment given (Figure 7E). The second example involves a 78-year-old female lung cancer patient who was treated with nintedanib in the WINTHER trial because of KIF5B-RET fusion but failed to respond to the therapy. SELECT assigns a low SL-score to nintedanib but suggests an alternative treatment option, olaratumab, that obtain much higher SL-score (Figure 7F). Overall, the drugs most frequently recommended by SELECT in this cohort are estramustine for their MAP1A/MAP2 inhibition followed by proteasome inhibitor (bortezomib) and MEK inhibitor (cobimetinib) (Figure 7G).
We further performed an SL-based drug coverage analysis in another independent transcriptomics-based trials dataset, from the TEMPUS cohort (Beaubier et al., 2019), focusing on the same cancer types and drugs as those studied in the WINTHER trial, shows a similar pattern of top recommended drugs (Figure 7H), pointing to the robustness of these predictions across a variety of patient cohorts. One of the top predicted drugs in both cohorts is the MEK inhibitor (cobimetinib) (>6% coverage in both cohorts), which is recommended for melanoma and lung cancer patients and now being clinically tested across different cancer types (Grimaldi et al., 2017). In addition to the WINTHER and TEMPUS cohorts, we analyzed the recently released POG570 cohort, where the post-treatment transcriptomics data together with treatment history is available for advanced or metastatic tumors of 570 patients (Pleasance et al., 2020). We first confirmed that the samples SL-scores are associated with longer treatment duration, which served as a proxy for therapeutic response in the original publication (Supplemental Figure 6A). We further confirmed that this trend holds true per individual cancer types (Supplemental Figure 6B) and across individual drugs (Supplemental Figure 6C).
Finally, we asked whether SELECT can successfully estimate the objective response rates (ORR) observed across different drug treatments in different clinical trials for a given cancer type. As these trials did measure and report the patients’ tumor transcriptomics, we estimated the coverage of each drug (the patients who are predicted to respond based on their SL-scores being larger than the 0.44 response threshold) in the TCGA cohort of the relevant cancer type (STAR Methods). We collected ORR data from multiple clinical trials in melanoma and non-small cell lung cancer (a total of 3,246 patients from 18 trials) (Supplemental Table 3). Reassuringly, we find that the resulting estimated coverage is significantly correlated with the observed ORR in each of these cancer types (Supplemental Figure 7).
Discussion
We have demonstrated that by mining large-scale “-omics” data from patients’ tumors one can computationally infer putative pairs of genetic interactions that can be used as predictive biomarkers for a variety of targeted and immunotherapy treatments, across multiple cancer types. The resulting prediction accuracy is considerable for many of the drugs tested. Furthermore, as shown in the analysis of the WINTHER trial, its application offers a promising way to increase the number of patients that could benefit from precision-based treatments, which should be further explored in prospective studies.
SELECT is fundamentally different from previous efforts for therapy response prediction in two important ways: (1) The SL/SR interactions underlying the prediction are inferred from analyzing pre-treatment data from the TCGA ensemble; they are further filtered using very limited training on a single treatment response dataset to set up a small number of classification hyper-parameters. This approach results in predictors that are likely to be less prone to the risk of overfitting, which frequently accompanies contemporary supervised predictors that are constructed by training on the relatively small clinical datasets. Furthermore, the SL/SR interactions used in this study are shared across many different cancer types making them less context-sensitive and more likely to be predictive in different cancer types. (2) Notably, the interactions enabling the predictions have clear biological meanings, as the functional interpretation of the arising SL/SR interactions and their scoring is simple and intuitive, differing from the typical “black-box” solutions characteristic of machine learning approaches.
The predictive performance of SELECT is superior to existing predictors. As far as we are aware, no single model has previously been shown to obtain such predictive accuracies across so many targeted and immunotherapy datasets. Reassuringly, while many of the currently available checkpoint cohorts are small, SELECT maintains its prediction accuracy when multiple cohorts are merged. The aggregate AUCs for melanoma, non-small cell lung cancer and renal cell carcinoma (where multiple cohorts are available) are also greater than 0.7. This suggests SL/SR-scores in multiple datasets in a given cancer type have a similar distribution (see Figure 4A compared to Figure 3A), enabling the use of a single uniform threshold, testifying to its generalizability. In computing the SL/SR-score, we considered the fraction of ‘down’-regulated partners rather than ‘up’-regulated partners. This is because it is less noisy to determine cases where a gene is under-expressed than over-expressed, since over-expression could result from a number of different factors, including the expression arising from a multitude of different cell types that are present in the tumor microenvironment.
SELECT opens a possibility for future prospective clinical studies based on tumor transcriptomics. For such prospective clinical trials, we suggest that initially a more conservative predictive threshold may be used, recommending treatments only where the SL/SR predictions have a very strong and clear support. Additionally, one should obviously be careful not to use SELECT as evidence for precluding other promising treatment options that may arise due to non-SL tumor vulnerabilities. The SELECT stratification signatures of each drug are focused on a small number of genes, suggesting that gene expression panels could be designed and readily incorporated in future trials in a cost-effective manner. Applying SELECT in the clinic would require overcoming two logistic challenges: (i) the need to obtain the genomics data and provide the analysis report in a timely manner and (ii) the need to address the noisy nature of the transcriptomics data, via careful standardization and normalization. Reassuringly, most of the transcriptomics data we analyzed in this study are from FFPE samples, which are the ones most commonly used in clinical practice. Notably, we are not relying on absolute expression values but rather on the genes’ relative ranking in the cohort, decreasing the sensitivity of our approach to potential transcriptomic measurement noise.
Importantly, we anticipate that the predictive performance of SELECT could be further improved in the foreseeable future. Such efforts will leverage on the increasing availability of accumulating tumor proteomic data and the development of computational approaches for inferring genetic interactions from single cell sequencing data or cell-type specific expression levels derived from deconvolving bulk RNAseq data, which will clarify the contribution of tumor heterogeneity and tumor microenvironment. Of note, the latter will enable the identification of genetic interactions that are grounded on specific cell types (e.g., between tumor cells and CD8+ T cell in their environment). As more data accumulates, we may learn how to combine SL and SR interactions together to further boost prediction performance.
In summary, this work introduces the first systematic transcriptomics-based precision oncology framework, SELECT. This framework prioritizes effective therapeutic options for cancer patients based on the biologically grounded concept of genetic interactions, which are validated on an unprecedented scale across many different treatments and cancer types. Our study provides both explicit stratification signatures and a computational pipeline, freely available for academic use, which lay a basis for further testing and improvement in future transcriptomics-based precision oncology clinical trials.
LIMITATIONS OF THE STUDY
Like other genome-wide computational prediction method, SELECT has several limitations that should be acknowledged and improved upon in the future: (1) First, the large scale screens and patient tumor data that are used for inferring the SL and SR interactions are obviously noisy, both on the molecular and on the phenotypic side. Given these data limitations, we took a conservative approach that is focused on inferring common core of ‘pan-cancer’ interactions that are shared across different tumor types, while obviously there are important cancer type-specific (and even sub-type) interactions that should be inferred in the future, as more data accumulates. (2) Second, SELECT works by inferring the SL/SR interactions of the targets of the drugs. To this end, it uses the currently best-established mappings of these targets, but those are noisy and imperfect too (and this may be one likely reason why SELECT performs poorly on chemotherapy drugs). Furthermore, while the mechanism of action of most cancer drugs does involve the inhibition of their targets, it is not the case for a few important cancer drugs, precluding the use of SELECT for such drugs; one such notable example are PARP inhibitors, whose mechanism of action results in the binding the PARP protein to the DNA, causing DNA damage-induced cell death (and furthermore, there is no PARP clinical trial data available with transcriptomics on which we could potentially test SL-based predictors, should one be able to infer them). (3) Thirdly, and possibly the most important limitation that should be stated, is that while SELECT has been retrospectively tested in a large collection of anti-cancer clinical trials, future prospective clinical trials are needed to carefully assess and further improve its potential translational benefits.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eytan Ruppin (eytan.ruppin@nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The transcriptomics profiles and treatment outcome information of the 23 publicly available clinical trials where SELECT is predictive and the custom code of our framework are available via ZENODO repository (https://zenodo.org/record/4540874). The data from the many of the remaining trials cannot be shared due to restrictions in the data sharing agreement. The new SMC dataset of pembrolizumab-treated lung adenocarcinoma cohort is available at GSE166449.
METHOD DETAILS
Data collection
We collected the cancer patients’ tumor transcriptomics data with therapy response information from public databases. Our search was focused on GEO and ArrayExpress but also included a general literature search using different combinations of the following search terms: ‘drug response, patient, cancer pre-treatment expression, transcriptomics, and therapy resistance’ performed by March 2019 and with additional incoming datasets added as they became available to us thereafter (Braun et al., 2020; Cho et al., 2020; Damotte et al., 2019; Liu et al., 2019; Roper et al., 2020; Thompson et al., 2020). We focused on those clinical trial cohorts that have at least 5 responders and non-responders with sample size greater than 15. The collected cohorts include 13 chemotherapy, 14 targeted therapy (drugs of specific targets) and 21 immune checkpoint therapy cohorts covering a total of 13 cancer types treated with 32 drugs. The immune checkpoint therapy cohorts include a new unpublished dataset of lung adenocarcinoma patients treated with pembrolizumab at Samsung Medical Center. Among the entire collection of the 48 clinical trial cohorts, 23 datasets are accessible via ZENODO (https://zenodo.org/record/4540874).
The SELECT pipeline
A. Identifying SL/SR interaction partners of drug targets
A.1. Generating an initial pool of SL drug target interactions for targeted therapy:
To identify clinically relevant SL interactions for targeted therapies, we followed the three-step procedure described in (Lee et al., 2018). (1) We created initial pool of SL pairs identified in cell lines via RNAi/CRISPR-Cas9 (Aguirre et al., 2016; Cheung et al., 2011; Cowley et al., 2014; Marcotte et al., 2012; Marcotte et al., 2016) or pharmacological screens (Barretina et al., 2012; Basu et al., 2013; Iorio et al., 2016). For drug target gene T and candidate SL partner gene P, we checked whether growth reduction induced by knocking out/down gene T or pharmacologically inhibiting gene T is stronger when gene P is inactive, via a Wilcoxon ranksum test. (2) Second, among the candidate gene pairs from the first step, we selected those gene pairs whose coinactivation is associated with better prognosis in patients, using a Cox proportional hazard model, testifying that they may thus hamper tumor progression. Third, we prioritized SL paired genes with similar phylogenetic profiles across different species. Because of the distinct distribution of P-values for the first two screens as shown in Supplemental Figure 2, we performed a false discovery correction with 1% for the in vitro screen (1st step) and 10% for the tumor screen (2nd step). We were able to identify significant SL partners with these FDR thresholds for most of the datasets. However, for the cases where we did not find any significant SL partners with this set of FDR thresholds, we relaxed the FDR thresholds in two step manner to find the most significant set of SL partners; we first relaxed the FDR for the in vitro screen to 5% while keeping the FDR threshold for the tumor screening step at 10% (when analyzing GSE8465 and GSE3964). If this did not provide any significant pairs, we further relaxed both FDRs to 20% (when analyzing GSE16391, GSE41994, GSE68871). If we did not identify any significant pairs even with 20% FDR, we declared the corresponding drug as non-predictable by SELECT, as we could not reliably identify its candidate SL partners.
A.2. Generating compact biomarker SL signatures for targeted therapy:
The number of SL partners that pass FDR ranges from 50 to 1,000 depending on the drugs and specific FDR thresholds. Among such candidate SL partners that pass above FDR threshold, we further filtered these SL partners to generate a small set that is used to make the drug response predictions. This further filtering has been motivated by the following three reasons: (1) Occam’s razor (regularization): predictor with a smaller number of variables are likely to generalize better. (2) Biomarker interpretability: Small sets of partners are more relevant for clinical use as predictive biomarkers. (3) Patient cohort comparative analysis: when comparing the SL-scores of different drugs to decide which would be the best fit for a given patient, using the same number of top predictors facilitates such an analysis on equal grounds. To determine the top significant SL partners, we performed a limited training on the BRAF inhibitor dataset (GSE50509) as shown in Supplemental Figure 1A. Following the training, the number of top significant set size was set to 25 where the SL partners were ranked with their patient survival significance in TCGA data.
A.3. Generating the initial pool of SR drug target interactions for immunotherapy:
To identify GIs for immunotherapy, we introduced a few modifications to our previously published GI inference pipelines (Lee et al., 2018; Sahu et al., 2019) to better capture the characteristics of immune checkpoint therapy. (1) For anti-PD1/PDL1 therapy, where the antibody blocks the physical interaction between PD1 and PDL1, we considered the interaction term (i.e. the product of PD1 and PDL1 gene expression values) to identify the SR partners of the treatment. For anti-CTLA4 therapy, where there the precise mechanism of action is less well characterized and involves several ligand/receptors interactions (Wei et al., 2017), we focused on the protein expression levels of CTLA4 (available via reverse phase protein lysate microarray (RPPA) values in TCGA data) as they are likely to better reflect the activity than the mRNA levels. (We have used the median CTLA4 protein expression for the samples where CTLA4 RPPA data is not available.) For anti-PD1/PDL1 therapy, we had to use gene expression rather than protein expression because protein expression of PD1 and PDL1 are not available for many samples. (2) For the GI partner levels, we resorted to their gene expression and somatic copy number alterations (SCNA) data as referenced in previous studies (Lee et al., 2018; Sahu et al., 2019) because protein expression was measured only for a small subset of genes. (3) Instead of considering all protein coding genes as candidates for SR partners, we focused on the genes that are covered by the NanoString panel (Supplemental Table 4) because (i) the gene expression of many of immune checkpoint therapy datasets was quantified by NanoString platform and (ii) NanoString panel is enriched with immune system related genes that are highly relevant to the response to immune checkpoint therapy. (4) We omitted the first step of the SL/SR inference procedure, which is aimed at identifying candidate genetic interactions from the cell line functional screening data, because these interactions are not relevant to immune checkpoint response. We could in principle use the genome-wide CRISPR screens in cancer cell/T-cell co-culture, but this data is limited to melanoma and the coverage is not fully genome-wide, where many genes included in the NanoString panel are missing. We kept all the remaining steps of the published SL/SR inference procedures. (5) We focused on the mediators of resistance to immune checkpoint therapies using synthetic rescue (SR) interactions, as no statistically significant SL interaction partners were identified via ISLE for either PD1/PDL1 or CTLA4. False discovery correction was done with FDR 10%.
A.4. Generating compact biomarker SR signatures for immunotherapy:
To determine the top significant SR partners, we performed a limited training on (Van Allen et al., 2015) as shown in Supplemental Figure 1B. Following the training, the number of top significant set size was set to 10 where the SR partners are ranked with their phylogenetic distances. These parameters were used in making all immune checkpoint therapy response predictions. We analyzed TCGA data applying this pipeline to identify pan cancer SR interactions that are more likely to be clinically relevant across many cancer types. In particular, we focused on the SR interactions where the inactivation of the target gene is compensated by the downregulation of the partner rescuer gene, as the other types of SR interactions introduced in (Sahu et al., 2019) were not predictive in the training dataset.
B. Predicting drug response in patients using SL/SR partners
We used the identified SL or SR partners for drug response prediction. We define SL-score for chemotherapy and targeted therapy as the fraction of inactive SL partners in a given sample out of all SL partners of that drug. The SL-score reflects the intuitive notion that a targeted drug would be more effective when a larger fraction of its SL partners is inactive in the tumor. In each patient drug response dataset, a gene is determined to be inactive if its expression is below bottom tertile across samples in the same dataset, adhering to our previously published approaches (Lee et al., 2018; Sahu et al., 2019). We made this choice of normalization (i) to account for the basal expression level of each gene in specific tumor type and (ii) to minimize batch effects occurring when different datasets are combined. We additionally multiplied the SL-score by a target gene factor to obtain the final SL-score. This has been motivated by the notion that an inhibitor will be not effective when its target gene is not expressed; thus, we set the target gene factor to be zero when the target gene is inactive (below bottom 30-percentile in the given sample), and we used mean expression of the targets genes when the given drug has more than one target gene. We used SR-score to predict response to immunotherapy, that quantifies the fraction f, SR partners that are inactive, and we used 1-f as the SR-score to predict responders. Higher SL- or SR-score is predictive of response to therapies.
Using the computed SL/SR-scores, we studied SELECT’s prediction accuracy in individual clinical trial cohorts. Overall, we used the entire relevant cohorts for our analysis to test the accuracy. There are several exceptions to this when we focus on a subset of the cohorts. For the cohorts that include both pre- and post-treatment biopsies, we naturally focused on pretreatment biopsies (GSE8465, GSE32603, GSE50509, GSE65185, GSE99898, GSE77750, GSE119262, and the immunotherapy cohort (Nathanson et al., 2017)); for the cohorts composed of multiple treatments or placebo arms, we focused on the patients receiving the specific targeted therapy of interest (GSE72970, GSE3264, TARGET AML, MMRF, GSE16391, and GSE66399). The pertaining detailed information is available in Supplemental Table 1 Column ‘Criteria for subsetting samples.’
To define the responders and non-responders, we aimed to faithfully use the criteria defined in the original clinical trials as much as possible. For the datasets where the response information is available in the form of RECIST criteria, we solved a classification problem, quantifying the AUC of the pertaining ROC curve (rounded up to the second digit after the decimal point). For the cohorts where RECIST information is available, we used CR, PR as responders and SD, PD as non-responders (GSE63885, GSE41998, GSE72970, GSE3264 and the immunotherapy cohorts (Damotte et al., 2019; Gide et al., 2018; Hugo et al., 2017; Mariathasan et al., 2018; Miao et al., 2018; Nathanson et al., 2017; Prat et al., 2017; Thompson et al., 2020; Van Allen et al., 2015), and New SMC dataset). For the Bortezomib cohort (GSE68871), we used CR, nCR as responders and PR, SD, VGPR as non-responders. For the cohorts where pathological clinical response (pCR) information is available, we used pCR as responders, and no-pCR as nonresponders (GSE22093, GSE32603, GSE20194, GSE66399, and GSE4779). For the cohorts where response and non-response are defined by specific individual clinical trial cohorts, we followed the definition of the original cohorts (GSE31811, GSE8465, GSE50509, and immunotherapy cohorts (Chen et al., 2016; Cho et al., 2020; Huang et al., 2019; Kim et al., 2018)). For GSE77750, we used ‘treatment’ as responders and ‘progressed’,’stable’ as nonresponders. For the immunotherapy cohort of (Hwang et al., 2020), we have used ‘no recurrence’ as responder and ‘recurrence’ as non-responder. For GSE119262, we have used a response metric based on Ki67, as used in the original study. For methotrexate (GSE10255), we have used the median white blood cell (WBC) change to define response, as was used in the original investigation. For (Braun et al., 2020), we used CR, PR, SD as responders (n=8) and PD as non-responders (n=8), which yields AUC=0.77, since the number of CR/PR only was too small (n=3); however, we confirmed that, even with taking CR/PR as responders (n=3) and SD/PD as non-responders (n=13) like other cohorts, SELECT is still predictive (AUC=0.81). For the cases where progression-free survival time is available for all patients (with no censoring event), we used the median progression-free survival from the relevant literature as cutoff to distinguish the responders from non-responders and solved a classification problem. For BRAF inhibitor cohorts, we have used 7-month progression-free survival as criteria for response following the literature (Sosman et al., 2012). For GSE16391, we have used 75-percentile of the relapse-free survival (=50 months; 6 responders and 17 non-responders) as threshold for response that is comparable with the reported values in the literature (Lohrisch et al., 2006); in this cohort, we also confirmed that the prediction accuracy is above 0.7 for a wide range of thresholds that separates responders vs non-responders including 4.8 years (the exact value reported in the literature). For the datasets where we only had the overall or progression-free survival with censoring information, we performed a Kaplan-Meier analysis. It is known that the response based on RECIST and patient survival does not necessarily correlate, and we thus evaluated SELECT’s predictive performance in either term according to the available data. The pertaining detailed information is available in Supplemental Table 1 Columns ‘Criteria for R,’ and ‘Criteria for NR.’ The mean SL-score is the mean value of the SL-scores across many different targeted therapies (the drugs that were used in the WINTHER trial (see Experimental Procedures section ‘Retrospective analysis of the WINTHER trial’ below)), which is assigned to each sample.
Samsung Medical Center anti-PD1-treated lung adenocarcinoma cohort
RNA was purified from formalin-fixed paraffin-embedded (FFPE) or fresh tumor samples using the AllPrep DNA/RNA Mini Kit (Qiagen, USA). The RNA concentration and purity were measured using the NanoDrop and Bioanalyzer (Agilent, USA). The library was prepared following the manufacturer’s instructions using the RNA Access Library Prep Kit (Illumina, USA). Tumor response was assessed by physicians using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1. 1 criteria. All 22 patients were treated with pembrolizumab, and all biopsy samples were obtained prior to the treatment.
TCGA anti-PD1 coverage analysis for predicting the cancer type-specific response to checkpoint therapy
To predict the objective response rates of anti-PD1 therapy in each cancer type in TCGA, we computed the SR scores of PD1 in each tumor sample based on each tumor’s transcriptomics. We chose the point of maximal F1-score as the response classification threshold, computed across all 9 immune checkpoint datasets where the SR-score is predictive. Using this uniform threshold, we computed the fraction of responders for each cancer type. We then compared these predicted fractions of responders with the actual response rates reported in 16 anti-PD1 clinical trials (Yarchoan et al., 2017) using a Spearman rank correlation.
Retrospective analysis of the WINTHER trial
(1) The data: The trial involved 10 different cancer types, mostly colon, lung and head and neck cancer. It had two arms, one recommending treatment based on actionable mutations in a panel of cancer driver genes and the other based on the patients’ transcriptomics data. We considered the Agilent microarray data of 71 patients with 50 different targeted treatments (single or combinations) that were available to us. One patient had a complete response, 7 had a partial response and 11 were reported to have stable disease (labeled as responders in our analysis), while 52 had a progressive disease (labeled as non-responders). To balance the size of responders vs non-responders in this cohort, we defined CR/PR/SD as responders and PD as non-responders. (2) Inferring the SL partners of the drugs given in the cohort: For each of the drug that was used in the trial, we first identified their target genes’ SL partners using SELECT. (3) Estimating the ability of SELECT to predict the patients’ response to the drugs given: Based on the SL signatures, we used SELECT to calculate the SL-score of each therapy and quantified the prediction accuracy of all therapies given via a ROC analysis. We focused on the patients who received a single treatment (with less than two drug targets), since our subsequent analysis has focused on monotherapies. We note that, as expected, as the number of targets of a drug increases, its prediction accuracy decreases. (4) Charting the landscape of SL cores of all drugs across all patients: Lastly, to study the landscape of alternative treatment options recommended by SL-based approach, we considered all FDA-approved targeted therapies obtained from the DrugBank database (Law et al., 2014) and the literature as potential treatment candidates. The drug-to-target mapping used in our analysis can be found in Supplemental Table 5. We then identified the SL partners of all the drugs’ target genes and computed their SL-scores in each of the patient’s based on their transcriptomic data and ranked them accordingly. The exact same procedure was used in the analysis of the TEMPUS cohort.
We additionally analyzed the post-treatment transcriptomics data together with treatment history from P0G570 cohort (Pleasance et al., 2020). We normalized the RNAseq data following the same procedure described above for WINTHER trial data, and calculated SL-score for each sample using the treatments that the patients have received. We associated them with treatment duration, following its utilization as a proxy for treatment response in the original publication. We also compared the median treatment duration of high vs low SL-score patients (adhering to the choice of SL-score of 0.44 as classification threshold) across all patient-treatment pairs, and calculated the ratio between the median treatment duration of high vs low SL-score patients for each cancer type and each treatment of sample size greater than 15 patients.
Coverage analysis on TCGA data for melanoma and non-small-cell lung cancer
We collected relevant objective response rates (ORRs) for various clinical trials in melanoma (8 drugs) and non-small cell lung cancer (10 drugs) (Supplemental Table 3) (Fruehauf et al., 2008; Daud et al., 2017; Sachdev et al., 2013; Decoster et al., 2010; Larkin et al., 2015; Ribas et al., 2016, Katakami et al., 2013; Wakelee et al., 2017; Han et al., 2016; Novello et al., 2009; Kiura et al., 2008; Mok et al., 2009; Shi et al., 2013; Brahmer et al., 2015; Reck et al., 2016; Antonia et al., 2017). We excluded those clinical trials of combination therapies or of a small sample size (like those with a particular driver mutation). The number of patients participating in each clinical trial and the trial details are provided in Supplemental Table 3.
We performed a coverage analysis on 356 TCGA melanoma or 981 TCGA non-small cell lung cancer patients using SELECT and predicted percentage of responders for each drug (using their drug targets). For predicting patient response, we used the threshold for SL/SR-scores to determine responders (i.e. a patient is responder if SL-score > 0.44 for chemotherapy or targeted-therapy; and SR-score > 0.9 for immunotherapy). Then across the drugs within a given cancer type, we computed the Spearman’s correlation between the fraction of responders in the respective TCGA cohorts (coverage) and the ORRs from clinical trials.
QUANTIFICATION AND STATISTICAL ANALYSIS
The receiver operating characteristics (ROC) and precision-recall analyses were performed using R library ROCR. Wilcoxon ranksum test and Student’s t-test were performed using R functions wilcox.test and t.test, respectively. Gene Ontology (GO) enrichment analysis was performed using R library topGO. Statistical details of each clinical trial dataset can be found in Supplemental Table 1. The Kaplan-Meier analysis was performed, where the effect size was measured by the median survival differences (or 80-percentile survival differences if survival exceeds 50% at the longest time point) and the corresponding significance was quantified by logrank test using R libraries, survival and survminer. All box plots follow the standard definition, where the box is drawn from top quartile (75 th percentile) to bottom quartile (25th percentile) of the data with a horizontal line drawn in the middle to denote the median value. The lowest point of the bottom whisker is the minimum of the data and the highest point of the top whisker is the maximum of the data.
ADDITIONAL RESOURCES
The clinical trial data and the source code are accessible for academic purposes via ZENODO repository: https://zenodo.org/record/4540874.
Supplementary Material
The top significant SL and SR partners used in the prediction of cytotoxic/targeted agents and immunotherapy were determined by varying the set sizes and ranking scheme in GSE50509 (Rizos et al., 2014) (cytotoxic and targeted) and Van Allen cohort (Van Allen et al., 2015) (immunotherapy), which are the datasets we initially started our analysis with, and determining the resulting prediction performance on these datasets. Based on this procedure, we selected 25 as the SL partners set size and patient survival P-values as the ranking scheme used in the analysis of all other cytotoxic/targeted agents and 10 as the size of the SR partners and phylogenetic score as the ranking scheme used for predicting the response for immune checkpoint immunotherapy.
This figure provides the rationale for determining the false discovery rates for SL inference. (A) The results of the first step of the SL screens. The volcano plot shows the point-biserial correlation coefficient (X-axis) and Wilcoxon ranksum P-values (Y-axis) of all protein coding genes from in vitro RNAi/CRISPR-Cas9 screen and pharmacological inhibitions. For each candidate SL pair between the drug target (gene T; BRAF in this case) and the partner (gene P), we checked whether the growth defect induced by gene knock-out/down or pharmacological inhibition of gene T is stronger when gene P is inactive either by mRNA expression or somatic copy number alteration (SCNA). (B) The results of the second step of SL screen from TCGA tumor data. The volcano plot shows the Cox hazard ratio (X-axis) and corresponding Wald P- values (Y-axis), where we checked whether co-inactivation of gene P and T leads to significantly enhanced patient survival in TCGA data as described in (Lee et al., 2018). Note that the significance level is considerably lower for tumor screen compared to the in vitro screen. We thus used more stringent FDR correction for in vitro screen (1%) than tumor screen (10%). (C) The results from phylogenetic screen showing the cumulative distribution of (rank-normalized) phylogenetic distances between BRAF and all protein coding genes. The cutoff of 50-percentile was used following (Lee et al., 2018). The 25 identified BRAF SL partners are denoted in the figures.
(A) Precision-recall curves depicting the prediction accuracy of the response to BRAF inhibitor using SL-scores in the three melanoma cohorts. (B) BRAF mutation status is not associated with SL-score. The SL-scores (Y-axis) of BRAF V600E mutated patients (red) vs BRAF V600K mutated patients (green) in three melanoma cohorts GSE50509, GSE65185, and GSE99898 (X-axis). Wilcoxon P-values are 0.16, 0.87, and 0.51, respectively. (C) Precision-recall curves of other cytotoxic agents and targeted therapies. The AUPRC of each precision-recall curve is denoted in the figure legend, and the circles denote the point of maximal F1-score. (D) Bar graphs showing the fraction of responders in the patients with high SL-scores (top tertile; green) and low SL-scores (bottom tertile; purple). The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SL group and non-responders in the low-SL group. (E) Precision-recall curves for the SR-scores across the 15 different datasets with AUCs > 0.7. The AUPRC of each precision-recall curve is denoted in the figure legend, and the circles denote the point of maximal F1-score. (F) Bar graphs showing the fraction of responders in the patients with high SR-scores (top tertile; green) and low SR-scores (bottom tertile; purple). The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SR group and non-responders in the low-SR group. (G) Bar graphs show the predictive accuracy in terms of AUC (Y-axis) of SR-based predictors and control predictors across the three glioma cohorts (X-axis). (control predictors are similar to those described in Figure 2C in the main text.) (H) Bar graphs showing the fraction of responders in the patients with high SR-scores (top tertile; green) and low SR-scores (bottom tertile; purple) in the three glioma datasets. The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SR group and non-responders in the low-SR group. (I) The bar graph of pathway enrichment analysis of 10 PD1-PDL1 and 10 CTLA4 SR partner genes in aggregate. X-axis shows the -log10 (Fisher exact test P) and Y-axis shows the enriched pathways.
(A) The ROC plots show that the SL-scores are most predictive of response to the different treatments prescribed at the trial (AUC of R0C=0.72), markedly higher than those of control predictors (as described in Figure 2C). (B) The parallel precision-recall, showing that the SL-score predictor outperforms an array of other control predictors (AUPRC=0.49). (C) The X-axis shows the SL-score classification threshold and Y-axis depicts the false positive rate (derived from the ROC curve presented in Figure 7B, (blue)), the overall coverage (the fraction of the patients in the WINTHER cohort for whom SELECT recommends higher SL-scoring alternate treatment options than those they have actually received, red), and the fraction of the non-responders in WINTHER cohort for whom SELECT recommends such treatment options (orange). As the SL-score decision threshold increases, less and less patients are denoted ‘positive’ (i.e., are assigned a treatment recommendation). At SL-score threshold of 0.44, SELECT recommends alternate treatments that are predicted to be effective for 65% of the entire cohort and 62% of non-responders (as marked by the stars).
SELECT’s prediction accuracy (AUCs) were compared between the cohorts where the mean SL-score is high (green) vs low (red, separated by median values of the mean SL-score). SELECT prediction accuracy is associated with the mean SL-score (A) for targeted therapy (Wilcoxon ranksum P<0.06) but (B) while a similar trend is also observed for immunotherapy, it is not significant (Wilcoxon ranksum P<0.15).
(A) The SL-score is associated with treatment duration in the P0G570 cohort (Wilcoxon P<0.023). (B-C) The fold change (FC) of median treatment duration in high SL-score group (SL-score>0.44) vs low SL-score group (SL- score<=0.44) for (B) each cancer type and (C) each drug, across all categories with more than 15 patients.
SELECT is predictive of coverage (fraction of the patients predicted to be responsive to the given treatment based on the criteria, SL-score>0.44 for targeted therapies and SR-score > 0.9 for immunotherapies) across different therapies in (A) melanoma and (B) non-small cell lung cancer. Spearman rank correlation and associated P-values are noted in the figures.
Supplemental Table 1. The clinical trial cohorts used in the manuscript, Related to Figure 1.
Supplemental Table 3. The clinical trial cohorts for coverage analysis, Related to Supplemental Figure 7.
Supplemental Table 4. Nanostring panel genes, Related to Figure 4, STAR Methods.
Supplemental Table 5. Drug to target gene mapping, Related to Figure 2,3,7, STAR Methods.
Highlights.
SELECT employs synthetic lethality and tumor transcriptome to advance precision oncology.
SELECT predicts therapy response in 28 out of the 35 targeted and immunotherapy trials.
SELECT facilitates cancer patient stratification in transcriptomic multi-arm trials.
Acknowledgements
This research is supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI), Center for Cancer Research (CCR). This work utilized the computational resources of the NIH HPC Biowulf cluster. We thank E. Michael Gertz, Alejandro A. Schäffer, Sanna Madan, Peng Jiang, Tom Misteli and Louis Staudt for providing many helpful comments. We thank all the patients and clinicians involved in the 48 clinical trials. We thank Elizabeth Jaffe, Ignacio Melero, Robert Prins, Raul Rabadan, Antoni Ribas, Jeffrey Thompson, Nitin Roper, Udayan Guha, and TEMPUS Inc. for sharing their data. Joo Sang Lee is partly supported by a grant of the National Research Foundation of Korea funded by the Korean Government (NRF-2020R1A2C2007652) and Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (No.2019-0-00421, AI Graduate School Support Program (Sungkyunkwan University)).
Footnotes
Declaration of Interests
Eytan Ruppin is a co-founder of Medaware Ltd, Metabomed Ltd and of Pangea Therapeutics, Ltd (divested from the latter). E.R. serves as a non-paid scientific consultant to Pangea Therapeutics, Ltd, a company developing a precision oncology SL-based multi-omics approach. Joo Sang Lee is a scientific consultant, Tuvik Beker is CEO and CTO, Gal Dinstag is head of R&D, Raanan Berger is a member of the Scientific Advisory Board, and Ze’ev Ronai is a co-founder and a scientific advisor at Pangea Therapeutics, Ltd. Razelle Kurzrock receives research funding from Genentech, Merck Serono, Pfizer, Boehringer Ingelheim, TopAlliance, Takeda, Incyte, Debiopharm, Medimmune, Sequenom, Foundation Medicine, Konica Minolta, Grifols, Omniseq, and Guardant, as well as consultant and/or speaker fees and/or advisory board for X-Biotech, Neomed, Pfizer, Actuate Therapeutics, Roche, Turning Point Therapeutics, TD2/Volastra, Bicara Therapeutics, Inc., has an equity interest in IDbyDNA and CureMatch Inc, serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch. A patent application associated with this manuscript is in process.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB., et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An Interactive Resource to Identify Cancer Genetic and Lineage Dependencies Targeted by Small Molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaubier N, Bontrager M, Huether R, Igartua C, Lau D, Tell R, Bobe AM, Bush S, Chang AL, Hoskinson DC, et al. (2019). Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol. [DOI] [PubMed] [Google Scholar]
- Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, et al. (2018). The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, Ross-Macdonald P, Berger AC, Jegede OA, Elagina L, et al. (2020). Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 26, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. (2011). Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A 108, 12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JW, Hong MH, Ha SJ, Kim YJ, Cho BC, Lee I, and Kim HR (2020). Genome-wide identification of differentially methylated promoters and enhancers associated with response to anti-PD-1 therapy in non-small cell lung cancer. Exp Mol Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE, et al. (2014). Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data 1, 140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damotte D, Warren S, Arrondeau J, Boudou-Rouquette P, Mansuet-Lupo A, Biton J, Ouakrim H, Alifano M, Gervais C, Bellesoeur A, et al. (2019). The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J Transl Med 17, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio M, Mollevi C, Bibeau F, Vie N, Selves J, Emile JF, Roger P, Gongora C, Robert J, Tubiana-Mathieu N, et al. (2017). Molecular subtypes of metastatic colorectal cancer are associated with patient response to irinotecan-based therapies. Eur J Cancer 76, 68–75. [DOI] [PubMed] [Google Scholar]
- Desmedt C, Giobbie-Hurder A, Neven P, Paridaens R, Christiaens MR, Smeets A, Lallemand F, Haibe-Kains B, Viale G, Gelber RD, et al. (2009). The Gene expression Grade Index: a potential predictor of relapse for endocrine-treated breast cancer patients in the BIG 1–98 trial. BMC Med Genomics 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Woo D, Roussel MF, and Cleveland JL (2001). Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol 21, 5063–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, Andre S, Piccart M, Campone M, Brain E, et al. (2009). A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15, 68–74. [DOI] [PubMed] [Google Scholar]
- Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, Lubrano S, Kishore A, Pachter JA, Konig GM, et al. (2019). A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 35, 457–472 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Wilmott JS, Scolyer RA, and Long GV (2018). Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin Cancer Res 24, 1260–1270. [DOI] [PubMed] [Google Scholar]
- Graudens E, Boulanger V, Mollard C, Mariage-Samson R, Barlet X, Gremy G, Couillault C, Lajemi M, Piatier-Tonneau D, Zaborski P, et al. (2006). Deciphering cellular states of innate tumor drug responses. Genome Biol 7, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi AM, Simeone E, Festino L, Vanella V, Strudel M, and Ascierto PA (2017). MEK Inhibitors in the Treatment of Metastatic Melanoma and Solid Tumors. Am J Clin Dermatol 18, 745–754. [DOI] [PubMed] [Google Scholar]
- Guarneri V, Dieci MV, Frassoldati A, Maiorana A, Ficarra G, Bettelli S, Tagliafico E, Bicciato S, Generali DG, Cagossi K, et al. (2015). Prospective Biomarker Analysis of the Randomized CHER-LOB Study Evaluating the Dual Anti-HER2 Treatment With Trastuzumab and Lapatinib Plus Chemotherapy as Neoadjuvant Therapy for HER2- Positive Breast Cancer. Oncologist 20, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, Vidaurre T, Holmes F, Souchon E, Wang HK, et al. (2011). A Genomic Predictor of Response and Survival Following Taxane-Anthracycline Chemotherapy for Invasive Breast Cancer. Jama-Journal of the American Medical Association 305, 1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Takiguchi Y, Minami H, Akiyoshi K, Segawa Y, Ueda H, Iwamoto Y, Kondoh C, Matsumoto K, Takahashi S, et al. (2020). Site-Specific and Targeted Therapy Based on Molecular Profiling by Next-Generation Sequencing for Cancer of Unknown Primary Site: A Nonrandomized Phase 2 Clinical Trial. JAMA Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak CE, Pusztai L, Xing G, Trifan OC, Saura C, Tseng LM, Chan S, Welcher R, and Liu D (2013). Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin Cancer Res 19, 1587–1595. [DOI] [PubMed] [Google Scholar]
- Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, Manne S, Kraya AA, Wubbenhorst B, Dorfman L, et al. (2019). A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 25, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. (2015). Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 162, 1271–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2017). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 168, 542. [DOI] [PubMed] [Google Scholar]
- Hwang S, Kwon AY, Jeong JY, Kim S, Kang H, Park J, Kim JH, Han OJ, Lim SM, and An HJ (2020). Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep 10, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Bianchini G, Booser D, Qi Y, Coutant C, Shiang CY, Santarpia L, Matsuoka J, Hortobagyi GN, Symmans WF, et al. (2011). Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst 103, 264–272. [DOI] [PubMed] [Google Scholar]
- Jansen MP, Knijnenburg T, Reijm EA, Simon I, Kerkhoven R, Droog M, Velds A, van Laere S, Dirix L, Alexi X, et al. (2013). Hallmarks of aromatase inhibitor drug resistance revealed by epigenetic profiling in breast cancer. Cancer Res 73, 6632–6641. [DOI] [PubMed] [Google Scholar]
- Julka PK, Chacko RT, Nag S, Parshad R, Nair A, Oh DS, Hu Z, Koppiker CB, Nair S,Dawar R, et al. (2008). A phase II study of sequential neoadjuvant gemcitabine plus doxorubicin followed by gemcitabine plus cisplatin in patients with operable breast cancer: prediction of response using molecular profiling. Br J Cancer 98, 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakavand H, Rawson RV, Pupo GM, Yang JYH, Menzies AM, Carlino MS, Kefford RF, Howle JR, Saw RPM, Thompson JF, et al. (2017). PD-L1 Expression and Immune Escape in Melanoma Resistance to MAPK Inhibitors. Clin Cancer Res 23, 6054–6061. [DOI] [PubMed] [Google Scholar]
- Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, Liu XQ, Sher X, Jung H, Lee M, et al. (2018). Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 24, 1449–1458. [DOI] [PubMed] [Google Scholar]
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. (2014). DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 42, D1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Das A, Jerby-Arnon L, Arafeh R, Auslander N, Davidson M, McGarry L, James D, Amzallag A, Park SG, et al. (2018). Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun 9, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, and Ruppin E (2019). Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowska KM, Olbryt M, Dudaladava V, Pamula-Pilat J, Kujawa K, Grzybowska E, Jarzab M, Student S, Rzepecka IK, Jarzab B, et al. (2014). Gene expression analysis in ovarian cancer - faults and hints from DNA microarray study. Front Oncol 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, et al. (2019). Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magbanua MJ, Wolf DM, Yau C, Davis SE, Crothers J, Au A, Haqq CM, Livasy C, Rugo HS, Investigators IST, et al. (2015). Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res 17, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manojlovic Z, Christofferson A, Liang WS, Aldrich J, Washington M, Wong S, Rohrer D,Jewell S, Kittles RA, Derome M, et al. (2017). Comprehensive molecular profiling of 718 Multiple Myelomas reveals significant differences in mutation frequencies between African and European descent cases. PLoS Genet 13, e1007087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, et al. (2012). Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2, 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, et al. (2016). Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 164, 293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monika Belickova M, Merkerova MD, Votavova H, Valka J, Vesela J, Pejsova B, Hajkova H, Klema J, Cermak J, and Jonasova A (2016). Up-regulation of ribosomal genes is associated with a poor response to azacitidine in myelodysplasia and related neoplasms. Int J Hematol 104, 566–573. [DOI] [PubMed] [Google Scholar]
- Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, Van Allen E, Merghoub T, Wolchok JD, Snyder A, et al. (2017). Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer Immunol Res 5, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Lee JS, Hasnis E, Tandoc K, Scott DA, Verma S, Feng Y, Larue L, Sahu AD, Topisirovic I, et al. (2019). Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinyol R, Montal R, Bassaganyas L, Sia D, Takayama T, Chau GY, Mazzaferro V, Roayaie S, Lee HC, Kokudo N, et al. (2019). Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 68, 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires da Silva I, Wang KYX, Wilmott JS, Holst J, Carlino MS, Park JJ, Quek C, Wongchenko M, Yan Y, Mann G, et al. (2019). Distinct Molecular Profiles and Immunotherapy Treatment Outcomes of V600E and V600K BRAF-Mutant Melanoma. Clin Cancer Res 25, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance E, Titmuss E, Williamson L, Kwan H, Culibrk L, Zhao EY, Dixon K, Fan K, Bowlby R, Jones MR., et al. (2020). Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nature Cancer 1, 452–468. [DOI] [PubMed] [Google Scholar]
- Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, Martinez A, Nuciforo P, Comerma L, Alos L, et al. (2017). Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res 77, 3540–3550. [DOI] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. (2014). BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 20, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, Tsimberidou A, Saintigny P, Ackerstein A, Brana I, et al. (2019). Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med 25, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper N, Brown AL, Wei JS, Pack S, Trindade C, Kim C, Restifo O, Gao S, Sindiri S, Mehrabadi F, et al. (2020). Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep Med 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabine VS, Sims AH, Macaskill EJ, Renshaw L, Thomas JS, Dixon JM, and Bartlett JM (2010). Gene expression profiling of response to mTOR inhibitor everolimus in pre-operatively treated post-menopausal women with oestrogen receptor-positive breast cancer. Breast Cancer Res Treat 122, 419–428. [DOI] [PubMed] [Google Scholar]
- Sahu AD, J SL, Wang Z, Zhang G, Iglesias-Bartolome R, Tian T, Wei Z, Miao B, Nair NU, Ponomarova O, et al. (2019). Genome-wide prediction of synthetic rescue mediators of resistance to targeted and immunotherapy. Mol Syst Biol 15, e8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, Inoges S, de Andrea C, Lopez-Diaz de Cerio A, Tejada S, et al. (2019). Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 25, 470–476. [DOI] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, et al. (2010). The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol 28, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD, Chang E, Aksoy BA, Al-Ahmadie H, Yusko E, et al. (2017). Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med 14, e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorich MJ, Pottier N, Pei D, Yang W, Kager L, Stocco G, Cheng C, Panetta JC, Pui CH, Relling MV, et al. (2008). In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med 5, e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spentzos D, Levine DA, Kolia S, Otu H, Boyd J, Libermann TA, and Cannistra SA (2005). Unique gene expression profile based on pathologic response in epithelial ovarian cancer. J Clin Oncol 23, 7911–7918. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanioka M, Fan C, Parker JS, Hoadley KA, Hu Z, Li Y, Hyslop TM, Pitcher BN, Soloway MG, Spears PA, et al. (2018). Integrated Analysis of RNA and DNA from the Phase III Trial CALGB 40601 Identifies Predictors of Response to Trastuzumab-Based Neoadjuvant Chemotherapy in HER2-Positive Breast Cancer. Clin Cancer Res 24, 5292–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terragna C, Remondini D, Martello M, Zamagni E, Pantani L, Patriarca F, Pezzi A, Levi G, Offidani M, Proserpio I, et al. (2016). The genetic and genomic background of multiple myeloma patients achieving complete response after induction therapy with bortezomib, thalidomide and dexamethasone (VTD). Oncotarget 7, 9666–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JC, Hwang WT, Davis C, Deshpande C, Jeffries S, Rajpurohit Y, Krishna V, Smirnov D, Verona R, Lorenzi MV, et al. (2020). Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer 139, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske OM, Bjork I, Salama SR, Beale H, Tayi Shah A, Sanders L, Pfeil J, Lam DL, Learned K, Durbin A, et al. (2019). Comparative Tumor RNA Sequencing Analysis for Difficult-to-Treat Pediatric and Young Adult Patients With Cancer. JAMA Netw Open 2, e1913968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133 e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, George LK, Grant GD, and Perou CM (2006). Common markers of proliferation. Nat Rev Cancer 6, 99–106. [DOI] [PubMed] [Google Scholar]
- Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, Barahona P, Wilkie EE, Sullivan P, Bowen-James R, et al. (2020). Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. [DOI] [PubMed] [Google Scholar]
- Wongchenko MJ, McArthur GA, Dreno B, Larkin J, Ascierto PA, Sosman J, Andries L, Kockx M, Hurst SD, Caro I, et al. (2017). Gene Expression Profiling in BRAF-Mutated Melanoma Reveals Patient Subgroups with Poor Outcomes to Vemurafenib That May Be Overcome by Cobimetinib Plus Vemurafenib. Clin Cancer Res 23, 5238–5245. [DOI] [PubMed] [Google Scholar]
- Yarchoan M, Hopkins A, and Jaffee EM (2017). Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 377, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 25, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The top significant SL and SR partners used in the prediction of cytotoxic/targeted agents and immunotherapy were determined by varying the set sizes and ranking scheme in GSE50509 (Rizos et al., 2014) (cytotoxic and targeted) and Van Allen cohort (Van Allen et al., 2015) (immunotherapy), which are the datasets we initially started our analysis with, and determining the resulting prediction performance on these datasets. Based on this procedure, we selected 25 as the SL partners set size and patient survival P-values as the ranking scheme used in the analysis of all other cytotoxic/targeted agents and 10 as the size of the SR partners and phylogenetic score as the ranking scheme used for predicting the response for immune checkpoint immunotherapy.
This figure provides the rationale for determining the false discovery rates for SL inference. (A) The results of the first step of the SL screens. The volcano plot shows the point-biserial correlation coefficient (X-axis) and Wilcoxon ranksum P-values (Y-axis) of all protein coding genes from in vitro RNAi/CRISPR-Cas9 screen and pharmacological inhibitions. For each candidate SL pair between the drug target (gene T; BRAF in this case) and the partner (gene P), we checked whether the growth defect induced by gene knock-out/down or pharmacological inhibition of gene T is stronger when gene P is inactive either by mRNA expression or somatic copy number alteration (SCNA). (B) The results of the second step of SL screen from TCGA tumor data. The volcano plot shows the Cox hazard ratio (X-axis) and corresponding Wald P- values (Y-axis), where we checked whether co-inactivation of gene P and T leads to significantly enhanced patient survival in TCGA data as described in (Lee et al., 2018). Note that the significance level is considerably lower for tumor screen compared to the in vitro screen. We thus used more stringent FDR correction for in vitro screen (1%) than tumor screen (10%). (C) The results from phylogenetic screen showing the cumulative distribution of (rank-normalized) phylogenetic distances between BRAF and all protein coding genes. The cutoff of 50-percentile was used following (Lee et al., 2018). The 25 identified BRAF SL partners are denoted in the figures.
(A) Precision-recall curves depicting the prediction accuracy of the response to BRAF inhibitor using SL-scores in the three melanoma cohorts. (B) BRAF mutation status is not associated with SL-score. The SL-scores (Y-axis) of BRAF V600E mutated patients (red) vs BRAF V600K mutated patients (green) in three melanoma cohorts GSE50509, GSE65185, and GSE99898 (X-axis). Wilcoxon P-values are 0.16, 0.87, and 0.51, respectively. (C) Precision-recall curves of other cytotoxic agents and targeted therapies. The AUPRC of each precision-recall curve is denoted in the figure legend, and the circles denote the point of maximal F1-score. (D) Bar graphs showing the fraction of responders in the patients with high SL-scores (top tertile; green) and low SL-scores (bottom tertile; purple). The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SL group and non-responders in the low-SL group. (E) Precision-recall curves for the SR-scores across the 15 different datasets with AUCs > 0.7. The AUPRC of each precision-recall curve is denoted in the figure legend, and the circles denote the point of maximal F1-score. (F) Bar graphs showing the fraction of responders in the patients with high SR-scores (top tertile; green) and low SR-scores (bottom tertile; purple). The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SR group and non-responders in the low-SR group. (G) Bar graphs show the predictive accuracy in terms of AUC (Y-axis) of SR-based predictors and control predictors across the three glioma cohorts (X-axis). (control predictors are similar to those described in Figure 2C in the main text.) (H) Bar graphs showing the fraction of responders in the patients with high SR-scores (top tertile; green) and low SR-scores (bottom tertile; purple) in the three glioma datasets. The grey line denotes the response rate in each cohort, and the stars denote the hypergeometric significance of enrichment of responders in the high-SR group and non-responders in the low-SR group. (I) The bar graph of pathway enrichment analysis of 10 PD1-PDL1 and 10 CTLA4 SR partner genes in aggregate. X-axis shows the -log10 (Fisher exact test P) and Y-axis shows the enriched pathways.
(A) The ROC plots show that the SL-scores are most predictive of response to the different treatments prescribed at the trial (AUC of R0C=0.72), markedly higher than those of control predictors (as described in Figure 2C). (B) The parallel precision-recall, showing that the SL-score predictor outperforms an array of other control predictors (AUPRC=0.49). (C) The X-axis shows the SL-score classification threshold and Y-axis depicts the false positive rate (derived from the ROC curve presented in Figure 7B, (blue)), the overall coverage (the fraction of the patients in the WINTHER cohort for whom SELECT recommends higher SL-scoring alternate treatment options than those they have actually received, red), and the fraction of the non-responders in WINTHER cohort for whom SELECT recommends such treatment options (orange). As the SL-score decision threshold increases, less and less patients are denoted ‘positive’ (i.e., are assigned a treatment recommendation). At SL-score threshold of 0.44, SELECT recommends alternate treatments that are predicted to be effective for 65% of the entire cohort and 62% of non-responders (as marked by the stars).
SELECT’s prediction accuracy (AUCs) were compared between the cohorts where the mean SL-score is high (green) vs low (red, separated by median values of the mean SL-score). SELECT prediction accuracy is associated with the mean SL-score (A) for targeted therapy (Wilcoxon ranksum P<0.06) but (B) while a similar trend is also observed for immunotherapy, it is not significant (Wilcoxon ranksum P<0.15).
(A) The SL-score is associated with treatment duration in the P0G570 cohort (Wilcoxon P<0.023). (B-C) The fold change (FC) of median treatment duration in high SL-score group (SL-score>0.44) vs low SL-score group (SL- score<=0.44) for (B) each cancer type and (C) each drug, across all categories with more than 15 patients.
SELECT is predictive of coverage (fraction of the patients predicted to be responsive to the given treatment based on the criteria, SL-score>0.44 for targeted therapies and SR-score > 0.9 for immunotherapies) across different therapies in (A) melanoma and (B) non-small cell lung cancer. Spearman rank correlation and associated P-values are noted in the figures.
Supplemental Table 1. The clinical trial cohorts used in the manuscript, Related to Figure 1.
Supplemental Table 3. The clinical trial cohorts for coverage analysis, Related to Supplemental Figure 7.
Supplemental Table 4. Nanostring panel genes, Related to Figure 4, STAR Methods.
Supplemental Table 5. Drug to target gene mapping, Related to Figure 2,3,7, STAR Methods.
Data Availability Statement
The transcriptomics profiles and treatment outcome information of the 23 publicly available clinical trials where SELECT is predictive and the custom code of our framework are available via ZENODO repository (https://zenodo.org/record/4540874). The data from the many of the remaining trials cannot be shared due to restrictions in the data sharing agreement. The new SMC dataset of pembrolizumab-treated lung adenocarcinoma cohort is available at GSE166449.