

Abstract
New appetite‐regulating antiobesity treatments such as semaglutide and agents under investigation such as tirzepatide show promise in achieving weight loss of 15% or more. Energy expenditure, fat oxidation, and lean mass preservation are important determinants of weight loss and weight‐loss maintenance beyond appetite regulation. This review discusses prior failures in clinical development of weight‐loss drugs targeting energy expenditure and explores novel strategies for targeting energy expenditure: mitochondrial proton leak, uncoupling, dynamics, and biogenesis; futile calcium and substrate cycling; leptin for weight maintenance; increased sympathetic nervous system activity; and browning of white fat. Relevant targets for preserving lean mass are also reviewed: growth hormone, activin type II receptor inhibition, and urocortin 2 and 3. We endorse moderate modulation of energy expenditure and preservation of lean mass in combination with efficient appetite reduction as a means of obtaining a significant, safe, and long‐lasting weight loss. Furthermore, we suggest that the regulatory guidelines should be revisited to focus more on the quality of weight loss and its maintenance rather than the absolute weight loss. Commitment to this research focus both from a scientific and from a regulatory point of view could signal the beginning of the next era in obesity therapies.
Study Importance.
What is already known?
-
►
Most available pharmacotherapies for obesity treatment target appetite regulation.
-
►
Beyond appetite regulation, energy expenditure and fat oxidation are important determinants of weight gain and weight‐loss maintenance.
-
►
Most of the drugs stimulating energy expenditure have failed in clinical development because of safety issues or lack of efficacy.
-
►
Maximizing the loss of fat mass while preserving lean mass and energy expenditure is a desirable target for obesity treatment.
What does this review add?
-
►
We propose that safe and durable high‐quality weight maintenance can be achieved by moderate modulation of energy expenditure and preservation of lean mass in combination with efficient appetite‐reducing compounds.
-
►
We discuss potential molecular targets to prevent the decrease in energy expenditure and lean mass observed during weight loss.
How might these results change the direction of research or the focus of clinical practice?
-
►
Commitment to this new research focus, including regulation of energy expenditure and fat oxidation and preservation of lean mass, could signal the beginning of the next era in obesity therapies.
-
►
We suggest that regulatory guidelines should be revisited to 1) recognize that weight‐loss induction and weight‐loss maintenance may require different approaches and 2) focus more on the quality of weight loss and its maintenance than the absolute weight loss.
INTRODUCTION
Lifestyle interventions can reliably produce and sustain only modest (~5%‐10%) weight loss (1), necessitating other types of antiobesity interventions, such as pharmacotherapy, to address obesity management. Antiobesity medication development initially relied on observations of weight loss in agents approved for other indications, which resulted in troubled first‐generation medications being used for weight management. However, new pharmacological treatments have been emerging based on an understanding of the biology of appetite regulation (2, 3, 4). These agents have been developed by taking advantage of their known effects on reducing energy intake and they appear promising in delivering more desirable amounts of weight loss of up to 15% from baseline after 6 to 10 months of treatment (2, 3, 4). Apart from weight loss per se, a more specific goal of weight management should be health improvement through a reduction of excess adipose tissue (which drives ill health) while at the same time preserving lean mass and healthy bone. And equally important, these improvements must be sustainable in the long term.
The purpose of this review was to look beyond appetite regulation as a target for pharmacological therapy and to explore the potential for targeting energy expenditure, fat oxidation, and preservation of lean mass as pathways to healthy weight loss and weight‐loss maintenance in combination with the newer, efficacious appetite‐regulating compounds. To do this, we discuss the importance of energy expenditure and fat oxidation as determinants of weight gain and of weight‐loss maintenance and explore the quality of weight loss versus simply the amount of weight lost. Our aim was to satisfactorily answer the question “How can we produce sustainable high‐quality weight loss through targeting energy expenditure, fat oxidation, and preservation of lean mass?”
ROLE OF ENERGY EXPENDITURE AND FAT OXIDATION IN BODY WEIGHT CONTROL
Energy balance is not only the reflection of energy in versus energy out but the sum of fat, carbohydrate, and protein balances (alcohol also needs to be considered if part of the diet) (5). Because of the physiological interactions between energy substrate availability and hormonal and enzymatic responses, body weight and body composition will remain stable only when macronutrient intakes are compensated by similar energy expenditure and substrate oxidation rates (Figure 1). As shown in Figure 1, over the short term, most energy surplus is stored as fat mass, whereas energy deficit is buffered by the loss of fat mass (5). However, if sustained, caloric restriction will lead to variable weight loss depending on the partitioning of these calories between lean mass and fat mass. In medical practice, most diets and current pharmacotherapies will lead to some degree of lean mass loss (Figure 2) (6, 7, 8).
FIGURE 1.
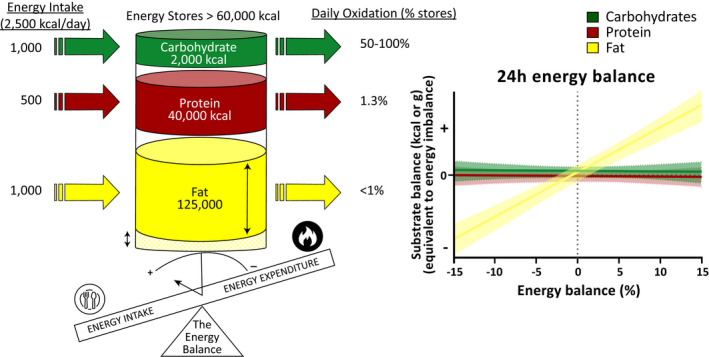
Short‐term substrate balance in response to perturbation of energy balance. Most of the energy reserves in the body are stored in the form of fat, whereas protein represents some stores while only a small fraction of energy reserves is stored as carbohydrates (left panel). In a state of energy balance, the daily turnover (intake and oxidation) of carbohydrates represents approximately 50% to 100% of the body carbohydrate stores (circulating glucose plus liver and muscle glycogen), whereas protein and fat turnovers typically represent approximately 1% of body reserves. Changes in daily carbohydrate and protein intakes are rapidly mirrored by proportional changes in their respective oxidation, whereas changes in fat intake are not compensated in the short term by changes in fat oxidation. Therefore, both carbohydrate and protein balances are tightly maintained in response to changes in energy intake (right panel). In contrast, a surplus or a deficit in energy intake will be buffered by changes in fat stores: a loss in case of energy restriction and a gain during a surplus in energy intake. Adapted from Flatt (163) and Abbott et al. (5)
FIGURE 2.
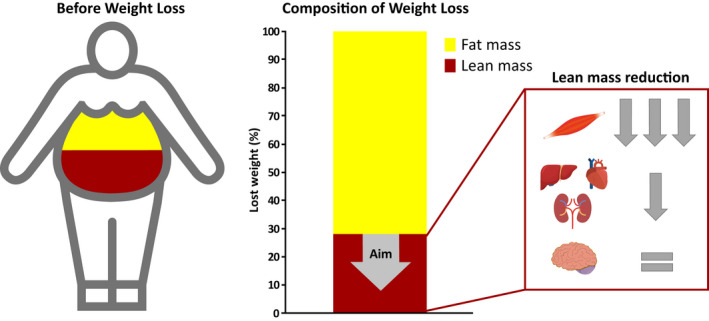
Long‐term change in body composition during sustained energy restriction. Body fat percentage is commonly higher than 25% in men with obesity and higher than 35% in women with obesity, and it increases with age. However, in many patients with severe obesity, fat mass can be up to 50% or more (left panel). The rest of the body (fat‐free mass) includes bone mass (3%‐5% body weight) and lean mass (with water). Lean mass is composed of large organs (including brain, liver, kidneys, and heart, 3%‐5% body weight), muscle mass (20%‐30%), and residual lean mass (digestive tract, small organs, connective tissue, tendons, etc.). In response to sustained energy restriction, most of the weight loss comes from loss of fat mass (60%‐85%) whereas 15% to 40% can come from a loss of lean mass. Most of the lost lean mass is a loss of skeletal muscle mass while other highly thermogenic organs, such as the liver, kidneys, and heart, are minimally impacted during weight loss (right panel). In addition, a small decrease in the residual lean mass (tendons, connective tissue, digestive tract, etc.) is also observed. Ideal pharmaceutical therapeutics for weight management should aim at preventing the decrease in lean mass, thus facilitating further loss of fat mass and weight‐loss maintenance
The best evidence for the role of energy expenditure and fat oxidation in the predisposition to weight gain has been from prospective studies among Pima Indians, a population with high rates of obesity and type 2 diabetes, although part of these findings has not been consistently replicated in other populations or studies conducted with non‐state‐of‐the‐art methods (9). At least three metabolic parameters have been found to be predictive of weight gain, when assessed under neutral energy balance conditions: a) low resting and 24‐hour energy expenditure while being sedentary (10); b) impaired fat oxidation, independently of energy expenditure (11); and c) reduced sympathetic activity, which might underlie the two previous factors (12). Moreover, recent studies have shown that changes in energy expenditure in response to acute 24‐hour overfeeding or 24‐hour complete fast are similarly predictive of body weight changes (9). Using this approach, two well‐differentiated phenotypes, “thrifty” and “spendthrift,” can be identified. Thrifty individuals are characterized by larger decreases in 24‐hour energy expenditure in response to fasting and smaller increases in 24‐hour energy expenditure in response to overfeeding (13). Conversely, spendthrift individuals exhibit attenuated decreases in 24‐hour energy expenditure in response to fasting and relatively large increases in 24‐hour energy expenditure in response to overfeeding. Importantly, the thrifty phenotype is associated with larger body weight gain during a free‐living follow‐up (14) or a 6‐week overfeeding intervention (15) and with lower weight loss during a tightly controlled calorie restriction intervention (16). In addition, acute overfeeding studies have shown that not only the increase in 24‐hour energy expenditure but also the changes in fat oxidation is predictive of body weight changes (17). This capacity to adapt substrate oxidation to its availability, known as metabolic flexibility, is also believed to play a central role in preventing ectopic fat accumulation and insulin resistance (18).
In summary, both low energy expenditure and impaired fat oxidation are risk factors for weight gain but they also cause resistance to weight loss. Therefore, any pharmacological treatment should keep in mind that a booster of either energy expenditure, fat oxidation, or both is likely to play a role in improved body weight management and favor loss of fat mass rather than lean mass.
CHALLENGES OF WEIGHT LOSS AND ITS MAINTENANCE WITH EMPHASIS ON LEAN MASS PRESERVATION AND METABOLIC ADAPTATION
Today, significant weight loss can be achieved in most patients living with obesity by intensive lifestyle, pharmacological, or surgical interventions. However, body weight often plateaus before reaching a medically or cosmetically desirable weight loss (Figure 3). Even more disturbing for many patients is the challenging struggle to maintain the lost weight. Following lifestyle interventions, 30% to 50% of the body weight loss is commonly recovered within a year (19) whereas more than half of patients recover their initial body weight within 5 years after the initiation of treatment (20). Even after bariatric surgery, weight regain is a common phenomenon (21). All in all, preventing weight regain after weight loss is commonly considered one of the biggest challenges in obesity treatment (22).
FIGURE 3.
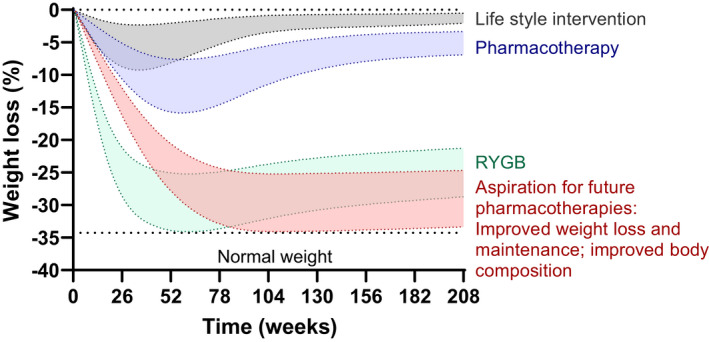
Weight‐loss trajectories with current and future therapies. Lifestyle interventions produce modest weight loss followed by weight regain. Most current pharmacotherapies induce additional but still insufficient weight loss, and weight regain typically occurs over time. RYGB is associated with substantially greater and more sustained weight loss, but nevertheless, some weight regain is observed after 2 years, and this therapy is not available for the majority of people living with obesity. The future of weight management must target not only weight loss but also quality of the weight loss and weight maintenance. To do that, medications must have effects beyond appetite suppression and must target preservation of lean mass and energy expenditure. RYGB, Roux‐en‐Y gastric bypass
Variability in physical activity and adherence to the specific treatment explain part of the variability in both the weight loss and its maintenance, but these are not the sole explanatory factors (23). Body weight (or more precisely, body energy stores) is under homeostatic regulation, which means that complex and interrelated physiological mechanisms are activated to counteract perturbations in energy balance so a stable body weight can be maintained (24) (Figure 4A). In people living with obesity, the homeostatic control of body weight is commonly altered so a higher body weight is often defended by alterations in the homeostatic control of both energy intake and energy expenditure (25). Consequently, energy expenditure is considerably reduced in parallel with weight loss (Figure 4A).
FIGURE 4.
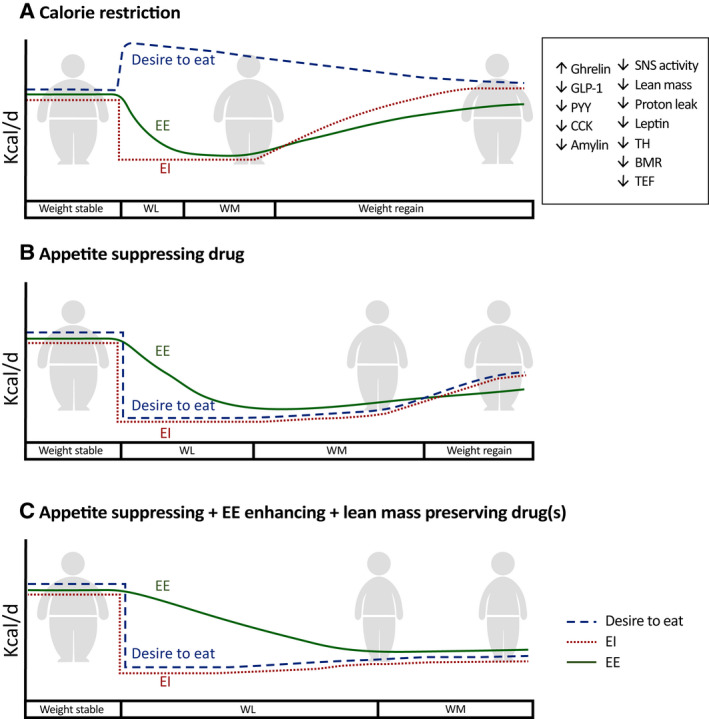
Trends in energy balance regulation in response to different weight‐loss regimens. Energy intake and energy expenditure are balanced in weight‐stable obesity. (A) The reduction in energy intake during calorie restriction results in increased hunger and decreased satiation, driven by the neurohormonal mechanisms shown in the insert. Furthermore, energy expenditure decreases because of changes in body mass/lean mass, neurohormonal changes, and metabolic adaptation. Together, the increased hunger, decreased satiation, and decreased energy expenditure can readily promote weight regain, and the decrease in energy expenditure has been shown in some cases to persist for years (37). (B) When the weight loss is induced by continuous treatment with an appetite‐reducing compound, the hunger is decreased and the satiation is increased, enabling a longer period with reduced energy intake. However, energy expenditure is typically still reduced owing to loss of body mass/lean mass, neurohormonal changes, and some degree of metabolic adaptation. Consequently, the reduction in body weight is still limited, and usually, over time, weight regain occurs despite continued treatment, albeit at a slower rate compared with the regain observed with calorie restriction in panel A. (C) The future aspiration for pharmacotherapy that combines appetite‐reducing, energy‐expenditure–boosting, and lean‐mass–preserving mechanisms. Such a combination will decrease hunger, increase satiety, and protect lean mass, resulting in less suppression of energy expenditure, which (together with an actual energy‐expenditure–boosting component) will have the potential to cause greater and more sustainable weight loss. BMR, basal metabolic rate; CCK, cholecystokinin; EE, energy expenditure; EI, energy intake; GLP‐1, glucagon‐like peptide‐1; PYY, peptide YY; SNS, sympathetic nervous system; TEF, thermic effect of food; TH, thyroid hormone; WL, weight loss; WM, weight maintenance
Lean mass preservation
In large part, the reduction in energy expenditure induced by weight loss is attributable to the decline in lean mass, the metabolically active tissues (26, 27, 28). Even if lifestyle modifications and currently available therapies for weight loss can deliver substantial decreases in fat mass, 15% to 40% of the weight loss represents reductions in lean mass (6, 7, 8) (Figure 2). Resting energy expenditure is reduced by approximately 13 kcal/d for each kilogram of muscle mass lost, in contrast with the much lower 4.5‐kcal/d reduction for each kilogram of fat mass lost (29). Moreover, besides reducing skeletal muscle mass, weight loss also decreases the mass of highly thermogenic organs such as liver, heart, and kidney (8) (Figure 2). The contribution of these thermogenic organs to basal metabolic rate is between 15‐ and 33‐fold higher, per mass unit, than the contribution of resting skeletal muscle (29); therefore, the decrease in resting energy expenditure because of the decreased lean mass can be substantial. Furthermore, after weight loss, most of the weight regained is typically fat mass, leading to a progressive decline in lean mass over several weight cycles, thereby further exacerbating the reduction in energy expenditure (30). Hence, drug candidates that either spare the loss of lean mass or even increase lean mass during weight loss (31) could potentially prevent part of the reduction in energy expenditure and thereby enable a more pronounced and sustainable weight loss.
Metabolic adaptation
Together, changes in fat mass, muscle mass, and organ size account for, on average, ~60% of the reductions in energy expenditure observed in response to weight loss, thus leaving ~40% of the energy expenditure decrease unexplained (8). The decrease in the energy necessary to sustain weight‐bearing activities at a reduced weight as well as the lower thermic effect of food owing to lower energy intake must also be considered. Nonetheless, part of the remaining ~40% is also explained by increased energy efficiency (i.e., energy expended per unit of metabolic mass), which is known as metabolic adaptation, adaptive thermogenesis, or metabolic slowing (28). Potential mechanisms underlying metabolic adaptation include decreases in plasma leptin concentrations (32), decreased activities of the thyroid (33) and sympathetic nervous system (SNS) axes (12), increased mitochondrial biogenesis (34), and improved mitochondrial coupling of oxidative phosphorylation (35). Metabolic adaptation to weight loss commonly accounts for reductions of 6% to 10% of total daily energy expenditure (33) and has been documented in response to varied behavioral interventions, including lifestyle, pharmacological, and surgical treatments of obesity (36). Metabolic adaptation may persist many years after weight loss (37), and it is believed to be a relevant barrier to weight‐loss maintenance. Nonetheless, evidence directly linking the degree of chronic metabolic adaptation to weight‐loss regain remains elusive (38). Some studies have failed to observe an association between metabolic adaptation in resting metabolic rate and later weight regain (37, 38). However, resting metabolic rate is only one of the components of total energy expenditure (39). Studies with an adequate assessment of all components of energy expenditure (sleeping metabolic rate, resting metabolic rate, nonexercise activity thermogenesis, exercise activity thermogenesis, and diet‐induced thermogenesis) and body composition, conducted in large sample sizes, are needed to elucidate the interindividual variability and the extent to which metabolic adaptation contributes to the variability in both weight loss and weight regain. Also, potential differences in metabolic adaptation after diet‐induced versus pharmacologically and surgically induced weight loss needs further investigation.
Energy expenditure and energy intake regulation
Energy expenditure has been proposed as a primary driver of energy intake (40). Changes in energy expenditure are usually matched by changes in energy intake, explaining why therapies that solely modify energy expenditure usually result in less‐than‐expected weight loss (41). However, even if energy intake generally matches energy expenditure in the long term, low levels of total energy expenditure seem to be linked to appetite dysregulation resulting in enhanced hunger and impaired satiation, favoring hyperphagia (40). This has led to thinking that the human body can better match energy intake to expenditure at high energy flux (42). This hypothesis suggests that, if energy expenditure is kept low enough, the drive to eat is not decreased proportionally. This seems to be corroborated by preliminary evidence showing that acute increases in energy flux improve appetite control and reduce the drive to eat (43). Moreover, the maintenance of high total energy expenditure by increasing voluntary or spontaneous physical activity (or targeting inactivity) is one of the few factors that has been shown to favor successful weight‐loss maintenance (42, 44). Consequently, the decreased energy expenditure induced by weight loss might, to some extent, oppose the effects exerted by appetite‐regulating drugs on energy intake. Indeed, a positive association between the increase in hunger and metabolic adaptation has been documented (36), which might explain why metabolic adaptation seems to be a weak predictor of weight‐loss maintenance. Furthermore, the decrease in lean mass in response to weight loss also seems to drive compensatory hyperphagia aiming at restoring the lost lean mass (30). Pharmacological strategies aimed at sparing lean mass or counteracting metabolic adaptation could therefore provide additional benefits for controlling energy intake, thus triggering larger and more sustainable weight loss.
FAILED PHARMACOLOGICAL TARGETS FOR INCREASING ENERGY EXPENDITURE OR FAT OXIDATION
Pharmacological targeting of energy expenditure has already been pursued for weight‐loss induction. In fact, some of the first antiobesity treatments used in the early nineteenth century, thyroid hormones and the mitochondrial uncoupler dinitrophenol (DNP), targeted energy expenditure (Table 1).
TABLE 1.
Targets explored in clinical trials for increased energy expenditure and induction of weight loss
| Target | Mode of action | Indication | Clinically relevant weight loss | If withdrawn, reasons for discontinuation; Remarks |
|---|---|---|---|---|
| TH mimetics | Increased REE via SNS activation, mitochondrial biogenesis and uncoupling | Currently, dyslipidemia; NASH | Yes | Lack of safety window on cardiac and bone parameters; thyrotoxicosis |
| Dinitrophenol | Mitochondrial uncoupling | Obesity, NASH | Yes | Steep dose response, hyperthermia, mortality |
| Monoaminergic system | ||||
| Fenfluramine/Fen‐Phen | Potentiation of thermic effect of food; appetite suppression | Obesity | Yes | Cardiac valvulopathies, increased blood pressure/heart rate |
| Sibutramine | Appetite suppression; increased REE in some trials | Obesity | Yes | Increased risk of heart attack and stroke |
| β3‐AR agonists | BAT activation | Obesity; diabetes | No | Lack of efficacy and off‐target effects on blood pressure and heart rate (β3‐AR not relevant for human BAT activation) |
| MC3/4 agonists Setmelanotide | Appetite suppression and presumed increase in SNS activity in animal models | Obesity | No (general obesity) | Several programs withdrawn for general obesity due to lack of efficacy (including setmelanotide), cardiovascular effects and/or hyperpigmentation. |
| Yes (genetic obesity) | Setmelanotide approved for rare genetic obesity | |||
| GLP‐1/glucagon coagonists (w/wo GIP) | Appetite suppression, fat oxidation, increased EE | Obesity, Diabetes, NASH | Yes | Some programs discontinued for obesity due to lack of glycemic control, ongoing trials for NASH |
| FGF21 analogues | Diverse effects on appetite, browning and/or increased REE in animal models | Obesity, Diabetes, NASH | No | Programs discontinued for obesity due to lack of efficacy, ongoing trials for NASH |
| MetAP2 inhibitors | Fat oxidation, lipolysis | Obesity | Yes | Venous thromboembolisms with beloranib; ZGN‐1061 in trials for obesity |
Abbreviations: AR, adrenergic receptor; BAT, brown adipose tissue; EE, energy expenditure; Fen‐phen, combination of fenfluramine and phentermine; FGF21, fibroblast growth factor 21; GIP, gastric inhibitory polypeptide; GLP‐1, glucagon‐like peptide 1; MC, melanocortin; MetAP2, methionine aminopeptidase 2; NASH, non‐alcoholic steatohepatitis; REE, resting energy expenditure; SNS, sympathetic nervous system; TH, Thyroid hormones; w/wo, with/without.
Thyroid hormones
Thyroid hormones as part of the hypothalamic‐pituitary‐thyroid axis increase energy expenditure by increasing SNS activity and mitochondrial uncoupling and/or biogenesis (45). Higher baseline free triiodothyronine, T3, was predictive of greater weight loss in the POUNDS LOST trial (46), whereas weight loss decreases free T3, an adaptation that contributes to the weight‐loss–induced reduction of energy expenditure (47). Thyroid extracts were once used for weight‐loss induction (48), and although a low dose of thyroid hormones may aid weight maintenance and the preservation of lean mass, hyperthyroidism or thyrotoxicosis in euthyroid patients (49) along with adverse effects on bone metabolism and cardiac mass and/or function are significant risks (50).
DNP
DNP is a chemical mitochondrial uncoupler that induces a proton leak that uncouples oxygen consumption from ATP production, leading to energy dissipation as heat and thereby increasing overall energy expenditure (51). DNP was discovered to induce weight loss in the 1920s, but despite good efficacy with a 30% to 40% increase in energy expenditure and a corresponding weight loss of 0.7 to 0.9 kg/wk (52), the dose‐response curve was very steep, and side effects from skin rashes, peripheral neuritis, cataract, and hyperthermia to sudden deaths led the US Food and Drug Administration to ban the use of DNP in 1938 (53).
Monoaminergic systems
Drugs that target monoaminergic systems cause appetite‐suppression–induced weight loss by inhibiting the reuptake or promoting the release of serotonin, dopamine, and/or noradrenaline or by stimulating the respective receptors (54). While some also increase energy expenditure in rodents (55), this effect is quite variable in humans. Sibutramine increased resting energy expenditure and attenuated the weight‐loss‐induced energy expenditure reduction in women with obesity (56, 57) but had no thermogenic effect in other obesity trials (58). Fenfluramine, in contrast, did not increase resting energy expenditure but enhanced the thermic effect of food in both rodents and humans (59). Many drugs in this class, including fenfluramine and sibutramine, failed because of detrimental cardiovascular or psychotropic effects (60).
β3 adrenergic receptor agonists
Modulation of energy expenditure via brown adipose tissue (BAT) activation has been long debated. The discovery that adults express significant amounts of BAT that can be recruited by cold exposure or appropriate pharmacological treatments has spurred interest (61). Selective β3 adrenergic receptor (AR) agonists have been extensively pursued based on promising data in rodents, but most human studies have resulted in limited efficacy and off‐target cardiovascular side effects (62). Recently, it has been shown that β2‐AR seems to be the only relevant β‐AR in human BAT (63), which could imply a potential for selective β2‐AR agonists for BAT activation, although selectivity and cardiovascular side effects remain a challenge (64).
Melanocortin receptor agonists
Melanocortin 4 receptor (MC4R) agonists suppress appetite and, in animal models, increase energy expenditure by increasing SNS activity (65, 66). Several MC4R agonists failed in the clinic because of increased blood pressure or lack of efficacy combined with hyperpigmentation owing to off‐target activity on MC1R (67, 68). Recently, setmelanotide successfully passed clinical development and is marketed for chronic weight management in patients with ultrarare genetic obesity linked to MC4R signaling. Treatment in those with pro‐opiomelanocortin deficiency demonstrated remarkable weight loss, unlike the minor weight loss observed with general obesity (69). Importantly, setmelanotide did not produce adverse cardiovascular effects (69). Although the primary factor driving weight loss was decreased appetite, an acute increase in resting energy expenditure (~6%, relative to placebo) was also observed in the clinic (70).
Glucagon and glucagon‐like peptide‐1 receptor co‐agonists
About a decade ago, the concept of using a dual glucagon‐like peptide‐1 (GLP‐1)/glucagon agonist for obesity emerged (71). The concept built on activation of both GLP‐1 receptors leading to decreased energy intake and glucagon receptors mainly causing increased energy expenditure, resulting in superior effects on body weight and glucose metabolism (72). Promising animal studies with potent GLP‐1/glucagon co‐agonists led to initiation of several clinical programs (73). Unfortunately, the challenge was much greater in humans, in which there is a narrow optimal ratio for getting additional weight loss on top of the GLP‐1 alone without affecting glycemia or causing adverse cardiovascular effects (74). Today, most obesity programs have been abandoned or turned to indications such as nonalcoholic steatohepatitis (NASH).
Fibroblast growth factor 21
In 2005, fibroblast growth factor 21 (FGF21), a hormone secreted from the liver, was described as a novel metabolic regulator (75). Treatment of animal models demonstrated impressive effects on body weight, glucose regulation, dyslipidemia, and NASH (76). Energy expenditure was markedly increased in rodents (77) in parallel to a browning of white adipocytes (78), suggesting that FGF21 analogues could be new powerful antiobesity agents. Several FGF21 analogues entered human clinical trials for the treatment of obesity and type 2 diabetes, but unfortunately, the impressive preclinical effects on energy expenditure did not translate to humans (76). However, there was robust lipid lowering in plasma and liver, and most of the FGF21 analogues have been transferred to the NASH indication.
Methionine aminopeptidase 2 inhibitors
Methionine aminopeptidase 2 (MetAP2) inhibitors induce weight loss in humans and were thought to increase energy expenditure because of observations of increased levels of ketones and FGF21 (79). Beloranib, an irreversible inhibitor, produced clinically relevant lean‐mass‐sparing weight loss but was terminated because of venous thromboembolic events (80). A second reversible inhibitor (ZGN‐1061) with an improved risk profile is currently in clinical development (79). MetAP2 inhibitors inhibit adipogenesis and enhance lipolysis and fat oxidation, although reduced hyperphagia has also been observed (81). Effects of MetAP2 inhibitors on energy expenditure in humans have not been reported, but in rodents, the inhibitor fumagillin actually decreased energy expenditure and sympathetic tone while suppressing appetite (82).
In summary, many of the drugs that have a relevant effect on energy expenditure have failed in clinical development or have been withdrawn from the market mainly because of safety issues or, in some cases, because of lack of efficacy (Table 1). Hence, there is a huge therapeutic gap with respect to safe targeting of this side of the energy balance equation, which, together with lean mass preservation, could significantly improve current weight‐management strategies.
RETHINKING APPROACHES FOR WEIGHT LOSS AND ITS MAINTENANCE
A general concern related to pharmacological targeting of increased energy expenditure is the concomitant increase in heart rate and cardiac workload needed to meet the body’s increased oxygen requirements, especially if energy expenditure is increased by more than 25% (83). In individuals with obesity who already have a challenged cardiovascular system, this additional burden may not be well tolerated. Based on predictive models, it can be calculated that an increase in energy expenditure of 5% to 10% will by itself provide a 5% weight loss over a year (84), and we suggest targeting this level of energy expenditure for safe promotion of weight loss and weight‐loss maintenance. In the same way, because skeletal muscle mass is typically already increased in (nonsarcopenic) individuals with obesity (85), the main aim should be to preserve or moderately increase muscle mass to counteract further decreases in energy expenditure thereby enabling sustained weight loss. Potential pathways that could be targeted with these aims in mind are shown in Figure 5 and are described in more detail subsequently.
FIGURE 5.
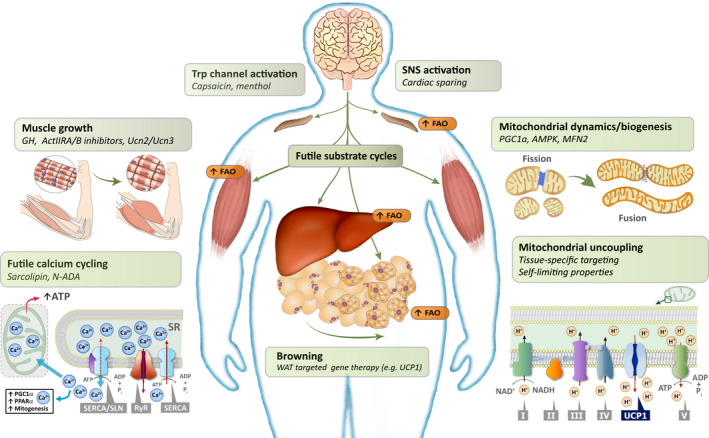
Rethinking approaches for weight maintenance by targeting energy expenditure and lean mass preservation. Potential ways of maintaining or increasing energy expenditure include a diversity of targets ranging from sympathetic nervous system and transient receptor potential channel activation over browning of white adipose tissue to increased nonshivering thermogenesis in various organs. Nonshivering thermogenesis in skeletal muscle and other tissues can be brought about by increasing mitochondrial proton leak, by pharmacologically induced futile calcium cycling or various other futile substrate cycles, all of which would increase fatty acid oxidation and energy expenditure. Mitochondrial biogenesis and improved mitochondrial function are needed to support this increased energy demand. Potential targets for maintaining or increasing muscle mass include growth hormone, activin type II receptors A/B, and urocortin 2 and 3, which will secondarily lead to maintenance of the energy expenditure. ActIIR A/B, activin type II receptors A/B; AMPK, AMP‐activated protein kinase; FAO, fatty acid oxidation; GH, growth hormone; MFN2, mitofusin 2; N‐ADA, N‐arachidonoyl dopamine; PGC1α, peroxisome proliferator‐activated receptor gamma coactivator 1α; PPARα, peroxisome proliferator‐activated receptor alpha; RyR, ryanodine receptor; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+‐dependent ATPase; SLN, sarcolipin; SNS, sympathetic nervous system; SR, sarcoplasmic reticulum; TRP, transient receptor potential; Ucn2/3, urocortin 2 and 3; UCP1, uncoupling protein 1; WAT, white adipose tissue
TARGETING ENERGY EXPENDITURE FOR INDUCTION OF WEIGHT LOSS AND WEIGHT‐LOSS MAINTENANCE
Thermogenesis occurs in a variety of tissues, such as skeletal muscle, BAT, white adipose tissue (WAT), and liver. Skeletal muscles compose up to 40% of body mass, are responsible for approximately 20% of resting energy expenditure (86), and contribute to cold‐induced thermogenesis by both shivering and nonshivering mechanisms. Muscle‐derived nonshivering thermogenesis (NST) may be the dominant source of heat production in animals in which BAT is either absent (such as in birds and pigs) or reduced (such as in large mammals, including adult humans) (87). Given the large mass of skeletal muscles, enhancement of NST in muscle may be relevant to increase energy expenditure sufficiently to impact weight management. Also, skeletal muscles have the vascularization needed to meet the increased demands for oxygen and substrate and dissipate the extra heat. Skeletal muscle NST can result from mitochondrial proton leak, sarcolipin (SLN)‐mediated futile calcium cycling, and various substrate cycles, which may also occur in other tissues (Figure 5) (88, 89, 90).
Mitochondrial proton leak
Most energy expenditure occurs through oxidative phosphorylation in mitochondria, leading to the production of ATP (86). However, the efficiency of the conversion of energy to ATP varies, owing to the naturally occurring mitochondrial proton leak, which uncouples part of the energy consumption from the ATP production and dissipates it as heat (89, 91). This proton leak is estimated to contribute 20% to 30% of the resting energy expenditure in rats, with the majority coming from the liver and skeletal muscles (91). Such variability in mitochondrial efficiency is associated with total energy expenditure and response to diet‐induced weight loss (92, 93). The mitochondrial proton leak consists of a basal component associated with the adenine nucleotide translocases (primarily ANT1 in muscles and ANT2 in other tissues) and uncoupling protein 1 (UCP1) in BAT, and an inducible part mediated by activation of ANT, UCP1, UCP2, and UCP3, with UCP3 being the most abundant UCP in skeletal muscle (89, 94). Although muscle‐specific overexpression of UCP3 in mice results in weight loss (95), in most species the contribution of both UCP2 and UCP3 to thermogenesis is thought to be negligible. Rather, the main function of UCP2/3 may be the regulation of reactive oxygen species (ROS), facilitation of mitochondrial fatty acid transport, and modulation of hormone secretion (89). In contrast, ANT1 catalyzes more than half of the basal mitochondrial proton leak in muscles (94) and thus may constitute a potential pharmacological target for increasing energy expenditure. However, so far, pharmacologically increased proton leak and decreased mitochondrial efficiency has primarily been targeted by chemical mitochondrial uncouplers.
Chemical mitochondrial uncouplers
The clinical efficacy of DNP is intriguing, and various ways of improving the narrow therapeutic window, by producing chemical uncouplers with different physical and chemical characteristics, have been explored (96, 97). A series of studies has shown that butylated hydroxytoluene utilizes the mitochondrial ANT to induce limited uncoupling at low concentration and conventional uncoupling at higher concentration resulting in a wide dynamic range of more than a millionfold in vitro (96). Tissue‐specific uncoupling has also been reported to avoid uncoupling of the more sensitive tissues such as heart, kidneys, and peripheral nerves (98). Furthermore, mitochondrial uncoupling in combination with pyruvate dehydrogenase activation has been shown to modulate DNP‐related side effects (99), and pro‐drugs and controlled‐release formulations of DNP may attenuate the peak plasma concentration, thereby improving the therapeutic window (100). However, whether it will be enough to confer these drugs a sufficient safety window is still questionable given the severity of the side effects. Despite the promising preclinical data and the many years of research in this area, none of the mitochondrial uncouplers has made it into clinical development for the treatment of obesity, underscoring the safety challenge with this mode of action.
Futile calcium cycling
Stimulation of skeletal muscle fibers leads to calcium (Ca2+) release into the cytoplasm via the ryanodine receptor 1 (RyR1). The released Ca2+ promotes heat generation from ATP hydrolysis during muscle contraction and when Ca2+ ions are pumped back into the sarcoplasmic reticulum by the sarcoplasmic/endoplasmic reticulum Ca2+‐dependent ATPase (SERCA) (90). Under resting conditions, there is evidence for continuous Ca2+ cycling via a partially open RyR1 channel and SERCA, leading to continuous ATP hydrolysis and heat generation (101). SERCA1a is expressed predominantly in fast‐twitch glycolytic muscle, whereas SERCA2a is expressed mostly in cardiac and slow‐twitch oxidative muscle fibers. SERCA2b is expressed in low levels in all tissues, including heart and muscle (90). SERCA gene expression and activity in heart and muscle are regulated by thyroid hormones (102) and by small proteins such as phospholamban, SLN, myoregulin, and dwarf open reading frame (90). Binding of SLN to SERCA reduces the efficiency of Ca2+ transport, resulting in more ATP being used to transport the same amount of Ca2+, thus leading to increased energy expenditure and heat production (103). In contrast to mitochondrial uncoupling that comes with a risk of severe ATP depletion, the uncoupling of SERCA seems to simultaneously improve mitochondrial biogenesis and increase oxidative metabolism to meet the increased ATP demand (90). Mice with skeletal‐muscle‐specific overexpression of SLN display reduced weight gain, indicating that SLN and potentially other compounds could pharmacologically activate this futile cycle to promote weight loss (104). A tissue‐targeted approach may be needed to avoid potential adverse effects in the heart as observed in mice overexpressing SLN selectively in the heart (105).
Futile substrate cycles
Substrate cycles may account for a significant fraction of ATP consumption, and modulation of their activity may increase heat production and energy expenditure (88, 106). An example is the triglyceride/fatty acid cycle, in which esterification of triglycerides is followed by hydrolysis (88). It occurs in WAT and muscle, is upregulated under several conditions, such as severe burns (107), cachexia (108), and exercise (109), and seems to be primarily mediated by catecholamines (107, 108). However, it can also be stimulated by peroxisome proliferator‐activated receptor alpha (PPARα) agonists in vitro in white adipocytes (110), indicating that pharmacological modulation of such substrate cycles is possible although the effect on whole‐body energy expenditure remains to be determined.
Mitochondrial dynamics and biogenesis
Enhancing mitochondrial biogenesis, thus providing structural integrity and oxidative capacity, might facilitate therapies aimed at increasing energy expenditure and/or fat oxidation. Obesity, and more specifically ectopic fat deposition, triggers mitochondrial impairments manifested by reduced oxidative capacity and fat oxidation, increased glucose dependence, increased ROS production, and a shift from mitochondrial fusion to fission (111, 112). Such mitochondrial fragmentation is accompanied by reduced mitochondrial biogenesis and structural changes resulting in swollen, more rounded mitochondria (111, 112). These impairments mimic deteriorations that occur with aging, sarcopenia, and some neurodegenerative diseases (113, 114). Importantly, exercise and caloric restriction induce opposite effects on mitochondrial integrity (115, 116). The defective mitochondrial function usually observed in obesity probably impacts the metabolic adaptation and the impaired fat oxidation triggered by weight loss. Therefore, a plethora of targets governing mitochondrial function may be used as an adjuvant therapy if the safety concerns of such an approach can be mitigated (117). Nonetheless, improved mitochondrial function and biogenesis need to be accompanied by increased mitochondrial activity in order to increase energy expenditure.
AMP‐activated protein kinase (AMPK), a heterotrimeric serine/threonine kinase, when activated, mimics the transcriptional signature induced by exercise in human skeletal muscle. AMPK is also implicated with peroxisome proliferator‐activated receptor gamma coactivator 1α (PGC1α) in increased mitochondrial biogenesis and fat oxidation (118). Currently, Betagenon’s O304 AMPK activator is in clinical development for diabetes with future potential for obesity (119, 120).
Mitofusin 2 (Mfn2) is a mitochondrial membrane‐associated guanidine triphosphate hydrolase that regulates mitochondrial fusion and mitochondrial–endoplasmic reticulum associations (121). Mfn2 expression is decreased with overfeeding, and Mfn2‐deficient mice have increased ROS production and mitochondrial dysfunction in muscle and liver (111). Targeted adipose‐specific Mfn2 deficiency results in an obesity phenotype in mice (122). Thus, Mfn2 activators present a potential therapeutic opportunity for enhancing energy expenditure if proven safe (123, 124).
Leptin for weight maintenance
When leptin was discovered in 1994 (125), hopes were high that a magic bullet had been found for the treatment of obesity. Leptin acts on neural circuits in the hypothalamus and other brain areas to regulate food intake and energy expenditure (125). The thermogenic effect is mediated by increased sympathetic efferent signaling to BAT and WAT, thereby increasing BAT activity and lipolysis (126). Several clinical trials have been conducted in people living with obesity (127), but most of them with disappointing outcomes. It turned out that the major physiological function of leptin is to signal states of negative energy balance and decreased energy stores rather than the opposite, explaining the lack of effect in individuals with obesity (127). However, in the hypometabolic state, which occurs after weight loss, there is a relative leptin insufficiency, and here low‐dose leptin can partially reverse the physiological and behavioral responses associated with weight loss, leading to reduced hunger and restoration of energy expenditure (127). Therefore, pharmacotherapies affecting the leptin system are likely to be effective for weight‐loss maintenance rather than weight loss itself, but currently, there is no regulatory pathway for a weight‐loss maintenance indication.
Increased SNS activity
The SNS plays a complex role in energy homeostasis and differentially regulates substrate mobilization in many innervated tissues (128, 129). Whereas SNS tone is inconsistently described as increased or decreased in obesity (12, 129), postsynaptic sympathetic signaling is consistently decreased in adipose tissue in obesity, leading to decreased lipolysis, BAT activity, and energy expenditure and to increased lipid deposition (130). This can be attributed to decreased β‐AR expression (131) and to increased uptake and degradation of norepinephrine in white adipocytes (132) and in specialized adipose‐tissue and sympathetic neuron‐associated macrophages (133, 134). Sympathetic neuron‐associated macrophage–specific silencing of solute carrier family 6 member 2 inhibits norepinephrine uptake and degradation in macrophages and induces weight loss via restoration of lipolysis and energy expenditure in mice (134). Whether this approach can produce clinically relevant effects on body weight is yet unclear.
Sympathomimetics are presumed to induce weight loss via central nervous system–mediated appetite suppression and SNS activation but also cause concomitant adverse cardiovascular effects. Spurring new interest in this area, a recent report using a pegylated amphetamine that did not cross the blood–brain barrier has shown increased lipolysis and thermogenesis resulting in weight loss in mice without affecting appetite or triggering cardiovascular side effects (135).
Transient receptor potential (TRP) channels detect environmental changes (e.g., temperature, touch, pain, taste) and regulate energy expenditure through SNS and BAT activation (reviewed by Saito (136)). Capsaicin, capsinoids, and menthol are examples of TRP channel activators suggested to affect energy expenditure and body weight, although only minor and somewhat BAT‐ and BMI‐dependent effects have been observed in humans (136).
Overall, without cell‐type–specific targeting, significant SNS activation bears the risk of cardiovascular side effects. Nonetheless, restoration of local sympathetic signaling will be important in preventing the metabolic adaptations that impede weight‐loss maintenance.
Browning of white adipocytes
The variable amount of BAT and the overall modest contribution of BAT to daily energy expenditure is challenging for activation of the existing BAT as an effective therapeutic concept (61, 137). In contrast, treatments that increase the overall adipose tissue thermogenic capacity by transforming the more abundant WAT into more mitochondria‐dense and metabolically active “beige” adipose tissue could result in relevant increases in energy expenditure (138). This process, termed “browning,” has been studied intensively in rodents but has also been observed in patients with norepinephrine‐producing tumors (139), severe burn injuries (140), and in humans treated with the phosphodiesterase type‐5 inhibitor sildenafil (141). Numerous pharmacological approaches for “browning” of WAT have been proposed (142, 143), although a sufficient capacity for pharmacological “browning” resulting in relevant effects on energy expenditure has yet to be proven in humans. Another way of increasing the thermogenic capacity could be by implantation of stem cell–derived or genetically engineered functional “beige” adipocytes as demonstrated in mice (144) or by gene‐based therapies aiming for "browning" of WAT as reviewed by Wang and Wei (145). The latter approach is supported by data from pigs with a WAT‐specific clustered regularly interspaced short palindromic repeats (CRISPR)‐based UCP‐1 reconstitution leading to a leaner phenotype (146). Common for both pharmacologically induced and transplanted “beige” adipose tissue is that pharmacological stimulation of thermogenesis in these cells is needed to obtain an effect on energy expenditure (142).
G protein–coupled receptor 75
Variants in the G protein–coupled receptor 75 (GPR75) have recently been shown to be associated with protection from obesity in humans, and knockout of the gene in high‐fat‐fed mice results in resistance to weight gain (147). Data from mouse studies with the GPR75 ligand 20‐HETE indicate that activation of the receptor decreases energy expenditure whereas there are no effects on energy intake (148). Together, this suggests that inhibition of GPR75 signaling may be a new therapeutic strategy for increasing energy expenditure and counteracting obesity, although cardiovascular safety must be considered.
PRESERVATION OF LEAN MASS
The decrease in lean mass and energy expenditure observed with weight loss can be somewhat prevented by resistance exercise (42, 44). In addition, drugs that favor lean mass preservation could have an important role in the weight maintenance phase (Figure 5).
Growth hormone
Growth hormone (GH) and its effector hormone insulin‐like growth factor 1 (IGF‐1) are key regulators of somatic growth, with major anabolic and lipolytic effects in muscle, liver, and adipose tissue (149). Besides its lipolytic effect, browning and fat oxidation has also been suggested to be involved in the GH regulated carbohydrate and lipid metabolism via interactions with insulin and IGF‐1 (150). GH is used for the treatment of GH deficiency in children and adults and has been tested in several studies in people with obesity (151), in whom a functional GH deficiency is often observed (149). However, GH treatment only has marginal effects on body weight per se but alters body composition in a favorable manner owing to simultaneous loss of fat mass and gain of lean mass (149, 151). GH has gained interest in the bariatric surgery field wherein the loss of lean mass can be significant (152). The addition of GH therapy in bariatric surgery patients with existing low GH and IGF‐1 levels results in a more favorable body composition with greater reductions in fat mass, preservation of lean mass, and similar improvements in metabolic parameters (153). In a similar manner, GH therapy may also be beneficial as adjunct to pharmacological antiobesity treatments in order to maintain lean mass.
Activin type II receptor inhibition
Bimagrumab (Novartis, Basel, Switzerland) is a human monoclonal antibody that binds the activin type II receptors (ActRII), thereby preventing binding of the natural ligands, including myostatin and activin A, that otherwise negatively regulate muscle growth (154). Treatment with bimagrumab effectively increases lean mass while at the same time decreasing fat mass in patients living with obesity and type 2 diabetes (31). Heymsfield et al. found that lean mass increased by 3.6%, with an impressive fat mass loss of 20.5% after 48 weeks of treatment, resulting in a total weight loss of 6.5% (31). Despite the modest weight loss, the superior quality of the weight loss gave rise to metabolic improvements on par with most current antidiabetic treatments (31). Interestingly, dietary intake did not differ from baseline to treatment week 48 in either of the two groups, suggesting lean mass preservation and increases in energy expenditure as part of the mechanism of action for the weight loss.
Urocortin 2 and 3
Many of the same beneficial effects on body composition and metabolism observed for the ActRII inhibitors are seen in preclinical animal models following treatment with urocortin (Ucn) 2 and Ucn3, which are selective agonists of the corticotrophin‐releasing hormone receptor 2 (CRHR2) (155). These urocortins have primarily been investigated for their beneficial effects on the cardiovascular system (156), but in addition, both Ucn2 and Ucn3 treatment or overexpression significantly increase muscle mass (157, 158) while at the same time modulating food intake, fat mass, glucose tolerance, and insulin sensitivity (157, 159, 160). The effect of urocortins on muscle seems to be partially mediated through a direct effect on CRHR2 receptors on the muscle fibers resulting in both muscle growth and increased glucose uptake via increased glucose transporter type 4 (GLUT4) translocation and increased insulin signaling (159, 160). Furthermore, ex vivo studies indicate a direct effect on thermogenesis in skeletal muscle potentially driven by substrate cycling between de novo lipogenesis and lipid oxidation (161) although increased whole‐body energy expenditure has not been observed (159). It remains to be seen whether the preclinical data translate to humans and whether the acute cardiovascular effects observed in the clinical trials can be mitigated.
IMPLICATIONS AND CONCLUDING REMARKS
Current evidence suggests that weight loss itself and weight‐loss maintenance may require different treatment strategies to be efficient. Although caloric restriction and appetite‐reducing treatments are very effective as the sole strategy for weight‐loss induction, long‐term weight‐loss maintenance requires additional modulation of energy expenditure and/or fat oxidation together with preservation of lean mass. Moreover, concomitantly targeting energy expenditure during appetite‐suppressant–driven weight loss might result in higher rates of weight loss (Figure 4). The primary efficacy criterion in the guidelines for weight‐management drugs for both the US Food and Drug Administration and the European Medicines Agency is the demonstration of a statistically significant, placebo‐corrected weight loss of at least 5% from baseline weight after 12 months of treatment, in addition to ensuring that the weight loss is caused primarily (>50%) by a reduction in fat content and not lean body mass. The current requirement for a 5% weight loss is inadequate for novel drugs that induce fat loss while preserving lean mass, as they might result in significant improvements in body composition and metabolic profile despite an overall lower weight loss (31). In addition, such a profile is likely to lead to better long‐term weight‐loss maintenance. However, it is currently not possible to obtain approval for drugs effective for weight‐loss maintenance even though this is often the greatest challenge for people who have lost weight. Weight regain is almost inevitable and it often leaves the patients in a worse position because of metabolic adaptation and a less favorable body composition caused by loss of lean mass and regain of primarily fat mass. Hopefully, more clinical data will be gathered to support a revision of regulatory guidance to 1) recognize that weight loss and weight‐loss maintenance may require different treatment strategies and 2) focus more on fat loss, lean mass preservation, and long‐term weight‐loss maintenance rather than absolute weight loss after 1 year.
In this review, we addressed the question “How can we produce sustainable high‐quality weight loss through targeting energy expenditure, fat oxidation, and preservation of lean mass?” and explored the biology and physiology underlying the sustainability of weight loss in the face of metabolic adaptation and the variability of the composition of the lost mass. Previous approaches targeting energy expenditure for weight loss have not been able to achieve an acceptable benefit/risk ratio, and currently, no pharmacological agents targeting energy expenditure are approved for obesity. By rethinking energy expenditure as a driver for weight‐loss maintenance rather than for significant weight loss, we propose that safe and durable high‐quality weight maintenance can be achieved. We suggest that moderate modulation of energy expenditure and preservation of lean mass in combination with efficient appetite‐reducing compounds are a prerequisite for a significant and long‐lasting healthy weight loss as depicted in Figures 3 and 4C. To achieve this goal, research activities need to look beyond appetite regulation to find safe, effective new modalities for weight‐loss enhancement and maintenance. Furthermore, it must be stressed that obesity is a chronic disease, and even with a multimodal treatment strategy, lifelong treatment is likely needed to prevent the weight regain that otherwise will occur when treatment is discontinued (162). Commitment to this research focus both from a scientific and regulatory point of view could signal the beginning of the next era in obesity therapies.O
CONFLICT OF INTEREST
BØC, KR, and LMJ are full‐time employees and minor stockholders at Novo Nordisk A/S. GS‐D is a stockholder at BuenaVida Centro Integral de Salud. ER serves on the Scientific Advisory Board to the Nutrilite Health Institute with Amway and YSOPIA (LNC Therapeutics); has a consultant contract with Merck, Kintai Therapeutics, Big Sky Health, and Generian; and is an advisor for The Center for Medical Weight Loss. ER has received research grants or unrestricted gifts from Amway, Nestle, the Nutrition Science Initiative (NuSI), Weight Watchers, Lilly, Ethicon Surgery, Novartis, and Sanofi‐Avantis. Although he is Editor‐in‐Chief of Obesity, he did not participate in the review or approval of the manuscript by the journal. DHR serves on the Scientific Advisory Board of Novo Nordisk, Wondr Health, Calibrate, Gila Therapeutics, Sanofi, Boeringer Ingelheim, Real Appeal, Phenomix, Epitomee, Xeno Bioscience, Lilly, and Ysopia; receives honoraria for lectures from Novo Nordisk, Bausch Health, and IFA Celtic; and is a stockholder in Gila Therapeutics, Epitomee, Calibrate, Roman, and Scientific Intake. Although she was Associate Editor‐in‐Chief of Obesity, she did not participate in the review or approval of this manuscript.
Christoffersen BØ, Sanchez‐Delgado G, John LM, Ryan DH, Raun K, Ravussin E. Beyond appetite regulation: Targeting energy expenditure, fat oxidation, and lean mass preservation for sustainable weight loss. Obesity (Silver Spring). 2022;30:841–857. doi: 10.1002/oby.23374
Funding information
GS‐D received funding from Fundación Alfonso Martin Escudero.
REFERENCES
- 1. Curry SJ, Krist AH, Owens DK, et al. Behavioral weight loss interventions to prevent obesity‐related morbidity and mortality in adults: US Preventive Services Task Force Recommendation Statement. JAMA. 2018;320:1163‐1171. [DOI] [PubMed] [Google Scholar]
- 2. Wilding JPH, Batterham RL, Calanna S, et al. Once‐weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384:989. doi: 10.1056/NEJMoa2032183 [DOI] [PubMed] [Google Scholar]
- 3. Baggio LL, Drucker DJ. Glucagon‐like peptide‐1 receptor co‐agonists for treating metabolic disease. Mol Metab. 2021;46:101090. doi: 10.1016/j.molmet.2020.101090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enebo LB, Berthelsen KK, Kankam M, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet. 2021;397:1736‐1748. [DOI] [PubMed] [Google Scholar]
- 5. Abbott WG, Howard BV, Christin L, et al. Short‐term energy balance: relationship with protein, carbohydrate, and fat balances. Am J Physiol. 1988;255:E332‐E337. [DOI] [PubMed] [Google Scholar]
- 6. Willoughby D, Hewlings S, Kalman D. Body composition changes in weight loss: strategies and supplementation for maintaining lean body mass, a brief review. Nutrients. 2018;10:1876. doi: 10.3390/nu10121876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ida S, Kaneko R, Imataka K, et al. Effects of antidiabetic drugs on muscle mass in type 2 diabetes mellitus. Curr Diabetes Rev. 2021;17:293‐303. [DOI] [PubMed] [Google Scholar]
- 8. Bosy‐Westphal A, Kossel E, Goele K, et al. Contribution of individual organ mass loss to weight loss‐associated decline in resting energy expenditure. Am J Clin Nutr. 2009;90:993‐1001. [DOI] [PubMed] [Google Scholar]
- 9. Piaggi P. Metabolic determinants of weight gain in humans. Obesity (Silver Spring). 2019;27:691‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ravussin E, Lillioja S, Knowler WC, et al. Reduced rate of energy expenditure as a risk factor for body‐weight gain. N Engl J Med. 1988;318:467‐472. [DOI] [PubMed] [Google Scholar]
- 11. Zurlo F, Lillioja S, Esposito‐Del Puente A, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24‐h RQ. Am J Physiol. 1990;259:E650‐E657. [DOI] [PubMed] [Google Scholar]
- 12. Spraul M, Ravussin E, Fontvieille AM, et al. Reduced sympathetic nervous activity. A potential mechanism predisposing to body weight gain. J Clin Invest. 1993;92:1730‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weyer C, Vozarova B, Ravussin E, Tataranni PA. Changes in energy metabolism in response to 48 h of overfeeding and fasting in Caucasians and Pima Indians. Int J Obes Relat Metab Disord. 2001;25:593‐600. [DOI] [PubMed] [Google Scholar]
- 14. Schlögl M, Piaggi P, Pannacciuli N, et al. Energy expenditure responses to fasting and overfeeding identify phenotypes associated with weight change. Diabetes. 2015;64:3680‐3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hollstein T, Ando T, Basolo A, et al. Metabolic response to fasting predicts weight gain during low‐protein overfeeding in lean men: further evidence for spendthrift and thrifty metabolic phenotypes. Am J Clin Nutr. 2019;110:593‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reinhardt M, Thearle MS, Ibrahim M, et al. A human thrifty phenotype associated with less weight loss during caloric restriction. Diabetes. 2015;64:2859‐2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Begaye B, Vinales KL, Hollstein T, et al. Impaired metabolic flexibility to high‐fat overfeeding predicts future weight gain in healthy adults. Diabetes. 2020;69:181‐ 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295:E1009‐E1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weiss EC, Galuska DA, Kettel Khan L, Gillespie C, Serdula MK. Weight regain in U.S. adults who experienced substantial weight loss, 1999‐2002. Am J Prev Med. 2007;33:34‐40. [DOI] [PubMed] [Google Scholar]
- 20. Wadden TA, Butryn ML, Byrne KJ. Efficacy of lifestyle modification for long‐term weight control. Obes Res. 2004;12:151S‐162S. [DOI] [PubMed] [Google Scholar]
- 21. Belligoli A, Bettini S, Segato G, Busetto L. Predicting responses to bariatric and metabolic surgery. Curr Obes Rep. 2020;9:373‐ 379. [DOI] [PubMed] [Google Scholar]
- 22. Aronne LJ, Hall KD, M. Jakicic J, et al. Describing the weight‐reduced state: physiology, behavior, and interventions. Obesity (Silver Spring). 2021;29(Suppl 1):S9‐S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dent R, McPherson R, Harper ME. Factors affecting weight loss variability in obesity. Metabolism. 2020;113:154388. doi: 10.1016/j.metabol.2020.154388 [DOI] [PubMed] [Google Scholar]
- 24. Manore MM, Larson‐Meyer DE, Lindsay AR, Hongu N, Houtkooper L. Dynamic energy balance: an integrated framework for discussing diet and physical activity in obesity prevention‐is it more than eating less and exercising more? Nutrients. 2017;9:905. doi: 10.3390/nu9080905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwartz MW, Seeley RJ, Zeltser LM, et al. Obesity pathogenesis: an Endocrine Society Scientific Statement. Endocr Rev. 2017;38:267‐ 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C. Determinants of 24‐hour energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest. 1986;78:1568‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weyer C, Snitker S, Rising R, Bogardus C, Ravussin E. Determinants of energy expenditure and fuel utilization in man: effects of body composition, age, sex, ethnicity and glucose tolerance in 916 subjects. Int J Obes Relat Metab Disord. 1999;23:715‐722. [DOI] [PubMed] [Google Scholar]
- 28. Müller MJ, Bosy‐Westphal A. Adaptive thermogenesis with weight loss in humans. Obesity (Silver Spring). 2013;21:218‐228. [DOI] [PubMed] [Google Scholar]
- 29. Elia M. Organ and tissue contribution to metabolic rate. In: Kinney J, Tucker HN, eds. Energy Metabolism: Tissue Determinants and Cellular Corollaries. Raven Press; 1992:61‐79. [Google Scholar]
- 30. Dulloo A, Jacquet J, Miles‐Chan JL, Schutz Y. Passive and active roles of fat‐free mass in the control of energy intake and body composition regulation. Eur J Clin Nutr. 2017;71:353‐357. [DOI] [PubMed] [Google Scholar]
- 31. Heymsfield SB, Coleman LA, Miller R, et al. Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity a phase 2 randomized clinical trial. JAMA Network Open. 2021;4:33457. doi: 10.1001/jamanetworkopen.2020.33457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knuth ND, Johannsen DL, Tamboli RA, et al. Metabolic adaptation following massive weight loss is related to the degree of energy imbalance and changes in circulating leptin. Obesity (Silver Spring). 2014;22:2563‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Redman LM, Smith SR, Burton JH, et al. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018;27:805‐815.e804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sparks LM, Redman LM, Conley KE, et al. Effects of 12 months of caloric restriction on muscle mitochondrial function in healthy individuals. J Clin Endocrinol Metab. 2017;102:111‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tremblay A, Royer MM, Chaput JP, Doucet E. Adaptive thermogenesis can make a difference in the ability of obese individuals to lose body weight. Int J Obes. 2013;37:759‐764. [DOI] [PubMed] [Google Scholar]
- 37. Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring). 2016;24:1612‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martins C, Dutton GR, Hunter GR, Gower BA. Revisiting the Compensatory Theory as an explanatory model for relapse in obesity management. Am J Clin Nutr. 2020;112:1170‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravussin E, Redman LM. Metabolic adaptation: is it really an illusion? Am J Clin Nutr. 2020;112:1653‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hopkins M, Blundell JE. Energy balance, body composition, sedentariness and appetite regulation: pathways to obesity. Clin Sci. 2016;130:1615‐1628. [DOI] [PubMed] [Google Scholar]
- 41. Martin CK, Johnson WD, Myers CA, et al. Effect of different doses of supervised exercise on food intake, metabolism, and non‐exercise physical activity: the E‐MECHANIC randomized controlled trial. Am J Clin Nutr. 2019;110:583‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foright RM, Presby DM, Sherk VD, et al. Is regular exercise an effective strategy for weight loss maintenance? Physiol Behav. 2018;188:86‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hägele FA, Büsing F, Nas A, et al. Appetite control is improved by acute increases in energy turnover at different levels of energy balance. J Clin Endocrinol Metab. 2019;104:4481‐4491. [DOI] [PubMed] [Google Scholar]
- 44. Ostendorf DM, Caldwell AE, Creasy SA, et al. Physical activity energy expenditure and total daily energy expenditure in successful weight loss maintainers. Obesity (Silver Spring). 2019;27:496‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu G, Liang L, Bray GA, et al. Thyroid hormones and changes in body weight and metabolic parameters in response to weight loss diets: the POUNDS LOST trial. Int J Obes. 2017;41:878‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reinehr T. Obesity and thyroid function. Mol Cell Endocrinol. 2010;316:165‐171. [DOI] [PubMed] [Google Scholar]
- 48. Aub JC, Stern NS. The influence of large doses of thyroid extract on the total metabolism and heart in a case of heart‐block. Arch Intern Med. 1918;XXI:130‐138. [Google Scholar]
- 49. Kaptein EM, Beale E, Chan LS. Thyroid hormone therapy for obesity and nonthyroidal illnesses: a systematic review. J Clin Endocrinol Metab. 2009;94:3663‐3675. [DOI] [PubMed] [Google Scholar]
- 50. Elbers LP, Kastelein JJ, Sjouke B. Thyroid Hormone mimetics: the past, current status and future challenges. Curr Atheroscler Rep. 2016;18:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ost M, Keipert S, Klaus S. Targeted mitochondrial uncoupling beyond UCP1‐The fine line between death and metabolic health. Biochimie. 2017;134:77‐85. [DOI] [PubMed] [Google Scholar]
- 52. Cutting WC, Mehrtens HG, Tainter ML. Actions and uses of dinitrophenol: promising metabolic applications. JAMA. 1933;101:193‐195. [Google Scholar]
- 53. Colman E. Dinitrophenol and obesity: an early twentieth‐century regulatory dilemma. Regul Toxicol Pharmacol. 2007;48:115‐117. [DOI] [PubMed] [Google Scholar]
- 54. Poulton AS, Hibbert EJ, Champion BL, Nanan RK. Stimulants for the control of hedonic appetite. Front Pharmacol. 2016;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hansen HH, Hansen G, Tang‐Christensen M, et al. The novel triple monoamine reuptake inhibitor tesofensine induces sustained weight loss and improves glycemic control in the diet‐induced obese rat: comparison to sibutramine and rimonabant. Eur J Pharmacol. 2010;636:88‐95. [DOI] [PubMed] [Google Scholar]
- 56. Saraç F, Pehlivan M, Çelebi G, et al. Effects of sibutramine on thermogenesis in obese patients assessed via immersion calorimetry. Adv Ther. 2006;23:1016‐1029. [DOI] [PubMed] [Google Scholar]
- 57. Walsh KM, Leen E, Lean ME. The effect of sibutramine on resting energy expenditure and adrenaline‐induced thermogenesis in obese females. Int J Obes Relat Metab Disord. 1999;23:1009‐1015. [DOI] [PubMed] [Google Scholar]
- 58. Halford J, Boyland EJ, Cooper SJ, et al. The effects of sibutramine on the microstructure of eating behaviour and energy expenditure in obese women. J Psychopharmacol. 2010;24:99‐109. [DOI] [PubMed] [Google Scholar]
- 59. Levitsky DA, Troiano R. Metabolic consequences of fenfluramine for the control of body weight. Am J Clin Nutr. 1992;55:167s‐172s. [DOI] [PubMed] [Google Scholar]
- 60. Di Giovanni G, Svob Strac D, Sole M, et al. Monoaminergic and histaminergic strategies and treatments in brain diseases. Front Neurosci. 2016;10:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O’Mara AE, Johnson JW, Linderman JD, et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest. 2020;130:2209‐2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blondin DP, Nielsen S, Kuipers EN, et al. Human brown adipocyte thermogenesis is driven by β2‐AR stimulation. Cell Metab. 2020;32:287‐300.e287. [DOI] [PubMed] [Google Scholar]
- 64. Lee P, Day RO, Greenfield JR, Ho KKY. Formoterol, a highly β2‐selective agonist, increases energy expenditure and fat utilisation in men. Int J Obes. 2013;37:593‐597. [DOI] [PubMed] [Google Scholar]
- 65. Kievit P, Halem H, Marks DL, et al. Chronic treatment with a melanocortin‐4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet‐induced obese rhesus macaques. Diabetes. 2013;62:490‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33:542‐547. [DOI] [PubMed] [Google Scholar]
- 67. Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol. 2011;23:186‐193. [DOI] [PubMed] [Google Scholar]
- 68. Fani L, Bak S, Delhanty P, van Rossum EF, van den Akker EL. The melanocortin‐4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int J Obes. 2014;38:163‐169. [DOI] [PubMed] [Google Scholar]
- 69. Clément K, van den Akker E, Argente J, et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single‐arm, open‐label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8:960‐970. [DOI] [PubMed] [Google Scholar]
- 70. Chen KY, Muniyappa R, Abel BS, et al. RM‐493, a melanocortin‐4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015;100:1639‐1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Day JW, Ottaway N, Patterson JT, et al. A new glucagon and GLP‐1 co‐agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749‐757. [DOI] [PubMed] [Google Scholar]
- 72. Tan TM, Field BCT, McCullough KA, et al. Coadministration of glucagon‐like peptide‐1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62:1131‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clemmensen C, Finan B, Müller TD, et al. Emerging hormonal‐based combination pharmacotherapies for the treatment of metabolic diseases. Nat Rev Endocrinol. 2019;15:90‐104. [DOI] [PubMed] [Google Scholar]
- 74. Drucker DJ. Mechanisms of action and therapeutic application of glucagon‐like peptide‐1. Cell Metab. 2018;27:740‐756. [DOI] [PubMed] [Google Scholar]
- 75. Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF‐21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kliewer SA, Mangelsdorf DJ. A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29:246‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coskun T, Bina HA, Schneider MA, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018‐6027. [DOI] [PubMed] [Google Scholar]
- 78. Cuevas‐Ramos D, Mehta R, Aguilar‐Salinas CA. Fibroblast growth factor 21 and browning of white adipose tissue. Front Physiol. 2019;10:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wentworth JM, Colman PG. The methionine aminopeptidase 2 inhibitor ZGN‐1061 improves glucose control and weight in overweight and obese individuals with type 2 diabetes: a randomized, placebo‐controlled trial. Diabetes Obes Metab. 2020;22:1215‐1219. [DOI] [PubMed] [Google Scholar]
- 80. Proietto J, Malloy J, Zhuang D, et al. Efficacy and safety of methionine aminopeptidase 2 inhibition in type 2 diabetes: a randomised, placebo‐controlled clinical trial. Diabetologia. 2018;61:1918‐1922. [DOI] [PubMed] [Google Scholar]
- 81. Hughes TE, Kim DD, Marjason J, et al. Ascending dose‐controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (Silver Spring). 2013;21:1782‐1788. [DOI] [PubMed] [Google Scholar]
- 82. An J, Wang L, Patnode ML, et al. Physiological mechanisms of sustained fumagillin‐induced weight loss. JCI Insight. 2018;3:e99453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dulloo AG. The search for compounds that stimulate thermogenesis in obesity management: from pharmaceuticals to functional food ingredients. Obes Rev. 2011;12:866‐883. [DOI] [PubMed] [Google Scholar]
- 84. Hall KD, Sacks G, Chandramohan D, et al. Quantification of the effect of energy imbalance on bodyweight. Lancet. 2011;378:826‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Webster JD, Hesp R, Garrow JS. The composition of excess weight in obese women estimated by body density, total body water and total body potassium. Hum Nutr Clin Nutr. 1984;38:299‐306. [PubMed] [Google Scholar]
- 86. Rolfe DFS, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731‐758. [DOI] [PubMed] [Google Scholar]
- 87. Rowland LA, Bal NC, Periasamy M. The role of skeletal‐muscle‐based thermogenic mechanisms in vertebrate endothermy. Biol Rev. 2015;90:1279‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Newsholme EA, Crabtree B. Substrate cycles in metabolic regulation and in heat generation. Biochem Soc Symp. 1976;61‐109. [PubMed] [Google Scholar]
- 89. Harper ME, Green K, Brand MD. The efficiency of cellular energy transduction and its implications for obesity. Annu Rev Nutr. 2008;28:13‐33. [DOI] [PubMed] [Google Scholar]
- 90. Periasamy M, Maurya SK, Sahoo SK, et al. Role of SERCA pump in muscle thermogenesis and metabolism. Compr Physiol. 2017;7:879‐890. [DOI] [PubMed] [Google Scholar]
- 91. Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta. 1994;1187:132‐139. [DOI] [PubMed] [Google Scholar]
- 92. Harper M‐E, Dent R, Monemdjou S, et al. Decreased mitochondrial proton leak and reduced expression of uncoupling protein 3 in skeletal muscle of obese diet‐resistant women. Diabetes. 2002;51:2459‐2466. [DOI] [PubMed] [Google Scholar]
- 93. Wijers SLJ, Schrauwen P, Saris WHM, van Marken Lichtenbelt WD. Human skeletal muscle mitochondrial uncoupling is associated with cold induced adaptive thermogenesis. PLoS One. 2008;3:e1777. doi: 10.1371/journal.pone.0001777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brand M, Pakay J, Ocloo A, et al. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392:353‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Clapham JC, Arch JRS, Chapman H, et al. Mice overexpressing human uncoupling protein‐3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415‐418. [DOI] [PubMed] [Google Scholar]
- 96. Lou P‐H, Hansen B, Olsen P, et al. Mitochondrial uncouplers with an extraordinary dynamic range. Biochem J. 2007;407:129‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Childress ES, Alexopoulos SJ, Hoehn KL, Santos WL. Small molecule mitochondrial uncouplers and their therapeutic potential. J Med Chem. 2018;61:4641‐4655. [DOI] [PubMed] [Google Scholar]
- 98. Fu Y‐Y, Zhang M, Turner N, et al. A novel chemical uncoupler ameliorates obesity and related phenotypes in mice with diet‐induced obesity by modulating energy expenditure and food intake. Diabetologia. 2013;56:2297‐2307. [DOI] [PubMed] [Google Scholar]
- 99. Jiang H, Jin J, Duan Y, et al. Mitochondrial uncoupling coordinated with PDH activation safely ameliorates hyperglycemia via promoting glucose oxidation. Diabetes. 2019;68:2197‐2209. [DOI] [PubMed] [Google Scholar]
- 100. Geisler JG. 2,4 Dinitrophenol as medicine. Cells. 2019;8:280. doi: 10.3390/cells8030280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Marie V, Silva JE. Calcium pool size modulates the sensitivity of the ryanodine receptor channel and calcium‐dependent ATPase of heavy sarcoplasmic reticulum to extravesicular free calcium concentration. J Cell Physiol. 1998;175:283‐294. [DOI] [PubMed] [Google Scholar]
- 102. Simonides WS, Thelen MHM, van der Linden CG, Muller A, van Hardeveld C. Mechanism of thyroid‐hormone regulated expression of the SERCA genes in skeletal muscle: implications for thermogenesis. Biosci Rep. 2001;21:139‐154. [DOI] [PubMed] [Google Scholar]
- 103. Bal NC, Maurya SK, Sopariwala DH, et al. Sarcolipin is a newly identified regulator of muscle‐based thermogenesis in mammals. Nat Med. 2012;18:1575‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Maurya SK, Bal NC, Sopariwala DH, et al. Sarcolipin is a key determinant of the basal metabolic rate, and its overexpression enhances energy expenditure and resistance against diet‐induced obesity. J Biol Chem. 2015;290:10840‐10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Babu GJ, Bhupathy P, Petrashevskaya NN, et al. Targeted overexpression of sarcolipin in the mouse heart decreases sarcoplasmic reticulum calcium transport and cardiac contractility. J Biol Chem. 2006;281:3972‐3979. [DOI] [PubMed] [Google Scholar]
- 106. Silva JE. Thermogenic mechanisms and their hormonal regulation. Physiol Rev. 2006;86:435‐464. [DOI] [PubMed] [Google Scholar]
- 107. Wolfe RR, Herndon DN, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med. 1987;317:403‐408. [DOI] [PubMed] [Google Scholar]
- 108. Klein S, Wolfe RR. Whole‐body lipolysis and triglyceride‐fatty acid cycling in cachectic patients with esophageal cancer. J Clin Invest. 1990;86:1403‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wolfe RR, Klein S, Carraro F, Weber JM. Role of triglyceride‐fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am J Physiol. 1990;258:E382‐E389. [DOI] [PubMed] [Google Scholar]
- 110. Mazzucotelli A, Viguerie N, Tiraby C, et al. The transcriptional coactivator peroxisome proliferator‐activated receptor (PPAR)γ coactivator‐1α and the nuclear receptor PPARα control the expression of glycerol kinase and metabolism genes independently of PPARγ activation in human white adipocytes. Diabetes. 2007;56:2467‐2475. [DOI] [PubMed] [Google Scholar]
- 111. de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci. 2018;192:26‐32. [DOI] [PubMed] [Google Scholar]
- 112. Dai W, Jiang L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Front Endocrinol (Lausanne). 2019;10:570. doi: 10.3389/fendo.2019.00570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim Y, Triolo M, Hood DA. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev. 2017;2017:3165396. doi: 10.1155/2017/3165396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Dorn GW II. Mitofusin 2 dysfunction and disease in mice and men. Front Physiol. 2020;11:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chimienti G, Picca A, Fracasso F, et al. The age‐sensitive efficacy of calorie restriction on mitochondrial biogenesis and mtDNA damage in rat liver. Int J Mol Sci. 2021;22:1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Perry CGR. Mitochondrial adaptations to exercise in human skeletal muscle: a possible role for cristae density as a determinant of muscle fitness. J Physiol. 2017;595:2773‐2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Steinberg GR, Carling D. AMP‐activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18:527‐551. [DOI] [PubMed] [Google Scholar]
- 119. Steneberg P, Lindahl E, Dahl U, et al. PAN‐AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight. 2018;3:e99114. doi: 10.1172/jci.insight.99114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Betagenon . Accessed April 1, 2021. https://betagenon.com/research‐development
- 121. Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mancini G, Pirruccio K, Yang X, et al. Mitofusin 2 in mature adipocytes controls adiposity and body weight. Cell Rep. 2019;26:2849‐2858.e2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dang X, Zhang L, Franco A, et al. Discovery of 6‐phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J Med Chem. 2020;63:7033‐7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Franco A, Dang X, Walton EK, et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. Elife. 2020;9:e61119. doi: 10.7554/eLife.61119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425‐432. [DOI] [PubMed] [Google Scholar]
- 126. Zeng W, Pirzgalska R, Pereira M, et al. Sympathetic neuro‐adipose connections mediate leptin‐driven lipolysis. Cell. 2015;163:84‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Rosenbaum M, Leibel RL. 20 years of leptin: role of leptin in energy homeostasis in humans. J Endocrinol. 2014;223:T83‐T96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bartness TJ, Liu Y, Shrestha YB, Ryu V. Neural innervation of white adipose tissue and the control of lipolysis. Front Neuroendocrinol. 2014;35:473‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. 2015;2015:341583. doi: 10.1155/2015/341583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mahu I, Domingos AI. The sympathetic neuro‐adipose connection and the control of body weight. Exp Cell Res. 2017;360:27‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Collins S, Daniel KW, Rohlfs EM. Depressed expression of adipocyte β‐adrenergic receptors is a common feature of congenital and diet‐induced obesity in rodents. Int J Obes (Lond). 1999;23:669‐677. [DOI] [PubMed] [Google Scholar]
- 132. Song W, Luo QI, Zhang Y, et al. Organic cation transporter 3 (Oct3) is a distinct catecholamines clearance route in adipocytes mediating the beiging of white adipose tissue. PLoS Biol. 2019;17:e2006571. doi: 10.1371/journal.pbio.2006571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Camell CD, Sander J, Spadaro O, et al. Inflammasome‐driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. 2017;550:119‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pirzgalska RM, Seixas E, Seidman JS, et al. Sympathetic neuron‐associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. 2017;23:1309‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mahú I, Barateiro A, Rial‐Pensado E, et al. Brain‐sparing sympathofacilitators mitigate obesity without adverse cardiovascular effects. Cell Metab. 2020;31:1120‐1135.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Saito M. Capsaicin and related food ingredients reducing body fat through the activation of TRP and brown fat thermogenesis. Adv Food Nutr Res. 2015;76:1‐28. [DOI] [PubMed] [Google Scholar]
- 137. Marlatt KL, Chen KY, Ravussin E. Is activation of human brown adipose tissue a viable target for weight management? Am J Physiol Regul Integr Comp Physiol. 2018;315:R479‐R483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Maurer S, Harms M, Boucher J. The colorful versatility of adipocytes: white‐to‐brown transdifferentiation and its therapeutic potential in humans. FEBS J. 2021;288:3628‐3646. [DOI] [PubMed] [Google Scholar]
- 139. Vergnes L, Davies GR, Lin JY, et al. Adipocyte browning and higher mitochondrial function in periadrenal but not SC fat in pheochromocytoma. J Clin Endocrinol Metab. 2016;101:4440‐4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sidossis L, Porter C, Saraf M, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Li S, Li Y, Xiang L, et al. Sildenafil induces browning of subcutaneous white adipose tissue in overweight adults. Metabolism. 2018;78:106‐117. [DOI] [PubMed] [Google Scholar]
- 142. Ruiz JR, Martinez‐Tellez B, Sanchez‐Delgado G, et al. Role of human brown fat in obesity, metabolism and cardiovascular disease: strategies to turn up the heat. Prog Cardiovasc Dis. 2018;61:232‐245. [DOI] [PubMed] [Google Scholar]
- 143. Mendez‐Gutierrez A, Osuna‐Prieto FJ, Aguilera CM, Ruiz JR, Sanchez‐Delgado G. Endocrine mechanisms connecting exercise to brown adipose tissue metabolism: a human perspective. Curr Diab Rep. 2020;20:40. [DOI] [PubMed] [Google Scholar]
- 144. Tharp KM, Jha AK, Kraiczy J, et al. Matrix‐assisted transplantation of functional beige adipose tissue. Diabetes. 2015;64:3713‐3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wang CH, Wei YH. Therapeutic perspectives of thermogenic adipocytes in obesity and related complications. Int J Mol Sci. 2021;22:7177. doi: 10.3390/ijms22137177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zheng Q, Lin J, Huang J, et al. Reconstitution of UCP1 using CRISPR/Cas9 in the white adipose tissue of pigs decreases fat deposition and improves thermogenic capacity. Proc Natl Acad Sci. 2017;114:E9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Akbari P, Gilani A, Sosina O, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. 2021;373:eabf8683. doi: 10.1126/science.abf8683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gilani A, Agostinucci K, Hossain S, et al. 20‐HETE interferes with insulin signaling and contributes to obesity‐driven insulin resistance. Prostaglandins Other Lipid Mediat. 2021;152:106485. doi: 10.1016/j.prostaglandins.2020.106485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Rasmussen MH. Obesity, growth hormone and weight loss. Mol Cell Endocrinol. 2010;316:147‐153. [DOI] [PubMed] [Google Scholar]
- 150. Vijayakumar A, Yakar S, Leroith D. The intricate role of growth hormone in metabolism. Front Endocrinol (Lausanne). 2011;2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Mekala KC, Tritos NA. Effects of recombinant human growth hormone therapy in obesity in adults: a metaanalysis. J Clin Endocrinol Metab. 2009;94:130‐137. [DOI] [PubMed] [Google Scholar]
- 152. Savastano S. Benefits from growth hormone therapy after bariatric surgery. Pediatr Endocrinol Rev. 2009;6:540‐544. [PubMed] [Google Scholar]
- 153. Savastano S, Di Somma C, Angrisani L, et al. Growth hormone treatment prevents loss of lean mass after bariatric surgery in morbidly obese patients: results of a pilot, open, prospective, randomized, controlled study. J Clin Endocrinol Metab. 2009;94:817‐826. [DOI] [PubMed] [Google Scholar]
- 154. Lach‐Trifilieff E, Minetti GC, Sheppard K, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014;34:606‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fekete EM, Zorrilla EP. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: ancient CRF paralogs. Front Neuroendocrinol. 2007;28:1‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Venkatasubramanian S, Newby DE, Lang NN. Urocortins in heart failure. Biochem Pharmacol. 2010;80:289‐296. [DOI] [PubMed] [Google Scholar]
- 157. Jamieson PM, Cleasby ME, Kuperman Y, et al. Urocortin 3 transgenic mice exhibit a metabolically favourable phenotype resisting obesity and hyperglycaemia on a high‐fat diet. Diabetologia. 2011;54:2392‐2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Borg ML, Massart J, De Castro Barbosa T, et al. Modified UCN2 peptide treatment improves skeletal muscle mass and function in mouse models of obesity‐induced insulin resistance. J Cachexia Sarcopenia Muscle. 2021;12:1232‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Gao MH, Giamouridis D, Lai NC, et al. One‐time injection of AAV8 encoding urocortin 2 provides long‐term resolution of insulin resistance. JCI Insight. 2016;1:e88322. doi: 10.1172/jci.insight.88322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Borg ML, Massart J, Schönke M, et al. Modified UCN2 peptide acts as an insulin sensitizer in skeletal muscle of obese mice. Diabetes. 2019;68:1403‐1414. [DOI] [PubMed] [Google Scholar]
- 161. Solinas G, Summermatter S, Mainieri D, et al. Corticotropin‐releasing hormone directly stimulates thermogenesis in skeletal muscle possibly through substrate cycling between de novo lipogenesis and lipid oxidation. Endocrinology. 2006;147:31‐38. [DOI] [PubMed] [Google Scholar]
- 162. Smith SR, Weissman NJ, Anderson CM, et al. Multicenter, placebo‐controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363:245‐256. [DOI] [PubMed] [Google Scholar]
- 163. Flatt JP. Issues and misconceptions about obesity. Obesity (Silver Spring). 2011;19:676‐686. [DOI] [PubMed] [Google Scholar]