

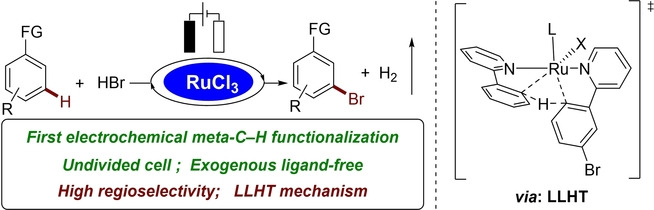
Abstract
While electrochemical ortho‐selective C−H activations are well established, distal C−H activations continue to be underdeveloped. In contrast, we herein describe the electrochemical meta‐C−H functionalization. The remote C−H bromination was accomplished in an undivided cell by RuCl3⋅3 H2O with aqueous HBr. The electrohalogenation proceeded under exogenous ligand‐ and electrolyte‐free conditions. Notably, pyrazolylarenes were meta‐selectively brominated at the benzenoid moiety, rather than on the electron‐rich pyrazole ring for the first time. Mechanistic studies were suggestive of an initial ruthenacycle formation, and a subsequent ligand‐to‐ligand hydrogen transfer (LLHT) process to liberate the brominated product.
Keywords: C−H Activation, Electrosynthesis, Halogenations, meta-Functionalization, Ruthenium
The first electrochemical transition‐metal‐catalyzed meta‐C−H functionalization has been accomplished, affording the positionally selective bromination product in an undivided cell by the catalysis of readily available RuCl3⋅3 H2O with aqueous HBr as the brominating agent.
Introduction
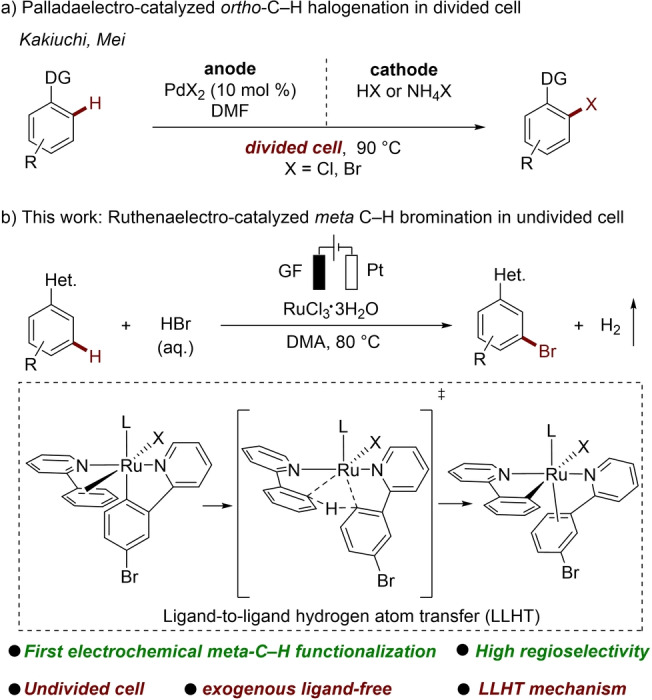
Electro‐organic synthesis has emerged as an increasingly powerful tool for molecular syntheses with electricity as an atom‐economic redox reagent, avoiding the use of traditional chemical redox agents.[ 1 , 2 ] Particularly, the merger of electrocatalysis with organometallic chemistry has set the stage for arene ortho‐C−H functionalizations, [3] with key contributions by the groups of Mei, [4] Kakiuchi, [5] Xu, [6] Lei [7] and Ackermann, [8] among others (Scheme 1a). [9] In contrast, while methods for distal C−H functionalization are in high demand,[ 10 , 11 , 12 , 13 , 14 ] the remote arene diversification by metallaelectro‐catalyzed C−H functionalization has thus far proven to be elusive. In addition, to avoid reduction of transition‐metal cation at the cathode, bidentate directing groups and/or divided cell setups were predominately exploited.
Scheme 1.
Transition‐metal‐catalyzed aryl C−H bromination.
Aryl halides serve as versatile functionalities to a rich array of value‐added chemicals and materials, including pharmaceutical intermediates and polymers. [15] Transition metal‐catalyzed regioselective C−H halogenations were developed to prepare aryl halides generally using corrosive reagents combined with stoichiometric external strong chemical oxidants. [16] With electricity as the oxidant, Kakiuchi [5] and Mei[ 4b , 4c ] reported palladaelectro‐catalyzed ortho‐C−H halogenation with divided cell setups being required (Scheme 1a). In sharp contrast, we herein report on a uniquely efficient electrochemical ruthenium‐catalyzed meta‐C−H bromination (Scheme 1b). Salient features of our strategy comprise a) the first metallaelectro‐catalyzed meta‐C−H functionalization, b) RuCl3 ⋅ 3H2O as the catalyst, c) user‐friendly aqueous HBr as the brominating source, d) an undivided cell set‐up, e) high regioselectivities for challenging pyrazolylarenes, and f) mechanistic studies supporting a new LLHT regime.
Results and Discussion
We initiated our studies by exploring the envisioned bromination of arene 1 a using graphite felt (GF) and platinum plate (Pt) as anode and cathode material, respectively (Table 1). After considerable experimentation, the desired bromination product 2 a was obtained in 83 % isolated yield with RuCl3⋅3 H2O as the catalyst and aqueous HBr as brominating agent in an undivided cell set‐up (Entry 1). Various solvents were tested, showing that N,N‐dimethylacetamide (DMA) was performing best (Entry 2). The bromination reaction was not viable with TBABr or LiBr, but occurred with HBr/HOAc, indicating that the acidic conditions played a key role for the electrocatalysis (Entries 3 and 4). Substrate 1 a was fully converted into meta‐bromo product 2 a in 20 h when increasing the current from 10 mA to 12 mA (Entry 6). Notably, ortho‐bromination and dibromination byproducts were not observed (see Figure S1). Control experiments showed that both the electricity and the ruthenium catalyst were essential for the electrochemical bromination (Entry 8). RuBr3 was also effective for the desired meta‐C−H bromination (Entry 9). Reactions with other transition metal catalysts, including OsCl3, RhCl3 and MnBr2, have also been probed, but none of these salts was effective for the C−H bromination (Entry 10). In addition, aqueous HCl and HI were not suitable for the halogenation reaction (See Figure S3).
Table 1.
Optimization of the ruthenaelectro‐catalyzed meta‐C−H bromination.[a]
|
| ||
|---|---|---|
|
Entry |
Variation from standard conditions |
2 a [%][b] |
|
1 |
no change |
83 |
|
2 |
CH3CN or DMSO as solvent |
0 |
|
3[c] |
LiBr or TBABr in place of HBr |
trace |
|
4 |
HBr in HOAc in place of aq. HBr |
81 |
|
5 |
CCE@5.0 mA |
58 |
|
6 |
CCE@12.0 mA, 20 h |
87 |
|
7 |
2.5 equiv HBr, 12 mA |
86 |
|
8 |
no electricity, no [Ru] |
trace, 0 |
|
9 |
RuBr3 as catalyst, 12 mA |
87 |
|
10 |
MnBr2, OsCl3 or RhCl3 as catalyst |
0 |
[a] Undivided cell, GF anode, Pt cathode, constant current=10.0 mA, 1 a (0.50 mmol), HBr (48 % aqueous solution, 2.0 mmol, 220 μL), RuCl3 ⋅ 3H2O (0.05 mmol), solvent (5.0 mL), N2, 24 h, work‐up with Et3N (2 mL) and pyridine (1.5 mL). [b] Yields of isolated product 2 a. [c] H2O (100 μL) as additive.
With the optimized reaction conditions in hand, the viable substrate scope of the ruthenaelectro‐catalyzed meta‐C−H bromination was explored next (Scheme 2). A range of pyridylarenes 1 a–1 n bearing various substituents could be selectively brominated using aqueous HBr as the bromination source. Functional groups, including ether, thioether, silyl and fluoro, chloro and bromo were tolerated (2 b–2 n). Generally, electron‐rich substrates reacted more efficiently than the electron‐deficient analog. Other heteroarenes, including phenylpyrimidine 1 o, benzo[h]quinoline 1 p and purine derivatives 1 q–1 r, were identified as amenable substrates for the electrochemical remote C−H bromination.
Scheme 2.
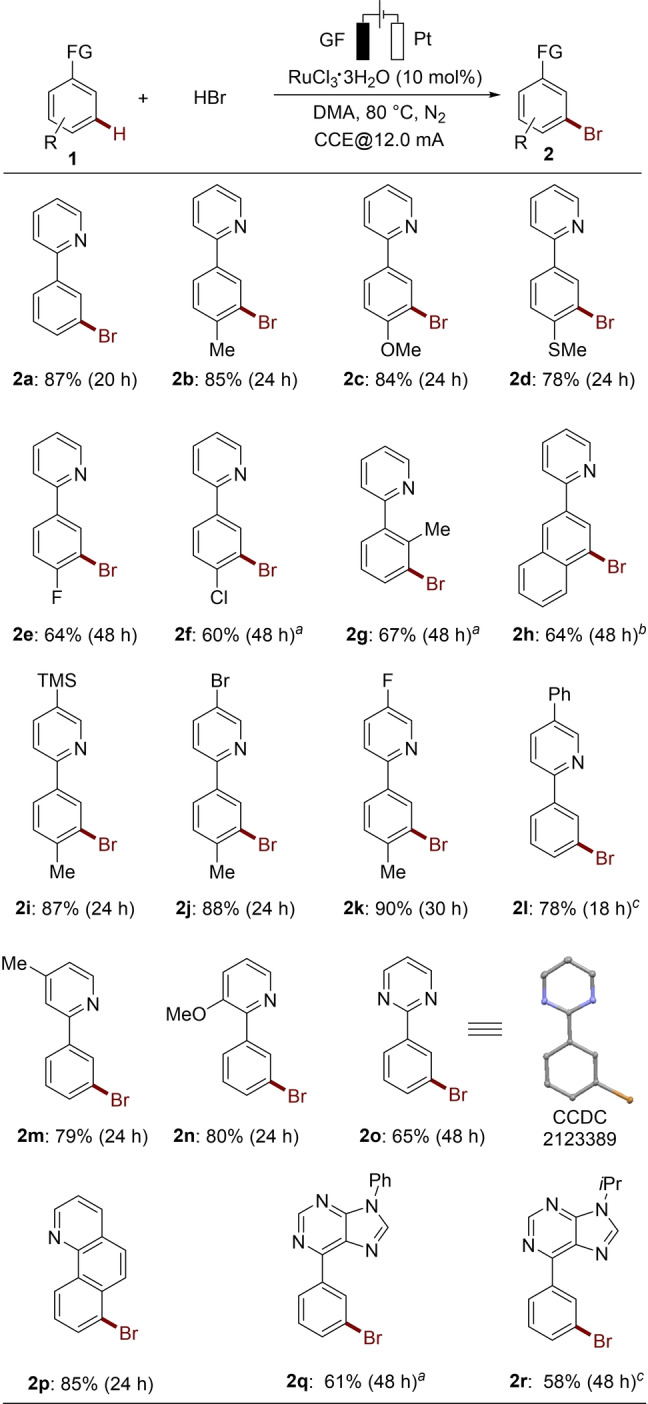
Ruthenaelectro‐catalyzed meta‐C−H bromination. [a] 90 °C. [b] RuBr3 as catalyst. [c] HBr (2.5 equiv), under air, 10 mA.
The selective bromination of pyrazolylarenes is challenging. Reported strategies under ruthenacatalysis[ 14c , 17 ] or electrochemical metal‐free [18] conditions were shown to selectively occur on the electron‐rich pyrazole heterocycle. In sharp contrast, by the ruthenaelectro‐catalysis the pyrazolylarenes 3 a–3 f were for the first time selectively brominated at the benzenoid moiety, rather than on the pyrazole motif (Scheme 3a). In sharp contrast, when the previously reported NBS/[RuCl2(p‐cymene)] catalysis was applied for substrates 3 a–3 d, the bromination only occurred on the pyrazole rings via a simple SEAr, and the desired meta‐C−H bromination was not observed (Scheme 3a). In addition, the RuCl3‐catalyzed bromination of pyrazolylarene 3 a with different brominating agents, such as NBS, TBABr3 and Br2, were further tested but failed to give the desired meta‐product (Scheme 3b and Figure S2).
Scheme 3.
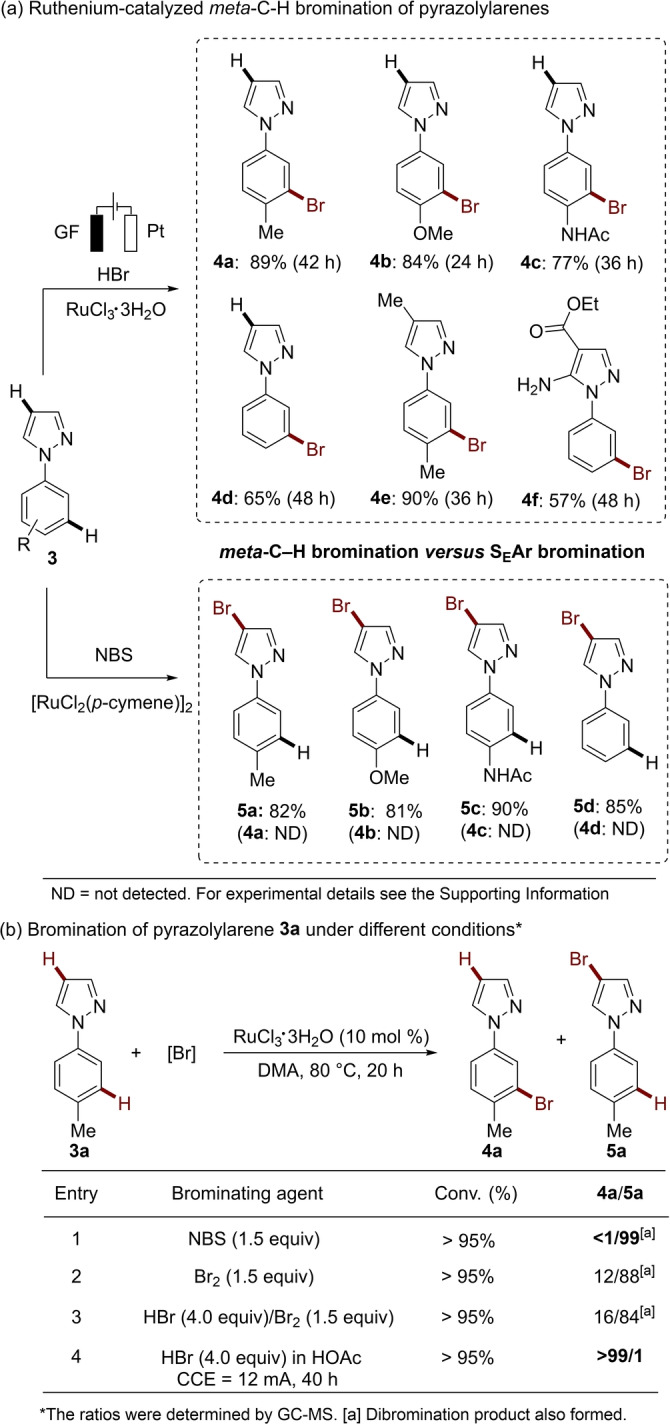
Ruthenaelectro‐catalyzed C−H bromination of pyrazolylarenes 3.
The practical utility of this ruthenaelectro‐catalyzed meta‐C−H bromination was illustrated by a gram‐scale preparation (Scheme 4a) as well as the subsequent diversification of the newly installed bromo group in various C−C couplings. Thereby, we successfully achieved the selective indirect C−H alkenylation and arylation (Scheme 4b). Furthermore, sequential twofold C−H functionalization involving meta‐bromination and ortho‐arylation was achieved in a one‐pot fashion (Scheme 4c). In addition, while the pyridine ring could be efficiently converted into piperidine and 2‐formylpyrrole (Scheme 4d), the pyrazolylmotif was removed to deliver synthetically meaningful anilines (Scheme 4e).
Scheme 4.
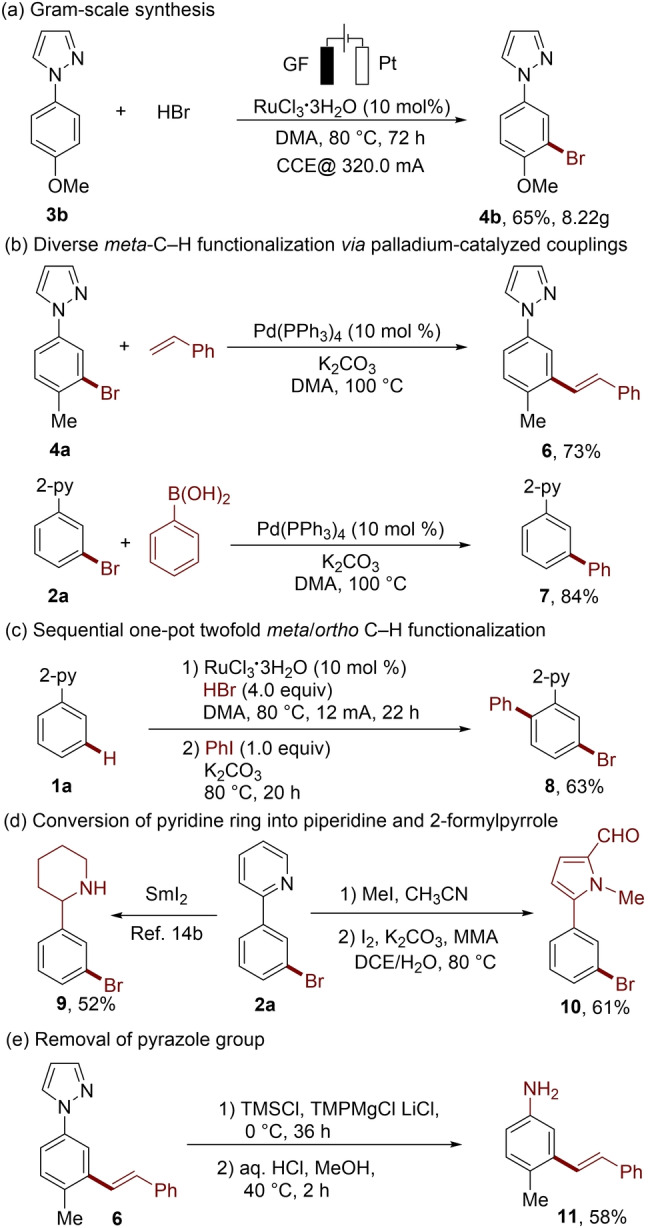
Gram‐scale synthesis and derivatization of the bromination products.
To gain insights into the reaction mechanism, we conducted a series of experiments. Cyclic voltammetry (CV) analysis showed two oxidation peaks of HBr (E p/2=0.65 and 1.17 V versus SCE in DMA), which are proposed to relate to the Br−/Br3 − and Br3 −/Br2 redox couples (Figure 1).[ 4c , 19 ] Additional studies of the reaction mixture exhibited oxidative peaks at ca. E p/2=0.70 V, suggesting that bromide anion easily undergoes anodic oxidation to generate Br2, which equilibrates with the tribromide anion Br3 − by combining/releasing a bromide ion under the standard conditions. In addition, when 1,3,5‐trimethoxybenzene 12, which can rapidly intercept bromine, was added into the electrochemical reaction, the meta‐C−H bromination process was fully inhibited, generating the brominated 1,3,5‐trimethoxybenzene 13 in 63 % yield (Scheme 5a). Protons were reduced at the cathode (E p/2=−0.44 V versus SCE, Figure S10) and H2 was observed by headspace GC analysis (see Figure S6). CV experiments showed that Br2 was easily to be reduced at the cathode (E p/2=−0.44 V versus SCE, Figure S10), which indicated Br2/Br3 − should exist in low concentration during the reaction. Indeed, addition of 1,3,5‐trimethoxybenzene 12 into the standard electrochemical reaction solution (CCE=12 mA, 3 h), cutting off the electricity and heating the mixture for another 3 h, the brominated product 13 was obtained only in 5 % yield (Scheme 5a). On‐off experiment further provided strong support for this conclusion (Scheme 5b). The Br2 produced by anodic oxidation was partially reduced at the cathode, resulting in low Faraday efficiency of the catalytic reaction. [20] In addition, the low concentration of Br2 during the reaction might be a major cause of the high regioselectivity for the electrochemical bromination of phenylpyrazoles (Scheme 3b). To prove this, the bromination of pyrazolylarene 3 a was conducted by sequential addition of Br2, the meta‐C−H brominated product 4 a was obtained selectively (see Figure S4). Increasing the electric current resulted in a higher initial reaction rate (Scheme 5c), indicating that the bromination step might be the rate determining step (RDS).
Figure 1.
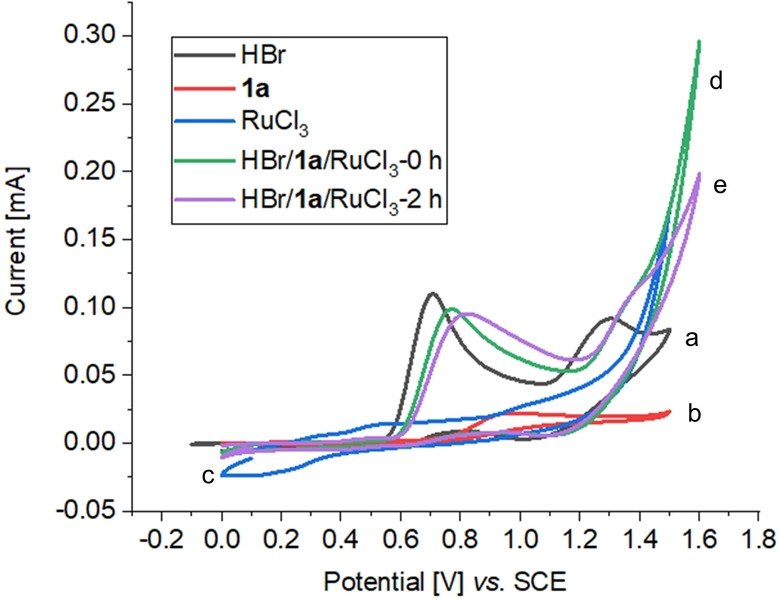
Cyclic voltammetry studies (DMA, 0.1 M nBu4NPF6, 100 mV s−1). a) 10 mM HBr; b) 10 mM 1 a; c) 10 mM RuCl3⋅3 H2O; d) 0.5 mL of the standard reaction (t=0 h) solution was diluted to 10 mL with DMA; e) 0.5 mL of the reaction (CCE=10 mA, t=2 h) solution was diluted to 10 mL with DMA.
Scheme 5.
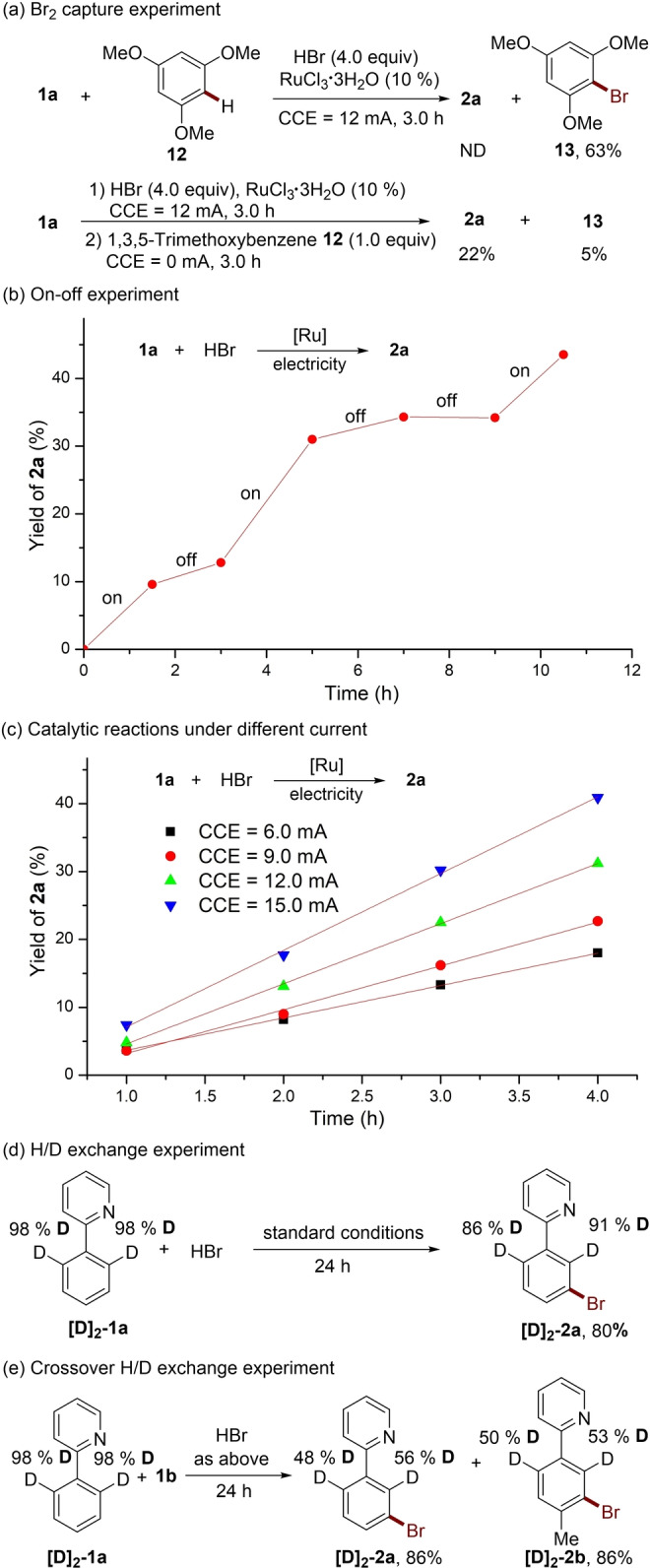
Summary of key mechanistic findings.
H/D exchange experiments with deuterated phenylpyridine [D]2 ‐1 a showed only a small proportion of D loss in the brominated product (Scheme 5d). However, crossover experiment between substrates [D]2 ‐1 a and 1 b indicated that H/D exchange occurred efficiently between different substrates (Scheme 5e). These results suggested that ruthenacycle complex involving ortho‐C−H activation was generated during the electrocatalysis, and protonolysis of the ruthenacycle was slow even under the acidic reaction conditions. Instead, a ligand‐to‐ligand hydrogen transfer (LLHT) process was considered. DFT calculations indicated that a LLHT process could readily occur via a σ‐bond metathesis pathway (Figure 2). [21]
Figure 2.
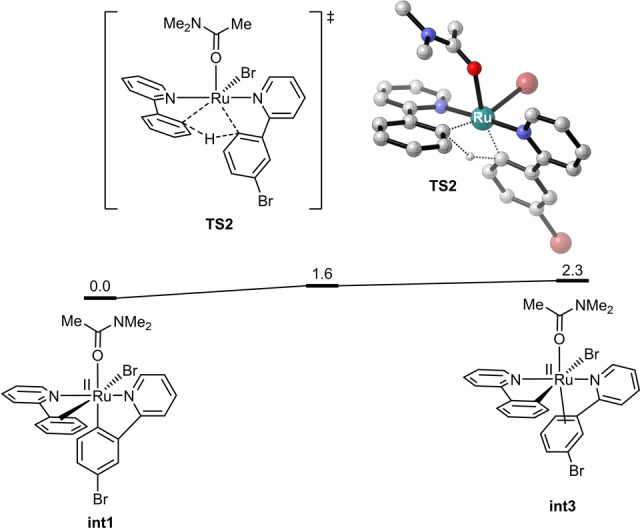
DFT‐computed free energy changes of ligand‐to‐ligand hydrogen transfer via a σ‐bond metathesis pathway from ruthenium (II) (int1). Computational methods: B3LYP‐D3(BJ)/6‐311+G(d,p)‐SDD‐SMD(n,n‐Dimethylacetamide)//B3LYP‐D3(BJ)/6‐31G(d)‐LANL2DZ.
Based on our experimental results and computational results, a plausible reaction mechanism is depicted in Figure 3. The bromide ion is directly oxidized to molecular Br2 at the anode. The ruthenium‐catalyzed bromination is initiated by the formation of the cycloruthenated complex A. [22] Then a single‐electron‐transfer process occurs between A and Br2 leading to intermediate B. Elimination of one molecule of HBr delivers intermediate C, which undergoes LLHT process and ligand exchange with 1 a to liberate the desired meta‐brominated product 2 a and regenerate complex A. An alternative pathway involving a ruthenium (III/III) regime may also be viable at the anode (Figure S8). In addition, the SEAr manifold between Br2 and intermediate A cannot be really ruled out. Notably, though the anodic oxidation was utilized, the balanced cathodic reaction also plays key role in the undivided cell synthesis. Too high cathodic reduction potential will cause the catalyst reduction at the cathode.
Figure 3.
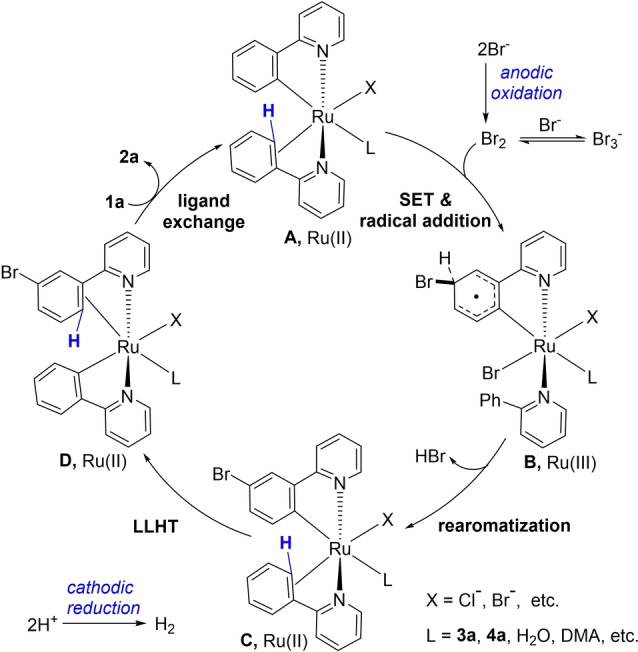
Proposed catalytic cycle.
Conclusion
In summary, we reported on the first electrochemical metal‐ catalyzed arene meta‐C−H functionalization. The distal bromination proceeded in an undivided cell using aqueous HBr as the brominating agent under exogenous ligand‐ and electrolyte‐free conditions. The versatility of the electrochemical remote bromination was reflected by unique regioselectivities for challenging pyrazolylarenes and gram‐scale electrosynthesis. The strategy avoided the use of external chemical oxidants and provided an approach for the synthesis of versatile meta‐substituted aryl bromide by HBr. Mechanistic studies indicated that a ruthenacycle complex is initially generated via ortho‐C−H activation, and a LLHT process occurs to liberate the brominated product.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
The authors gratefully acknowledge support from the ERC Advanced Grant no. 101021358 (L.A.), the Alexander‐von‐Humboldt Foundation (fellowship to Y.W.) and the CSC (fellowships to Z. L. and X. C) is gratefully acknowledged. We thank Dr. Christopher Golz (Göttingen University) for assistance with the X‐ray diffraction analysis. Open Access funding enabled and organized by Projekt DEAL.
Y. Wang, H. Simon, X. Chen, Z. Lin, S. Chen, L. Ackermann, Angew. Chem. Int. Ed. 2022, 61, e202201595; Angew. Chem. 2022, 134, e202201595.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.For selected reviews of electro-organic synthesis, see:
- 1a. Siu J. C., Fu N., Lin S., Acc. Chem. Res. 2020, 53, 547–560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Kingston C., Palkowitz M. D., Takahira Y., Vantourout J. C., Peters B. K., Kawamata Y., Baran P. S., Acc. Chem. Res. 2020, 53, 72–78; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Meyer T. H., Choi I., Tian C., Ackermann L., Chem 2020, 6, 2484–2496; [Google Scholar]
- 1d. Xiong P., Xu H.-C., Acc. Chem. Res. 2019, 52, 3339–3350; [DOI] [PubMed] [Google Scholar]
- 1e. Yuan Y., Lei A., Acc. Chem. Res. 2019, 52, 3309–3324; [DOI] [PubMed] [Google Scholar]
- 1f. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 1g. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Feng R., Smith J. A., Moeller K. D., Acc. Chem. Res. 2017, 50, 2346–2352. [DOI] [PubMed] [Google Scholar]
- 2.For selected recent examples of electro-organic synthesis, see:
- 2a. Dong X., Roeckl J. L., Waldvogel S. R., Morandi B., Science 2021, 371, 507–514; [DOI] [PubMed] [Google Scholar]
- 2b. Yan H., Hou Z.-W., Xu H.-C., Angew. Chem. Int. Ed. 2019, 58, 4592–4595; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4640–4643; [Google Scholar]
- 2c. Liang Y., Lin F., Adeli Y., Jin R., Jiao N., Angew. Chem. Int. Ed. 2019, 58, 4566–4570; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4614–4618; [Google Scholar]
- 2d. Rafiee M., Wang F., Hruszkewycz D. P., Stahl S. S., J. Am. Chem. Soc. 2018, 140, 22–25; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Horn E. J., Rosen B. R., Chen Y., Tang J., Chen K., Eastgate M. D., Baran P. S., Nature 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews of metallaelectro-catalyzed C−H functionalization, see:
- 3a. Goebel J. F., Zeng Z., Gooßen L. J., Synthesis 2022, 54, 565–569; [Google Scholar]
- 3b. Zhu C., Ang N. W. J., Meyer T. H., Qiu Y., Ackermann L., ACS Cent. Sci. 2021, 7, 415–431; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Ma C., Fang P., Liu Z.-R., Xu S.-S., Xu K., Cheng X., Lei A.-W., Xu H.-C., Zeng C.-C., Mei T.-S., Sci. Bull. 2021, 66, 2412–2429; [DOI] [PubMed] [Google Scholar]
- 3d. Gandeepan P., Finger L. H., Meyer T. H., Ackermann L., Chem. Soc. Rev. 2020, 49, 4254–4272; [DOI] [PubMed] [Google Scholar]
- 3e. Ackermann L., Acc. Chem. Res. 2020, 53, 84–104; [DOI] [PubMed] [Google Scholar]
- 3f. Jiao K.-J., Xing Y.-K., Yang Q.-L., Qiu H., Mei T.-S., Acc. Chem. Res. 2020, 53, 300–310; [DOI] [PubMed] [Google Scholar]
- 3g. Sauermann N., Meyer T. H., Qiu Y., Ackermann L., ACS Catal. 2018, 8, 7086–7103. [Google Scholar]
- 4.
- 4a. Yang Q.-L., Xing Y.-K., Wang X.-Y., Ma H.-X., Weng X.-J., Yang X., Guo H.-M., Mei T.-S., J. Am. Chem. Soc. 2019, 141, 18970–18976; [DOI] [PubMed] [Google Scholar]
- 4b. Yang Q.-L., Wang X.-Y., Weng X.-J., Yang X., Xu X.-T., Tong X., Fang P., Wu X.-Y., Mei T.-S., Acta Chim. Sin. 2019, 77, 866–873; [Google Scholar]
- 4c. Yang Q.-L., Wang X.-Y., Wang T.-L., Yang X., Liu D., Tong X.-F., Wu X.-Y., Mei T.-S., Org. Lett. 2019, 21, 2645–2649; [DOI] [PubMed] [Google Scholar]
- 4d. Yang Q.-L., Wang X.-Y., Lu J.-Y., Zhang L.-P., Fang P., Mei T.-S., J. Am. Chem. Soc. 2018, 140, 11487–11494; [DOI] [PubMed] [Google Scholar]
- 4e. Yang Q.-L., Li Y.-Q., Ma C., Fang P., Zhang X.-J., Mei T.-S., J. Am. Chem. Soc. 2017, 139, 3293–3298. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Kakiuchi F., Kochi T., Mutsutani H., Kobayashi N., Urano S., Sato M., Nishiyama S., Tanabe T., J. Am. Chem. Soc. 2009, 131, 11310–11311; [DOI] [PubMed] [Google Scholar]
- 5b. Konishi M., Tsuchida K., Sano K., Kochi T., Kakiuchi F., J. Org. Chem. 2017, 82, 8716–8724. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wu Z.-J., Su F., Lin W., Song J., Wen W.-B., Zhang H.-J., Xu H.-C., Angew. Chem. Int. Ed. 2019, 58, 16770–16774; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16926–16930; [Google Scholar]
- 6b. Xu F., Li Y.-J., Huang C., Xu H.-C., ACS Catal. 2018, 8, 3820–3824. [Google Scholar]
- 7.
- 7a. Gao X., Wang P., Zeng L., Tang S., Lei A., J. Am. Chem. Soc. 2018, 140, 4195–4199; [DOI] [PubMed] [Google Scholar]
- 7b. Tang S., Wang D., Liu Y., Zeng L., Lei A., Nat. Commun. 2018, 9, 798–803; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Huang P., Wang P., Wang S., Tang S., Lei A., Green Chem. 2018, 20, 4870–4874; [Google Scholar]
- 7d. Zeng L., Li H., Tang S., Gao X., Deng Y., Zhang G., Pao C.-W., Chen J.-L., Lee J.-F., Lei A., ACS Catal. 2018, 8, 5448–5453. [Google Scholar]
- 8.
- 8a. Zhang S.-K., Del Vecchio A., Kuniyil R., Messinis A. M., Lin Z., Ackermann L., Chem 2021, 7, 1379–1392; [Google Scholar]
- 8b. Tan X., Hou X., Rogge T., Ackermann L., Angew. Chem. Int. Ed. 2021, 60, 4619–4624; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4669–4674; [Google Scholar]
- 8c. Wang Y., Oliveira J. C. A., Lin Z., Ackermann L., Angew. Chem. Int. Ed. 2021, 60, 6419–6424; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 6490–6495; [Google Scholar]
- 8d. Dhawa U., Tian C., Wdowik T., Oliveira J. C. A., Hao J., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 13451–13457; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13553–13559; [Google Scholar]
- 8e. Meyer T. H., Oliveira J. C. A., Ghorai D., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 10955–10960; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 11048–11053; [Google Scholar]
- 8f. Samanta R. C., Struwe J., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 14154–14159; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 14258–14263; [Google Scholar]
- 8g. Qiu Y., Zhu C., Stangier M., Struwe J., Ackermann L., CCS Chem. 2020, 2, 1529–1552; [Google Scholar]
- 8h. Kong W.-J., Finger L. H., Messinis A. M., Kuniyil R., Oliveira J. C. A., Ackermann L., J. Am. Chem. Soc. 2019, 141, 17198–17206; [DOI] [PubMed] [Google Scholar]
- 8i. Qiu Y., Stangier M., Meyer T. H., Oliveira J. C. A., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 14179–14183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14375–14379; [Google Scholar]
- 8j. Sauermann N., Meyer T. H., Tian C., Ackermann L., J. Am. Chem. Soc. 2017, 139, 18452–18455. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Jin S., Kim J., Kim D., Park J.-W., Chang S., ACS Catal. 2021, 11, 6590–6595; [Google Scholar]
- 9b. Zeng Z., Goebel J. F., Liu X., Gooßen L. J., ACS Catal. 2021, 11, 6626–6632; [Google Scholar]
- 9c. Luo M.-J., Hu M., Song R.-J., He D.-L., Li J.-H., Chem. Commun. 2019, 55, 1124–1127; [DOI] [PubMed] [Google Scholar]
- 9d. Amatore C., Cammoun C., Jutand A., Adv. Synth. Catal. 2007, 349, 292–296. [Google Scholar]
- 10.
- 10a. Dutta U., Maiti S., Bhattacharya T., Maiti D., Science 2021, 372, eabd5992; [DOI] [PubMed] [Google Scholar]
- 10b. Meng G., Lam N. Y. S., Lucas E. L., Saint-Denis T. G., Verma P., Chekshin N., Yu J. Q., J. Am. Chem. Soc. 2020, 142, 10571–10591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Mihai M. T., Genov G. R., Phipps R. J., Chem. Soc. Rev. 2018, 47, 149–171; [DOI] [PubMed] [Google Scholar]
- 10d. Wang J., Dong G., Chem. Rev. 2019, 119, 7478–7528; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145–7153; [DOI] [PubMed] [Google Scholar]
- 10f. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Porey S., Zhang X., Bhowmick S., Kumar Singh V., Guin S., Paton R. S., Maiti D., J. Am. Chem. Soc. 2020, 142, 3762–3774; [DOI] [PubMed] [Google Scholar]
- 11b. Bag S., Surya K., Mondal A., Jayarajan R., Dutta U., Porey S., Sunoj R. B., Maiti D., J. Am. Chem. Soc. 2020, 142, 12453–12466; [DOI] [PubMed] [Google Scholar]
- 11c. Shi H., Herron A. N., Shao Y., Shao Q., Yu J.-Q., Nature 2018, 558, 581–585; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Simonetti M., Larrosa I., Nat. Chem. 2016, 8, 1086–1088; [DOI] [PubMed] [Google Scholar]
- 11e. Kuninobu Y., Ida H., Nishi M., Kanai M., Nat. Chem. 2015, 7, 712–717; [DOI] [PubMed] [Google Scholar]
- 11f. Leow D., Li G., Mei T.-S., Yu J.-Q., Nature 2012, 486, 518–522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11g. Phipps R. J., Gaunt M. J., Science 2009, 323, 1593–1597. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Sagadevan A., Greaney M. F., Angew. Chem. Int. Ed. 2019, 58, 9826–9830; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9931–9935; [Google Scholar]
- 12b. Wang X.-G., Li Y., Liu H.-C., Zhang B.-S., Gou X.-Y., Wang Q., Ma J.-W., Liang Y.-M., J. Am. Chem. Soc. 2019, 141, 13914–13922; [DOI] [PubMed] [Google Scholar]
- 12c. Leitch J. A., McMullin C. L., Mahon M. F., Bhonoah Y., Frost C. G., ACS Catal. 2017, 7, 2616–2623; [Google Scholar]
- 12d. Saidi O., Marafie J., Ledger A. E. W., Liu P. M., Mahon M. F., Kociok-Köhn G., Whittlesey M. K., Frost C. G., J. Am. Chem. Soc. 2011, 133, 19298–19301. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Wei W., Yu H., Zangarelli A., Ackermann L., Chem. Sci. 2021, 12, 8073–8078; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Korvorapun K., Moselage M., Struwe J., Rogge T., Messinis A. M., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 18795–18803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18956–18965; [Google Scholar]
- 13c. Gandeepan P., Koeller J., Korvorapun K., Mohr J., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 9820–9825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9925–9930; [Google Scholar]
- 13d. Ruan Z., Zhang S.-K., Zhu C., Ruth P. N., Stalke D., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 2045–2049; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2077–2081; [Google Scholar]
- 13e. Li J., Warratz S., Zell D., De Sarkar S., Ishikawa E. E., Ackermann L., J. Am. Chem. Soc. 2015, 137, 13894–13901; [DOI] [PubMed] [Google Scholar]
- 13f. Hofmann N., Ackermann L., J. Am. Chem. Soc. 2013, 135, 5877–5884. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Warratz S., Burns D. J., Zhu C., Korvorapun K., Rogge T., Scholz J., Jooss C., Gelman D., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 1557–1560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1579–1582; [Google Scholar]
- 14b. Teskey C. J., Lui A. Y. W., Greaney M. F., Angew. Chem. Int. Ed. 2015, 54, 11677–11680; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11843–11846; [Google Scholar]
- 14c. Yu Q., Hu L., Wang Y., Zheng S., Huang J., Angew. Chem. Int. Ed. 2015, 54, 15284–15288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15499–15503. [Google Scholar]
- 15. Hassan J., Sévignon M., Gozzi C., Schulz E., Lemaire M., Chem. Rev. 2002, 102, 1359–1470. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Petrone D. A., Ye J., Lautens M., Chem. Rev. 2016, 116, 8003–8104; [DOI] [PubMed] [Google Scholar]
- 16b. Lyons T. W., Sanford M. S., Chem. Rev. 2010, 110, 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Wang L., Ackermann L., Chem. Commun. 2014, 50, 1083–1085; [DOI] [PubMed] [Google Scholar]
- 16d. Sun X., Shan G., Sun Y., Rao Y., Angew. Chem. Int. Ed. 2013, 52, 4440–4444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4536–4540; [Google Scholar]
- 16e. Mei T.-S., Giri R., Maugel N., Yu J.-Q., Angew. Chem. Int. Ed. 2008, 47, 5215–5219; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5293–5297; [Google Scholar]
- 16f. Whitfield S. R., Sanford M. S., J. Am. Chem. Soc. 2007, 129, 15142–15143. [DOI] [PubMed] [Google Scholar]
- 17.Ru(C5H5)(CO)2]2/PhI(OCOCF3)2/NBS were reported to achieve the meta-bromination of 2-phenylpyridine, but the bromination of phenylpyrazoles occurred on the electron-rich pyrazole rings. See the Supporting Information of the following reference: Reddy G. M., Rao N. S., Maheswaran H., Org. Chem. Front. 2018, 5, 1118–1123. [Google Scholar]
- 18. Yuan Y., Yao A., Zheng Y., Gao M., Zhou Z., Qiao J., Hu J., Ye B., Zhao J., Wen H., Lei A., iScience 2019, 12, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu L., Jin X., Chen G. Z., J. Electroanal. Chem. 2013, 688, 371–378. [Google Scholar]
- 20.Since some cycloruthenated complexes were reported to feature reversible redox potentials between 0–1.0 V (see ref. [13c] for example), ruthenacycle was oxidized at the anode (Ru n →Ru n+1) and reduced at the cathode (Ru n+1→Ru n ) might be another reason for the low Faraday efficiency.
- 21.Computations were performed with the Gaussian 16 software package; computational details are included in the Supporting Information. For the evaluation of possible cyclometalated ruthenium(II) species, see Figure S11 in the Supporting Information.
- 22.RuCl3 could convert to ruthenium(II) even under electro-free conditions via an oxidation–reduction process, see:
- 22a. Yanga S., Yana B., Zhonga L., Jiab C., Yaoa D., Yanga C., Suna K., Li G., Org. Chem. Front. 2020, 7, 2474–2479; [Google Scholar]
- 22b. Ackermann L., Althammer A., Born R., Synlett 2007, 2833–2836; [Google Scholar]
- 22c. Ackermann L., Althammer A., Born R., Tetrahedron 2008, 64, 6115–6124. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.