

Abstract
The broad differential diagnosis of neonatal erythroderma often poses a diagnostic challenge. Mortality of neonatal erythroderma is high due to complications of the erythroderma itself and the occasionally severe and life‐threatening underlying disease. Early correct recognition of the underlying cause leads to better treatment and prognosis. Currently, neonatal erythroderma is approached on a case‐by‐case basis. The purpose of this scoping review was to develop a diagnostic approach in neonatal erythroderma. After a systematic literature search in Embase (January 1990 – May 2020, 74 cases of neonatal erythroderma were identified, and 50+ diagnoses could be extracted. Main causes were the ichthyoses (40%) and primary immunodeficiencies (35%). Congenital erythroderma was present in 64% (47/74) of the cases, predominantly with congenital ichthyosis (11/11; 100%), Netherton syndrome (12/14, 86%) and Omenn syndrome (11/23, 48%). Time until diagnosis ranged from 102 days to 116 days for cases of non‐congenital erythroderma and congenital erythroderma respectively. Among the 74 identified cases a total of 17 patients (23%) died within a mean of 158 days and were related to Omenn syndrome (35%), graft‐versus‐host disease (67%) and Netherton syndrome (18%). Disease history and physical examination are summarized in this paper. Age of onset and a collodion membrane can help to narrow the differential diagnoses. Investigations of blood, histology, hair analysis, genetic analysis and clinical imaging are summarized and discussed. A standard blood investigation is proposed, and the need for skin biopsies with lympho‐epithelial Kazal‐type related Inhibitor staining is highlighted. Overall, this review shows that diagnostic procedures narrow the differential diagnosis in neonatal erythroderma. A 6‐step flowchart for the diagnostic approach for neonatal erythroderma during the first month of life is proposed. The approach was made with the support of expert leaders from international multidisciplinary collaborations in the European Reference Network Skin‐subthematic group Ichthyosis.
Background
Neonatal erythroderma (NE) is erythema or generalized dermatitis covering at least 90% of the body surface, present at birth or appearing during the first 4 weeks after delivery. 1 Incidence rate of NE patients in the Netherlands is estimated among dermatologists to be 10 patients per 100 000 newborns. 2 Erythroderma itself is not a diagnosis, but a phenotype. Proper diagnosis is a challenge due to low incidence, phenotypical heterogeneity and a wide range of possible underlying diagnoses (Fig. 1). Etiologically, six main categories are known: congenital ichthyoses (including Netherton Syndrome (NS)), primary immunodeficiencies (PIDs), metabolic disorders, drug use, cutaneous infections and ‘other’. 3 , 4 , 5 The ‘other’ group contains common dermatoses, such as atopic dermatitis, psoriasis and seborrheic dermatitis, which mainly occur after 3 months of age. 1 , 3 , 4 , 6 Congenital ichthyosis (including NS) (46%) and PIDs (30%) were the main causes in two studies, although erythrodermic cases from both neonates and infants were included. 3 , 4 Previously reported mortality rate is high (16% after a mean of 17 months) due to complications of the erythroderma such as dehydration, electrolyte imbalance and infections, or due to severe and life‐threatening underlying diseases such as PID and metabolic disorders. 4 , 7 An optimal diagnostic approach and awareness of underlying causes is mandatory to prevent diagnostic delay, thereby decreasing the rate of complications and death. Since a differential diagnosis is often made on a case‐by‐case basis, an algorithmic approach would be recommended. Several authors proposed a multidisciplinary/diagnostic approach, which is mainly based on a differential diagnosis perspective and not on the clinical presentation. 1 , 6 , 8 , 9 , 10 , 11 The present systematic literature search provides a scoping overview of diagnostic characteristics in cases of neonatal erythroderma during the neonatal period specifically that can lead to a timely diagnosis. Based on these results and the opinion of experts, a protocol for NE using an algorithmic approach is proposed.
Figure 1.
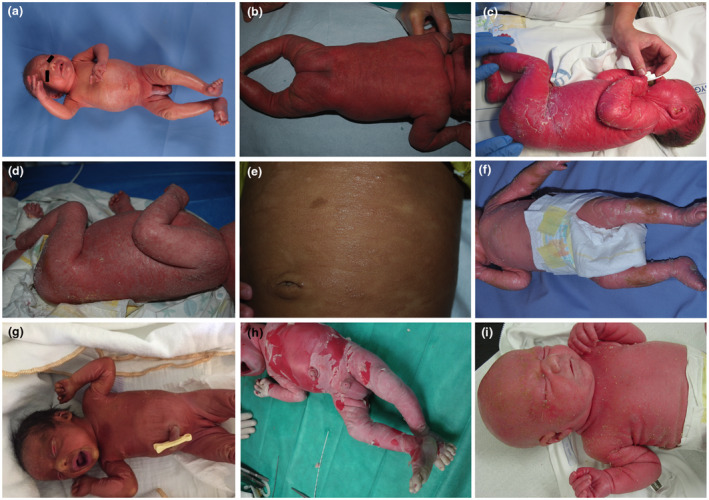
Clinical pictures of patients with neonatal erythroderma. (a) Self‐healing collodion baby, (b) Omenn syndrome (courtesy by Iria Neri), (c) Ichthyosis variegata (courtesy by Anette Bygum), (d) Netherton syndrom (courtesy by Iria Neri), (e) diffuse cutaneous mastrocytosis (courtesy by Iria Neri), (f) Harlequin ichthyosis (courtesy by Cristina Has), (g) Ichthyosis NOS, (h) Epidermolytic ichthyosis (courtesy by Cristina Has), (i) Omenn syndrome.
Methods
A scoping review was performed based on a systematic literature search.
Search criteria and strategies
All publications which described diagnostic procedures in NE within the first year were included. A search strategy was developed by an experienced information specialist. First search (April 2020) was followed by a more extensive search (May 2020). The search was carried out in EMBASE, using specific search terms (Appendix S1). Publications were selected based on title and/or abstract. When abstracts were unavailable, full text was consulted. Reference lists of eligible articles were checked for other relevant publications. Finally, the included articles were analysed for study parameters, focused on diagnostic procedures, clinical symptoms, numbers of patients, age range and diagnosis.
Inclusion and exclusion criteria
Scientific publications (January 1990 – May 2020) were screened. English publications with clinical information (clinical and neonatal characteristics, extracutaneous and systemic symptoms) and/or diagnostic procedures (e.g. laboratory blood analysis, bacterial culture swabs, hair analysis, histopathology or genetics) on NE before the age of 1 year were included. Publications on NE that contained information about family (consanguinity and family history) and pregnancy (premature birth and complications) were also included. Review articles, expert opinions, case reports and case series were eligible. Additionally, textbooks on (paediatric) dermatology were screened. Publications with erythroderma, starting after the 1st month of age, were excluded. Publications were excluded when diagnostics or specific details of the clinical onset of NE were absent. Poster presentations, conference abstracts and publications with unavailable full text were excluded. As underlying diagnoses, congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome has been excluded due to an unilateral erythroderma, and Leiner disease has been excluded because in former cases the disease seemed to be related to NS and Omenn syndrome (OS). 11 , 12 , 13
Group experts
Members of the European Reference Network (ERN)‐SKIN subthematic group Ichthyosis provided their expert opinion on the review and algorithmic diagnostic approach.
Results
The literature search identified 2127 publications. Finally, 183 publications were screened for further analysis leaving 98 publications for qualitative analysis (Appendix S2) in which eventually 74 cases were identified. The level of evidence was low (level 4) (59 case reports and 13 case series). 14
Underlying diagnoses in neonatal erythroderma
Diagnoses of the 74 case reports, including age of onset of the erythroderma and the mean delay until the final diagnosis, are summarized in Appendix S3. Main causes were subtypes of ichthyoses (30/74; 40%) and PIDs (26/74; 35%). Syndromic ichthyoses subtypes were more prevalent (23/30; 77%). Ectodermal dysplasia (AEC) was described in 4 cases (6%). Other causes were infections (4/74; 5%, all SSSS), metabolic disorders (2/74; 3%) and drug‐induced erythroderma (DIHS) (1/74; 1%). A complete overview of reported underlying diagnoses of congenital erythroderma and NE are summarized in Table 1. Underlying diagnoses described in reviews or textbooks are marked with an asterisk. Diagnoses that can be accompanied by a collodion membrane are indicated. Relatively new diagnoses such as severe dermatitis, multiple Allergies and metabolic wasting (SAM) syndrome (DSG1 gene), which can resemble NS, or SAM‐like phenotypes (DSP gene) 63 and autosomal recessive keratitis ichthyosis deafness (KIDAR) syndrome (AP1B1 gene) must be considered. 36 , 62
Table 1.
Overview of differential diagnoses in neonatal erythroderma
|
NON‐SYNDROMAL ICHTHYOSIS
SYNDROMAL ICHTHYOSIS
ECTODERMAL DYSPLASIA
METABOLIC DISORDERS
INFECTIONS
|
Reference(s) |
IMMUNODEFICIENCIES
DRUGS
OTHER
|
Reference(s) |
AD, autosomal dominant; AR, autosomal recessive; ARCI, Autosomal recessive congenial ichthyosis; COFS, Cerebro‐oculo‐facio‐skeletal syndrome; CVID, combined variable immunodeficiency; EB, epidermolysis bullosa; IFAP, Ichthyosis Follicularis Atrichia and Photophobia; SCID, severe combined immunodeficiency.
Diagnoses are marked when found in textbooks or reviews (*) or when associated with a collodion membrane ©
Congenital erythroderma was described in a total of 47/74 cases (64%), predominantly in congenital ichthyoses (11/11; 100%), NS (12/14; 86%) and OS (11/23; 48%). In a study on PIDs, congenital erythroderma was frequently seen in cases of autosomal dominant hyper IgE syndrome (AD‐HIES) (65‐80%), autosomal recessive hyper IgE syndrome (AR‐HIES) (24%) and Wiskott‐Aldrich syndrome (WAS) (rare). 67 , 79 NS often shows erythroderma within 1‐2 days after birth. 79 Metabolic disorders and Sjögren‐Larsson syndrome (SLS) should be considered as differential diagnoses when erythroderma develops in the first month of life. 6 , 80 Although one case of immunodysregulation polyendocrinopathy enteropathy X‐linked (IPEX) syndrome was described in the first month, erythroderma in IPEX usually occurs after the first month. 67 Although congenital psoriasis was observed in two cases, it is rare in the neonatal phase. 1 , 6 , 80 Erythroderma due to infections and drug reactions can occur at any age.
Time until final diagnosis differs with age of onset (Appendix S3). Mean age of diagnosis after the onset of the erythroderma in cases of congenital erythroderma vs non‐congenital NE is 116 days (range 1 day–5 years, median 20 days) and 102 days (range 1 day–5 years, median 15 days) (P < 0.4303) respectively. Overall, the longest delay was observed in NS, IPEX, OS and non‐syndromic forms of ichthyoses such as ichthyosis with confetti (IWC), lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE).
History features
Pregnancy, family history, consanguinity and gender
No details are known about complications during pregnancy as a possible underlying cause of NE, and less is known about the correlation between birth weight or pregnancy period and diagnostic outcome. From the 74 cases, details were extracted about pregnancy, drug use postpartum, family history, consanguinity and neonatal characteristics for example, e.g. gender. Details of the pregnancy period were described in 39/74 of cases. Prematurity would be more likely in cases of ichthyosis prematurity syndrome (IPS) and collodion babies. 21 , 81 Preterm birth was described in 15/74 cases (median 35 weeks; range 26–36 weeks), mainly in neonates with NS (n = 5) (Appendix S3), and one collodion baby (out of 2) was born prematurely (33 weeks). Family history was positive in 13/74 cases and negative in 28/74 cases. In the present case series, a positive family history for atopy, PID, ichthyosis, psoriasis and NS increased the risk of the same diagnosis in the newborn. Besides NS and OS, family history of atopy was also reported in cases of seborrheic dermatitis, atopic dermatitis and selective IgA deficiency. 4 Familial erythroderma was reported in cases of OS and NS. 4 Spontaneous abortion was reported in a case of Harlequin ichthyosis (HI), while unexplained death of siblings was reported in families with PID and NS. 4 Consanguinity was reported as a risk factor for NE in cases of ichthyosis ((25%), NS (88%), PID (33%) and in metabolic disorders. 4 , 47 Variations exist due to cultural and regional backgrounds, for example in Egypt consanguinity was reported in the majority of congenital ichthyoses (79%), of which LI constituted 32%. 80 Chanarin‐Dorfman syndrome (CDS) was found in patients with consanguine parents from the Mediterranean, Middle‐east, Saudi Arabia, India and Japan. 82 Consanguinity was reported in 9/74 cases of which seven had lethal complications (Appendix S4). The majority of 14 NS cases were male (57%), whereas in 23 cases of OS the majority (57%) were female. One study reported a female predominance in NS, 83 however, not confirmed by others. 84 Male predominance (ratio 2,5:1) was suggested in diffuse cutaneous mastocytosis (DCM). 73
Medication and nutrition
Ceftriaxone, vancomycin and phenytoin can induce NE. 6 , 10 , 69 Although treatment with penicillin, aminoglycosides and cephalosporin can cause an erythematous rash, it rarely results in erythroderma. 10 Zinc deficiency is a significant finding in acrodermatitis enteropathica, although herein the erythroderma rarely occurs in the neonatal period.
Specifically related to NE, holocarboxylase synthetase deficiency manifests with alopecia, dehydration, secondary cutaneous candidiasis and ketoacidosis. Patchy alopecia and acrodermatitis enteropathica‐like skin lesions are suggestive for biotinidase deficiency. Psoriasis‐like scaling appears periorificial before it generalizes. As human milk contains biotins, clinical manifestations in biotinidase deficient neonates occur earlier in breastfed than in formula‐fed babies. 10
Physical examination
Collodion membrane and scaling
A collodion membrane is a descriptive term for a transient condition in newborns, without implicating a specific diagnosis or disorder. 85 It can occur with erythroderma or the erythroderma appears when the collodion membrane disappears. 1 , 4 , 6 The collodion membrane usually sheds within 3–4 weeks after birth. 85 In 10% of cases a collodion membrane sheds, without leaving any sign of ichthyosis and is known as self‐healing collodion baby (SHCB, also known as self‐improving congenital ichthyosis or lamellar ichthyosis of the newborn). 86 The most common related disorders associated with the collodion membrane are inherited ichthyoses (Table 1). 85 , 86 , 87 , 88 Inherited ichthyosis has been divided in non‐syndromic ichthyoses, syndromic ichthyoses and unspecified. 86 , 89 , 90 From other studies, in 90% of cases of collodion babies, non‐bullous ichthyoses were the underlying diagnosis. 1 , 80 From the case series, 4/74 cases reported a collodion membrane: IWC, LI, Gaucher disease and holocarboxylase deficiency.
Rubio‐Gomez et al. described a severity score for collodion baby in order to predict the underlying diagnosis, based on clinical characteristics, such as an ectropion and eclabium. 21 A generalized collodion membrane was more common in autosomal recessive congenital ichthyosis (ARCI), while partial involvement was more frequent in unspecified congenital ichthyosis and syndromic ichthyosis. 21 A 15‐year retrospective study on collodion babies in the Netherlands could not confirm this severity score. 91
The presence and type of scaling in patients with NE can help in the differential diagnosis. 6 Neonatal scaling and exfoliative erythroderma are suggestive for ichthyoses and NS. 6 In ichthyosis, rough, dry, scaly skin is commonly seen; brown, dark scaling (frequently with collodion membrane) is typical for LI, while brown and fine white scaling with erythroderma is suggestive for CIE. 3 , 6 Annular scaling is suggestive for NS, but the typical ichthyosis linearis circumscripta is not always present. 79 Psoriasis can resemble ARCI, but usually more areas of unaffected skin can be seen. 6 Recalcitrant diaper dermatitis with generalization and pustules, sometimes with fever, could suggest erythrodermic psoriasis, while greasy scales on scalp (cradle cap) and skin folds (axilla, neck, retro auricular and diaper areas) suggests seborrheic dermatitis. 10 Perioral scale crusts in combination with desquamation, peeling and a characteristic tenderness of the skin suggest SSSS. Periorificial psoriasiform scaling should also suggest metabolic diseases such as holocarboxylase deficiency, biotinidase deficiency or acrodermatitis enteropathica. Due to maternal zinc suppletion, signs of acrodermatitis enteropathica in the neonatal period are rare, especially an erythrodermic phenotype. Collaret scales are typical for congenital cutaneous candidiasis (CCC). Skin induration and pruritus is suggestive for a PID, 4 although pruritus can also manifest in NS, scabies or desmosomal disorders, such as SAM syndrome. 62
Alopecia, bullae and pustules
Alopecia is common (45‐46% of cases) and is considered as a complication of any severe type of NE. 3 , 4 It frequently occurs in PIDs, particularly in OS, 6 but also presents in metabolic disorders. 4 , 10 Alopecia was mentioned in 12/74 cases: trichothiodystrophy (TTD) (1/1; 100%), NS (4/13; 31%), OS (6/23; 26%) and AEC syndrome (1/4; 25%). Alopecia totalis or alopecia of eyebrows and eyelashes is suggestive for a PID. 4 , 6 Bullae can be seen in epidermolytic ichthyosis (EI) (formerly known as bullous ichthyosis, keratinopathic ichthyosis or epidermolytic hyperkeratosis), staphylococcal scalded skin syndrome (SSSS) and DCM. In EI, the bullae will diminish over time and are replaced by diffuse hyperkeratosis. 86 In SSSS, the skin is usually painful, and the child can present with fever, general malaise and irritability. 6 , 92 The erythroderma starts abruptly with bullae developing within the erythematous area within 24–48 h. 92 There can be desquamation with scalded appearance, mainly in friction zones, periorificial cracking and a positive Nikolsky sign. 92 Mucosae are usually not affected and the erythema often starts in the umbilical area (omphalitis) and diaper area (e.g. pustules, impetigo and cellulitis). 92 In neonatal DCM, a red skin and blistering was reported in 17/22 cases (79%). 73 Diffuse erythroderma is unusual in DCM that may show an indurated, thickening of the skin (peau d’orange). 6 Extensive blistering with positive Darier sign is a more frequent presentation. 6 In CCC, papules and pustules may evolve into NE and will usually spare the oral cavity and diaper area but may include palms and soles. 6 Widely scattered spots on placenta and umbilical cord are a clue to the diagnosis. 6 , 9 In CCC, as being a possible opportunistic infection, one must be aware of an underlying PID such as severe combined immunodeficiency (SCID). 93 Systemic infections are seen especially in premature infants. 6
Extracutaneous involvement and systemic symptoms
Extracutaneous symptoms can occur in other organ systems and are often present in syndromic ichthyoses, PIDs and metabolic disorders. 12 , 94 Common systemic symptoms in NS are failure to thrive (FTT) and hypernatremic dehydration. 95 FTT is absent in atopic dermatitis, minimal in ichthyosis but was described in 69% of erythrodermas, predominantly in NS and all PIDs. 4 , 10 , 55 However, due to a protective effect of maternal antibodies, PIDs rarely give systemic symptoms at birth. 10 NS is highly suspected when FTT is combined with a mild to severe erythroderma, sepsis and hypernatremic dehydration. 6 A PID (OS or severe combined immunodeficiency (SCID)) should be suspected with lymphadenopathy and hepatosplenomegaly in combination with diarrhoea and/or FTT. 3 , 4 , 6 Opportunistic infections and candidiasis are mainly seen in PIDs. 4
Specific combinations of cutaneous, extracutaneous and systemic symptoms can be found in NS, CDS, KID and DCM as showed in Appendix S5. 79 , 93 , 95 , 96 Metabolic diseases are suspected when desquamative erythroderma is accompanied with neurological manifestations such as coma. 6
Systemic symptoms and presence of syndromic symptoms of the 74 cases are summarized in Appendix S4. From these 74 cases, 17 died (23%) (range 3 days – 19 months; mean 158 days; median 60 days), with high individual mortality rates for OS (2/8; 35%), graft‐versus‐host disease (GvHD) (2/3; 67%), metabolic disorders (1/3; 33%), AEC syndrome (1/4; 25%) and NS (2/14; 18%). Complications described are septic shock, renal or hepatic failure, multi‐organ failure, secondary hemophagocytic syndrome, failure of the central nervous system, respiratory failure, malnutrition, anaemia, thrombocytopenia and extensive diarrhoea.
Additional testing
Skin smears and culturing
If CCC or another fungal infection is suspected, a potassium hydroxide smear is recommended. A fungal culture and eventually polymerase chain reaction (PCR) can support the diagnosis. 97 If a local S. aureus or another infection such as SSSS is suspected, skin swabs from infected sites or from eyes, nose, umbilicus and the vagina of the mother should be sent for bacterial culture. 7 , 92 , 97
Blood tests
Sarkar stated that laboratory analysis in NE would contribute minimally and advice to start laboratory analysis after exclusion of atopic dermatitis, seborrheic dermatitis, ichthyosis and drug use. 97 We summarized the individual elements of laboratory tests as recommended by other authors (Table 2). These can be categorized in NE in general or specifically when certain diagnoses are suspected and are stratified as ‘standard test’ or as ‘on indication’. Hypernatremic dehydration and hypoalbuminemia can occur in any case of NE but hypernatremia was more frequently reported in NS. 4 , 6 Eosinophilia is common in NS 4 , 83 , 97 although no relation between eosinophilia and severity of NE or underlying diagnosis in NE could be found. 3 , 4 In DCM, elevated levels of serum tryptase in adults are correlated with systemic involvement in cutaneous mastocytosis, but this correlation has not been found in children. 101 Laboratory results and other diagnostics in the 74 patients are summarized in Appendix S6.
Table 2.
Laboratory tests performed in patients with neonatal erythroderma
| Deviation expected | Altered in specific diagnosis or in common NE | Reference(s) | |
|---|---|---|---|
| Laboratory test in general | |||
| Sodium/potassium | ↓↑ | Common | 1, 3, 4, 5, 7, 9 |
| Serum albumin | ↓ | Common | 1, 3, 4, 5, 7, 9 |
| Complete blood count, leuco diff and platelets | ↓↑ | Common, NS, OS | 1, 4, 9, 83, 97 |
| Haemoglobin | ↓ | Common | 5 |
| Erythrocyte sedimentation rate | ↑ | Common | 5 |
| Serum creatinine and serum urea | ↑ | Common | 7 |
| C‐reactive protein | ↑ | Common | 9 |
| Laboratory test on indication | |||
| Capillary blood gas | ketoacidosis | Common | 1 |
| Natural Killer (NK‐) cells | ↓ | NS | 98 |
| Immunoglobulins IgE, IgG, and IgA | ↓↑ | NS, Omenn, AD, WAS, IPEX, IgA deficiency | 12, 84, 97, 98 |
| T and B lymphocytes | ↓ | PID | 99, 100 |
| Serum zinc and alkaline phosphatase | ↓ | Acrodermatitis enteropathica | 97 |
| Biotinidase and holocarboxylase essays | ↓ | Biotinidase and holocarboxylase deficiency | 97 |
| Serum tryptase | ↑ | Diffuse Cutaneous Mastocytosis | 73, 101 |
| Serum creatin kinase | ↑ | CDS | 82 |
| Ceruloplasmin and serum copper | ↑ | Menkes disease | 43 |
| Glucose | ↓ | Common | 7, 9 |
| Ammino acids (urine) | ↑ | NS | 83 |
| Serum ammonia | ↑ | Metabolic diseases | 7, 9 |
| Serum Calcium | ↓ | DiGeorge | 102 |
AD, atopic dermatitis; CDS, Chanarin‐Dorfman syndrome; IPEX, Immunodysregulation polyendocrinopathy enteropathy X‐linked; NS, Netherton syndrome; OS, Omenn syndrome; PID, primary immunodeficiency; WAS, Wiskott‐Aldrich syndrome.
Identifying SCID‐PIDs, newborn screening can be facilitated by quantifying T‐cell receptor excision circles (TRECs) and kappa deleting‐recombination excision circles (KRECs). 99 , 100 A quantitative assay of immunoglobulin subsets with flow cytometry is recommended. 97 , 102 However, immunoglobulins in neonates are not fully produced yet. Why immunoglobulin‐E (IgE) is increased in neonates is unknown. IgE in newborns is well researched in relation to the risk of developing asthma and allergies. 103 , 104 Atopy in the mother seems to be the highest risk factor, 103 , 104 and this can contribute to a higher IgE level in the newborn. 105 However, no relation between family atopy and level of IgE level has been reported. 103 Chang et al. revealed that several genes are related to an increase of IgE production for diseases such as IPEX, OS and WAS syndrome. 106 This could be an explanation of increased IgE in newborns in cases besides atopy. Levels of IgE vary in cases of CDS (>1000 IU/mL), 79 NS (982–15302 IU/mL), 83 , 95 WAS, 106 atopic dermatitis (0–1000 IU/mL), 83 , 95 OS (0–45 000 IU/mL) 83 , 95 and ichthyosis. IgA can be reduced in NS 98 or selective IgA deficiency. 102 IgG levels can be reduced in cases of immunodeficiencies, or NS but can also be normal due to maternal IgG. 98 Although not considered as a routine investigation, elevation of activated IL17/IL‐22 is mainly seen in ichthyosis. 107 Additional tests are also recommended in cases of ichthyosis combined with alopecia and neurological signs.
A blood smear is strongly advised when syndromic ichthyosis is suspected. Vacuolated leucocytes called lipid droplets (Jordan’s anomaly) are mainly but not only found in CDS. 6 , 82 , 108 , 109 However, the severity of the disease is not related to the presence of lipid droplets. 108 When present, investigation of other organ systems, such as a liver ultrasound is recommended. 82
Skin biopsies
Histology of skin biopsies in NE is strongly recommended in several studies. 6 , 67 , 97 , 110 A biopsy can confirm the diagnosis in ~40% of cases. 3 , 4 , 7 Especially high specificity and sensitivity is reported in NS and PIDs, of which in NS due to the sensitive staining of lymho‐epithelial Kazal‐type related inhibitor (LEKTI) which is absent in NS. 6 , 83 , 110 LEKTI staining is recommended particularly in psoriasiform epidermal hyperplasia to rule out NS, 111 but in rare cases can also differentiate from desmosomal disorders such as severe dermatitis, atopic diatheses and metabolic (SAM) syndrome, that can resemble NS. 62 In NS, different histology patterns are seen, based on different phases within the spectrum. 67 In PID/OS, specific features are observed in the majority of cases. 4 , 67 The diagnosis of PID is made based on a spongiotic reactions pattern with keratinocytic necrosis, satellite cell lymphocytes and infiltrates of lymphocytes with or without eosinophils. 4 , 110 A biopsy is determinative in OS to differentiate with SCID and maternal GVHD 1 ; however, publications exist revealing no histological difference between OS and GVHD. 110 In ichthyoses, a biopsy is helpful in ~50% of cases. 4 Skin biopsies were taken in cases of psoriasis (2/2), OS (9/24), NS (7/14), GvHD (2/3), DCM (1/1), AEC (3/4), non‐syndromic ichthyosis (4/10), syndromic ichthyosis (1/2), metabolic disorder (1/3) and other (1/4). Besides classical H&E staining, electron microscopy examination may be helpful, for example, in NS. 28 Ultrastructural examination is not frequently performed nowadays. Skin histology is also helpful in SSSS 6 and EI. 1 , 6 In bullous skin diseases, EI can be differentiated from SSSS and hereditary epidermolysis bullosa (EB). 1 Also, in cases of skin detachment, to determine the level of detachment, immunofluorescence on skin biopsies is most valuable on narrowing the diagnosis. The recommended number of biopsies range from 2 110 to 3 97 although original literature herein refers to obtain multiple biopsies in adult erythroderma, in which diagnosis confirmation could better be determined compared to a single biopsy. 112
When a punch biopsy is not feasible or permitted, an alternative approach is described to differentiate between EI, toxic epidermal necrolysis (TEN) and SSSS: the ‘jelly roll’ frozen technique was described and proposed for cases of vesiculo‐bullous diseases of the newborn, wherein sloughed skin is wrapped around a cotton tipped applicator and subjected to frozen section analysis. 113
Hair analysis
Hair abnormalities are known in genetic disorders, such as NS, Menkes disease, TTD, uncombable hair syndrome and loose anagen hair syndrome. NS, Menkes disease and TTD are herein associated with NE. Hairs can be investigated through dermoscopy, reflectance confocal microscopy, optical microscopy and electron microscopy. 114
In the published cases, hair analysis was limited to NS, Menkes disease and TTD, which showed characteristic features such as trichorrhexis invaginata, pili torti and trichorrhexis nodosa. In NS, also samples of pili torti and trichorrhexis nodosa were found. Hairs of scalp, eyebrows or eyelashes seems suitable for investigation. 83 One study advises to cut the hairs instead of plucking them. 83 , 115 In TTD, hair shaft abnormalities will be profoundly seen in area of repeated trauma such as the occipital area. 115 In NS, hair from eyelashes are easier to investigate than from the scalp. 116 Trichorrhexis invaginata (bamboo hair) is specific for NS. 11 , 117 Trichorrhexis nodosa seems nonspecific, while seen in ectodermal dysplasia, TTD, NS and Menkes disease. 83 , 115 , 118 In Menkes disease, due to mutations in the copper transporting ATPase, all the copper enzymes are defective and the cross‐linking of collagen, elastin and keratin is impaired. Macroscopically, the hair is sparse, fine, slow growing, depigmented and lusterless with a peculiar steely consistency. On microscopic examination, pili torti or kinky hair is observed 43 , 117 , 118 but this can also be found in NS. 79 , 83 Trichoschisis, characterized by linear fracture of the hair shaft, given it the appearance of a ‘tiger tail’ is specific for TTD under polarized light microscopy. 117 , 118 Although not seen in the current literature case series, the majority of patients with keratosis ichthyosis deafness (KID) syndrome suffer from alopecia due to hair deformities. 119 In a study on trichoscopy in erythroderma, hair deformities were found in 41% (mainly in NS), 31% in cases with neurological impairment and in 33% of cases with immunological defects. 119 Hair abnormalities are not always present at the onset of NE, but many details are unknown. In cases of NS, trichorrhexis invaginata was found after an average of 10 months 4 with a range from 7 months 3 to several years. 79 Therefore, repeated hair samples may be required to confirm the diagnosis.
Molecular genetic analysis
Genetic mutation analysis is available for almost the complete differential spectrum of NE, but features prominently in diagnosing primary immunodeficiencies, metabolic disorders and all types of ichthyoses (syndromic and non‐syndromic). In ichthyoses, 13 genes are currently associated with ARCI: ABCA12, ALOX12B, ALOXE3, CASP14, CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7, SLC27A4 and TGM1). 1 However, in not all patients a known mutation is found. 6 TGM1 mutations was most frequently (30‐75%) found in ARCI patients but varied due to geographical and cultural origin. 80 , 120 , 121 , 122 A correlation between phenotype and genotype variations has been described in SPINK5, associated with NS. 3 , 98
Genetic analysis was performed in 39/74 cases and found positive in 30/74 (41%) cases; in OS (16/24; 67%), NS (4/14; 29%), AEC (3/4; 75%), HI (2/4; 50%), EI (2/2; 100%) and non‐EI (3/4; 75%). In cases of OS, mutations were found in RAG1 (14/16; 88%), RAG2 and Artemis. Mutation analysis was also diagnostic in single cases of TTD, IPEX, Menkes disease, KID, holocarboxylase deficiency and kindler epidermolysis bullosa (EB). Remarkably, in a case of HI, DNA was extracted from hairs. 14 No mutation was found in cases of SSSS, DCM and CDS.
Recently in a prospective study, in 70% of patients with NE, a mutation could be detected by next generation sequencing (NGS), with a selected gene panel of 60 genes 123 that will be adjusted according to this review (Appendix S7). Based on the diagnostic delay in cases of NS, OS, IPEX and non‐syndromic ichthyosis in this review, one can assume that performing mutation analysis in an early phase is important to limit complications. Although in most cases in this review genetic analysis was performed on blood samples, collecting material via non‐invasive methods are important to consider as a future alternative in patients with NE. Collecting saliva in neonates with buccal swabs for DNA and RNA sampling seem very promising, but different techniques herein determine optimal outcome. 124
Imaging investigations
Several imaging investigations are described as useful for the diagnosis in NE, especially in PIDs related to NE. 102 In X‐linked agammaglobulinemia, chest X‐rays shows sparse lymphoid tissue (tonsillar and adenoidal), while in common variable immunodeficiency (CVID), bronchiectasis, bronchial wall thickness and atelectasis are seen. 125 , 126 In CVID, a magnetic resonance imaging (MRI) of the central nervous system can display diffuse leptomeningeal thickness and enlargement with cerebral atrophy and abscesses. 125 In SCID and DiGeorge syndrome (DGS), chest X‐ray can show a narrow upper mediastinal contour and retrosternal lucency, due to the absence of thymus. 125 , 127 In SCID, DGS and WAS recurrent pneumonias can be seen. 128 As is shown in Supplemental S5, skeletal deformities can be visualized with X‐rays in SLS, Conradi‐Hünermann‐Happle syndrome (CHH) and DGS. Echocardiography can be performed when suspicion arises for DGS, SLS or KID. In our cases found, investigations such as X‐rays (NS, Menkes disease and DCM), ultrasonography (AEC), EEG (Menkes disease) and MRI (DCM) were performed, though in the majority did not lead to the diagnosis. Considering that imaging can be useful in typical cases, imaging is predominantly performed after the neonatal period.
Assessment of diagnostic approach
A multidisciplinary approach is highly recommended by previous publications wherein an alternative approach in patients with NE was reported. 1 , 6 , 9 , 10 , 11 , 90 , 94 According to this scoping review, many aspects of diagnostics contribute in finding a highly suggestive or definite diagnosis in patients with NE. A multidisciplinary 6‐step diagnostic approach is proposed (Fig. 2).
Step 1. A combined approach of dermatologists and paediatricians is recommended with a complete medical history (pregnancy, complications and miscarriages, recent medication use, consanguinity, family history of PID, ichthyosis, atopy and psoriasis) and physical examination with special attention to the presence of collodion membrane, alopecia, blisters and pustules. The collodion membrane suggests a subtype of ichthyoses. When blisters are present, it is recommended to perform an additional skin biopsy (in addition to step 3) for immunofluorescence microscopy to detect the level of blister formation, and have the possibility to detect absence/reduction in desmosomal proteins. Exclude bacterial or fungal infections such as SSSS or CCC, by taking swabs.
Step 2. In all cases of NE a baseline laboratory blood test should be started for ‘NE in general’ (Table 2). Evaluate findings of step 1 and blood test results to specify a diagnosis.
Step 3. If no diagnosis is found, blood tests should be expanded (Table 2), and histology of skin performed (including LEKTI staining) eventually also trichoscopy. A single skin biopsy is sufficient for evaluation by a dermatopathologist. After genetic counselling and consent, whole blood samples (or DNA) can be sent for molecular genetic testing (e.g. Sanger sequencing, targeted NGS panel, WES and WGS) (Appendix S7). Genetic testing in an early phase is recommended.
Step 4. Investigate for extracutaneous, syndromic and/or systemic symptoms. If present, consult a paediatric immunologist, neurologist, cardiologist, ENT specialist or ophthalmologist. The combination of neurological signs, hair abnormalities and specific extracutaneous symptoms can narrow down the possible diagnosis towards types of syndromic ichthyosis such as NS. PIDs and metabolic diseases should be excluded, especially in cases with failure to thrive, superimposed infections or lack of response to therapy. On indication, further research can be done.
Step 5. When step 4 shows no abnormalities, consider an inherited ichthyosis. Also consider benign inflammatory skin disorders, such as atopic dermatitis, psoriasis and seborrheic dermatitis, which are rare in the neonatal period and when erythrodermic, mainly manifest after three months of age.
Step 6. No recommendations on follow‐up are known, but we recommend first check within 1‐2 weeks after first clinical evaluation, if possible by a neonatologist or paediatrician and a dermatologist in an academic hospital. Then, a complete physical examination and re‐evaluation of systemic or extracutaneous symptoms if indicated.
Figure 2.
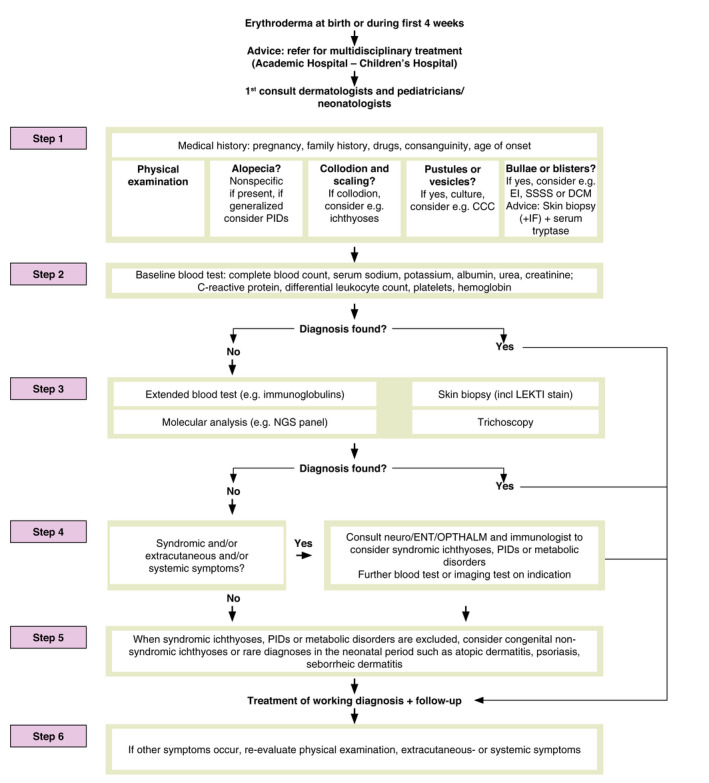
A 6‐step approach of neonatal erythroderma.
Discussion and conclusions
NE comprises a heterogeneous group of underlying potentially life‐threatening diagnoses. This literature review on NE systematically describes the many characteristics based on the medical history, family history, physical characteristics and additional diagnostic tests that can aid the clinician in narrowing the differential diagnoses. The aid of the ERN‐SKIN‐subthematic group Ichthyosis have resulted in an expert opinioned diagnostical approach for practical use.
Additional testing is recommended and helpful in the majority of cases wherein molecular genetics is preferred to start in an early stage of the diagnostic process. However, they are less feasible in cases of (financial) restrictions or lack of (familial) consent and genetic testing is a time‐consuming process in which the results are not always known on a short notice. In those cases, the multidisciplinary diagnostic proposed can be very helpful. Molecular genetics is an evolving area nowadays though, wherein establishing a diagnosis can be done more quickly in time to come. In specific cases, such as bullous characteristics, PIDs and NS, histology of skin biopsies (including LEKTI staining) is a critical element that can easily exclude other causes of NE. Blood tests are needed in NE in general, to reduce and monitor common complications as hypernatremia, dehydration and infections. Specific blood tests can be initiated on indication when metabolic disorders, syndromic ichthyoses or PIDs are suspected and infectious diseases are excluded. In collodion babies, the approach with diagnostic tests could be limited because the etiological entities in congenital ichthyoses, wherein metabolic diseases and PIDs are not obvious. Imaging studies seems to have minimal added value in most cases of NE.
The strengths of this review are the systematic literature search, the assistance of a library expert and the expert opinion by the ERN‐SKIN group. Limitations are the design of a scoping review, and included cases are prone to publication bias. Future recommendations in the approach of NE include a high sensitivity of skin biopsies, short‐time delay of molecular genetics, and an ongoing multidisciplinary approach in these rare cases. In the Netherlands based on the literature, a NGS NE panel has been developed that is available internationally.
In conclusion, this scoping review with a systematic literature search highlights the characteristics all around NE and proposes a systematic diagnostic approach for all patients with NE, with help from expert opinion of the ERN‐SKIN group‐subthematic Ichthyosis.
Supporting information
Appendix S1. EMBASE search terms.
Appendix S2. Flowchart literature review of neonatal erythroderma.
Appendix S3. Characteristics, age of onset and diagnosis delay in 74 patients.
Appendix S4. Alopecia, extracutaneous, systemic symptoms and mortality in 74 patients.
Appendix S5. Overview of syndromic and extracutaneous symptoms in neonatal erythroderma.
Appendix S6. Laboratory results and diagnostics performed in 74 patients with neonatal erythroderma.
Appendix S7. Suggestion NGS panel neonatal erythroderma.
Acknowledgements
No conflicts of interest. This study is part of the Academic Centers of Excellence of Congenital Anatomical Abnormalities and Primary Immunodeficiency Center of the Erasmus MC University Medical Center‐Sophia Children’s Hospital and of the European Reference Network‐SKIN‐Ichthyosis. The (parents of the) patients in this manuscript have given written informed consent to the publication of their case details. We thank Wichor Bramer, PhD, information technologist of the Erasmus MC University Medical Center Rotterdam, The Netherlands, for his contribution to the systematic literature search.
Conflicts of interest
None to declare.
Funding source
None to declare.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.
References
- 1. Ott H. Guidance for assessment of erythroderma in neonates and infants for the pediatric immunologist. Pediatr Allergy Immunol 2019; 30: 259–268. [DOI] [PubMed] [Google Scholar]
- 2. Cuperus E, Pasmans SGMA, Sigurdsson V. The incidence of neonatal erythroderma: results of a survey among all dermatologists in The Netherlands. (2016, unpublished data).
- 3. Al‐Dahlimi MAA. Neonatal and infantile erythroderma: a clinical and follow‐up study of 42 cases. J Dermatol 2007; 34: 302–307. [DOI] [PubMed] [Google Scholar]
- 4. Pruzskowski A, Bodemer C, Fraitag S, Teillac‐Hamel D, Amoric JC, de Prost Y. Neonatal and infantile erythrodermas. A retrospective study of 51 patients. Arch Dermatol 2000; 136: 875–880. [DOI] [PubMed] [Google Scholar]
- 5. Sarkar R, Sharma RC, Koranne RV, Sardana K. Erythroderma in children: a clinico‐etiological study. J Dermatol 1999; 26: 507–511. [DOI] [PubMed] [Google Scholar]
- 6. Fraitag S, Bodemer C. Neonatal erythroderma. Curr Opin Pediatr 2010; 22: 438–444. [DOI] [PubMed] [Google Scholar]
- 7. Ragunatha S, Inamader AC. Neonatal dermatological emergencies. Indian J Dermatol Venereol Leprol 2010; 76: 328–340. [DOI] [PubMed] [Google Scholar]
- 8. Ott H, Hoeger PH. Differential diagnosis of neonatal erythroderma. In Hoeger PH, Yan AC, Irvine AD, eds. Harper’s Textbook of Pediatric Dermatology, 3rd edn. Wiley‐Blackwell Ltd, West‐Sussex, UK, 2011: 11.1–11.13. [Google Scholar]
- 9. Ott H, Hütten M, Baron JM, Merk HF, Folster‐Holst R. Neonatal and infantile erythrodermas. J Dtsch Dermatol Ges 2008; 6: 1070–1085. [DOI] [PubMed] [Google Scholar]
- 10. Dhar S, Banerjee R, Malakar R. Neonatal erythroderma: diagnostic and therapeutic challenges. Indian J Dermatol 2012; 57: 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoeger PH, Harper JI. Neonatal erythroderma: differential diagnosis and management of the ‘red baby’. Arch Dis Child 1998; 79: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoneda K. Inherited ichthyosis: syndromic forms. J Dermatol 2016; 43: 252–263. [DOI] [PubMed] [Google Scholar]
- 13. Ramphul K, Kota V, Mejias SG. Child syndrome. Statpearls [Internet], 2019. [PubMed]
- 14. Burns PB, Rohroch RJ, Chung KC. The levels of evidence and their role in evidence‐based medicine. Plast Reconstr Surg 2011; 128: 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takeichi T, Sugiura K, Matsuda K. Novel ABCA12 splice site deletion mutation and ABCA12 mRNA analysis of pulled hair samplex in harlequin ichthyosis. J Dermatol Science 2013; 69: 259–265. [DOI] [PubMed] [Google Scholar]
- 16. Tanahashi K, Sugiura K, Sato T, Akiyama M. Noteworthy clinical findings of harlequin ichthyosis: digital autoamputation caused by cutaneous constriction bands in a case with novel ABCA12 mutations. Br J Dermatol 2016; 174: 689–691. [DOI] [PubMed] [Google Scholar]
- 17. Hashemzadeh A, Heydarian F. Harlequin ichthyosis. Acta Medica Iranica 2009; 47: 81–82. [Google Scholar]
- 18. Long MC. Ichthyosis with confetti: a rare diagnosis and treatment plan. BMJ Case Rep 2014; 10: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zvulunov A. Life‐threatening cutaneous conditions in neonates. Clin Dermatol 2005; 23: 134–143. [DOI] [PubMed] [Google Scholar]
- 20. Claus S, Terliesner N, Simon JC, Treudler R. Congenital erythroderma. J Dtsch Dermatol Ges 2016; 14: 435–437. [DOI] [PubMed] [Google Scholar]
- 21. Rubio‐Gomez GA, WeinsteinM PE. Development of a disease severity score for newborns with collodion membrane. J Am Acad Dermatol 2013; 70: 506–511. [DOI] [PubMed] [Google Scholar]
- 22. Scheimberg I, Hoeger PH, Harper JI, Lake B, Malone M. Omenn’s syndrome: differential diagnosis in infants with erythroderma and immunodeficiency. Pediatr Dev Pathol 2001; 4: 237–245. [DOI] [PubMed] [Google Scholar]
- 23. Ansai S, Mitsuhashi Y, Sasai K. Netherton’s syndrome in siblings. Br J Dermatol 1999; 141: 1097–1100. [DOI] [PubMed] [Google Scholar]
- 24. Chao SC, Richard G, Lee JYY. Netherton syndrome: report of two Taiwanese siblings with staphylococcal scalded skinsyndrome and mutation of SPINK5. Br J Dermatol 2005; 152: 159–165. [DOI] [PubMed] [Google Scholar]
- 25. Okulu E, Tunc G, Erdeve O et al. Netherton syndrome: a neonatal case with respiratory insufficiency. Arch Argent Pediatr 2018; 116: e609–e611. [DOI] [PubMed] [Google Scholar]
- 26. Itoh K, Kako T, Suzuki N, Sakurai N, Sugiyama K, Yamanishi K. Severe lethal phenotype of a Japanese case of Netherton with homozygous founder mutations of SPINK5 c.375_376delAT. J Dermatology 2015; 42:1212–1214. [DOI] [PubMed] [Google Scholar]
- 27. Mizuno Y, Suga Y, Haruna K et al. A case of a Japanese neonate with congenital ichthyosiform erythroderma diagnosed as Netherton syndrome. Clin Exp Dermatol 2006; 31: 677–680. [DOI] [PubMed] [Google Scholar]
- 28. Muller FB, Hausser I, Berg D et al. Genetic analysis of a severe case of Netherton syndrome and application for prenatal testing. Br J Dermatol 2002; 146: 495–499. [DOI] [PubMed] [Google Scholar]
- 29. Shwayder T, Banerjee S. Netherton syndrome presenting as congenital psoriasis. Pediatr Dermatol 1997; 14: 473–476. [DOI] [PubMed] [Google Scholar]
- 30. Lo YP, Snehal D, Shih CJ, Wu PY. Erythroderma and bamboo hair at birth: a case of Netherton syndrome and literature review. Dermatologica Sinica 2018; 36: 159–160. [Google Scholar]
- 31. El Khoury RA, Maalouf EH, Kechichian E, Tomb R. Steroid‐resistant erythroderma and alopecia in newborn. Indian J Dermatol 2018; 84: 304. [DOI] [PubMed] [Google Scholar]
- 32. Mendiratta V, Yadav P, Chander R, Aggarwal S. Recurrent pustular eruption masquerading as pustular psoriasis in Netherton syndrome. Pediatr Dermatol 2014; 32: 147–157. [DOI] [PubMed] [Google Scholar]
- 33. Magnani C, Bertolini P, Tondelli T, Bassissi GIP, Ricci R, Bevilacqua G. Early skeletal signs in Netherton syndrome. Pediatr Dermatol 2006; 23: 590–591. [DOI] [PubMed] [Google Scholar]
- 34. Aksu G, Kalkan Ucar S, Bulut Y et al. Renal involvement as a rare complication of Dorfman‐Chanarin Syndrome: a case report. Pediatr Dermatol 2008; 25: 326–331. [DOI] [PubMed] [Google Scholar]
- 35. Kayahashi K, Mizumoto Y, Myojo S, Mitani Y, Tajima A, Fujiwara H. A successful case of neoadjuvant chemotherapy and radical hysterectomy during pregnancy for advanced uterine cervical cancer accompanied by neonatal erythroderma. J Obstet Gynaecol Res 2018; 44: 2003–2007. [DOI] [PubMed] [Google Scholar]
- 36. Cremers CWRJ, Philipsen VMJG, Mali JWH. Deafness, ichthyosiform erythroderma, corneal involvement, photophobia and dental dysplasia. J Laryng 1977; 91: 585–589. [DOI] [PubMed] [Google Scholar]
- 37. Vilas Boas P, Sanchez‐Herrero A, Súarez‐Fernández R, Campos‐Dominguez M. Neonatal ichthyosis and hypotrichosis. Pediatr Dermatol 2019; 36: e77–e78. [DOI] [PubMed] [Google Scholar]
- 38. Naiki M, Mizuno S, Yamada K et al. MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet 2012; 158A: 97–102. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Z, Cheng R, Liang J et al. Ankyloblepharon‐ectodermal dysplasia‐clefting syndrome misdiagnosed as epidermolysis bullosa and congenital ichthyosiform erythroderma: case report and review of published work. J Dermatol 2019; 46: 422–425. [DOI] [PubMed] [Google Scholar]
- 40. Yoo J, Berk DR, Fabre E, Lind AC, Mallory SB. Ankyloblepharon‐ectodermal dysplasia‐clefting (AEC) syndrome with neonatal erythroderma: report of two cases. Int J Dermatol 2007; 46: 1196–1197. [DOI] [PubMed] [Google Scholar]
- 41. Berk DR, Crone K, Bayliss SJ. AEC syndrome caused by a novel P63 mutation and demonstrating erythroderma followed by extensive depigmentation. Pediatr Dermatol 2009; 26: 617–618. [DOI] [PubMed] [Google Scholar]
- 42. Zhang G, Chen J, Yan LX. Image gallery: rapidly spontaneous onset of erythroderma in a neonate. Br J Dermatol 2018; 178: 142. [DOI] [PubMed] [Google Scholar]
- 43. Galve J, Vicente A, González‐Enseñat MA et al. Neonatal erythroderma as a first manifestation of Menkes disease. Pediatrics 2012; 130: e239–e242. [DOI] [PubMed] [Google Scholar]
- 44. Fertitta L, Welfringer‐Morin A, Rigourd V et al. Neonatal staphylococcal scalded skin syndrome in a breastfed neonate. J Eur Acad Dermatol Venereol 2020; 34: e36–e38. [DOI] [PubMed] [Google Scholar]
- 45. Gupta A, Jacobs N. Visual diagnosis: 2‐week‐old has a red, peeling rash. Pediatr Rev 2013; 34: e9–e12. [DOI] [PubMed] [Google Scholar]
- 46. Satyapal S, Mehta J, Dhurat R, Jerajani H, Vaidya M. Staphylococcal scalded skin syndrome. Indian J Pediatr 2002; 69: 899–901. [DOI] [PubMed] [Google Scholar]
- 47. Boull CL, Hook KP. Neonatal erythroderma‐clinical perspective. Res Rep Neonatol 2017; 7: 1–9. [Google Scholar]
- 48. Greenberg‐Kushnir N, Lee YN, Simon AJ et al. A large cohort of RAG1/2‐deficient SCID patients‐clinical, immunological, and prognostic analysis. J Clin Immunol 2020; 40: 211–222. [DOI] [PubMed] [Google Scholar]
- 49. Sharapova SO, Guryanova IE, Pashchenko OE et al. Molecular characteristics, clinical and immunologic manifestations of 11 children with Omenn syndrome in East Slavs (Russia, Belarus, Ukraine). J Clin Immunol 2016; 36: 46–55. [DOI] [PubMed] [Google Scholar]
- 50. Meshaal SS, El Hawary RE, Abd Elaziz DS et al. Phenotypical heterogeneity in RAG‐deficient patients from a highly consanguineous population. Clin Exp Immunol 2019; 195: 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faletra F, Bruno I, Berti I, Pastore S, Pirrone A, Tommasini A. A red baby should not be taken too lightly. Acta Paediatr 2012; 101: e573–e577. [DOI] [PubMed] [Google Scholar]
- 52. Katugampola RP, Morgan G, Khetan R, Williams N, Blackford S. Omenn’s syndrome: lessons from a red baby. Clin Exp Dermatol 2008; 33: 425–428. [DOI] [PubMed] [Google Scholar]
- 53. Tallar M, Routes J. Omenn syndrome identified by newborn screening. Clin Perinatol 2020; 47: 77–86. [DOI] [PubMed] [Google Scholar]
- 54. Cotter CL, Rivers E, Salisbury J, Duarte Williamson E, Tewari A. Neonatal erythroderma. Clin Exp Dermatol 2020; 45: 646–649. [DOI] [PubMed] [Google Scholar]
- 55. Cuperus E, van Montfrans JM, van Gijn ME et al. Congenital erythroderma should be considered as an urgent warning sign of immuodeficiency: a case of Omenn syndrome. Eur J Dermatol 2017; 27: 313–314. [DOI] [PubMed] [Google Scholar]
- 56. Zafar R, Ver Heul A, Beigelman A et al. Omenn syndrome presenting with striking erythroderma and extreme lymphocytosis in a newborn. Pediatr Dermatol 2017; 34: e37–e39. [DOI] [PubMed] [Google Scholar]
- 57. Mansur Tatli MM, Sarraoglu S, Shermatov K, Salih Gurel M, Karadag A. Exfoliative erythroderma, recurrent infections, generalized lymphadenopathy and hepatosplenomegaly in a newborn: Omenn syndrome. Australas J Dermatol 2007; 48: 133–134. [DOI] [PubMed] [Google Scholar]
- 58. Dyke MP, Marlow N, Berry PJ. Omenn’s disease. Arch Dis Child 1991; 66: 1247–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jaouad IC, Ouldim K, Ali Ou Alla S, Kriouile Y, Villa A, Sefiani A. Omenn syndrome with mutation in RAG1 gene. Indian J Pediatr 2008; 74: 944–946. [DOI] [PubMed] [Google Scholar]
- 60. Flidel O, Barak Y, Lipschitz‐Mercer B, Frumkin A, Mogilner BM. Graft versus Host disease in extremely low birth weight neonate. Pediatrics 1992; 89: 689–690. [PubMed] [Google Scholar]
- 61. Ozdoğan T, Metin F, Doğu A, Timur C, Yildiz E. Transfusion‐associated graft‐versus‐host disease in a newborn. J Matern Fetal Neonatal Med 2007; 20: 271–272. [DOI] [PubMed] [Google Scholar]
- 62. Samuelov L, Sarig O, Harmon RM et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013; 45: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vakkilainen S, Puhakka L, Klemetti P et al. Novel DSP Spectrin 6 region variant causes neonatal erythroderma, failure to thrive, severe herpes simplex infections and brain lesions. Derm Venereol 2019; 99: 789–796. [DOI] [PubMed] [Google Scholar]
- 64. Eblan MJ, Goker‐Alpan O, Sidransky E. Perinatal lethal Gaucher disease: a distinct phenotype along the neuronopathic continuum. Fetal Pediatr Pathol 2005; 24: 205–222. [DOI] [PubMed] [Google Scholar]
- 65. Stone DL, Carey WF, Christodoulou J et al. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Netonatal Ed 2000; 82: F163–F166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gniadecki R. The Skin and disorders of the haematopoietic and immune systemis. In Griffiths C, Barker J, Bleiker T, Chalmers R, Creamer D eds. Rooks’s Textbook of Dermatology, Volume 4, 9th edn. Wiley Blackwell, Oxford, 2016: 148.1–148.19. [Google Scholar]
- 67. Sillevis Smitt JH, Kuijpers TW. Cutaneous manifestations of primary immunodeficiency. Curr Opin Pediatr 2013; 25: 492–497. [DOI] [PubMed] [Google Scholar]
- 68. Kucukguclu S, Tuncok Y, Ozkan H, Guven H, Uguz A, Maltepe F. Multiple‐dose activated charcoal in an accidental vancomycin overdose. J Toxicol Clin Toxicol 1996; 34: 83–86. [DOI] [PubMed] [Google Scholar]
- 69. Yigit S, Korkmaz A, Sekerel B. Drug‐induced hypersensitivity syndrome in a premature infant. Pediatr Dermatol 2005; 22: 71–74. [DOI] [PubMed] [Google Scholar]
- 70. Cadoz M, Denis F, Guerma T, Prince‐David M, Diop MI. Bacteriological, pharmacological and clinical comparison between amoxycillin and ceftriaxone in the treatment of 300 purulent meningitis. Pathol Biol (Paris) 1982; 30: 522–525. [PubMed] [Google Scholar]
- 71. Chang SE, Choi JH, Koh JK. Congenital erythrodermic psoriasis. Br. J. Dermatol. 1999; 140: 538–539. [DOI] [PubMed] [Google Scholar]
- 72. Parimalam K, Thomas J. Congenital erythrodermic psoriasis with atopic dermatitis: an example of immunogenetic spinoff. Indian J. Pathol. Microbiol. 2013; 56: 72. [DOI] [PubMed] [Google Scholar]
- 73. Koga H, Kokubo T, Akaishi M, Iida K, Korematsu S. Neonatal onset diffuse cutaneous mastocytosis: a case report and review of the literature. Pediatr Dermatol 2011; 28: 542–546. [DOI] [PubMed] [Google Scholar]
- 74. Suzumura H, Nitta A, Arisaka O. Cerebro‐oculo‐facio‐skeletal syndrome complicated by congenital ichthyosis. Clin Dysmorphol 2006; 15: 39–40. [DOI] [PubMed] [Google Scholar]
- 75. Fassihi H, Wessagowit V, Jones C et al. Neonatal diagnosis of Kindler syndrome. J Dermatol Sci 2005; 39: 183–185. [DOI] [PubMed] [Google Scholar]
- 76. Aguilera BA, González SR. High prolactin levels in a newborn with erythroderma. Pediatr Infect Dis J 2016; 35: 593–594. [DOI] [PubMed] [Google Scholar]
- 77. Haim A, Grunwald MH, Kapelushnik K, Moser AM, Beigelman A, Reuveni H. Hypereosinophilia in red scaly infants with scabies. J Pediatr 2005; 146: 712. [DOI] [PubMed] [Google Scholar]
- 78. Inamadar AD, Palit A. Acrodermatitis enteropathica‐like eruptions: many faces, one expression. In Inamadar AC, Palit A, eds. Advances in Pediatric Dermatology. Jaypee Brothers Medical Publishers, New Delhi, 2011: 234–248. [Google Scholar]
- 79. Van Gysel D, Koning H, Baert MRM, Savelkoul HF, Neijens HJ, Oranje AP. Clinico‐immunological heterogeneity in Comèl‐Netherton syndrome. Dermatology 2001; 202: 99–107. [DOI] [PubMed] [Google Scholar]
- 80. El‐Sayed N, Seifeldin NS, Gobrial CKT. High frequency of primary hereditary ichthyoses in the North‐East region of Cairo, Egypt. Postepy Dermatol Alergol 2018; 35: 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saral S, Vural A, Wollenberg A, Ruzicka T. A practical approach to ichthyoses with systemic manifestations. Clin Genet 2017; 91: 799–812. [DOI] [PubMed] [Google Scholar]
- 82. Mogahed EA, El‐Hennawy A, El‐Sayed R, El‐Karaksy H. Chanarin‐Dorfman syndrome: a case report and review of the literature. Arab J Gastroenterol 2015; 16: 142–144. [DOI] [PubMed] [Google Scholar]
- 83. Sun JD, Linden KG. Netherton syndrome: a case report and review of the literature. Int J Dermatol 2006; 45: 693–697. [DOI] [PubMed] [Google Scholar]
- 84. Smith DL, Smith JG, Wong SW, deShazo RD. Netherton’s syndrome. a syndrome of elevated IgE and characteristic skin and hair findings. J Allergy Clin Immunol 1995; 95: 116–123. [DOI] [PubMed] [Google Scholar]
- 85. Van Gysel GD, Lijnen RL, Moekti SS, de Laat PC, Oranje AP. Collodion baby: a follow‐up study of 17 cases. J Eur Acad Dermatol Venereol 2002; 16: 472–475. [DOI] [PubMed] [Google Scholar]
- 86. Oji V, Tadini G, Akiyama M et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in Soreze 2009. J Am Acad Dermatol 2010; 63: 607–641. [DOI] [PubMed] [Google Scholar]
- 87. Larrègue M, Ottavy N, Bressieux JM, Lorette J. Collodion baby: 32 new case reports. Ann Dermatol Venereol 1986; 113: 773–785. [PubMed] [Google Scholar]
- 88. Prado R, Ellis LZ, Gamble R, Funk T, Alan Arbuckle H, Bruckner AL. Collodion baby: an update with a focus on practical management. J Am Acad Dermatol 2012; 67: 1362–1374. [DOI] [PubMed] [Google Scholar]
- 89. Schmuth M, Martinz V, Janecke AR et al. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur J Hum Genetics 2013; 21: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oji V, Preil ML, Kleinow B et al. S1 Guidelines for the diagnosis and treatment if ichthyoses – update. J Dtsch Dermatol Ges 2017; 15: 1053–1065. [DOI] [PubMed] [Google Scholar]
- 91. Cuperus E, Bolling MC, de Graaf M et al. Collodion babies: A 15‐year retrospective multicenter study in The Netherlands—Evaluation of severity scores to predict the underlying disease. J Am Acad Dermatol 2021; 84: 1111–1113. [DOI] [PubMed] [Google Scholar]
- 92. Leung AKC, Barankin B, Leong KF. Staphylococcal‐scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr 2018; 14: 116–120. [DOI] [PubMed] [Google Scholar]
- 93. Bruno C, Bertini E, Di Rocco M et al. Clinical and genetic characterization of Chanarin‐Dorfman syndrome. Biochem Biophys Res Commun 2008; 369: 1125–1128. [DOI] [PubMed] [Google Scholar]
- 94. Inamadar AC, Ragunatha S. The rash that becomes an erythroderma. Clin Dermatol 2019; 37: 88–98. [DOI] [PubMed] [Google Scholar]
- 95. Saleem HMK, Shadid MF, Shahbaz A, Sohail A, Arslan Shadid M, Sachmechi I. Netherton syndrome: a case report and review of the literature. Cureus 2018; 10: e3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Caceres‐Rios H, Tamayo‐Sanchez K, Duran‐Mckinster C, de la Luz OM, Ruiz‐Maldonado R. Keratitis, ichthyosis, and deafness (KID syndrome): review of the literature and proposal of a new terminology. Pediatr Dermatol 1996; 13: 105–113. [DOI] [PubMed] [Google Scholar]
- 97. Sarkar R, Garg VK. Erythroderma in children. Indian J Dermatol Venereol Leprol 2010; 76: 341–347. [DOI] [PubMed] [Google Scholar]
- 98. Hannula‐Jouppi K, Laasanen SL, Ilander M et al. Intrafamily and interfamilial phenotype variation and immature immunity in patients with Netherton syndrome and Finnish SPINK5 founder mutation. JAMA Dermatol 2016; 152: 435–442. [DOI] [PubMed] [Google Scholar]
- 99. El‐Sayed Z, Radwan N. Newborn screening for primary immunodeficiencies: the gaps, challenges, and outlook for developing countries. Front Immunol. 2020; 10: 2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jiang T, Li Z, Zhang Q. Advances in neonatal screening for primary immune deficiencies. Exp Ther Med 2016; 11: 1542–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Brockow K, Akin C, Huber M, Metcalfe DD. Assessment of the extent of cutaneous involvement in children and adults with mastocytosis: relationship to symptomatology, tryptase levels, and bone marrow pathology. J Am Acad Dermatol 2003; 48: 508–516. [DOI] [PubMed] [Google Scholar]
- 102. Wu EY, Ehrlich L, Handly B, Frush DP, Buckley RH. Clinical and imaging considerations in primary immunodeficiency disorders: an update. Pediatr Radiol 2016; 46: 1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Koning H, Baert MRM, Oranje AP, Savelkoul HF, Neijens HJ. Development of immune functions related to allergic mechanisms in young children. Pediatr Res 1996; 40: 363–375. [DOI] [PubMed] [Google Scholar]
- 104. Mohammadzadeh I, Haghshenas M, Asefi S, Alizadeh‐Navaei R. IgE levels in newborn umbilical cord and its relationship with some maternal factors. Clin Mol Allergy 2019; 17: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Peters JL, Cohen S, Staudenmayer J, Hosen J, Platts‐Mills TAE, Wright RJ. Prenatal negative life events increases cord blood IgE: interactions with dust mite allergen and maternal atopy. Allergy 2012; 67: 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chang J‐C, Kuo H‐C, Hsu T‐Y et al. Different genetic associations of the IgE production among fetus, infancy and childhood. PLoS One 2013; 8: e70362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Czarnowicki T, He H, Leonard A et al. The Major orphan forms of ichthyosis are characterized by systemic T‐Cell activation and Th‐17/Tc‐17/Th‐22/Tc‐22 polarization in blood. J Invest Dermatol 2018; 138: 2157–2167. [DOI] [PubMed] [Google Scholar]
- 108. Gomez‐Moyano E, Godoy‐Diaz DJ, Ponce‐Verdugo L, Sanz‐Trelles A, Vera‐Casaño A, Sierra‐Salinas C. Chanarin‐Dorfman syndrome in three siblings in a non‐consanguineous family. J Eur Acad Dermatol Venereol 2016; 30: 157–159. [DOI] [PubMed] [Google Scholar]
- 109. Mitra S, Samanta M, Sarkar M, Chatterjee S. Dorfman‐Chanarin syndrom: a rare neutral lipid storage disease. Indian J Pathol Microbiol 2010;53:799–801. [DOI] [PubMed] [Google Scholar]
- 110. Leclerc‐Mercier S, Bodemer C, Bourdon‐Lanoy E et al. Early skin biopsy is helpful for the diagnosis and management of neonatal and infantile erythrodermas. J Cutan Pathol 2010; 37: 249–255. [DOI] [PubMed] [Google Scholar]
- 111. Bitoun E, Micheloni A, Lamant L et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum Mol Genet 2003; 12: 2417–2430. [DOI] [PubMed] [Google Scholar]
- 112. Walsh NMG, Prokopetz R, Tron VA et al. Histopathology in erythroderma: review of a series of cases by multiple observers. J Cutan Pathol 1994; 21: 419–423. [DOI] [PubMed] [Google Scholar]
- 113. Galler B, Bowen C, Arnold J, Kobayashi T, Dalton SR. Use of frozen section ‘jelly‐roll’ rechnique to aid in the diagnosis of bullous congenital ichthyosiform erythroderma (epidermolytic hyperkeratosis). J Cutan Pathol 2016; 43: 434–437. [DOI] [PubMed] [Google Scholar]
- 114. Chen L, Yang Y, Tian X et al. Dermatoscopy of the hair compared to three alternatives for the diagnosis of pediatric Netherton syndrome. J Dermatol 2020; 47: e195–e196. [DOI] [PubMed] [Google Scholar]
- 115. Itin PH, Fistarol SK. Hair shaft abnormalities – clues to diagnosis and treatment. Dermatology 2005; 211: 63–71. [DOI] [PubMed] [Google Scholar]
- 116. Powell J, Dawber RPR, Ferguson DJP, Griffiths WAD. Netherton’s syndrome: Increased likelihood of diagnosis by examining eyebrow hairs. Br J Dermatol 1999; 141: 544–546. [DOI] [PubMed] [Google Scholar]
- 117. Silengo M, Valenzise M, Sorasio L, Ferrero GB. Hair as a diagnostic tool in dysmorphology. Clin Genet 2002; 62: 270–272. [DOI] [PubMed] [Google Scholar]
- 118. Smith VV, Anderson G, Malone M, Sebire NJ. Light microscopic examination of scalp hair samples as an aid in the diagnosis of paediatric disorders: retrospective review of more than 300 cases from a single centre. J Clin Pathol 2005; 58: 1294–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Itin P. KID syndrome. In Hansen RC, Schachner LA, eds. Pediatric Dermatology. Mosby, Edinburgh, 2003: 416–418. [Google Scholar]
- 120. Israeli S, Goldberg I, Fuchs‐Telem D et al. Non‐syndromic autosomal recessive congenital ichthyosis in the Israeli population. Clin. Exp. Dermatol. 2013; 28: 911–916. [DOI] [PubMed] [Google Scholar]
- 121. Rodriguez‐Pazos L, Fachal GL, Toribio J, Carracedo A, Vega A. Analysis of TGM1, ALOX12B, ALOXE3, NIPAL4 and CYP4F22 in autosomal recessive congenital ichthyosis from Galicia (NW Spain): evidence of founder effects. Br J Dermatol 2011; 165: 906–911. [DOI] [PubMed] [Google Scholar]
- 122. Youssefian L, Vahidnezhad H, Saeidian AH et al. Autosomal recessive congenital ichthyosis: genomic landscape and phenotypic spectrum in a cohort of 125 consanguineous families. Hum Mutat 2019; 40: 288–298. [DOI] [PubMed] [Google Scholar]
- 123. Cuperus E, Sigurdsson V, van den Akker PC, Bolling MC, van Gijn ME, Pasmans SGMA. Diagnostic next generation sequencing in neonatal erythroderma. J Dtsch Dermatol Ges 2021; 19: 612–614. [DOI] [PubMed] [Google Scholar]
- 124. Yen E, Kaneko‐Tarui T, Maron JL. Technical considerations and protocol optimization for neonatal salivary biomarker discovery and analysis. Front Pediatr 2021; 8: 618553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yin EZ, Frush DP, Donnelly LA, Buckley RH. Primary immunodeficiency disorders in pediatric patients: clinical features and imaging findings. Am J Roentgenol 2001; 176: 1541–1552. [DOI] [PubMed] [Google Scholar]
- 126. Curtin JJ, Webster AD, Farrant J, Katz D. Bronchiectasis in hypogammaglobulinaemia – a computed tomography assessment. Clin Radiol 1991; 44: 82–84. [DOI] [PubMed] [Google Scholar]
- 127. Hedlund GL, Griscom NT. Respiratory system. In Kirks DR, ed., Practical Pediatric Imaging. Lippincott‐Raven, Cleveland, Philadelphia, 1998: 756–757. [Google Scholar]
- 128. Kuhn JP, Slovis TL, Silverman FN, Kuhns LR. The neck and respiratory system. In Silverman FN, Kuhn JP, eds. Caffey’s Pediatric x‐ray Diagnosis, 9th edn. CV Mosby, St. Louis, 1993: 563–566. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. EMBASE search terms.
Appendix S2. Flowchart literature review of neonatal erythroderma.
Appendix S3. Characteristics, age of onset and diagnosis delay in 74 patients.
Appendix S4. Alopecia, extracutaneous, systemic symptoms and mortality in 74 patients.
Appendix S5. Overview of syndromic and extracutaneous symptoms in neonatal erythroderma.
Appendix S6. Laboratory results and diagnostics performed in 74 patients with neonatal erythroderma.
Appendix S7. Suggestion NGS panel neonatal erythroderma.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.