

Abstract
Hereditary transthyretin‐mediated (hATTR) amyloidosis, or ATTRv amyloidosis, is a progressive disease, for which liver transplantation (LT) has been a long‐standing treatment. However, disease progression continues post‐LT. This Phase 3b, open‐label trial evaluated efficacy and safety of patisiran in patients with ATTRv amyloidosis with polyneuropathy progression post‐LT. Primary endpoint was median transthyretin (TTR) reduction from baseline. Twenty‐three patients received patisiran for 12 months alongside immunosuppression regimens. Patisiran elicited a rapid, sustained TTR reduction (median reduction [Months 6 and 12 average], 91.0%; 95% CI: 86.1%–92.3%); improved neuropathy, quality of life, and autonomic symptoms from baseline to Month 12 (mean change [SEM], Neuropathy Impairment Score, −3.7 [2.7]; Norfolk Quality of Life‐Diabetic Neuropathy questionnaire, −6.5 [4.9]; least‐squares mean [SEM], Composite Autonomic Symptom Score‐31, −5.0 [2.6]); and stabilized disability (Rasch‐built Overall Disability Scale) and nutritional status (modified body mass index). Adverse events were mild or moderate; five patients experienced ≥1 serious adverse event. Most patients had normal liver function tests. One patient experienced transplant rejection consistent with inadequate immunosuppression, remained on patisiran, and completed the study. In conclusion, patisiran reduced serum TTR, was well tolerated, and improved or stabilized key disease impairment measures in patients with ATTRv amyloidosis with polyneuropathy progression post‐LT (www.clinicaltrials.gov NCT03862807).
Keywords: clinical research/practice, clinical trial, liver allograft function/dysfunction, liver transplantation/hepatology, molecular biology: small interfering RNA, neurology, patient survival, pharmacology
Short abstract
Patisiran, a small‐interfering RNA therapeutic, is well‐tolerated and stabilizes or improves multiple disease symptoms for patients with hereditary transthyretin‐mediated amyloidosis with polyneuropathy who experience disease progression after liver transplantation.
Abbreviations
- AE
adverse event
- ATTRv
hereditary transthyretin (v for variant)
- CI
confidence interval
- COMPASS‐31
Composite Autonomic Symptom Score‐31
- FAP
familial amyloidotic polyneuropathy
- hATTR
hereditary transthyretin‐mediated
- IRR
infusion‐related reaction
- LFT
liver function test
- LS
least‐squares
- LT
liver transplantation
- mBMI
modified body mass index
- NIS
Neuropathy Impairment Score
- Norfolk QOL‐DN
Norfolk Quality of Life‐Diabetic Neuropathy questionnaire
- NYHA
New York Heart Association
- OLE
open‐label extension
- PND
polyneuropathy disability
- QOL
quality of life
- RNAi
ribonucleic acid interference
- R‐ODS
Rasch‐built Overall Disability Scale
- SAE
serious adverse event
- SAS
Statistical Analysis System
- SD
standard deviation
- SEM
standard error of the mean
- TTR
transthyretin
- ULN
upper limit of normal
- V30M
valine to methionine substitution at position 30
- wt
wild‐type
1. INTRODUCTION
Hereditary transthyretin‐mediated (hATTR) amyloidosis, also known as ATTRv (hereditary transthyretin [v for variant]) amyloidosis, is a rare, underdiagnosed, rapidly progressive, debilitating, and fatal disease caused by variants in the transthyretin (TTR) gene which cause misfolded TTR to accumulate as amyloid fibrils that lead to damage in multiple organs and tissues. 1 , 2 , 3 The disease has a heterogeneous clinical presentation; patients often develop a mixed phenotype of polyneuropathy (sensory, motor, or autonomic) and/or cardiomyopathy. 4 , 5 , 6 , 7 , 8 , 9 , 10 ATTRv amyloidosis is associated with progressive deterioration in quality of life (QOL) and significant morbidity and mortality, with a median survival of 4.7 years following diagnosis, reducing to 3.4 years in patients presenting with cardiomyopathy. 11 , 12 , 13 , 14
The liver is the primary source of circulating TTR. Liver transplantation (LT) eliminates hepatic production of variant TTR and was first utilized in the 1990s to manage ATTRv amyloidosis (Figure 1). 15 , 16 By 2018, over 2 200 patients worldwide had undergone LT for ATTRv amyloidosis. 17 However, the benefits of LT vary between patients according to their disease characteristics. Better prognoses are associated with early‐stage disease, early‐onset disease (age <50 years), V30M variant, higher modified body mass index (mBMI) (≥600 kg/m2 g/L) at transplant, short disease duration before transplantation, and an absence of cardiac involvement. 15 , 18 , 19 , 20 , 21 , 22 Poor outcomes in patients with late‐onset disease and/or a non‐V30M genotype may be related to differences in the amyloid fibril type (A or B). 23
FIGURE 1.
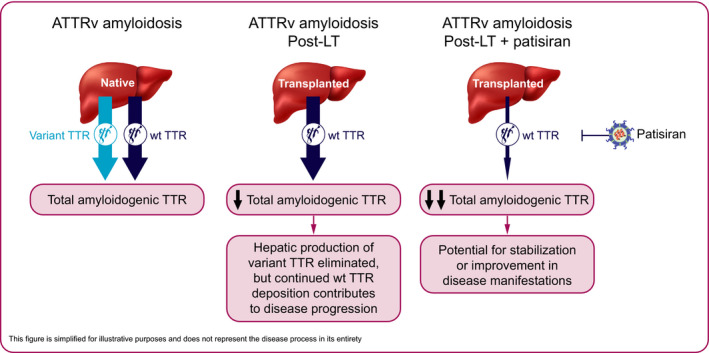
Treatment of ATTRv amyloidosis with LT and with patisiran post‐LT. The liver is the predominant source of circulating TTR protein. While unstable, variant TTR protein drives the pathogenesis of ATTRv amyloidosis, both variant and wt TTR protein form the amyloid deposits in multiple tissues. LT eliminates the hepatic production of variant TTR. However, ongoing deposition of wt TTR contributes to disease progression. Patisiran suppresses the production of variant and wt TTR, whether as first‐line therapy (native) or after LT (transplanted), thus allowing for the stabilization or improvement in the manifestations of ATTRv amyloidosis. ATTRv, hereditary transthyretin (v for variant); LT, liver transplantation; TTR, transthyretin; wt, wild‐type
Notably, a progressive decline in both cardiac and neurologic function can occur post‐LT 24 , 25 due to continued deposition of amyloid consisting of wild‐type (wt) TTR. 26 , 27 QOL can thus be significantly impaired. 28 Additionally, patients who undergo LT require lifelong immunosuppression to prevent allograft rejection. Patients are also at risk of death, with a 1‐year mortality rate of 7%–30% post‐LT reported in some studies. 25 , 29 , 30 , 31 , 32 A long‐term retrospective study demonstrated that deaths in the first year post‐LT were mainly due to worsening amyloidosis associated with progression of prior neuropathy or cardiac disease, and cardiac events were the leading cause of death post‐LT. 33
New pharmacotherapies for ATTRv amyloidosis may benefit patients who show disease progression post‐LT. TTR stabilizers, such as tafamidis and diflunisal, stabilize the TTR protein complex, slowing dissociation of the TTR tetramer into amyloidogenic monomers. TTR gene‐silencing agents, such as patisiran (a ribonucleic acid interference [RNAi] therapeutic) and inotersen (an antisense oligonucleotide), reduce the production of both variant and wt TTR. 34
Patisiran is a hepatocyte‐directed therapeutic that utilizes endogenous mechanisms of RNAi to reduce circulating levels of variant and wt TTR protein. As disease progression post‐LT is associated with wt TTR deposition, reducing levels of wt TTR is a particularly important consideration for this patient population (Figure 1). In the Phase 3 APOLLO study of patients with ATTRv amyloidosis with polyneuropathy, patisiran demonstrated the capacity to halt or reverse polyneuropathy and improve QOL from baseline in the majority of patients. Compared with placebo, patisiran also improved motor strength, disability, gait speed, nutritional status, and autonomic symptoms. 4 As a result, patisiran has been approved in >30 countries for the treatment of hATTR amyloidosis with polyneuropathy; specific indications vary by country and region. Here, we present the 12‐month efficacy and safety results of a prospective study of patisiran in patients with ATTRv amyloidosis who have had polyneuropathy progression post‐LT.
2. METHODS
2.1. Study oversight
This was a Phase 3b, global, open‐label study (www.clinicaltrials.gov NCT03862807; first submitted February 28, 2019) to evaluate the safety, efficacy, and pharmacokinetics of patisiran in patients with ATTRv amyloidosis with polyneuropathy progression post‐LT. Patients were enrolled at 10 centers in France, Germany, Italy, Portugal, Spain, Sweden, and the UK, between March 27, 2019, and August 5, 2019.
2.2. Patients
Eligible patients were aged ≥18 years, had received a LT for treatment of ATTRv amyloidosis ≥12 months before study entry, and had experienced polyneuropathy progression post‐LT. Polyneuropathy progression was defined as a documented increase in polyneuropathy disability (PND) score compared with the pre‐LT assessment or a documented increase in PND score between any two assessments post‐LT. 21 Other inclusion criteria included: Karnofsky Performance Status of ≥70%; and maintenance on a stable immunosuppression regimen for ≥3 months prior to study entry. Key exclusion criteria included: New York Heart Association (NYHA) class >II; PND score IV; serum levels of aspartate transaminase, alanine transaminase, or total bilirubin greater than the upper limit of normal (ULN); previous liver allograft rejection episodes or abnormal liver function tests (LFTs) suggestive of possible allograft rejection ≤6 months prior to the study; estimated glomerular filtration rate ≤30 ml/min/1.73 m2 at screening; any other organ transplant; unable to comply with required premedications. On‐study use of TTR stabilizers (tafamidis or diflunisal), tauroursodeoxycholic acid, doxycycline, or other investigational agents was prohibited, as was past use of patisiran and current or past use of inotersen.
2.3. Study design
Eligible patients received an intravenous infusion of 0.3 mg/kg patisiran once every 3 weeks for 12 months. All patients received premedications to minimize risk of infusion‐related reactions (IRRs) (10 mg dexamethasone or equivalent, 500 mg paracetamol/acetaminophen, 50 mg diphenhydramine or equivalent H1 blocker, and 50 mg ranitidine or equivalent H2 blocker). Patients who tolerated their infusions were eligible for a stepwise taper of the corticosteroid. All participants were advised to take the recommended daily allowance of vitamin A supplements to mitigate against potential deficiency due to TTR reduction by patisiran.
The study protocol was approved by central and local institutional review boards and ethics committees (Data S1). The study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All participants provided written informed consent.
2.4. Study endpoints and assessments
The primary endpoint was the average of Month 6 and Month 12 serum TTR percent reduction, assessed using a validated enzyme‐linked immunosorbent assay, which detects both wt and variant TTR. 35 However, since continued deposition of wt TTR contributes to disease progression post‐LT, the assay predominantly detects wt TTR in this study.
Secondary efficacy endpoints included the change from baseline to Month 12 in: neuropathy impairment; patient‐reported outcomes of QOL, disability, and autonomic symptoms; and nutritional status. Secondary endpoint outcomes were also assessed at Month 6. Neuropathy was determined using the Neuropathy Impairment Score (NIS), which assesses motor weakness, sensation, and reflexes (range, 0–244; higher scores reflect worse polyneuropathy). 36 QOL was assessed using the Norfolk QOL‐Diabetic Neuropathy questionnaire (Norfolk QOL‐DN), which evaluates physical functioning/large‐fiber neuropathy, activities of daily living, symptoms, small‐fiber neuropathy, and autonomic neuropathy (range, −4 to 136; higher scores represent greater impairment). 37 Disability was assessed using the Rasch‐built Overall Disability Scale (R‐ODS), a 24‐item questionnaire with a linearly weighted scale that captures activity and social participation limitations (range, 0–48; lower scores represent worse disability). 38 Autonomic symptoms were assessed using the Composite Autonomic Symptom Score‐31 (COMPASS‐31), a 31‐item questionnaire that measures orthostatic intolerance, vasomotor, secretomotor, gastrointestinal, bladder, and pupillomotor (range, 0–100; higher scores indicate greater autonomic impairment). 39 Nutritional status was assessed by mBMI (body mass index × albumin concentration; lower mBMI indicates worse nutritional status). Exploratory endpoints included change in disease stage (PND score, familial amyloidotic polyneuropathy stage), which were assessed at 6 and 12 months.
Frequency and severity of adverse events (AEs; classified according to MedDRA [version 23.0]) were assessed throughout the study. AEs of special clinical interest were transaminase elevations >3 × ULN and potential or confirmed events of liver transplant rejection. Monitoring for possible allograft rejection included repeat LFTs in the event of transaminase elevation >3 × ULN, checking and recording immunosuppressive drug levels every 3 months, and biopsy if possible allograft rejection was suspected. Electrocardiogram findings were collected at the screening visit. Patients were also assessed every 3 months at minimum for vital signs and laboratory tests, including LFTs.
2.5. Statistical analysis
Assuming a normally distributed TTR reduction from baseline of 80% with a standard deviation (SD) of 18%, a sample size of 16 patients would yield a 95% confidence interval (CI) with half‐width of approximately 10%. Enrollment of approximately 20 patients was planned, assuming a 20% premature discontinuation rate.
The median percent reduction in serum TTR and two‐sided 95% distribution‐free CI were summarized, with the p‐value obtained using the Wilcoxon signed‐rank test. Absolute values and change from baseline were summarized for the efficacy parameters of NIS, Norfolk QOL‐DN, R‐ODS, and mBMI. The analysis of these parameters was based on the last‐observation‐carried‐forward method: if a patient missed a visit during the study, their last post‐baseline assessment result was used for the subsequent missed visit(s). COMPASS‐31 was analyzed using a mixed‐effects model for repeated measures approach to account for potential missingness in this score; this approach makes use of fully and partially observed data sequences from individual patients by estimating the covariance between data at different time points and assumes data are missing at random. 40 Least‐squares (LS) mean estimates and standard error of the mean (SEM) for change from baseline at Month 6 and Month 12 in COMPASS‐31 score were presented. The study was not powered to assess the statistical significance of the secondary or exploratory endpoints; therefore, only the pre‐specified, descriptive analyses are presented for these endpoints. Descriptive statistical analyses were performed using SAS statistical software (version 9.4 or later) unless otherwise noted.
The patient populations analyzed in this study were the Safety Analysis Set (all patients who received any amount of patisiran) and the Per Protocol Analysis Set (all patients in the Safety Analysis Set who missed ≤2 doses of patisiran due to COVID‐19, such as COVID‐19 infection in the patient or the inability to receive dose due to local restrictions). The Safety Analysis Set was used for the primary efficacy and safety analyses, and the Per Protocol Analysis Set was used for the secondary efficacy analyses.
3. RESULTS
3.1. Patient population and disposition
Twenty‐four patients with ATTRv amyloidosis and post‐LT polyneuropathy progression from seven countries were enrolled in the study; 23 received patisiran and were included in the Safety Analysis Set; one patient stopped participation in the trial prior to first dose (see Figure S1). Twenty‐one patients were included in the Per Protocol Analysis Set. All 23 patients completed the study; one patient discontinued treatment due to patient decision but completed the study (see Figure S1). The study was performed during the COVID‐19 pandemic; no patients discontinued treatment with patisiran or stopped study participation due to COVID‐19.
Baseline demographics and characteristics are shown in Table 1. Median (range) age at ATTRv amyloidosis diagnosis was 50.0 (25.0–63.0) years, at LT was 54.0 (32–66) years, and at study inclusion was 58.0 (43–75) years; 13 (56.5%) patients were male. Fifteen (65.2%) patients had the V30M genotype. Over half of the patients had previously received a TTR stabilizer (13 [56.5%]; 11 [47.8%] tafamidis, two [8.7%] diflunisal). Ten (43.5%) patients had NYHA class I/II, indicating some degree of symptomatic cardiac involvement. Patients underwent LT a median (range) of 2.7 (0.4–10.3) years after diagnosis of ATTRv amyloidosis and received their first dose of patisiran a median (range) of 9.2 (1.4–20.8) years after the LT. Tacrolimus was the most frequently used immunosuppressant; other immunosuppressants included azathioprine, ciclosporin, everolimus, and mycophenolate (Table 1).
TABLE 1.
Baseline demographics and characteristics
| Demographic/characteristic | Safety analysis set (n = 23) |
|---|---|
| Age, years | |
| Mean (SD) | 58.1 (9.9) |
| Median (range) | 58.0 (43–75) |
| Male, n (%) | 13 (56.5) |
| Race, n (%) | |
| White | 22 (95.7) |
| Asian | 1 (4.3) |
| Country, n (%) | |
| Spain | 7 (30.4) |
| France | 5 (21.7) |
| Germany | 3 (13.0) |
| Portugal | 3 (13.0) |
| Italy | 2 (8.7) |
| Sweden | 2 (8.7) |
| UK | 1 (4.3) |
| Age <50 years at onset of ATTRv amyloidosis symptoms, n (%) | 13 (56.5) |
| Age at ATTRv amyloidosis diagnosis, years | |
| Mean (SD) | 46.7 (11.7) |
| Median (range) | 50.0 (25–63) |
| V30M genotype, a n (%) | 15 (65.2) |
| Previous TTR stabilizer use, b n (%) | 13 (56.5) |
| Median (range) duration of prior TTR stabilizer use, years | 2.1 (0.2–7.0) |
| Age at LT, years | |
| Mean (SD) | 50.1 (10.8) |
| Median (range) | 54.0 (32–66) |
| Time from ATTRv amyloidosis diagnosis to LT, years | |
| Mean (SD) | 3.7 (3.0) |
| Median (range) | 2.7 (0.4–10.3) |
| Time from LT to first patisiran dose, years | |
| Mean (SD) | 9.4 (5.1) |
| Median (range) | 9.2 (1.4–20.8) |
| Immunosuppression regimen at baseline, n (%) | |
| Tacrolimus | 10 (43.5) |
| Tacrolimus + mycophenolate | 7 (30.4) |
| Everolimus | 1 (4.3) |
| Ciclosporin | 1 (4.3) |
| Tacrolimus + everolimus | 1 (4.3) |
| Tacrolimus + azathioprine | 1 (4.3) |
| Ciclosporin + everolimus | 1 (4.3) |
| Ciclosporin + mycophenolate | 1 (4.3) |
| BMI, kg/m2 | |
| Mean (SD) | 23.5 (3.6) |
| Median (range) | 23.2 (18.0–30.5) |
| Serum TTR level, mg/L | |
| Mean (SD) | 202.1 (54.1) |
| Median (range) | 192.1 (123.7–315.1) |
| NIS total score | |
| Mean (SD) | 60.3 (39.0) |
| Median (range) | 59.5 (7.0–136.5) |
| Norfolk QOL‐DN score | |
| Mean (SD) | 66.7 (24.5) |
| Median (range) | 75.0 (16.0–98.0) |
| Karnofsky Performance Status, n (%) | |
| 70%−80% | 17 (73.9) |
| 90%−100% | 6 (26.1) |
| PND score, n (%) | |
| I: preserved walking, sensory disturbances | 1 (4.3) |
| II: impaired walking but can walk without stick/crutch | 9 (39.1) |
| IIIA: walk with one stick/crutch | 7 (30.4) |
| IIIB: walk with two sticks/crutches | 6 (26.1) |
| IV: confined to wheelchair/bedridden | 0 |
| FAP stage, n (%) | |
| 1: unimpaired ambulation | 10 (43.5) |
| 2: assistance with ambulation required | 13 (56.5) |
| 3: wheelchair‐bound or bedridden | 0 |
| NYHA class, n (%) | |
| 0: no heart failure | 13 (56.5) |
| I | 5 (21.7) |
| II | 5 (21.7) |
Abbreviations: ATTRv, hereditary transthyretin (v for variant); BMI, body mass index; FAP, familial amyloidotic polyneuropathy; LT, liver transplantation; NIS, Neuropathy Impairment Score; Norfolk QOL‐DN, Norfolk Quality of Life‐Diabetic Neuropathy questionnaire; NYHA, New York Heart Association; PND, polyneuropathy disability; SD, standard deviation; TTR, transthyretin; V30M, valine to methionine substitution at position 30.
Other genotypes included: S77Y (3), G47A (1), G47V (1), L12V (1), F64L (1), and Y116S (1).
Tafamidis in 11 (47.8%) patients; diflunisal in 2 (8.7%) patients.
At study baseline, 13 (56.5%) patients had a PND score of IIIA/B (IIIA, seven [30.4%]; IIIB, six [26.1%]; Table 1). Sixteen patients (69.6%) had experienced a 1‐unit increase from the earliest historic PND score to study baseline; the remainder experienced a 2‐ or 3‐unit increase in PND (Table 2). Fourteen patients (60.9%), who previously had preserved walking ability (i.e., PND score of I at first documentation), developed difficulties in ambulation over time prior to study baseline (i.e., progressed to a PND score of II‐IIIB). The median (range) total NIS and Norfolk QOL‐DN scores at baseline were 59.5 (7.0–136.5) and 75.0 (16.0–98.0) points, respectively (Table 1), indicating notable neuropathy and QOL impairment.
TABLE 2.
Shift from first documented PND score to PND score at study baseline in the safety analysis set
| First documented PND score a | PND score at study baseline, n (%) b | ||||||
|---|---|---|---|---|---|---|---|
| 0 | I | II | IIIA | IIIB | IV | Total | |
| 0 | 0 | 1 (4.3) | 0 | 0 | 0 | 0 | 1 (4.3) |
| I | 0 | 0 | 9 (39.1) | 2 (8.7) | 3 (13.0) | 0 | 14 (60.9) |
| II | 0 | 0 | 0 | 5 (21.7) | 2 (8.7) | 0 | 7 (30.4) |
| IIIA | 0 | 0 | 0 | 0 | 1 (4.3) | 0 | 1 (4.3) |
| IIIB | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IV | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 0 | 1 (4.3) | 9 (39.1) | 7 (30.4) | 6 (26.1) | 0 | 23 (100.0) |
Abbreviations: LT, liver transplantation; PND, polyneuropathy disability.
First documented PND score was either the most recent PND score prior to LT, or first post‐LT PND score if no PND score prior to LT.
Percentages are based on total number of patients in the safety analysis set (n = 23).
3.2. Serum TTR reduction (primary endpoint)
Patisiran elicited a rapid and sustained reduction in serum TTR levels (Figure 2). The median percent reduction from baseline in serum TTR (average of Month 6 and Month 12) was 91.0% (95% CI: 86.1–92.3; p = 4.5 × 10−8). The median TTR reduction from baseline was >80.0% at all post‐baseline timepoints.
FIGURE 2.
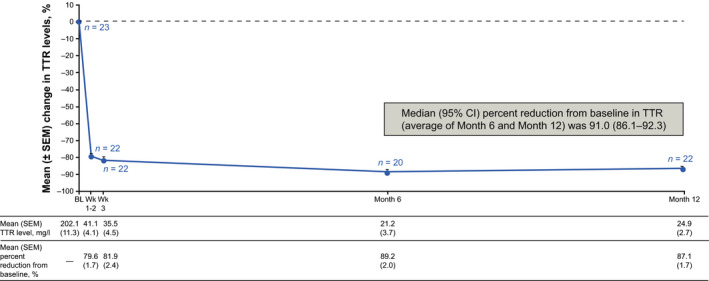
Percent change in serum TTR through Month 12 (Safety analysis set). BL, baseline; CI, confidence interval; SEM, standard error of the mean; TTR, transthyretin; Wk, week
3.3. Secondary and exploratory efficacy endpoints
At Month 12, there was an improvement in neuropathy, QOL, and autonomic symptoms with patisiran treatment, demonstrated by a decrease from baseline in mean total NIS score (mean [SEM] change from baseline of −3.7 [2.7]), Norfolk QOL‐DN score (mean [SEM] change from baseline of −6.5 [4.9]), and LS mean total COMPASS‐31 score (LS mean [SEM] change from baseline of −5.0 [2.6]) (Figure 3). Through 12 months of patisiran treatment, measures of disability (total R‐ODS score) and nutritional status (mBMI) appeared to be generally stable compared with baseline (mean [SEM] change from baseline of −0.1 [1.1] for R‐ODS, and +4.4 [21.8] for mBMI; Figure 3). At Month 12, the PND score remained unchanged relative to baseline in 20 of 21 patients in the per protocol analysis set and progressed from IIIA to IIIB in one patient.
FIGURE 3.
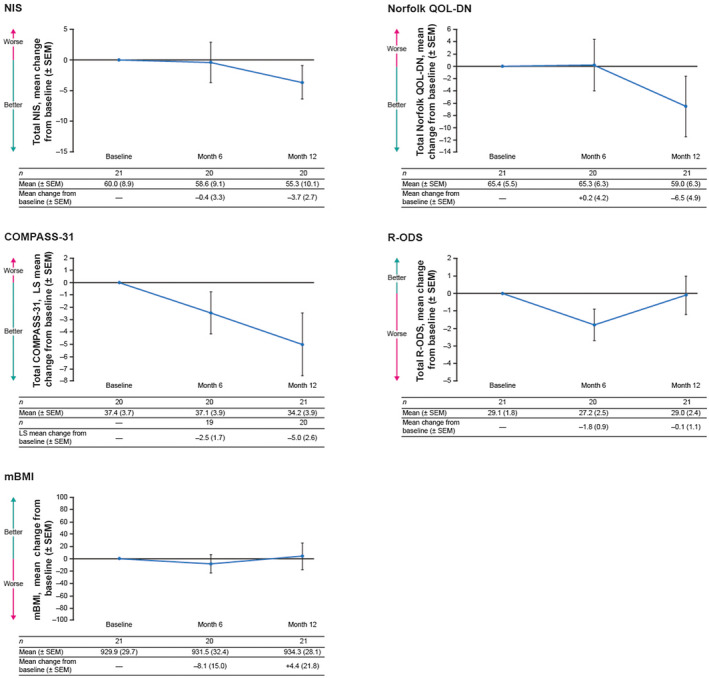
Mean change from baseline through Month 12 in secondary efficacy endpoints of NIS, Norfolk QOL‐DN, COMPASS‐31, R‐ODS, and mBMI. Graphs show mean score ± SEM for the per protocol analysis set. COMPASS‐31 graph shows LS mean score ± SEM for the per protocol analysis set and was analyzed using an MMRM approach. LOCF was used to impute missing assessments at or after Month 6 for mBMI. LOCF was not used for Month 12 assessment summaries for NIS, Norfolk QOL‐DN, and R‐ODS since all patients either had complete data at Month 12 or had no available post‐baseline data for LOCF imputation. COMPASS‐31, Composite Autonomic Symptom Score‐31; LOCF, last observation carried forward; LS, least‐squares; mBMI, modified body mass index; MMRM, mixed‐effects model for repeated measures; NIS, Neuropathy Impairment Score; Norfolk QOL‐DN, Norfolk Quality of Life‐Diabetic Neuropathy questionnaire; R‐ODS, Rasch‐built Overall Disability Scale; SEM, standard error of the mean
3.4. Safety
All 23 patients completed the full 12‐month study. The mean (SD) duration of patisiran exposure was 12.6 (2.6) months, with 24.1 cumulative person‐years of exposure. All patients experienced ≤1 AE (Table 3). The majority of AEs were mild or moderate in severity. There were no deaths, and no patients discontinued study treatment due to an AE.
TABLE 3.
Safety summary
| Category, n (%) | Safety analysis set (n = 23) |
|---|---|
| Any AE | 23 (100) |
| AEs reported in ≥10% of patients | |
| Diarrhea | 8 (34.8) |
| Infusion‐related reaction | 6 (26.1) |
| Peripheral edema | 5 (21.7) |
| Back pain | 5 (21.7) |
| Cardiac failure | 3 (13.0) |
| Fall | 3 (13.0) |
| Fatigue | 3 (13.0) |
| Headache | 3 (13.0) |
| Pyrexia | 3 (13.0) |
| Urinary tract infection | 3 (13.0) |
| AE related to study drug | 8 (34.8) |
| Any serious AE | 5 (21.7) |
| Serious AE related to study drug | 1 (4.3) |
| AE leading to discontinuation | 0 |
| AE leading to study drug interruption | 5 (21.7) |
| AE leading to death | 0 |
Abbreviation: AE, adverse event.
The most common AEs were diarrhea (eight patients [34.8%]) and IRRs (six patients [26.1%]) (Table 3). AEs of IRRs were all mild or moderate in severity. Back pain was the only IRR sign or symptom that occurred in >1 patient (n = 4 [17.4%]). Four patients had an infusion interruption due to an IRR, but the entire dose was completed in each case. One IRR considered related to patisiran was classified as a serious AE (SAE) (dizziness). This event occurred with the patient's first patisiran infusion, resolved by the following day without intervention and without a change in patisiran treatment, and did not recur at subsequent infusions.
In addition to the one SAE of IRR, four other patients experienced an SAE, which were all considered to be unrelated to patisiran treatment; there were 13 SAEs in total. Cardiac failure was the only SAE in >1 patient; all three patients with this SAE had a history of cardiomyopathy.
There was one case of liver transplant rejection, which was deemed unrelated to patisiran by the investigator. Histopathologic examination showed evidence of mild acute cellular rejection characterized by focal lymphocytic inflammation, some showing activation, within scattered portal triads and the subendothelium of a few portal venules. No central endothelial or bile duct inflammation was identified. The findings were consistent with mild acute cellular rejection, which was deemed by the investigator to be due to inadequate immunosuppression. The patient's immunosuppression regimen was modified, and the patient remained on the study drug, completing the study. The patient's transaminases subsequently remained stable, ranging from 1 to 2 × ULN.
LFTs were normal in the majority of patients. Mild and transient transaminase elevations (<3 × ULN) were incidentally observed in 11 (47.8%) patients. The elevations resolved without any change in dose or interruption of patisiran dosing. One patient experienced a transient alanine aminotransferase elevation >3 × ULN in association with cholangitis, which was deemed unrelated to patisiran by the investigator. No clinically relevant changes in other laboratory abnormalities, including platelet counts, were considered related to patisiran. There were no cases of platelet count <50 000/mm3, and mean platelet counts remained normal throughout the study.
No apparent drug interactions were observed between patisiran and the immunosuppressant treatments. In 14 of the 19 patients receiving tacrolimus at baseline, the dose remained constant throughout the study, while the dose was increased in two patients and decreased in three patients at study completion.
4. DISCUSSION
This Phase 3b prospective clinical trial evaluated the safety and efficacy of a therapeutic agent in patients with ATTRv amyloidosis with polyneuropathy progression post‐LT. In this 12‐month study, patisiran demonstrated a positive benefit:risk profile and was able to improve neuropathy, QOL, and autonomic symptoms, and to stabilize other key outcome measures in patients who had previously experienced disease worsening.
The rapid and sustained reduction in serum TTR levels, the improvements in neuropathy, QOL, and autonomic symptoms, and the stabilization of disability and nutritional status measures observed here are consistent with previous patisiran studies. 4 , 41 These results contrast with the worsening reported in studies of patients with ATTRv amyloidosis post‐LT, 25 , 26 and with the expected worsening seen in patients without LT from natural history studies and placebo groups in clinical studies. 1 , 4 , 5 , 42 , 43 The improvement and stabilization of disease endpoints seen here are particularly notable given that this study population was enrolled based on their progressive disease.
Patisiran was generally well tolerated, with no deaths or discontinuations due to AEs. All patients who received patisiran completed this year‐long trial. The safety profile was consistent with that established in the APOLLO and Phase 2 open‐label extension (OLE) studies. 4 , 41 , 44 No new safety concerns were identified in this population of patients who had undergone LT and were receiving a wide range of immunosuppression regimens; no evidence of drug interactions between patisiran and immunosuppressants was observed. Importantly, no signs of allograft rejection, including hepatotoxicity, due to patisiran were noted. Mild and transient elevations of liver enzymes were observed in almost half of the patients; all cases resolved without any change in dose or interruption of patisiran dosing. Of note, no clinically relevant transaminase elevations have been associated with patisiran in previous studies. As in prior studies, 4 , 44 diarrhea and IRRs were among the most common AEs in this study. Diarrhea was also the most frequent AE in the APOLLO placebo group, presumably as gastrointestinal symptoms are a common manifestation of ATTRv amyloidosis. 43 No thrombocytopenia or renal dysfunction due to patisiran was observed.
The efficacy outcomes from this study are consistent with those from the 18‐month APOLLO study. 4 APOLLO demonstrated that patisiran had the ability to halt or reverse polyneuropathy progression and improve QOL, when compared with baseline, in the majority of patients. Patisiran also improved autonomic dysfunction and stabilized disability and nutritional status of patients from baseline through Month 18 in APOLLO. Notably, all endpoints favored the patisiran arm when compared with placebo. While the patient populations and timing of endpoint assessments differ between APOLLO and the current study, the totality and consistency of the data suggest that the benefits of patisiran treatment extend to a wide population of patients with ATTRv amyloidosis, including those who have experienced progressive decline after LT.
The ongoing deposition of wt TTR causes disease progression after LT. 24 , 26 , 27 , 45 For example, the PND score worsened by ≤1 point at a median 79.5 months’ follow‐up post‐LT in 21% of patients in a single‐center study, 25 with worsening symptoms due to autonomic, sensory, and motor neuropathy; cardiomyopathy was also commonly seen. Likewise, all patients in the present study had a decline in neurologic function, as demonstrated by a documented increase in PND score after LT. Yet through 12 months of patisiran treatment in this study, the patients showed improvements in neuropathy, QOL, and autonomic symptoms, and stabilization in measures of disability and nutritional status, which is consistent with the results of the patisiran Global OLE study of patients who had not undergone LT. 4 , 41 The clinical course of the post‐LT patients in this study is reminiscent of patients who received placebo during APOLLO. While the APOLLO‐placebo group accumulated a greater disease burden than those who had received patisiran during the double‐blind portion of the study, the rapid progression was halted upon initiation of patisiran treatment in the Global OLE study. Indeed, the patients showed improvements in measures of neuropathy, QOL, autonomic symptoms, and nutritional status, as assessed by mean change from Global OLE baseline to 12 months of patisiran treatment. While patients in the APOLLO‐placebo group benefited from patisiran treatment in the Global OLE period, their level of disease impairment remained greater due to their notable polyneuropathy progression while on placebo, compared with those who received patisiran at the start of APOLLO, indicating the importance of early treatment.
There are few data concerning other therapeutic agents in patients with ATTRv amyloidosis after LT because prior clinical trials have excluded this population. In a retrospective review of nine patients with ATTRv amyloidosis who had disease progression after LT, inotersen improved or stabilized NIS compared with the pretreatment score. 46 Treatment was discontinued in five patients (56%) due to thrombocytopenia (n = 3) or reversible liver transplant rejection (n = 2). Similarly, rejection episodes after starting inotersen treatment have been reported as part of regulatory assessments. 47 , 48 Published data about tafamidis in patients with ATTRv amyloidosis progression following LT are limited to a case report that showed tafamidis was well tolerated and may be beneficial, as the patient showed no further clinical deterioration over the 18‐month follow‐up. 49 Due to a lack of data, European Medicines Agency regulatory guidelines state that tafamidis or inotersen should be discontinued in individuals undergoing LT. 48 , 50 US regulatory guidelines propose LFT monitoring in patients who have had a liver transplant and discontinuation of inotersen in patients who develop signs of liver transplant rejection. The lack of prospective studies of other therapeutic agents in patients with ATTRv amyloidosis and polyneuropathy progression post‐LT highlight the unmet need of this population.
In addition to being prospective and multicentered, the diversity in the baseline disease characteristics of the present study population, such as the range of time since LT, the variety of immunosuppression regimens, and the range of PND progression, is a strength of this study. The diversity allows for greater confidence in extrapolating the findings to the general population of LT patients with ATTRv amyloidosis. Furthermore, the multiple endpoints measured reflect the spectrum of impairment that is observed in this disease, allowing for a fuller assessment of the positive impact of patisiran on polyneuropathy progression. One potential limitation of this study is that other manifestations of ATTRv amyloidosis that can be observed following LT, such as ocular symptoms, were not assessed. However, due to the liver‐targeted mechanism of action of patisiran, it may not impact manifestations resulting from TTR produced locally in the eye or central nervous system. Other limitations of this study include the open‐label design, lack of a comparator arm, and limited study duration. While it is the largest study of a therapeutic agent in patients with ATTRv amyloidosis with polyneuropathy progression post‐LT to date, the study size did not allow inferences on the statistical significance of the change from baseline in secondary efficacy endpoints. Nevertheless, the totality of the results suggest a positive benefit:risk profile of patisiran in this patient population.
In conclusion, patients with ATTRv amyloidosis with polyneuropathy progression post‐LT remain in need of an effective treatment option. To our knowledge, this study is the first trial of a liver‐targeted RNAi therapeutic used in a transplant population. The data support the benefit of patisiran treatment in improving or stabilizing measures of disease impairment post‐LT and suggest the potential application of RNAi therapeutics to other conditions after organ transplantation.
Continued, long‐term follow‐up will be important to understand the impact of patisiran on neuropathy, cardiomyopathy, and survival in the post‐LT population. The potential of patisiran treatment is also of interest for the management of ATTRv amyloidosis in other transplanted populations, such as in patients with combination organ transplants (e.g., heart–liver or heart–kidney transplantation), domino LT, as well as heart transplantation alone.
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. PYJ, XL, and SA are employees of Alnylam Pharmaceuticals and hold stock or stock options. DA has received consulting fees from Alnylam Pharmaceuticals and from Pfizer Inc. JDG received institutional grants during the conduct of the study and honoraria for expert advisory boards outside the submitted work from Alnylam Pharmaceuticals, and honoraria for expert advisory boards outside the submitted work from Akcea Therapeutics and Eidos. JDG was the co‐chair of the steering committee for the ATTRibute‐CM (Eidos) trial. AM has received speaker fees and consulting honoraria from Alnylam Pharmaceuticals, Akcea Therapeutics, and Pfizer Inc. VP‐B has received symposia speaker fees and principal investigator fees from Ionis/Akcea Therapeutics, and principal investigator and advisory board fees from Alnylam Pharmaceuticals. JW has received institutional fees outside of the submitted work for lectures and advisory boards, from Alnylam Pharmaceuticals, Pfizer Inc, and Akcea Therapeutics. HHS and LL have no competing interests. FM‐B has received honoraria for speaker bureaus from Alnylam Pharmaceuticals and Pfizer Inc and for participating in advisory boards for Pfizer Inc and Akcea Therapeutics.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank all the patients who participated in the study and contributed to its results. The authors thank Professor Teresa Coelho (Hospital de Santo António, Centro Hospitalar Universitário do Porto, Porto, Portugal) for her contribution as an investigator in this study, including acquisition of data and support of the trial, its participants, and their families. The authors also thank Dr. Ilias Kounis (AP‐HP Hôpital Paul‐Brousse, Centre Hépato‐Biliaire; Inserm, Université Paris‐Saclay, UMR‐S 1193, France) for his contribution in this study, including acquisition of data, data interpretation, and support of the trial, its participants, and their families. The authors thank Dr. Alba Cachero (Hospital Universitari de Bellvitge, Barcelona, Spain), Dr. Cecile Cauquil (Neurology Department, AP‐HP, Hôpital Bicêtre, Le Kremlin Bicêtre, France), and Dr. Maeva Stephant (Université Paris‐Saclay, U1195, INSERM, Neurology Department, AP‐HP, Hôpital Bicêtre, Le Kremlin Bicêtre, France). The authors would like to thank all collaborators who are part of the patisiran post‐LT study group for their contribution to this study and support of the trial, its participants, and their families. The members of the patisiran post‐LT study group are: Hartmut Schmidt, Christel Langestroer, Anna Huesing‐Kabar, Matthias Schilling, and Iyad Kabar from Universitätsklinikum Münster, Münster, Germany; Jonas Wixner, Rolf Backlund, Intissar Anan, Erik Nordh, Erika Uneus, Björn Pilebro, and Ulrika Englund from Umeå University Hospital, Umeå, Sweden; Teresa Coelho, Marta Novais, Javier Perez, Ana Martins da Silva, Helena Pesseguerio Miranda, Joana Ramalho, Raquel Monte, Cristina Alves, Ines Cardaso, and Nádia Guimaraes from Centro Hospitalar Universitário do Porto, Porto, Portugal; Anna Mazzeo from University of Messina and A.O.U. Policlinico “G. Martino” Messina, Italy, and Luca Gentile, Massimo Russo, and Gianluca Di Bella from A.O.U. Policlinico “G. Martino” Messina, Italy; David Adams, Amina Gaouar, Cécile Cauquil‐Michon, Ilias Kounis, Andoni Echaniz‐Laguna, Maëva Stéphant, Fetra Rakotondratafika, Yasmine Boubrit, and Celine Labeyrie from CHU Bicêtre, Paris, France; Violaine Plante‐Bordenueve, Cecile Focsenaunu, Phillippe Le Corvoisier, Samar S. Ayache, Thierry Gendre, Laetitia Vervoitte, and Raphaele Arrouasse from CHU Henri Mondor, Créteil, Paris, France; Francisco Munoz Beamud, Alvaro Gragera Martinez, Cristina Borrachero, Ana Manovel, and Eusebio Diaz Rodriguez from Hospital Universitario Juan Ramón Jimenez, Huelva, Spain; Laura Llado, Marta Gutiérrez Gándara, Elena Fabra Jiménez, Patricia Valentina Vélez Santamaría, Yurema Martínez Vilar, and Alba Cachero from Hospital Universitari de Bellvitge, Barcelona, Spain; Julian D. Gillmore, Lisa Rannigan, Marianna Fontana, Richard Orrell, Sarah Louth, Liza Chacko, Sindhu Varughese, Douglas Throburn, Oliver Cohen, Steven Law, Angelique Smit, and Svetla Strehina from University College London Medical School, London, UK. Editorial support was provided by Kristen Brown, PhD, of Adelphi Communications Ltd, Macclesfield, UK, in accordance with Good Publication Practice (GPP3) guidelines, funded by Alnylam Pharmaceuticals, MA, USA. This study was sponsored by Alnylam Pharmaceuticals, MA, USA. The authors funded by Alnylam Pharmaceuticals had a role in the study design, in the collection, analysis, and interpretation of the data, in the writing of the report, and in the decision to submit the article for publication. The views and opinions expressed in this manuscript are those of the authors. Open Access funding enabled and organized by Projekt DEAL.
Schmidt HH, Wixner J, Planté‐Bordeneuve V, et al; the patisiran post‐LT study group . Patisiran treatment in patients with hereditary transthyretin‐mediated amyloidosis with polyneuropathy after liver transplantation. Am J Transplant. 2022;22:1646–1657. doi: 10.1111/ajt.17009
Contributor Information
Hartmut H. Schmidt, Email: hepar@ume.de.
the Patisiran Post‐LT Study Group:
Christel Langestroer, Anna Huesing‐Kabar, Matthias Schilling, Iyad Kabar, Rolf Backlund, Intissar Anan, Erik Nordh, Erika Uneus, Björn Pilebro, Ulrika Englund, Teresa Coelho, Marta Novais, Javier Perez, Ana Martins da Silva, Helena Pesseguerio Miranda, Joana Ramalho, Raquel Monte, Cristina Alves, Ines Cardaso, Nádia Guimaraes, Luca Gentile, Massimo Russo, Gianluca Di Bella, Amina Gaouar, Cécile Cauquil‐Michon, Ilias Kounis, Andoni Echaniz‐Laguna, Maëva Stéphant, Fetra Rakotondratafika, Yasmine Boubrit, Celine Labeyrie, Cecile Focsenaunu, Phillippe Le Corvoisier, Samar S. Ayache, Thierry Gendre, Laetitia Vervoitte, Raphaele Arrouasse, Alvaro Gragera Martinez, Cristina Borrachero, Ana Manovel, Eusebio Diaz Rodriguez, Marta Gutiérrez Gándara, Elena Fabra Jiménez, Patricia Valentina Vélez Santamaría, Yurema Martínez Vilar, Alba Cachero, Lisa Rannigan, Marianna Fontana, Richard Orrell, Sarah Louth, Liza Chacko, Sindhu Varughese, Douglas Throburn, Oliver Cohen, Steven Law, Angelique Smit, and Svetla Strehina
DATA AVAILABILITY STATEMENT
Data presented will not be available to share.
REFERENCES
- 1. Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50‐57. [DOI] [PubMed] [Google Scholar]
- 3. Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528‐540. [DOI] [PubMed] [Google Scholar]
- 4. Adams D, Gonzalez‐Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 5. Benson MD, Waddington‐Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22‐31. [DOI] [PubMed] [Google Scholar]
- 6. Coelho T, Maurer MS, Suhr OB. THAOS – The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild‐type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63‐76. [DOI] [PubMed] [Google Scholar]
- 7. Conceição I, González‐Duarte A, Obici L, et al. "Red‐flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin‐related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520‐528. [DOI] [PubMed] [Google Scholar]
- 9. Shin SC, Robinson‐Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79(6):733‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolte FJ, Langenstroer C, Friebel F, et al. Patient‐reported outcomes on familial amyloid polyneuropathy (FAP). Orphanet J Rare Dis. 2020;15(1):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gertz MA, Kyle RA, Thibodeau SN. Familial amyloidosis: a study of 52 North American‐born patients examined during a 30‐year period. Mayo Clin Proc. 1992;67(5):428‐440. [DOI] [PubMed] [Google Scholar]
- 13. Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120‐1127. [DOI] [PubMed] [Google Scholar]
- 14. Swiecicki PL, Zhen DB, Mauermann ML, et al. Hereditary ATTR amyloidosis: a single‐institution experience with 266 patients. Amyloid. 2015;22(2):123‐131. [DOI] [PubMed] [Google Scholar]
- 15. Ericzon B‐G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847‐1854. [DOI] [PubMed] [Google Scholar]
- 16. Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP‐met30). Clin Genet. 1991;40(3):242‐246. [DOI] [PubMed] [Google Scholar]
- 17. FAPWTR Results from the Familial World Transplant Registry. http://www.FAPWTR.org. Published 2018. Accessed December 22, 2021.
- 18. Adams D, Samuel D, Goulon‐Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7):1495‐1504. [DOI] [PubMed] [Google Scholar]
- 19. Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015;21(3):282‐292. [DOI] [PubMed] [Google Scholar]
- 20. Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl. 2009;15(10):1229‐1235. [DOI] [PubMed] [Google Scholar]
- 21. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coelho T, Inês M, Conceição I, et al. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):e1999‐e2009. [DOI] [PubMed] [Google Scholar]
- 23. Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281(4):337‐347. [DOI] [PubMed] [Google Scholar]
- 24. Olofsson BO, Backman C, Karp K, et al. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002;73(5):745‐751. [DOI] [PubMed] [Google Scholar]
- 25. Yamamoto S, Wilczek HE, Nowak G, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single‐center experience over 16 years. Am J Transplant. 2007;7(11):2597‐2604. [DOI] [PubMed] [Google Scholar]
- 26. Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yazaki M, Tokuda T, Nakamura A, et al. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild‐type transthyretin. Biochem Biophys Res Commun. 2000;274(3):702‐706. [DOI] [PubMed] [Google Scholar]
- 28. Drent G, Graveland CW, Hazenberg BPC, et al. Quality of life in patients with familial amyloidotic polyneuropathy long‐term after liver transplantation. Amyloid. 2009;16(3):133‐141. [DOI] [PubMed] [Google Scholar]
- 29. Sa Couto P, Barros F, Esteves S, et al. Comparison of outcome data after liver transplant: familial amyloid polyneuropathy (Portuguese type) recipients versus classical indications for liver transplant [abstract]. Eur J Anaesthesiol. 2010;27(Suppl. 1 47):13;Abstract 11AP13‐16. [Google Scholar]
- 30. Takei Y‐I, Ikeda S‐I, Ikegami T, et al. Ten years of experience with liver transplantation for familial amyloid polyneuropathy in Japan: outcomes of living donor liver transplantations. Intern Med. 2005;44(11):1151‐1156. [DOI] [PubMed] [Google Scholar]
- 31. Barreiros A‐P, Post F, Hoppe‐Lotichius M, et al. Liver transplantation and combined liver‐heart transplantation in patients with familial amyloid polyneuropathy: a single‐center experience. Liver Transpl. 2010;16(3):314‐323. [DOI] [PubMed] [Google Scholar]
- 32. Algalarrondo V, Antonini T, Théaudin M, et al. Prediction of long‐term survival after liver transplantation for familial transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(19):2154‐2156. [DOI] [PubMed] [Google Scholar]
- 33. Algalarrondo V, Antonini T, Théaudin M, et al. Cause of death analysis and temporal trends in survival after liver transplantation for transthyretin familial amyloid polyneuropathy. Amyloid. 2018;25(4):253‐260. [DOI] [PubMed] [Google Scholar]
- 34. Adams D, Koike H, Slama M, et al. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387‐404. [DOI] [PubMed] [Google Scholar]
- 35. Zhang X, Goel V, Attarwala H, et al. Patisiran pharmacokinetics, pharmacodynamics, and exposure‐response analyses in the phase 3 APOLLO trial in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis. J Clin Pharmacol. 2020;60(1):37‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dyck PJB, González‐Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS+7. J Neurol Sci. 2019;405:116424. [DOI] [PubMed] [Google Scholar]
- 37. Vinik EJ, Vinik AI, Paulson JF, et al. Norfolk QOL‐DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2014;19(2):104‐114. [DOI] [PubMed] [Google Scholar]
- 38. van Nes SI, Vanhoutte EK, van Doorn PA, et al. Rasch‐built Overall Disability Scale (R‐ODS) for immune‐mediated peripheral neuropathies. Neurology. 2011;76(4):337‐345. [DOI] [PubMed] [Google Scholar]
- 39. Sletten DM, Suarez GA, Low PA, et al. COMPASS 31: a refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc. 2012;87(12):1196‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mallinckrodt CH, Lane PW, Schnell D, et al. Recommendations for the primary analysis of continuous endpoints in longitudinal clinical trials. Drug Info J. 2008;42(4):303‐319. [Google Scholar]
- 41. Adams D, Polydefkis M, González‐Duarte A, et al. Long‐term safety and efficacy of patisiran for hereditary transthyretin‐mediated amyloidosis with polyneuropathy: 12‐month results of an open‐label extension study. Lancet Neurol. 2021;20(1):49‐59. [DOI] [PubMed] [Google Scholar]
- 42. Obici L, Berk JL, González‐Duarte A, et al. Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin‐mediated amyloidosis. Amyloid. 2020;27(3):153‐162. [DOI] [PubMed] [Google Scholar]
- 43. Wixner J, Mundayat R, Karayal ON, et al. THAOS: gastrointestinal manifestations of transthyretin amyloidosis ‐ common complications of a rare disease. Orphanet J Rare Dis. 2014;9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coelho T, Adams D, Conceição I, et al. A phase II, open‐label, extension study of long‐term patisiran treatment in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277‐282. [DOI] [PubMed] [Google Scholar]
- 46. Moshe‐Lilie O, Dimitrova D, Heitner SB, et al. TTR gene silencing therapy in post liver transplant hereditary ATTR amyloidosis patients. Amyloid. 2020;27(4):250‐253. [DOI] [PubMed] [Google Scholar]
- 47. Akcea Therapeutics US prescribing information: TEGSEDI (inotersen) injection, for subcutaneous use – October 2019 update. https://tegsedi.com/prescribing‐information.pdf. Published 2019. Accessed December 22, 2021.
- 48. European Medicines Agency Summary of product characteristics: Tegsedi 284 mg solution for injection in pre‐filled syringe 2019. https://www.ema.europa.eu/en/documents/product‐information/tegsedi‐epar‐product‐information_en.pdf. Accessed December 22, 2021.
- 49. Romero‐Imbroda J, Sagrario‐Fustero T, Del Canto‐Pérez C. Tafamidis for a transplant patient with transthyretin amyloid polyneuropathy. J Clin Neurol. 2017;13(4):444‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. European Medicines Agency Summary of product characteristics: Vyndaqel 20 mg soft capsules. emc: DataPharm. https://www.medicines.org.uk/emc/product/2837/smpc. Published 2020. Accessed December 22, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Data presented will not be available to share.