

Abstract
Beckwith‐Wiedemann syndrome (BWS) and Temple syndrome (TS) are classical imprinting disorders (IDs) with nonconfluent clinical features. We report here on a patient with clinical features of both syndromes, in whom epimutations were found at the BWS and TS imprinted regions, consistent with multilocus imprinting disturbance (MLID). This is the first case report of a patient with clinical features of both conditions who was found to have loss of methylation (LOM) of KCNQ1OT1: TSS‐DMR (ICR2) in the 11p15 imprinted region associated with BWS and LOM of MEG3: TSS‐DMR in the 14q32 imprinted region associated with TS. The report draws attention to the importance of testing for MLID as a cause of atypical clinical presentations of patients with IDs.
Keywords: Beckwith‐Wiedemann, imprinting, multilocus imprinting disturbance, Silver‐Russell, Temple
1. INTRODUCTION
Imprinting is a phenomenon by which epigenetic markers such as DNA methylation are maintained from one generation to another, ensuring parent of origin‐specific monoallelic expression of genes critical for growth, development, and metabolism (Mackay & Temple, 2017; Monk et al., 2019). Disturbances to this process are associated with deleterious developmental effects in pregnancy and beyond, including disturbance of pre‐ and postnatal growth, development, metabolism, and behavior.
Imprinting disorders (IDs) may be caused by cis‐acting genetic (copy number variants [CNV], uniparental disomy [UPD], chromosomal rearrangement) or epigenetic errors of disease‐associated loci. However, a subset of affected individuals has methylation abnormalities at multiple imprinted loci (multilocus imprinting disturbance [MLID]). In MLID, cis‐acting genetic changes are not found (Elbracht et al., 2020). Implicated instead are environmental, iatrogenic, and trans‐acting genetic factors affecting oocyte development or early developmental epigenetic reprogramming, including trans‐acting mutations found either in the affected person or their mother (Begemann et al., 2018; Mackay et al., 2008; Sanchez‐Delgado et al., 2016).
Determining the prevalence of MLID across IDs is challenging. Firstly, it is almost always mosaic, which complicates clinical diagnosis; secondly, its heterogeneous phenotype means that patients may not meet the clinical criteria for molecular testing, thus escaping diagnosis; thirdly, comprehensive epigenomic analysis is not routinely performed in individuals with molecular imprinting disturbance, so MLID may be missed. It is therefore probable that cases of MLID, whether or not associated with classical phenotypes, are going undetected. There is currently no standardized protocol for MLID diagnosis, nor has the impact of a positive diagnosis on clinical management been formally defined (Eggermann et al., 2014).
There is some evidence that individuals with MLID may exhibit a more diverse phenotype than cases with isolated imprinting errors (Poole et al., 2013). Testing a more extensive number of imprinted loci therefore may blur the boundaries between individual IDs which may have overlapping phenotypes. The concept of a network of imprinted genes that interact with each other further undermines any notion that molecular or clinical findings can be neatly compartmentalized (Arima et al., 2005).
Here, we report on a patient with imprinting disturbances at loci associated with two distinct classical IDs: one at chromosome 11p15 in the imprinted region associated with Beckwith‐Wiedemann syndrome (BWS: OMIM #130650) and one at chromosome 14q32 in the imprinted region associated with Temple syndrome (TS: OMIM #616222).
BWS is clinically characterized by macrosomia, macroglossia, visceromegaly, hyperinsulinism, exomphalos, and elevated risk of pediatric tumors; its incidence is estimated as 1:10,500 live births (Mussa et al., 2013). Fifty percentage of affected individuals have KCNQ1OT1: TSS‐DMR loss of methylation (LOM), of whom 30% have MLID (Brioude et al., 2018; Elbracht et al., 2020). TS is characterized by low birth weight, hypotonia, feeding difficulties, early puberty, and short stature (Gillessen‐Kaesbach et al., 2018); it has phenotypic overlap with Silver‐Russell syndrome (SRS) but is rarer than SRS, with unknown incidence and no agreed clinical scoring system. Only around 12% of cases of TS are due to an imprinting error (Ioannides et al., 2014; Kagami, Nagasaki, et al., 2017), and MLID has rarely been reported in individuals clinically characterized with TS (Kagami, Matsubara, et al., 2017). While several individuals have been described with MLID involving numerous loci, including KCNQ1OT1 and MEG3 (Begemann et al., 2018; Caliebe et al., 2014; Cubellis et al., 2020), to date only one case has been reported where the only disease‐associated loci involved were KCNQ1OT1 and MEG3 (Bens et al., 2016), for which no clinical data were provided.
1.1. Clinical case report
The child was the first born to nonconsanguineous parents. He was conceived naturally when his mother was 37 years of age, 1 month after she had undergone a fallopian tube cannulation following several years of infertility. The pregnancy was complicated by cholestasis but growth in utero was normal. Following an emergency cesarean section for fetal distress, he was born at 39 weeks of gestation, weighing 2.73 kg (−1.54 standard deviation score [SDS]). Concerns were raised regarding his head size; his occipitofrontal circumference [OFC] was not recorded at birth, but in his first year it was consistently >98th centile (Figure 1a).
FIGURE 1.
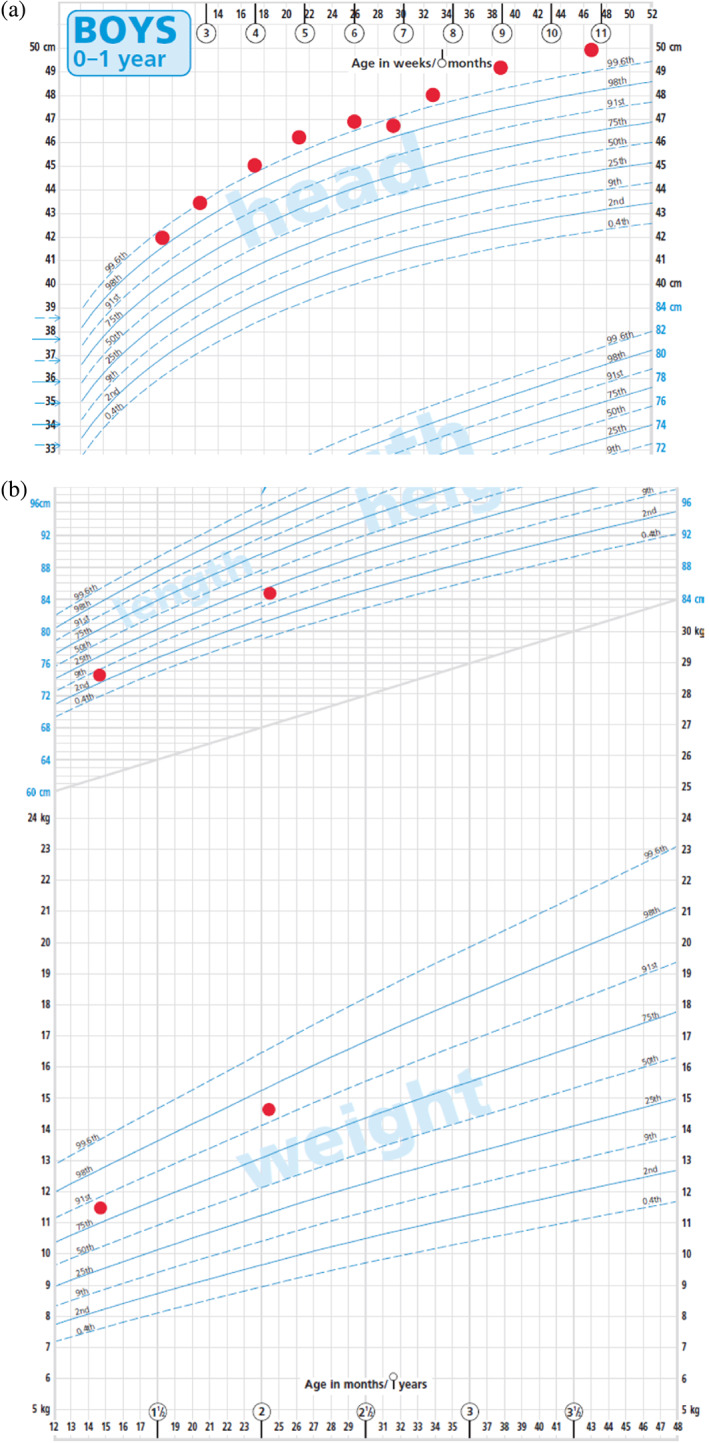
(a) Head circumference chart at 2–11 months. (b) Growth chart (height and weight) at 15 months and 24 months
At 8 months of age, he was referred because of macrocephaly, facial and body asymmetry, and nevus flammeus. His right side appeared smaller than his left, but both sides were functionally normal, so it was unclear which was the abnormal side. In addition to facial asymmetry, he was noted to have a prominent forehead, broad nasal tip, and flat nasal bridge (see Figure 2). No macroglossia, exomphalos, or organomegaly was identified. Hands and feet were of normal size.
FIGURE 2.
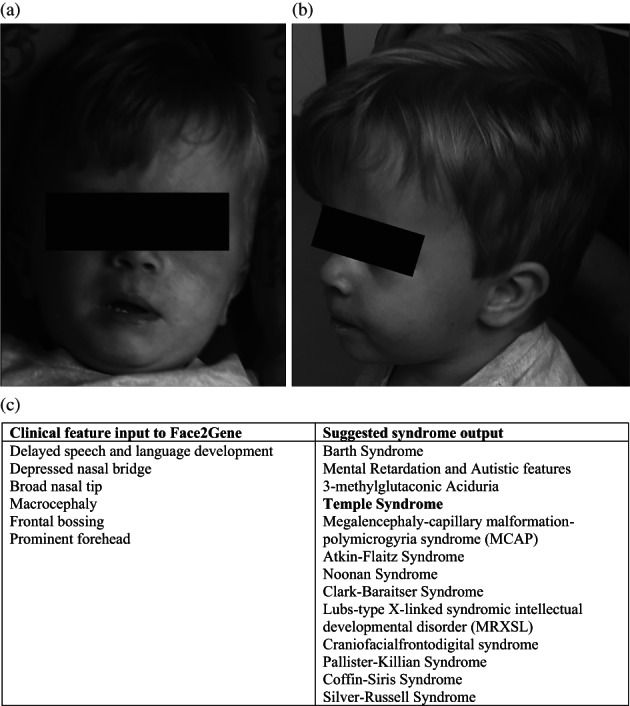
(a, b) Clinical images of the proband aged 2 years, demonstrating facial asymmetry, protruding forehead, and macrocephaly. (c) Clinical features included in Face2Gene analysis and suggested syndrome outputs
There was no formal history of feeding difficulties, although his mother described him as a “fussy eater.” Despite this, his weight was disproportionate to his height at 15 months (height SDS: −1.39, weight SDS: +1.22, and OFC SDS: +3.78 SDS) and 2 years of age (height SDS: −0.89, weight SDS: +1.66, and OFC SDS: +3.71 SDS) (Figure 1b).
He sat at 6 months of age and walked independently at 17 months. Mild speech and language delay was evident at age 2.
There was no family history of obvious significance. The mother's sibling had four children, all of whom were healthy. The index case's father had two healthy children from a previous relationship.
The initial differential included hypochondroplasia, Noonan's syndrome, and PTEN‐related disorders. Plain radiographs of the pelvis and leg were normal, as were parathyroid, calcium, and phosphate levels. Head ultrasound scan showed no evidence of hydrocephalus.
In view of the macrocephaly, body asymmetry, and nevus flammeus, BWS was also suspected, although his BWS score was <4 (Table 1). He exhibited some features atypical of BWS, such as relatively small stature in comparison with most cases with the syndrome, protruding forehead, and relative macrocephaly in infancy; these features are more consistent with other IDs such as Silver Russell syndrome [SRS]. His pre‐ and postnatal growth were not consistent with SRS, although he scored 3/6 using the Netchine‐Harbison scoring system (see Table 1, and assuming head size at birth on similar centiles to those recorded through the first year of life). His relatively short stature and disproportionately high weight were more suggestive of TS. Clinical features were assessed using Face2Gene analysis (https://www.face2gene.com): this assessment placed TS in the top four potential syndromes (Figure 2). As his phenotype did not align clearly with a single ID, he was tested for MLID at 2 years of age.
TABLE 1.
Results of targeted DNA methylation analysis of imprinted loci by methylation‐specific multiplex ligation‐dependent probe amplification (MLPA) and methylation‐specific PCR
| Beckwith‐Wiedemann syndrome | Temple syndrome | Silver‐Russell syndrome |
|---|---|---|
| Macroglossia (2) | SGA (prenatal growth failure) | SGA (birth weight and/or birth length) (1) |
| Exomphalos (2) | Postnatal growth failure | Postnatal growth failure (1) |
| Hyperinsulinism (lasting >1 week and requiring escalated treatment) (2) | Feeding difficulties | Feeding difficulties and/or low BMI (1) |
| Multifocal and/or bilateral Wilms tumor or nephroblastomatosis (2) | Prominent forehead in infancy | Protruding forehead (1) |
| Pathology findings: adrenal cortex cytomegaly, placental mesenchymal dysplasia, or pancreatic adenomatosis (2) | Relative macrocephaly at birth | Relative macrocephaly at birth (1) |
| Lateralized overgrowth (2) | Body asymmetry (1) | |
| Facial nevus simplex (1) | Small hands and feet | |
| Umbilical hernia and/or diastasis recti (1) | Hypotonia | |
| Birthweight >2 SDS above the mean (1) | Precocious puberty | |
| Ear creases and/or pits (1) | ||
| Transient hypoglycemia (lasting <1 week) (1) | ||
| Typical BWS tumors (neuroblastoma, rhabdomyosarcoma, unilateral Wilms tumor, hepatoblastoma, adrenocortical carcinoma, or phaeochromocytoma) (1) | ||
| Nephromegaly and/or hepatomegaly (1) | ||
| Polyhydramnios and/or placentomegaly (1) |
Note: The table presents the clinical features of BWS, TS, and SRS, with scores indicated in brackets for clinical features, and the features of the patient are highlighted in bold text. BWS clinical scoring criteria are adapted from Brioude et al. (2018), with 2 points for major features, 1 point for minor features, and a clinical diagnosis of BWS in persons scoring ≥4 points. Netchine‐Harbison clinical scoring system for SRS is adapted from Wakeling et al. (2017), 2017 with one point per feature and a clinical diagnosis of SRS in persons scoring ≥4 points. Clinical features found in >60% of TS cases are adapted from Kagami, Nagasaki et al. (2017).
Abbreviations: BMI, body mass index; BWS, Beckwith‐Wiedemann syndrome; nd, not done; SDs, standard deviations; SGA, small for gestational age; SRS, Silver‐Russell syndrome; TS, Temple Syndrome.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The patient was recruited into the research study “Imprinting disorders—finding out why” (IDFOW: Southampton and South West Hampshire Research Ethics approval 07/H0502/85) through the UK Comprehensive Local Research network (www.southampton.ac.uk/geneticimprinting/informationpatients/imprintingfindingoutwhy.page). Written consent was obtained from the parents of the proband, including consent to access medical records and for the publication of patient images.
2.2. Molecular studies
Initial determination of DNA methylation was performed on blood‐derived DNA by methylation‐specific MLPA (MS‐MLPA; MRC‐Holland b.v.) according to the manufacturer's specification, using kits for BWS/SRS (ME030‐C3); GRB10, MEST, PLAGL1, and MEG3 (ME032‐A1); Prader‐Willi/Angelman syndrome (ME028‐C1); pseudohypoparathyroidism (ME031‐B2); and MLID (ME034‐C1).
Methylation‐specific PCR (MS‐PCR) analysis was performed using previously described primers and protocol (Poole et al., 2013): loci analyzed were DIRAS3 Ex2 differentially methylated region (DMR) (DIRAS3, 1p31); PLAGL1 TSS alt‐DMR (PLAGL1, 6q24); IGF2R Int2 DMR (IGF2R, 6q27); GRB10 alt‐TSS DMR (GRB10, 7q32); MEST alt‐TSS DMR (MEST, 7q32); H19 TSS DMR (H19, 11p15.5); IGF2 DMR0 (11p15.5); KCNQ1OT1 TSS DMR (KCNQ1OT1 or IC2, chr11); MEG3 TSS DMR (DLK1 or MEG3, 14q32); SNRPN alt‐TSS DMR (SNRPN, 15q11); IGF1R Int2 DMR (IGF1R, 15q26); PEG3 TSS DMR (PEG3, 19q32); GNAS‐AS1 TSS DMR (NESPAS/GNAS, 20q13); WRB alt‐TSS DMR (WRB/GET1, 21q22); and SNU13 alt‐TSS DMR (SNU13/NHP2L1, 22q13).
Array CGH reported a karyotype 46:XY with benign CNVs only. PTEN testing by gene panel and MLPA analysis, and Noonan syndrome testing by gene panel analysis, reported no pathogenic variants. Results of imprinting analysis are presented in Table 2. MS‐MLPA detected partial loss of DNA methylation at the KCNQ1OT1 TSS DMR and MEG3 TSS DMR, with no copy number change at either locus. MS‐PCR confirmed these results and additionally detected partial hypomethylation of DIRAS3 Ex2 DMR, IGF2R Int2 DMR, and WRB alt‐TSS DMR (WRB/GET1), as well as complete hypomethylation of SNU13 alt‐TSS DMR. These results were consistent with a diagnosis of MLID.
TABLE 2.
Results of DNA methylation analysis at imprinted loci
| Proper name | Informal name | Methylation index (SD) MS‐MLPA | Methylation index (SD) MS‐PCR |
|---|---|---|---|
| DIRAS3 TSS DMR | DIRAS3 | nd | 0.2 (−20) |
| PLAGL1 TSS alt‐DMR | PLAGL1 | 0.57 (0.1) | 0.53 (0.7) |
| IGF2R Int2 DMR | IGF2R | nd | 0.33 (−4.5) |
| GRB10 alt‐TSS DMR | GRB10 | 0.54 (−0.7) | 0.45 (−2.5) |
| MEST alt‐TSS DMR | MEST | 0.56 (0.1) | 0.5 (0) |
| H19 TSS DMR | H19 | 0.51 (−1.3) | 0.47 (−1.4) |
| IGF2 alt‐TSS DMR | IGF2 DMR0 | 0.51 (0.47) | |
| KCNQ1OT1 TSS DMR | KCNQ1OT1 | 0.42 (−6.8) | 0.32 (−26) |
| MEG3 TSS DMR | MEG3 | 0.37 (−7.5) | 0.32 (−9.5) |
| MEG8 Int2 DMR | MEG8 | 0.72 (4.7) | nd |
| SNURF TSS DMR | SNRPN | 0.57 (−1) | 0.52 (1.4) |
| IGF1R Int2 DMR | IGF1R | nd | 0.51 (0.16) |
| PEG3 TSS DMR | PEG3 | 0.55 (0.5) | 0.47 (−1.5) |
| GNAS‐NESP TSS DMR | NESP55 | 0.59 (0.7) | nd |
| GNAS‐AS1 TSS DMR | GNAS‐AS1 | 0.56 (0.2) | 0.54 (1.55) |
| GNAS‐XL Ex1 DMR | GNAS‐XL | 0.55 (1.3) | nd |
| GNAS A/B TSS DMR | GNAS A/B | 0.54 (0.6) | nd |
| WRB alt‐TSS DMR | WRB | nd | 0.32 (−8.5) |
| NHP2L1 alt‐TSS DMR | NHP2L1 | nd | 0 (–) |
Note: The table presents the results of DNA methylation analysis by MS‐MLPA and MS‐PCR. The MS‐MLPA results presented are those from ME034‐C1. Results are presented as methylation indices, and SD represents the number of standard deviations from five normal controls assayed in the same experiment. Bold values indicate results >3SD from normal controls.
Abbreviations: MS‐MLPA, methylation‐specific MLPA; MS‐PCR, methylation‐specific PCR; nd, not done.
3. DISCUSSION
Previous studies of patients found to have hypomethylation affecting multiple loci suggest that clinical phenotypes of patients with MLID do not map neatly onto a single clinical disorder, and the involvement of additional loci may alter the extent or severity of the phenotype (Poole et al., 2013). There is some evidence that the dominant phenotype may be determined by the locus with the most severe methylation abnormality or its prevalence in a target organ where mosaicism occurs (Azzi et al., 2014). Epigenotypes conferring clinically opposing effects could theoretically “balance” each other in the resultant phenotype, but this has not been proved, and can only be deduced from epigenotype‐phenotype correlations.
This is the first clinical case report of an individual with clinical features of two IDs, BWS and TS, and MLID involving the loci associated with these disorders: KCNQ1OT1: TSS‐DMR (ICR2) and MEG3: TSS‐DMR. Although LOM at these two loci has been described previously (Begemann et al., 2018; Caliebe et al., 2014; Cubellis et al., 2020), the affected patients had MLID involving loci of numerous IDs, and therefore the contribution of each locus to the phenotype was unclear. Furthermore, clinical features reported were minimal. In the patient reported here, the only disease‐associated loci are ICR2 and MEG3 and the role, if any, of the other epimutations remains to be found.
The proband has phenotypic features of both BWS and TS (Figure 2, Table 1). Relative macrocephaly is seen in both TS and BWS, which could explain the patient's excessive head circumference. His body mass index was over the 99th centile at age 2: this may be the consequence of BWS in a person with relatively short stature, or conversely, it could reflect excessive food intake for someone with TS. He had a prominent nevus flammeus, a minor sign of BWS (Table 1), but few of the signs of overgrowth classically seen in BWS (e.g., no macroglossia and a normal birth weight). His facial features were more typical of TS than BWS, but his hands and feet were not as small as typically found in TS.
The idea that different phenotypic features could “offset” each other raises intriguing questions about whether the resultant phenotype might not meet clinical scoring criteria and thereby elude diagnosis. Furthermore, the involvement of additional epigenetic mutations at loci with no known phenotype may contribute to the overall clinical presentation. As an example, our patient walked at 17 months, arguably on the cusp of mild developmental delay, and had confirmed speech and language delay. While this is insufficient to be considered unambiguous evidence of learning difficulties, it is interesting given that a previous cohort of patients with BWS in combination with MLID were more likely to have developmental delay than those with “isolated” BWS (Poole et al., 2013).
There was no evidence of a cis‐acting genetic defect underlying any of the epimutations detected. The fact that there had been no prior pregnancies, and several years of infertility, raises the possibility that oocyte quality was compromised, either through maternal mutations or environmental factors. Notably, the epigenetic features of the patient suggested that the imprinting errors occurred post‐zygotically: both maternally and paternally methylated germline DMRs were affected, ruling out a developmental defect specific to either the oocyte or the sperm, while the patient's mosaicism was inconsistent with an imprinting error present in the germline or the one‐cell zygote. These features have been described in cases of MLID associated with maternal‐effect mutations (e.g., Begemann, 2018; Caliebe et al., 2014; Cubellis et al., 2020).
As the core set of tested loci expands and more data are gathered regarding epigenotype‐phenotype correlations, the conventional paradigm of a clinical diagnosis confirmed with a molecular test may no longer be the best way to capture patients whose clinical features are nuanced or conflicting. This case illustrates how a molecular diagnosis of MLID can alter the clinical outcome and inform the health surveillance required for the patient. Clinical features such as excessive weight gain and small stature appear to counterbalance each other, and the patient will be managed for both BWS and TS. Continued surveillance of individuals with MLID will determine whether the affected loci influence their phenotype over the long term.
In summary, we describe a patient with clinical and epigenetic features of both TS and BWS, who illustrates how the clinical phenotype of MLID may depend on the imprinted loci involved. We contend that patients whose emergent phenotype is not associated with classical phenotypes may escape diagnosis and suggest that such patients may benefit from molecular testing of multiple loci, agnostic to clinical definitions of classical IDs.
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
ACKNOWLEDGMENTS
The authors thank the patient and his family for their cooperation with the preparation of this manuscript. They are grateful to Dr Gabriella Gazdagh, Wessex Clinical Genetics Service, for performing Face2Gene analysis. Sarah E. Grosvenor acknowledges the support of the MSc Genomic Medicine Programme at Southampton University. NIHR CRN: Wessex and the NIHR Southampton Clinical Research Facility supported the IDFOW study. I. Karen Temple was supported in part by the Southampton NIHR Biomedical Research Centre, UK (2017–2022; IS‐BRC‐1215‐20004). This work was supported in part by the Child Growth Foundation (grant 519026101), University of Southampton.
Grosvenor, S. E. , Davies, J. H. , Lever, M. , Sillibourne, J. , Mackay, D. J. G. , & Temple, I. K. (2022). A patient with multilocus imprinting disturbance involving hypomethylation at 11p15 and 14q32, and phenotypic features of Beckwith‐Wiedemann and Temple syndromes. American Journal of Medical Genetics Part A, 188A:1896–1903. 10.1002/ajmg.a.62717
Funding information Child Growth Foundation, UK, Grant/Award Number: 519026101; NIHR Southampton Clinical Research Facility and CRN Wessex; Southampton NIHR Biomedical Research Centre (2017‐2022), Grant/Award Number: IS‐BRC‐1215‐20004
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- Arima, T. , Kamikihara, T. , Hayashida, T. , Kato, K. , Inoue, T. , Shirayoshi, Y. , Oshimura, M. , Soejima, H. , Mukai, T. , & Wake, N. (2005). ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith‐Wiedemann syndrome. Nucleic Acids Research, 33(8), 2650–2660. 10.1093/nar/gki555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi, S. , Blaise, A. , Steunou, V. , Harbison, M. D. , Salem, J. , Brioude, F. , Rossignol, S. , Habib, W. A. , Thibaud, N. , Neves, C. D. , Jule, M. L. , Brachet, C. , Heinrichs, C. , Bouc, Y. L. , & Netchine, I. (2014). Complex tissue‐specific epigenotypes in Russell‐Silver syndrome associated with 11p15 ICR1 hypomethylation. Human Mutation, 35(10), 1211–1220. 10.1002/humu.22623 [DOI] [PubMed] [Google Scholar]
- Begemann, M. , Rezwan, F. I. , Beygo, J. , Docherty, L. E. , Kolarova, J. , Schroeder, C. , Buiting, K. , Chokkalingam, K. , Degenhardt, F. , Wakeling, E. L. , Kleinle, S. , González Fassrainer, D. , Oehl‐Jaschkowitz, B. , Turner, C. L. S. , Patalan, M. , Gizewska, M. , Binder, G. , Bich Ngoc, C. T. , Chi Dung, V. , … Mackay, D. J. G. (2018). Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. Journal of Medical Genetics, 55, 497–504. 10.1136/jmedgenet-2017-105190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bens, S. , Kolarova, J. , Beygo, J. , Buiting, K. , Caliebe, A. , Eggermann, T. , Gillessen‐Kaesbach, G. , Prawitt, D. , Thiele‐Schmitz, S. , Begemann, M. , Enklaar, T. , Gutwein, J. , Haake, A. , Paul, U. , Richter, J. , Soellner, L. , Vater, I. , Monk, D. , Horsthemke, B. , … Siebert, R. (2016). Phenotypic spectrum and extent of DNA methylation defects associated with multilocus imprinting disturbances. Epigenomics, 8(6), 801–816. 10.2217/epi-2016-0007 [DOI] [PubMed] [Google Scholar]
- Brioude, F. , Kalish, J. M. , Mussa, A. , Foster, A. C. , Bliek, J. , Ferrero, G. B. , Boonen, S. E. , Cole, T. , Baker, R. , Bertoletti, M. , Cocchi, G. , Coze, C. , De Pellegrin, M. , Hussain, K. , Ibrahim, A. , Kilby, M. D. , Krajewska‐Walasek, M. , Kratz, C. P. , Ladusans, E. J. , … Maher, E. R. (2018). Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nature Reviews Endocrinology, 14(4), 229–249. 10.1038/nrendo.2017.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliebe, A. , Richter, J. , Ammerpohl, O. , Kanber, D. , Beygo, J. , Bens, S. , Haake, A. , Jüttner, E. , Korn, B. , Mackay, D. J. G. , Martin‐Subero, J. I. , Nagel, I. , Sebire, N. J. , Seidmann, L. , Vater, I. , von Kaisenberg, C. S. , Temple, I. K. , Horsthemke, B. , Buiting, K. , & Siebert, R. (2014). A familial disorder of altered DNA‐methylation. Journal of Medical Genetics, 51(6), 407–412. 10.1136/jmedgenet-2013-102149 [DOI] [PubMed] [Google Scholar]
- Cubellis, M. V. , Pignata, L. , Verma, A. , Sparago, A. , del Prete, R. , Monticelli, M. , Calzari, L. , Antona, V. , Melis, D. , Tenconi, R. , Russo, S. , Cerrato, F. , & Riccio, A. (2020). Loss‐of‐function maternal‐effect mutations of PADI6 are associated with familial and sporadic Beckwith‐Wiedemann syndrome with multi‐locus imprinting disturbance. Clinical epigenetics., 12(1), 139. 10.1186/s13148-020-00925-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann, T. , Heilsberg, A.‐K. , Bens, S. , Siebert, R. , Beygo, J. , Buiting, K. , Begemann, M. , & Soellner, L. (2014). Additional molecular findings in 11p15‐associated imprinting disorders: An urgent need for multi‐locus testing. Journal of Molecular Medicine (Berlin, Germany), 92(7), 769–777. 10.1007/s00109-014-1141-6 [DOI] [PubMed] [Google Scholar]
- Elbracht, M. , Mackay, D. , Begemann, M. , Kagan, K. O. , & Eggermann, T. (2020). Disturbed genomic imprinting and its relevance for human reproduction: Causes and clinical consequences. Human Reproduction Update, 26(2), 197–213. 10.1093/humupd/dmz045 [DOI] [PubMed] [Google Scholar]
- Gillessen‐Kaesbach, G. , Albrecht, B. , Eggermann, T. , Elbracht, M. , Mitter, D. , Morlot, S. , van Ravenswaaij‐Arts, C. , Schulz, S. , Strobl‐Wildemann, G. , Buiting, K. , & Beygo, J. (2018). Molecular and clinical studies in 8 patients with Temple syndrome. Clinical Genetics, 93(6), 1179–1188. 10.1111/cge.13244 [DOI] [PubMed] [Google Scholar]
- Ioannides, Y. , Lokulo‐Sodipe, K. , Mackay, D. J. G. , Davies, J. H. , & Temple, I. K. (2014). Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. Journal of Medical Genetics, 51(8), 495–501. 10.1136/jmedgenet-2014-102396 [DOI] [PubMed] [Google Scholar]
- Kagami, M. , Matsubara, K. , Nakabayashi, K. , Nakamura, A. , Sano, S. , Okamura, K. , Hata, K. , Fukami, M. , & Ogata, T. (2017). Genome‐wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami–Ogata syndrome. Genetics in Medicine, 19(4), 476–482. 10.1038/gim.2016.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami, M. , Nagasaki, K. , Kosaki, R. , Horikawa, R. , Naiki, Y. , Saitoh, S. , Tajima, T. , Yorifuji, T. , Numakura, C. , Mizuno, S. , Nakamura, A. , Matsubara, K. , Fukami, M. , & Ogata, T. (2017). Temple syndrome: Comprehensive molecular and clinical findings in 32 Japanese patients. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(12), 1356–1366. 10.1038/gim.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, D. J. , Callway, J. L. , Marks, S. M. , White, H. E. , Acerini, C. L. , Boonen, S. E. , Dayanikli, P. , Firth, H. V. , Goodship, J. A. , Haemers, A. P. , Hahnemann, J. M. D. , Kordonouri, O. , Masoud, A. F. , Oestergaard, E. , Storr, J. , Ellard, S. , Hattersley, A. T. , Robinson, D. O. , & Temple, I. K. (2008). Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nature Genetics, 40, 949–951. [DOI] [PubMed] [Google Scholar]
- Mackay, D. J. G. , & Temple, I. K. (2017). Human imprinting disorders: Principles, practice, problems and progress. European Journal of Medical Genetics, 60(11), 618–626. 10.1016/j.ejmg.2017.08.014 [DOI] [PubMed] [Google Scholar]
- Monk, D. , Mackay, D. J. G. , Eggermann, T. , Maher, E. R. , & Riccio, A. (2019). Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nature Reviews Genetics, 20(4), 235–248. 10.1038/s41576-018-0092-0 [DOI] [PubMed] [Google Scholar]
- Mussa, A. , Russo, S. , De Crescenzo, A. , Chiesa, N. , Molinatto, C. , Selicorni, A. , Richiardi, L. , Larizza, L. , Silengo, M. C. , Riccio, A. , & Ferrero, G. B. (2013). Prevalence of Beckwith–Wiedemann syndrome in north west of Italy. American Journal of Medical Genetics: Part A, 161A, 2481–2486. 10.1002/ajmg.a.36080 [DOI] [PubMed] [Google Scholar]
- Poole, R. L. , Docherty, L. E. , Al Sayegh, A. , Caliebe, A. , Turner, C. , Baple, E. , Wakeling, E. , Harrison, L. , Lehmann, A. , Temple, I. K. , Mackay, D. J. G. , & International Clinical Imprinting Consortium . (2013). Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. American Journal of Medical Genetics: Part A, 161A(9), 2174–2182. 10.1002/ajmg.a.36049 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Delgado, M. , Riccio, A. , Eggermann, T. , Maher, E. R. , Lapunzina, P. , Mackay, D. , & Monk, D. (2016). Causes and consequences of multi‐locus imprinting disturbances in humans. Trends in Genetics, 32(7), 444–455. 10.1016/j.tig.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Wakeling, E. L. , Brioude, F. , Lokulo‐Sodipe, O. , O'Connell, S. M. , Salem, J. , Bliek, J. , Canton, A. P. M. , Chrzanowska, K. H. , Davies, J. H. , Dias, R. P. , Dubern, B. , Elbracht, M. , Giabicani, E. , Grimberg, A. , Grønskov, K. , Hokken‐Koelega, A. C. S. , Jorge, A. A. , Kagami, M. , … Netchine, I. (2017). Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nature Reviews Endocrinology, 13(2), 105–124. 10.1038/nrendo.2016.138 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors.