

Abstract
Mathematical models, such as physiologically‐based pharmacokinetic models, are used to predict, for example, drug disposition and toxicity. However, populations differ in the abundance of proteins involved in these processes. To improve the building and refinement of such models, they must take into account these interindividual variabilities. In this study, we used global proteomics to characterize the protein composition of jejunum and liver from 37 donors with obesity enrolled in the COCKTAIL study. Liver protein levels from the 37 donors were further compared with those from donors without obesity. We quantified thousands of proteins and could present the expression of several drug‐metabolizing enzymes, for the first time, in jejunum, many of which belong to the cytochrome P450 (CYP) (e.g., CYP2U1) and the amine oxidase (flavin‐containing) (e.g., monoamine oxidase A (MAOA)) families. Although we show that many metabolizing enzymes had greater expression in liver, others had higher expression in jejunum (such as, MAOA and CES2), indicating the role of the small intestine in extrahepatic drug metabolism. We further show that proteins involved in drug disposition are not correlated in the two donor‐matched tissues. These proteins also do not correlate with physiological factors such as body mass index, age, and inflammation status in either tissue. Furthermore, the majority of these proteins are not differently expressed in donors with or without obesity. Nonetheless, interindividual differences were considerable, with implications for personalized prediction models and systems pharmacology.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Protein quantifications in human liver and small intestine have targeted specific proteins, in small sample sizes and used membrane enrichment. These data are inadequate for building and refining accurate physiologically‐based prediction models.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We compared the proteomes of matched human jejunum and liver from 37 donors, at a global level—comparing all quantified proteins—and at an absorption, distribution, metabolism, and excretion–specific level.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ We provide a large data set of donor‐matched protein levels for human jejunum (6,398) and liver (5,773) from 37 donors. Expression levels of several drug‐metabolizing enzymes are presented for the first time, for human jejunum, and compared with the levels in liver from the same donors. The high expression of several of these jejunal enzymes suggests this organ has an important role in extrahepatic drug metabolism.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The proteomics data from the 37 donors revealed large interindividual differences in protein levels. Such differences can greatly influence models used for predicting drug disposition or toxicity. Knowledge of these differences will contribute to developing personalized systems pharmacology.
Mathematical models, such as physiologically‐based pharmacokinetic models, are frequently applied to predict drug disposition, efficacy, and toxicity in humans. These models rely on accurate translation of experimental in vitro data to in vivo. 1 Such translation has been successfully performed with scaling factors based on specific protein concentrations in the in vitro model and target tissue. 2 , 3 To build and refine predictive models, they must take into account interindividual variability. 1 Information regarding the protein composition of tissues involved in drug disposition across different populations is therefore imperative.
The small intestine—comprised of the duodenum, jejunum, and ileum—and liver are important organs for nutritional absorption and digestion, as well as drug disposition. Recent advancements in mass spectrometry–based proteomics analysis have mapped proteomes in many tissues, including the small intestine and liver. 4 However, these studies have focused on the comparison of many different tissue types from few and unmatched donors, rather than the protein profiles of specific tissues with respect to interindividual variability. Furthermore, when proteins have been quantified from the small intestine and liver of small sets of matching donors (n = 9), focus has been on a set of enzymes 5 and transporters 6 that is currently considered clinically relevant for drug disposition.
Other proteomics studies on the jejunum—the major drug absorption site—have quantified selected proteins involved in drug disposition 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 and, again, in a small number of donors. 7 , 8 , 10 , 11 , 12 , 14 Furthermore, many of these studies used membrane enrichment procedures, 9 , 10 , 11 , 12 , 13 which introduces variability to the protein quantification. 15 , 16 , 17 For example, a global proteomics study of mucosal fractions of jejunum identified up to 5,700 proteins, 7 but it only included four donors, which does not capture the interindividual variability. Meanwhile, studies of the jejunum from larger sample donor sets (up to 28 donors), have only targeted specific drug‐metabolizing enzymes and transporters. 9
The liver proteome has been more studied. Although membrane enrichment has also been used for liver protein quantification, 13 , 18 , 19 , 20 other studies have quantified proteins in whole liver tissue lysates, 5 , 6 , 15 , 16 , 21 both on a global level 15 , 16 and for selected drug disposition proteins. 5 , 6 , 21 However, as with the jejunum studies, sample sizes have been small (up to 15 donors) in whole liver lysate. 5 , 6 , 15 , 16 , 21 Larger sample sets (up to 39 donors) have been analyzed in membrane‐enriched liver microsomes. 13 , 19
In this study, we quantified the global proteomes of matched whole lysate jejunum and liver obtained from 37 donors undergoing gastric bypass surgery. These patients were enrolled in the COCKTAIL (Impact of Body Weight and Weight Loss on Drug Bioavailability, Cardiovascular Risk Factors and Metabolic Biomarkers) study. 22 A primary objective in COCKTAIL was to investigate the relationship between body composition and jejunum and liver protein expression and activity of proteins involved in the absorption, distribution, metabolism, and excretion (ADME) of drugs. The proteomics data on these donor‐matched tissues enabled us to (i) perform an individual comparison of the global protein composition of the two organs; (ii) capture interindividual variability of protein levels involved in ADME in each tissue; and (iii) study how these levels relate to individual biological factors, such as body mass index (BMI), inflammation, age, and sex.
METHODS
Human jejunal and liver tissue
Jejunal and liver biopsies were obtained from 38 patients with obesity undergoing gastric bypass surgery. Additionally, liver biopsies were also obtained from 17 lower weight controls undergoing cholecystectomy as part of the study. Patients gave informed consent as part of the COCKTAIL study approved by the Regional Committee for Medical and Health Research Ethics (Ref: 2013/2379/REKsørøst A). For jejunum, pinch biopsies of the intestinal mucosa were collected 60 cm distal to the ligament of Treitz. For liver, true‐cut biopsies were obtained from parenchyma close to the edge of the right liver lobule. Biopsies were snap frozen in liquid nitrogen directly upon sampling and stored at −80°C until analysis. 22
Protein quantification
Biopsies were thawed and lysed in 2% sodium dodecyl sulfate, and the proteins were denatured at 95°C. Samples were prepared for proteomic analysis with the multienzyme digestion filter‐aided sample preparation protocol, using endoproteinase LysC and trypsin. 23 Total protein and peptide amounts were determined based on tryptophan fluorescence. 24 Peptides were separated on an EASY‐spray C18‐column (50 cm, 75 µm inner diameter), using an acetonitrile/water gradient (0.1% formic acid) at 300 nL/min. The nano–liquid chromatography (nLC) was coupled to a Q Exactive HF or Q Exactive HF‐X (Thermo Fisher Scientific, Waltham, MA). Mass spectrometry (MS) data were processed with MaxQuant (https://www.maxquant.org/), 25 using the human UniProtKB (https://www.uniprot.org/). Spectral raw intensities were normalized with variance stabilization. 26 Protein abundances (fmol/µg protein) were calculated with the total protein approach. 27 See Supplementary Methods for details.
Clinical parameters
Body weight and body composition were determined using the Inbody 720, Body Composition Analyzer (Biospace, Seoul, South Korea). Clinical laboratory analyses were performed at the Central Laboratory, Vestfold Hospital Trust and at the Hormone Laboratory, Oslo University Hospital (Norway). Plasma concentrations of high‐sensitivity C‐reactive protein (hs‐CRP) were measured with immunoturbidimetry (Advia Chemistry XPT systems, Siemens, Munich, Germany) at Fürst Medical Laboratory (Oslo, Norway). Nonalcoholic fatty liver disease (NAFLD) scores were calculated based on fasting serum levels of aspartate aminotransferase, alanine amino transferase, and insulin, together with if the donors had type 2 diabetes (T2D) and/or metabolic syndrome. 28
Statistical analysis
Biopsies were collected over 2 years and analyzed in three batches (on average 10 months after collection). Batch effects were removed by geometric mean‐centering of protein levels. Initial evaluation of the proteomics data with principal component analysis showed deviating results in the jejunum of one patient (most likely due to temporary technical fluctuation in the nLC‐MS/MS analysis). Therefore, both jejunum and liver data from this patient were excluded from the pairwise analysis of the proteomes, leaving data from 37 donor‐matched jejunum and liver samples for further analysis. To ensure high‐quality quantification, only proteins identified with at least three unique + razor peptides were considered to be quantified. Those that were quantified in at least 50% of the samples (n = 19) were included in the global comparative analyses. Functional annotation clustering of gene ontology (GO) enrichments of GO terms molecular function and biological processes was performed in R (https://www.r‐project.org/) with the clusterProfiler‐package. 29 The background set of proteins from which GO‐enrichments were calculated was set to all the proteins identified in the jejunum and liver samples. Spearman rank correlation coefficients were determined from protein abundances in at least 50% of the 37 donors (n = 19) for all proteins in the respective tissues (used for between‐tissue intercorrelation of proteins, and protein‐clinical parameter correlation). P values were adjusted for multiple comparisons with Benjamini‐Hochberg. Significance differences in protein abundances due to sex and glucose tolerance were performed in GraphPad Prism (version 9.0.0; GraphPad Software, San Diego, CA) with the Mann‐Whitney nonparametric test, adjusted for multiple comparison with False Discovery Rate and two‐way analysis of variance followed by the Tukey multiple comparison test, respectively.
RESULTS
Patient demographics
Inclusion criteria for COCKTAIL are described elsewhere. 22 At the time of surgery, the 37 donors included in the proteomics analysis (see the Method section for exclusion) had a median (range) weight of 123 (79–155) kg, and corresponding median (range) BMI of 43 (30–51) kg/m2 (Figure 1a ). All donors were White with a median (range) age of 48 (23–63) years. Two‐thirds of the donors were female (n = 25), and 35% of donors (n = 13) had T2D (Table S1 ).
Figure 1.
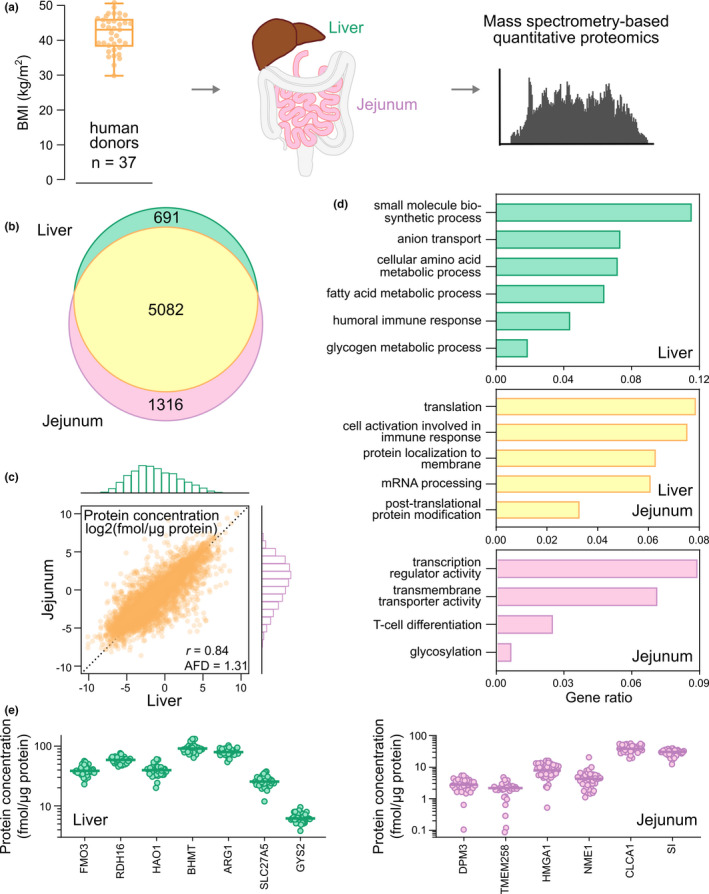
Global proteomics analysis of liver and jejunal biopsies from 37 donors undergoing gastric bypass surgery. (a) BMI distribution of the 37 donors and schematic overview of quantitative proteomics analysis. (b) Venn diagram of jejunal and liver proteins. (c) Correlation and distribution of protein levels in jejunal and liver biopsies (geometric mean protein level from the 37 donors). (d) Molecular function and biological processes from ClusterProfiler 29 of proteins specific to jejunum and liver, or overlapping proteins in the two tissues. (e) Protein level distribution of representative proteins found specifically in jejunal or liver biopsies from the 37 donors. AFD, average fold difference (jejunal protein levels/liver levels); 59 BMI, body mass index; mRNA, messenger RNA; r: Pearson’s correlation coefficient calculated from log2‐transformed protein levels.
Global proteomes of human jejunum and liver
In total, 6,398 jejunal proteins and 5,773 liver proteins were quantified with an overlap of 5,082 proteins between the two tissues (88% of the liver proteins and 79% of the jejunal ones; Figure 1b ). There were slightly higher levels of the overlapping proteins in jejunum than in liver, with an average fold difference (AFD) of 1.3 (95% confidence interval (CI), 1.2–1.4; Figure 1c ). These matching proteins were involved in basic cellular processes, such as transcription (e.g., “nucleoporin proteins” and “heterogeneous nuclear ribonucleoproteins”), protein translation and localization (e.g., proteins from the small and large subunits of the ribosome, and “eukaryotic initiation factor proteins”), and post‐translational protein modification (e.g., “proteasome subunit proteins”) (Figure 1d ; Data S1 ).
Proteins found only in jejunum (n = 1,316), were involved in biological processes connected to transcription (e.g., HMGA1 and NME1), protein glycosylation (e.g., DPM3 and TMEM258), and absorption (e.g., CLCA1 and SI). These proteins had median abundances ranging from 1.7 (TMEM258) to 38.4 (CLCA1) fmol/µg protein (Figure 1e ) for the 37 donors.
In contrast, proteins found only in liver (n = 691), were involved in liver specific functions, such as small molecule and drug metabolism (e.g., FMO3 and CYP2A6), fatty acid and lipid metabolism (e.g., RDH16 and HAO1), amino acid metabolism (e.g., BHMT and ARG1), glycogen metabolism (e.g., GYS2), and transport (e.g., SLC27A5 and SLCO1B1). Their median abundances ranged from 2.6 (SLCO1B1) to 91.6 (BHMT) fmol/µg protein (Figure 1e ).
ADME‐related proteins in human jejunum and liver
As the jejunum and liver are key contributors to drug disposition, we next focused the analysis on 682 selected proteins related to ADME (a list compiled by Schröder et al., 30 ). With our set criteria, 321 ADME‐related proteins were quantified in jejunum, and 328 in liver. These proteins overlapped to 80% (272) for the two tissues (Figure 2a ), and constituted many enzymes and transcription factors (Figure 2c ).
Figure 2.
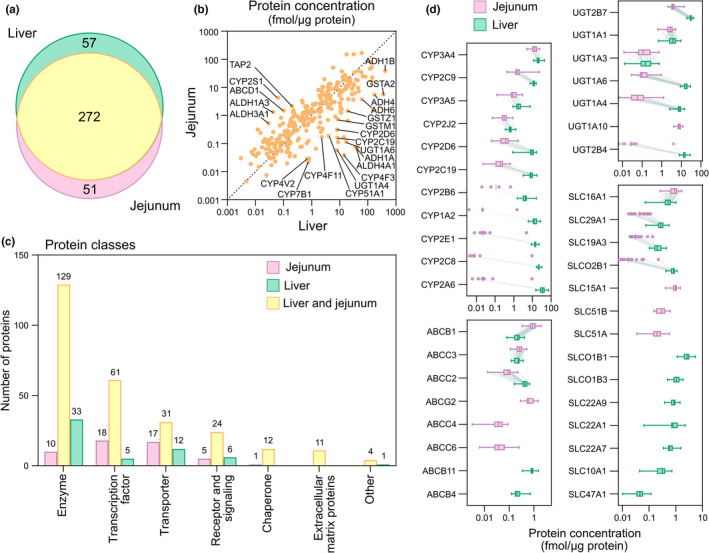
Proteomics analysis focused on proteins involved in drug disposition (ADME (absorption, distribution, metabolism, and elimination)) in human jejunal and liver biopsies from 37 donors undergoing gastric bypass surgery. (a) Venn diagram of jejunal and liver proteins. (b) Correlation of proteins quantified in jejunal and liver biopsies (geometric mean protein levels from the donors). (c) Protein classes of drug disposition proteins specific to jejunum and liver, or overlapping proteins in the two tissues. (d) Protein levels of major drug‐metabolizing enzymes and transporting proteins in the jejunal and liver biopsies. Box plot displays median, 25th, and 75th percentile, and whiskers denote minimum and maximum values. Dots are displayed instead of box plot for proteins represented in fewer than 50% (n = 19) of the donors.
Abundances of ADME‐related proteins were similar in jejunum and liver with an AFD of 0.88 (95% CI, 0.3–1.5), and generally higher levels in liver (Data S1 ). Notably, several enzymes had more than 10‐fold higher levels in liver. These included cytochromes P450 (CYPs) (CYP2C19, CYP4F3, CYP4F11, CYP4V2, CYP7B1, and CYP51A1), alcohol dehydrogenases (ADH1A, ADH1B, ADH4, ADH6), an aldehyde dehydrogenase (ALDH4A1), uridine diphosphate (UDP)–glucuronosyltransferases (UGT1A4, UGT1A6), and glutathione S‐transferases (GSTM1, GSTZ1; Figure 2b ). In contrast, the enzymes CYP2S1, ALDH1A3, and ALDH3A1, and the adenosine triphosphate (ATP)–binding cassette transporters, ABCD1 and TAP2 (ABCB3), were more than 10‐fold higher in jejunum (Figure 2b ).
In general, no correlations were found between ADME‐related protein levels expressed in the two tissues. Among the 272 matching proteins, only six were significantly correlated (GSTM4, SULT1A1, CYP4F12, GSTT1, CYP2D6, and GSTM3; with Rs > 0.54 and P value < 0.05 after Benjamini‐Hochberg adjustment for multiple comparisons; Figure S1a,b; Data S1 ).
Phase I drug‐metabolizing enzymes
The ADME‐related proteins contained several groups of important phase I drug‐metabolizing enzymes in both tissues. In jejunum, these included, among others, 27 CYPs, 6 ADHs, 17 ALDHs, 4 dimethylaniline monooxygenases (N‐oxide‐forming) (previously known as flavin‐containing monooxygenases; FMOs), 5 arylamine N‐acetyltransferases (NAT), and the 2 amine oxidases (flavin‐containing), MAOA and MAOB. In liver, 32 CYPs (26 of the 27 also found in jejunum, and 6 additional ones), 7 ADHs (1 additional to those found in jejunum), 16 ALDHs, 4 FMOs, 3 NATs, and MAOA and MAOB were quantified (Table 1 ; Data S1 ).
Table 1.
Protein levels of CYP enzymes and auxiliary proteins in jejunum and liver
| Jejunum | Liver | |||
|---|---|---|---|---|
| Median (min–max) | N | Median (min–max) | N | |
| CYP3A4 | 13.4 (5–25.3) | 37 | 21.2 (11.8–44.3) | 37 |
| CYP27A1 | 5.9 (4–10.2) | 37 | 10.2 (7.8–12.2) | 37 |
| CYP4F2 | 5.4 (1.6–15.5) | 37 | 1.9 (0.6–4.2) | 37 |
| CYP2S1 | 4.5 (1.6–7.5) | 37 | 0.1 (0–0.2) | 37 |
| CYP2C9 | 1.5 (0.4–23.5) | 37 | 12.3 (7.5–16.2) | 37 |
| CYP3A5 | 0.9 (0.1–3) | 37 | 1.8 (0.9–7.6) | 37 |
| CYP20A1 | 0.8 (0.5–1.4) | 37 | 1.2 (0.7–2.6) | 37 |
| CYP2C18 | 0.5 (0.2–6) | 35 | 0.2 (0–0.5) | 37 |
| CYP4F12 | 0.5 (0.1–3.8) | 37 | 0.6 (0.1–1.4) | 37 |
| CYP2D6; CYP2D7 | 0.4 (0.1–1.7) | 32 | 9.6 (0.9–17) | 37 |
| CYP2J2 | 0.3 (0.1–0.9) | 37 | 0.6 (0.4–1.3) | 37 |
| CYP1A1 | 0.2 (0–3.1) | 35 | 0.1 (0–0.7) | 36 |
| CYP51A1 | 0.2 (0–2.9) | 36 | 4 (2.2–9.8) | 37 |
| CYP4F11 | 0.2 (0–0.6) | 30 | 2.4 (1–4.3) | 37 |
| CYP2C19 | 0.2 (0–0.6) | 34 | 8.1 (3.3–17.8) | 37 |
| CYP2B6 | 0.1 (0–0.7) | 4 | 4.1 (1.6–16.5) | 37 |
| CYP2U1 | 0.1 (0–0.9) | 28 | 0.1 (0–0.4) | 37 |
| CYP1A2 | 0.02 (0–1.5) | 3 | 12.4 (5.9–27.7) | 37 |
| CYP2A6 | 0.02 (0.01–9.6) | 7 | 35.3 (15.2–74.9) | 37 |
| CYP2E1 | 0.02 (0.01–4.8) | 7 | 14.7 (8.6–23.1) | 37 |
| CYP4F3 | 0.04 (0–0.4) | 21 | 14.2 (9.6–21.5) | 37 |
| CYP8B1 | 0 (0–2.2) | 3 | 9.4 (5.8–13.9) | 37 |
| CYP2C8 | 0.01 (0–9.5) | 5 | 22.3 (16.5–31.9) | 37 |
| CYP4V2 | 0.03 (0.01–0.1) | 23 | 0.7 (0.3–1.3) | 37 |
| CYP2W1 | 0.03 (0–0.1) | 19 | ND | |
| CYP7B1 | 0.02 (0.01–0.1) | 20 | 0.7 (0.2–1.8) | 37 |
| CYP4A11 | 0.01 (0–4.5) | 8 | 20.3 (11.8–32.3) | 37 |
| CYP4A22 | ND | 0.4 (0.2–1) | 37 | |
| CYP39A1 | ND | 0.1 (0–0.6) | 37 | |
| CYP3A7 | ND | 0.1 (0–1.2) | 32 | |
| CYP4F22 | ND | 0.03 (0.01–0.1) | 33 | |
| CYP7A1 | ND | 0.03 (0.01–0.1) | 18 | |
| CYP2A7 | ND | 0.01 (0–0.2) | 14 | |
| Auxiliary proteins | ||||
| CYB5A | 48.9 (11.2–86.5) | 37 | 118.1 (94.9–166.4) | 37 |
| POR | 6.7 (4.4–10.5) | 37 | 11.4 (8.1–16.4) | 37 |
Protein levels of CYP enzymes and auxiliary proteins in jejunal and liver biopsies from the 37 donors. Protein levels are given in fmol/µg protein. For proteins separated with “;” the specific isoforms could not be distinguished by the MaxQuant search engine due to large sequence overlap.
max, maximum; min, minimum; N , number of donors for which the protein was quantified. ND, not detected.
All of the major drug‐metabolizing CYP enzymes 31 were quantified in both tissues, and were usually higher in liver (AFD = 3.7; 95% CI, 2.7–4.8; Figure 2d ). Importantly, the least expressed CYP2B6, CYP1A2, CYP2A6, CYP2E1, and CYP2C8 were quantified only in less than 7 (18%) of the 37 jejunal biopsies, but in all 37 liver biopsies (Table 1 ). The auxiliary proteins NADPH‐cytochrome P450 reductase (POR) and cytochrome b5 (CYB5A) were found at twofold higher concentrations in liver. Similarly, ADH enzymes and ALDH enzymes were expressed 15‐fold and 2‐fold higher, respectively, in liver (Table S2 ; Table S3 ).
The FMO and NAT enzymes were low abundant in both jejunum and liver, with the exception of FMO3 and FMO5 in liver (38.6 and 10.9 fmol/µg protein). MAOA and MAOB had median levels of 29.3 and 9.0 fmol/µg protein in jejunum, respectively, and 15.1 and 27.1 fmol/µg protein in liver (Table S4 ).
Phase II drug‐metabolizing enzymes
In terms of phase II drug‐metabolizing enzymes, we found 14 UDP‐glucuronosyltransferase (UGT) isoforms, 16 glutathione S‐transferases (GSTs), 3 carboxylesterases (CESs), and 6 sulfotransferases (SULTs) in jejunum. Furthermore, 11 UGT isoforms, 16 GSTs (of which 15 were the same as in jejunum), 3 CESs, and 6 SULTs were quantified in liver (Table 2 ; Data S1 ).
Table 2.
Protein levels of UGT enzymes in jejunum and liver
| Jejunum | Liver | |||
|---|---|---|---|---|
| Median (min–max) | N | Median (min–max) | N | |
| UGT1A1 | 2.6 (0.6–5.1) | 37 | 3.8 (0.7–9.4) | 37 |
| UGT1A10 | 7.7 (4–12.1) | 37 | ND | |
| UGT1A3 | 0.1 (0–0.7) | 25 | 0.1 (0.01–0.7) | 37 |
| UGT1A4 | 0.1 (0–1.2) | 22 | 7.8 (2.6–14.8) | 37 |
| UGT1A6 | 0.1 (0–0.9) | 31 | 17.9 (9–29.2) | 37 |
| UGT2B4 | 0.03 (0.002–3.9) | 9 | 13.5 (7.9–28.4) | 37 |
| UGT2B7 | 3.6 (2.1–14) | 37 | 30.1 (19.2–42.9) | 37 |
| UGT1A8 | 0.02 (0.02–0.02) | 3 | ND | ND |
| UGT1A9; UGT1A6 | 2.1 (2.1–2.1) | 1 | ND | ND |
|
UGT1A9; UGT1A8; UGT1A10; UGT1A6 |
ND | ND | 2.5 (1.3–4.3) | 37 |
| UGT2A1 | 1.1 (0.4–2.9) | 37 | ND | ND |
| UGT2A3 | 7.7 (3.3–13.3) | 37 | 1.2 (0.5–2.4) | 37 |
| UGT2B10 | 0.1 (0.01–2.5) | 6 | 15.2 (9.1–23.5) | 37 |
| UGT2B15 | 0.04 (0.003–0.3) | 7 | 6.3 (3.9–9) | 37 |
| UGT2B17 | 18.5 (0.5–53.1) | 37 | 17.7 (11.8–32) | 37 |
| UGT3A1 | 0.02 (0.01–0.1) | 6 | 0.2 (0.03–0.5) | 37 |
Protein levels of UGT enzymes in jejunal and liver biopsies from the 37 donors. Protein levels are given in fmol/µg protein. For proteins separated with “;” the specific isoforms could not be distinguished by the MaxQuant search engine due to large sequence overlap.
max, maximum; min, minimum; N, number of donors for which the protein was quantified; ND, not detected; UGT, uridine diphosphate (UDP)–glucuronosyltransferase.
The quantified UGT isoforms 32 , 33 were 11‐fold higher in liver than jejunum (Figure 2d ; Table 2 ; Table S5 ). Furthermore, CES1 had 100‐fold higher levels in liver, while CES2 and CES3 had 3‐fold and 2‐fold higher levels in jejunum (Table S7 ). In contrast, nine GSTs had fourfold higher levels in liver, while six GSTs were fivefold higher in jejunum (Table S6 ). The SULTs had twofold higher levels in jejunum. Importantly, SULT1C2 and SULT2B1 were found only in jejunum (0.14 and 0.7 fmol/µg protein, respectively; Table S7 ). Moreover, thioredoxin (TXN) was 67.8 fmol/µg protein in jejunum and 35.7 fmol/µg protein in liver; intestinal‐type alkaline phosphatase (ALPI) was 8.1 fmol/µg protein in jejunum (Table S7 ).
Drug‐transporting proteins
Transporter proteins, such as ATP‐binding cassette (ABC) and solute carrier (SLC) transporter families were found in both tissues. Twenty of the 26 ABCs in jejunum overlapped with 23 in liver. For the SLC transporters, 158 were quantified in jejunum and 97 in liver. Of these, 76 were found only in jejunum, and 15 only in liver (Table 3 ; Data S1 ; Tables S8 and S9 ).
Table 3.
Protein levels of the major drug transporting ABC and SLC proteins in jejunum and liver
| Jejunum | Liver | |||
|---|---|---|---|---|
| Median (min–max) | N | Median (min–max) | N | |
| TAP2 (ABCB3) | 2.2 (0.7–4.3) | 37 | 0.2 (0.1–0.7) | 37 |
| TAP1 (ABCB2) | 1.5 (0.8–3.4) | 37 | 0.2 (0.1–0.8) | 37 |
| ABCB1 | 0.9 (0.3–1.9) | 37 | 0.2 (0.1–0.4) | 37 |
| ABCG2 | 0.6 (0.3–1.5) | 37 | ND | |
| ABCC3 | 0.3 (0.1–0.5) | 37 | 0.2 (0.1–0.4) | 37 |
| ABCC2 | 0.1 (0–0.2) | 37 | 0.4 (0.2–0.7) | 37 |
| ABCC4 | 0.04 (0.003–0.1) | 37 | ND | |
| ABCC6 | 0.04 (0.01–0.3) | 34 | ND | |
| ABCB11 | ND | 0.8 (0.3–1.5) | 37 | |
| ABCB4 | ND | 0.2 (0.1–0.7) | 37 | |
| SLC15A1 | 0.9 (0.4–1.5) | 37 | ND | |
| SLC16A1 | 0.8 (0.3–1.7) | 37 | 0.5 (0.1–1) | 37 |
| SLC19A3 | 0.04 (0.02–0.1) | 13 | ND | |
| SLC29A1 | 0.04 (0.02–0.1) | 17 | 0.3 (0.1–0.6) | 37 |
| SLC51A | 0.2 (0.03–0.6) | 37 | ND | |
| SLC51B | 0.3 (0.2–0.6) | 21 | ND | |
| SLCO2B1 | 0.02 (0.01–0.2) | 14 | 0.8 (0.4–1.1) | 37 |
| SLC22A1 | 0.5 (0.5–0.5) | 1 | 0.8 (0.1–2.3) | 37 |
| SLC22A7 | ND | 0.6 (0.3–1.5) | 37 | |
| SLC47A1 | ND | 0.05 (0.01–0.1) | 32 | |
| SLCO1B1 | ND | 2.6 (1.1–5.5) | 37 | |
| SLCO1B3 | ND | 1.1 (0.5–1.9) | 37 | |
Protein levels of the major drug transporting ABC and SLC proteins in jejunal and liver biopsies from the 37 donors. Protein levels are given in fmol/µg protein.
ABC, adenosine triphosphate (ATP)–binding cassette; max, maximum; min, minimum; N , number of donors for which the protein was quantified; ND, not detected; SLC, solute carrier.
The four most important enterocyte ABC drug transporters 34 (ABCC3, ABCB1, ABCC2, and ABCG2) were quantified to 0.26–0.60 fmol/µg protein in jejunum (Figure 2d ; Table 3 ). Of the eight most important liver ABC drug transporters, 34 five (ABCB1, ABCC2, ABCB11, ABCB4, and ABCC3) were quantified with median levels of 0.21–0.84 fmol/µg protein (Figure 2d ; Table 3 ). Importantly, the three nonquantified ABC transporters (ABCC4, ABCC6, and ABCG2) are known to be low abundant and thus difficult to quantify. 34 In line with this, ABCC6 was detected in only eight of the 37 liver biopsies, and then identified by only one peptide.
Of the 11 major enterocyte SLC drug transporters, 34 7 were quantified in jejunum (SLC15A1, SLC16A1, SLC19A3, SLCO2B1, SLC29A1, SLC51A, and SLC51B) with median levels ranging 0.02–0.91 fmol/µg protein (Figure 2d ; Table 3 ). Similarly, 8 of the 12 main liver SLC drug transporters 34 were quantified (SLC22A1, SLC22A7, SLC22A9, SLCO1B1, SLCO1B3, SLCO2B1, SLC29A1, and SLC47A1) to median levels of 0.05–2.6 fmol/µg protein. Importantly, the uptake transporter, SLC10A1, was identified with only two unique + razor peptides in liver (and thus below our stringent quantification criterion), but in all 37 donors, at a median level of 0.3 fmol/mg protein (Figure 2d ; Table 3 ).
Interindividual variability
In general, larger interindividual variability of drug‐metabolizing enzymes were found in jejunum than in liver. For example, CYP enzymes showed a median 34‐fold difference between maximum and minimum levels in jejunum and only 5‐fold difference in liver. Importantly, the polymorphic CYP2D6 showed a 27‐fold difference in jejunum and 19‐fold in liver (Figure 2d ; Table 1 ; Table S2 ). Similarly, in jejunum, ADH and ALDH showed on average 25‐fold and 17‐fold variabilities, respectively, and only on average 3‐fold and 7‐fold variability, respectively, in liver (Table S3 ). Although interindividual variability of phase II enzymes was also larger in jejunum, the differences between the two tissues were smaller: The interindividual variability for UGTs was 7‐fold in jejunum and 3‐fold in liver (Table 2 ; Table S5 ). Similarly, SULT‐differences were 11‐fold in jejunum and 6‐fold in liver (Table S7 ). However, CES showed larger differences between tissues: 24‐fold in jejunum and 2‐fold in liver (Table S7 ). In contrast, GST enzymes varied more in liver (10‐fold) than in jejunum (6‐fold; Table S6 ).
Similar interindividual variabilities were observed between the two tissues for both ABC and SLC transporters. ABC transporters showed on average 9‐fold and 6‐fold differences in jejunum and liver, respectively, and SLC transporters had average fold differences of 11 and 9 in jejunum and liver, respectively (Table 3 ; Tables S8 and S9 ).
Comparison with donors without obesity
We further compared our results of drug‐metabolizing enzymes and transporters (252 selected proteins) with levels from donors without obesity.
For liver, we compared with data from 17 lower weight donors undergoing cholecystectomy (Table S1 )—included as controls in the COCKTAIL study—combined with published data, 15 , 16 , 21 to increase the number of donors without obesity (N ≤ 49). We found 74 proteins with significantly different liver levels (P < 0.05 Mann‐Whitney test corrected for multiple comparisons with false discovery rate) between donors with and without obesity. However, the differences between the two groups (AFD of 1.2; 95% CI, 1–1.4) were smaller than the interindividual variability within each group (AFDs of 7.9 (95% CI, 7.4–8.5) and 17.4 (95% CI, 16.7–18.1) for donors with and without obesity, respectively). For example, while CYP2C9 levels were 1.6‐fold lower (P < 0.05) in donors with obesity, the interindividual variabilities were 2.2‐fold and 15‐fold in donors with and without obesity, respectively. Similarly, SLCO1B1 levels were 1.5‐fold (P < 0.05) higher in donors with obesity, and interindividual variabilities were 4.9‐fold and 21‐fold in donors with and without obesity, respectively (Figure 3a–d ; Data S1 ).
Figure 3.

Protein levels in liver from donors with obesity (n ≤ 37) and without obesity (from lower weight control group in COCKTAIL study and published data; 15 , 16 , 21 n ≤ 49) (a) CYP enzymes and auxiliary proteins, (b) UGT enzymes, (c) ABC transporters, and (d) SLC transporters important for drug metabolism and transport. Line, box, and whiskers represent median, 25th–75th percentile, and minimum‐maximum. The * represents significance P < 0.05 Mann‐Whitney test corrected for multiple comparisons with false discovery rate, between the two groups. ABC, adenosine triphosphate (ATP)–binding cassette; CYP, cytochrome P450; SLC, solute carrier; UGT, uridine diphosphate (UDP)–glucuronosyltransferase.
For jejunum, we also compared the levels of ABCs, CYPs, UGTs, and CESs (selection based on availability in the literature) with published data, 8 , 13 , 14 and found an AFD of 1.7 (95% CI, 0.9–2.5). As for the liver, the AFD between our and published data was lower than the interindividual variability in jejunum from donors with obesity for these proteins (AFD 9.6; 95% CI, 8.8–10.3; Tables S2 – S9 ).
Comparison with clinical data
Interindividual variations may alter levels of ADME‐related proteins, so we finally compared protein levels in jejunum and liver with sex, age, BMI, plasma levels of the inflammation marker hs‐CRP, and glycated hemoglobin A1c (HbA1c), the latter being a proxy for glucose tolerance/metabolic control. Liver protein levels were also compared with NAFLD scores. In the liver comparisons, data from both the 37 donors with obesity and the 17 lower weight controls were used.
In general, correlations were poor between the physiological parameters and protein concentrations in both tissues. In jejunum, the median rank order correlations for age, BMI, HbA1c, and hs‐CRP comparisons were Rs = −0.02, 0.02, 0.002, and 0.03, respectively (Rs 25th and 75th percentile: −0.10–0.16; Figure 4a ). Similarly, in liver, the median Rs for age, BMI, HbA1c, hs‐CRP, and NAFLD score comparisons were Rs = −0.02, 0.02, −0.01, 0.04, and 0.004, respectively (Rs 25th and 75th percentile: −0.13–0.15; Figure 4a ). Out of 1,639 comparisons, only 10 significant correlations were found: NAFLD‐score and liver proteins COL18A1, UGT1A3, GSR, PON1, SLC22A7, and TPMT; hs‐CRP and STAT3; Age and ABCB11 and PSMC5; and BMI and HMOX1 with absolute Rs between 0.48–0.58 and adjusted P values 0.014–0.036 (Figure S2 ). For more correlations see Data S1 .
Figure 4.

Correlation between clinical parameters and protein levels in jejunum and liver. (a) Distribution of Spearman rank correlation coefficients between clinical parameters and drug disposition proteins quantified in the jejunal biopsies from the 37 donors and liver biopsies from the 37 donors and 17 lower weight controls. The panel to the far right describes the comparisons between protein level and clinical parameter for the correlation calculations. (b) Distribution of drug disposition protein levels in donors with normal glucose tolerance (jejunum: n = 12; liver: n = 27), with prediabetes (jejunum: n = 12; liver: n = 14), and type 2 diabetes (jejunum and liver n = 13). (c) Distribution of drug disposition protein levels in males (jejunum: n = 12; liver: n = 15) and females (jejunum: n = 25; liver: n = 39). b,c, Line, box, and whiskers represent median, 25th–75th percentile, and minimum‐maximum. BMI, body mass index; HbA1c, glycated hemoglobin A1c; hs‐CRP, high‐sensitivity C‐reactive protein; NAFLD, nonalcoholic fatty liver disease; Rs , Spearman rank correlation coefficient.
In agreement with poor correlations between HbA1c values and protein levels, no significant differences in ADME‐related proteins were found in either tissue type for donors with “normal glucose tolerance” (HbA1c < 37.8 mmol/mol), “prediabetes” (HbA1c 37.8–46.5 mmol/mol), and “T2D” (HbA1c > 46.5 mmol/mol), (Figure 4b ).
Finally, no significantly different protein levels were found between sexes (n = 12 males and 25 females) in either tissue (Figure 4c ). However, jejunal proteins with the largest differences were ALDH1A2, CYP2W1, and the protein kinase CDK9, with 10‐fold, 2‐fold, and 2‐fold higher levels, respectively, in males. In contrast, FMO1, SULT1C2, GCLM, and SLC7A5 had threefold, threefold, twofold, and twofold higher jejunal levels, respectively, in females. In liver, ALDH1A2, BZ1B, and SLC47A1 had 1.8‐fold, 1.7‐fold, and 1.7‐fold higher levels in males, whereas CYP3A7 and CYP2U1 were 1.7‐fold and 1.6‐fold higher in females.
DISCUSSION
In this study, we examined the interindividual variability in protein composition of the two major organs involved in drug disposition. To this end, we compared the proteomes of matched jejunal and liver biopsies from 37 donors enrolled in the COCKTAIL study. We compared all quantified proteins at a global level, and, at a specific level, the ones implicated in ADME. To our knowledge, this is the first study of this size—in terms of the number of quantified proteins (6,398) and number of donor‐matched jejunal and liver biopsies (n = 37).
Global proteomics of jejunum and liver
The small intestine is the major site for absorption of nutrients and drugs. In line with this, proteins found only in the jejunal biopsies were involved in biological processes such as absorption, for example SI, which is important for carbohydrate digestion and subsequent absorption. 35 Further, the constant turnover, proliferation, and differentiation of enterocytes in the jejunum 36 likely explains the enrichment of transcription processes among the proteins found only there (e.g., HMGA1 37 ). Moreover, the mucus lining the intestinal epithelium contains glycosylated proteins important for physiological protection and immunological processes. 38 Thus, proteins involved in glycosylation processes, such as DPM3, 39 were also found only in the jejunal biopsies.
The liver is the major organ for synthesis, metabolism, and storage of carbohydrates, proteins, and lipids, and plays an important role in the maintenance of metabolic homeostasis. Not surprisingly, proteins quantified only in the liver biopsies were involved in processes such as lipid and fatty acid metabolism—exemplified by HAO1 that metabolizes glycolate and hydroxyl fatty acids 40 —as well as glycogen metabolism (e.g., GYS2 41 ). We also quantified proteins important for drug transport (SLCO1B1) 34 and hepatic fatty acid uptake and bile acid recycling (SLC27A5, or FATP5). 42 Another important process in the liver is amino acid metabolism. Here proteins found only in the liver biopsies (e.g., ARG1 and BHMT) play important roles. BHMT is primarily located in the liver 43 and have been associated with diet‐induced NAFLD and inflammatory response. 44 Partly in line with this, BHMT liver concentrations significantly correlated with the inflammatory marker hs‐CRP (Rs = −0.35, P = 0.038), but not with the NAFLD score (Rs = −0.21; Figure S3 ).
ADME‐related proteins
To our knowledge, we report for the first time the presence in human jejunum of several enzymes related to drug disposition: e.g., 1 ADH, 15 ALDHs, 7 CYPs, 2 FMOs, 5 NATs, 2 MAOs, 9 GSTs, 3 CESs, and 4 SULTs. Although most of these were at low levels, MAOA and CES2 were expressed at levels that could influence intestinal drug metabolism (Tables 1 and 2 ; Tables S2 – S7 ).
Importantly, we quantified seven new CYP enzymes in jejunum: CYP2U1, CYP20A1, CYP4F3, CYP4F11, CYP4F12, CYP4V2, and CYP7B1. These have not been previously reported in MS‐based proteomics studies, 5 , 7 , 14 but were all confirmed to high levels in small intestine with antibody staining. 43 We also, for the first time, identified CYP2A6, CYP8B1, and CYP2W1 in human jejunum, although CYP2A6 was only found in 7 and CYP8B1 in 3 of the 37 biopsies. The levels of these three proteins were low in jejunum, which may be why they had not been identified by antibody staining or previous proteomics studies. CYP1A2, CYP2B6, CYP2E1, and CYP2C8 (all of which are considered among the major drug‐metabolizing CYPs 31 ) have previously not been possible to quantify in jejunum with proteomics 5 without membrane enrichment (only CYP1A2 9 , 10 ). Correspondingly, we only managed to quantify these four, low abundant proteins in a small subset of the jejunal biopsies.
In agreement with previous smaller scale proteomics studies on donor‐matched jejunum and liver (n = 9), 5 , 6 many drug‐metabolizing enzymes and transporters showed higher liver levels. The exceptions were the major metabolizing enzyme CYP3A4 and efflux transporter ABCB1; both showed similar or higher jejunal levels compared with liver levels in our study and previous ones. 5 , 6 These higher liver levels are not surprising since the major fraction of drug metabolism occurs in the liver. Nonetheless, drug‐metabolizing enzymes from families other than CYP enzymes were also found in our jejunal biopsies, indicating that the small intestine overall is involved in extrahepatic drug biotransformation. 45
As membrane‐bound transporters have hydrophobic domains, their extraction, digestion, and peptide identification complicate quantification. 46 In addition, peptides from low abundant transporters can be “masked” if co‐eluting with those from high abundant proteins in global proteomics MS analysis. 46 These are likely contributors to why we could not quantify some low abundant drug transporters, including a few of those identified by the International Transporter Consortium (ITC) 34 as relevant for human pharmacokinetics, or other potentially important drug transporters. 47 For example, we could not detect the liver drug transporters ABCC4 and ABCG2, and only identified ABCC6 in few donors, and then with only one peptide. Importantly, ABCG2 has also previously only been found at very low levels with global proteomics in whole tissue lysates, 15 while others could not quantify the protein with global proteomics even after membrane enrichment. 13 , 18 , 19 Targeted approaches, when selecting specific proteins, have been more successful in liver ABCG2 quantification, but required membrane enrichment. 15 , 48 Furthermore, ABCC6 has previously only been quantified to low levels in membrane‐enriched liver samples, 13 , 19 whereas ABCC4 could not be quantified in either whole tissue lysates or after membrane enrichment. 6 , 13 These differences in sample preparation and MS analysis methods 15 , 46 may affect the results. We therefore compared our data with studies using whole cell lysates, excluding data from membrane‐enriched samples as this introduces further variability. 15 , 16 , 17 Furthermore, mucosal isolation of jejunal samples (as used by refs. 5, 6, 7, 8, 13) may lead to protein degradation during the time until samples are frozen 49 and give different results than biopsies snap‐frozen upon excision (as used in our study).
Biological factors can also affect protein levels. For example, body weight, diabetes, and inflammation are reported to alter levels of drug‐metabolizing enzymes, e.g., those in the CYP family. 50 , 51 , 52 However, the results are conflicting, possibly due to differences in study design. Furthermore, it is difficult to study individual biological factors in a physiologically relevant manner. 50 , 53 , 54 We recently showed correlations between liver CYP3A4 and body weight for our 37 donors with obesity together with the lower weight control group. 55 In contrast, we found no difference in hepatic uptake clearance of rosuvastatin and body weight in the same donors. 3 Furthermore, using the published model 3 and the protein levels of the transporters SLCO1B1, SLCO2B1, SLCO1B3, and SLC10A1, we found no difference in predicted area under the curve vs. concentration (AUC; ng/mL/h) of rosuvastatin in the 37 donors with obesity and the 17 lower weight donors (median AUC (25th–75th percentiles): 3159.6 (2,659.7–4,932.8) ng/mL/h and 3,497.8 (3,027.7–4,462.5) ng/mL/h), respectively.
Although we found significant differences in some liver drug‐metabolizing enzymes and transporters in our donors compared with those in donors without obesity (from the lower weight control group and published data 15 , 16 , 21 ), the differences between the groups were markedly smaller than the interindividual variability within each group. This suggests that for most of these proteins obesity plays a minor role in their regulation. The small differences between the groups could be an effect of the 3‐week low‐calorie diet that our donors with obesity underwent prior to biopsy collection. Notably, it has been shown that chronic cholestatic disease recipients (primary biliary cholangitis and primary sclerosing cholangitis) in liver transplant recipients may affect certain liver protein levels. 56 However, the 17 control donors who underwent planned elective cholecystectomy in our study were otherwise healthy, with no clinical or biochemical signs of cholestatic liver disease. Accordingly, all liver‐related laboratory measures of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and albumin were within the reference ranges (ALT: females < 45 units/litre (U/L), males < 70 U/L; AST: females < 35 and males < 45 U/L; ALP: 35–105 U/L, and albumin: 34–48 g/L). 22 Nevertheless, the observed larger interindividual variability stresses the importance of modeling each patient individually regardless of whether they have obesity or not.
Fewer data are available for the jejunum. Selected drug‐metabolizing enzymes and transporters have previously been quantified in jejunum in a large set of morbidly obese patients (n = 28, ref. 9). Unfortunately, the use of microsomal (membrane) enrichment in that study prevents proper comparison with our data. Nevertheless, we found similar levels of jejunal enzymes and transporters in studies of mucosal isolates. 8 , 13 , 14 However, these studies were limited to fewer donors and the body weights were not specified. Therefore, it is difficult to draw conclusions about the effect of obesity on the jejunal proteins.
Although our study demonstrates that protein concentrations are not conclusively correlated to physiological factors, there were large interindividual differences across the 37 donors, as in our previous study. 56 For some of these proteins, such as CYP2C19 and CYP2D6, genetic polymorphisms resulting in low expression could contribute to the variability. 57 This was observed for CYP2D6 in liver where the lowest protein level was found in a donor with *4/*4 (11‐fold lower than the median protein level; Figure S4 ). However, most proteins quantified in our study do not have known genetic variants that influence expression, and thus polymorphisms would not explain the interindividual differences. The larger interindividual differences found in jejunum compared with liver could be because jejunum is a more heterogeneous tissue with several distinct tissue layers. Therefore, the sampling depth of the pinch‐biopsy technique could result in slightly different sample composition of mucosa and submucosa. This, in turn, could lead to different dilution effects of the quantified proteins calculated with the total protein approach. 56 We attempted to correct for differences in sampling by using enterocyte‐specific proteins (SI and VIL1) for normalizing jejunal protein levels in each donor. This approach assumes that these proteins are expressed at constant levels in all donors, which might not be true, as even housekeeping genes have been shown to vary in protein levels across individuals. 56 Indeed, correcting for SI and VIL1 resulted in higher interindividual variability (Figure S5 ), suggesting that more sophisticated algorithms for cell type composition are required. 58 As differences in protein levels can greatly influence mathematical prediction models for drug disposition or toxicity, they should be taken into account when personalizing systems pharmacology. 1 However, care should be taken in using the concentrations provided for multimeric assemblies (e.g., ABCG2 (BCRP) is active as a dimer). Furthermore, expression levels of, e.g., polymorphic proteins do not always correlate to activity.
In summary, we used global proteomics analysis to establish the protein profiles of matched jejunum and liver obtained from 37 donors. Although most of the drug‐metabolizing enzymes had greater expression in liver, jejunum contained levels of enzymes from many important families that would influence drug disposition. We did not find correlations between ADME‐related protein concentrations in the two donor‐matched tissues, or between protein levels and physiological factors, such as age, BMI, and inflammation. Nonetheless, our data demonstrate large interindividual differences in protein levels for both tissues. As these differences can influence drug disposition, knowledge of them is a prerequisite for building and refining accurate prediction models. Here we examined a subset of the proteins quantified in jejunum (n = 6,398) and liver (n = 5,773). Further exploration in the protein composition of the two tissues from the 37 donors can be made using Data S1 .
FUNDING
The research of first and last authors were sponsored by Vetenskapsrådet. This study was supported by the Swedish Research Council (approval numbers 5715, 01951, and 01586).
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
C.W. and P.A. wrote the manuscript. C.W., H.C., J.K.H., J.H., R.J.‐L., A.Å., T.B.A., and P.A. designed research. C.W., J.R.W., I.R., H.C., J.K.H., J.H., R.J‐L., A.Å., T.B.A., and P.A. performed research. C.W., J.R.W., and I.R. analyzed data. J.R.W. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
Data
ACKNOWLEDGMENTS
We thank André Mateus and Nils Kurzawa at EMBL, Heidelberg, Germany for valuable discussions and help with statistical analysis. We also thank Katharina Zettl at MaxPlanck Institute of Biochemistry, Martinsreid, Germany for technical assistance. We thank Lars Thomas Seeberg with colleagues at the Department of Gastrointestinal Surgery, Vestfold Hospital Trust in Norway, and the COCKTAIL study staff for the jejunal and liver biopsies. We are grateful to the patients at the Morbid Obesity Center, Vestfold Hospital Trust, for participating in the COCKTAIL study and providing biopsies.
- 1. Taskar, K.S. , Harada, I. & Alluri, R.V. Physiologically‐based pharmacokinetic (PBPK) modelling of transporter mediated drug absorption, clearance and drug‐drug interactions. Curr. Drug Metab. 22, 523–531 (2021). [DOI] [PubMed] [Google Scholar]
- 2. Bosgra, S. et al. Predicting carrier‐mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell‐lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur. J. Pharm. Sci. 65, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Wegler, C. et al. Proteomics‐informed prediction of rosuvastatin plasma profiles in patients with a wide range of body weight. Clin. Pharmacol. Ther. 109, 762–771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Di Meo, A. et al. Proteomic profiling of the human tissue and biological fluid proteome. J. Proteome Res. 20, 444–452 (2021). [DOI] [PubMed] [Google Scholar]
- 5. Drozdzik, M. et al. Protein abundance of clinically relevant drug‐metabolizing enzymes in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 104, 515–524 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Drozdzik, M. et al. Protein abundance of clinically relevant drug transporters in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 105, 1204–1212 (2019). [DOI] [PubMed] [Google Scholar]
- 7. Al‐Majdoub, Z.M. et al. Quantification of proteins involved in intestinal epithelial handling of xenobiotics. Clin. Pharmacol. Ther. 109, 1136–1146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang, H. et al. Regional proteomic quantification of clinically relevant non‐cytochrome P450 enzymes along the human small intestine. Drug Metab. Dispos. 48, 528–536 (2020). [DOI] [PubMed] [Google Scholar]
- 9. Miyauchi, E. et al. Quantitative atlas of cytochrome P450, UDP‐glucuronosyltransferase, and transporter proteins in jejunum of morbidly obese subjects. Mol. Pharm. 13, 2631–2640 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Gröer, C. et al. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP‐glucuronosyltransferases by mass spectrometry‐based targeted proteomics. J. Pharm. Biomed. Anal. 100, 393–401 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Akazawa, T. , Uchida, Y. , Miyauchi, E. , Tachikawa, M. , Ohtsuki, S. & Terasaki, T. High expression of UGT1A1/1A6 in monkey small intestine: comparison of protein expression levels of cytochromes P450, UDP‐glucuronosyltransferases, and transporters in small intestine of cynomolgus monkey and human. Mol. Pharm. 15, 127–140 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Brück, S. , Strohmeier, J. , Busch, D. , Drozkzik, M. & Oswald, S. Caco‐2 cells – expression, regulation and function of drug transporters compared with human jejunal tissue. Biopharm. Drug Dispos. 38, 115–126 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Al‐Majdoub, Z.M. et al. Mass spectrometry‐based abundance atlas of ABC transporters in human liver, gut, kidney, brain and skin. FEBS Lett. 594, 4134–4150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Couto, N. et al. Quantitative proteomics of clinically relevant drug‐metabolizing enzymes and drug transporters and their intercorrelations in the human small intestine. Drug Metab. Dispos. 48, 245–254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wegler, C. et al. Variability in mass spectrometry‐based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol. Pharm. 14, 3142–3151 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Wegler, C. et al. Influence of proteome profiles and intracellular drug exposure on differences in CYP activity in donor‐matched human liver microsomes and hepatocytes. Mol. Pharm. 18, 1792–1805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wiśniewski, J.R. , Wegler, C. & Artursson, P. Subcellular fractionation of human liver reveals limits in global proteomic quantification from isolated fractions. Anal. Biochem. 509, 82–88 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Ahire, D.S. , Basit, A. , Karasu, M. & Prasad, B. Ultrasensitive quantification of drug‐metabolizing enzymes and transporters in small sample volume by microflow LC‐MS/MS. J. Pharm. Sci. 110, 2833–2840 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Couto, N. , Al‐Majdoub, Z.M. , Achour, B. , Wright, P.C. , Rostami‐Hodjegan, A. & Barber, J. Quantification of proteins involved in drug metabolism and disposition in the human liver using label‐free global proteomics. Mol. Pharm. 16, 632–647 (2019). [DOI] [PubMed] [Google Scholar]
- 20. Vildhede, A. , Wiśniewski, J.R. , Norén, A. , Karlgren, M. & Artursson, P. Comparative proteomic analysis of human liver tissue and isolated hepatocytes with a focus on proteins determining drug exposure. J. Proteome Res. 14, 3305–3314 (2015). [DOI] [PubMed] [Google Scholar]
- 21. Weiß, F. et al. Direct quantification of cytochromes P450 and drug transporters—a rapid, targeted mass spectrometry‐based immunoassay panel for tissues and cell culture lysates. Drug Metab. Dispos. 46, 387–396 (2018). [DOI] [PubMed] [Google Scholar]
- 22. Hjelmesæth, J. et al. Impact of body weight, low energy diet and gastric bypass on drug bioavailability, cardiovascular risk factors and metabolic biomarkers: protocol for an open, non‐randomised, three‐armed single centre study (COCKTAIL). BMJ Open 8, e021878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wiśniewski, J.R. & Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 84, 2631–2637 (2012). [DOI] [PubMed] [Google Scholar]
- 24. Wiśniewski, J.R. & Gaugaz, F.Z. Fast and sensitive total protein and peptide assays for proteomic analysis. Anal. Chem. 87, 4110–4116 (2015). [DOI] [PubMed] [Google Scholar]
- 25. Tyanova, S. , Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Huber, W. , Heydebreck, A.V. , Sültmann, H. , Poustka, A. & Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 (suppl. 1), S96–S104 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Wiśniewski, J.R. & Rakus, D. Multi‐enzyme digestion FASP and the ‘Total Protein Approach’‐based absolute quantification of the Escherichia coli proteome. J. Proteomics. 109, 322–331 (2014). [DOI] [PubMed] [Google Scholar]
- 28. Kotronen, A. et al. Prediction of non‐alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology 137, 865–872 (2009). [DOI] [PubMed] [Google Scholar]
- 29. Yu, G. , Wang, L.G. , Han, Y. & He, Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schröder, A. et al. Genomics of ADME gene expression: mapping expression quantitative trait loci relevant for absorption, distribution, metabolism and excretion of drugs in human liver. Pharmacogenomics J. 13, 12–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zanger, U.M. & Schwab, M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141 (2013). [DOI] [PubMed] [Google Scholar]
- 32. Mackenzie, P.I. et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenet. Genom, 7, 255–269 (1997). [DOI] [PubMed] [Google Scholar]
- 33. Guillemette, C. Pharmacogenomics of human UDP‐glucuronosyltransferase enzymes. Pharmacogenomics J. 3, 136–158 (2003). [DOI] [PubMed] [Google Scholar]
- 34. Zamek‐Gliszczynski, M.J. et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 104, 890–899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gericke, B. , Amiri, M. & Naim, H.Y. The multiple roles of sucrase‐isomaltase in the intestinal physiology. Mol. Cell. Pediatr. 3, 2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moor, A.E. et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175, 1156–1167.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Wisniewski, J.R. & Schwanbeck, R. High mobility group I/Y: multifunctional chromosomal proteins causally involved in tumor progression and malignant transformation (review). Int. J. Mol. Med. 6, 409–419 (2000). [DOI] [PubMed] [Google Scholar]
- 38. Goto, Y. , Uematsu, S. & Kiyono, H. Epithelial glycosylation in gut homeostasis and inflammation. Nat. Immunol. 17, 1244–1251 (2016). [DOI] [PubMed] [Google Scholar]
- 39. Ashida, H. , Maeda, Y. & Kinoshita, T. DPM1, the catalytic subunit of dolichol‐phosphate mannose synthase, is tethered to and stabilized on the endoplasmic reticulum membrane by DPM3*. J. Biol. Chem. 281, 896–904 (2006). [DOI] [PubMed] [Google Scholar]
- 40. Wang, M. et al. High throughput cell‐based assay for identification of glycolate oxidase inhibitors as a potential treatment for Primary Hyperoxaluria Type 1. Sci. Rep. 6, 34060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kamenets, E.A. et al. Hepatic glycogen synthase (GYS2) deficiency: seven novel patients and seven novel variants. JIMD Rep. 53, 39–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hubbard, B. et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology 130, 1259–1269 (2006). [DOI] [PubMed] [Google Scholar]
- 43. Uhlén, M. et al. Proteomics. Tissue‐based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 44. Pacana, T. et al. Dysregulated hepatic methionine metabolism drives homocysteine elevation in diet‐induced nonalcoholic fatty liver disease. PLoS One 10, e0136822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xie, F. , Ding, X. & Zhang, Q.‐Y. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm. Sin. B 6, 374–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prasad, B. et al. Toward a consensus on applying quantitative liquid chromatography‐tandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin. Pharmacol. Ther. 106, 525–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wenzel, C. , Drozdzik, M. & Oswald, S. Organic cation transporter 1 an intestinal uptake transporter: fact or fiction? Front. Pharmacol. 12, 648388 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prasad, B. , Lai, Y. , Lin, Y. & Unadkat, J.D. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J. Pharm. Sci. 102, 787–793 (2013). [DOI] [PubMed] [Google Scholar]
- 49. Espina, V. et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol. Cell. Proteomics 7, 1998–2018 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Jong, L.M. , Jiskoot, W. , Swen, J.J. & Manson, M.L. Distinct effects of inflammation on cytochrome P450 regulation and drug metabolism: lessons from experimental models and a potential role for pharmacogenetics. Genes (Basel) 11, 1509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodríguez‐Morató, J. et al. Short‐ and medium‐term impact of bariatric surgery on the activities of CYP2D6, CYP3A4, CYP2C9, and CYP1A2 in morbid obesity. Sci. Rep. 9, 20405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dostalek, M. , Akhlaghi, F. & Puzanovova, M. Effect of diabetes mellitus on pharmacokinetic and pharmacodynamic properties of drugs. Clin. Pharmacokinet. 51, 481–499 (2012). [DOI] [PubMed] [Google Scholar]
- 53. Cobbina, E. & Akhlaghi, F. Non‐alcoholic fatty liver disease (NAFLD) — pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 49, 197–211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stanke‐Labesque, F. , Gautier‐Veyret, E. , Chhun, S. , Guilhaumou, R. & French Society of Pharmacology and Therapeutics . Inflammation is a major regulator of drug metabolizing enzymes and transporters: consequences for the personalization of drug treatment. Pharmacol. Ther. 215, 107627 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kvitne, K.E. et al. Short‐ and long‐term effects of body weight loss following calorie restriction and gastric bypass on CYP3A‐activity ‐ a non‐randomized three‐armed controlled trial. Clin. Transl. Sci. 15, 221–233 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wegler, C. et al. Global variability analysis of mRNA and protein concentrations across and within human tissues. NAR Genom. Bioinform. 2, lqz010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sim, S.C. & Ingelman‐Sundberg, M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer‐reviewed database of CYP variants and their associated effects. Human Genomics 4, 278–281 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang, X. , Park, J. , Susztak, K. , Zhang, N.R. & Li, M. Bulk tissue cell type deconvolution with multi‐subject single‐cell expression reference. Nat. Commun. 10, 380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tang, H. , Hussain, A. , Leal, M. , Mayersohn, M. & Fluhler, E. Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metab. Dispos. 35, 1886–1893 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data