

Abstract
GPNMB (glycoprotein nonmetastatic B) and other TFE3/TFEB transcriptional targets have been proposed as markers for microphthalmia (MiT) translocation renal cell carcinomas (tRCCs). We recently demonstrated that constitutive mTORC1 activation via TSC1/2 loss leads to increased activity of TFE3/TFEB, suggesting that the pathogenesis and molecular markers for tRCCs and TSC1/2‐associated tumors may be overlapping. We examined GPNMB expression in human kidney and angiomyolipoma (AML) cell lines with TSC2 and/or TFE3/TFEB loss produced using CRISPR–Cas9 genome editing as well as in a mouse model of Tsc2 inactivation‐driven renal tumorigenesis. Using an automated immunohistochemistry (IHC) assay for GPNMB, digital image analysis was employed to quantitatively score expression in clear cell RCC (ccRCC, n = 87), papillary RCC (papRCC, n = 53), chromophobe RCC (chRCC, n = 34), oncocytoma (n = 4), TFE3‐ or TFEB‐driven tRCC (n = 56), eosinophilic solid and cystic RCC (ESC, n = 6), eosinophilic vacuolated tumor (EVT, n = 4), and low‐grade oncocytic tumor (LOT, n = 3), as well as AML (n = 29) and perivascular epithelioid cell tumors (PEComas, n = 8). In cell lines, GPNMB was upregulated following TSC2 loss in a MiT/TFE‐ and mTORC1‐dependent fashion. Renal tumors in Tsc2 +/− A/J mice showed upregulation of GPNMB compared with normal kidney. Mean GPNMB expression was significantly higher in tRCC than in ccRCC (p < 0.0001), papRCC (p < 0.0001), and chRCC (p < 0.0001). GPNMB expression in TSC1/2/MTOR alteration‐associated renal tumors (including ESC, LOT, AML, and PEComa) was comparable to that in tRCC. The immunophenotype of tRCC and TSC1/2/MTOR alteration‐associated renal tumors is highly overlapping, likely due to the increased activity of TFE3/TFEB in both, revealing an important caveat regarding the use of TFE3/TFEB‐transcriptional targets as diagnostic markers. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: renal cell carcinoma, angiomyolipoma, TSC1/2, translocation renal cell carcinoma, TFE3, TFEB, GPNMB
Introduction
Renal cell carcinoma (RCC) classification has historically been based on morphology, with clear cell RCC (ccRCC), papillary RCC (papRCC), and chromophobe RCC (chRCC) identified by their distinctive histopathologic features under the microscope. However, advances in molecular phenotyping have challenged this purely morphological classification of renal tumors. Although there is a strong association between morphology and genotype for the most common categories of RCC listed above, morphologically unclassified RCC cases – many of which have eosinophilic or oncocytic features – have demonstrated a much weaker correlation between their appearance under the microscope and their underlying molecular landscape. Accordingly, several newer molecular subtypes of RCC have been defined with overlapping or heterogeneous morphologic features. For example, translocation RCC (tRCC) – harboring genomic rearrangements that drive expression of the microphthalmia (MiT) family of transcription factors including TFE3 and TFEB – can assume a wide spectrum of clear cell, papillary, and eosinophilic morphologies, making identification based on H&E difficult or impossible in some cases [1, 2, 3, 4]. Similarly, several novel or emerging subtypes of renal tumors associated with underlying TSC1/2 loss or MTOR activating mutations have recently been described with overlapping histologic features, including eosinophilic or oncocytic morphology [5, 6, 7, 8, 9, 10, 11, 12, 13, 14].
In contrast to succinate dehydrogenase‐ or fumarate hydratase‐deficient RCC, where pathologists can screen for the genomic driver alteration using an immunohistochemistry (IHC) assay, IHC assays to sensitively screen for tRCC or TSC1/2/MTOR alteration‐associated RCC cases remain problematic. IHC panels including markers such as CK7, CK20 or CD117 have been reported for TSC1/2/mTOR‐associated cases [6, 10, 12, 14] but are not specific. TFE3 and TFEB overexpression by IHC [2, 15] is technically challenging for diagnosis of tRCC. Fluorescence in situ hybridization (FISH) can be used to examine for common TFEB/TFE3 rearrangements [16, 17, 18]; however, FISH assays are expensive screening tools and not widely available. Intriguingly, recent work shows that MiT family transcriptional targets – such as melan A, cathepsin K, or TRIM63 – identify both tRCC and TSC1/2/MTOR alteration‐associated renal tumors [7, 19, 20]. Although initially puzzling, this finding is entirely consistent with a recent study from our group demonstrating that constitutive mTORC1 activation via TSC2 loss leads to increased activity of TFE3 and TFEB [21]. Taken together, this work suggests that a common underlying molecular pathogenesis may explain the overlapping morphology and immunophenotype for tRCC and TSC1/2/MTOR alteration‐associated RCC cases.
In the current study, we explore the expression and regulation of GPNMB (glycoprotein nonmetastatic B), a transmembrane protein and MITF/TFE3 transcriptional target recently proposed as a specific marker for tRCC in mouse models [22]. Similar to the other TFE3/TFEB targets described above, we demonstrate that GPNMB is also highly expressed in human kidney cell lines and murine kidney tumors with TSC2 deletion and regulated by mTOR kinase and TFE3/TFEB in vitro. Clinically, GPNMB identifies tRCC and TSC1/2/MTOR alteration‐associated renal tumors, distinguishing them from most conventional morphologic subtypes.
Materials and methods
Patients and tissue samples
With Johns Hopkins institutional review board approval, this study included a total of 11 tissue microarray (TMA) cohorts comprising evaluable renal tumors from nephrectomies dating from 1998 to 2012, including the common subtypes: clear cell RCC (ccRCC; n = 90; 87 with evaluable GPNMB staining), papillary RCC (papRCC; n = 54; 53 with evaluable GPNMB staining), and chromophobe RCC (chRCC; n = 36; 34 with evaluable GPNMB staining). A detailed description of these cohorts has been previously published [23, 24, 25]. In addition, we studied TMAs and standard sections including predominantly previously published [1] TFE3 tRCC cases confirmed by fluorescence in situ hybridization (FISH) (n = 16; 15 with evaluable GPNMB staining), TFEB tRCC confirmed by FISH (n = 18), tRCC confirmed by IHC (n = 18), and five cases on standard section of cathepsin K‐negative tRCC. We also studied standard sections from four cases of oncocytoma. In addition to these cohorts, we created a new TMA of conventional angiomyolipoma (AML) cases (n = 24); used a previously published TMA containing four cases of metastatic epithelioid AML and one case on standard section (n = 5) [23]; and obtained additional cases of perivascular epithelioid cell tumors (PEComa) (n = 8, from seven unique patients). In addition, we searched the Johns Hopkins pathology archives for emerging or new renal tumor diagnoses during the time period of 2008–2021 and obtained standard sections of eosinophilic solid and cystic RCC (ESC) (n = 8; 6 with evaluable GPNMB staining), eosinophilic and vacuolated tumor (EVT) (n = 4), and low‐grade oncocytic tumor of the kidney (LOT) (n = 3).
Immunohistochemistry
GPNMB immunohistochemistry (IHC) on human tissues was performed using the Ventana Discovery ULTRA platform (Ventana/Roche, Oro Valley, AZ, USA) using hand‐applied GPNMB primary antibody (E4D7P XP® Rabbit mAb #38313, 1:1500; Cell Signaling Technology, Danvers, MA, USA). Positive staining in histiocytes was considered as an internal positive control in all cases (see supplementary material, Figure S1). Additional validation was performed using an independent GPNMB antibody (#20338‐1‐AP; Proteintech, Rosemont, IL, USA) on the Ventana Discovery ULTRA (Ventana/Roche) (see supplementary material, Figure S1).
Because the E4D7P antibody clone is specific for human GPNMB and does not detect mouse GPNMB, GPNMB IHC on murine tissues was performed on the Ventana Discovery ULTRA (Ventana/Roche) using a hand‐applied mouse‐specific anti‐GPNMB clone (E7U1Z, #90205, 1:100; Cell Signaling Technology).
Renal cystadenoma and carcinoma tissue from Tsc2 +/− mice was immunostained for p‐S6 (#4858; Cell Signaling Technology; 1:100), p‐4E‐BP1 (#2855; Cell Signaling Technology; 1:200), and TSC2 (#4308; Cell Signaling Technology; 1:100) on the Ventana Discovery ULTRA (Ventana/Roche).
IHC analysis
Visual scoring was performed by a trained uropathologist (DCS) using a simple scoring rubric of 1–3+ staining, with 0+ representing no visible color reaction, 1+: <10% of cells showing strong intensity membranous staining, 2+: 10–90% of cells showing strong intensity membranous staining, and 3+: >90% of cells showing strong intensity membranous staining. For digital scoring, immunostained slides were scanned, and the whole‐slide images were loaded into HALO v3.1.1076.433 (Indica Labs, Albuquerque, NM, USA). A trained uropathologist (DCS) manually annotated the tumor epithelium in all cases, in four regions of each standard section slide measuring 100 × 100 μm each, and in all available tumor tissue from the TMAs. TMA cores with no available tissue or no available tumor in the core were considered non‐contributory. The Membrane algorithm (v1.7; Indica Labs) was optimized to correspond to visually detected GPNMB intensity and applied uniformly to all cases described above to derive the digital H‐score.
Cell lines
Human embryonic kidney HEK293T cells with or without somatic genomic deletion (KO) of TSC2 using CRISPR–Cas9 genome editing were a kind gift of the TSC Alliance and Dr Nellist [26]. TRI102 cells derived from a TSC2‐null human AML and TRI103 [27, 28] cells derived from TRI102 cells stably transfected with wild‐type TSC2 (pcDNA3.1 TSC2‐zeo) were obtained from the ATCC (Manassas, VI, USA). RCC cell lines from human tRCC (PRCC–TFE3; UOK124) and sporadic clear cell RCC (ccRCC; UOK111) were a kind gift from Dr W Marston Linehan [22]). All cell lines were maintained in DMEM high glucose medium (Gibco/Thermo Fisher Scientific, Waltham, MA, USA) with l‐glutamine, 10% FBS, and 1% penicillin/streptomycin stock solution at 37 °C in 5% CO2. RCC cell lines were cultured as above in DMEM containing additional 1X essential amino acids. Rapamycin (200 nm) and Torin 1 (1 μm) (LC Laboratories, Woburn, MA, USA) treatment was performed for 72 h prior to cell lysis for immunoblotting.
Xenograft studies
UOK111 and UOK124 cells were used for xenograft studies. Three million cells were mixed with growth‐factor reduced Matrigel (356231; Corning, Corning, NY, USA) 1:1 (v/v) in a final volume of 100 μl and injected subcutaneously into the flanks of NSG (NOD scid gamma) mice. Tumor volume and body weight were measured every 3 days by using calipers and calculated using the formula (length × width2/2). Mice were euthanized 21 days after inoculation, and tumors were harvested for histological examination and immunoblotting.
siRNA‐mediated gene silencing
siRNAs [# M‐011741‐01‐0005: siGENOME Human GPNMB (10457) siRNA – SMARTpool] for human GPNMB and negative control were purchased from Dharmacon (GE Healthcare, Lafayette, CO, USA). HEK293T TSC2 KO cells were transfected with 100 nm siRNA using Lipofectamine RNAiMAX reagent according to the manufacturer's transfection guidelines (Thermo Fisher Scientific).
CRISPR–Cas9 genome editing for TFEB and TFE3
We designed single‐guide RNA (sgRNA) for three target sequences in the human TFE3 gene (GGCGATTCAACATTAACGACAGG, GCGACGCTCAACTTTGGAGAGGG, TCGCCTGCGACGCTCAACTTTGG) and four target sequences in the human TFEB gene (CAACCCTATGCGTGACGCCATGG, GCGGTAGCAGTGAGTCGTCCAGG, TGCCTAGCGAAGAGGGCCCAGGG, and GAGTACCTGTCCGAGACCTA) and cloned these into the lentiCRISPR v2 vector (#52961; Addgene, Watertown, MA, USA). Lentivirus was produced as previously described [29] and TSC2 KO HEK293T cells were infected for 48 h and selected with puromycin (1 μg/ml) for 10 days, and monoclonal cell colonies established. Immunoblotting was used for confirmation of TFEB and TFE3 KO.
Transgenic mice
Tsc2 +/− A/J mice have been described previously [30] and were the kind gift of David Kwiatkowski and the TSC Alliance. Mice were sacrificed at 14 months of age, and kidneys were removed, then formalin‐fixed and paraffin‐embedded, followed by histologic sectioning prior to immunohistochemistry. In parallel, fresh tissue from the contralateral kidney was snap‐frozen in liquid nitrogen and cryosectioned for laser capture microdissection as described below.
Lysosomal fractionation
Lysosomal fractionation assays were carried out as described previously [31]. Cells grown on 150‐mm‐diameter dishes were harvested and lysed in 750 μl of cold fractionation buffer (50 mm KCl, 90 mm potassium gluconate, 1 mm EGTA, 50 mm sucrose, 5 mm glucose, protease inhibitor cocktail tablet, and 20 mm HEPES, pH 7.4). The cells were then lysed by syringing, and the nuclear fraction was removed by centrifugation at 1000 × g for 10 min at 4 °C. The supernatant was then centrifuged at 20 000 × g for 30 min at 4 °C. The precipitated lysosome‐enriched fraction was resuspended in the fractionation buffer, and the supernatant was separated as the cytosolic fraction.
Immunoblotting
Whole‐cell or lysosome‐enriched fractions were utilized for immunoblotting as described previously [32]. The primary antibodies used were as follows (all were used at 1:1000 unless otherwise noted): GPNMB [#38313, Cell Signaling Technology (CST); 1:500], TSC2 (#4308, CST), Phospho‐p70 S6 Kinase (Thr389) (#9205, CST), p70 S6 Kinase (#9202, CST), Phospho‐S6 Ribosomal Protein (Ser235/236) (#4858, CST), S6 Ribosomal Protein (#2317, CST), Phospho‐4E‐BP1 (Ser65) (#9451, CST), total 4E‐BP1 (#9644, CST), TFE3 (#14779, CST), TFEB (#4240, CST), Phospho‐Akt (S473) (#4060, CST), Phospho‐Akt (T308) (#2965, CST), Akt (pan) (#4691, CST), Phospho‐FoxO1(Thr24)/FoxO3a(Thr32)/FoxO4(Thr28) (#2599, CST), FoxO1 (#2880, CST), BECN1 (#3495, CST), beta‐actin (#4970, CST), GAPDH (#5174, CST).
Laser capture microdissection (LCM)
Frozen, OCT‐embedded kidney sections from Tsc2 +/− A/J mice were prepared for LCM processing as follows: tissues were cut in 10 μm‐thick sections on 4 μm polyethylene naphthalate (PEN) membrane slides (11600288; Leica, Wetzlar, Germany) for LCM and 4 μm on a regular slide for hematoxylin and eosin (H&E) staining. Before using for sectioning, PEN slides were sterilized overnight under ultraviolet radiation to degrade RNases. Sections were stained with hematoxylin and LCM was performed using a Leica DM7000 microscope. Tumor regions were isolated and stored in Buffer RLT lysis buffer (Qiagen, Hilden, Germany). Respective H&E/p‐S6‐stained sections were used as a reference to identify the morphology of the tissue and renal tumors. LCM‐sorted cells were then homogenized using a QIAshredder (Qiagen) and total RNA was extracted using an RNeasy kit (Qiagen). RT‐qPCR was performed as described below.
RNA extraction, cDNA synthesis, and qPCR analysis
Total RNA from cell pellets/xenografts was extracted using an RNeasy kit (Qiagen), following the manufacturer's instructions. Using the SuperScript III First‐Strand Synthesis System for RT‐PCR (Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) and oligo(dT)50 primers, cDNA synthesis was realized from 2 μg of total RNA. qPCR reactions were performed using the QuantStudio 12K Flex Real‐Time PCR System (Life Technologies/Thermo Fisher Scientific, Waltham, MA, USA), using primer/probes for GPNMB (human: Hs01095669_m1 and mouse: Mm01328587_m1).
Statistical analyses
Comparisons between groups were made using Student's t‐test or one‐way ANOVA (Gpnmb relative level) or Mann–Whitney or Kruskal–Wallis tests (comparisons of H‐scores or visual scores between tumor groups). GraphPad Prism 9.1.2 (GraphPad Software Inc, San Diego, CA, USA) was used for all analyses.
Results
GPNMB is upregulated with TSC2 inactivation in a TFE3/TFEB‐ and mTORC1‐dependent fashion
We first examined HEK293T cells with TSC2 deletion (KO) by CRISPR–Cas9 genome editing, where mTORC1 signaling is increased by immunoblotting compared with parental cells (Figure 1A). Notably, GPNMB expression was increased in cells with TSC2 KO compared with parental cells (Figure 1B). There were two bands detected with the anti‐GPNMB antibody: one at approximately 95 kDa (the predicted molecular weight for the precursor form of GPNMB) and one below the 75 kDa marker (which has not been described for GPNMB previously). The upper band at 95 kDa increased dramatically in cells with TSC2 KO compared with parental cells (Figure 1B) and was decreased by treatment of TSC2 KO cells with siRNA specific for GPNMB (supplementary material, Figure S2), confirming its specificity. In contrast, the lower band did not change upon TSC2 KO or with GPNMB siRNA, suggesting it was likely nonspecific. GPNMB localizes to plasma membranes, including melanosomes or lysosomal/endosomal vesicles in non‐melanocytes, and we confirmed that GPNMB is upregulated in lysosome‐enriched fractions of HEK293T lysates with TSC2 KO compared with parental cells (Figure 1C). To determine whether GPNMB expression in the setting of TSC2 deletion was mTOR‐dependent or ‐independent, we inhibited mTOR with the allosteric inhibitor rapamycin in HEK293T TSC2 KO cells and found that this was sufficient to downregulate GPNMB protein levels by immunoblotting (Figure 1D). GPNMB is a transcriptional target of MITF and TFE3 as well as tRCC‐associated TFE3 fusion proteins [22, 33, 34], and in other systems, TFE3 and TFEB have cooperative rather than redundant effects [35, 36]; however, whether MiT/TFE factors regulate GPNMB in TSC2 KO cells has not been determined. We used CRISPR–Cas9 genome editing with multiple gRNAs to generate clones of HEK293T TSC2 KO cells with TFE3 deletion, TFEB deletion, and dual deletion of TFE3 and TFEB, and found that either TFE3 or TFEB deletion was sufficient to variably downregulate GPNMB gene and protein expression, with the dual knockdown showing more consistent suppression across clones (Figure 1E,F).
Figure 1.
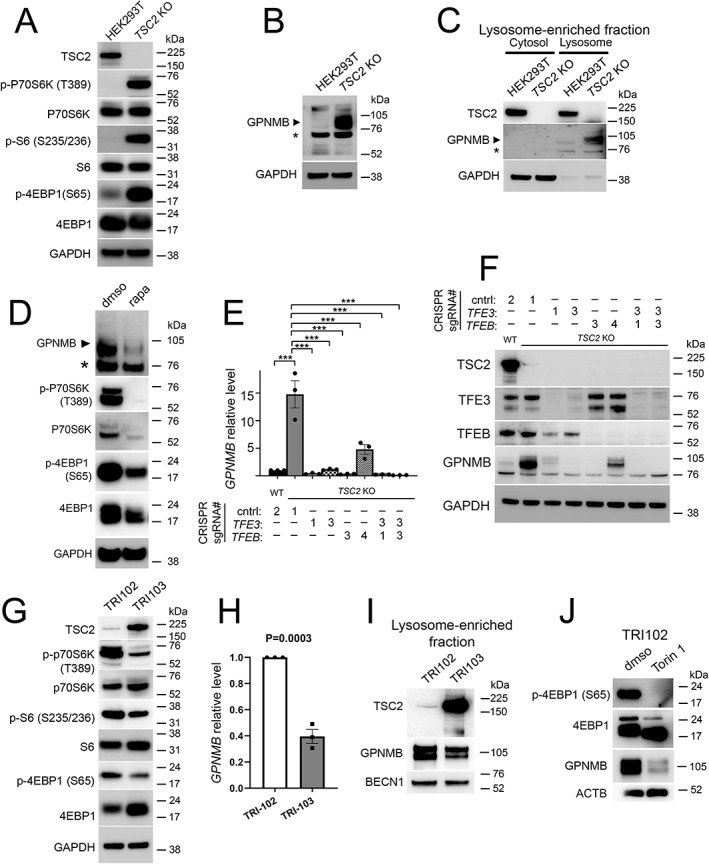
GPNMB is upregulated following TSC2 loss in a TFE3/TFEB‐ and mTORC1‐dependent fashion. (A) Immunoblotting of whole‐cell lysates from parental and TSC2 KO HEK293T cells for TSC2 and mTORC1 activation markers. (B) Immunoblotting of whole‐cell lysates from parental and TSC2 KO HEK293T cells for GPNMB. A non‐specific band (*) based on the siRNA experiment in supplementary material, Figure S2 is present in this cell line by immunoblotting. (C) Immunoblotting of cell lysates enriched for lysosomal content from parental and TSC2 KO HEK293T cells. (D) Immunoblotting of whole‐cell lysates from TSC2 KO HEK293T cells with or without 72‐h treatment with mTOR inhibitor rapamycin (200 nm). (E) RT‐qPCR for relative GPNMB gene expression in TSC2 KO HEK293T cells with or without genomic deletion of TFE3, TFEB, or TFEB and TFE3 (r = 3; ***p < 0.0001 by one‐way ANOVA). (F) Immunoblotting of whole‐cell lysates from TSC2 KO HEK293T cells with or without genomic deletion of TFE3, TFEB, or TFEB and TFE3. (G) Immunoblotting of whole‐cell lysates from TRI‐102 parental cells (TSC2 −/−) and TRI‐103 cells (TSC2 −/− with stable transfection of wild‐type TSC2) for TSC2 and mTORC1 activation markers. (H) RT‐qPCR for relative GPNMB gene expression in TRI‐102 and TRI‐103 cells. (I) Immunoblotting of cell lysates from TRI102 and TRI103 cells enriched for lysosomal content. (J) Immunoblotting of whole‐cell lysates from TRI‐102 cells with or without 72‐h treatment with mTOR kinase inhibitor Torin 1 (1 μm).
For independent validation, we queried TRI‐102 cells, a human angiomyolipoma (AML) cell line with TSC2 deletion, and compared these with cells stably transfected with wild‐type TSC2 (TRI‐103 cells). Expression of exogenous TSC2 was sufficient to inhibit mTORC1 signaling (Figure 1G), to significantly downregulate GPNMB gene expression in this cell line (Figure 1H), and to decrease lysosome‐associated GPNMB protein levels (Figure 1I). Similarly, treatment of TRI‐102 cells with the mTOR kinase inhibitor Torin 1 was sufficient to reduce GPNMB protein expression (Figure 1J).
GPNMB is upregulated in murine models of renal tumorigenesis with Tsc2 inactivation
We next examined whether these in vitro findings could be replicated in vivo. Heterozygous deletion of Tsc2 in murine models followed by spontaneous inactivation of the second allele leads to the development of renal cystadenomas (Figure 2A), and a subset of mice develop oncocytic high‐grade renal cell carcinomas with aging [37]. We used laser capture microdissection (LCM) to isolate RNA from microscopic adenoma lesions (Figure 2B) and found that Gpnmb gene expression was upregulated in these precursor lesions compared with surrounding normal renal parenchyma (Figure 2C). By immunohistochemistry (IHC) using an independent antibody clone from that used in human cell lines, there was a similar upregulation of GPNMB protein in the oncocytic carcinoma cells (Figure 2D). Taken together, our in vitro and in vivo data suggest that GPNMB is upregulated with TSC2 inactivation in a TFE3/TFEB‐ and mTORC1‐dependent fashion.
Figure 2.
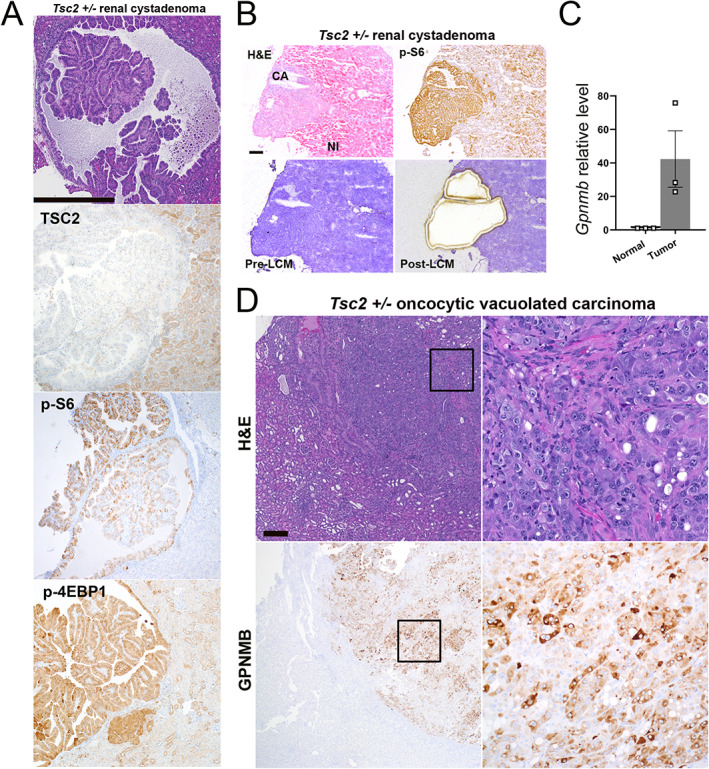
Renal tumors from A/J Tsc2 +/− mice show increased GPNMB expression. (A) Hematoxylin and eosin (H&E) and TSC2, p‐S6, and p‐4EBP1 IHC staining of cystadenoma lesions occurring in 14‐month‐old A/J Tsc2 +/− mice. Scale bar: 500 μm. (B) Laser capture microdissection (LCM) of renal cystadenomas from 14‐month‐old A/J Tsc2 +/− mice guided by p‐S6 immunostaining for mTORC1 activation. Images reduced from 100× magnification. Scale bar: 250 μm. (C) RT‐qPCR for relative GPNMB gene expression in normal kidney parenchyma and LCM‐captured renal cystadenoma tissue. (D) H&E and GPNMB IHC staining of oncocytic vacuolated carcinomas occurring in 14‐month‐old A/J Tsc2 +/− mice. Scale bar: 500 μm. Right panels are higher‐magnification views of the outlined area in each image.
GPNMB is upregulated in tRCC and TSC1/2/mTOR alteration‐associated renal tumors compared with more common RCC subtypes
Next, we assessed GPNMB expression across a wide spectrum of human renal tumors using an automated assay with digital quantification (Figures 3, 4, 5, 6 and supplementary material, Figure S3). GPNMB was expressed at negligible levels in normal renal tissue (Figure 3A, left panels). However, it was highly expressed in histocytes, consistent with their high lysosomal content and the initial cloning of GPNMB from cells in the mononuclear phagocytic system [38]. GPNMB was only infrequently expressed in clear cell RCC (ccRCC) (median H‐score 0; range 0–83; Figure 3A, middle panels), with confirmation of this result using a second, independent antibody clone (supplementary material, Figure S1). There was similarly low GPNMB expression in papillary RCC (papRCC; Figure 3A, right panels) with rare exceptions, likely due in part to difficulty in excluding admixed histocytes during quantification (median H‐score 0.02; range 0–228). Interestingly, a larger subset of chromophobe RCC cases had GPNMB expression (chRCC; Figure 3B, right panels; Figure 6; and supplementary material, Figure S3), though the median expression was low (median H‐score 2; range 0–218). In contrast to chRCC, the limited number of oncocytomas included in the current study had uniformly low expression of GPNMB (median H‐score 10; range 1–16; Figure 3B, left panels).
Figure 3.
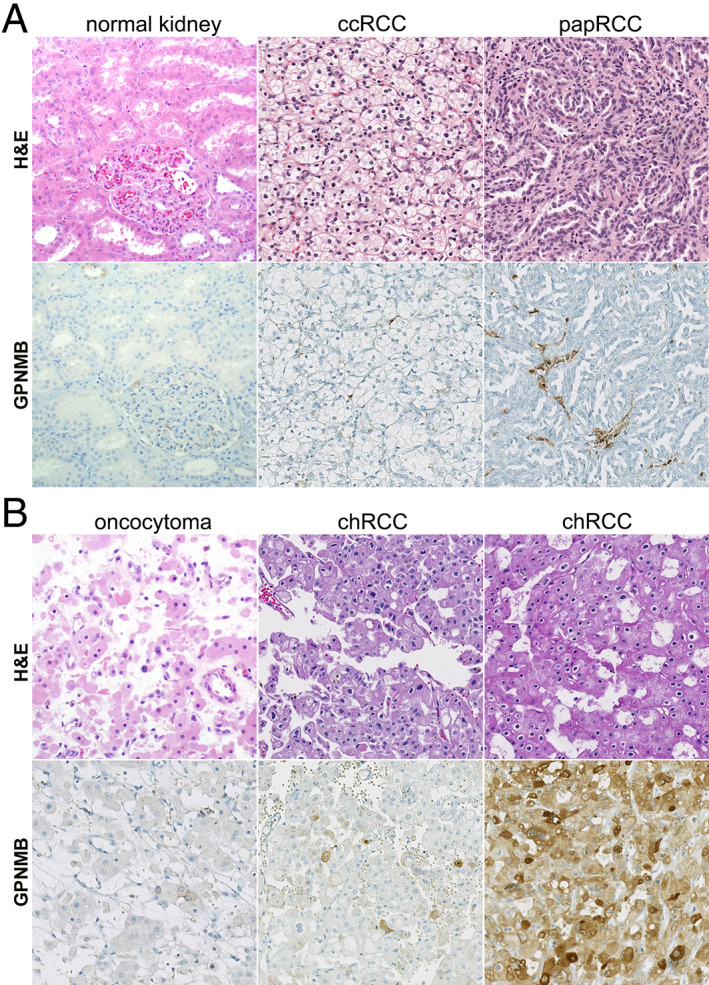
Representative GPNMB immunohistochemical (IHC) staining in common renal cell carcinoma (RCC) subtypes. (A) Representative H&E and GPNMB IHC images for normal renal parenchyma, clear cell RCC (ccRCC), and papillary RCC (papRCC). All images were reduced from 200× magnification. (B) Representative H&E and GPNMB IHC images for oncocytoma and chromophobe RCC (chRCC). Note variable staining in two different chRCC cases. All images were reduced from 200× magnification.
Figure 4.
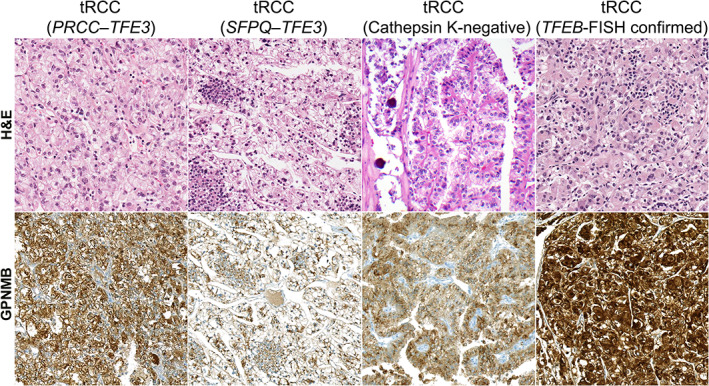
Representative GPNMB staining in translocation RCC (tRCC) cases. Representative H&E and GPNMB IHC images for TFE3‐driven tRCC [PRCC–TFE3 and SFPQ–TFE3 cases confirmed by fluorescence in situ hybridization (FISH)] as well as a tRCC negative for cathepsin K immunostaining and a TFEB‐driven (FISH‐confirmed) tRCC case. All images were reduced from 200× magnification.
Figure 5.
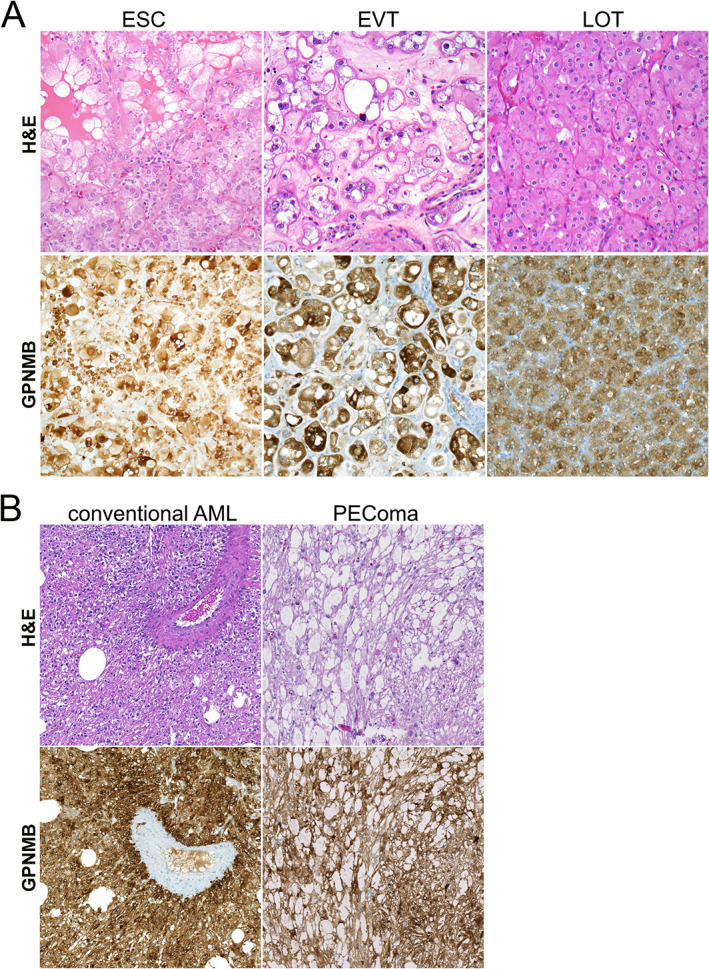
Representative GPNMB immunohistochemical (IHC) staining in TSC1/2/mTOR‐related tumors. (A) Representative H&E and GPNMB IHC images for eosinophilic solid and cystic carcinoma (ESC), eosinophilic vacuolated tumor (EVT), and low‐grade oncocytic tumor (LOT). All images were reduced from 200× magnification. (B) Representative H&E and GPNMB IHC images for conventional angiomyolipoma (AML) and perivascular epithelioid cell tumor (PEComa). All images were reduced from 200× magnification.
Figure 6.
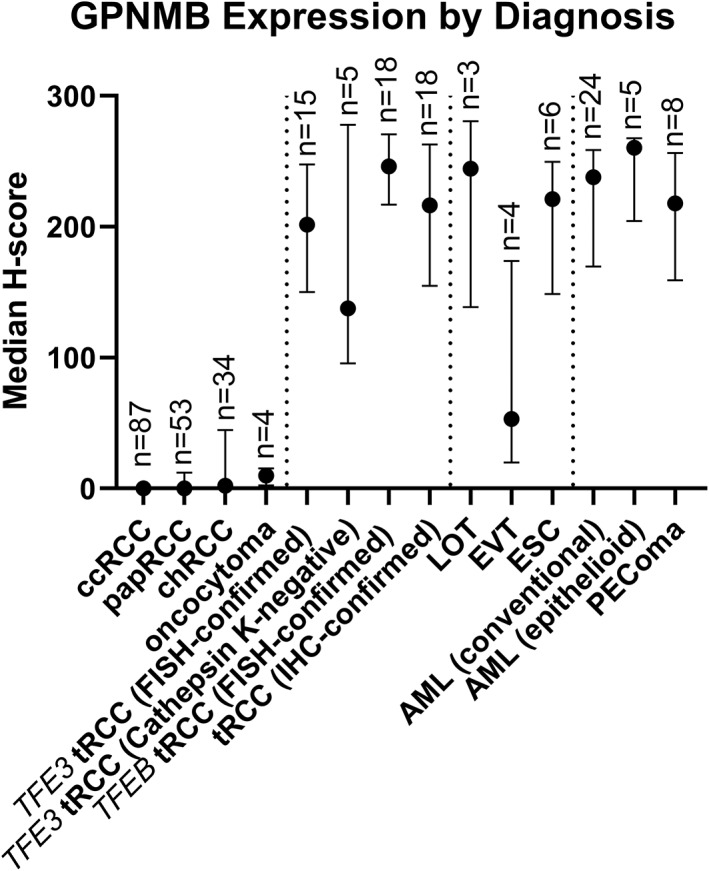
Digitally quantified GPNMB expression by renal tumor diagnosis. The point represents the median H‐score, and bars represent the interquartile range for each diagnostic group.
Although these differences in GPNMB protein expression among the common RCC subtypes were not statistically significant, the trends in these data parallel the statistically significant differences in GPNMB mRNA expression observed in the TCGA cohorts, where ccRCC cases have the lowest normalized counts, followed by papRCC and chRCC cases (supplementary material, Figure S4) [39, 40, 41]. The higher GPNMB expression among at least a subset of chRCCs compared with other common subtypes could potentially be consistent with the previously reported increased frequency of TSC1/TSC2/MTOR alterations among eosinophilic chRCC cases, ranging from 6% of cases in TCGA to 17% in a metastatic chRCC cohort [41, 42, 43]. Within the TCGA chRCC cohort (KICH) [41], a previous study has demonstrated that high FOXI1 mRNA expression is characteristic of chRCC, and outlier cases lacking FOXI1 expression (suggestive of misclassification) are enriched for TSC1/TSC2/MTOR alterations (present in 4/6 cases with low FOXI1 expression) [42]. Interestingly, we found that these FOXI1‐negative outlier tumors showed increased levels of GPNMB expression at the high end of the spectrum seen in chRCC, consistent with their underlying TSC1/TSC2/MTOR alterations (supplementary material, Figure S5).
Though sequencing data were not available in our chRCC cohort, we used genetically validated immunohistochemistry assays for TSC2, p‐S6, and p‐4EBP1 (Figure 2A) to assess potential loss of TSC2 within our chRCC cohort. Using this assay, only 1/34 chRCCs expressed detectable p‐S6; only 4/34 chRCCs expressed detectable p‐4EBP1; and 5/34 chRCCs had loss of TSC2 protein relative to background normal kidney. Since all of these markers (particularly the phospho‐epitopes) may show false‐negative staining with poor or delayed fixation (as is common with human tissue), we required both loss of TSC2 protein and detectable staining of p‐S6 and p‐4EBP1 to consider a case likely to have TSC2 genomic loss based on immunostaining. Only one case met these stringent criteria (supplementary material, Figure S6). Strikingly, this case was the chRCC case with the highest GPNMB expression in the entire cohort (mean H‐score 218). On morphologic re‐review, this case has many features of LOT, an entity which was not described at the time this case was originally reviewed.
GPNMB expression in tRCC was significantly higher than that seen in the more common RCC subtypes in most cases, consistent with the fact that GPNMB is a TFE3/TFEB‐target gene. We tested a wide spectrum of tRCC cases (n = 56 in total), including TFE3‐ and TFEB‐driven cases, some with FISH and some with IHC confirmation (Figures 4 and 6 and supplementary material, Figure S3). Overall, GPNMB expression was highest in TFEB tRCC with FISH confirmation (median H‐score 246; range 67–283; Figure 4, right panels), with similar results in TFE3 FISH‐confirmed tRCC (median H‐score 202; range 19–277; Figure 4, left panels) and IHC‐confirmed tRCC (median H‐score 216; range: 5–280). GPNMB scores in these tRCC subgroups were significantly higher than those seen in ccRCC (p < 0.0001 for all comparisons), papRCC (p ≤ 0.0001 for all comparisons), and chRCC (p = 0.0001 for TFEB FISH‐confirmed tRCC; p = 0.03 for TFE3 FISH‐confirmed tRCC; and p = 0.002 for IHC‐confirmed tRCC cases). Notably, a subset of tRCC cases that were cathepsin K‐negative also had high GPNMB H‐scores compared with other tRCC cases (median H‐score 138; range 64–279; Figure 4, second panels from right), with significantly higher scores than ccRCC (p = 0.002) and a similar trend for papRCC (p = 0.12), though this was not seen in comparison to chRCC (p > 0.99).
Similar to tRCC and as predicted by our in vitro data, GPNMB expression in renal tumor subtypes associated with TSC1/2/MTOR alterations was generally significantly higher than in the more common renal tumor subtypes (Figures 5 and 6 and supplementary material, Figure S3). For example, low‐grade oncocytic tumors (LOT; median H‐score 244; range 139–281; Figure 5A, right panels) and eosinophilic solid and cystic carcinomas (ESC; median H‐score 221; range 68–266; Figure 5A, left panels) showed significantly higher expression of GPNMB compared with ccRCC (p = 0.02 and 0.0003, respectively), with a similar trend compared with papRCC (p = 0.36 and 0.04, respectively), though no significant difference was seen when these tumors were compared with chRCC. Similarly, conventional and epithelioid AML had significantly higher GPNMB expression (median H‐scores 238 and 260; ranges 22–282 and 184–270, respectively; Figure 5B) when compared with ccRCC (p < 0.0001 and p = 0.0001, respectively), papRCC (p < 0.0001 and p = 0.01, respectively), and chRCC (p = 0.0001 and p = 0.18, respectively). The only exception to this pattern was for the small number of eosinophilic vacuolated tumors (EVT) tested (n = 4), where there was a wide range in GPNMB expression and no significant differences when compared with more common RCC subtypes (Figure 5A, middle panels). In total, eight PEComas from seven patients were analyzed, including three positive for TFE3 gene rearrangements (median H‐score 229) and two lacking TFE3 gene rearrangements (median H‐score 164), without a significant difference in GPNMB expression between these groups (p = 0.2).
Performance characteristics of GPNMB immunohistochemistry
Quantitative H‐scores are not typically used for clinical diagnosis, but we assessed the performance characteristics of the GPNMB assay using an H‐score cutoff of 150 to define positive and negative cases, analogous to previous work with other biomarkers for tRCC [19]. With this cutoff, GPNMB IHC identified 82% (27/33) of FISH‐confirmed tRCC cases, 83% (15/18) of IHC‐confirmed tRCC cases versus 0% (0/87) of ccRCCs, 6% (3/53) of papRCCs, 3% (1/34) of chRCCs, and 0% (0/4) of oncocytomas. Among TSC1/2/mTOR‐related tumors, this threshold identified 67% (2/3) of LOT cases, 83% (5/6) of ESC cases, 86% (25/29) of AMLs, and 88% (7/8) of PEComas, but only 25% (1/4) of EVT cases.
To make this study more relevant to clinical practice, we also tested a simple visual scoring system from 0 to 3+ (supplementary material, Figure S7). Digital H‐scores were significantly different between all visual score groups (p ≤ 0.001 for all comparisons), validating the digital scoring system. When we used a cutoff of 3+ by visual score (>90% of cells with strong intensity staining) to identify GPNMB‐positive cases, GPNMB IHC identified 70% (39/56) of all tRCC cases [including 40% of (or 2/5) cathepsin K‐negative cases] versus 0% (0/87) of ccRCCs, 2% (1/53) of papRCCs, 3% (1/34) of chRCCs, and 0% (0/4) of oncocytomas. Among TSC1/2/mTOR‐related tumors, this threshold identified 67% (2/3) of LOT cases, 67% (4/6) of ESC cases, 86% (25/29) of AMLs, and 63% (5/8) of PEComas, but none (0/4) of EVT cases.
Discussion
The MiT transcription factor family includes TFEB and TFE3. While these factors are key regulators of lysosomal gene expression in normal cells, they may become oncogenic drivers when dysregulated by involvement in gene rearrangements or amplifications [18]. In current models, physiologic mTORC1 activity phosphorylates MiT/TFE proteins, resulting in their cytoplasmic retention and inactivation [44]. In contrast, short‐term pharmacological mTORC1 inactivation with Torin 1 promotes their dephosphorylation, nuclear localization, and activity [45, 46, 47, 48, 49, 50]. However, a number of different lines of evidence have emerged recently to suggest that the role of mTORC1 in TFE3/TFEB regulation is likely much more complex than previously appreciated. In a small number of previous studies, including one by our group, constitutive mTORC1 hyperactivity (via TSC1/2 loss) paradoxically positively regulated TFEB‐dependent lysosomal v‐ATPase expression [51] and promoted TFE3/TFEB nuclear localization in an mTORC1‐dependent manner [21, 52, 53, 54].
While paradoxical, these results are highly consistent with genomic data from human tumors. TFE3/TFEB gene rearrangements or TSC1/2 loss‐of‐function alterations are established and mutually exclusive molecular drivers of PEComas, suggesting that the biological consequences of these two alterations may be overlapping [55, 56, 57, 58]. There is also emerging evidence that rare PEComas may be driven by folliculin inactivation [59], an alteration that also results in increased TFE3/TFEB activity [33, 60]. Supporting the concept that TFE3/TFEB rearrangements and TSC1/2 inactivation have biologically overlapping effects, MiT gene targets such as melanocyte markers (PMEL17 or Melan A) [61], cathepsin K (a lysosomal protease) [62], and TRIM63 [63] are all diagnostic biomarkers of PEComas [19, 58, 64] and tRCC [19, 65, 66], in addition to identifying RCC associated with TSC1/2 loss [7, 19]. Though it would be of interest to quantify TFE3 or TFEB nuclear protein expression in RCC associated with TSC1/2/MTOR alterations, technical issues with these IHC assays have been prevalent [67] and they remain insufficiently sensitive and specific for tRCC, further motivating the search for additional markers of their activity.
In this context, it should not come as a surprise that GPNMB – a well‐characterized TFE3/TFEB gene target [33, 34] and marker of tRCC [22] – is also highly expressed in human kidney cell lines and murine kidney tumors with TSC2 deletion and regulated by mTOR kinase and TFEB/TFE3 in vitro. GPNMB is expressed at relatively low levels in most normal tissues (with a prominent exception being histiocytes) but highly expressed in aggressive cancers such as melanoma (where it was first described), triple‐negative breast cancer (TNBC), and invasive gliomas, where it is generally associated with poor prognosis [68]. There is emerging evidence for the direct role of GPNMB in tumor progression [69], cellular intravasation, invasion, metastases [70], and angiogenesis [71]. Consistent with its regulation by MiTF/TFEB/TFE3, GPNMB expression is upregulated in mouse models of numerous MiT‐family neoplasms, including tRCC [22] and alveolar soft part sarcoma [70], with a previous small study also documenting increased expression in a series of nine TFE3‐driven human tRCC cases [22].
Here, we confirm these data in a much larger series of 56 tRCC cases, encompassing a spectrum of TFE3‐ and TFEB‐driven tumors. In tuberous sclerosis‐associated neoplasms, previous work has also demonstrated GPNMB overexpression in angiomyolipomas [72, 73], lymphangioleiomyomatosis [74], and subependymal giant cell astrocytomas (SEGAs) [75] compared with their respective normal tissues, findings that we confirm with a different assay in both sporadic conventional and epithelioid variants of AML as well as in PEComas. Notably, no prior studies have examined GPNMB expression in TSC1/2/MTOR alteration‐associated RCC cases, including LOT, ESC or EVT, likely because these have only recently been recognized [14]. In the current study, despite a relatively small sample size, we find that GPNMB expression is similar among ESC and LOT tumors to that seen in tRCCs and considerably lower in ccRCCs, papRCCs, chRCCs, and oncocytomas. Of note, we found that GPNMB expression is more variable among a small series of EVTs. Future, more extensive studies will be necessary to confirm these findings as these entities are increasingly recognized and large case series are accrued. Nevertheless, our work reveals an important caveat surrounding the use of TFE3/TFEB‐transcriptional targets when used as diagnostic markers in renal tumors.
Among conventional RCC subtypes, it is notable that GPNMB expression in a subset of chRCCs is somewhat higher than that seen in oncocytoma, papRCC or ccRCC, while lower than tRCC and other TSC1/2/MTOR alteration‐associated RCC subtypes. The higher expression could reflect the fact that some eosinophilic chRCC cases may harbor underlying alterations in TSC1/2 or mTOR signaling pathways [41, 42]; indeed, we show evidence using genetically validated IHC assays that the chRCC case in our series with the highest GPNMB expression levels had concurrent TSC2 protein loss and mTORC1 signaling activation, with morphologic features potentially consistent with LOT. Similarly, expression outlier cases from the TCGA KICH cohort which are enriched for TSC1/2/MTOR alterations [42] also showed high GPNMB expression levels (supplementary material, Figure S5), and future sequencing studies may be helpful to definitively determine whether GPNMB positivity could be an indicator of underlying TSC1/2/MTOR alterations in chRCC cases. GPNMB positivity in a subset of chRCCs is also interesting in light of the fact that one prior study suggested that GPNMB expression might distinguish Birt–Hogg–Dubé‐associated hybrid oncocytic renal tumors from sporadic chRCC [76], consistent with emerging data implicating folliculin in the negative regulation of TFE3/TFEB activity [33, 60]. Given the rarity of Birt–Hogg–Dubé, it seems unlikely that the positivity that we observed in a subset of chRCCs could be due to underlying sporadic or inherited FLCN mutations; however, future sequencing studies may specifically address this question. Curiously, we did not find evidence of GPNMB expression in a substantial proportion of ccRCC cases (using two independent antibodies), despite prior evidence of non‐genomic mTOR activation in a large proportion of ccRCCs. It is possible that the cellular context and the underlying mechanism of mTORC1 activation may further modulate the effects of mTOR on GPNMB expression in different RCC subtypes.
Molecularly, TFE3/TFEB activity and mTOR activity may exist in a reciprocal feed‐forward signaling loop, each driving the other. tRCC demonstrates increased mTOR activity compared with ccRCC [1], and recent work suggests that the mechanism for this may be increased RAGD transcription downstream of TFEB activity [77]. Reciprocally, we have shown previously [21], and herein, that mTOR activity can drive TFEB/TFE3 activity, as evidenced by GPNMB expression. Given the overlapping molecular pathways active in tRCC and TSC1/2/mTOR‐associated renal tumors, it is notable that the clinical behavior of these tumors is highly variable. While tRCC [4] and subsets of ESC [14] can behave aggressively, many of the other TSC1/2/MTOR alteration‐associated tumors including AML, LOT, and EVT are almost all indolent in the initial descriptions [14]. Future work may identify additional molecular alterations or signaling perturbations that drive aggressive behavior in the context of TSC1/2/MTOR alterations and/or TFE3/TFEB activity during renal tumorigenesis.
Clinically, we propose that GPNMB may be useful to add to our arsenal of IHC screening tools for tRCC and TSC1/2/MTOR alteration‐associated renal tumors, distinguishing them from common morphologic subtypes. While screening all renal tumors with GPNMB would be neither practical nor necessary given the ease of diagnosis of conventional subtypes by morphologic analysis alone, GPNMB could be useful as an adjunct marker, similar to cathepsin K, for screening unclassified RCC cases. A GPNMB H‐score of 150 effectively distinguished most common RCC subtypes from tRCC and TSC1/2/MTOR‐alteration associated RCC. Since most clinical IHC assays are interpreted dichotomously, in practice it may be most beneficial to only consider cases with >90% of cells showing intense membrane staining by visual scoring as GPNMB‐positive, and histiocytes are important to assess as an internal positive control in each case. In our small sample of five cathepsin K‐negative cases, for example, this cutoff still detected 40% of cases as positive, suggesting that GPNMB could be a useful marker in addition to cathepsin K in unclassified cases. It is notable that some tRCC cases in each diagnostic category had lower expression of GPNMB than others, and the source of this variability is unclear. Some effect of sample fixation and/or storage prior to staining may contribute, as we anecdotally saw a higher fraction of GPNMB‐negative cases among those that were also cathepsin K‐negative. Alternatively, it is possible that a subset of tRCC cases does not maintain a TFE3/TFEB‐regulated gene expression program, either due to co‐occurring genomic alterations or due to peculiarities of their underlying specific gene fusion event. Finally, we cannot exclude non‐MiT/TFE‐driven mechanisms that may affect GPNMB expression. Limitations of the current study include the relatively small sample sizes of emerging RCC diagnostic entities and the lack of associated genomic data for correlation with GPNMB expression.
In addition to its utility as a biomarker, characterizing GPNMB expression in renal tumors is clinically significant since it may serve as a future clinical diagnostic biomarker or even as a therapeutic target for tRCC. As GPNMB is a transmembrane protein on the cell surface, radiolabeled ligands or antibodies could be used for diagnostic imaging of tRCC or TSC1/2/MTOR alteration‐associated RCC. Although most TSC1/2/MTOR alteration‐related tumors are relatively indolent, tRCC may be aggressive and effective targeted therapies have yet to be identified. Notably, an antibody–drug conjugate (ADC) specific for GPNMB – glembatumumab vedotin (CDX‐011) – was well tolerated in clinical trials for breast cancer [68, 78]. Though these trials were disappointing for breast tumors [79], future work may examine the efficacy of similar ADC strategies in tRCC.
In conclusion, we have demonstrated that GPNMB is highly expressed in human kidney cell lines and murine kidney tumors with TSC2 deletion and regulated by mTOR kinase and TFEB/TFE3 in vitro, similar to other MiT/TFE targets. This study adds to emerging data suggesting that tumors harboring TFE3/TFEB rearrangements and those with TSC1/2/MTOR alterations may overlap phenotypically due to high underlying TFE3/TFEB activity, revealing a major caveat for using TFE3/TFEB targets as diagnostic biomarkers. Clinically, GPNMB may be useful as a sensitive screening tool for tRCC and TSC1/2/MTOR alteration‐associated renal tumors when paired with confirmatory genomic assays and may help to distinguish these tumors from more common subtypes.
Author contributions statement
TLL, PA and KA conceived the study. TLL, DCS and KA drafted the manuscript. PA, DCS, JW, TV, HBL, IV, AM, GJN and KA completed the data collection and analysis. All the authors critically reviewed the manuscript and agreed to submit for publication.
Supporting information
Figure S1. Comparison of GPNMB immunohistochemistry using two independent antibody clones
Figure S2. Immunoblotting of HEK293T TSC2 KO cells treated with GPNMB siRNA confirms that the higher molecular weight band (~95 kDa, arrow) is the correct one and the lower band (denoted by *) is non‐specific
Figure S3. Digitally quantified GPNMB expression by renal tumor diagnosis
Figure S4. GPNMB mRNA expression by TCGA RCC cohort
Figure S5. GPNMB versus FOXI1 expression in the TCGA chRCC (KICH) cohort, with annotated TSC1/TSC2/MTOR mutations
Figure S6. GPNMB, TSC2, p‐S6, and p‐4EBP1 expression in benign and tumor tissue from a case classified as chRCC
Figure S7. Digitally quantified GPNMB expression compared with visually scored expression
Acknowledgements
This research was supported in part by the CDMRP KCRP grant W81XWH‐20‐1‐0843 (to KA and TLL), as well as W81XWH‐19‐1‐0781 (to KA and TLL) and the NCI Cancer Center Support Grant 5P30CA006973‐52. Additional funding was provided by Joey's Wings Foundation and Dahan Translocation Carcinoma Fund, and cell line and mouse models were obtained from the TSC Alliance.
Conflict of interest statement: TLL has received research support from Roche/Ventana, DeepBio, and Myriad Genetics for other studies. No other conflicts of interest were declared.
References
- 1. Argani P, Hicks J, De Marzo AM, et al. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol 2010; 34: 1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Argani P, Laé M, Hutchinson B, et al. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific Alpha‐TFEB gene fusion by immunohistochemistry, RT‐PCR, and DNA PCR. Am J Surg Pathol 2005; 29: 230–240. [DOI] [PubMed] [Google Scholar]
- 3. Ross H, Argani P. Xp11 translocation renal cell carcinoma. Pathology 2010; 42: 369–373. [DOI] [PubMed] [Google Scholar]
- 4. Gandhi JS, Malik F, Amin MB, et al. MiT family translocation renal cell carcinomas: a 15th anniversary update. Histol Histopathol 2020; 35: 125–136. [DOI] [PubMed] [Google Scholar]
- 5. Trpkov K, Hes O, Bonert M, et al. Eosinophilic, solid, and cystic renal cell carcinoma: clinicopathologic study of 16 unique, sporadic neoplasms occurring in women. Am J Surg Pathol 2016; 40: 60–71. [DOI] [PubMed] [Google Scholar]
- 6. Trpkov K, Abou‐Ouf H, Hes O, et al. Eosinophilic solid and cystic renal cell carcinoma (ESC RCC): further morphologic and molecular characterization of ESC RCC as a distinct entity. Am J Surg Pathol 2017; 41: 1299–1308. [DOI] [PubMed] [Google Scholar]
- 7. Palsgrove DN, Li Y, Pratilas CA, et al. Eosinophilic solid and cystic (ESC) renal cell carcinomas harbor TSC mutations: molecular analysis supports an expanding clinicopathologic spectrum. Am J Surg Pathol 2018; 42: 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mehra R, Vats P, Cao X, et al. Somatic bi‐allelic loss of TSC genes in eosinophilic solid and cystic renal cell carcinoma. Eur Urol 2018; 74: 483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trpkov K, Bonert M, Gao Y, et al. High‐grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology 2019; 75: 440–442. [DOI] [PubMed] [Google Scholar]
- 10. He H, Trpkov K, Martinek P, et al. “High‐grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch 2018; 473: 725–738. [DOI] [PubMed] [Google Scholar]
- 11. Chen YB, Mirsadraei L, Jayakumaran G, et al. Somatic mutations of TSC2 or MTOR characterize a morphologically distinct subset of sporadic renal cell carcinoma with eosinophilic and vacuolated cytoplasm. Am J Surg Pathol 2019; 43: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trpkov K, Williamson SR, Gao Y, et al. Low‐grade oncocytic tumour of kidney (CD117‐negative, cytokeratin 7‐positive): a distinct entity? Histopathology 2019; 75: 174–184. [DOI] [PubMed] [Google Scholar]
- 13. Tjota M, Chen H, Parilla M, et al. Eosinophilic renal cell tumors with a TSC and MTOR gene mutations are morphologically and immunohistochemically heterogenous: clinicopathologic and molecular study. Am J Surg Pathol 2020; 44: 943–954. [DOI] [PubMed] [Google Scholar]
- 14. Trpkov K, Williamson SR, Gill AJ, et al. Novel, emerging and provisional renal entities: the Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 2021; 34: 1167–1184. [DOI] [PubMed] [Google Scholar]
- 15. Argani P, Lal P, Hutchinson B, et al. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003; 27: 750–761. [DOI] [PubMed] [Google Scholar]
- 16. Green WM, Yonescu R, Morsberger L, et al. Utilization of a TFE3 break‐apart FISH assay in a renal tumor consultation service. Am J Surg Pathol 2013; 37: 1150–1163. [DOI] [PubMed] [Google Scholar]
- 17. Argani P, Yonescu R, Morsberger L, et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin‐embedded material using a break‐apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol 2012; 36: 1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kauffman EC, Ricketts CJ, Rais‐Bahrami S, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol 2014; 11: 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang XM, Zhang Y, Mannan R, et al. TRIM63 is a sensitive and specific biomarker for MiT family aberration‐associated renal cell carcinoma. Mod Pathol 2021; 34: 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tretiakova MS. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: series of 4 primary tumors and 2 metastases. Hum Pathol 2018; 80: 65–75. [DOI] [PubMed] [Google Scholar]
- 21. Asrani K, Murali S, Lam B, et al. mTORC1 feedback to AKT modulates lysosomal biogenesis through MiT/TFE regulation. J Clin Invest 2019; 129: 5584–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baba M, Furuya M, Motoshima T, et al. TFE3 Xp11.2 translocation renal cell carcinoma mouse model reveals novel therapeutic targets and identifies GPNMB as a diagnostic marker for human disease. Mol Cancer Res 2019; 17: 1613–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schultz L, Chaux A, Albadine R, et al. Immunoexpression status and prognostic value of mTOR and hypoxia‐induced pathway members in primary and metastatic clear cell renal cell carcinomas. Am J Surg Pathol 2011; 35: 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chaux A, Schultz L, Albadine R, et al. Immunoexpression status and prognostic value of mammalian target of rapamycin and hypoxia‐induced pathway members in papillary cell renal cell carcinomas. Hum Pathol 2012; 43: 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaux A, Albadine R, Schultz L, et al. Dysregulation of the mammalian target of rapamycin pathway in chromophobe renal cell carcinomas. Hum Pathol 2013; 44: 2323–2330. [DOI] [PubMed] [Google Scholar]
- 26. Overwater IE, Swenker R, van der Ende EL, et al. Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet 2016; 24: 1688–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hong F, Larrea MD, Doughty C, et al. mTOR‐raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell 2008; 30: 701–711. [DOI] [PubMed] [Google Scholar]
- 28. Yu J, Astrinidis A, Howard S, et al. Estradiol and tamoxifen stimulate LAM‐associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am J Physiol Lung Cell Mol Physiol 2004; 286: L694–700. [DOI] [PubMed] [Google Scholar]
- 29. Walker A, Singh A, Tully E, et al. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic Biol Med 2018; 120: 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodrum C, Nobil A, Dabora SL. Comparison of three rapamycin dosing schedules in A/J Tsc2 +/− mice and improved survival with angiogenesis inhibitor or asparaginase treatment in mice with subcutaneous tuberous sclerosis related tumors. J Transl Med 2010; 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim YC, Park HW, Sciarretta S, et al. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun 2014; 5: 4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asrani K, Sood A, Torres A, et al. mTORC1 loss impairs epidermal adhesion via TGF‐β/Rho kinase activation. J Clin Invest 2017; 127: 4001–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hong SB, Oh H, Valera VA, et al. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One 2010; 5: e15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ripoll VM, Meadows NA, Raggatt LJ, et al. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene 2008; 413: 32–41. [DOI] [PubMed] [Google Scholar]
- 35. Pastore N, Vainshtein A, Klisch TJ, et al. TFE3 regulates whole‐body energy metabolism in cooperation with TFEB. EMBO Mol Med 2017; 9: 605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pastore N, Brady OA, Diab HI, et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016; 12: 1240–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Onda H, Lueck A, Marks PW, et al. Tsc2 +/− mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 1999; 104: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shikano S, Bonkobara M, Zukas PK, et al. Molecular cloning of a dendritic cell‐associated transmembrane protein, DC‐HIL, that promotes RGD‐dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem 2001; 276: 8125–8134. [DOI] [PubMed] [Google Scholar]
- 39. Cancer Genome Atlas Research Network , Linehan WM, Spellman PT, et al. Comprehensive molecular characterization of papillary renal‐cell carcinoma. N Engl J Med 2016; 374: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. The Cancer Genome Atlas Research Network . Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014; 26: 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tong K, Hu Z. FOXI1 expression in chromophobe renal cell carcinoma and renal oncocytoma: a study of The Cancer Genome Atlas transcriptome‐based outlier mining and immunohistochemistry. Virchows Arch 2021; 478: 647–658. [DOI] [PubMed] [Google Scholar]
- 43. Roldan‐Romero JM, Santos M, Lanillos J, et al. Molecular characterization of chromophobe renal cell carcinoma reveals mTOR pathway alterations in patients with poor outcome. Mod Pathol 2020; 33: 2580–2590. [DOI] [PubMed] [Google Scholar]
- 44. Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 2018; 37: e98804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roczniak‐Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Settembre C, Zoncu R, Medina DL, et al. A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31: 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martina JA, Diab HI, Lishu L, et al. The nutrient‐responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 2014; 7: ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Napolitano G, Esposito A, Choi H, et al. mTOR‐dependent phosphorylation controls TFEB nuclear export. Nat Commun 2018; 9: 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sha Y, Rao L, Settembre C, et al. STUB1 regulates TFEB‐induced autophagy–lysosome pathway. EMBO J 2017; 36: 2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peña‐Llopis S, Vega‐Rubin‐de‐Celis S, Schwartz JC, et al. Regulation of TFEB and V‐ATPases by mTORC1. EMBO J 2011; 30: 3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Betschinger J, Nichols J, Dietmann S, et al. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 2013; 153: 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kawano H, Ito Y, Kanai F, et al. Aberrant differentiation of Tsc2‐deficient teratomas associated with activation of the mTORC1‐TFE3 pathway. Oncol Rep 2015; 34: 2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Villegas F, Lehalle D, Mayer D, et al. Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 2019; 24: 257–270.e8. [DOI] [PubMed] [Google Scholar]
- 55. Malinowska I, Kwiatkowski DJ, Weiss S, et al. Perivascular epithelioid cell tumors (PEComas) harboring TFE3 gene rearrangements lack the TSC2 alterations characteristic of conventional PEComas: further evidence for a biological distinction. Am J Surg Pathol 2012; 36: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Agaram NP, Sung YS, Zhang L, et al. Dichotomy of genetic abnormalities in PEComas with therapeutic implications. Am J Surg Pathol 2015; 39: 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giannikou K, Malinowska IA, Pugh TJ, et al. Whole exome sequencing identifies TSC1/TSC2 biallelic loss as the primary and sufficient driver event for renal angiomyolipoma development. PLoS Genet 2016; 12: e1006242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol 2010; 34: 1395–1406. [DOI] [PubMed] [Google Scholar]
- 59. Akumalla S, Madison R, Lin DI, et al. Characterization of clinical cases of malignant PEComa via comprehensive genomic profiling of DNA and RNA. Oncology 2020; 98: 905–912. [DOI] [PubMed] [Google Scholar]
- 60. Napolitano G, Di Malta C, Esposito A, et al. A substrate‐specific mTORC1 pathway underlies Birt–Hogg–Dubé syndrome. Nature 2020; 585: 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Du J, Miller AJ, Widlund HR, et al. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol 2003; 163: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Motyckova G, Weilbaecher KN, Horstmann M, et al. Linking osteopetrosis and pycnodysostosis: regulation of cathepsin K expression by the microphthalmia transcription factor family. Proc Natl Acad Sci U S A 2001; 98: 5798–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pablo Tortola C, Fielitz B, Li Y, et al. Activation of tripartite motif containing 63 expression by transcription factor EB and transcription factor binding to immunoglobulin heavy chain enhancer 3 is regulated by protein kinase D and class IIa histone deacetylases. Front Physiol 2020; 11: 550506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29: 1558–1575. [DOI] [PubMed] [Google Scholar]
- 65. Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001; 158: 2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martignoni G, Pea M, Gobbo S, et al. Cathepsin‐K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol 2009; 22: 1016–1022. [DOI] [PubMed] [Google Scholar]
- 67. Sharain RF, Gown AM, Greipp PT, et al. Immunohistochemistry for TFE3 lacks specificity and sensitivity in the diagnosis of TFE3‐rearranged neoplasms: a comparative, 2‐laboratory study. Hum Pathol 2019; 87: 65–74. [DOI] [PubMed] [Google Scholar]
- 68. Rose AAN, Biondini M, Curiel R, et al. Targeting GPNMB with glembatumumab vedotin: current developments and future opportunities for the treatment of cancer. Pharmacol Ther 2017; 179: 127–141. [DOI] [PubMed] [Google Scholar]
- 69. Maric G, Annis MG, MacDonald PA, et al. GPNMB augments Wnt‐1 mediated breast tumor initiation and growth by enhancing PI3K/AKT/mTOR pathway signaling and β‐catenin activity. Oncogene 2019; 38: 5294–5307. [DOI] [PubMed] [Google Scholar]
- 70. Tanaka M, Homme M, Yamazaki Y, et al. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res 2017; 77: 897–907. [DOI] [PubMed] [Google Scholar]
- 71. Rose AAN, Annis MG, Dong Z, et al. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS One 2010; 5: e12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Siroky BJ, Yin H, Dixon BP, et al. Evidence for pericyte origin of TSC‐associated renal angiomyolipomas and implications for angiotensin receptor inhibition therapy. Am J Physiol Renal Physiol 2014; 307: F560–F570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martin KR, Zhou W, Bowman MJ, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun 2017; 8: 15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Prizant H, Taya M, Lerman I, et al. Estrogen maintains myometrial tumors in a lymphangioleiomyomatosis model. Endocr Relat Cancer 2016; 23: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tyburczy ME, Kotulska K, Pokarowski P, et al. Novel proteins regulated by mTOR in subependymal giant cell astrocytomas of patients with tuberous sclerosis complex and new therapeutic implications. Am J Pathol 2010; 176: 1878–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Furuya M, Hong SB, Tanaka R, et al. Distinctive expression patterns of glycoprotein non‐metastatic B and folliculin in renal tumors in patients with Birt–Hogg–Dubé syndrome. Cancer Sci 2015; 106: 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017; 356: 1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bendell J, Saleh M, Rose AAN, et al. Phase I/II study of the antibody–drug conjugate glembatumumab vedotin in patients with locally advanced or metastatic breast cancer. J Clin Oncol 2014; 32: 3619–3625. [DOI] [PubMed] [Google Scholar]
- 79. Yardley DA, Weaver R, Melisko ME, et al. EMERGE: a randomized phase II study of the antibody–drug conjugate glembatumumab vedotin in advanced glycoprotein NMB‐expressing breast cancer. J Clin Oncol 2015; 33: 1609–1619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of GPNMB immunohistochemistry using two independent antibody clones
Figure S2. Immunoblotting of HEK293T TSC2 KO cells treated with GPNMB siRNA confirms that the higher molecular weight band (~95 kDa, arrow) is the correct one and the lower band (denoted by *) is non‐specific
Figure S3. Digitally quantified GPNMB expression by renal tumor diagnosis
Figure S4. GPNMB mRNA expression by TCGA RCC cohort
Figure S5. GPNMB versus FOXI1 expression in the TCGA chRCC (KICH) cohort, with annotated TSC1/TSC2/MTOR mutations
Figure S6. GPNMB, TSC2, p‐S6, and p‐4EBP1 expression in benign and tumor tissue from a case classified as chRCC
Figure S7. Digitally quantified GPNMB expression compared with visually scored expression