

Summary
Although the impressive clinical responses seen with modern cancer immunotherapy are currently limited to a subset of patients, the underlying paradigm shift has resulted in now hardly a segment in oncology that has not been touched by the immuno‐oncology revolution. A growing body of data indicates that radiation therapy (RT) can modulate the tumour immune microenvironment and complement cancer immunotherapy via non‐overlapping mechanisms to reinvigorate immunity against cancer. Thus, increasingly RT is viewed as a highly unique partner for immunotherapy across the spectrum of cancer settings, as radiobiology and cancer immunology foreseeably become more intertwined. Considering these developments, this review summarises the key concepts and terminology in immunology for the radiation oncologist, with a focus on the cancer setting and with reference to important recent advances. These concepts will provide a starting point for understanding the strategies that underlie current and emerging immunotherapy trials, as well as the indirect effects of RT by which immune responses against cancer are shaped.
Keywords: cancer immunology, cancer biology, immunotherapy, Radiotherapy
Introduction
A solid grasp of cancer biology is fundamental for the knowledge base of a radiation oncologist. While the study of cancer has in large part centred on its cell‐intrinsic and cell‐autonomous effects, the concept of the host immune system influencing its initiation and progression has been debated for over a century. In the late 1990’s, this question was put to rest when advances in animal models and in our knowledge of immunology made possible the unequivocal demonstration of the immune system as a critical extrinsic tumour suppressor. 1 The clinical importance of this paradigm shift was significantly spotlighted by breakthroughs of modern cancer immunotherapy, namely that of immune checkpoint blockade (ICB) and chimeric antigen receptor (CAR) T‐cell therapy. Together, the impressive and durable clinical responses produced by these therapies highlight the potential of harnessing the patient's immune system to recognise and eradicate tumour cells. 2 Thus, modern cancer immunotherapy represents a major cancer treatment modality of distinct primary mechanism of action compared to that of surgery, radiation therapy (RT), chemotherapy and targeted therapy.
Concurrently, the lack of response to cancer immunotherapy in a considerable proportion of patients has brought into focus resistance mechanisms that preclude cancer immunity. These can be classified as primary, whereby the cancer lacks a pre‐existing contexture that is permissive for intervention; adaptive, whereby the cancer is recognised by the immune system but is able to protect itself from immune attack through modifications; or acquired, whereby the cancer had initially responded to immunotherapy for a period of time before relapsing. 3 In all three contexts, an increasing body of pre‐clinical and clinical data have indicated that RT can complement cancer immunotherapy by modulating immune processes and overcoming barriers within the tumour microenvironment, thus providing an opportunity for radiation oncologists to play a revitalised role in the multi‐disciplinary care of the cancer patient in the immuno‐oncology era. 4 , 5 To this end, this review aims to serve as a refresher on the key concepts and terminology in immunology for the radiation oncologist, with a focus on recent advances in this area and their relevance to the cancer setting. These concepts will provide a starting point for understanding the rationale behind current and emerging immunotherapy trials and the role that RT can play in shaping immune responses against cancer, the latter of which will form the focus of a separate article in this Special Issue. Key terms are highlighted bold in this review.
An overview of the immune system
Innate and adaptive immunity
The human immune system is a complex and tightly regulated defence system against pathogens. It is classically categorised into the innate and adaptive arms, distinguished by their response kinetics as well as antigen specificity. Innate immune responses are rapid and recognise common molecular signatures associated with microbial pathogens (pathogen‐associated molecular patterns, PAMPs) or host cell damage (danger‐associated molecular patterns, DAMPs). In contrast, adaptive immunity has slower response kinetics but is marked by clonal specificity for antigens and immunological memory. Innate immune cell types are diverse; those most relevant to cancer immunology include dendritic cells (DCs), natural killer (NK) cells, neutrophils, macrophages and a group of poorly defined immature myeloid cells called myeloid‐derived suppressor cells (MDSCs) (Fig. 1). T and B cells constitute the cellular subsets of adaptive immunity (Fig. 1). Linking these two arms to form a highly coordinated network of inter‐dependent components is a myriad of soluble mediators such as cytokines and growth factors. At the heart of cancer‐directed immune responses are arguably CD8+ T cells due to their unique specificity for endogenous antigens as expressed by tumour cells (Box 1). Importantly, CD8+ T cells are heavily regulated by other immune cells from both innate and adaptive arms. The major immune cell subsets are described below, with brief mention of associated cytokines and other molecular components where relevant.
Fig. 1.
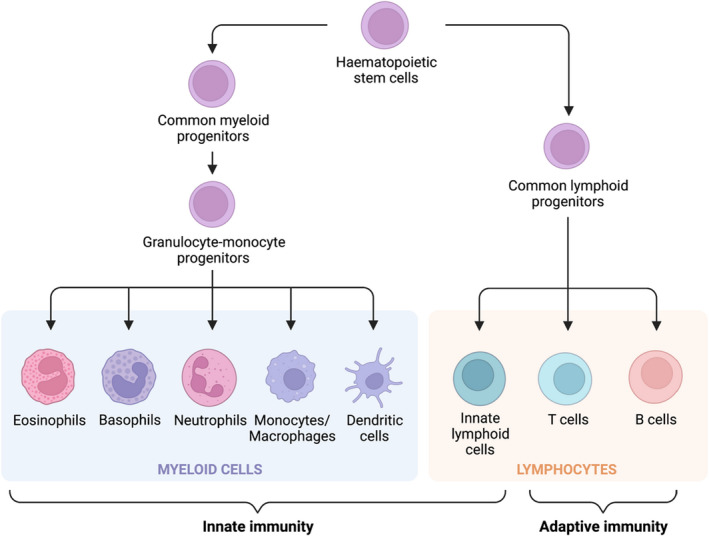
The major immune cell subsets and their lineages that make up our innate and adaptive immune defences. Although these cells are categorised as either elements of innate or adaptive immunity, there is significant interplay between the two arms of the immune system. Important subsets not in this diagram are natural killer (NK) cells, which are a member of innate lymphoid cells, and myeloid‐derived suppressor cells (MDSCs), which are a poorly defined group of immature myeloid cells that include progenitors of neutrophils and monocytes/macrophages. Illustration created with Biorender.com. [Colour figure can be viewed at wileyonlinelibrary.com]
Box 1. Core concepts for T cells in relation to cancer.
CD8+ T cells are central to cancer immunology due to their specificity for endogenous antigens.
T‐cell priming requires three signals: (i) T‐cell receptor (TCR) engagement of an antigen presented on a major histocompatibility complex (MHC), (ii) co‐stimulation of CD28 on the T cell and (iii) the presence of appropriate inflammatory cytokines.
T‐cell priming is most commonly performed by dendritic cells (DCs). DCs undergo maturation and gain the ability to prime T cells upon phagocytosis of antigens and detection of danger signals.
Activation of T cells induces the expression of immune checkpoint receptors as an in‐built breaking mechanism. Persistent T‐cell stimulation leads to persistently high expression of checkpoint receptors and a terminally exhausted state.
Conventional cancer therapies can evoke T‐cell priming by releasing tumour antigens and danger signals.
Tissue‐resident memory T cells (TRM cells) are important in controlling cancer growth and predicting response to immunotherapy.
Regulatory T cells (TREG cells) are an important subset of CD4+ T cells that hamper anti‐tumour immune responses.
T cells: Classification, activation and memory
T cells are descended from the lymphocyte lineage, which also includes B cells and innate lymphoid cells (ILCs; Fig. 1). The most well‐studied T cells (‘conventional T cells’) have T‐cell receptors (TCRs) comprised of α and β chains that recognise peptide antigens processed within the target cell into linear oligopeptides and loaded onto major histocompatibility complexes (MHC) for expression on the cell surface. 6 Conventional T cells are further categorised by the expression of one of a pair of TCR accessory molecules – CD8 or CD4 – which bind to class I MHC (MHC‐I) and class II MHC (MHC‐II) respectively. Unconventional T cells, comprising of invariant natural killer T cells (iNKT cells), mucosal‐associated invariant T (MAIT) cells and γδ T cells, do not recognise peptide–MHC complexes but rather lipids, vitamin B metabolites and phosphoantigens respectively. 7 Our understanding of unconventional T cells and their role in cancer control is an area of progress and not elaborated here. 8
Naïve conventional T cells do not survey peripheral tissues but circulate between lymphoid organs where they continuously scan antigens acquired and presented by DCs and other antigen presenting cells (APCs). T cells are activated, or primed, when three signals are present: (i) recognition by the TCR of a cognate peptide–MHC complex, (ii) engagement of the co‐stimulatory receptor CD28 by CD80 or CD86 and (iii) inflammatory cytokines such as interleukin (IL)‐2, IL‐12 and type I interferon (IFN; Fig. 2). 9 In most circumstances, all three signals are provided by DCs. The degree of requirement for each signal is influenced by the strength of the TCR‐peptide–MHC interaction, and insufficient signalling may lead to suboptimal activation or even unresponsiveness to subsequent antigen stimulation (T‐cell anergy). 6 When naïve T cells are activated, they undergo rapid clonal expansion into a large pool of effector T cells and change their expression pattern of chemokine receptors, which enables their exit from lymphoid organs and migration to sites of inflammation. Like many other immune cells, trafficking of T cells is guided by chemokine gradients, which are secreted by a large variety of cells. The functions of activated CD8+ and CD4+ T cells at peripheral sites are later described in their respective sections below.
Fig. 2.
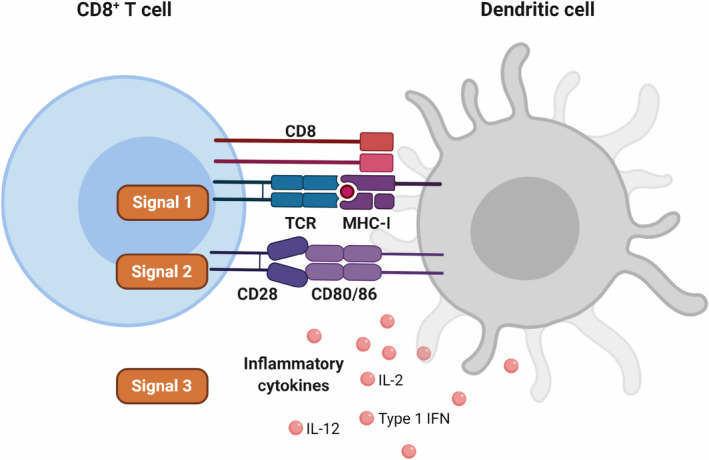
Priming of naïve T cells. Three signals are required for the efficient activation, or priming, of naïve T cells. These signals are generally provided by dendritic cells (DCs), although inflammatory cytokines such as interleukin (IL)‐2, IL‐12 and type I interferon (IFN) can originate from other sources. Here, the priming of a naïve CD8+ T cell is illustrated. For CD4+ T cells, Signal 1 requires a synapse between the T‐cell receptor (TCR) and a cognate antigen bound on class II major histocompatibility complex (MHC). Illustration created with Biorender.com. [Colour figure can be viewed at wileyonlinelibrary.com]
In the normal setting, following clearance of the source of inflammation the contraction phase occurs, in which the majority of activated, effector T cells undergo apoptosis. Conventionally, it has been thought that a small subset (5–10%) of effector T cells escape death to subsequently differentiate into memory T cells. More recently, a separate lineage model in which memory T cells precede effector T cells in the differentiation pathway has been put forward. 10 Memory T cells are long‐lived, less reliant on co‐stimulation and exhibit more rapid activation compared to the original priming event, resulting in significantly heightened kinetics and amplitude of response following subsequent exposure to the same antigen – a hallmark of adaptive immunity. Two aspects of memory T cells deserve specific mention in relation to cancer immunology. Firstly, there is increasing awareness of the critical role tissue‐resident memory T cells (T RM cells) play in the control of solid tumours. TRM cells are a specialised subset of memory T cells that are non‐migratory but take up permanent residency in peripheral tissues, mediated by their expression of CD103 and CD49a, which bind to E‐cadherin and collagen respectively. In various human cancers, the degree of infiltration by CD8+ TRM cells correlate better with clinical outcomes than infiltration by total CD8+ T cells, supporting the hypothesis that TRM cells are especially adapted for important functions within the specific tumour and tissue microenvironment (‘quality over quantity’). 11 These observations are further corroborated by functional studies involving cancer vaccination and isolation of tumour‐infiltrating lymphocytes. 11 Secondly, while ICB can reinvigorate exhausted effector T cells to a degree (immune checkpoint receptors are described below), recent studies suggest it is in fact a self‐renewing, ‘stem‐like’ population of memory T cells, also referred to as precursor exhausted T cells (TPEX cells), that selectively proliferates to expand the pool of antigen‐specific T cells after such therapy. 12 , 13
T cells: Immune checkpoint receptors
Delicately balancing immune activation are immune tolerance processes, which avoid harmful states of autoimmunity. Central tolerance refers to clonal deletion of high‐affinity self‐reactive clones during T‐cell development in the thymus. Beyond the thymus, peripheral tolerance is regulated by cell‐extrinsic and cell‐intrinsic mechanisms. Cell‐extrinsic peripheral immune tolerance is mediated by external factors such as regulatory T cells, MDSCs and associated molecules (described in later sections). In contrast, a key cell‐intrinsic mechanism of peripheral tolerance is the expression of immune checkpoint receptors on effector T cells (Fig. 3). With progressive antigen exposure of the TCR, these negative co‐stimulatory receptors are also increasingly expressed, thus serving as an in‐built breaking mechanism. Persistent antigen exposure can lead to T‐cell exhaustion, the terminal stage of which is an irreversible epigenetic state with significantly dampened effector function and high expression of immune checkpoint receptors. 14 , 15
Fig. 3.
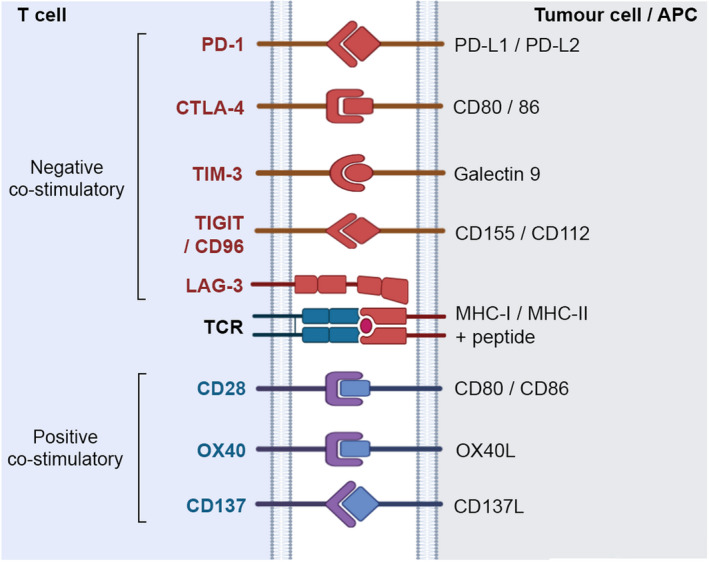
Positive and negative co‐stimulatory receptors regulate T‐cell activation. The optimal activation of T cells requires engagement of positive co‐stimulatory receptors with their cognate ligands. In general, negative co‐stimulatory receptors, also known as immune checkpoint receptors, are upregulated upon T‐cell activation as a homeostatic mechanism to regulate the activation status of T cells. Listed are some of the best characterised receptors and their ligands. These receptors each have distinct signalling pathways and are not all expressed on the same cell. Illustration created with Biorender.com. [Colour figure can be viewed at wileyonlinelibrary.com]
The two most well‐known immune checkpoint receptors are cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) and programmed cell death protein 1 (PD‐1), of which blocking antibodies are now in routine clinical use for treatment of various solid cancers. 16 CTLA‐4 restricts T‐cell activation primarily by competing with the co‐stimulatory receptor CD28 for binding to CD80 and CD86 on APCs, thus reducing the intensity of T‐cell‐activating signals. Hence, the CTLA‐4 axis is thought to be most relevant in secondary lymphoid organs, where T‐cell priming predominantly occurs. By contrast, PD‐1 binds to programmed death‐ligand 1 (PD‐L1) and programmed death‐ligand 2 (PD‐L2), which are highly expressed on a range of immune and non‐immune cells at sites of inflammation, such as on macrophages and tumour cells. Therefore, there are general distinctions in tissue location and molecular mechanism of regulation by CTLA‐4 and PD‐1. Nonetheless, reflecting the complexity of the immune system, recent data has also revealed non‐canonical facets of these receptors that are still being uncovered. 17 For example, the surprising and perplexing role of PD‐1 on myeloid cells (rather than T cells) has been inferred using pre‐clinical models. 18 In this rapidly moving translational space, the next wave of T‐cell checkpoint receptors against which therapeutic antibodies are in clinical trials include lymphocyte‐activation gene 3 (LAG‐3), T‐cell immunoglobulin and mucin domain‐3 (TIM‐3), T‐cell immunoreceptor with Ig and ITIM domains (TIGIT), and CD96 (Fig. 3). 19 , 20 Just recently, the added efficacy of combining relatlimab (a first‐in‐class human LAG‐3 blocking antibody) with nivolumab (a PD‐1 blocking antibody) in patients with advanced melanoma, compared to nivolumab alone, was established in a randomised phase 2–3 study. 21 The mechanisms of these ‘newer’ immune checkpoints are not elaborated here.
T‐cell subsets: CD8 + T cells
Of conventional T cells, CD8 + T cells are most well known for their cytotoxic activity. When activated CD8+ T cells reach their destination, they kill their targets in an antigen‐specific fashion and secrete inflammatory cytokines such as IFN‐γ, which among its many effects further enhances the cytotoxic function of CD8+ T cells. The two mechanisms of cell killing by CD8+ T cells are the transferring of cytotoxic enzymes (granzymes) into the target cell via perforin pores inserted on the target cell membrane, and secretion of molecules (such as the tumour necrosis factor (TNF) superfamily of ligands) that bind to death receptors on the target cell. Recognition of targets by CD8+ T cells depends on the display of cognate antigens bound to MHC‐I on the target cell surface. Notably, MHC‐I is expressed on all nucleated cells in the normal setting and carries endogenous peptides. Loss of MHC‐I expression is, therefore, a major immune evasion mechanism of cancer cells, although this paradoxically increases their susceptibility to being targeted by NK cells (discussed in below sections). 22
T‐cell subsets: CD4 + T cells, including regulatory T cells
CD4 + T cells are also called helper T cells (Th cells) because they assist in the initiation and regulation of a broad range of immune responses, mediated primarily through the secretion of cytokines. The cytokine profiles of Th cells further allow their functional classification. Th1 cells are most pertinent to cancer immunology as they secrete cytokines involved in cell‐mediated immunity, such as IL‐2 and IFN‐γ, while Th2 cells control antibody responses. In rarer cases, CD4+ T cells can exert direct cytotoxic activity via the granzyme/perforin and death receptor pathways, in which case killing is restricted to recognition of cognate MHC‐II‐bound peptides. 23 Typically, MHC‐II is expressed by mature APCs and displays peptides acquired from extracellular sources, but it is now appreciated that a variety of human cancers can surprisingly also express MHC‐II on tumour cells due to aberrant MHC‐II regulation and can present endogenous antigens via non‐classical MHC‐II processing pathways. 24 Thus, the roles of Th cells in cancer immunology are multi‐faceted and continue to be elucidated. 25
Regulatory T cells (T REG cells) are a prominent subset of CD4+ T cells that play a key immunosuppressive function and are often major culprits behind ineffectual anti‐tumour immune responses. 26 , 27 TREG cells are identified by concurrent expression of CD4 and the transcription factor Forkhead box P3 (Foxp3), the latter a master regulator of TREG cells. 28 A hallmark of TREG cells is their constitutive expression of CTLA‐4 under Foxp3 instruction. TREG cells express TCRs and homing receptors that are similar to effector T cells, and are found in a variety of tissues, including lymph nodes, mucosal barriers, sites of inflammation, as well as tumours. 29 There, they suppress the proliferation, differentiation, and effector functions of a range of immune subsets, such as non‐regulatory effector T cells, DCs and NK cells, via indirect and direct mechanisms. 29 Indirect mechanisms include generation of immunosuppressive molecules such as IL‐10 and TGF‐β, as well as competitive consumption of IL‐2. Direct cell contact‐dependent pathways include granzyme/perforin‐mediated killing of effector T cells and CTLA‐4‐mediated regulation of T‐cell activation.
Dendritic cells (DCs)
Antigen presenting cells, as their names would suggest, play a vital role in supporting antigen‐specific adaptive immunity. DCs are considered ‘professional’ APCs, given their ability to acquire, process and present antigens on both MHC‐I and MHC‐II, respond to danger signals, secrete cytokines and express co‐stimulatory molecules for priming of T cells. Immature DCs have high phagocytic but low T‐cell priming ability. After ingestion of a pathogen or foreign antigen, DCs undergo maturation when danger signals, in the form of DAMPs and PAMPs, are detected by pattern recognition receptors (PRRs). Mature DCs migrate from the location of the antigen to secondary lymphoid organs where they engage and activate T cells. Notably, the secretion of inflammatory cytokines and expression of the co‐stimulatory ligands CD80 and CD86 are tightly limited to mature DCs. Cross‐presentation of antigens (the special ability to load acquired antigens onto MHC‐I) to CD8+ T cells is performed by type 1 conventional DCs (cDC1s) and selectively requires type I IFN. 30 , 31 , 32
A PRR that has recently attracted significant attention in the oncology setting is the cGAS‐STING axis, which responds to cytoplasmic double‐stranded DNA as a DAMP enriched in damaged tumour cells, leading to downstream type I IFN production, cDC1 maturation and anti‐tumour CD8+ T‐cell priming. 33 , 34 Significantly, this pathway is a putative major mechanism by which cancer therapies that do not inherently target the immune compartment, such as RT, can induce cancer immunity. 33 , 35 Other PRRs relevant to cancer therapy are also under investigation. 36 cDC1‐CD8+ T‐cell priming interactions have been thought to predominantly occur in lymphoid organs, but the importance of tumour‐associated cDC1s is now recognised, with their frequency being correlated with response to anti‐PD‐1 therapy and improved cancer patient survival. 37 , 38 , 39 There are multiple other important DC subsets, which collectively constitute a complex field in immunology. 6 While DCs are the predominant professional APCs, other cells such as macrophages, monocytes and B cells can also play the role of APCs. 40
Natural killer (NK) cells
NK cells are the most well‐known member of ILCs (Fig. 1). In contrast to T cells, NK cells were originally identified by their ability to spontaneously kill cells in vitro without prior activation. 41 , 42 NK cells are categorised as innate immune cells because they lack clonally diverse antigen receptors for activation but rely on an array of germline‐encoded, invariant activating and inhibitory receptors, the sum of signals from which determine their reactivity. In general, NK cell‐activating receptors recognise stress‐induced ligands and viral molecules, while NK cell‐inhibitory receptors recognise self MHC‐I. This pattern of activation is called the ‘altered‐self’ and ‘missing‐self’ hypothesis. 6 In this model, NK cells target rogue cells that express surface ligands suggesting transformation, damage or viral infection (leading to gain of activating signalling for NK cells) and/or have downregulated MHC‐I expression to escape recognition by adaptive immunity (resulting in loss of inhibitory signalling for NK cells). A subset of NK cells also express Fcγ receptors, which bind to the Fc portion of class‐G immunoglobulins (IgGs) to transmit an activating signal, thus mediating antibody‐dependent cellular cytotoxicity (ADCC). Once activated, NK cells kill via the granzyme/perforin pathway. Additionally, the cytokine secretion profile and transcription factor requirements of NK cells are remarkably similar to those of T cells. 43 Because NK cells are early responders to sites of inflammation and tumours, NK cells can shape subsequent adaptive immune responses, including facilitating the recruitment of cDC1s into the local microenvironment. 39 , 44 Furthermore, it has become apparent that targeted therapies such as anti‐EGFR and HER2 antibodies achieve their full therapeutic potential in part via engaging NK cells for ADCC. 45 Tumour infiltration of NK cells is prognostic in a wide range of human cancers. 46 , 47 , 48 , 49 , 50 , 51
Myeloid‐derived suppressor cells (MDSCs), neutrophils and macrophages
MDSCs are a heterogeneous group of developmentally immature cells of myeloid lineage, collectively characterised by their ability to suppress T‐cell function. These cells were only recognised in pathological conditions relatively recently. 52 , 53 In the cancer setting, persistent stimulation of the myeloid cell compartment from abnormal production of growth factors and inflammatory signals results in aberrant myelopoiesis and systemic expansion of immature myeloid cells that are distinct to differentiated myeloid cells (monocytes/macrophages, neutrophils and DCs). Although MDSCs can be grouped into polymorphonuclear (PMN‐MDSCs) and monocytic MDSCs (M‐MDSCs) based on their granulocytic and monocytic myeloid cell lineages, respectively, their nomenclature and classification are complex and still to be clearly defined. 53
Myeloid‐derived suppressor cells play important tumour‐promoting roles by their immunosuppressive function, mediated through cell‐to‐cell contact with T and NK cells as well as via secretion of soluble mediators. 54 In addition, PMN‐MDSCs together with neutrophils (the two subsets are often difficult to distinguish) are heavily implicated in various steps of the metastatic cascade, including preparing the pre‐metastatic niche for engraftment of tumour cells, escorting and promoting the proliferative capacity of circulating tumour cells, and recruiting tumour cells to metastatic sites via neutrophil extracellular traps (NETs). 55 Unsurprisingly, the neutrophil‐to‐lymphocyte ratio is a prognostic factor for survival in many tumour types. 56 While PMN‐MDSCs are unequivocally tumour‐promoting, neutrophils can exert anti‐tumour effects in certain circumstances. 57 This functional plasticity in response to microenvironmental cues partly underlies the longstanding confusion surrounding their role in cancers.
Macrophages are differentiated from circulating monocytes upon their entry into tissue. In healthy tissues, ‘resident’ macrophages take on specific names (e.g. Kupffer cells, microglia and Langerhans cells in the liver, brain and skin respectively) and in the current paradigm are thought to be seeded before birth and maintained independently of monocytes throughout adulthood. 58 , 59 In contrast, ‘passenger’ macrophages originate from infiltrating monocytes during inflammation. Whether resident and passenger macrophages are functionally distinct remains unclear. A significant proportion of tumour‐associated macrophages (TAMs) are differentiated rapidly from M‐MDSCs that infiltrate the tumour. 53 TAMs also exhibit a dual capacity for anti‐ or pro‐tumour roles, respectively, called M1 and M2 macrophages, although this bipolar characterisation of TAMs is most likely an oversimplification of what might better resemble a spectrum of phenotypes. 60 Altogether, MDSCs, neutrophils and macrophages are highly complex cells to study due to their short lifespan, diversity and plasticity, and our understanding of these subsets in relation to cancer is constantly evolving.
B cells
In contrast to T (and to an extent NK) cell‐mediated immunity, which protects against intracellular antigens and alterations, host defences in the extracellular milieu are predominantly served by humoral immunity. Here, antibodies (or immunoglobulins) secreted by B cells play a significant role in controlling the spread of pathogens by (i) neutralisation of their toxic effects and infectivity, (ii) allowing binding with the complement system (opsonisation) to enhance uptake and destruction by phagocytes, (iii) and ADCC, which is the marking for killing by cytotoxic cells through Fcγ receptors (described briefly in NK cell section). B cells differentiate into antibody‐producing plasma cells and long‐lasting memory B cells upon recognition of an antigen, in a process that is also facilitated by Th2 cells (refer section on CD4+ T cells). Because of their principal activity in the extracellular space, attention given to B cells in cancer has taken a backstage to T cells. However, clinical responses to ICB and patient survival for a range of cancers but especially for melanoma have been correlated with levels of circulating tumour‐directed immunoglobulins and tumour‐infiltrating B and plasma cells, altogether suggesting that humoral immunity is not inert in tumour control. 61 , 62 , 63 , 64 It has been proposed that B cells can play the role of APCs, especially in light of the recognition of tertiary lymphoid structures (TLS) within tumours where B and T cells are in close association. 65 B cells can also induce tumour cell killing indirectly via antibody‐mediated mechanisms and directly via expression of death receptor ligands. 66 On the other hand, a poorly characterised but important subset of B cells that exert T‐cell‐inhibitory effects via production of immunosuppressive cytokines has been described, collectively termed regulatory B cells. 67 Overall, the identification of key B cell subsets in oncology as well as factors that influence their anti‐ and pro‐tumour effects remain to be clarified.
Concepts in cancer immunology
Tumour antigens and immunosurveillance
The process of accumulating oncogenic mutations and transcriptional aberrations in a tumour cell results in the expression of tumour antigens, which forms the basis behind immune recognition of cancers, also termed immunosurveillance. The relevance of this process in humans is supported by three key threads of evidence: (i) the correlation between intra‐tumoural immune responses and the positive prognosis of the cancer patient, (ii) the higher relative rates of cancer incidence in immunosuppressed individuals, and (iii) the development of spontaneous immune responses associated with cancer, including that of paraneoplastic syndromes. 68 Advances in DNA sequencing technology have greatly expanded the study of tumour antigens from cancer‐testis and differentiation antigens (which are respectively expressed in germ cells and in specific differentiation phases of cell types, but otherwise suppressed in non‐cancerous mature somatic tissue) to neoantigens, which are bona fide novel proteins entirely absent from normal tissues. However, not all cancer genomic mutations are exonic or result in neoantigens that efficiently elicit an immune response. Determinants of a peptide's immunogenicity include having the appropriate MHC‐I binding motifs for expression on the cell surface, as well as epitopes that are sufficiently visible to TCRs and divergent from self‐antigens. Modelling and prediction tools in this context is an area of significant research. 69 Furthermore, it is now understood that most solid cancers develop through branching evolution, resulting in extensive intra‐ and inter‐tumoural heterogeneity, which can limit effective immune‐mediated eradication of the disease. 70 Thus, the identification and targeting of truncal mutations, present near the origin of the cancer evolutionary tree, are highly sought after in cancer immunotherapy.
Cancer immunoediting
Another overarching tenet in cancer immunology is that the immune system not only controls tumour initiation but also shapes tumour immunogenicity via a process termed cancer immunoediting. 68 According to this model, immune cells and effector molecules recognise and destroy tumour cells, but in a Darwinian fashion paradoxically favours growth of variants that are more capable of surviving immune pressure. The three phases of elimination, equilibrium and escape have been described in this process. 68 An important implication of this model is that all clinically apparent tumours in the immunocompetent patient have been sculpted by the host immune system and enriched for pathways for immune evasion. Any point in the immune cascade could be hijacked to escape immune pressure. Briefly, tumour cell mechanisms of evasion include selecting for antigen‐loss variants, suppression of MHC‐I expression, upregulation of immune checkpoint ligands, expression of mutated non‐functional forms of death receptors, secretion of immunosuppressive molecules and induction of fibroblast proliferation to block effective infiltration of immune cells into tumours. 71 Critically, this relatively recent shift in recognition of the pervasive influence of the host immune system on tumour evolution was reflected in its inclusion in Hanahan and Weinberg's update on the hallmarks of cancer, 72 and has underpinned the development of modern immunotherapeutic strategies to overcome barriers in the immune control of cancer.
Cancer‐immunity cycle
Following from the above, a mechanistic model for the immune control of cancer further allows for therapeutic focus on the specific points of failure. To this end, a sequence of seven events occurring in a self‐propagating cyclic fashion has been put forward and referred to as the cancer‐immunity cycle (Fig. 4). 73 These steps are: (i) release of tumour antigens in a suitable inflammatory milieu, (ii) capture and presentation of antigens by DCs to T cells in secondary lymphoid organs, (iii) priming and activation of tumour antigen‐specific T‐cell responses, (iv) trafficking of activated T cells through blood vessels, (v) infiltration of T cells into the tumour, (vi) recognition of tumour cells by T cells and (vii) T cell‐mediated killing of tumour cells. Tumour cell death releases further tumour antigens, which restarts the cycle, resulting in an iterative expansion of both the repertoire and amplitude of T‐cell responses. Each step of the cycle is regulated by stimulatory and inhibitory factors and can be subverted by the tumour for immune evasion.
Fig. 4.
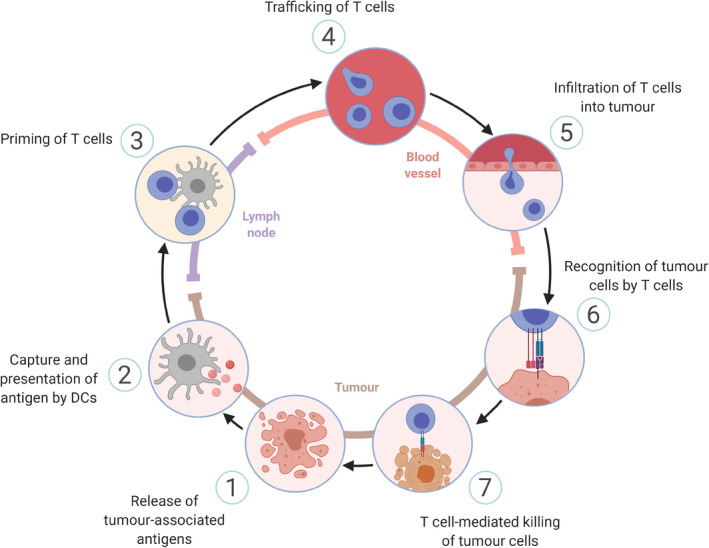
The cancer‐immunity cycle. A sequence of events occurs in a self‐propagating cyclic process in the generation of an optimal anti‐tumour immune response. The cycle is divided in seven major steps, as illustrated above. Each step occurs at a particular anatomical site and is carefully regulated by a balance of stimulatory and inhibitory factors. Figure adapted from Chen et al. 73 Illustration created with Biorender.com. [Colour figure can be viewed at wileyonlinelibrary.com]
Importantly, points of failure can potentially be reversed to enable the immune system to regain control of the tumour. For example, the mechanisms of action underlying CTLA‐4 and PD‐1/PD‐L1 inhibition in cancer therapy are canonically assigned to the overcoming of barriers in steps 3 (priming of T cells in secondary lymphoid organs) and 7 (engagement of T cells in the tumour microenvironment) respectively. By contrast, CAR‐T cell therapy, which is the genetic engineering of a cancer patient's autologous T cells to express synthetic activation receptors, followed by expansion and infusion back into the patient, is designed to effectuate steps 6 and 7. 74 Notably, RT can potentially address blockades in steps 1 and 2 (by evoking an immunogenic cell death), 5 (by promoting T‐cell recruitment) and 6 (by increasing tumour cell expression of MHC‐I), as will be elaborated elsewhere in this Special Issue. Whereas in some cancers monotherapy is sufficient to (re‐)establish the cancer‐immunity cycle, in others a combined modality approach is required. For example, anti‐CTLA‐4 alone is effective in patients with chemotherapy‐refractory advanced melanoma but not non‐small cell lung cancer, 75 , 76 yet in the latter group the combination of anti‐CTLA‐4 therapy and RT appeared to be able to activate systemic cancer immunity. 77 Biomarkers to predict the effectiveness of a treatment modality in promoting the cancer‐immunity cycle is a highly active area of immuno‐oncology research.
Cancer‐immune set point, immune phenotypes and gut microbiome
Where the cancer‐immunity cycle is stalled, the equilibrium between the stimulatory and inhibitory pressures for a particular patient is referred to as the cancer‐immune set point, which also represents the threshold that must be exceeded to achieve cancer immunity. 78 As can be inferred from the heterogeneity of immunotherapy responses observed in clinic, this set point is highly variable across tumours and even between patients with overtly similar tumours, therefore, conceivably dependent on a multitude of tumour and host‐related factors. At a basic tumour level, the cancer‐immune set point is reflected in the observation of the three broad immune phenotypes that correlate with response to anti‐PD‐1/PD‐L1 therapy: immune‐inflamed, immune‐excluded and immune‐desert. 78 Briefly, in immune‐inflamed tumours, the tumour parenchyma is characterised by the infiltration of T cells in proximity to tumour cells as well as expression of PD‐L1 on tumour and immune cells. Altogether, this phenotype suggests pre‐existing anti‐tumour immune responses that are arrested within the tumour. In immune‐excluded tumours, immune cells are present but do not penetrate the tumour parenchyma. Rather, they are retained in the tumour stroma surrounding tumour cell nests, suggesting that T‐cell migration and penetration into the tumour is the rate‐limiting step. In immune‐desert tumours, there is a dearth of T cells in either the tumour parenchyma or stroma, which points to a poorly immunogenic tumour that has not alerted a T‐cell response. Unsurprisingly, immune‐inflamed tumours often, but not always, respond to anti‐PD‐1/PD‐L1 therapy (reflecting a lower cancer‐immune set point), while immune‐excluded and immune‐desert tumours rarely do (reflecting a higher cancer‐immune set point).
Factors that determine the cancer‐immune set point are vast and are being continuously elucidated. Tumour mutational burden, which dictates tumour antigenicity, is a major element, but the secretory phenotype of the tumour, presence of infectious agents, use of previous chemotherapy, as well as host age, susceptibility to inflammatory stimuli and overall health are also important considerations. 78 , 79 More recently, the gut microbiome has been established as an important host‐related factor, whereby certain species of gut bacteria intriguingly activate host innate immune responses better than others, thus heightening baseline immunosurveillance and shifting the cancer‐immune set point. 80 Indeed, gut microbiome diversity is correlated with response to immunotherapy across a range of cancers as well as rates of immune‐related adverse events. 81 , 82 , 83 More impressively, a randomised Phase 1 trial testing live bacterial supplementation in conjunction with ICB in patients with metastatic renal cell carcinoma have reported a significant improvement in progression‐free survival. 84 However, emerging larger‐scale data caution about substantial cohort‐dependency in gut microbiome associations and that no single species is consistently predictive across studies, which reflect the complex interactions between gut microbial abundances, clinical factors and even geographical location. 85
In conclusion, the impact of the immuno‐oncology revolution is inarguably far‐reaching and the proliferation of immunotherapy trials across the spectrum of cancer types and settings is tremendous. In this backdrop and especially given the growing interest in the use of RT for synergy with immunotherapeutic agents, a working knowledge of the immune system and its role in shaping and controlling cancer growth should no longer be peripheral for the radiation oncologist. The reader is directed to other articles in this Special Issue for the mechanisms of and clinical evidence for RT as an immune adjuvant. From a larger perspective, just as how the oncology landscape has shifted dramatically over the decades, it is both sobering and stimulating that unexpected discoveries are continually being made in our understanding of the immune system. How its intersection with oncology will continue to evolve will be exciting to observe.
Funding Statement
J.S. has received funding from the Australian Government Research Training Program Scholarship and the RANZCR Withers and Peters Grant.
Acknowledgements
Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
J Sia FRANZCR, PhD; PJ Neeson PhD; NM Haynes PhD.
Conflict of interest: None.
References
- 1. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol 2002; 3: 991–8. [DOI] [PubMed] [Google Scholar]
- 2. Couzin‐Frankel J. Breakthrough of the year 2013. Cancer Immunother Sci 2013; 342: 1432–3. [DOI] [PubMed] [Google Scholar]
- 3. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168: 707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haikerwal SJ, Hagekyriakou J, MacManus M, Martin OA, Haynes NM. Building immunity to cancer with radiation therapy. Cancer Lett 2015; 368: 198–208. [DOI] [PubMed] [Google Scholar]
- 5. Weichselbaum RR, Liang H, Deng L, Fu YX. Radiotherapy and immunotherapy: A beneficial liaison? Nat Rev Clin Oncol 2017; 14: 365–79. [DOI] [PubMed] [Google Scholar]
- 6. Paul WE. Fundamental Immunology. 7th edn. Wolters Kluwer, Health/Lippincott Williams & Wilkins, Philadelphia, 2013: xviii, 1283 pp. [Google Scholar]
- 7. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol 2015; 16: 1114–23. [DOI] [PubMed] [Google Scholar]
- 8. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T cell targets for cancer immunotherapy. Immunity 2018; 48: 453–73. [DOI] [PubMed] [Google Scholar]
- 9. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 2010; 22: 333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol 2018; 18: 340–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amsen D, van Gisbergen KPJM, Hombrink P, van Lier RAW. Tissue‐resident memory T cells at the center of immunity to solid tumors. Nat Immunol 2018; 19: 538–46. [DOI] [PubMed] [Google Scholar]
- 12. Im SJ, Hashimoto M, Gerner MY et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature 2016; 537: 417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siddiqui I, Schaeuble K, Chennupati V et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019; 50: 195–211. [DOI] [PubMed] [Google Scholar]
- 14. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abdel‐Hakeem MS, Manne S, Beltra J‐C et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol 2021; 22: 1008–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8: 1069–86. [DOI] [PubMed] [Google Scholar]
- 18. Strauss L, Mahmoud MAA, Weaver JD et al. Targeted deletion of PD‐1 in myeloid cells induces antitumor immunity. Sci Immunol 2020; 5. doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson AC, Joller N, Kuchroo VK. Lag‐3, Tim‐3, and TIGIT: Co‐inhibitory receptors with specialized functions in immune regulation. Immunity 2016; 44: 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017; 276: 112–20. [DOI] [PubMed] [Google Scholar]
- 21. Tawbi HA, Schadendorf D, Lipson EJ et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med 2022; 386: 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Freeman AJ, Vervoort SJ, Ramsbottom KM et al. Natural killer cells suppress T cell‐associated tumor immune evasion. Cell Rep 2019; 28: 2784–94. [DOI] [PubMed] [Google Scholar]
- 23. Takeuchi A, Saito T. CD4 CTL, a cytotoxic subset of CD4+ T cells, their differentiation and function. Front Immunol 2017; 8. doi: 10.3389/fimmu.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC‐II expression by tumor cells in cancer. Clin Cancer Res 2019; 25: 2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther 2021; 28: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression — Implications for anticancer therapy. Nat Rev Clin Oncol 2019; 16: 356–71. [DOI] [PubMed] [Google Scholar]
- 27. Sia J, Hagekyriakou J, Chindris I et al. Regulatory T cells shape the differential impact of radiation dose‐fractionation schedules on host innate and adaptive antitumor immune defenses. Int J Radiat Oncol Biol Phys 2021; 111: 502–14. [DOI] [PubMed] [Google Scholar]
- 28. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol 2017; 17: 703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133: 775–87. [DOI] [PubMed] [Google Scholar]
- 30. Bottcher JP, Reis ESC. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 2018; 4: 784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diamond MS, Kinder M, Matsushita H et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011; 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fuertes MB, Kacha AK, Kline J et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011; 208: 2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deng L, Liang H, Xu M et al. STING‐dependent cytosolic DNA sensing promotes radiation‐induced type I interferon‐dependent antitumor immunity in immunogenic tumors. Immunity 2014; 41: 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sia J, Szmyd R, Hau E, Gee HE. Molecular mechanisms of radiation‐induced cancer cell death: A primer. Front Cell Dev Biol 2020; 8: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanpouille‐Box C, Formenti SC, Demaria S. TREX1 dictates the immune fate of irradiated cancer cells. Onco Targets Ther 2017; 6: e1339857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goedegebuure RSA, Kleibeuker EA, Buffa FM et al. Interferon‐ and STING‐independent induction of type I interferon stimulated genes during fractionated irradiation. J Exp Clin Cancer Res 2021; 40: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Broz ML, Binnewies M, Boldajipour B et al. Dissecting the tumor myeloid compartment reveals rare activating antigen‐presenting cells critical for T cell immunity. Cancer Cell 2014; 26: 638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barry KC, Hsu J, Broz ML et al. A natural killer‐dendritic cell axis defines checkpoint therapy‐responsive tumor microenvironments. Nat Med 2018; 24: 1178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bottcher JP, Bonavita E, Chakravarty P et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018; 172: 1022–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kambayashi T, Laufer TM. Atypical MHC class II‐expressing antigen‐presenting cells: Can anything replace a dendritic cell? Nat Rev Immunol 2014; 14: 719–30. [DOI] [PubMed] [Google Scholar]
- 41. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer 1975; 16: 230–9. [DOI] [PubMed] [Google Scholar]
- 42. Kiessling R, Klein E, Pross H, Wigzell H. "Natural" killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol 1975; 5: 117–21. [DOI] [PubMed] [Google Scholar]
- 43. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat Immunol 2016; 17: 758–64. [DOI] [PubMed] [Google Scholar]
- 44. Pallmer K, Oxenius A. Recognition and regulation of T cells by NK cells. Front Immunol 2016; 7: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferris RL, Lenz HJ, Trotta AM et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev 2018; 63: 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cursons J, Souza‐Fonseca‐Guimaraes F, Foroutan M et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol Res 2019; 7: 1162–74. [DOI] [PubMed] [Google Scholar]
- 47. Soo RA, Chen Z, Yan Teng RS et al. Prognostic significance of immune cells in non‐small cell lung cancer: Meta‐analysis. Oncotarget 2018; 9: 24801–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ishigami S, Natsugoe S, Tokuda K et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000; 88: 577–83. [PubMed] [Google Scholar]
- 49. Coppola A, Arriga R, Lauro D et al. NK cell inflammation in the clinical outcome of colorectal carcinoma. Front Med (Lausanne) 2015; 2: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Donskov F, von der Maase H. Impact of immune parameters on long‐term survival in metastatic renal cell carcinoma. J Clin Oncol 2006; 24: 1997–2005. [DOI] [PubMed] [Google Scholar]
- 51. Gannon PO, Poisson AO, Delvoye N, Lapointe R, Mes‐Masson AM, Saad F. Characterization of the intra‐prostatic immune cell infiltration in androgen‐deprived prostate cancer patients. J Immunol Methods 2009; 348: 9–17. [DOI] [PubMed] [Google Scholar]
- 52. Gabrilovich DI, Bronte V, Chen SH et al. The terminology issue for myeloid‐derived suppressor cells. Cancer Res 2007; 67: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Veglia F, Perego M, Gabrilovich D. Myeloid‐derived suppressor cells coming of age. Nat Immunol 2018; 19: 108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ostrand‐Rosenberg S, Fenselau C. Myeloid‐derived suppressor cells: Immune‐suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 2018; 200: 422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hedrick CC, Malanchi I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat Rev Immunol 2022; 22: 173–87. [DOI] [PubMed] [Google Scholar]
- 56. Templeton AJ, McNamara MG, Šeruga B et al. Prognostic role of neutrophil‐to‐lymphocyte ratio in solid tumors: A systematic review and meta‐analysis. J Natl Cancer Inst 2014; 106: dju124. [DOI] [PubMed] [Google Scholar]
- 57. Shaul ME, Fridlender ZG. Tumour‐associated neutrophils in patients with cancer. Nat Rev Clin Oncol 2019; 16: 601–20. [DOI] [PubMed] [Google Scholar]
- 58. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol 2016; 17: 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ginhoux F, Guilliams M. Tissue‐resident macrophage ontogeny and homeostasis. Immunity 2016; 44: 439–49. [DOI] [PubMed] [Google Scholar]
- 60. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 2016; 17: 34–40. [DOI] [PubMed] [Google Scholar]
- 61. Fässler M, Diem S, Mangana J et al. Antibodies as biomarker candidates for response and survival to checkpoint inhibitors in melanoma patients. J Immunother Cancer 2019; 7: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti‐MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer 2003; 103: 97–100. [DOI] [PubMed] [Google Scholar]
- 63. Fremd C, Stefanovic S, Beckhove P et al. Mucin 1‐specific B cell immune responses and their impact on overall survival in breast cancer patients. Onco Targets Ther 2016; 5: e1057387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wouters MCA, Nelson BH. Prognostic significance of tumor‐infiltrating B cells and plasma cells in human cancer. Clin Cancer Res 2018; 24: 6125–35. [DOI] [PubMed] [Google Scholar]
- 65. Dieu‐Nosjean MC, Goc J, Giraldo NA, Sautès‐Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol 2014; 35: 571–80. [DOI] [PubMed] [Google Scholar]
- 66. Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B‐side of cancer immunity: the underrated tune. Cell 2019; 8: 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chekol Abebe E, Asmamaw Dejenie T, Mengie Ayele T, Dagnew Baye N, Agegnehu Teshome A, Tilahun Muche Z. The role of regulatory B cells in health and diseases: a systemic review. J Inflamm Res 2021; 14: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011; 331: 1565–70. [DOI] [PubMed] [Google Scholar]
- 69. Peters B, Nielsen M, Sette A. T cell epitope predictions. Annu Rev Immunol 2020; 38: 123–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Levine AJ, Jenkins NA, Copeland NG. The roles of initiating truncal mutations in human cancers: The order of mutations and tumor cell type matters. Cancer Cell 2019; 35: 10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol 2006; 90: 51–81. [DOI] [PubMed] [Google Scholar]
- 72. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 73. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 2013; 39: 1–10. [DOI] [PubMed] [Google Scholar]
- 74. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T‐cell immunotherapy for human cancer. Am J Hematol 2019; 94 (S1): S3–9. [DOI] [PubMed] [Google Scholar]
- 75. Hodi FS, O'Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zatloukal P, Heo DS, Park K et al. Randomized phase II clinical trial comparing tremelimumab (CP‐675,206) with best supportive care (BSC) following first‐line platinum‐based therapy in patients (pts) with advanced non‐small cell lung cancer (NSCLC). J Clin Oncol 2009; 27(15_suppl): 8071. [Google Scholar]
- 77. Formenti SC, Rudqvist NP, Golden E et al. Radiotherapy induces responses of lung cancer to CTLA‐4 blockade. Nat Med 2018; 24: 1845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature 2017; 541: 321–30. [DOI] [PubMed] [Google Scholar]
- 79. Chowell D, Yoo S‐K, Valero C et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat Biotechnol 2021; doi: 10.1038/s41587-021-01070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dart A. Tumour microenvironment: Microbes matter. Nat Rev Cancer 2018; 18: 1. [DOI] [PubMed] [Google Scholar]
- 81. Gopalakrishnan V, Spencer CN, Nezi L et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 2018; 359: 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Routy B, Chatelier EL, Derosa L et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science 2018; 359: 91–7. [DOI] [PubMed] [Google Scholar]
- 83. Dubin K, Callahan MK, Ren B et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint‐blockade‐induced colitis. Nat Commun 2016; 7: 10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dizman N, Meza L, Bergerot P et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med 2022; doi: 10.1038/s41591-022-01694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lee KA, Thomas AM, Bolte LA et al. Cross‐cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med 2022; 28: 535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]