

Abstract
Mucinous tubular and spindle cell carcinoma (MTSCC) is a relatively rare renal epithelial neoplasm resembling type 1 papillary renal cell carcinoma (PRCC) morphologically and immunohistochemically. The accurate diagnosis of MTSCC remains a challenge. Here, by using proteomic profiling, we characterized MTSCC and PRCC to identify diagnostic biomarkers. We found that the MTSCC tumor proteome was significantly enriched in B‐cell‐mediated immunity when compared with the proteome of adjacent normal tissues of MTSCC or tumors of PRCC. Importantly, we identified MZB1, VCAN, and SOSTDC1 as diagnostic biomarkers to distinguish MTSCC from the solid variant of type 1 PRCC, with an AUC of 0.985 when combined. MZB1 was inversely correlated with tumor clinical stage and may play an anti‐tumor role by activating the complement system. Finally, unsupervised clustering revealed two molecular subtypes of MTSCC, displaying different morphology, expression signatures of oxidative phosphorylation, and aggravation. In summary, our analyses identified a three‐protein diagnostic panel and molecular subtypes for MTSCC. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: kidney, mucinous tubular carcinoma, spindle cell carcinoma, proteomic, immunohistochemistry
Introduction
Mucinous tubular and spindle cell carcinoma (MTSCC) of the kidney is a relatively rare renal epithelial neoplasm characterized by tubular formations merging with bland spindle cells and a myxoid stroma, which was first recognized as a distinct entity in the 2004 World Health Organization (WHO) tumor classification as well as in the newly revised 4th edition published in 2016 [1]. It occurs more frequently in females, with a female‐to‐male ratio of 3–4:1. Most tumors have an indolent clinical behavior; a small but distinct subset with high‐grade transformation (mainly sarcomatoid differentiation) may show distant metastasis and can be fatal [2, 3, 4, 5, 6, 7, 8, 9, 10]. There is a lack of comprehensive understanding of this cancer because of its rare occurrence (only about 100 cases reported in the English language literature).
The main differential diagnostic consideration for MTSCC is type 1 papillary renal cell carcinoma (PRCC), particularly the solid variant, which has predominantly solid or tubular architectural patterns or contains low‐grade spindle cell areas [3, 4, 11]. Immunohistochemical profiles of these two types of tumors also show significant overlap, with a majority of cases exhibiting immunoreactivity to CK7, AMACR, and EMA [3, 12]. Therefore, it remains unclear whether MTSCC is molecularly distinct from PRCC variants and how pathologists should practically manage those cases with overlapping histologic features.
Recent studies have shown that MTSCC could be a genetically distinctive entity different from PRCC. Genomic investigations for MTSCC have demonstrated multiple chromosomal numerical aberrations in these tumors, with losses of all or parts of chromosomes 1, 4, 6, 8, 9, 11, 13, 14, 15, 18, 22, and X, as well as gains of all or parts of chromosomes 2, 3, 4, 5, 7, 9, 10, 12, 15, 16, 17, 18, 19, 20, 22, and Y by comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), and high‐resolution single nucleotide polymorphism (SNP) array [5, 13, 14, 15]. However, type 1 PRCC cases were observed to have typical gains on chromosomes 7 and 17, and losses on chromosome Y [16, 17]. Using whole‐exome sequencing, a recent study of MTSCC with classic morphology also revealed monosomy of chromosomes 1, 6, 9, 14, 15, and 22 in 100% of 22 cases, and frequent loss of chromosomes 4, 8, and 13 in 80–90% [18] (supplementary material, Table S1). However, new technologies for genetic tests including CGH, FISH, SNP, and next‐generation sequencing (NGS) require elaborate equipment as well as experienced technicians, which makes them hard to gain popularity in most hospitals. Immunohistochemistry remains a widely used method in pathological diagnosis. However, it is still hard for pathologists in basic hospitals to practically identify them and it is urgent to find new diagnostic markers for immunohistochemical detection to clinically distinguish the two kinds of tumors.
Mass spectrometry (MS)‐based proteomics is a powerful and unbiased technique for characterizing complex biological systems [19]. A large‐scale study of 110 treatment‐naive clear cell renal cell carcinoma (ccRCC) and 84 paired‐matched normal adjacent tissues (NATs) identified the proteomic landscape in ccRCC, and microenvironment cell signatures that delineated four immune‐based ccRCC subtypes using proteomic profiling [20]. Another study used isobaric labeling‐based quantitative proteomics to assess HBV‐related acute‐on‐chronic liver failure (ACLF)‐associated proteomic changes. Wu et al identified several candidate biomarkers that allowed differentiation between ACLF non‐survivors and survivors and developed a prognostic score (the P5 score) as a high‐performance prognostic score for HBV‐ACLF [21]. Since proteins are ultimately the functional effectors of biological activity in cancer cells, we hypothesized that global proteomic analysis may be an extremely sensitive method to comprehensively understand the molecular mechanism of MTSCC and identify potential diagnostic markers to distinguish MTSCC from PRCC.
Here, we present the first proteomic analysis of 18 pairs of MTSCC and 6 pairs of type 1 PRCC. Based on proteomic results, selected candidates were validated by IHC on a cohort of 32 cases of MTSCC and 36 cases of type 1 PRCC to assess their clinical potential for discriminating these tumors. Our study guides clinical differential diagnosis to distinguish MTSCC from type 1 PRCC and provides rich resources for data mining toward altered pathways in MTSCC.
Materials and methods
Patient samples
The study was performed under Institutional Review Board‐approved protocols of The First Hospital of Sun Yat‐sen University (No. [2021]404), and procedures involving human subjects were in accordance with the Helsinki Declaration of 1975, as revised in 1983. Patient samples used in this study were obtained from multiple institutions in PR China including The First Affiliated Hospital of Sun Yat‐sen University, Sun Yat‐sen University Cancer Center, The Third Affiliated Hospital of Sun Yat‐sen University, Affiliated Hospital of Jining Medical University, The Affiliated Hospital of Qingdao University, and The Seventh Affiliated Hospital of Sun Yat‐sen University. Details are provided in Supplementary materials and methods.
Tandem mass tagging (TMT) proteomics analysis
The experimental procedures for TMT proteomics analysis include protein extraction, trypsin digestion, TMT labeling, HPLC fractionation, LC–MS/MS analysis, and database searching. The detailed procedure as well as the pre‐processing methods for the proteomics analyses is presented in Supplementary materials and methods. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [22] partner repository with the dataset identifier PXD027972. The sample‐channel mappings of the mass spectrometry proteomics data are listed in supplementary material, Table S2.
Bioinformatics analyses
Principal component analysis, differential expression analysis, Gene Ontology analysis, gene set enrichment analysis, and consensus clustering analysis were performed for proteome profiling and details are presented in Supplementary materials and methods.
Immunohistochemistry and immunofluorescence
For immunohistochemistry, the slides were incubated with anti‐SOSTDC1 (Abcam, Cambridge, UK), anti‐MZB1 (Proteintech, Rosemont, IL, USA), anti‐VCAN (Abcam), anti‐CD138 (LBP, Guangzhou, PR China), anti‐C3/C3b (Abcam), anti‐CD20 (LBP), anti‐CD3 (LBP), anti‐CD5 (LBP), anti‐CD4 (Origene, Beijing, PR China), anti‐CD8 (Origene), and anti‐CD21 (LBP). For immunofluorescence, the sections were incubated with anti‐MZB1 and anti‐CD138. Detailed methods, antibody staining information, scoring criteria, and cut‐offs are provided in Supplementary materials and methods and supplementary material, Table S3.
Construction of combined diagnosis models
Binary logistic regression was performed for constructing combined diagnosis models. Subsequently, five‐fold cross‐validation was performed on the entire dataset. Details are provided in Supplementary materials and methods.
Statistical analyses
Mann–Whitney U‐tests and Kruskal–Wallis tests were used to analyze quantitative and categorical traits. Differences with a P value less than 0.05 were considered significant or as otherwise indicated. A chi‐squared test was used to determine the statistical significance of differences between the proportions of MTSCC and type 1 PRCC tumors staining for each marker.
Results
Clinical, morphologic, and immunohistochemical features of MTSCC and PRCC
We enrolled 32 cases of MTSCC and 36 cases of type 1 PRCC from six hospitals in China. The clinicopathologic features of the tumors in the 68 cases are summarized in Table 1. The ages of the patients were similar in the MTSCC and PRCC cohorts. The median patient age in our MTSCC cohort was 55 years (range 32–77 years), and patient age in the PRCC cohort ranged from 31 to 77 years (median age 54.7 years). There was a female predilection (2.6:1) in the MTSCC group, distinct from the male predominance (4.1:1) observed in the PRCC group. There was no significant difference between tumor sizes among the two groups. The mean follow‐up of the cohorts for overall survival was 62 (range 10–126) months for MTSCC and 33 (range 13–87) months for PRCC, without any patients suffering recurrence or metastasis.
Table 1.
Clinicopathological features of the MTSCC and PRCC cases
| MTSCC (n = 32) | PRCC (n = 36) | P value | |
|---|---|---|---|
| Age* | 55.0 (32–79) | 54.7 (31–77) | 0.909 |
| Gender | |||
| Female | 23 (71.9%) | 7 (19.4%) | <0.001 |
| Male | 9 (28.1%) | 29 (80.6%) | |
| Nephrectomy | |||
| Partial | 14 (43.8%) | 27 (75.0%) | 0.009 |
| Total | 18 (56.2%) | 9 (25.0%) | |
| Tumor size (cm)* | 4.8 (2.0–10.0) | 4.4 (1.8–11.0) | 0.423 |
| T stage (at nephrectomy) | |||
| pT1 | 27 (84.4%) | 32 (88.9%) | 0.584 |
| pT2 | 5 (15.6%) | 4 (11.1%) | |
| pT3 | 0 | 0 | |
| Regional lymph nodes (at nephrectomy) | |||
| N0 | 32 (100%) | 36 (100%) | † |
| N1 | 0 | 0 | |
| Distant metastasis (during follow‐up) | |||
| M0 | 32 (100%) | 36 (100%) | † |
| M1 | 0 | 0 | |
| Recurrence | |||
| Positive | 0 | 0 | † |
| Negative | 32 (100%) | 36 (100%) | |
| Follow‐up (months)* | 62 (10–126) | 33 (13–87) | <0.001 |
| Deaths (n) | 0 | 0 | † |
Median (range).
No statistics were computed.
Histologically, the tumors of the MTSCC cases consisted of a mixture of tubular and spindle cell components, which were separated by variable amounts of mucinous stroma. The tubules were short (31/32, 97%) or elongated (28/32, 88%), and sometimes formed a solid growth pattern (18/32, 56%) (Figure 1A, a–c). The spindle cells (32/32, 100%) were usually arranged in sheets (Figure 1A, d). Papillary structure with tumor cell tufts protruding into the tubular lumen (10/32, 31%) was common (Figure 1A, e). True papilla with well‐shaped fibrovascular cores (4/32, 13%) was rare (Figure 1A, f). Among the 32 cases of MTSCC, two cases (6%) had sarcomatoid differentiation (SMTS) regions, with high‐grade spindle cell proliferation, marked cytologic atypia, and tumor necrosis (Figure 1B, a–c). Besides, compared with the other typical MTSCCs, the immunohistochemical expression of vimentin in sarcomatoid differentiation lesions was stronger and the Ki‐67 index was higher (Figure 1B, d–g).
Figure 1.
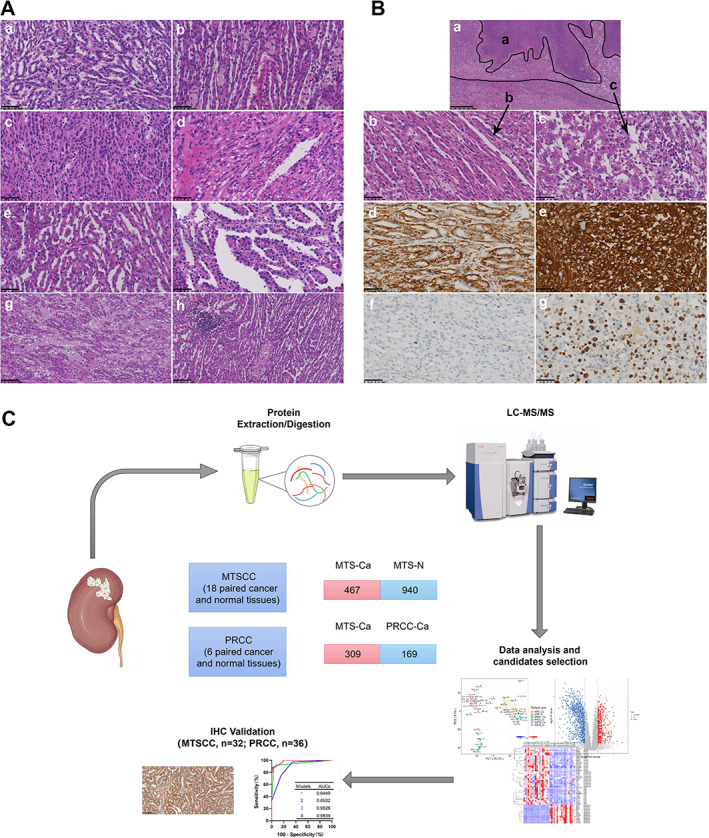
Morphologic and immunohistochemical features of MTSCC. (A) Morphologic spectrum of MTSCC. (a) Short tubules; (b) elongated tubules; (c) solid areas; (d) spindle cells; (e) micronodulous papillae; (f) well‐formed papillae; (g) stromal mucin; (h) scattered foci of foamy macrophages and lymphoplasmacytic cells. (a–f) 400× magnification; scale bar: 50 μm; (g, h) 200× magnification; scale bar: 100 μm. (B) Histologic and immunohistochemical features of MTSCC with sarcomatoid transformation. (a) Microscopic features of MTSCC with sarcomatoid transformation (black a, tumor necrosis; black b, typical MTSCC component; black c, sarcomatoid component). (b) Typical MTSCC areas. (c) Sarcomatoid regions characterized with significant cytologic atypia, coarse chromatin, and prominent nucleoli. (d–g) Immunohistochemical features of MTSCC with different components. Vimentin (d) and Ki‐67 (f) immunohistochemical staining in the typical MTSCC component. Immunohistochemical staining for vimentin (e) and Ki‐67 (g) in the sarcomatoid component. (a) 40× magnification; scale bar: 625 μm; (b–g) 400× magnification; scale bar: 50 μm). (C) Schematic of the proteome profiling of MTSCC and PRCC.
In addition, 25 cases (78%) of MTSCC exhibited apparent stromal mucin, whereas seven cases (22%) were mucin‐poor (Figure 1A, g). Other stromal changes that could be seen in the MTSCC cases included scattered focal foamy macrophages (14/32, 44%) and cuffing infiltrations of lymphoplasmacytic cells (20/32, 63%) surrounding the tumor cell nests (Figure 1A, h).
The solid variant of type 1 PRCC (15 cases), sharing similar morphologic features with MTSCC, is composed of compressed abortive papillae (micronodules/abortive papillae) (15/15,100%), ill‐defined tubules (short tubules) (15/15, 100%), true papillae (11/15, 73%), evenly elongated tubules (8/15, 53%), spindle cells (3/15, 20%), and solid sheets (7/15, 47%) with scant cytoplasm and small nuclei of monomorphic epithelial cells (supplementary material, Figure S1 and Table S4). Abundant stromal foamy macrophages were found in 11 of 15 (73%) cases (supplementary material, Figure S1, g). Stromal mucin and psammoma bodies were found in a few cases (2/15, 13%). Lymphoplasmacytic cells were absent in 13 of 15 cases (87%). Classic type 1 PRCC (21 cases) was mainly composed of true papillae formed by delicate fibrovascular cores that often contained foamy macrophages (supplementary material, Figure S1, h).
Immunohistochemically, both the tubules and the spindle cells in the MTSCC cases stained consistently positively for CK (31/31), vimentin (25/30), CK7 (17/21), EMA (15/18), E‐cadherin (7/8), AMACR (13/14), PAX8 (13/16), and RCC (9/12). A few cases displayed expression of CD10 (5/30). In agreement with previous literature, the results of CK7, AMACR, PAX8, and EMA were similar between the MTSCC and PRCC groups (supplementary material, Table S5).
In conclusion, MTSCC and type 1 PRCC, especially solid variants of type 1 PRCC, showed significant morphologic and immunophenotypic overlap. Given the difference in postoperative treatment and prognosis between the two tumors, there is an urgent need for new effective IHC biomarkers to distinguish MTSCC from type 1 PRCC.
Proteomic profiling of tumors and normal specimens of MTSCC and PRCC
To identify proteins elevated in or exclusive to MTSCC versus PRCC, we developed a proteomic analysis pipeline with random selection of surgically resected formalin‐fixed, paraffin‐embedded (FFPE) tissues from 18 MTSCC and six type 1 PRCC cases, including matched tumor and normal tissues (Figure 1C). Proteomic analysis was performed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) using tandem mass tag (TMT) chemical labeling experiments. The quality of protein extraction met the requirements of the study (supplementary material, Figure S2). A total of 48 samples were measured and the results showed good consistency in proteome identification and quantification (supplementary material, Figure S3A).
During the discovery stage, a total of 8163 proteins were identified, among which 6874 proteins were quantified (supplementary material, Table S6). Multivariate principal component analysis (PCA) and Pearson's correlation coefficient showed that most tumors of MTSCC (MTS‐Ca), adjacent normal tissues of MTSCC (MTS‐N), tumors of PRCC (PRCC‐Ca), and adjacent normal tissues of PRCC (PRCC‐N) clustered separately, suggesting that the samples selected in the experiment have good repeatability (Figure 2A and supplementary material, Figure S3B).
Figure 2.
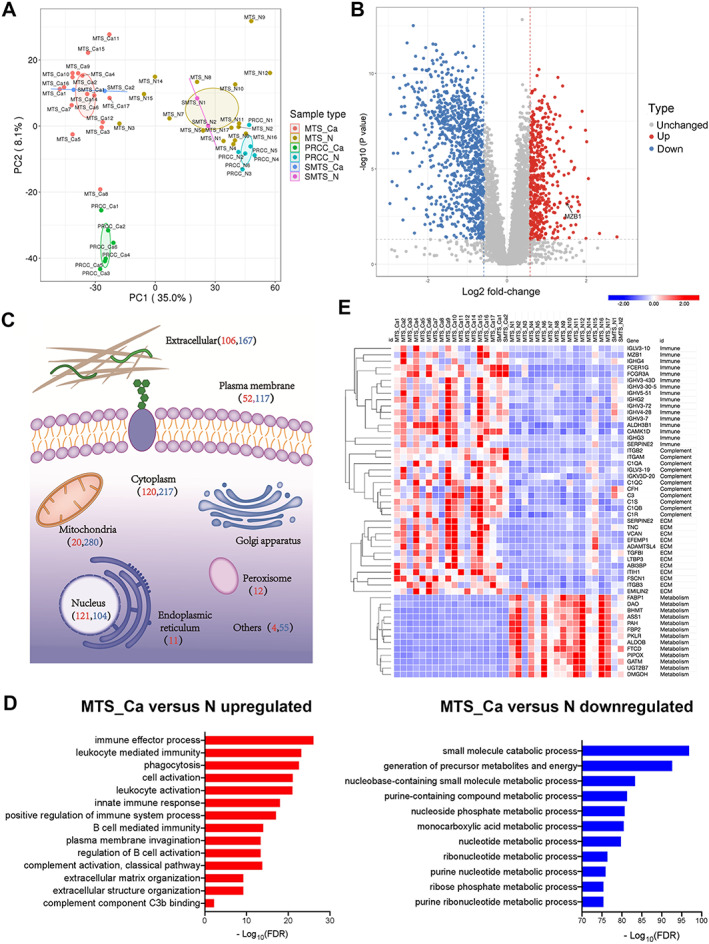
Overview of the proteome profiling of tumors and normal specimens. (A) Principal component analysis illustrating the moderate clustering of samples within each subtype. (B) Volcano plot highlighting the differentially expressed proteins in MTS‐Ca versus MTS‐N (red: upregulated proteins; blue: downregulated proteins). (C) Subcellular location of the differentially expressed proteins in MTS‐Ca versus MTS‐N (red numbers: number of upregulated proteins; blue numbers: number of downregulated proteins). (D) The differentially expressed proteins between MTS‐Ca and MTS‐N were classified by Gene Ontology (GO) annotation. (E) Heat map of differentially expressed proteins between MTS‐Ca and MTS‐N.
In addition, previous studies have identified different chromosomal alterations between MTSCC and PRCC [15, 16]. We reasoned that the protein level could be an indicator of chromosomal alteration. By plotting the fold‐change of proteins between tumor and normal samples by chromosome location, we found decreased expression of proteins in chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22 for MTSCC, and increased protein expression in chromosomes 7 and 17 for PRCC (supplementary material, Figure S3C,D). These data are consistent with chromosomal alterations in previous studies [15, 16].
To obtain a global view of the difference in protein expression, we next performed differential protein expression analysis. By using an adjusted P value less than 0.05 with a fold‐change of 1.5 or more, we identified 1407 differentially expressed proteins between MTS‐Ca and MTS‐N, including 467 upregulated proteins and 940 downregulated proteins (Figure 2B and supplementary material, Table S7). As shown in Figure 2C, a large number of differentially expressed proteins were of cytoplasmic (337 proteins, 23.95%), mitochondrial (300 proteins, 21.32%), and extracellular (273 proteins, 19.4%) origin. Biological process annotation indicated that the tumor proteome was significantly enriched in immune biology, complement activation, and extracellular matrix, including immune effector process, B‐cell‐mediated immunity, plasma membrane invagination, complement component C3b binding, and extracellular matrix organization (Figure 2D), while the matched normal tissue proteome was enriched in the metabolism process (Figure 2D). We observed a similar enrichment pattern in the gene set enrichment analysis (supplementary material, Figure S4A). As shown in Figure 2E and supplementary material, Table S8, differentially expressed proteins were involved in B‐cell‐mediated immune biology (FCER1G, SERPINE2, IGHG3, IGHV1‐69, IGHV6‐1, IGHV3‐43D, IGHV4‐34, GREM1, MZB1), complement activation (C3, C1QA, C1QB, C1QC, C1R, C1S), extracellular matrix (VCAN, SERPINE2, ABI3BP, EFEMP1, EMILIN2, LOX, GREM1, C1QA), and metabolism process (AASDH, ASS1, UGT2B7, FABP1, PAH, DAO, PTGDS, PIPOX, PKLR).
Proteomic comparisons between MTSCC and PRCC revealed activation of B‐cell immunity, complement system, and extracellular matrix formation but reduction of metabolism in MTSCC
We thoroughly explored the biological processes that differed between MTSCC and PRCC. As shown in Figure 2A, PCA demonstrated a clear distinction between MTS‐Ca and PRCC‐Ca tissues. Using an adjusted P value less than 0.05 with a difference of 1.5‐fold or more, we identified 478 differentially expressed proteins between MTS‐Ca and PRCC‐Ca tissues, including 309 upregulated proteins and 169 downregulated proteins (Figure 3A and supplementary material, Table S7). The differentially expressed proteins were mainly located in the extracellular matrix (33.05%), nucleus (22.59%), and cytoplasm (19.67%) (Figure 3B). Gene ontology enrichment analysis revealed that B‐cell‐mediated immune biology (MZB1, IGHG3, IGKC, IGHV3‐72, IGHV3‐43D, IGHG2) such as B‐cell‐mediated immunity and humoral immune response, complement activation (CAV1, C1QA, C5, CFH, C8A, C3), and extracellular matrix (VCAN, FBN1, CAVIN1, APOA2, MFAP5, APOA1) related proteins were upregulated and that metabolism pathways (FAAH, SLC2A9, SHMT1, ALDH3A2, NMNAT1, ASRGL1) were downregulated in MTS‐Ca compared with PRCC‐Ca (Figure 3C,D and supplementary material, Table S8). We observed a similar enrichment pattern in the gene set enrichment analysis (supplementary material, Figure S4B). These data together suggested that B‐cell immunity was enriched in MTSCC.
Figure 3.
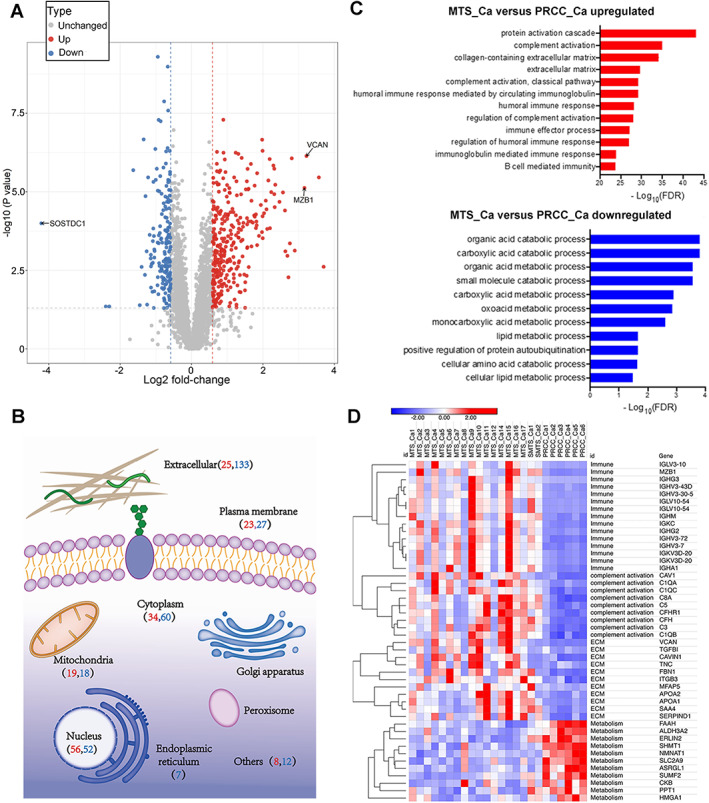
Comparison of MTSCC and PRCC tumor proteomics. (A) Volcano plot highlighting the differentially expressed proteins in MTS‐Ca versus PRCC‐Ca. (B) Subcellular location of the differentially expressed proteins in MTS‐Ca versus PRCC‐Ca (red numbers: number of upregulated proteins; blue numbers: number of downregulated proteins). (C) The differentially expressed proteins between MTS‐Ca and PRCC‐Ca were classified by GO annotation. (D) Heat map of differentially expressed proteins between MTS‐Ca and PRCC‐Ca.
Validating a three‐marker panel for differential diagnosis of MTSCC and PRCC
To filter candidates of MTSCC or PRCC for validation by IHC, we applied the following criteria in the selection: (1) they had to be proteins with peptide evidence in more than 67% of tumor samples from one and less than 33% of samples from the other histotype; (2) the MTS_Ca/PRCC_Ca ratio of differentially expressed proteins had to be greater than 6 or the PRCC_Ca/MTS_Ca ratio of differentially expressed proteins had to be greater than 2; and (3) the protein had to have corresponding commercial antibodies by IHC. Through the above criteria, we selected three markers highly expressed in MTSCC and four markers highly expressed in PRCC (supplementary material, Table S9). Sixty‐eight independent tumor sections (32 MTSCC and 36 PRCC) performed by IHC were scored using a three‐tier system as described in Supplementary materials and methods.
For these candidates, we confirmed that expression of VCAN (versican) and MZB1 (marginal zone B and B1 cell‐specific protein) was significantly higher in MTSCC than in PRCC, whereas expression of SOSTDC1 (sclerostin domain‐containing‐1) showed the contrary (p < 0.001; Figure 4A,B and supplementary material, Figure S5A–D and Table S10). Pie charts in supplementary material, Figure S5E show the expressed consistent percentages of MTSCC and type 1 PRCC samples for these biomarkers.
Figure 4.

Immunohistochemistry staining and diagnostic performance of three candidates in MTSCC and type 1 PRCC. (A) Images (200× magnification; scale bar: 100 μm) taken from MTSCC and PRCC tumor sections showing the morphological characteristics and expression of VCAN, MZB1, and SOSTDC1 using IHC. (B) Distribution of the staining scores of VCAN, MZB1, and SOSTDC1 between MTSCC and PRCC by IHC. (C) ROC analysis for distinguishing MTSCC from the solid variant of type 1 PRCC using each of the candidate biomarkers. (D) ROC curves for multi‐marker combination models to distinguish MTSCC from the solid variant of type 1 PRCC. (E) The mean ROC‐AUCs of five‐fold cross‐validation for each of the candidate biomarkers and combination models.
To examine the ability of each of the markers to distinguish MTSCC from PRCC, we performed binary logistic regression as a measure of performance. As shown in supplementary material, Figure S6A, the ROC‐AUCs of VCAN, MZB1, and SOSTDC1 for detecting MTSCC or PRCC were 0.937 [95% confidence interval (CI) 0.871–1.000], 0.867 (95% CI 0.775–0.960), and 0.667 (95% CI 0.538–0.795). VCAN was mainly expressed in the extracellular matrix of MTSCC tumor tissue (31/32, 96.87%) and yielded the highest AUC value with fairly high sensitivity (93.8%) and specificity (91.7%) at the threshold point of 0.598 for distinguishing MTSCC from type 1 PRCC (supplementary material, Figures S5A and S6A). MZB1 is a B‐cell‐specific and ER‐localized protein [23], which is most abundantly expressed in the marginal zone (MZ) B and B‐1 cells [24] and is upregulated during B‐cell differentiation into plasma cells, playing an important role in promoting plasma cell differentiation and antibody secretion [25]. In this study, MZB1 was mainly expressed in immune cells in the stroma of MTSCC (27/32, 84.38%). The sensitivity and specificity of MZB1 were 97.2% and 65.6%, with an ROC of 0.867 at an optimal cut‐off value of 0.735 (supplementary material, Figures S5B and S6A). SOSTDC1 was overexpressed in only 33.33% (12/36) of type 1 PRCC cases (AUC = 0.667, sensitivity = 33.3%, specificity = 66.7%), while the solid variant of type 1 PRCC had significantly higher expression of cell membrane SOSTDC1 (66.67%, 10/15) (supplementary material, Figures S5C and S6A). The AUC of SOSTDC1 for the solid variant of type 1 PRCC was 0.833, with a sensitivity of 66.7% and a specificity of 100.0%, presenting a significant diagnostic value for distinguishing MTSCC from the solid variant of type 1 PRCC (Figure 4C).
Given that individual markers did not perform very well, we also evaluated multi‐marker combinations. Using our candidates, we established four models. Model 1 (SOSTDC1 plus VCAN) and Model 2 (SOSTDC1 plus MZB1) achieved AUCs of 0.945 and 0.893. Model 3 (VCAN plus MZB1) had an improved AUC of 0.983 (supplementary material, Figure S6B), showing greater diagnostic value than Model 1 or 2. Model 4 (VCAN, MZB1, and SOSTDC1), which achieved an AUC of 0.984, with a sensitivity of 100.0% and a specificity of 86.1% at an optimal cut‐off value of 0.093, showed the greatest diagnostic value for differential diagnosis of MTSCC and type 1 PRCC. Since the main differential diagnostic consideration for MTSCC is the solid variant of type 1 PRCC, we also analyzed the diagnostic performance of our four models for detection between MTSCC and the solid variant of type 1 PRCC. The diagnostic value was further improved, with Model 1 achieving AUC = 0.991, Model 2 AUC = 0.923, Model 3 AUC = 0.987, and Model 4 AUC = 0.993 (Figure 4D). Comparing the performances among all the combined models, the three‐protein combination (VCAN, MZB1, and SOSTDC1) was found to be better in terms of differentiating MTSCC from type 1 PRCC, especially from the solid variant of type 1 PRCC (supplementary material, Table S11).
To avoid overfitting, five‐fold cross‐validation was employed to obtain more reliable performance estimates (supplementary material, Figure S6D). The mean ROC‐AUCs (mAUCs) of the above individual markers and combined models for distinguishing between MTSCC and PRCC are shown in supplementary material, Figure S6C–F (Model 1 mAUC = 0.939, Model 2 mAUC = 0.873, Model 3 mAUC = 0.970, and Model 4 mAUC = 0.972). mAUCs were further calculated to distinguish MTSCC from the solid variant of type 1 PRCC, achieving values of 0.980, 0.961, 0.981, and 0.985 in each of the models, respectively (Figure 4E). Model 4, which combined all three markers, had the highest mAUC – 0.985 – indicating the greatest diagnostic value in distinguishing MTSCC from the solid variant of type 1 PRCC.
Finally, we found that there was high consistency of protein abundance between LC–MS/MS and IHC (supplementary material, Figure S7 and Table S12). In addition, we explored the expression of these biomarkers in the pan‐RCC RNA expression data in the TCGA (supplementary material, Figure S8). We observed significantly higher SOSTDC1 expression and slightly higher VCAN expression in KIRP (kidney renal papillary cell carcinoma), compared with KIRC (kidney renal clear cell carcinoma) and KICH (kidney chromophobe cell carcinoma). Interestingly, MZB1 mRNA increased moderately in KIRC compared with the other RCC subtypes, which is worthy of further study in the future.
MZB1 may play an anti‐tumor role by activating the complement system
According to our proteomic results, the MTSCC tumor proteome was significantly enriched in B‐cell‐mediated immunity, and MZB1, a significantly and differentially expressed protein, was involved. Therefore, we further explored the expression of MZB1 and its possible roles in MTSCC.
In our study, immunohistochemistry showed that MZB1 was positive in 27 of 32 MTSCC tumors; MZB1‐positive cells were scattered both in the stroma of the tumor and in adjacent normal tissues. The distribution of MZB1‐positive cells was consistent with that of CD138 (plasma cell marker)‐positive cells both in the tumor and in adjacent normal tissues, and partially overlapped with CD20‐positive cells but did not overlap with CD3‐ and CD5‐positive cells (Figure 5A and supplementary material, Figure S9). Immunofluorescence showed that MZB1 signal co‐localized with CD138 signal, suggesting that MZB1 was mainly expressed in plasma cells (Figure 5B). Further analysis found that MZB1 was positive in the typical MTSCC component, and negative in the sarcomatoid transformation component in cases of MTSCC with sarcomatoid differentiation (SMTS) (Figure 5C). MZB1 was positively correlated with the expression of CD4 but not with the expression of CD8 in MTSCC (Figure 5D). Besides, the expression of MZB1 in MTSCC was negatively correlated with tumor clinical stage (Table 2). The above results suggested that MZB1 might play an anti‐tumor role in the tumor immunity of MTSCC.
Figure 5.
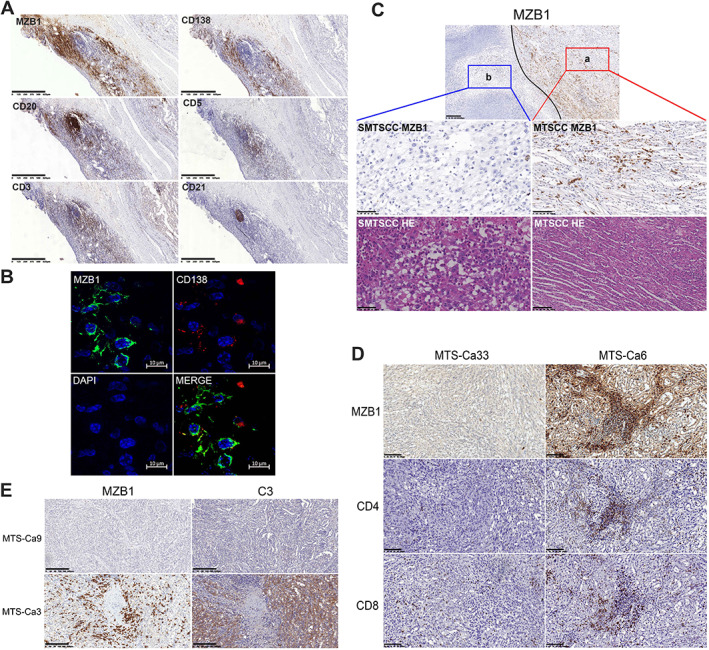
MZB1 may play an anti‐tumor role by activating the complement system. (A) Images (60× magnification; scale bar: 625 μm) taken from adjacent normal tissues of MTSCC showing the expression of MZB1, CD138, CD20, CD5, CD3, and CD21 using IHC. (B) Representative confocal image of three‐color immunostaining with antibodies to the indicated proteins in a formalin‐fixed, paraffin‐embedded tissue section of a patient with MTSCC (scale bar: 10 μm). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI), and CD138 stained plasma cells. (C) Images (MTSCC: 200× magnification; scale bar: 100 μm; SMTSCC: 400× magnification; scale bar: 50 μm) taken from adjacent normal tissues of MTSCC with sarcomatoid showing the morphological characteristics and the expression of MZB1 in different components using IHC (a, typical MTSCC component; b, sarcomatoid component; 100× magnification; scale bar: 200 μm). (D) Images (200× magnification; scale bar: 100 μm) taken from different MTSCC cases showing the expression of MZB1, CD4, and CD8 using IHC. (E) Images (100× magnification; scale bar: 200 μm) taken from different MTSCC cases showing the expression of MZB1 and C3 using IHC.
Table 2.
Correlation between MZB1 expression and the clinicopathological characteristics of the MTSCC patients
| Characteristics | No. of cases (%) | MZB1 | Fisher's exact test P value | |
|---|---|---|---|---|
| Low expression No. (%) | High expression No. (%) | |||
| Gender | ||||
| Female | 23 (71.9) | 8 (25.0) | 15 (46.9) | 1.000 |
| Male | 9 (28.1) | 3 (9.4) | 6 (18.7) | |
| T stage | ||||
| pT1 | 27 (84.4) | 7 (21.9) | 20 (62.5) | 0.037 |
| pT2 | 5 (15.6) | 4 (12.5) | 1 (3.1) | |
| pT3 | 0 | 0 | 0 | |
| Regional lymph nodes | ||||
| N0 | 32 (100) | 11 (34.4) | 21 (65.6) | * |
| N1 | 0 | 0 | 0 | |
| Distant metastasis | ||||
| M0 | 32 (100) | 11 (34.4) | 21 (65.6) | * |
| M1 | 0 | 0 | 0 | |
| Recurrence | ||||
| Positive | 32 (100) | 11 (34.4) | 21 (65.6) | * |
| Negative | 0 | 0 | 0 | |
| Clinical stage | ||||
| I | 27 (84.4) | 7 (21.9) | 20 (62.5) | 0.037 |
| II | 5 (15.6) | 4 (12.5) | 1 (3.1) | |
| III | 0 | 0 | 0 | |
No statistics were computed.
Furthermore, the proteomics analysis showed that tumor B‐cell‐mediated immunity (especially immunoglobulin, such as IGHG3, IGHV1‐69, IGHV6‐1, IGHV3‐43D, and IGHV4‐34) and complement system signaling pathway proteins (in particular, the complement intrinsic components involved in the classical pathway, including C3, C1QA, C1QB, and C1QC) were upregulated in MTSCC (Figure 2E). Immunohistochemistry also confirmed that the expression of C3 was increased in MTSCC with high expression of MZB1, while C3 was negative in MTSCC with low expression of MZB1 (Figure 5E). It is well known that the specific antibody binds with the corresponding antigen on the cell membrane surface to form an immune complex and activate the classical pathway of complement, which plays a role in tumor cell lysis through complement‐dependent cytotoxicity (CDC). Therefore, we speculate that MZB1 may regulate the differentiation and maturation of plasma cells, promote the secretion of antibodies, and play an anti‐tumor role by activating the complement system through tumor antigen–antibody immune complexes.
Unsupervised consensus clustering identified two molecular subtypes with different levels of oxidative phosphorylation for MTSCC
Lastly, we employed unsupervised consensus clustering to identify tumor subtypes. Clustering of all 48 samples revealed three tumor subgroups (groups 1, 2, and 3) and one adjacent normal group (group 4), which contained 14, 5, 7, and 22 samples, respectively (Figure 6A and supplementary material, Table S13). Groups 1 and 2 had most of the MTSCC samples, while group 3 contained all the PRCC samples. The tumor subtypes were not associated with differences in the overall rate of survival and clinical variables between group 1 and group 2 (p > 0.05; supplementary material, Table S14).
Figure 6.
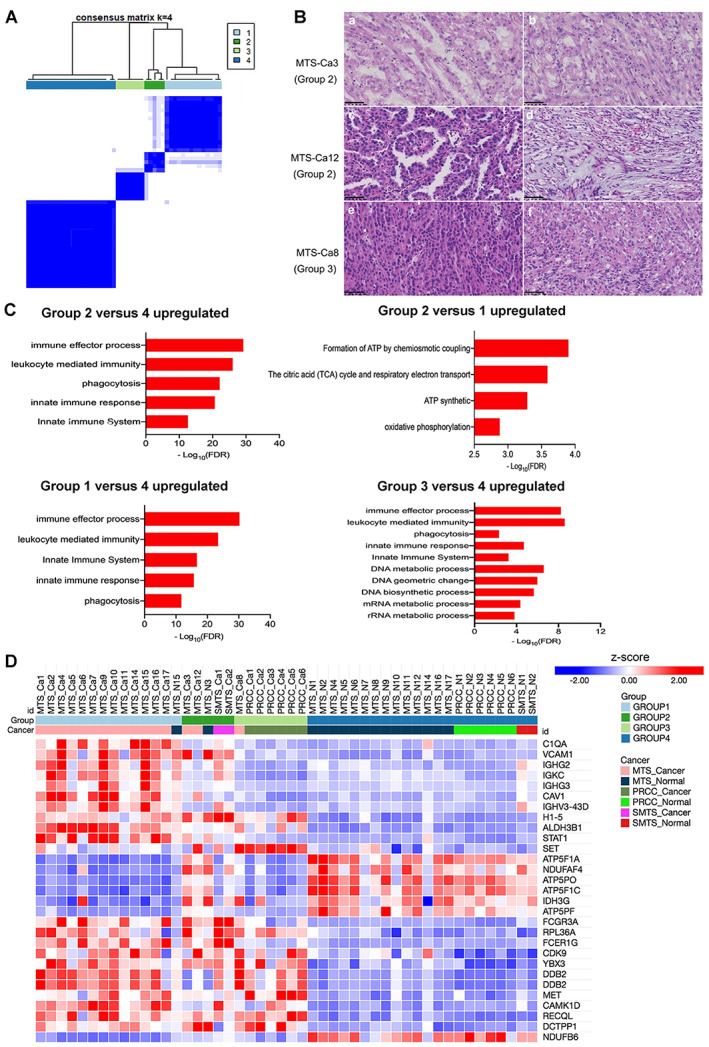
Identification of the molecular subtypes for MTSCC. (A) Similarity matrix for the four groups of samples. (B) Representative histological morphology for MTS‐Ca3 (group 2, a: micronodules papillae; b: typical MTSCC areas), MTS‐Ca12 (group 2, c: well‐formed papillae; d: stromal mucin), and MTS‐Ca8 (group 3, e: solid areas; f: stromal mucin). 400× magnification; scale bar: 50 μm. (C) The differentially expressed proteins for four subtypes were classified by GO annotation. (D) Heat map of the differentially expressed proteins for four subtypes.
We found that each subtype has unique histological and biological characteristics. Group 1 included 13 cases of MTSCC with classic morphology features and revealed a high level of B‐cell‐mediated immune biologies such as regulation of immune effector process and innate immune response (IGHG3, FCER1G, C1QA, CAV1, STAT1, ALDH3B1, IGHG2, and IGHV3) but a low level of oxidative phosphorylation; thus, we defined group 1 as the low oxidative phosphorylation group (Figure 6C,D and supplementary material, Table S13). In contrast, group 2 consisted of two cases of MTSCC with sarcomatoid differentiation and another two cases of MTSCC with 50% area of classic morphology features of MTSCC and 50% area of papillary structure (Figure 6B). Although MTSCC is typically indolent, a small subset of cases with high‐grade transformation has been reported to exhibit aggressive clinical behavior [5, 6, 9, 10]. These results suggest that when MTSCC cases have more papillary structures, they may have similar expression signatures, and aggravation to MTSCC cases with sarcomatoid differentiation. Group 2 was enriched not only for ‘immune biology’ but also for genes involved in oxidative phosphorylation (ATP5PF, NDUFAF4, ATP5PO, IDH3G, NDUFB6, ATP5F1A, and ATP5F1C); thus, we named it the high oxidative phosphorylation group (Figure 6C,D and supplementary material, Table S13). Group 3 included six cases of classical PRCC and one case of MTSCC (MTS‐Ca8) with a mainly solid growth pattern (Figure 6B). It was characterized with enrichment of DNA and RNA metabolic processes (H1‐5, DDB2, DCTPP1, YBX3, MET, RECQL, SET, RPL36A, and CDK9) (Figure 6C,D and supplementary material, Table S13), which related with MET protein, consistent with previous studies reporting that altered MET status, in combination with a frequent gain of chromosome 7, was identified in 81.3% of type 1 PRCCs [17, 26]. Group 4 consisted of 24 cases of normal tissue adjacent to cancer.
Discussion
MTSCC and type 1 PRCC show significant overlap both in morphology and in immunohistochemical profiles. We identified and validated a three‐marker panel which achieved an ROC‐AUC of 0.985 for distinguishing MTSCC from the solid variant of type 1 PRCC. We found molecular markers for the differential diagnosis of these tumors on IHC for the first time, which have high clinical utility for an accurate diagnosis. Moreover, we revealed two molecular subtypes of MTSCC, group 1 and group 2.
In this study, we found that lymphoplasmacytic cells commonly appeared in the stroma of MTSCC tumors and that these lymphoplasmacytic cells were positive for CD3 (T‐cell marker), CD5 (T‐cell marker), CD20 (B‐cell marker), and CD21 (follicular dendritic cell marker). Besides, there were different numbers of high endothelial venules in these lymphoplasmacytic cells morphologically. Therefore, it is likely that these lymphoplasmacytic cells are tertiary lymphoid structures (TLSs) in the tumor microenvironment (TME). TLSs are ectopic lymphoid aggregates that develop in non‐lymphoid tissues at sites of chronic inflammation including tumors [27]. TLSs have been detected in the tumor invasive margin and/or in the stroma within a wide range of human cancers at all stages of the disease, in primary as well as metastatic lesions [28]. They display an overall organization similar to that observed in canonical secondary lymphoid organs (SLOs), such as lymph nodes (LNs) [29]. In this study, the tumor proteome of MTSCC was significantly enriched in B‐cell‐mediated immune biology. We speculate that the phenomenon of rich TLSs in the tumor was the morphological basis of the upregulation of immune biology proteins in MTSCC. Large‐scale studies found that the presence of TLS is a prognostic and predictive marker in various primary cancers, including melanoma, breast, gastric, pancreatic, non‐small cell lung, oral squamous cell, and renal cell cancer [30]. Therefore, it was suggested that the rich TLSs in MTSCC might be related to the better prognosis of the tumor.
Our study identified three candidates – VCAN, MZB1, and SOSTDC1– which may serve as sensitive and specific markers to clinically distinguish MTSCC from PRCC. VCAN is one of the main components of the ECM, and plays an essential role in cell adhesion, proliferation, migration, and angiogenesis [31, 32]. Studies have demonstrated that VCAN is upregulated in several tumors such as renal carcinoma, gastric cancer, pancreatic cancer, and colon cancer [33, 34, 35, 36, 37], and is expressed predominantly in cancer stroma rather than in epithelial components [36, 37]. Our study found that VCAN was highly expressed in 96.88% of MTSCCs, which is consistent with previous findings [34, 36]. The role of SOSTDC1 as a bone morphogenetic protein antagonist [38] in malignant tumors has not been fully clarified. A previous report has indicated that, compared with normal kidney, SOSTDC1 is downregulated in renal cancer, particularly ccRCC, while PRCC and chromophobe renal cell carcinoma (chRCC) are slightly upregulated [39]. In our study, SOSTDC1 was negative in all MTSCCs, suggesting it as a specific marker for the solid variant of type 1 PRCC. Combination of the three candidates could further improve sensitivity and specificity for the differential diagnosis. However, given the relatively small size of this cohort, further studies using larger independent cohorts are needed to better assess their utility.
Two main pathways are transducing energy in cancer cells: the anaerobic pathway and the oxidative pathway. Aerobic glycolysis has long been considered as the major metabolic process for energy production and anabolic growth in cancer cells – the so‐called ‘Warburg effect’ [40]. However, more and more evidence supports the notion that mitochondria play a key role in oncogenesis. In our study, there existed excessive oxidative phosphorylation of group 2 compared with group 1, and respiratory electron transport was upregulated, both of which support the theory that oxidative phosphorylation of mitochondria is further restored at a certain stage of oncogenesis and progression, reducing the dependence of cells on aerobic glycolysis [41]. Oxidative phosphorylation was the main route to produce reactive oxygen species (ROS), and ROS could promote the occurrence and development of tumors in a variety of ways. One of the major mechanisms is that mitochondrial ROS favors the accumulation of potentially oncogenic DNA defects and the activation of potentially oncogenic signaling pathways [42, 43]. Therefore, we hypothesized that with the progression of MTSCC, the tumor cells' metabolism undergoes transformation, reprogramming from glycolysis to oxidative phosphorylation, then producing many ROS to induce the malignant transformation of tumors.
In summary, we studied a multi‐institutional cohort of 32 cases of MTSCC and 36 cases of type 1 PRCC through quantitative proteomics and IHC. The analysis of these data will provide a deeper understanding of the oncology of these cancers and identify new biomarker panels for distinguishing MTSCC from type 1 PRCC in the era of precision oncology.
Author contributions statement
FW, ZL and LW conceived and designed the study. FW and HX acquired data. ZL, HX and WL analyzed and interpreted data. FW, HX and WL drafted the manuscript. ZL critically revised the manuscript for important intellectual content. HX performed statistical analysis. CZ and LH were responsible for methodology, and NC, XL, FH, JZ and RW for resources.
Supporting information
Supplementary materials and methods
Figure S1. Morphologic spectrum of type 1 PRCC
Figure S2. SDS‐PAGE electrophoretogram of protein extracted from paraffin tissue of each sample
Figure S3. Quality control for the mass spectrometry platform
Figure S4. Gene set enrichment analysis
Figure S5. Representative staining and scoring of validated biomarkers
Figure S6. Three‐candidate model construction and diagnostic performance
Figure S7. Correlation analysis between LC–MS/MS and immunohistochemistry for each of the validated biomarkers
Figure S8. The pan‐RCC RNA expression data in TCGA
Figure S9. Images (40× magnification; scale bar: 400 μm) taken from MTSCC tumor areas showing the expression of MZB1, CD138, CD20, CD5, CD3, and CD21 using IHC
Table S1. Summary of the chromosomal aberrations of MTSCC and type 1 PRCC
Table S2. The sample‐channel mappings of the mass spectrometry proteomics data deposited in PRIDE
Table S3. Antibody staining information
Table S4. Morphologic features of MTSCC and the solid variant of type 1 PRCC cases
Table S5. Summary of the immunohistochemical staining profile of MTSCC and PRCC
Table S6. Proteins quantified and detected by TMT labeling quantitative proteomics in all samples
Table S7. Differentially expressed proteins in the whole cohort
Table S8. Protein abundance of differentially expressed proteins
Table S9. Proteins enriched in MTSCC and PRCC
Table S10. Immunohistochemistry scoring of VCAN, MZB1, and SOSTDC1
Table S11. Results of ROC curves in different combined models for differential diagnosis between MTSCC and PRCC
Table S12. The concordance of protein abundance between LC–MS/MS and IHC in the discovery cohort
Table S13. Differentially expressed proteins of altered pathways in each subtype
Table S14. Correlation between subtypes and the clinicopathological characteristics of the MTSCC patients
Acknowledgements
This research was supported by the National Natural Science Foundation of China (NSFC) (FW, No. 81972884 and ZL, No. 82001589); Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515110832) to ZL; The Fundamental Research Funds for the Central Universities, Sun Yat‐sen University (No. 19ykpy52) to FW; and the Natural Science Foundation of Guangdong Province (2017A030310457) to RW.
No conflicts of interest were declared.
Contributor Information
Liantang Wang, Email: wanglt@mail.sysu.edu.cn.
Zhenhua Luo, Email: luozhh35@mail.sysu.edu.cn.
Fen Wang, Email: wangfen5@mail.sysu.edu.cn.
Data availability statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [22] partner repository with the dataset identifier PXD027972.
References
- 1. Moch HHP, Ulbright TM, Reuter VE. Tumours of the kidney. In WHO Classification of Tumours of the Urinary System and Male Genital Organs (4th edn), Moch H, Humphrey PA, Ulbright TM, et al. (eds). IARC: Lyon, 2016; 37. [Google Scholar]
- 2. Ferlicot S, Allory Y, Compérat E, et al. Mucinous tubular and spindle cell carcinoma: a report of 15 cases and a review of the literature. Virchows Arch 2005; 447: 978–983. [DOI] [PubMed] [Google Scholar]
- 3. Paner GP, Srigley JR, Radhakrishnan A, et al. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney: significant immunophenotypic overlap warrants diagnostic caution. Am J Surg Pathol 2006; 30: 13–19. [DOI] [PubMed] [Google Scholar]
- 4. Fine SW, Argani P, DeMarzo AM, et al. Expanding the histologic spectrum of mucinous tubular and spindle cell carcinoma of the kidney. Am J Surg Pathol 2006; 30: 1554–1560. [DOI] [PubMed] [Google Scholar]
- 5. Dhillon J, Amin MB, Selbs E, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am J Surg Pathol 2009; 33: 44–49. [DOI] [PubMed] [Google Scholar]
- 6. Bulimbasic S, Ljubanovic D, Sima R, et al. Aggressive high‐grade mucinous tubular and spindle cell carcinoma. Hum Pathol 2009; 40: 906–907. [DOI] [PubMed] [Google Scholar]
- 7. Kenney PA, Vikram R, Prasad SR, et al. Mucinous tubular and spindle cell carcinoma (MTSCC) of the kidney: a detailed study of radiological, pathological and clinical outcomes. BJU Int 2015; 116: 85–92. [DOI] [PubMed] [Google Scholar]
- 8. Kuroda N, Hes O, Michal M, et al. Mucinous tubular and spindle cell carcinoma with Fuhrman nuclear grade 3: a histological, immunohistochemical, ultrastructural and FISH study. Histol Histopathol 2008; 23: 1517–1523. [DOI] [PubMed] [Google Scholar]
- 9. Ursani NA, Robertson AR, Schieman SM, et al. Mucinous tubular and spindle cell carcinoma of kidney without sarcomatoid change showing metastases to liver and retroperitoneal lymph node. Hum Pathol 2011; 42: 444–448. [DOI] [PubMed] [Google Scholar]
- 10. Qi XL, Zhao M, He XL, et al. Mucin‐poor mucinous tubular and spindle cell renal cell carcinoma with sarcomatoid transformation and multiple metastases: report of a unique case and review of literature. Int J Clin Exp Pathol 2016; 9: 2451–2458. [Google Scholar]
- 11. Sokolakis I, Kalogirou C, Frey L, et al. Mucin‐poor mucinous tubular and spindle cell carcinoma of the kidney presented with multiple metastases two years after nephrectomy: an atypical behaviour of a rare, indolent tumour. Case Rep Urol 2017; 2017: 6597592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen SS, Ro JY, Tamboli P, et al. Mucinous tubular and spindle cell carcinoma of kidney is probably a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol 2007; 11: 13–21. [DOI] [PubMed] [Google Scholar]
- 13. Kuroda N, Naroda T, Tamura M, et al. High‐grade mucinous tubular and spindle cell carcinoma: comparative genomic hybridization study. Ann Diagn Pathol 2011; 15: 472–475. [DOI] [PubMed] [Google Scholar]
- 14. Brandal P, Lie AK, Bassarova A, et al. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol 2006; 19: 186–194. [DOI] [PubMed] [Google Scholar]
- 15. Ren Q, Wang L, Al‐Ahmadie HA, et al. Distinct genomic copy number alterations distinguish mucinous tubular and spindle cell carcinoma of the kidney from papillary renal cell carcinoma with overlapping histologic features. Am J Surg Pathol 2018; 42: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cossu‐Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006; 19: 488–493. [DOI] [PubMed] [Google Scholar]
- 17. Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive molecular characterization of papillary renal‐cell carcinoma. N Engl J Med 2016; 374: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mehra R, Vats P, Cieslik M, et al. Biallelic alteration and dysregulation of the Hippo pathway in mucinous tubular and spindle cell carcinoma of the kidney. Cancer Discov 2016; 6: 1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics 2010; 10: 1886–1890. [DOI] [PubMed] [Google Scholar]
- 20. Clark DJ, Dhanasekaran SM, Petralia F, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 2019; 179: 964–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu D, Zhang S, Xie Z, et al. Plasminogen as a prognostic biomarker for HBV‐related acute‐on‐chronic liver failure. J Clin Invest 2020; 130: 2069–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez‐Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019; 47: D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Anken E, Pena F, Hafkemeijer N, et al. Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc Natl Acad Sci U S A 2009; 106: 17019–17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flach H, Rosenbaum M, Duchniewicz M, et al. Mzb1 protein regulates calcium homeostasis, antibody secretion, and integrin activation in innate‐like B cells. Immunity 2010; 33: 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimizu Y, Meunier L, Hendershot LM. pERp1 is significantly up‐regulated during plasma cell differentiation and contributes to the oxidative folding of immunoglobulin. Proc Natl Acad Sci U S A 2009; 106: 17013–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Linehan WM, Ricketts CJ. The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol 2019; 16: 539–552. [DOI] [PubMed] [Google Scholar]
- 27. Colbeck EJ, Ager A, Gallimore A, et al. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or bystander sentinels in disease? Front Immunol 2017; 8: 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sautès‐Fridman C, Petitprez F, Calderaro J, et al. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 2019; 19: 307–325. [DOI] [PubMed] [Google Scholar]
- 29. Germain C, Gnjatic S, Tamzalit F, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 2014; 189: 832–844. [DOI] [PubMed] [Google Scholar]
- 30. Sautès‐Fridman C, Lawand M, Giraldo NA, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol 2016; 7: 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinsella MG, Bressler SL, Wight TN. The regulated synthesis of versican, decorin, and biglycan: extracellular matrix proteoglycans that influence cellular phenotype. Crit Rev Eukaryot Gene Expr 2004; 14: 203–234. [DOI] [PubMed] [Google Scholar]
- 32. Rahmani M, Wong BW, Ang L, et al. Versican: signaling to transcriptional control pathways. Can J Physiol Pharmacol 2006; 84: 77–92. [DOI] [PubMed] [Google Scholar]
- 33. Mitsui Y, Shiina H, Kato T, et al. Versican promotes tumor progression, metastasis and predicts poor prognosis in renal carcinoma. Mol Cancer Res 2017; 15: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng Y, Sun H, Wu L, et al. VUp‐regulation of VCAN promotes the proliferation, invasion and migration and serves as a biomarker in gastric cancer. Onco Targets Ther 2020; 13: 8665–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Q, Yu D, Zhao Y, et al. Screening and identification of hub genes in pancreatic cancer by integrated bioinformatics analysis. J Cell Biochem 2019; 120: 19496–19508. [DOI] [PubMed] [Google Scholar]
- 36. Chida S, Okayama H, Noda M, et al. Stromal VCAN expression as a potential prognostic biomarker for disease recurrence in stage II–III colon cancer. Carcinogenesis 2016; 37: 878–887. [DOI] [PubMed] [Google Scholar]
- 37. Long X, Deng Z, Li G, et al. Identification of critical genes to predict recurrence and death in colon cancer: integrating gene expression and bioinformatics analysis. Cancer Cell Int 2018; 18: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yanagita M, Oka M, Watabe T, et al. USAG‐1: a bone morphogenetic protein antagonist abundantly expressed in the kidney. Biochem Biophys Res Commun 2004; 316: 490–500. [DOI] [PubMed] [Google Scholar]
- 39. Blish KR, Wang W, Willingham MC, et al. A human bone morphogenetic protein antagonist is down‐regulated in renal cancer. Mol Biol Cell 2008; 19: 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smolková K, Plecitá‐Hlavatá L, Bellance N, et al. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol 2011; 43: 950–968. [DOI] [PubMed] [Google Scholar]
- 42. Porporato PE, Filigheddu N, Pedro JMB, et al. Mitochondrial metabolism and cancer. Cell Res 2018; 28: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell 2016; 61: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Morphologic spectrum of type 1 PRCC
Figure S2. SDS‐PAGE electrophoretogram of protein extracted from paraffin tissue of each sample
Figure S3. Quality control for the mass spectrometry platform
Figure S4. Gene set enrichment analysis
Figure S5. Representative staining and scoring of validated biomarkers
Figure S6. Three‐candidate model construction and diagnostic performance
Figure S7. Correlation analysis between LC–MS/MS and immunohistochemistry for each of the validated biomarkers
Figure S8. The pan‐RCC RNA expression data in TCGA
Figure S9. Images (40× magnification; scale bar: 400 μm) taken from MTSCC tumor areas showing the expression of MZB1, CD138, CD20, CD5, CD3, and CD21 using IHC
Table S1. Summary of the chromosomal aberrations of MTSCC and type 1 PRCC
Table S2. The sample‐channel mappings of the mass spectrometry proteomics data deposited in PRIDE
Table S3. Antibody staining information
Table S4. Morphologic features of MTSCC and the solid variant of type 1 PRCC cases
Table S5. Summary of the immunohistochemical staining profile of MTSCC and PRCC
Table S6. Proteins quantified and detected by TMT labeling quantitative proteomics in all samples
Table S7. Differentially expressed proteins in the whole cohort
Table S8. Protein abundance of differentially expressed proteins
Table S9. Proteins enriched in MTSCC and PRCC
Table S10. Immunohistochemistry scoring of VCAN, MZB1, and SOSTDC1
Table S11. Results of ROC curves in different combined models for differential diagnosis between MTSCC and PRCC
Table S12. The concordance of protein abundance between LC–MS/MS and IHC in the discovery cohort
Table S13. Differentially expressed proteins of altered pathways in each subtype
Table S14. Correlation between subtypes and the clinicopathological characteristics of the MTSCC patients
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [22] partner repository with the dataset identifier PXD027972.