

Abstract
Aim
To evaluate BAY 2433334, an oral activated factor XI (FXIa) inhibitor, in volunteers.
Methods
Phase 1 study of healthy men at a German centre. Part A: randomized, single‐blind, multiple dose‐escalation study of BAY 2433334 (25/50/100 mg once daily [OD]) vs. placebo. Part B: similar design to Part A; evaluated BAY 2433334 25 mg twice daily. Part C: nonrandomized, open‐label study; evaluated potential interactions between BAY 2433334 (25/75 mg OD) and midazolam (7.5 mg), a CYP3A4 index substrate. Primary variables: treatment‐emergent adverse events (TEAEs; Parts A and B); area under the plasma concentration–time curve (AUC) and maximum plasma concentration of midazolam and α‐hydroxymidazolam (Part C). Study period: 18 days plus follow‐up visit.
Results
Parts A and B: 36 participants randomized to BAY 2433334; 12 to placebo. Part C: 48 participants assigned to BAY 2433334 plus midazolam. BAY 2433334 was well tolerated in all study parts. AUC and maximum plasma concentration of BAY 2433334 in plasma appeared dose proportional over 25–100 mg OD, with low‐to‐moderate variability in pharmacokinetic parameters. Multiple dosing caused minor‐to‐moderate accumulation and a mean terminal half‐life (15.8–17.8 h) supporting once‐daily dosing. Dose‐dependent FXIa activity inhibition and activated partial thromboplastin time prolongation were observed. BAY 2433334 appeared to have a minor effect on AUC for midazolam (ratio [90% confidence interval]: 1.1736 [1.0963–1.2564]) and α‐hydroxymidazolam (0.9864 [0.9169–1.0612]) only for BAY 2433334 75 mg OD on day 10.
Conclusion
Multiple dosing of BAY 2433334 in healthy volunteers was well tolerated, with a predictable pharmacokinetic/pharmacodynamic profile and no clinically relevant CYP3A4 induction or inhibition.
Keywords: BAY 2433334, clinical trial, factor XIa, pharmacodynamics, pharmacokinetics, phase 1, safety, tolerability

What is already known about this subject
Thromboembolic diseases are a major cause of mortality and morbidity worldwide.
Inhibiting activated factor XI (FXIa) has the potential to reduce thrombus formation without substantially increasing the risk of bleeding; this merits further study.
BAY 2433334 is a novel, oral, small‐molecule inhibitor of FXIa.
What this study adds
In this phase 1 multiple‐dose study of healthy volunteers, BAY 2433334 had a predictable pharmacokinetic and pharmacodynamic profile and was well tolerated.
There were no clinically relevant bleeding‐related adverse events.
There was no indication of any clinically relevant inhibition and/or induction of CYP3A4 by BAY 2433334.
1. INTRODUCTION
Thromboembolic diseases are a major cause of mortality and morbidity worldwide. Venous thromboembolism (VTE) is the third most common cardiovascular condition worldwide, with an annual incidence of 10–20 cases per 10 000 individuals. 1 , 2
Although anticoagulation forms a mainstay of antithrombotic therapy, the usefulness of current therapies is limited by an increased risk of bleeding, which is lessened but not eliminated with direct oral anticoagulants compared with vitamin K antagonists. 3 Hence, a need remains for new therapies, particularly for patients at high risk of thromboembolic events, bleeding events or both.
Inhibition of activated factor XI (FXIa) represents an alternative therapeutic antithrombotic target. In the intrinsic pathway of the coagulation cascade, FXIa is produced through activation of factor XI (FXI) by activated factor XII (FXIIa), and in turn initiates a series of enzymatic reactions leading to thrombin generation and consequently clot formation. 4 , 5 , 6 It also forms part of a feedback loop that maintains the generation of thrombin to consolidate coagulation. 5
Evidence that lowering FXIa reduces thrombus formation, without markedly increasing bleeding risk, comes from many sources. Individuals with lower FXI levels have a lower risk of venous thrombosis and ischaemic stroke, 7 but because the extrinsic pathway remains intact, the normal haemostatic response to injury remains relatively unaffected 6 , 7 , 8 ; indeed, congenital FXI deficiency in humans produces only a mild‐to‐moderate bleeding disorder. 5 Additionally, in animal models, deficiencies in intrinsic coagulation pathway zymogens reduce experimentally‐induced thrombogenesis, without affecting haemostasis. 4 , 9 , 10
Phase 2 proof‐of‐concept studies in patients undergoing total knee arthroplasty demonstrated that subcutaneous administration of a FXI antisense oligonucleotide, 11 or intravenous administration of an antibody that inhibits FXIa, 12 reduced the risk of VTE with a numerically lower rate of bleeding events than enoxaparin. However, antisense oligonucleotides require a prolonged treatment period to sufficiently decrease FXI levels, while intravenous administration of antibodies is invasive in routine clinical practice.
BAY 2433334 is a novel, oral, direct, small molecule inhibitor of FXIa. In in vitro and ex vivo studies, 13 it produced concentration‐dependent prolongation of the activated partial thromboplastin time (aPTT) in whole blood or plasma and dose‐dependent reductions in thrombus weight in animal models. This study investigated the safety, pharmacokinetics and pharmacodynamics of BAY 2433334 after multiple dosing in healthy volunteers.
2. METHODS
All drug/molecular target nomenclature conforms to the IUPHAR/BPS Guide to PHARMACOLOGY nomenclature classification. 14
2.1. Study design
This randomized, phase 1 study in healthy men (registration not required) was approved by the independent ethics committee of the North‐Rhine Medical Council (Ethikkommission bei der Aerztekammer Nordrhein) and the competent authority BfArM (Bundesinstitut für Arzneimittel und Medizinprodukte). It was conducted in accordance with the principles of the Declaration of Helsinki, the International Council for Harmonisation guideline on Good Clinical Practice, and German Drug Law (Arzneimittelgesetz).
The study consisted of 3 parts (Figure 1). Part A was a randomized, single‐blind, placebo‐controlled, multiple dose‐escalation study evaluating the safety, pharmacokinetics and pharmacodynamics of once‐daily dosing with BAY 2433334. Part B had a similar design to Part A, but evaluated twice‐daily dosing of 1 dose strength. Part C was a nonrandomized, open‐label study evaluating potential influence of BAY 2433334 to inhibit or induce CYP3A4 using the recommended index drug midazolam. 15 , 16
FIGURE 1.
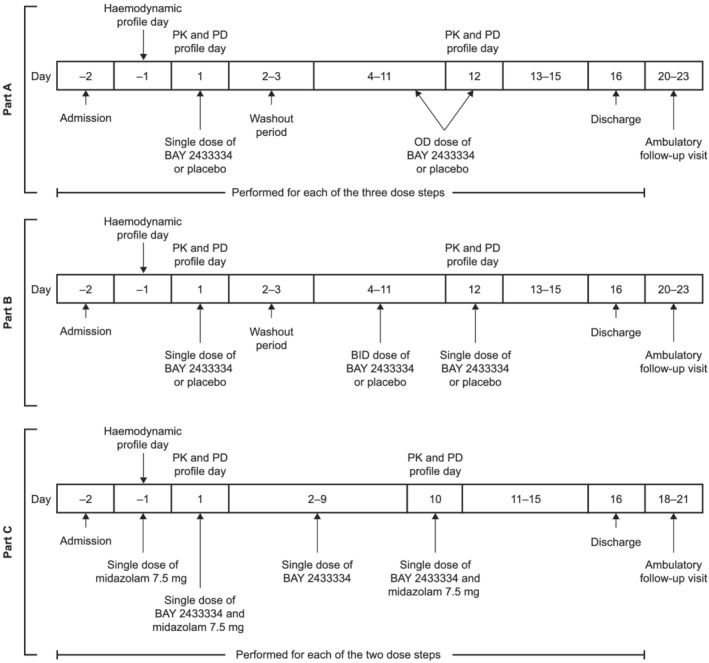
Overview of study design. BID, twice daily; OD, once daily; PD, pharmacodynamic; PK, pharmacokinetic
The primary aims of Parts A and B were to investigate the safety and tolerability of multiple doses of BAY 2433334, and the secondary objectives were to investigate the pharmacokinetics and pharmacodynamics following multiple dosing. In Part C, the primary objective was to investigate the effect of single and multiple once‐daily doses of BAY 2433334 on the pharmacokinetics of single oral doses of midazolam. The potential of BAY 2433334 to inhibit and/or induce CYP3A4 over time was investigated by comparing the area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax) of midazolam and α‐hydroxymidazolam before and after coadministration of midazolam with a single dose of BAY 2433334, and after multiple doses of BAY 2433334. 17
The study was conducted in 1 clinical trial unit in Germany between November 2018 and August 2019. Participants remained on a ward throughout the study period.
2.2. Participants
Healthy adult white men aged 18–45 with a body mass index ≥18.0 and ≤29.9 kg m−2 were enrolled into 3 nonoverlapping cohorts, 1 for each part of the study. The full inclusion and exclusion criteria are listed in the Supporting Information.
Written informed consent was obtained from all participants prior to enrolment.
2.3. Study treatment
BAY 2433334 treatment regimens per study part are shown in Figure 1. Ambulatory follow‐up visits occurred on days 20–23 (Parts A and B) or days 18–21 (Part C).
In Parts A and B, participants were randomized to receive BAY 2433334 25, 50 or 100 mg once daily, 25 mg twice daily, or placebo. A single dose of BAY 2433334 or placebo was administered on day 1 with a washout period on days 2–3, followed by once‐daily or twice‐daily treatment with BAY 2433334 or placebo on days 4–11. On day 12, BAY 2433334 was administered once daily. Dose escalation only occurred after the previous dose had been assessed to have acceptable safety and tolerability. All randomizations in Parts A and B were performed using a computer‐generated randomization list.
In Part C, participants received BAY 2433334 25 or 75 mg once daily on days 1–10. In addition, single doses of midazolam 7.5 mg (Dormicum, Roche Pharma AG, Germany) were administered on the day before the first dose of BAY 2433334 and on days 1 and 10 of treatment with BAY 2433334.
All doses of BAY 2433334 were given with approximately 240 mL noncarbonated water in the morning and in the evening for participants in Part B. On the days when pharmacokinetic and pharmacodynamic profiling was scheduled, BAY 2433334 was administered following an overnight fast of at least 10 h; on other days, meals were served at predefined times.
2.4. Safety analysis
Safety and tolerability were assessed by recording adverse events. Adverse events were considered treatment‐emergent if they started or worsened after first administration of any study medication, up to 7 days after the end of treatment with BAY 2433334.
Other safety assessments included physical examination, vital signs, 12‐lead electrocardiogram (ECG) and clinical laboratory evaluation.
2.5. Pharmacokinetic analyses
2.5.1. BAY 2433334 pharmacokinetics
Blood and urine samples were obtained as outlined in Figure S1 and stored at or below −15°C. BAY 2433334 concentrations in urine were assessed only in the 50‐ and 100‐mg dose steps in Parts A and B. Blood samples were analysed within 50 days and urine samples within 120 days after sampling. Concentrations of BAY 2433334 in plasma and urine were measured by high‐pressure liquid chromatography–tandem mass spectrometry, as described in the Supporting Information.
Pharmacokinetic parameters (Table S1) were calculated by noncompartmental analysis using WinNonlin (version 5.3; Pharsight Corporation, USA) in conjunction with Automation Extension (version 2.90; Bayer AG, Germany). Steady‐state conditions were assessed by comparing the trough concentrations drawn on 9 consecutive days of dosing.
2.5.2. Midazolam pharmacokinetics
In Part C, blood samples were obtained and analysed as outlined in Figure S1 and the Supporting Information. Cmax, AUC and AUC to time of last measurement above the lower limit of quantification, AUC(0–tlast), were among the pharmacokinetic parameters calculated for midazolam and α‐hydroxymidazolam. The molecular weight (MW)‐corrected metabolic AUC ratio (MR AUC ) for α‐hydroxymidazolam to midazolam on days −1, 1 and 10 were calculated using the formula:
2.6. Pharmacodynamic analysis
Blood samples for pharmacodynamic evaluation were obtained as outlined in Figure S1.
Pharmacodynamic markers were measured using validated techniques according to the manufacturers' instructions. aPTT was assayed using a kaolin trigger reagent (STA C.K. Prest, Diagnostica Stago, France) on a STA coagulation analyser. A prothrombin time (PT) assay was performed using STA‐Neoplastine Cl Plus reagent (Diagnostica Stago, France) on a STA coagulation analyser, with results expressed as absolute and International Normalized Ratio (INR) values. FXIa activity was assessed using a proprietary (Bayer) fluorogenic substrate assay (FXIa activity assay). FXII activity was measured using an aPTT‐based coagulation test in FXII‐deficient plasma, using reagents and hardware from Instrumentation Laboratories (USA). FXI and FXII concentrations were measured by microtitre plate‐based enzyme‐linked immunosorbent assay, with polyclonal antibodies from Affinity Biologicals, Inc. (USA). Activity of C1 esterase inhibitor (one of the endogenous inhibitors of FXIa) was assessed using a chromogenic substrate assay (Berichrom C1 inhibitor kit) on a Sysmex CS‐5100 instrument according to the manufacturer's instructions (Siemens Healthcare GmbH, Germany). Von Willebrand factor (vWF) antigen level and vWF ristocetin cofactor activity were analysed using turbidimetric assays (Diagnostica Stago, France).
aPTT data were presented as ratio to the individual baseline for each volunteer, which showed the prolongation of aPTT. FXIa activity was also presented as ratio to the individual baseline for each volunteer.
2.7. Data and statistical analysis
All statistical analyses were performed using SAS version 9.2 software (SAS Institute Inc., Cary, NC, USA).
Pharmacokinetic data were analysed for the Pharmacokinetic analysis set, which included all participants on active treatment with a valid on‐treatment pharmacokinetic profile (Parts A and B) or valid midazolam pharmacokinetic profile on day −1, and on day 1 and/or day 10, without findings that would have affected BAY 2433334 dosing (Part C). Pharmacodynamic data were analysed for the Pharmacodynamic analysis set, which included all participants who received all scheduled doses of study medication, and had evaluable pharmacodynamic data after the last scheduled dose. For Parts A and B, all participants who had a valid on‐treatment pharmacodynamic profile were included in the Pharmacokinetic analysis set. Safety data were analysed in the Safety analysis set, which included all participants who received at least 1 dose of study medication.
Demographic and quantitative data were summarized using descriptive statistics.
2.7.1. Pharmacokinetic data
In Part A, dose proportionality for the pharmacokinetic parameters of BAY 2433334 (day 1: Cmax divided by dose [Cmax/D] and AUC divided by dose [AUC/D]; day 12: Cmax/D and AUC within the dosing interval τ [AUCτ] divided by dose [AUCτ/D]) was assessed by analysis of variance (ANOVA) on log‐transformed data, with treatment as a factor. In Part C, pharmacokinetic parameters of midazolam were analysed by ANOVA of log‐transformed data, with participant and treatment effects. For these variables, 90% confidence intervals (CIs) for the ratios of [BAY 2433334 + midazolam at day 1]/[midazolam alone at day −1] and [BAY 2433334 + midazolam at day 10]/[midazolam alone at day −1] were evaluated. A lack of interaction was confirmed if the 90% CI was within the equivalence range of 80–125% for the ratio of [BAY 2433334 + midazolam at day 10]/[midazolam alone at day −1] (with regard to potential induction) or for the ratio of [BAY 2433334 + midazolam at day 1]/[midazolam alone at day −1] (with regard to potential inhibition).
2.7.2. Pharmacodynamic data
In Part A, dose steps were compared sequentially with placebo starting with the highest dose, using a Wilcoxon rank‐sum test with a 1‐sided level of significance (α = 0.05). The Hodges–Lehmann estimator of location shift (Δ) was presented, together with the associated 2‐sided 90% CI, in addition to the P‐value of the Wilcoxon test.
2.7.3. Sample size determination
No formal sample size estimation was performed for Parts A and B, because the primary objective of these parts was to evaluate safety. With a sample size of at least 6 participants in each active treatment group, a doubling of the principal pharmacokinetic parameters after the highest dose could be detected with α = 0.05 and a power of 80%, assuming that the expected interindividual coefficient of variation (CV) was ≤41%, and the expected dose‐normalized pharmacokinetic parameters with the other 2 doses were identical (SAS software, version 9.4, Proc GLMPOWER, SAS Institute Inc.).
In Part C, a sample size of 19 participants per treatment was estimated to be sufficient to achieve the study objectives. Based on the anticipated variation in AUC, this sample size would give 80% power to confirm that the 90% CI for the [BAY 2433334 + midazolam at day 10]/[midazolam alone at day −1] ratio would be contained within the range of 80–125%.
3. RESULTS
In total, 112 participants were enrolled in Parts A and B, of whom 48 were randomized; 36 received BAY 2433334 (n = 9 per treatment group) and 12 received placebo (n = 3 per treatment group). One participant randomized to receive 100 mg BAY 2433334 in Part A discontinued treatment after the third dose for personal reasons. In Part C, 79 participants were enrolled, of whom 48 received BAY 2433334 25 or 75 mg once daily (n = 24 per treatment group) and midazolam. Two participants assigned to 75 mg discontinued treatment because of treatment‐emergent adverse events (TEAEs).
Demographic and baseline characteristics of the participants are summarized in Table 1. In general, the treatment groups in each part of the study were comparable in terms of demographics.
TABLE 1.
Demographics and baseline characteristics of participants who received study medication. Data are presented as mean (standard deviation)
| Parts A and B | Part C | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BAY 2433334 25 mg OD (n = 9) | BAY 2433334 50 mg OD (n = 9) | BAY 2433334 100 mg OD (n = 9) | Placebo OD (n = 9) | BAY 2433334 25 mg BID (n = 9) | Placebo BID (n = 3) | Total (n = 48) | BAY 2433334 25 mg OD + midazolam 7.5 mg (n = 24) | BAY 2433334 75 mg OD + midazolam 7.5 mg (n = 24) | Total (n = 48) | |
| Age (y) | 34.0 (8.4) | 36.7 (5.0) | 29.9 (6.9) | 29.0 (4.7) | 31.2 (4.8) | 34.3 (10.0) | 32.3 (6.7) | 28.7 (7.0) | 31.2 (6.7) | 30.0 (6.9) |
| Weight (kg) | 77.82 (9.97) | 79.49 (9.02) | 71.22 (8.01) | 77.21 (6.75) | 81.57 (12.08) | 83.67 (7.38) | 77.85 (9.51) | 81.69 (7.97) | 78.58 (10.84) | 80.13 (9.54) |
| Height (cm) | 178.2 (5.3) | 181.9 (8.2) | 178.8 (8.1) | 180.2 (4.6) | 180.1 (3.7) | 180.3 (5.5) | 179.9 (6.0) | 179.8 (6.1) | 179.9 (6.3) | 179.9 (6.1) |
| Body mass index (kg m−2) | 24.50 (3.01) | 24.01 (1.93) | 22.30 (2.37) | 23.81 (2.18) | 25.09 (3.20) | 25.73 (1.86) | 24.05 (2.61) | 25.29 (2.40) | 24.23 (2.62) | 24.76 (2.54) |
OD, once daily; BID, twice daily.
3.1. Safety and tolerability in healthy volunteers
The incidence of TEAEs by study part is summarized in Tables 2 and 3. All events were mild; no serious adverse events were reported.
TABLE 2.
Number (%) of healthy volunteers with treatment‐emergent adverse events during Parts A and B
| BAY 2433334 25 mg OD (n = 9) | BAY 2433334 50 mg OD (n = 9) | BAY 2433334 100 mg OD (n = 9) | Placebo OD (n = 9) | BAY 2433334 25 mg BID (n = 9) | Placebo BID (n = 3) | Total BAY 2433334 (n = 36) | Total placebo (n = 12) | |
|---|---|---|---|---|---|---|---|---|
| Any adverse event | 3 (33.3%) | 5 (55.6%) | 5 (55.6%) | 3 (33.3%) | 4 (44.4%) | 2 (66.7%) | 17 (47.2%) | 5 (41.7%) |
| Most frequently reported adverse events a | ||||||||
| Headache | 1 (11.1%) | 2 (22.2%) | 3 (33.3%) | 0 | 1 (11.1%) | 1 (33.3%) | 7 (19.4%) | 1 (8.3%) |
| Nasopharyngitis | 0 | 1 (11.1%) | 0 | 1 (11.1%) | 0 | 1 (33.3%) | 1 (2.8%) | 2 (16.7%) |
| Oropharyngeal pain | 0 | 1 (11.1%) | 0 | 0 | 1 (11.1%) | 0 | 2 (5.6%) | 0 |
| Toothache | 1 (11.1%) | 1 (11.1%) | 0 | 0 | 0 | 0 | 2 (5.6%) | 0 |
| Medical device site urticaria | 0 | 0 | 1 (11.1%) | 0 | 1 (11.1%) | 0 | 2 (5.6%) | 0 |
| Glutamate dehydrogenase increased | 0 | 1 (11.1%) | 0 | 0 | 1 (11.1%) | 0 | 2 (5.6%) | 0 |
| Lipase increased | 0 | 1 (11.1%) | 1 (11.1%) | 0 | 0 | 0 | 2 (5.6%) | 0 |
| Back pain | 0 | 0 | 1 (11.1%) | 0 | 0 | 1 (33.3%) | 1 (2.8%) | 1 (8.3%) |
| Epistaxis | 0 | 0 | 0 | 1 (11.1%) | 1 (11.1%) | 0 | 1 (2.8%) | 1 (8.3%) |
| Any study drug‐related adverse event(s) | 0 | 0 | 2 (22.2%) b | 0 | 0 | 0 | 2 (5.6%) | 0 |
| Any study procedure‐related adverse event(s) | 1 (11.1%) | 3 (33.3%) | 1 (11.1%) | 2 (22.2%) | 2 (22.2%) | 1 (33.3%) | 7 (19.4%) | 3 (25.0%) |
| Any adverse event(s) of special interest | 1 (11.1%) | 1 (11.1%) | 1 (11.1%) | 0 | 1 (11.1%) | 0 | 4 (11.1%) | 0 |
| Bleeding | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Investigations | 1 (11.1%) c | 1 (11.1%) d | 1 (11.1%) d | 0 | 1 (11.1%) e | 0 | 4 (11.1%) | 0 |
BID, twice daily; OD, once daily.
Treatment‐emergent adverse events reported (by preferred term) in 2 or more volunteers.
Headache.
Amylase increased.
Lipase increased.
Glutamate dehydrogenase increased.
TABLE 3.
Number (%) of healthy volunteers with treatment‐emergent adverse events during Part C
| BAY 2433334 25 mg treatment sequence | BAY 2433334 75 mg treatment sequence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Midazolam alone (n = 24) | BAY 2433334 25 mg single dose + midazolam (n = 24) | BAY 2433334 25 mg multiple dose alone (n = 24) | BAY 2433334 25 mg multiple dose + midazolam (n = 24) | Total (N = 24) | Midazolam alone (n = 24) | BAY 2433334 75 mg single dose + midazolam (n = 24) | BAY 2433334 75 mg multiple dose alone (n = 24) | BAY 2433334 75 mg multiple dose + midazolam (n = 24) | Total (N = 24) | |
| Any adverse event | 3 (12.5%) | 0 | 7 (29.2%) | 1 (4.2%) | 9 (37.5%) | 3 (12.5%) | 1 (4.2%) | 4 (16.7%) | 2 (8.3%) | 8 (33.3%) |
| Most frequently reported adverse events a | ||||||||||
| Headache | 0 | 0 | 0 | 0 | 0 | 1 (4.2%) | 0 | 3 (12.5%) | 2 (8.3%) | 6 (25.0%) |
| Study drug‐related adverse event(s) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Study procedure‐related adverse event(s) | 3 (12.5%) | 0 | 1 (4.2%) | 0 | 4 (16.7%) | 1 (4.2%) | 0 | 0 | 1 (4.2%) | 2 (8.3%) |
| Adverse event(s) of special interest b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
The Total columns count the volunteers across all treatment periods. Total is not necessarily the sum of the previous columns.
Treatment‐emergent adverse events reported (by preferred term) in 2 or more volunteers.
Includes: haemorrhage, particularly fatal bleeding or symptomatic bleeding in a critical area or organ and/or symptomatic bleeding causing a clinically significant fall in haemoglobin level; all adverse events related to hepatobiliary dysfunction; and all adverse events possibly related to pancreatic disorders.
No adverse events leading to discontinuation were reported in Parts A or B. In Part C, 2 participants (BAY 2433334 75 mg) discontinued study medication because of TEAEs (intermittent, second‐degree [Wenckebach] atrioventricular block; elevation of C‐reactive protein due to trochanteric bursitis).
The most frequently reported TEAEs were headaches (n = 8 in Parts A and B; n = 6 in Part C), followed by nasopharyngitis (n = 3 in Parts A and B; n = 1 in Part C). No other TEAEs were reported by >2 participants in Parts A and B, or >1 participant in Part C.
In Parts A and B, study drug‐related TEAEs (headache) were reported in 2 participants, both of whom received BAY 2433334 100 mg. No study drug‐related TEAEs were reported in Part C.
No clinically relevant effects of BAY 2433334 were observed in vital signs, ECG or clinical laboratory investigations, other than the anticipated prolongation of aPTT.
3.2. Pharmacokinetic analysis
Figure 2 shows geometric mean plasma concentration–time data of BAY 2433334 in Parts A and B of the study. Pharmacokinetic parameters are summarized in Tables 4 and 5.
FIGURE 2.

Geometric mean (standard deviation) plasma concentration of BAY 2433334 versus time in Parts A (once‐daily dosing with BAY 2433334 25, 50 or 100 mg) and B (twice‐daily dosing with BAY 2433334 25 mg on days 4–11, with once‐daily dosing on days 1 and 12) on a semi‐logarithmic scale. BID, twice daily; OD, once daily
TABLE 4.
Pharmacokinetic parameters of BAY 2433334 in plasma and urine on day 1 in Parts A and B of the study. Data are presented as geometric mean/geometric coefficient of variation (%), unless otherwise indicated
| BAY 2433334 25 mg OD n = 9 | BAY 2433334 50 mg OD n = 9 | BAY 2433334 100 mg OD n = 9 | BAY 2433334 25 mg BID n = 9 | |
|---|---|---|---|---|
| AUC (μg h L−1) | 6560/21.0 | 13 400/27.2 | 27 200/28.8 | 6120/25.5 |
| AUC/D (h L−1) | 0.263/21.0 | 0.268/27.2 | 0.272/28.8 | 0.245/25.5 |
| Cmax (μg L−1) | 358/23.6 | 675/25.7 | 1230/23.9 | 341/13.7 |
| Cmax/D (10−3 L−1) | 14.3/23.6 | 13.5/25.7 | 12.3/23.9 | 13.6/13.7 |
| tmax a (h) | 2.00 (1.48–5.98) | 2.00 (0.750–4.00) | 3.97 (1.47–6.00) | 3.00 (1.00–5.98) |
| t1/2 (h) | 14.0/15.0 | 15.9/13.6 | 15.8/18.6 | 14.6/23.5 |
| %AE,ur b (%) | – | 9.10/1.77 | 7.61/3.61 | 13.4/13.4 |
| CLR (L h−1) | – | 0.361/24.5 | 0.267/64.2 | 0.440/67.4 |
%AE,ur, percentage amount of drug excreted in urine; AUC, area under the plasma concentration–time curve; AUC/D, area under the plasma concentration–time curve divided by dose; BID, twice daily; CLR, renal clearance; Cmax, maximum plasma concentration; Cmax/D, maximum plasma concentration divided by dose; OD, once daily; t1/2, terminal half‐life; tmax, time to Cmax.
Median (range).
Arithmetic mean/standard deviation.
TABLE 5.
Pharmacokinetic parameters of BAY 2433334 in plasma and urine on day 12 in Parts A and B of the study. Data are presented as geometric mean/geometric coefficient of variation (%), unless otherwise indicated
| BAY 2433334 25 mg OD n = 9 | BAY 2433334 50 mg OD n = 9 | BAY 2433334 100 mg OD n = 8 | BAY 2433334 25 mg BID n = 9 | |
|---|---|---|---|---|
| AUCτ (μg h L−1) | 6910/11.0 c | 13 800/25.0 c | 29 500/32.7 c | 6180/32.2 d |
| AUCτ/D (h L−1) | 0.277/11.0 c | 0.276/25.0 c | 0.295/32.7 c | 0.247/32.2 d |
| Cmax (μg L−1) | 507/12.7 c | 963/24.8 c | 1950/28.1 c | 646/30.5 d |
| Cmax/D (10−3 L−1) | 20.3/12.7 c | 19.3/24.8 c | 19.5/28.1 c | 25.8/30.5 d |
| tmax a (h) | 4.00 (1.00–6.03) | 3.00 (0.750–6.05) | 4.00 (1.50–6.00) | 1.50 (1.00–6.00) |
| t1/2 (h) | 15.8/18.7 | 17.8/17.0 | 17.7/22.8 | 15.8/24.1 |
| RAAUC | 1.50/8.69 c | 1.54/10.0 c | 1.72/16.6 c | 2.22/18.5 d |
| RACmax | 1.41/13.4 c | 1.43/24.9 c | 1.63/14.3 c | 1.89/18.8 d |
| RLIN | 1.05/11.9 c | 1.03/7.86 c | 1.08/7.58 c | 1.01/10.1 d |
| %AE,ur,τ b (%) | – | 13.4/3.69 c | 10.1/4.21 c | 13.5/3.42 d |
| CLR (L h−1) | – | 0.469/31.5 c | 0.317/69.9 c | 0.528/40.8 d |
Median (range).
Arithmetic mean/standard deviation (range).
τ = 24 h.
τ = 12 h.
%AE,ur,τ, percentage amount of drug excreted in urine within the dosing interval; AUCτ, area under the plasma concentration–time curve within the dosing interval; AUCτ/D, area under the plasma concentration–time curve within the dosing interval divided by dose; CLR, renal clearance; Cmax, maximum plasma concentration; Cmax/D, maximum plasma concentration divided by dose; RAAUC, accumulation ratio calculated from area under the plasma concentration–time curve within the dosing interval after multiple dosing and after single dosing; RACmax, accumulation ratio calculated from maximum plasma concentration after multiple dosing and after single dosing; RLIN, linearity factor calculated from area under the plasma concentration–time curve within the dosing interval after multiple dosing and area under the plasma concentration–time curve after single dosing; t1/2, terminal half‐life; tmax, time to maximum plasma concentration.
On day 1 of Parts A and B, the first dose of BAY 2433334 was absorbed with a median time to Cmax of 2.00–3.97 h in all treatment groups (Table 4). After multiple dosing, median time to Cmax for BAY 2433334 on day 12 was 3.00–4.00 h following once‐daily dosing, and was 1.50 h following twice‐daily dosing (Table 5). BAY 2433334 was eliminated with a geometric mean terminal half‐life (t1/2) of 14.0–15.9 h across all treatment groups on day 1 (Table 4). On day 12, geometric mean t1/2 was within a similar range (15.8–17.8 h; Table 5). The degree of accumulation of BAY 2433334 at steady state was reflected by geometric mean accumulation ratio values of 1.50–1.72 and 1.41–1.63 for AUCτ(0–24) or Cmax after multiple once‐daily dosing for 9 consecutive days, compared with AUC or Cmax after single‐dose administration, respectively (RAAUC and RACmax; Table 5). Geometric mean RAAUC and RACmax values were higher after twice‐daily administration (2.22 and 1.89, respectively) than after once‐daily administration (Table 5). Following repeated dosing for 9 consecutive days, BAY 2433334 pharmacokinetics appeared to be linear with time, with a linearity factor calculated from AUCτ after multiple dosing and AUC after single dosing (RLIN) of 1.03–1.08 after once‐daily administration and 1.01 after twice‐daily administration (Table 5). The interindividual variability in AUCτ(0–24) and AUCτ(0–12) was low to moderate, with CVs of 11.0–32.7% across treatment groups. The apparent oral clearance did not show any relevant differences between the treatment groups and after single or multiple dosing, with geometric means of 3.68–4.09 L h−1 on day 1 and 3.39–4.04 L h−1 on day 12. The arithmetic mean renal elimination (%AE,ur) of unchanged drug accounted for approximately 7.61–13.4% of the dose after single‐dose administration and for approximately 10.1–13.5% after multiple dosing (Tables 4 and 5). AUC and Cmax of BAY 2433334 in plasma appeared to increase dose proportionally over the dose range of 25–100 mg after single and multiple dosing in Part A. Respective point estimates for the ratios of AUC/D, AUCτ/D and Cmax/D were close to 1.
Maximum plasma concentrations of midazolam and α‐hydroxymidazolam were rapidly reached (0.583–0.750 h) and similar for both compounds and across the treatments with and without BAY 2433334. Further, AUC, Cmax and t1/2 did not seem to be meaningfully different between the treatments. Acceptable extrapolated AUC when comparing AUC and AUC(0‐tlast) justified the 24‐hour sampling period as expected by the observed t1/2 (Table 6). On day 1, coadministration of BAY 2433334 25 mg resulted in an increase in the AUC for midazolam and α‐hydroxymidazolam by 4.2 and 4.4%, respectively. Coadministration of BAY 2433334 75 mg resulted in an increase in the AUC for midazolam by 4.5% and a decrease in the AUC for α‐hydroxymidazolam by 1.5%. The metabolic AUC ratios of α‐hydroxymidazolam to midazolam were comparable between day 1 (geometric mean, 0.335) and day −1 (geometric mean, 0.330) for BAY 2433334 25 mg, and slightly lower on day 1 (geometric mean, 0.369) than day −1 (geometric mean, 0.391) for BAY 2433334 75 mg. Following repeated dosing with BAY 2433334 25 mg once daily for 9 consecutive days and coadministration of BAY 2433334 25 mg on day 10, there were increases in the AUCs for midazolam and α‐hydroxymidazolam of 6.3% and 6.0%, respectively. After pre‐ and coadministration of BAY 2433334 75 mg, there was an increase in the AUC for midazolam of 17% and a decrease in the AUC for α‐hydroxymidazolam of 7.2%. The metabolic AUC ratios were slightly lower on day 10 (geometric mean, 0.319 [25 mg]; 0.310 [75 mg]) than day −1 (geometric mean, 0.330 [25 mg]; 0.391 [75 mg]) for both treatments. Based on the results of the ANOVA for both treatments, BAY 2433334 appeared to have no relevant effect on the AUC of midazolam and α‐hydroxymidazolam on days 1 and 10, because the point estimates for the ratios were close to 1 (Table 7). The 90% CIs of the AUC ratio for both midazolam and α‐hydroxymidazolam were within the no effect boundary of 0.80 to 1.25, except for the upper 90% CI for AUC ratio for midazolam (1.0963; 1.2564) on day 10 in the 75 mg group, which was numerically only marginally above the 1.25 boundary (third decimal place).
TABLE 6.
Pharmacokinetic parameters for midazolam and α‐hydroxymidazolam in plasma in Part C of the study. Data are presented as geometric mean/geometric coefficient of variation (%), unless otherwise indicated
| Day −1 | Day 1 | Day 10 | ||||
|---|---|---|---|---|---|---|
| Dose of BAY 2433334 | 25 mg OD (n = 24) | 75 mg OD (n = 24) | 25 mg OD (n = 24) | 75 mg OD (n = 24) | 25 mg OD (n = 24) | 75 mg OD (n = 22) |
| Midazolam | ||||||
| AUC (μg h L−1) | 86.0/38.6 | 94.8/31.6 | 89.6/42.4 | 99.1/31.4 | 91.4/38.8 | 111/29.2 |
| AUC(0–tlast; μg h L−1) | 84.1/38.0 | 93.0/31.3 | 87.4/42.5 | 96.6/31.3 | 89.3/38.5 | 108/28.9 |
| Cmax (μg L−1) | 32.7/33.7 | 41.7/44.8 | 36.9/59.8 | 37.2/49.6 | 34.4/59.7 | 38.8/55.0 |
| tmax a (h) | 0.583 (0.483–4.00) | 0.750 (0.500–3.00) | 0.742 (0.500–4.00) | 0.750 (0.300–4.00) | 0.750 (0.483–4.00) | 0.750 (0.250–2.00) |
| t1/2 (h) | 4.78 b (2.29–6.84) | 4.79 b (2.49–7.91) | 4.97/26.1 | 4.62/32.7 | 5.01/27.6 | 5.04/26.7 |
| α‐Hydroxymidazolam | ||||||
| AUC (μg h L−1) | 29.8/31.0 | 38.9/33.2 | 31.1/33.2 c | 38.3/30.9 | 31.6/41.2 d | 36.1/22.8 c |
| AUC(0–tlast; μg h L−1) | 27.7/29.4 | 34.5/25.8 | 29.8/39.2 | 35.7/30.5 | 29.0/38.1 | 34.7/25.7 |
| Metabolic AUC ratio e | 0.330/44.6 | 0.391/46.1 | 0.335/31.1 c | 0.369/42.9 | 0.319/33.1 d | 0.310/36.8 c |
| Cmax (μg L−1) | 13.0/60.5 | 18.0/59.0 | 15.6/71.6 | 15.8/85.7 | 13.8/77.7 | 16.1/56.0 |
| tmax a (h) | 0.708 (0.483–4.00) | 0.750 (0.500–3.00) | 0.742 (0.500–4.00) | 0.750 (0.483–4.00) | 0.750 (0.483–4.00) | 0.750 (0.483–2.00) |
| t1/2 (h) | 7.36/69.4 | 12.6/94.8 | 8.81/85.0 c | 7.96/71.8 | 8.48/78.0 d | 7.99/59.3 c |
AUC, area under the plasma concentration–time curve; AUC(0–tlast), AUC to time of last measurement above the lower limit of quantification; Cmax, maximum plasma concentration; OD, once daily; tmax, time to Cmax; t1/2, terminal half‐life.
Median (range).
Median (range) presented; geometric mean not calculated.
n = 20.
n = 23.
α‐Hydroxymidazolam AUC/midazolam AUC, corrected for molecular weight.
TABLE 7.
ANOVA analysis of AUC of midazolam and α‐hydroxymidazolam following treatment with BAY 2433334 in Part C
| AUC ratio day 1/day−1 | AUC ratio day 10/day−1 | ||||
|---|---|---|---|---|---|
| Treatment | Analyte | n | Point estimate geometric LS means (geometric 90% CI) | n | Point estimate geometric LS means (geometric 90% CI) |
| BAY 2433334 25 mg OD + Midazolam | Midazolam | 24 | 1.0415 (0.9659–1.1230) | 24 | 1.0630 (0.9859–1.1461) |
| α‐Hydroxymidazolam | 22 | 1.0650 (0.9661–1.1741) | 24 | 1.0560 (0.9595–1.1622) | |
| BAY 2433334 75 mg OD + Midazolam | Midazolam | 24 | 1.0451 (0.9785–1.1164) | 22 | 1.1736 (1.0963–1.2564) |
| α‐Hydroxymidazolam | 24 | 0.9856 (0.9206–1.0551) | 20 | 0.9864 (0.9169–1.0612) |
ANOVA, analysis of variance; AUC, area under the plasma concentration–time curve; CI, confidence interval; LS, least squares; OD, once daily.
3.3. Pharmacodynamic analysis
In Parts A and B, BAY 2433334 produced a dose‐dependent prolongation of aPTT (Figure 3). Statistical evaluation of the data from Part A showed that all doses of BAY 2433334 produced significant prolongations of aPTT, compared with placebo (P < .05 for all dose comparisons; BAY 2433334 25–100 mg once daily in Part A; Figure 3). Prolongation of aPTT was apparent at the first time point (30 min after dosing), with maximal prolongation observed approximately 4 h after dosing. Statistically significant aPTT prolongation was still present 24 h after dosing for all doses tested. Comparison of the maximal relative increases in aPTT (geometric mean) after 50 mg once‐daily and 25 mg twice‐daily dosing showed a statistically significant difference (P = .0005; Kruskal–Wallis test), with more pronounced effects for the maximal effect, and lower effects at 12 h postdose after once‐daily dosing than twice‐daily dosing at steady state.
FIGURE 3.
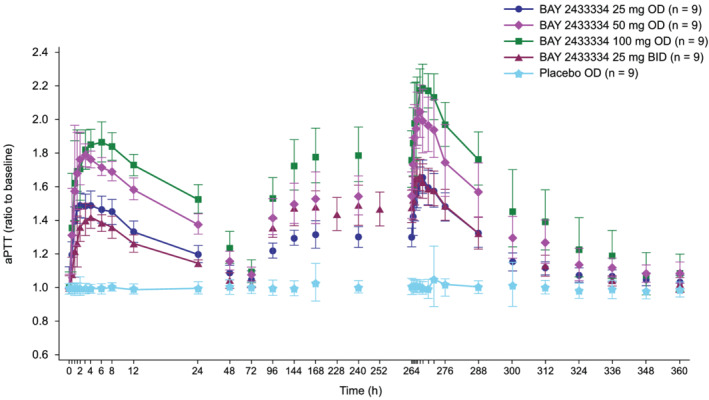
Geometric mean (standard deviation) effect of BAY 2433334 on ratio to baseline of aPTT following once‐daily treatment with BAY 2433334 25–100 mg in Part A, and twice‐daily treatment with BAY 2433334 25 mg in Part B. aPTT, activated partial thromboplastin time; BID, twice daily; OD, once daily
BAY 2433334 produced rapid and dose‐dependent inhibition of FXIa activity, whereas placebo had no effect (Figure 4). Maximum inhibition of FXIa activity occurred 1.5–4.0 h after a single administration of BAY 2433334 (residual FXIa activity levels of 10.6 and 6.7% for 25 mg once and twice daily, respectively). Higher doses (50 and 100 mg) showed complete inhibition of FXIa activity, with 6 and 9 volunteers assigned to these doses, respectively, showing at least 1 measurement below the lower limit of quantification. Degree of FXIa inhibition for all doses were different from placebo and indicated dose‐dependent effects 24 h after the first dose on day 1. On day 12 at steady state, minimum residual FXIa activity of 5.52% was observed around 4 h after administration of 25 mg once daily, whilst complete inhibition was sustained for approximately 11 and 24 h with 50 and 100 mg of BAY 2433334, respectively. Normalization of FXIa activity occurred in a linear manner after the last dose.
FIGURE 4.
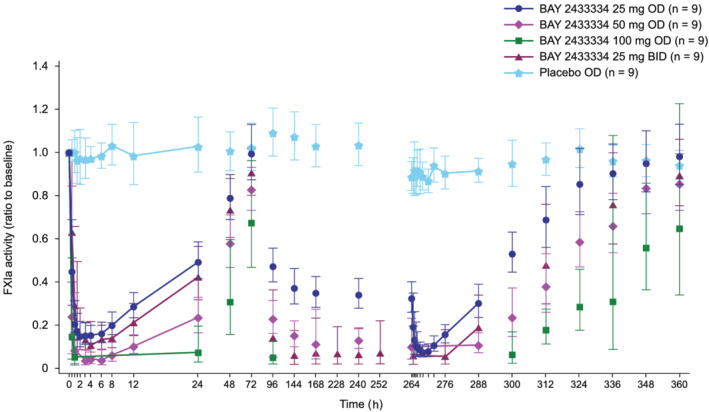
Geometric mean (standard deviation) effect of BAY 2433334 on ratio to baseline of FXIa activity. BAY 2433334 25–100 mg was given once daily in Part A. In Part B, BAY 2433334 25 mg was given twice daily (once daily on days 1 and 12). BID, twice daily; FXIa, activated factor XI; OD, once daily
Comparison of the maximal relative inhibition of FXIa activity at steady state (geometric mean) between 50 mg once‐daily and 25 mg twice‐daily doses 24 h after administration on day 12 did not reveal a significant difference (P = .0576).
There were close correlations between plasma concentrations of BAY 2433334 and both aPTT prolongation and inhibition of FXIa activity (Figure 5). The maximal relative changes in aPTT and FXIa activity following treatment with BAY 2433334 in Part A are summarized in Tables 8 and 9, respectively.
FIGURE 5.
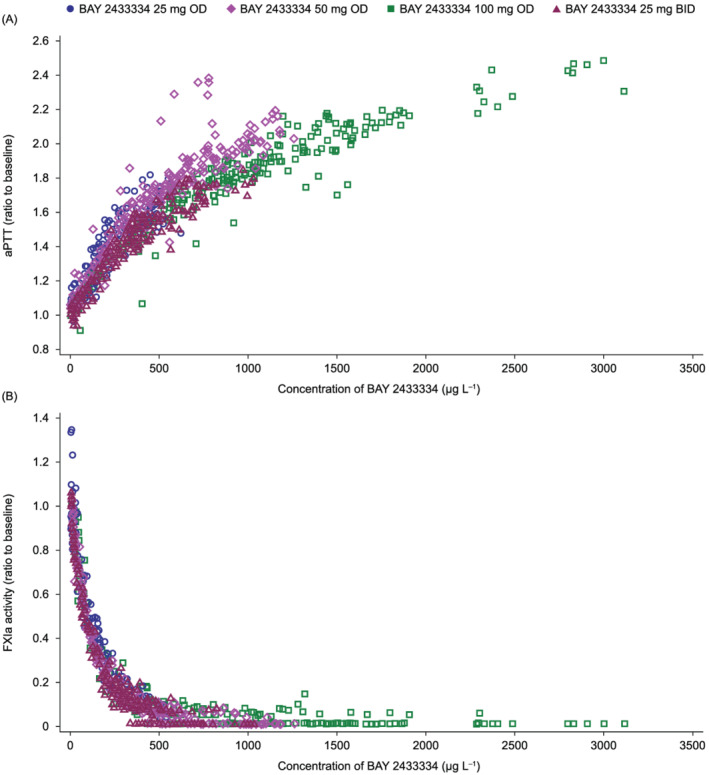
Correlations between plasma concentrations of BAY 2433334 and (A) prolongation of aPTT (ratio to baseline) and (B) inhibition of FXIa activity in plasma (ratio to baseline). Data are derived from the Pharmacokinetic analysis set and Pharmacodynamic analysis set (n = 48). aPTT, activated partial thromboplastin time; BID, twice daily; FXIa, activated factor XI; OD, once daily
TABLE 8.
Summary of changes in activated partial thromboplastin time following treatment with BAY 2433334 in Part A
| Characteristic | Day | Treatment | Geometric mean | Geometric coefficient of variation (%) | P vs. placebo a |
|---|---|---|---|---|---|
| Maximal relative increase | 1 | BAY 2433334 100 mg (n = 9) | 1.92 | 5.37 | .0002 |
| BAY 2433334 50 mg (n = 9) | 1.84 | 5.25 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.53 | 4.96 | .0002 | ||
| Placebo (n = 9) | 1.04 | 3.98 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | 2.22 | 5.96 | .0003 | |
| BAY 2433334 50 mg (n = 9) | 2.08 | 7.24 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.69 | 5.36 | .0005 | ||
| Placebo (n = 9) | 1.08 | 15.82 | n.a. | ||
| Ratio to baseline at 12 h | 1 | BAY 2433334 100 mg (n = 9) | 1.73 | 3.64 | .0002 |
| BAY 2433334 50 mg (n = 9) | 1.59 | 4.06 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.33 | 4.80 | .0002 | ||
| Placebo (n = 9) | 0.99 | 2.93 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | 1.97 | 6.61 | .0003 | |
| BAY 2433334 50 mg (n = 9) | 1.75 | 12.24 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.48 | 5.73 | .0002 | ||
| Placebo (n = 9) | 1.02 | 3.38 | n.a. | ||
| Ratio to baseline at 24 h | 1 | BAY 2433334 100 mg (n = 9) | 1.53 | 5.60 | .0002 |
| BAY 2433334 50 mg (n = 9) | 1.38 | 4.36 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.20 | 4.26 | .0002 | ||
| Placebo (n = 9) | 1.00 | 3.69 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | 1.76 | 9.03 | .0003 | |
| BAY 2433334 50 mg (n = 9) | 1.57 | 10.28 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 1.33 | 6.88 | .0002 | ||
| Placebo (n = 9) | 1.00 | 3.78 | n.a. |
n.a., not applicable.
Wilcoxon test.
TABLE 9.
Summary of changes in FXIa activity following treatment with BAY 2433334 in Part A
| Characteristic | Day | Treatment | Geometric mean | Geometric coefficient of variation (%) | P vs. placebo a |
|---|---|---|---|---|---|
| Maximal relative increase | 1 | BAY 2433334 100 mg (n = 9) | n.c. | n.c. | n.c. |
| BAY 2433334 50 mg (n = 9) | n.c. | n.c. | n.c. | ||
| BAY 2433334 25 mg (n = 9) | 0.133 | 35.24 | .0002 | ||
| Placebo (n = 9) | 0.896 | 9.38 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | n.c. | n.c. | n.c. | |
| BAY 2433334 50 mg (n = 9) | n.c. | n.c. | n.c. | ||
| BAY 2433334 25 mg (n = 9) | 0.065 | 45.42 | .0002 | ||
| Placebo (n = 9) | 0.825 | 9.24 | n.a. | ||
| Ratio to baseline at 12 h | 1 | BAY 2433334 100 mg (n = 9) | n.c. | n.c. | n.c. |
| BAY 2433334 50 mg (n = 9) | 0.105 | 41.88 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 0.287 | 20.58 | .0002 | ||
| Placebo (n = 9) | 0.986 | 14.69 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | n.c. | n.c. | n.c. | |
| BAY 2433334 50 mg (n = 9) | n.c. | n.c. | n.c. | ||
| BAY 2433334 25 mg (n = 9) | 0.157 | 26.04 | .0002 | ||
| Placebo (n = 9) | 0.906 | 8.47 | n.a. | ||
| Ratio to baseline at 24 h | 1 | BAY 2433334 100 mg (n = 9) | 0.076 | 121.32 | .0002 |
| BAY 2433334 50 mg (n = 9) | 0.234 | 35.35 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 0.495 | 17.21 | .0002 | ||
| Placebo (n = 9) | 1.030 | 12.44 | n.a. | ||
| 12 | BAY 2433334 100 mg (n = 8) | n.c. | n.c. | n.c. | |
| BAY 2433334 50 mg (n = 9) | 0.110 | 42.53 | .0002 | ||
| BAY 2433334 25 mg (n = 9) | 0.304 | 25.76 | .0002 | ||
| Placebo (n = 9) | 0.917 | 6.04 | n.a. |
FXIa, activated factor XI; n.a., not applicable; n.c., not calculated (statistics were only calculated if at least 2/3 of the individual data were measured, and were above the lower limit of quantification).
Wilcoxon test.
BAY 2433334 had no effect, compared with placebo, on PT (both absolute and INR values), FXI concentrations or on complement C1 esterase inhibitor activity; no formal statistical analysis of these variables was prespecified. Similarly, there were no correlations between plasma BAY 2433334 concentrations and either PT, FXI concentrations or C1 esterase inhibitor activity.
Plasma FXII concentrations and activity, vWF antigen levels and vWF ristocetin cofactor activity were generally within reference ranges at baseline.
4. DISCUSSION
This multiple‐dose escalation study evaluated the safety, pharmacokinetics and pharmacodynamics of BAY 2433334, an oral, direct inhibitor of FXIa, in healthy volunteers.
BAY 2433334 was well tolerated in this study. All TEAEs were mild in severity and, apart from headache and nasopharyngitis, occurred in no more than 1 or 2 participants. There were no clinically relevant adverse events related to bleeding, changes in vital signs, ECG or clinical laboratory evaluations.
Systemic exposure to BAY 2433334, as measured by AUC and Cmax after the first dose and AUCτ and Cmax at steady state, appeared to increase in a dose‐proportional manner between doses of 25 and 100 mg once daily, with low‐to‐moderate variability in pharmacokinetic parameters. Mean t1/2 values for BAY 2433334 after multiple doses (range, 15.8–17.8 h) were supportive of a once‐daily dosing schedule. As would be predicted based on its t1/2, minor‐to‐moderate accumulation of BAY 2433334 (as assessed by RAAUC and RACmax) was observed after repeated once‐ and twice‐daily dosing at steady state. Furthermore, the data suggested that the pharmacokinetics of BAY 2433334 were time independent. The low apparent oral clearance observed suggests that BAY 2433334 is a low‐clearance drug. Renal elimination of unchanged BAY 2433334 was only a minor route of elimination. The mean renal clearance ranging from 0.267 to 0.440 L h−1 after single‐dose administration is consistent with the fraction unbound (6.4%, unpublished data on file) in plasma and the glomerular filtration rate (0.064*120 mL/min = 0.4608 L h−1), suggesting that the main mechanism for BAY 2433334 renal elimination may be glomerular filtration, which might explain the observed low‐to‐high interindividual variability (geometric CV between 24.5 and 67.4%). The impact of renal function on the pharmacokinetics of BAY 2433334 will be explored within the ongoing clinical development programme (NCT04510987).
Based on preclinical in vitro data from human hepatocytes and clinically relevant therapeutic plasma concentrations, BAY 2433334 was predicted to be a weak‐to‐moderate inducer toward sensitive CYP3A4 substrates (no‐observed‐effect level of 370 μg L−1). However, BAY 2433334 25 mg or 75 mg administered together with midazolam showed no indication of any clinically relevant inhibition and/or induction of CYP3A4 in healthy volunteers. Further, there was no evidence of any inductive potential of BAY 2433334 25 or 75 mg once daily on CYP3A4, as no decrease in midazolam AUC in parallel with an increase of α‐hydroxymidazolam AUC (as expected for CYP3A4 inducers) was observed. Clinically relevant pharmacokinetic drug–drug interaction due to CYP3A4 induction or inhibition by BAY 2433334 can therefore be regarded as unlikely.
In the pharmacodynamic evaluation, BAY 2433334 treatment resulted in dose‐dependent aPTT prolongation compared with placebo, with maximum prolongation observed approximately 4 h after administration, and still detectable 12 and 24 h after single and multiple dosing, respectively, at all doses. Correlation analyses showed a close curvilinear correlation of aPTT prolongation with increasing BAY 2433334 plasma concentrations supporting a predictable PK/PD effect. BAY 2433334 had no effect on PT, indicating its specificity for the intrinsic coagulation pathway. 18
Maximal inhibition of FXIa activity was observed approximately 2–4 h after administration of a single 25 mg dose of BAY 2433334, while higher doses resulted in complete inhibition of FXIa activity. Inhibition of FXIa activity persisted for all doses at 12 and 24 h after first and steady‐state dosing, and normalization of FXIa activity occurred in a linear manner after the last dose. Complete inhibition of FXIa was observed with 50 and 100 mg once daily and 25 mg twice daily at steady state, the duration of which was dose‐dependent, consistent with previous animal and ex vivo studies. 13 Given that this FXIa assay is newly developed, further studies are required to allow an assessment of the clinical relevance of this readout.
BAY 2433334 had no significant effect on FXI concentrations or C1 esterase inhibitor activity. Plasma FXII concentrations, FXII activity, vWF antigen levels or vWF ristocetin cofactor activity measurements prior to BAY 2433334 administration showed no abnormal findings that might have influenced the pharmacodynamic evaluations.
FXIa inhibitors offer the potential for blockade of thrombus formation, mediated by the intrinsic coagulation pathway, without affecting the tissue factor‐driven haemostatic response to injury; as a result, bleeding complications would be expected to be lower with FXIa inhibitors than with conventional anticoagulants. 18 , 19 However, more data, specifically in patients, are required to evaluate this hypothesis further. The present study confirmed the safety and predictable pharmacokinetic and pharmacodynamic properties of BAY 2433334 in healthy male volunteers. A large phase 2 programme for BAY 2433334 is underway in patients with atrial fibrillation (NCT04218266), after an acute noncardioembolic stroke (NCT04304508), and after an acute myocardial infarction (NCT04304534). In addition, several potential FXI(a) inhibitors are currently under development, including small molecules, polypeptides, and polyclonal or monoclonal antibodies. 19
This study does have some limitations. Some of these, notably the small numbers of participants, are inherent in phase 1 clinical trials. Similarly, the lack of safety data from reproductive studies restricted recruitment to healthy men. As a result, further studies are needed to confirm the generalizability of the findings to other populations. In addition, the clinical relevance of the FXIa activity changes observed using the newly developed fluorogenic substrate assay needs to be established. While the observed changes in pharmacodynamic parameters are consistent with the mechanism of action of BAY 2433334, further studies will be required to correlate these changes with clinical outcomes, such as the risk of thrombotic events or bleeding. The clinical trial experience to date with an antisense oligonucleotide 11 and an anti‐FXIa antibody 12 suggests that an association between changes in aPTT and FXI clotting activity and the risk of thrombotic events may be anticipated.
In conclusion, this study in healthy volunteers has shown that BAY 2433334, an oral FXIa inhibitor, was well tolerated over a wide dose range, with a predictable pharmacokinetic and pharmacodynamic profile that supports once‐daily dosing. These findings support further evaluation of BAY 2433334 in phase 2 clinical trials.
COMPETING INTERESTS
S.S. is an employee of Bayer AG and holds stocks in Bayer AG; D.K., F.K. and M.H. are employees of Bayer AG; J.D. is an employee of ClinStat GmbH on behalf of Bayer AG; A.K. is an employee of CRS Clinical Research Services Wuppertal GmbH.
CONTRIBUTORS
Study concept and design: All authors. Acquisition of data: All authors. Analysis or interpretation of data: All authors. Drafting of the manuscript: All authors. Critical revision of the manuscript for important intellectual content: All authors.
Supporting information
TABLE S1 Pharmacokinetic parameters calculated for BAY 2433334.
TABLE S2 Study flow chart: screening visit.
FIGURE S1 Blood and urine sample collection flow schematic
ACKNOWLEDGEMENTS
Editorial support was provided by Gemma Rogers PhD of Oxford PharmaGenesis, Oxford, UK, with funding from Bayer AG. Bioanalysis of BAY 2433334 was performed by Lukas Fiebig PhD of Bayer AG, Wuppertal. Bioanalysis of midazolam and α‐hydroxymidazolam was performed by Guenter Michl, PhD and Richard Abbott, PhD, Bayer AG and Johanne Lefebvre, D.E.C. of Syneos Health Clinique. The expert technical assistance of Anja Schäfer BSc and Angelika Roth BSc of Bayer AG, Assay Technologies Wuppertal is gratefully acknowledged for the analysis of FXIa activity and FXI concentration was performed at Bayer AG, Wuppertal. Analysis of FXII activity and concentration, vWF level and vWF ristocetin cofactor activity was performed at MLM Medical Labs, Mönchengladbach.
This study was funded by Bayer AG, Berlin, Germany.
Kubitza D, Heckmann M, Distler J, Koechel A, Schwers S, Kanefendt F. Pharmacokinetics, pharmacodynamics and safety of BAY 2433334, a novel activated factor XI inhibitor, in healthy volunteers: A randomized phase 1 multiple‐dose study. Br J Clin Pharmacol. 2022;88(7):3447-3462. doi: 10.1111/bcp.15230
The authors confirm that the Principal Investigator for this paper is Annemone Koechel and that she had direct clinical responsibility for healthy volunteers.
Funding information Bayer AG
DATA AVAILABILITY STATEMENT
The data are not available.
REFERENCES
- 1. Adelborg K, Sundbøll J, Sørensen HT. Arterial cardiovascular events and mortality following venous thromboembolism. Ann Transl Med. 2015;3(9):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piazza G, Goldhaber SZ. Venous thromboembolism and atherothrombosis: an integrated approach. Circulation. 2010;121(19):2146‐2150. [DOI] [PubMed] [Google Scholar]
- 3. Marano G, Vaglio S, Pupella S, Liumbruno GM, Franchini M. How we treat bleeding associated with direct oral anticoagulants. Blood Transfusion = Trasfusione del Sangue. 2016;14:465‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa‐mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116(19):3981‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115(13):2569‐2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gailani D. Future prospects for contact factors as therapeutic targets. Hematol Am Soc Hematol Educ Program. 2014;2014(1):52‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Georgi B, Mielke J, Chaffin M, et al. Leveraging Human Genetics to Estimate Clinical Risk Reductions Achievable by Inhibiting Factor XI. Stroke. 2019;50(11):3004‐3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gailani D, Smith SB. Structural and functional features of factor XI. J Thromb Haemost: JTH. 2009;7(Suppl 1):75‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride‐induced carotid artery occlusion in mice. J Thromb Haemost: JTH. 2005;3(4):695‐702. [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Smith PL, Hsu MY, et al. Effects of factor XI deficiency on ferric chloride‐induced vena cava thrombosis in mice. J Thromb Haemost: JTH. 2006;4(9):1982‐1988. [DOI] [PubMed] [Google Scholar]
- 11. Büller HR, Bethune C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weitz JI, Bauersachs R, Becker B, et al. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. Jama. 2020;323(2):130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heitmeier S, Visser M, Gäfke A, et al. Preclinical Pharmacology of BAY 2433334, a Small Molecule Inhibitor of Coagulation Factor XIa [abstract]. Res Pract Thromb Haemost. 2020;4(Suppl 1):83. [Google Scholar]
- 14. Alexander SP, Kelly E, Marrion NV, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Overview. Br J Pharmacol. 2017;174(Suppl 1):S1‐s16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guideline on the investigation of drug interactions. In: European Medicines Agency, 2012.
- 16. U.S. Food and Drug Administration . Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. In, 2020.
- 17. Kapetas AJ, Sorich MJ, Rodrigues AD, Rowland A. Guidance for Rifampin and Midazolam Dosing Protocols To Study Intestinal and Hepatic Cytochrome P450 (CYP) 3A4 Induction and De‐induction. AAPS J. 2019;21(5):78. [DOI] [PubMed] [Google Scholar]
- 18. Thomas D, Thelen K, Kraff S, et al. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: First evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thromb Haemost. 2019;3(2):242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al‐Horani RA, Afosah DK. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med Res Rev. 2018;38(6):1974‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Pharmacokinetic parameters calculated for BAY 2433334.
TABLE S2 Study flow chart: screening visit.
FIGURE S1 Blood and urine sample collection flow schematic
Data Availability Statement
The data are not available.