

SUMMARY
Mutations in the catalytic subunit of protein kinase A (PKAc) drive the stress hormone disorder adrenal Cushing’s syndrome. We define mechanisms of action for the PKAc-L205R and W196R variants. Proximity proteomic techniques demonstrate that both Cushing’s mutants are excluded from A kinase-anchoring protein (AKAP)-signaling islands, whereas live-cell photoactivation microscopy reveals that these kinase mutants indiscriminately diffuse throughout the cell. Only cAMP analog drugs that displace native PKAc from AKAPs enhance cortisol release. Rescue experiments that incorporate PKAc mutants into AKAP complexes abolish cortisol overproduction, indicating that kinase anchoring restores normal endocrine function. Analyses of adrenal-specific PKAc-W196R knockin mice and Cushing’s syndrome patient tissue reveal defective signaling mechanisms of the disease. Surprisingly each Cushing’s mutant engages a different mitogenic-signaling pathway, with upregulation of YAP/TAZ by PKAc-L205R and ERK kinase activation by PKAc-W196R. Thus, aberrant spatiotemporal regulation of each Cushing’s variant promotes the transmission of distinct downstream pathogenic signals.
Graphical Abstract
In brief
Mutations in the catalytic subunit of protein kinase A (PKAc) cause a stress hormone disorder called adrenal Cushing’s syndrome. Omar et al. uncover mislocalization as a required component of mutant-kinase pathology. Remarkably, two seemingly similar disease-causing PKAc variants diverge in subcellular localization, physiochemical properties, and downstream signaling pathways.
INTRODUCTION
Cushing’s syndrome is an endocrine disorder caused by chronically elevated levels of the stress hormone cortisol. Patients classically present with weight gain, chronic fatigue, hypertension, and depression (Lacroix et al., 2015). Adrenal Cushing’s syndrome is often a consequence of defective cAMP signaling in the adrenal cortex (Hernandez-Ramirez and Stratakis, 2018; Rosenberg et al., 2002). Whole-exome sequencing of adrenal tissue from patients has identified mutations and insertions in protein kinase A catalytic subunits (PKAc) that are drivers of Cushing’s syndrome (Beuschlein et al., 2014; Cao et al., 2014; Di Dalmazi et al., 2014; Espiard et al., 2018; Goh et al., 2014; Ronchi et al., 2016; Sato et al., 2014). Most PKAc mutations cluster where the kinase domain interfaces with its regulatory subunits (R) (Bathon et al., 2019). This protein-protein interaction is not only necessary for autoinhibition of kinase activity, but it also directs the subcellular targeting of PKA holoenzymes through association with A kinase-anchoring proteins (AKAPs) (Scott and Pawson, 2009; Tasken and Aandahl, 2004; Wong and Scott, 2004).
A traditional view of cAMP signaling proposes that this diffusible second messenger unilaterally activates PKA throughout the cell. Yet, contrary to this dogma, elegant fluorescent spectroscopy studies show that under physiological parameters cAMP accumulation occurs within nanometer-sized domains (Bock et al., 2021; Musheshe et al., 2018). The existence of cAMP nanodomains argues that the local actions of PKA, EPAC guanine-nucleotide-exchange factors, and Popeye-domain-containing proteins are more intricately organized than was originally considered (Amunjela et al., 2019; Bauman et al., 2006; Viña et al., 2021; Yarwood, 2020). This cAMP nanodomain hypothesis is consistent with our own molecular evidence that anchored, intact, and active PKA holoenzymes operate within a 200- to 400-Å radius of action (Langeberg and Scott, 2015; Smith et al., 2013, 2017). Anchoring of PKA within cAMP nanodomains creates discrete and autonomous AKAP “signaling islands” where this kinase is sequestered near its preferred substrates (Figure 1A) (Bock et al., 2021; Smith et al., 2017). This concept also explains how the ubiquitous second-messenger cAMP can simultaneously propagate distinct signaling events within the same cell (Omar and Scott, 2020).
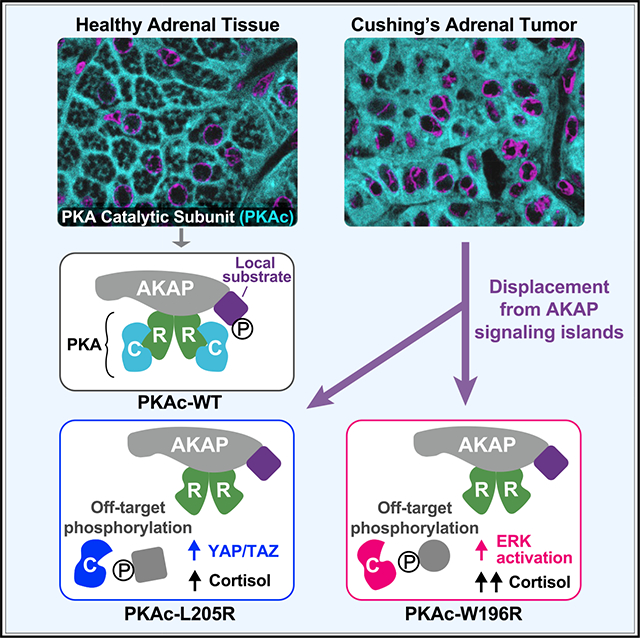
Figure 1. Cushing’s mutants are excluded from AKAP signaling islands.
(A) Model of AKAP signaling island. Regulatory subunits, R; catalytic subunits, C.
(B) Structure of PKAc bound to the inhibitory region of RIIβ (green). The W196R (C) and L205R (D) mutations fall within the red region.
(E) Immunoprecipitation of AKAP79 from H295R adrenal cells.
(F) Immunoprecipitation of PKAc from H295R cells.
(G) AKAP79 (297–427)-PKA complex formation in vitro ± cAMP.
(H) Changes in PKA-dependent phosphorylation of a fluorescently labeled peptide substrate upon increasing concentrations of RIIα. Mean ± SD; n = 4.
(I) Immunoblots of CRISPR PKAcα−/− U2OS cells expressing RII-GFP with either WT or mutant PKAc-V5. Mean ± 95% CI.
(J) Cortisol measurements from H295R cells infected with PKAc variants. Mean ± SE. *p ≤ 0.05, Student’s t tests after one-way ANOVA; n = 6.
(K) Photoactivation time courses in H295R cells. AKAP79-YFP, RII-iRFP, and PKAc tagged with photoactivatable mCherry were expressed. Scale bar, 10 μm.
(L) Quantitation of (K). Three experimental replicates.
(M) Integration of (L). Mean ± SE. ****p ≤ 0.0001, one-way ANOVA with Dunnett’s correction. See also Figure S1.
PKA is a broad-spectrum kinase that phosphorylates hundreds of cellular substrates (Shabb, 2001; Taylor et al., 2012; Turnham and Scott, 2016). Accordingly, lesions in PRKAC and PRKAR genes are linked to a growing number of diseases (Ma et al., 2019; Palencia-Campos et al., 2020; Stratakis, 2018; Turnham et al., 2019). Herein, we define the mechanism of action for PKAc variants that occur in patients suffering from adrenal (adrenocorticotropic hormone-independent) Cushing’s syndrome (Figures 1B–1D). The most prevalent mutant identified is L205R, which is found in ~45% of Cushing’s patients (Bathon et al., 2019). Seven other PKAc mutations including the W196R variant have been identified in adenomas from Cushing’s patients (Bathon et al., 2019; Espiard et al., 2018). We report that the Cushing’s variants PKAc-L205R and PKAc-W196R are physiochemically distinct protein kinases that are chronically mislocalized inside adrenal cells and surprisingly engage distinct downstream signaling pathways.
RESULTS
Cushing’s mutants are excluded from AKAP signaling islands
Initially, we assessed whether Cushing’s syndrome mutations in PKAc disrupt association with AKAP signaling islands in NCI-H295R human adrenocortical carcinoma cells. Immunoprecipitation of AKAP79 pulled down type II regulatory subunits (RII) and wild-type (WT) PKAc (Figure 1E, lane 1). In contrast, PKAc-L205R and PKAc-W196R were not detected in AKAP79 immune complexes (Figure 1E, lanes 2 and 3). Next, we tested the association between R subunits and each PKAc form. Immunoprecipitation of WT PKAc pulled down type I and type II regulatory subunits (Figure 1F, lane 1). Neither L205R nor W196R co-precipitated the R subunits (Figure 1F, lanes 2 and 3). Recombinant proteins were utilized to further evaluate effects of L205R or W196R on interactions with AKAPs. Upon pull-down of an AKAP79 fragment (aa 297–427), RII was detected in all conditions (Figure 1G, top). WT PKAc and PKAc-R133A, a variant known to be deficient in PKI binding, retained the ability to bind RII (Figure 1G, lanes 1 and 3). In contrast, RII failed to pull down the PKAc-L205R or W196R variants (Figure 1G, lanes 5–8). Loading controls verify that equivalent levels of AKAP79, RII, and C subunits were present in each experimental condition (Figure 1G, middle and bottom). Other controls confirmed that incubation with supraphysiological levels of cAMP disrupted all PKAc interactions (Figure 1G, lanes 2 and 4).
Regulatory subunits autoinhibit PKAc activity. Accordingly, titration of RII causes a precipitous reduction in WT PKA activity between RII concentrations of 0.83 and 8.3 nM (Figure 1H, black). In contrast, neither PKAc-L205R (blue) nor PKAc-W196R (red) is autoinhibited by RII (Figures 1H and S1A). A more rigorous test examined the phosphorylation of the canonical PKA substrates CREB and ATF1 in a genetic background depleted of native kinase. CRISPR-Cas9 gene editing was used to remove PKAcα from U2OS cells (Smith et al., 2017). Rescue experiments reintroduced PKAc-L205R or PKAc-W196R. Immunoblot detection of phospho-CREB and phospho-ATF1 served as an index of kinase activity (Figure 1I). Under basal conditions the WT kinase minimally phosphorylated either substrate (Figure 1I, lane 1). In contrast, PKAc-L205R enhanced phosphorylation 1.9 ± 0.2-fold and PKAc-W196R enhanced phosphorylation 2.1 ± 0.2-fold compared with WT. (Figure 1I, lanes 2 and 3, and Figure S1B). Detection of PKA substrates, with an antibody against phosphorylated target sequence RRXS/T, was also increased by Cushing’s variants (Figure 1I, fifth panel).
Next, we measured human stress hormone (cortisol) release from H295R cells. Initially we established baseline 1-h cortisol release from H295R adrenal cells expressing WT PKAc (Figure 1J, gray). Cortisol release was elevated upon expression of PKAc-L205R (Figure 1J, blue). Interestingly, expression of PKAc-W196R had the most robust effect, showing over 3-fold production of stress hormone compared with WT kinase (Figure 1J, red). Collectively, these structural, biochemical, and cell-based data suggest that both L205R and W196R mutations disrupt inhibition of PKAc by its regulatory subunits and displace the kinase from AKAP signaling islands.
To monitor how Cushing’s mutations impact localization and mobility, we used live-cell photoactivation microscopy. Upon activation of a 2 μM circle on the membrane of H295R cells, WT PKAc tagged with photoactivatable mCherry maintained its localization relative to fluorescently tagged AKAP79 (Figure 1K, top row, and Figure 1L, gray trace; Figure S1C and Video S1). This is characteristic of PKAc retention within AKAP signaling islands. In contrast, PKAc-L205R and W196R variants rapidly diffused away from membrane-associated AKAP79, filling the cytosol (Figure 1K, middle and bottom rows; Figure S1C and Videos S2 and S3). Quantification of 15 cells from three experiments detected significant differences in the mobility of PKAc variants, including greater movement of PKAc-L205R compared with W196R (Figures 1L and 1M). Thus, Cushing’s syndrome PKAc variants are not compartmentalized by AKAPs.
Proximity proteomics identify distinct associations among PKAc variants
To gain further insight into the molecular action of PKAc-L205 and W196R, we conducted proximity proteomic screens using the biotin ligase miniTurbo (Figure 2A) (Branon et al., 2018; Smith et al., 2018). We generated stable H295R cell lines expressing low levels (~20% of endogenous) of WT or mutant PKAc variants tagged with miniTurbo (Figure 2, bottom, and Figures S2A–S2C). Live biotinylation for 1 h labeled proteins proximal to catalytic subunits. Distinct patterns of labeled proteins associated with each PKAc-miniTurbo variant were detected by neutravidin-HRP western blot (Figure 2B, top, and Figures S2A and S2B). This strategy generated a “proximitome” rather than an “interactome” for each PKAc variant.
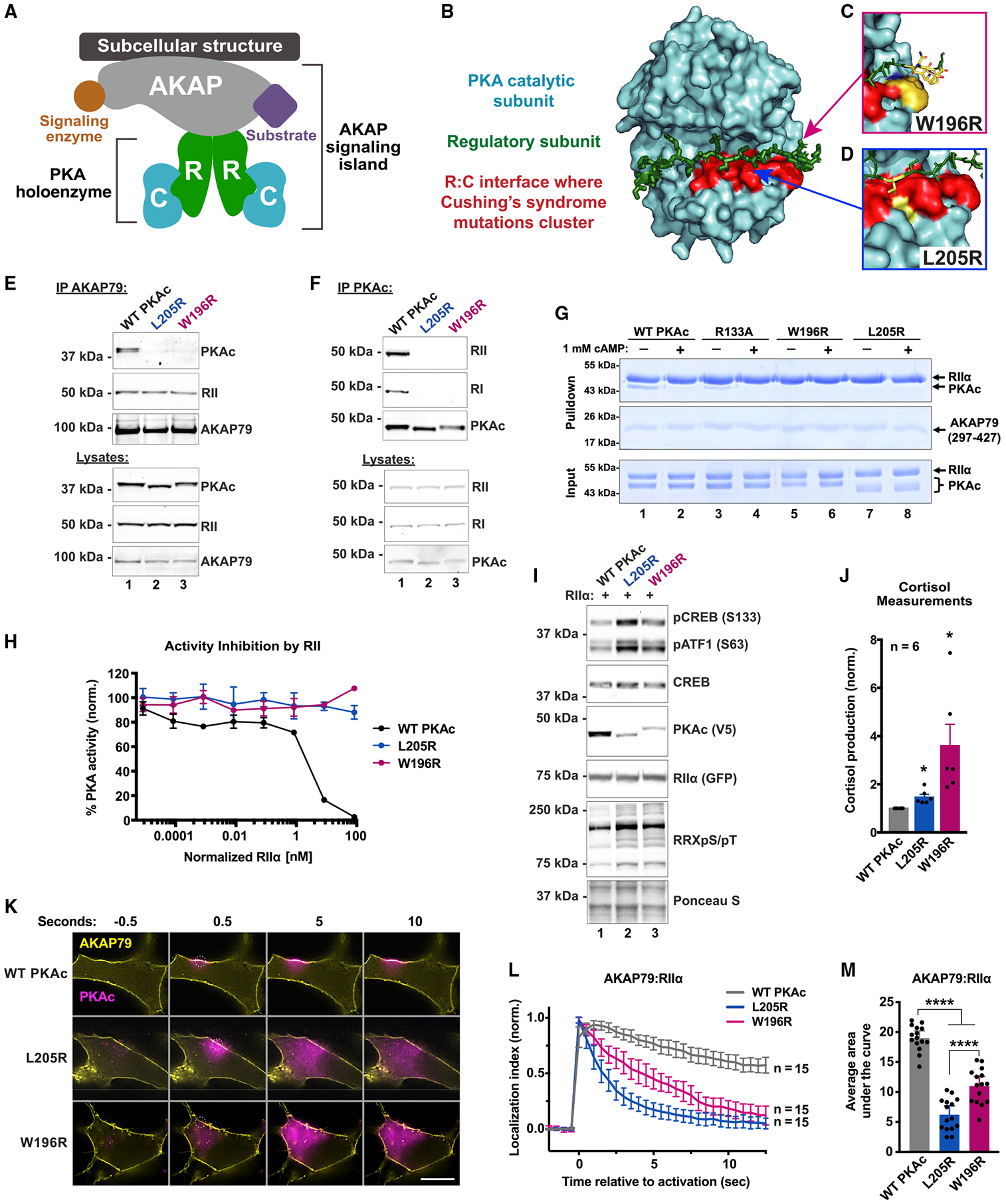
Figure 2. Proximity proteomics identify distinct associations among PKAc variants.
(A) Model of PKAc tagged with miniTurbo biotin ligase at an AKAP signaling island. Upon application of biotin, proteins in proximity (5–10 nm) to PKAc are biotinylated.
(B) Immunoblots of lysates from stable H295R lines after proximity biotinylation. Neutravidin-HRP signal (top) shows banding differences among conditions. PKAc signal (bottom) shows expression of tagged kinase at low levels versus endogenous PKAc.
(C and D) Volcano plots of proximity proteomics. Black, proteins underrepresented versus WT. Blue (L205R) and Red (W196R), proteins enriched in the mutant conditions. Gray, proteins with a corrected p value lower than 0.05. Four biological replicates.
(E–G) STRING network depictions of selected enriched proteins in L205R (E) and W196R (G). Between is a list (F) of proteins identified as enriched in both mutant conditions.
(H) Quantitation of association with PKA holoenzyme components.
(I) Association with AKAPs as determined by peptides identified versus WT condition.
(J) Gene ontology enrichment scores for cell component relative to WT.
(K and L) Graphical depictions of (J). See also Figure S2.
Proteomic analysis identified both shared and distinct pools of proximal proteins among PKAc variants (Figures 2C and 2D). Quantitative bioinformatic analyses of each proximitome revealed reduced association with AKAPs and R subunits (Figures 2C and 2D, black dots). Proteins enriched in the mutant conditions (Figure 2C, blue dots, and Figure 2D, red dots) were analyzed using STRING to identify network enrichments and gene ontology (GO) cell component annotations (Szklarczyk et al., 2019). Scrutiny of the PKAc-L205R dataset identified subnetworks for nuclear pore export, protein translation, and spliceosome functions (Figure 2E). The PKAc-W196R proximitome also included proteins involved in nuclear pore function and translation, as well histone modification and melanosome protein subnetworks (Figure 2G). Nineteen significantly enriched proteins were present in both PKAc-L205R and W196R proximitomes (Figure 2F). Equivalent self-labeling among PKAc variants served as a positive control, whereas deficient labeling of R subunits by each mutant kinase was a negative control (Figure 2H). Loss of association with AKAPs provides further evidence for displacement from AKAP signaling islands in both PKAc-L205R (blue) and PKAc-W196R (red) datasets (Figure 2I).
By comparing GO enrichment scores for cell component association, we were able to quantify differences in the subcellular distribution of PKAc variants. Whereas both Cushing’s variants exhibit increased proximity to mitochondria, PKAc-L205R has greater access to nuclear components (Figures 2J and 2K). Cytoskeletal and membrane association are reduced for both PKAc-L205R and W196R (Figures 2J and 2K). Hence, decreased association with AKAPs favors redistribution of Cushing’s PKAc variants to different subcellular compartments.
Cushing’s mutants exhibit distinct physiochemical profiles
Our data imply that PKAc-L205R and PKAc-W196R have distinct mechanisms of action. In vitro kinase activity measurements (using Kemptide as a substrate) confirm that PKAc-L205R is less catalytically efficient than WT, requiring 20-fold more enzyme to achieve 50% substrate phosphorylation (Figure 3A, blue, and Figure S3A). In contrast, activity curves for PKAc-W196R were identical to the WT kinase (Figure 3A, red and black). ATP incorporation was similar among the tested PKAc forms (Figure S3B). Quantitative mass spectrometry analysis revealed stark differences in the autophosphorylation status of each PKA variant (Figure 3B). Up to 11 sites were fully occupied on WT PKAc, with W196R showing decreased phosphate incorporation at most sites (Figure 3B). Surprisingly, PKAc-L205R was effectively unphosphorylated (Figure 3B). This provides molecular context to explain the faster migration of PKAc-L205R in gel electrophoresis (Figure S3A). We then used thermal stability to measure the structural integrity of the variants. Both Cushing’s mutants were compromised, with unfolding occurring at 2°–3°C lower than WT kinase (Figure 3C). Such physiochemical differences between PKAc-L205R and W196R offer possible explanations for the differential behavior of these Cushing’s variants.
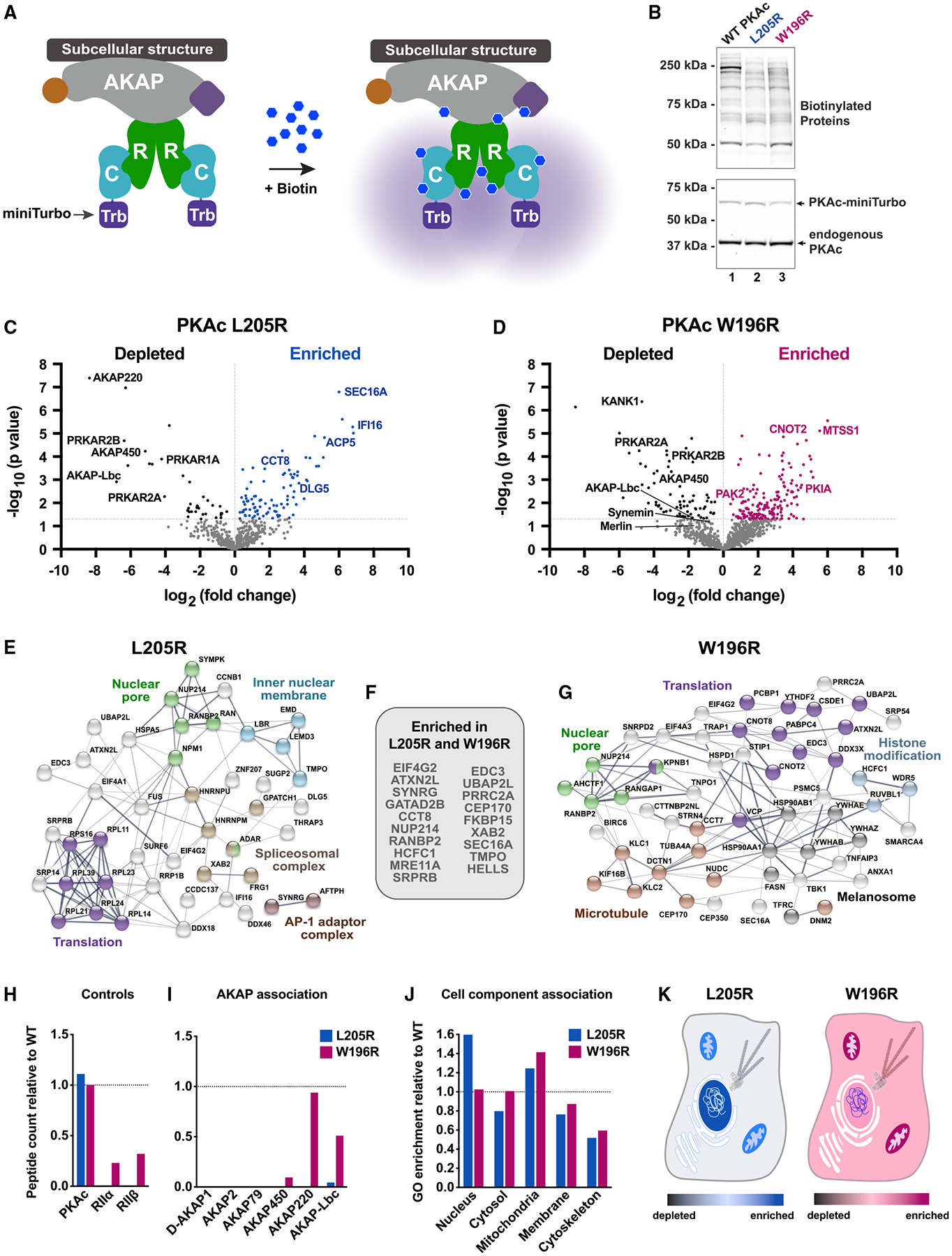
Figure 3. Cushing’s mutants exhibit distinct physiochemical profiles.
(A) Catalytic activity of recombinant WT and mutant PKAc toward a peptide substrate. Data are %substrate phosphorylation (mean ± SD) after 5 min; n = 4. Relative rates were determined per ng of enzyme as pmol phosphate incorporation per min. Calculated from assays containing 0.8 ng WT and W196R PKAc and 19.5 ng L205R at linear rates of phosphorylation.
(B) Structural depictions and quantification of relative abundance of phosphorylated sites compared with WT PKAc as measured by mass spectrometry. Red ≥66.7% WT levels. Gold, 1–66.6% WT levels; n = 3.
(C) Normalized thermal melt curves for WT and mutant PKAc.
(D) Orthogonal depictions of PKAc immunofluorescence in H295R cells. Side, zy planes; bottom, zx planes; scale bar, 10 μm.
(E) Line plots of PKAc signals in (D) as indicated by yellow dotted lines.
(F) Quantitation of line plots using DAPI signal to define nucleus; n ≥ 11; mean ± SE.****p ≤ 0.0001, one-way ANOVA with Dunnett’s correction.
(G) H295R cells expressing V5-tagged PKAc variants were stained for V5 and pATF1. Scale bar, 20 μm.
(H) Immunoblot of H295R lysates infected with PKAc variants as listed. Represents three replicate experiments.
(I) Cortisol measurements from H295R cells. Kinase activity is necessary for the mutant’s effect on cortisol production. Mean ± SE. **p ≤ 0.01 versus WT and ###p ≤ 0.001 versus W196R; one-way ANOVA with Sidak correction; n = 3.
(J) Immunoblot of H295R lysates after 1-h incubation with vehicle or H89. Represents three experimental replicates.
(K) Cortisol measurements from H295R cells after 1-h vehicle or H89. Mean ± SE. ***p ≤ 0.001, one-way ANOVA with Sidak correction. See also Figure S3.
High-resolution immunofluorescent imaging examined the subcellular location of each PKAc variant in H295R adrenal cells. The L205R variant displayed a more pronounced nuclear localization than PKAc-W196R or the WT kinase (Figures 3D and 3E). Line plot analyses quantitatively validated these observations (Figures 3E and 3F). Expression of either Cushing’s variant enhanced phosphorylation of a canonical PKA substrate, serine 63, on ATF1 (Figure 3G, middle). Importantly, pATF1 detection was attenuated when control studies were conducted in H295R adrenal cells expressing K72A kinase-dead mutants (Figure S3C). Similar effects were observed in the mouse adrenocortical tumor cell line ATC7L when the same antibodies were used to detect phospho-CREB (Figures S3D and S3E). Immunoblot analysis demonstrated that PKAc-W196R enhanced PKA substrate phosphorylation 1.37 ± 0.07-fold compared with the WT. This Cushing’s variant also up-regulated levels of the mitochondrial cholesterol importer steroidogenic acute regulatory protein (StAR), a cAMP-responsive gene that controls the rate-limiting step in steroidogenesis, by 2.9 ± 0.4-fold. (Figure 3H, lane 2, and Figures S3F and S3G) (Selvaraj et al., 2018). Introduction of a kinase-dead mutation abolished both effects (Figures 3H, S3F, and S3G). Hormone measurements further revealed that expression of kinase-dead PKAc-W196R rescued overproduction of cortisol (Figure 3I). Pharmacological inhibition of native PKA with H89 for 1 h caused a modest reduction in pATF1 levels and decreased autophosphorylation of RII (Figures 3J, S3H, and S3I). This acute treatment was also sufficient to decrease cortisol production (Figure 3K). Similar effects were noted in ATC7L cells but with greater sensitivity to H89 (Figures S3J and S3K). Together, these data demonstrate differences between the activity and compartmentalization of PKAc-L205R and -W196R Cushing’s variants. Despite these mutations altering PKAc activity, escape of the kinase from AKAP signaling islands promotes aberrant substrate hyperphosphorylation and cortisol overproduction.
PKA activation is not sufficient for stress hormone overproduction
Cell-permeable analogs of cAMP that differentially occupy the cyclic nucleotide-binding pockets of R subunits are useful tools to manipulate the activity and location of native PKAc. Two pharmacological regimens were utilized to ensure experimental rigor: (1) a cocktail of Sp-5,6-DCI-cBIMPS and 6-benzyl cAMP (BIMPS/Bnz) and (2) the less cell-permeable analog Sp-cAMPS (Figure 4A). The concentrations of cAMP analogs used in our studies were based on previously published values (Dostmann et al., 1990; Poppe et al., 2008; Sandberg et al., 1991). Immunoblot detection of phosphorylated ATF1 demonstrated that both drug treatments activated the kinase (BIMPS/Bnz 1 μM/3 μM = 1.45 ± 0.16 versus WT, Sp-cAMPS 100 μM = 1.41 ± 0.16 versus WT; Figure 4B, top, and Figure 4C). Detection of phospho-RII, an index of PKAc activity within the anchored holoenzyme, was prominent at all concentrations of BIMPS/Bnz (Figure 4B, lanes 2–4, and Figure 4D, teal, 1 μM/3 μM = 1.15 ± 0.09 versus WT). Conversely, increasing the dosage of Sp-cAMPS decreased RII phosphorylation (Figure 4B, middle, lanes 5–7, and Figure 4D, purple, 100 μM = 0.45 ± 0.09 versus WT). This latter observation is consistent with release of PKAc from anchored holoenzymes and diffusion into the cell. As a positive control, the adenylyl cyclase activator forskolin and phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (Fsk/IBMX) were used to boost supraphysiological accumulation of intracellular cAMP. This drug regimen promoted the unilateral release of anchored PKA and maximally enhanced cortisol production (Figure 4B, lane 8). Parallel studies in mouse ATC7L cells gave the same pattern but with heightened sensitivity to cAMP analogs (Figure S4A). These pharmacological studies argue that BIMPS/Bnz cocktails selectively activate PKAc immobilized within anchored holoenzymes, whereas Sp-cAMPS releases active kinase into the cytosol.
Figure 4. PKA activation is not sufficient for stress hormone overproduction.
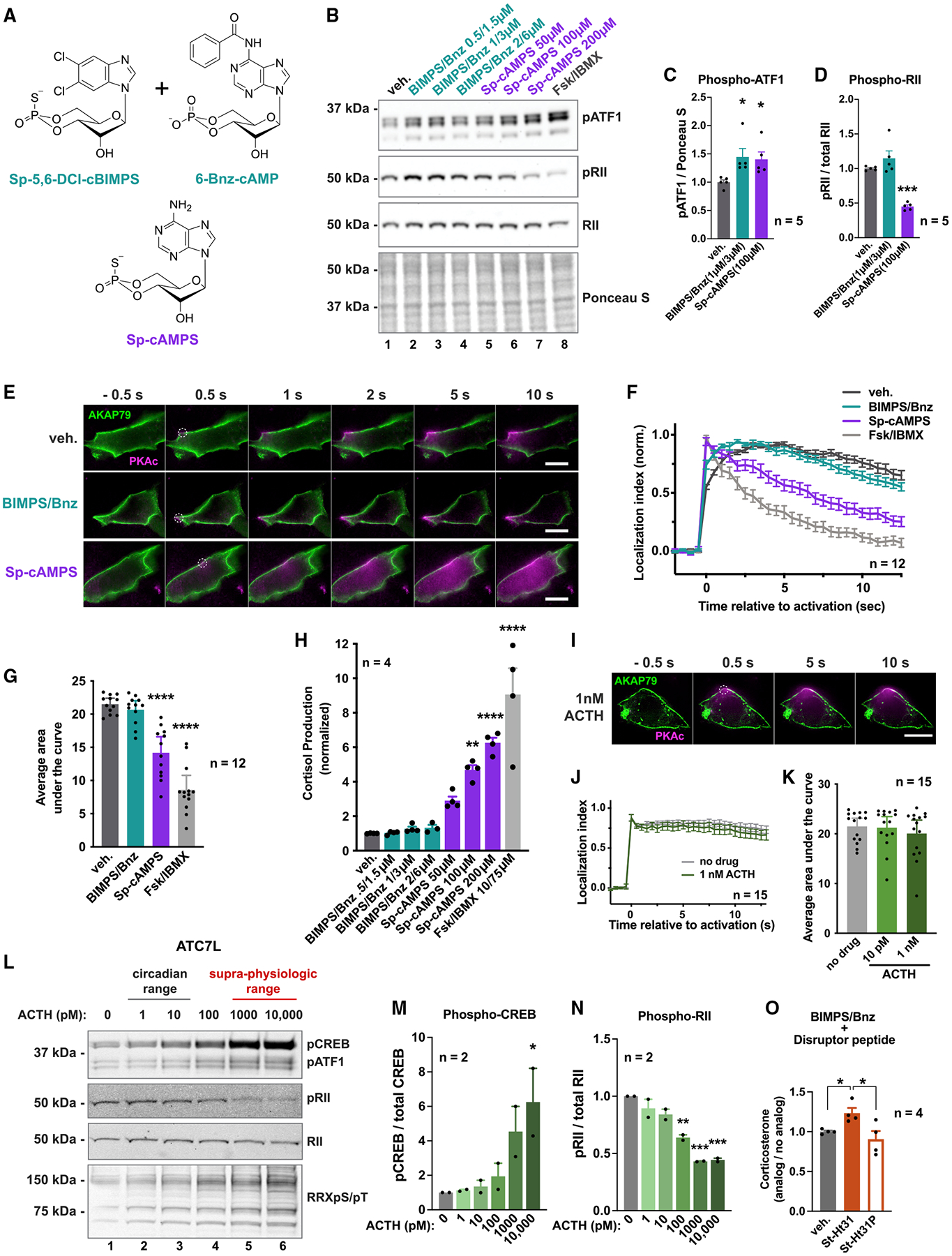
(A) cAMP analogs used.
(B) Immunoblot of H295R lysates after 1 h with vehicle or increasing concentrations of PKA-activating drugs.
(C and D) Quantification of phospho-ATF1 signal normalized to total protein (C) and phospho-RIIβ signal divided by total RIIβ (D). Teal, 1 μM Sp-5,6-DCl-cBIMPS + μM 6-Bnz-cAMP. Purple, 100 μM Sp-cAMPS. Mean ± SE. *p < 0.05, one-way ANOVA with Holm-Sidak correction; n = 5.
(E) Photoactivation time course of H295R cells expressing AKAP79-YFP, RII-iRFP, and WT PKAc tagged with photoactivatable mCherry. Top, vehicle only; middle, 1 μM Sp-5,6-DCl-cBIMPS + 3 μM 6-Bnz-cAMP; bottom, 100 μM Sp-cAMPS. Scale bar, 10 μm.
(F) Amalgamated photoactivation data from three experimental replicates. Mean ± SE.
(G) Integration of values from (F). Mean ± SE. ****p ≤ 0.0001, one-way ANOVA with Dunnett’s correction.
(H) Cortisol measurements from H295R cells treated for 1 h with vehicle or increasing concentrations of PKA-activating drugs. Mean ± SE. ***p ≤ 0.001, ****p ≤ 0.0001, one-way ANOVA with Dunnett’s correction; n = 4.
(I) Photoactivation time course after exposure to 1 nM ACTH. Scale bar, 10 μm.
(J) Photoactivation quantitation of H295R cells with and without ACTH. Data from two experimental replicates. Mean ± SE.
(K) Integration of curves from (J). No significant differences by one-way ANOVA. Mean ± SE.
(L) Immunoblot of ATC7L cells after 1-h ACTH treatment.
(M and N) Quantification of (K). Phospho-CREB signal divided by total CREB (M) and phospho-RIIβ signal divided by total RIIβ (N). Mean ± SE. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, one-way ANOVA with Holm-Sidak correction; n = 2 full dosage curves.
(O) Two-hour corticosterone production of ATC7L cells with no peptide (gray), stearated Ht31 (25 μM; filled orange) or proline-mutated stearated Ht31 (25 μM; open orange). Quantification normalizes hormone production after 2 μM Sp-5,6-DCl-cBIMPS/6 μM 6-Bnz-cAMP treatment to no analog treatment. Mean ± SE. *p < 0.05, one-way ANOVA with Holm-Sidak correction; n = 4. See also Figure S4.
To further test this postulate, we utilized live-cell photoactivation imaging assays (Figure 4E). H295R cells were transfected with AKAP79-YFP (green) to sequester PKA holoenzymes at the plasma membrane. Diffusion of photoactivated PKAc (magenta) was minimal over a time course of 10 s in vehicle-treated cells (Figure 4E, top; Video S4). Similar effects were observed in cells treated with the BIMPS/Bnz cocktail (Figure 4E, middle; Video S5). Conversely, application of Sp-cAMPS promoted the release of WT PKAc from sites of photoactivation (Figure 4E, bottom; Video S6). Supraphysiological stimulation of cAMP accumulation with Fsk/IBMX induced a rapid loss of PKAc localization to AKAP79 signaling islands (Figure S4B; Video S7). Quantification from multiple experiments confirmed the results (Figures 4G and 4F). Thus, BIMPS/Bnz stimulates PKA in the context of AKAP signaling islands, whereas Sp-cAMPS promotes release of the active kinase, thereby replicating the mechanistic effect of Cushing’s mutants. Stress hormone measurements validated this hypothesis, detecting remarkable differences between the cAMP analog regimens in H295R (Figure 4H) and ATC7L (Figure S4C) cells. BIMPS/Bnz cAMP activation of PKA in the context of AKAP signaling islands had no effect on stress hormone levels (Figure 4H, teal, and Figure S4C). In contrast, releasing the kinase upon application of Sp-cAMPS induced stress hormone production in a dose-dependent manner (Figure 4H, purple, and Figure S4C). Controls confirmed that supraphysiological cAMP stimulation with Fsk/IBMX promoted maximal cortisol release (Figure 4H, gray, and Figure S4C).
In healthy individuals, adrenocorticotropic hormone (ACTH) is released from the pituitary gland to stimulate cortisol production in the adrenal glands (Rosenberg et al., 2002). We tested how ACTH impacts activation and localization of native PKAc. Normal circadian concentrations of ACTH (1–10 pM) caused modest elevation of phospho-ATF1 in H295R cells (Figure S4D). Photoactivation experiments showed minimal changes in mobility of the kinase upon hormonal stimulation (Figures 4I–4K). ATC7L cells exhibited dosage-dependent increases to both phospho-CREB and phospho-ATF1 (Figure 4L, top, and Figures 4M, S4E, and S4F). Detection of the phospho-RII signal was used as an index of kinase activity within the confines of the PKA holoenzyme (Isensee et al., 2018). Immunoblot detection of phospho-RII was prominent upon stimulation of cells with physiological levels of ACTH and was markedly reduced by supraphysiological application of the hormone (Figure 4L, middle, and Figure 4N). Next, we used cell-permeable anchoring disruptor peptides to independently test whether overproduction of stress hormone required mislocalization of PKA activity in ATC7L cells. Application of stearated (St)-Ht31 increased hormone production when combined with BIMPS/Bnz treatment (Figure 4O). In contrast, the St-Ht31P control peptide had no effect (Figure 4O). Hence, Figure 4 suggests that activation of PKAc alone is not sufficient to strongly elevate stress hormone production. Rather, displacement of PKAc from AKAPs is required for this event.
Tethering Cushing’s mutants restores normal stress hormone production
Our working hypothesis is that displacement of active PKAc from AKAP signaling islands underlies Cushing’s syndrome (Figure 5A). This presupposes that correcting the displacement of Cushing’s PKAc variants should abolish their effects on stress hormone overproduction. Rescue experiments were accomplished by tethering PKAc-L205R or W196R via a peptide linker to the RII subunit (Figure 5B). We established proof of principle by immunofluorescence detection of PKAc variants (magenta) coexpressed with plasma-membrane-targeted AKAP79 (Figures 5C and 5D, green). Control experiments evaluated PKA fusions linked by a P2A self-cleaving peptide (Figure 5C). This technology ensures equivalent production of both protein components during translation (Liu et al., 2017). Only native PKAc colocalized with AKAP79 (Figure 5C, top row), whereas free PKAc-L205R and W196R filled the cytosol (Figure 5C, middle and bottom rows). In contrast, each RII-PKAc fusion (magenta) co-localized with AKAP79 (Figure 5D, green).
Figure 5. Tethering Cushing’s mutants restores normal stress hormone production.
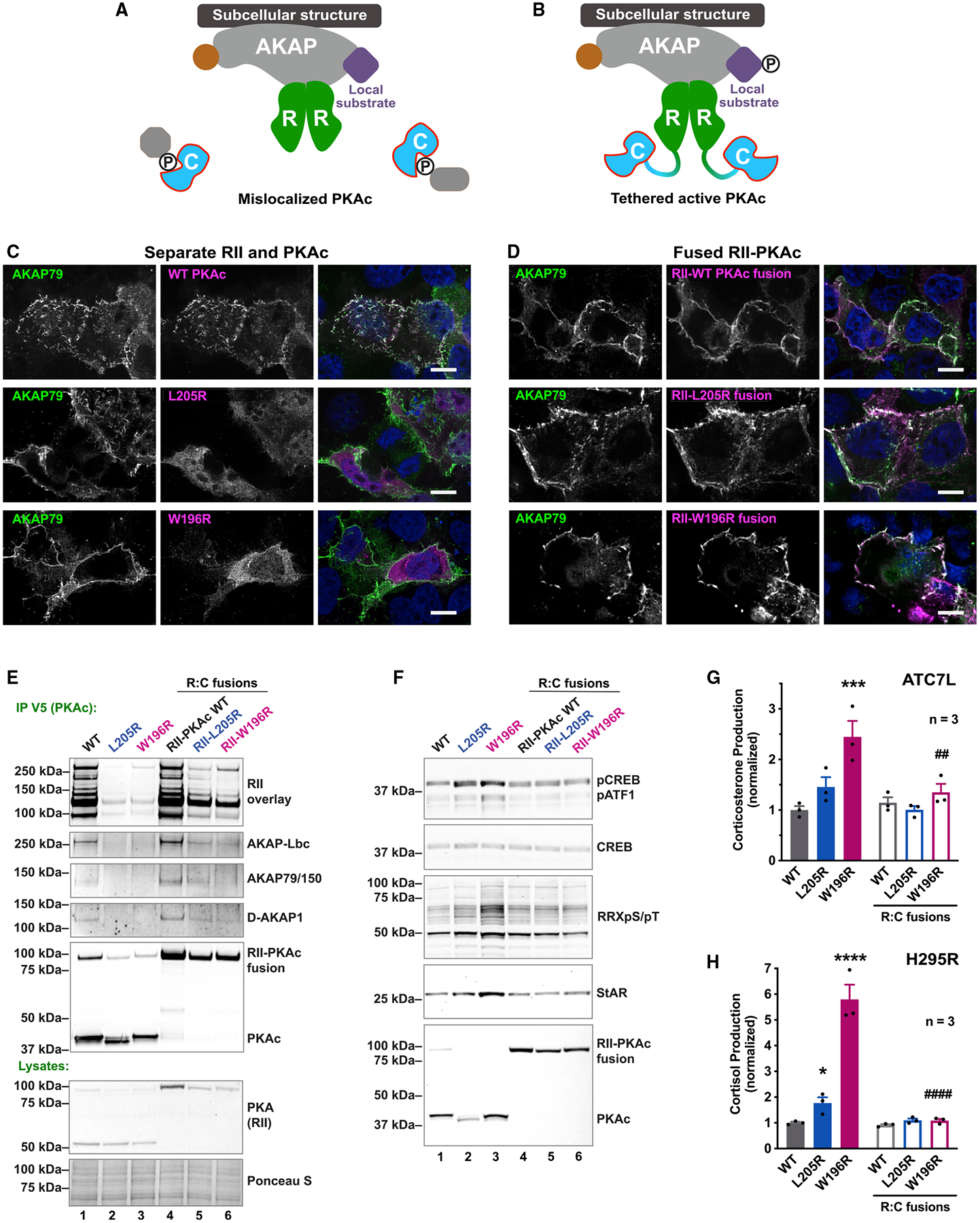
(A) Model of PKAc Cushing’s mutant displaced from AKAPs.
(B) Our strategy to tether active kinase mutants to AKAPs by fusing RII with PKAc-L205R or W196R.
(C) U2OS cells expressing AKAP79 (green) along with a self-cleaving RII-P2A-PKAc-V5 (PKAc in magenta). Scale bar, 10 μm.
(D) Same as (C) but with fused constructs of RII-PKAc (magenta). Scale bar, 10 μm.
(E) Western blots evaluating AKAP association after immunoprecipitating P2A (lanes 1–3) or fusion (lanes 4–6) constructs of PKAc. RII overlay detects multiple type II AKAPs. Represents three replicates.
(F) Immunoblot of ATC7L lysates. P2A constructs, lanes 1–3; fused RII-PKAc, lanes 4–6. Represents three replicates.
(G) Corticosterone measurements from ATC7L cells expressing separate or fused RII and PKAc variants. Mean ± SE. ***p ≤ 0.001 versus WT, ##p ≤ 0.01 versus W196R unfused condition. One-way ANOVA with Sidak correction; n = 3.
(H) Cortisol measurements from H295R cells expressing separate or fused RII and PKAc. Mean ± SE. ****p ≤ 0.0001 versus WT, ####p ≤ 0.0001 versus W196R unfused condition. One-way ANOVA with Sidak correction; n = 3. See also Figure S5.
This experimental strategy was used to test whether reconstitution of AKAP signaling islands in adrenal cells corrects stress hormone overproduction. Few AKAPs were detected when PKAc-L205R or W196R immune complexes were probed by RII overlay (Figure 5E, top, lanes 2 and 3). Yet, parallel analyses of RII-L205R or RII-W196R fusions detected multiple AKAPs, including AKAP-Lbc, AKAP79/150, and D-AKAP1 (Figure 5E, top, lanes 5 and 6). Additional biochemical analyses found that free Cushing’s mutants stimulate phosphorylation of CREB, ATF1, and PKA substrates (Figure 5F, top three panels, lanes 1–3, and Figure S5A). Similarly, expression of free PKAc-L205R or W196R elevated levels of StAR compared with native kinase (Figure 5F, fourth panel, lanes 1–3, and Figure S5B). Fusion of each mutant kinase to RII returned these cAMP-signaling events back to baseline levels (Figure 5F, lanes 4–6). Importantly, fusion of PKAc-L205R or W196R to RII rescued overproduction of stress hormone in mouse ATC7L and human H295R adrenal cells (Figures 5G and 5H). Collectively, the data in Figure 5 show that displacement of PKA from AKAP signaling islands is a pathological event in Cushing’s syndrome.
Proximity phosphoproteomic analyses reveal disruption of ERK and Hippo signaling
A logical extension of our working premise is that mislocalization of Cushing’s variants alters phosphorylation of substrates. Therefore, we conducted phosphoproteomic screens to identify signaling mechanisms implicated in the disease. Proximity-biotinylated phosphoproteins were isolated from H295R cells expressing miniTurbo-tagged versions of PKAc-L205R or W196R (Figure 6A). Volcano plot visualization of mass spectrometry data illustrates the phosphopeptides depleted and enriched for each mutant (Figures 6B and 6C). Deciphering the PKAc-L205R and W196R phosphoproximitomes revealed 48 and 30 hits, respectively, that were significantly depleted compared with WT (Figures 6B and 6C, black dots, and Figure S5A). Reduced detection of phosphopeptides from AKAP-Lbc and AKAP220 occurred in both screens. This is consistent with evidence that Cushing’s PKAc variants are displaced from AKAP signaling islands (Figures 1E and 1K).
Figure 6. Proximity phosphoproteomic analyses reveal disruption of ERK and Hippo signaling.
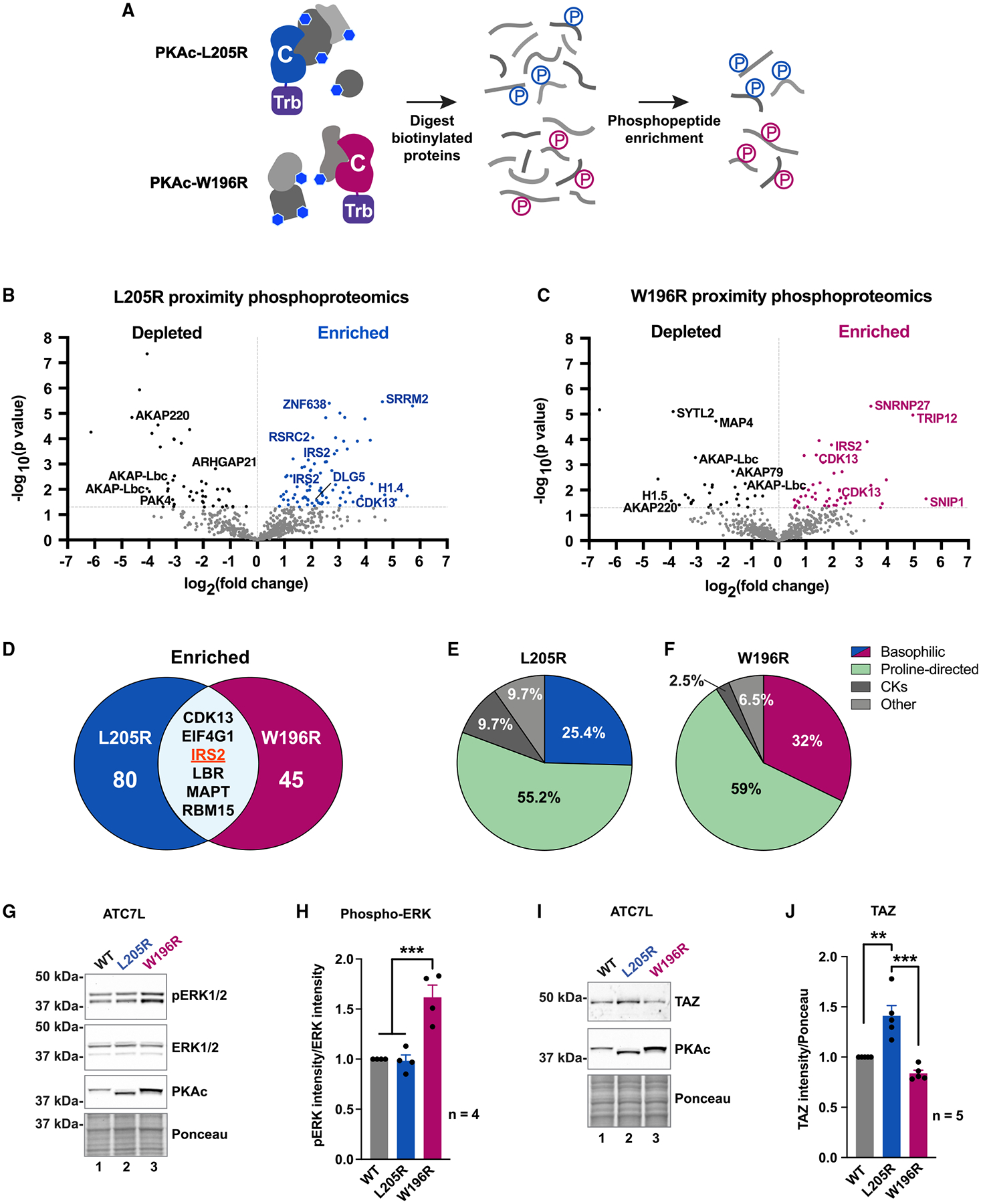
(A) Locally biotinylated proteins are isolated, digested, and subjected to phosphoenrichment.
(B and C) Proximity phosphoproteomics of PKAc-miniTurbo-biotinylated H295R samples from L205R (B) and W196R (C) versus WT. Phosphopeptides depleted in the mutant conditions, black; enriched phosphopeptides, blue (L205R) or red (W196R).
(D) Venn diagram of significantly enriched phosphopeptides identified in mutant conditions.
(E and F) NetworKIN kinase prediction for phosphosites enriched in L205R (E) and W196R (F) conditions.
(G) Immunoblot of ATC7L cells expressing PKAc variants.
(H) Quantitation of (G). Mean ± SE. ***p ≤ 0.001, one-way ANOVA with Sidak correction; n = 4.
(I) Immunoblot of ATC7L cells expressing PKAc variants.
(J) Quantitation of (I). Mean ± SE. ***p ≤ 0.001, **p ≤ 0.01, one-way ANOVA with Sidak correction; n = 5. See also Figure S6.
Eighty phosphopeptides were enriched in the PKAc-L205R phospho-proximitome (Figure 6B, blue dots, and 6D, left), and 45 phosphopeptides were enriched in the PKAc-W196R dataset (Figure 6C, red dots, and 6D, right). Six enriched phosphopeptides were common to both datasets, including sites on the insulin receptor substrate 2 (IRS2; Figures 6D and S5A). This scaffolding protein couples insulin action to MAP kinase signaling (De Meyts, 2000). Hence, we hypothesized that PKAc variants could impact ERK signaling to drive proliferation of Cushing’s adenomas. Additional motivation to pursue this postulate was provided by NetworKIN analysis showing that PKAc-L205R and W196R phosphoproximitomes were enriched with substrates for mitogen activated kinases (Figures 6E and 6F) (Horn et al., 2014).
Immunoblot detection of phospho-ERK in lysates from ATC7L adrenal cells served as an index to test if Cushing’s PKAc variants impact mitogen activated signaling. Although phospho-ERK levels were similar in WT and PKAc-L205R, the phospho-ERK signal was elevated in the W196R condition (Figure 6G, top). Densiometric analyses of data from independent experiments supported this unanticipated result (Figure 6H, magenta; n = 4). Immunoblot detection of total ERK confirmed equivalent levels of this kinase in all experimental conditions (Figure 6G, upper middle). Supplemental studies revealed that fusion of PKAc-W196R to RII returned phospho-ERK levels to baseline (Figure S5B, top, lane 6). Fusion of native PKAc or L205R to RII had no effect (Figure S5B, lanes 4 and 5).
As previously stated, phospho-ERK levels were unchanged in adrenal cells expressing PKAc-L205R (Figure 6H). This suggested that the RAF-MEK-ERK kinase cascade is not engaged by this Cushing’s variant. Upon further scrutiny of proteomic screens, we noted that discs large homolog 5 (DLG5) is enriched only in the PKAc-L205R condition (Figure 2C, blue, and 6A, blue). Three DLG5 phosphopeptides were identified including the consensus PKA site (R-R-L-S) at residues 1663–1666 (Figures 6B and S5C). DLG5 is a scaffolding protein that coordinates Hippo signaling, an evolutionarily conserved enzyme cascade that impinges on YAP/TAZ transcription factors to control cell survival and proliferation (Kwan et al., 2016; Yu and Guan, 2013). Immunoblot analysis detected upregulation of TAZ in ATC7L cells expressing PKAc-L205R (Figure 6I, lane 2, and 6J, blue). Additional studies demonstrated that fusion of PKAc-L205R to RII returned TAZ expression back to baseline (Figure S5D). Neither effect was observed in cells expressing PKAc-W196R (Figure 6I, lane 3, and 6J, red, and Figure S5D). Remarkably, these structurally alike Cushing’s PKAc variants appear to potentiate distinct downstream mitogenic-signaling pathways.
In vivo and clinical studies of Cushing’s PKAc mutants
It was imperative to investigate this concept in vivo. C57BL/6 mice harboring a conditional allele of PKAcα-W196R were crossed to an adrenal cortex-specific AS-Cre line (Figure 7A) (Freedman et al., 2013; Niswender et al., 2005). This yielded adult mice heterozygous for the PKA-W196R Cushing’s mutation in adrenal zona fasciculata (Figure 7B). These animals exhibited pathological features of adrenal Cushing’s syndrome. Female heterozygote PKAcWT/W196R mice displayed a 6.2 ± 2.0 g (SEM, n = 3) increase in body weight compared with littermate controls (n = 5; Figure 7C). Differences in weight were not detected in male mice (Figure S6A). Adrenal glands were larger in both sexes of PKAcWT/W196R mice, with a 1.57 ± 0.09-fold increase (SEM, n = 6) in protein content versus littermate controls (n = 9; Figures 7D–7F). Endocrine hallmarks of Cushing’s syndrome include elevated cortisol that triggers a negative-feedback response to block ACTH release from the pituitary (Figure 7G). Enzyme-linked immunosorbent assays measured a 1.61 ± 0.19-fold increase (SEM, n = 6) in the murine stress hormone corticosterone in serum from PKAcWT/W196R mice compared with littermate controls (n = 9; Figure 7G). Serum ACTH levels were reduced by 76% ± 17 SEM in the same cohort of animals (Figure 7I).
Figure 7. In vivo and clinical studies of Cushing’s PKAc mutants.
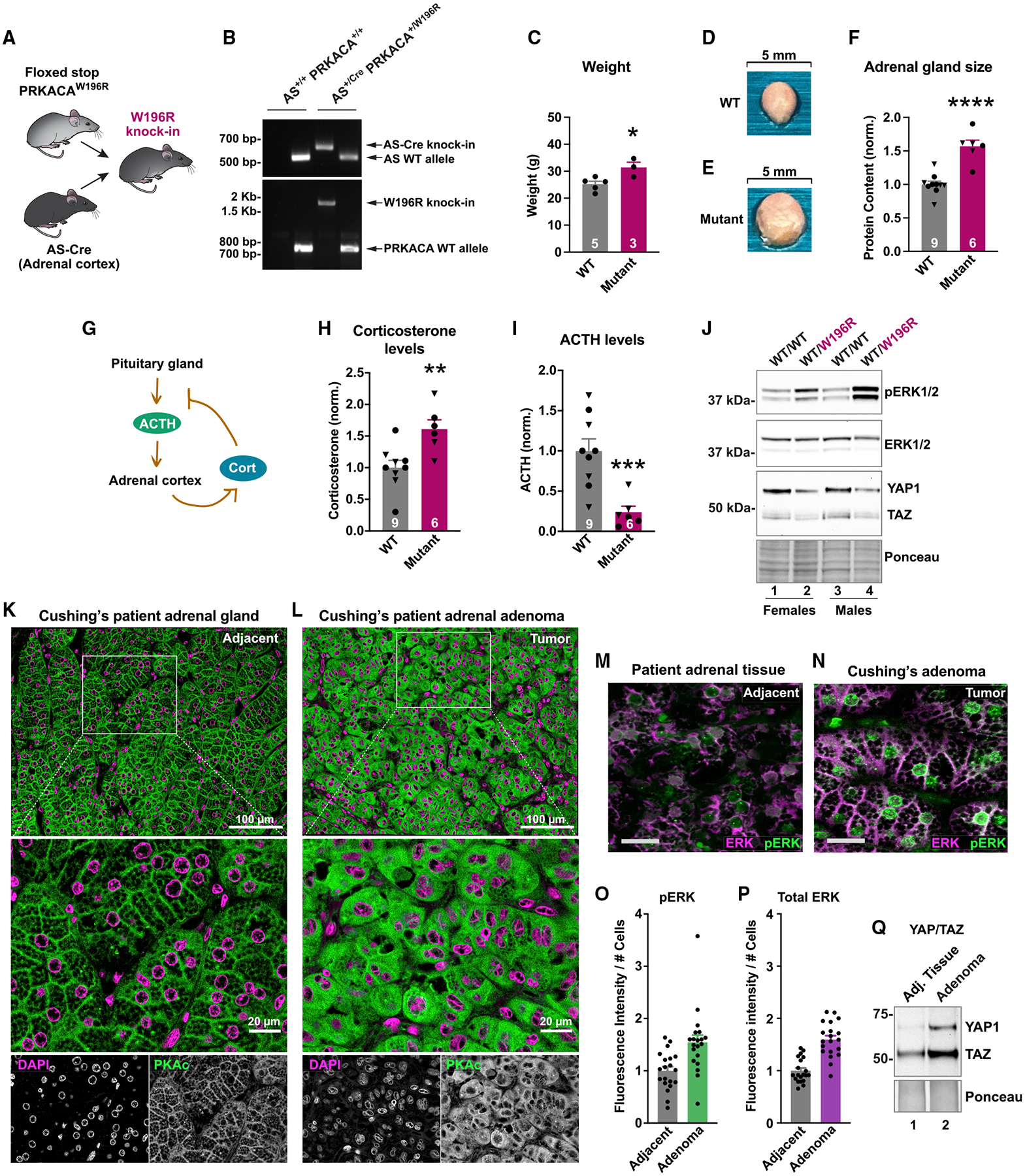
(A) Strategy to make a mouse with adrenal cortex-specific heterozygous expression of PKAc-W196R.
(B) Gel of genotyping PCR for littermate males.
(C) Weight of 1-year female mutant mice (red; n = 3) compared with littermate controls (gray; n = 5). Mean ± SE. *p ≤ 0.05 by Student’s t test.
(D and E) Images of adrenal glands from WT (D) and PKAc-W196R heterozygotes (E).
(F) Adrenal gland lysate protein concentration from male (triangles) and female (circles) mice. Mean ± SE. ****p ≤ 0.0001 by Student’s t test.
(G) Negative feedback mechanism wherein stress hormone downregulates ACTH.
(H and I) Serum corticosterone (H) and ACTH (I) levels from male (triangles) and female (circles) mice. **p ≤ 0.01 and ***p ≤ 0.001 by Welch’s t test. Mean ± SE.
(J) Immunoblot of adrenal gland lysates from female (lanes 1 and 2) and male (lanes 3 and 4) littermate mice.
(K and L) Resected tissue from a Cushing’s patient. PKAc (green) and DAPI (magenta) were stained in normal adjacent adrenal tissue (K) and adenoma (L). (M and N) Immunostaining for ERK (magenta) and phospho-ERK (green) in adjacent adrenal gland
(M) and adenoma (N) of a Cushing’s patient. Scale bar, 20 μm.
(O and P) Quantitation of fluorescence for phospho-ERK (O) and total ERK (P). Ten images each per condition from two patients. Mean ± SE.
(Q) Immunoblot detecting YAP1 and TAZ levels in normal adrenal and adenoma from a Cushing’s patient. Represents two patients. See also Figure S7.
Tissue-specific expression of a PKAc-W196R allele correlates with adrenal hyperplasia (Figures 7D and 7E). Therefore, immunoblot detection of phospho-ERK in adrenal lysates from sibling pairs examined whether enhanced cell proliferation involves mobilization of the ERK-signaling cascade. Phospho-ERK signals were elevated 2.6 ± 0.6-fold in both sexes of PKAcWT/W196R mice compared with littermate controls (Figure 7J, top, lanes 2 and 4, and Figure S7B). Total ERK levels were similar in both phenotypes and both sexes (Figure 7J, upper middle). Conversely, immunoblot detection of the Hippo-signaling elements YAP1 and TAZ was reduced 0.56 ± 0.14-fold and 0.55 ± 0.18-fold, respectively, in PKAcWT/W196R mice (Figure 7J, lower middle, and Figures S7C and S7D). Thus, our in vivo profiling establishes that the PKAcWT/W196R mouse model recapitulates pathological features of Cushing’s syndrome, and adrenal expression of a single PKAc-W196R allele enhances ERK signaling.
Finally, we investigated defective signaling mechanisms in tissue from Cushing’s patients. Paraffin-embedded sections of adrenal adenoma were stained for PKAc (green) and nuclei (magenta) (Figures 7K and 7L). High-resolution imaging of normal adrenal tissue revealed accumulation of the native kinase at cell-cell junctions (Figure 7K, green). This highly organized pattern of PKAc localization was most evident at higher magnifications (Figure 7K, bottom). In contrast, PKAc staining was markedly different in sections of Cushing’s adenoma, displaying a diffuse cytoplasmic distribution (Figure 7L, green). Similar patterns of PKAc mislocalization were observed in multiple Cushing’s patients (Figures S6E and S6F). Immunofluorescence detection of phospho-ERK evaluated the activation status of ERK kinase in adjacent and adenoma tissue sections (Figures 7M and 7N). Analyses of 10 images from each patient revealed a 1.55 ± 0.14-fold (SEM) increase in phospho-ERK (Figure 7O). Total ERK levels were also elevated (Figure 7P). Interestingly, immunoblots showed a marked upregulation of both YAP1 and TAZ in adenoma versus adjacent tissue (Figure 7Q). These patient studies corroborate our model of mislocalized PKA catalytic subunits and altered downstream signaling in adrenal Cushing’ syndrome.
DISCUSSION
We have discovered fascinating new insights about the molecular pathology of adrenal Cushing’s syndrome. The PKAc-L205R and W196R variants display different physiochemical properties, are differentially compartmentalized, and engage distinct downstream signaling pathways. At first glance, the PKAc-L205R and PKAc-W196R variants appear virtually identical. Both disease-causing mutations occur within the same exon of the PRKACA gene and promote amino acid changes only nine residues apart. However, protein melting curves in Figure 3C indicate that both Cushing’s kinases are more susceptible to temperature-dependent unfolding than native PKAc. Thermal instability of PKAc-L205R or W196R could play a part in the etiology of Cushing’s, since factors that influence protein folding and degradation have been implicated in other PKA pathologies (Rinaldi et al., 2019; Turnham et al., 2019). Likewise, phosphorylation of the catalytic core contributes to the stability, maturation, and activity of AGC kinases (Baffi et al., 2021; Newton, 2003). The V600E phosphomimetic mutation in BRAF, a common oncogenic driver in melanoma, dramatically elevates basal kinase activity (Davies et al., 2002; Holderfield et al., 2014). Similarly, multisite phosphorylation of PKC is necessary for a fully mature kinase (Van et al., 2021). As indicated in Figures 1E and S3A, PKAc-L205R has a faster electrophoretic mobility than its counterparts, which is a hallmark of reduced phosphate incorporation. Quantitative mass spectroscopy data in Figure 3B provides compelling proof of this phenomenon. Whereas the native kinase is fully phosphorylated on 11 sites, the L205R variant is almost totally devoid of phosphate. Accordingly, PKAc-L205R displays a 20-fold lower rate of catalysis toward conventional in vitro substrates. This could explain why PKAc-L205R induces lower levels of cortisol production than the PKAc-W196R form. Thus, subtle changes in physiochemical properties of the kinase catalytic core can be amplified by environmental factors into more profound effects on PKA physiology.
A key discovery of this report is that cortisol overproduction is a consequence of the exclusion of mutant kinases from AKAP signaling islands. This is suggested by three lines of evidence. First, live-cell imaging data in Figure 1K show that the PKAc-L205R and W196R variants have greater intracellular mobility than the native kinase. Analyses of tissue sections from Cushing’s syndrome patients in Figures 7K and 7L consolidate this finding by demonstrating a dramatic mislocalization of PKAc in resected Cushing’s adenomas. Second, quantitative proteomic screening of the PKAc-L205R and PKAc-W196R proximitomes in Figure 2 reveal that both Cushing’s variants have reduced interaction with AKAPs. Third, pharmacological studies with cAMP analogs in Figure 4 demonstrate that release of native PKAc from AKAPs, but not activation alone, enhances stress hormone production. These results converge on the notion that incorrect subcellular targeting of PKAc contributes to hypersecretion of cortisol. This new model is substantiated by rescue experiments in Figure 5, wherein sequestration of Cushing’s variants within AKAP signaling islands restores stress hormone production back to baseline levels. Hence, while it is accepted that localization of PKAc governs its biological action, our findings now show that chronic mislocalization of this kinase is a pathological driver of Cushing’s syndrome.
An additional contribution of this study is a mouse model for PKAc-driven adrenal Cushing’s syndrome. The phenotypic characterization of our adrenal-specific PKAc-W196R mice not only uncovers remarkable parallels to the human endocrine disorder but also illustrates how a point mutation in a single allele of PRKACA is sufficient to cause this debilitating disease. Interestingly, female mice exhibit more pronounced indicators of Cushing’s, including a 20% increase in weight. This mirrors the clinical state, where adrenal Cushing’s syndrome occurs up to five times more often in women (Di Dalmazi et al., 2014). Such sexual dimorphism in mice could result from more comprehensive AS-Cre expression in the zona fasciculata of females, due to higher rates of adrenocortical cell turnover (Dumontet et al., 2018). Consequently, our mouse model creates a valuable new tool for future in vivo, sex-based, and therapeutic studies on Cushing’s syndrome.
Aberrant phosphorylation events that ensue upon mislocalization of active Cushing’s kinases are the most likely drivers of this pathology. Modification to substrate selectivity via mutations to the substrate recognition loop of the kinase may be a pathological factor. The potential for altered substrate interactions was originally shown by Lubner et al. using yeast (Lubner et al., 2017). Since then, a pair of studies has demonstrated that L205R impacts the kinase’s substrate preference toward targets with a negative charge at the P+1,2,3 sites (Bathon et al., 2019; Walker et al., 2019). In addition, the phosphorylation of atypical substrates would be favored by changes in the spatial distribution of mutant kinases as detailed in this report. One interesting observation from our proximity phosphoproteomic screening presented in Figure 6 is the upregulation of histone H1.4 in the L205R condition. This corroborates an earlier report of nuclear H1.4 hyperphosphorylation in Cushing’s (Bathon et al., 2019). Thus, changes in the intracellular pattern of phosphorylation events can be linked to the action of distinct Cushing’s PKAc variants.
Another interesting feature of the proximity phosphoproteomics and cell-based validation in Figure 6 is the evidence of up-regulated ERK activity in adrenal cells expressing PKAc-W196R. This effect was also seen in Figure 7, both in mice harboring an adrenal-specific knockin of PKAc-W196R and in Cushing’s syndrome patient adenomas. Indirect support for this finding comes from recent evidence that the MEK inhibitor PD184352 reduces basal cortisol production from H295R cells (Pereira et al., 2019). Furthermore, AKAP-Lbc, a prevalent AKAP identified in our proteomic experiments, is a known regulator of MAP kinase signaling (Smith et al., 2010). This tallies with recent whole-exome sequencing of a Cushing’s syndrome patient that identified an allele of AKAP-Lbc fused to the phosphodiesterase PDE8A (Di Dalmazi et al., 2020). At the molecular level, this chimeric protein could efficiently terminate local PKA signaling by metabolizing cAMP within AKAP-Lbc signaling islands.
Perhaps one of the most unexpected findings of this report is that signals emanating from PKAc-L205R are markedly different than cellular events downstream of PKAc-W196R. As indicated in Figure 6J, this Cushing’s mutant does not potentiate MAP kinases but instead regulates the Hippo pathway, an evolutionarily conserved signaling cascade that controls organ size and development (Yu and Guan, 2013). Analyses of Cushing’s patient adenomas confirmed this finding, showing a strong increase in both YAP1 and TAZ levels in the tumor. Intriguingly, data in Figure 7J show that PKAc-W196R downregulates YAP/TAZ levels in our mouse model, suggesting possible cross-talk between different pathways. This could proceed through increased ERK activation in these mice, as a recent clinical study reported an inverse correlation between ERK1 and YAP1 levels in breast cancer patients (Yu et al., 2019). Although we have discovered differences between both PKAc mutants studied here, their impact in adrenal Cushing’s syndrome remains poorly understood. Although the mutation is less prevalent, our data consistently show that PKAc-W196R causes more cortisol production than PKAc-L205R. This is accompanied by higher levels of the steroidogenic protein StAR, as shown in Figure 5F. Likewise, phospho-ERK levels are elevated in PKAcWT/W196R mice, which is consistent with evidence of adrenal hyperplasia in these animals. Such pathological disparities imply that patients carrying a mutation at position 196 in PKAc may experience a more advanced form of the disease. In conclusion, we have discovered that adrenal Cushing’s syndrome is a disease of aberrant kinase location and that point mutations in the same subdomain of the PKAc catalytic core manifest distinct pathological signaling outputs.
Limitations of the study
Both NCI-H295R and ATC7L cells were derived from tumors and do not fully represent adrenal cell physiology. Although we demonstrate substantial impairment of in vitro PKAc-L205R activity, how this mutant impacts in vivo signaling is less clear and likely affected by environmental factors such as the action of other kinases. Substrate rewiring caused by active site mutations of PKAc may impact catalytic activity using the canonical PKA substrate Kemptide. Future studies with other PKA substrates would expand this work. The biophysical mechanisms of cAMP analogs are not fully understood. These compounds may have unrecognized effects on ACTH signaling through native PKAc. Finally, StAR levels and stress hormone production show that PKAc-W196R displays a more severe phenotype than PKAc-L205R. This observation would benefit from direct comparison with an L205R mouse model or with tissue samples from W196R patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John D. Scott (scottjdw@uw.edu).
Materials availability
Plasmids and cell lines generated for this study are available upon request.
Data and code availability
The autophosphorylation mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030888 and https://doi.org/10.6019/PXD030888. The proximity biotinylation mass spectrometry proteomics data have been deposited to MassIVE with the identifier MSV000088654, ftp://massive.ucsd.edu/MSV000088654/. These mass spectrometry datasets and identifiers are also listed in the key resources table. All data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PKAc | BD Biosciences | 610981; RRID: AB_398294 |
| PKARIIα | BD Biosciences | 612243; RRID: AB_399566 |
| GFP | Rockland | 600-101-215; RRID: AB_218182 |
| AKAP79 | custom rabbit polyclonal, Scott lab | VO89 |
| PKARIβ | Santa Cruz | sc-907; RRID: AB_2237411 |
| V5-tag | Thermo Fisher | R96025; RRID: AB_2556564 |
| phospho-CREB/phospho-ATF1 | CST | 9198; RRID: AB_2561044 |
| CREB | CST | 9104; RRID: AB_490881 |
| phospho-PKA substrate | CST | 9624; RRID: AB_331817 |
| StAR | CST | 8449; RRID: AB_10889737 |
| phospho-RIIβ | Santa Cruz | sc-136460; RRID: AB_10655524 |
| RIIβ | BD Biosciences | 610626; RRID: AB_397958 |
| phospho-ERK1/2 | CST | 9101; RRID: AB_331646 |
| ERK1/2 | CST | 9102; RRID: AB_330744 |
| pan ERK | BD Biosciences | 610123; RRID: AB_397529 |
| YAP/TAZ | CST | 8418; RRID: AB_10950494 |
| Bacterial and virus strains | ||
| BL21 (DE3) Competent Cells | Novagen | 70235-M |
| GC10 Competent Cells | Genesee | 42–659 |
| One Shot Stbl3 Competent Cells | Invitrogen | C737303 |
| Stable Competent Cells | NEB | C3040I |
| Biological samples | ||
| Human adrenal gland tissue | NW BioSpecimen | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Adenosine-3′,5′-cyclic monophosphate (cAMP), free acid | Biolog | A 001–500 H |
| IPTG | Sigma | I5502 |
| Glutathione Sepharose | GE Healthcare | 17075605 |
| Biotin | Sigma | B4501 |
| NeutrAvidin-HRP | Pierce | 31030 |
| Nanolink magnetic streptavidin beads | Tri-link Biotechnologies, Vector Labs | M-1002–020 |
| Endoproteinase LysC | NEB | P8109S |
| Pierce Trypsin protease, MS grade | Thermo Fisher | 90057 |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | Thermo Scientific | A32992 |
| Reprosil-Pur 120 C18-AQ, 3 mm | Dr. Maisch | R13.AQ.0001 |
| H-89 | Enzo Life Sciences | BML-EI196–0005 |
| DAPI | Life Technologies | 62248 |
| Sp-5,6-DCl-cBIMPS [5,6-Dichlorobenzimidazole riboside-3′,5′-cyclic monophosphorothioate, Sp- isomer (Sp-5,6-DCl-cBIMPS), sodium salt] | Biolog | D 014–05 |
| 6-Bnz-cAMP [N6-Benzoyladenosine-3′,5′-cyclic monophosphate (6-Bnz-cAMP), sodium salt] | Biolog | B 009–10 |
| Sp-cAMPS [Adenosine-3′,5′-cyclic monophosphorothioate, Sp-isomer (Sp-cAMPS), sodium salt | Biolog | A 003–10 S |
| Forskolin | Santa Cruz | sc-3562 |
| IBMX [3-isobutyl-1-methylxanthine] | Sigma | I7018 |
| ACTH, human | Sigma | A0423 |
| St-Ht31 disruptor peptide | Tocris | 6286 |
| St-Ht31P control peptide | Tocris | 6287 |
| Qproteome FFPE Tissue | Qiagen | 37623 |
| Critical commercial assays | ||
| Mouse/Rat ACTH ELISA kit | Abcam | ab263880 |
| Cortisol ELISA kit | Enzo Life Sciences | ADI-900-071 |
| Corticosterone ELISA kit | Enzo Life Sciences | ADI-900-097 |
| Deposited data | ||
| Autophosphorylation mass spectrometry proteomics | ProteomeXchange Consortium via the PRIDE partner repository (deposited) | 10.6019/PXD030888 |
| Proximity biotinylation mass spectrometry proteomics | MassIVE (deposited) | MSV000088654 |
| Experimental models: Cell lines | ||
| HEK293T cell line | GE Lifesciences | HCL4517 |
| U2OS PKA KO cell line | Smith et al., (2017) | N/A |
| NCI-H295R cell line | ATCC | CRL-2128 |
| ATC7L cell line | Ragazzon et al., (2006) | N/A |
| H295R-PKAcWT-miniTurbo stable cell line | This paper | N/A |
| H295R-PKAcL205R-miniTurbo stable cell line | This paper | N/A |
| H295R-PKAcW196R-miniTurbo stable cell line | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: AS-Cre: Cyp11b2tm1.1(cre)Brlt/+ | Freedman et al., (2013) | N/A |
| Mouse: Floxed stop PRKACAW196R | Niswender et al., (2005) | MMRRC: 032825-MU |
| (CαR): B6.129X1-Prkacatm3Gsm/Mmmh | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain #:000664; RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| DNA primers | Table S1 | N/A |
| Recombinant DNA | ||
| pcDNA3.1_PKAcWT_v5 | This paper | N/A |
| pcDNA3.1_PKAcL205R_v5 | This paper | N/A |
| pcDNA3.1_PKAcW196R_V5 | This paper | N/A |
| pEYFP-N1_AKAP79 | This paper | N/A |
| pEGFP-N1_RIIα | This paper | N/A |
| pEGFP_smAKAP | This paper | N/A |
| piRFP-N1_RIα | This paper | N/A |
| piRFP-N1_RIIα | This paper | N/A |
| pPAmCherry-N1_PKAcWT | This paper | N/A |
| pPAmCherry-N1_PKAcL205R | This paper | N/A |
| pPAmCherry-N1_PKAcW196R | This paper | N/A |
| pCW57-PKAcWT-miniTurbo | This paper | N/A |
| pCW57-PKAcL205R-miniTurbo | This paper | N/A |
| pCW57-PKAcW196R-miniTurbo | This paper | N/A |
| pSMALB-PKAcWT-V5 | This paper | N/A |
| pSMALB-PKAcL205R-V5 | This paper | N/A |
| pSMALB-PKAcW196R-V5 | This paper | N/A |
| pSMALB-PKAcK72A-V5 | This paper | N/A |
| pSMALB-PKAcL205R; K72A-V5 | This paper | N/A |
| pSMALB-PKAcW196R; K72A-V5 | This paper | N/A |
| pSMALB-RIIα-PKAcWT-V5_fusion | This paper | N/A |
| pSMALB-RIIα-PKAcL205R-V5_fusion | This paper | N/A |
| pSMALB-RIIα-PKAcW196R-V5_fusion | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcWT-V5 | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcL205R-V5 | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcW196R-V5 | This paper | N/A |
| pCW57-MCS1-P2A-MCS2 (Blast) | Adam Karpf; http://n2t.net/addgene:80921 | 80921; RRID: Addgene_80921 |
| psPAX2 | DidierTrono; http://n2t.net/addgene:12260 | 12260; RRID: Addgene_12260 |
| pMD2.G | DidierTrono; http://n2t.net/addgene:12259 | 12259; RRID: Addgene_12259 |
| pSMALB | John Dick & Peter van Galen; http://n2t.net/addgene:161786 | 161786; RRID: Addgene_161786 |
| Software and algorithms | ||
| ImageJ (FIJI) | https://imagej.net/downloads | N/A |
| GraphPad Prism 9 | www.graphpad.com | N/A |
| MaxQuant | https://www.maxquant.org/ | N/A |
| Perseus | https://www.maxquant.org/ | N/A |
| STRING database | https://string-db.org/ | N/A |
| The Gene Ontology Resource powered by PANTHER | http://geneontology.org/ | N/A |
| NetworKIN | https://kinomexplorer.info/ | N/A |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The PKA-CαR (W196R conditional knock-in) mice (Niswender et al., 2005) were made by Stanley McKnight (U Washington) and gifted to us by Diwakar Pattabiraman (Dartmouth). These mice carry a PRKACA allele encoding the W196R variant in a Cre-dependent manner. To make adrenal cortex-specific mutant mice, we crossed these to AS-Cre mice (Freedman et al., 2013), which were a gift from David Breault. Adult (11–13 month) mice of both sexes were used for our studies. PKA-CαR mice were maintained on a C57BL/6J background. AS-Cre mice were backcrossed to C57BL/6J a minimum of three times and all experiments used randomly assigned littermates as controls. Control mice included fully WT and singly heterozygous littermates for either floxed W196R or AS-Cre. Mice were housed in groups of 2 or more with littermates of the same sex until the night before the procedure. To minimize stress responses, mice were singly housed the night before the procedure and cages were left on racks until euthanasia. On the day of the procedure, mice were quickly moved to a separate room and immediately euthanized by isoflurane and cervical dislocation. Weight was then measured and blood was taken by cardiac puncture and kept on ice until all samples were collected. Blood was allowed to clot at room temperature for 1 h. Adrenal glands were dissected from acutely euthanized mice and frozen on dry ice for immunoblot analysis. All mice were cared for and euthanized in accordance with University of Washington regulatory standards and approved IACUC protocols.
Human tissue
De-identified, formalin-fixed, paraffin-embedded human adrenal cortical adenoma samples were obtained from the UW Northwest Biospecimen resource. Patient age and sex were not provided. Adrenal tissue for immunostaining was formalin fixed and paraffin embedded. Samples were deparaffinized by placing slides in 100% xylenes once for ten minutes and once for five minutes. Samples were then rehydrated by placing slides in 100% ethanol twice for ten minutes each, followed by 95% ethanol for ten minutes, 80% ethanol for ten minutes, and deionized water two times for five minutes each. Antigen retrieval was performed by placing slides in a chamber with pre-boiled 10 mM sodium citrate buffer (pH6.0). The chamber was then placed inside of a vegetable steamer for one hour. Slides were placed under cold running water for ten minutes before permeabilization in 0.4% Triton X-100/PBS for 7 min. Blocking was carried out in 5% BSA and 10% donkey serum in PBST (containing 0.05% Tween) for two hours at room temperature. For immunofluorescence, slides were incubated with primary antibodies in 5% BSA in PBST overnight at 4°C. Cells were washed 3x in PBST for ten minutes each and incubated with Alexa Fluor conjugated secondary antibodies and DAPI in 3% BSA in PBST for 1 h at room temperature. Slides were then washed six times for ten minutes each in PBST. Samples were mounted on glass slides using ProLongTM Diamond anti-fade mountant (Thermo Fisher) and cured overnight. Images were acquired using a Leica DMI6000B inverted microscope with a spinning disk confocal head (Yokagawa) and a CoolSnap HQ camera (Photometrics) controlled by MetaMorph 7.6.4 (Molecular Devices). Tissue for immunoblotting was processed using Qproteome FFPE Tissue (Qiagen 37,623).
Cell lines
All mammalian cells were grown under 37°C, 5% CO2 incubation conditions. Female NCI-H295R cells were purchased from ATCC September 2019 (CVCL_0458) and were maintained in ATCC DMEM:F12 medium containing 2.5% Corning NuSerum I and 1% Corning ITS + supplement. Female U2OS CRISPR PRKACA−/−cells (Smith et al., 2017) were maintained in DMEM medium with 10% fetal bovine serum. Male ATC7L cells (Ragazzon et al., 2006) were maintained in Gibco DMEM:F12 with added 5 mL L-Glutamine (200 mM) and 5 mL Gibco ITS supplement. Virus was produced in female HEK293T cells purchased from Dharmacon in 2015 and grown in DMEM medium with 10% fetal bovine serum.
Microbe strains
All purified proteins were produced in BL21 (DE3) pLysS E. coli cells (Novagen) and expression induced with 0.5 mM IPTG for 18 h at 18°C. Amplification of non-viral mammalian expression plasmids was performed in GC10 competent cells (Genesee) and grown at 37°C. Amplification of viral vectors was performed in either Stbl3 (Invitrogen) or Stable (NEB) competent cells and grown at 30°C.
METHOD DETAILS
Structural models
Models of PKA catalytic subunit in Figures 1B and 3B were made using PYMOL database file 3 (Zhang et al., 2012).
Antibodies
The following antibodies were used in our studies: PKAc BD 610981 (IF, WB); PKA RIIα BD 612243 (WB); GFP Rockland 600-101-215 (IF, IP, WB); AKAP79 custom V089 (WB); PKA RIβ Santa Cruz sc-907 (WB); V5-tag Thermo Fisher R96025 (IF, IP); phospho-CREB/phospho-ATF1 CST 9198 (IF, WB); CREB CST 9104 (IF, WB); phospho-PKA Substrate CST 9624 (WB); NeutrAvidin-HRP Pierce 31,030 (WB); StAR CST 8449 (WB); phospho-RIIβ Santa Cruz sc-136460 (WB); RIIβ BD 610626; phospho-ERK1/2 CST 9101 (IF, WB); ERK1/2 CST 9102 (WB); pan ERK BD 610123 (IF); YAP/TAZ CST 8418.
Plasmid generation
Specific plasmids are listed in the key resources table. Standard cloning was performed using PCR (36.2 μL ddH2O, 10 μL 5x HF Phusion buffer (NEB), 1 μL of 10 mM mixed dNTPs, 1.5 μL combined primers at 10 μM each, 1 μL template DNA at 5 ng/μL, and 0.3 μL Hot-Start Phusion polymerase (NEB)) in a Bio-Rad thermocycler. Thermocycling protocols varied depending on primer conditions and length of target region. For mutagenesis protocols, DpnI restriction enzyme and polynucleotide kinase were used (NEB). Some constructs were made using Gateway cloning system (Thermo Fisher). Ligation was performed with T4 DNA ligase (NEB) for 10–20 min at RT using manufacturer’s recommendations. Transformation into competent DNA (see Microbe strains above) was performed on ice for 15–30 min before heat shock for 30 s at 37°C.
Immunoprecipitations
Cell lysates were made using lysis buffer containing 1% Triton X-100, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 50 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 μM leupeptin, and 1 mM benzamidine. Lysates were incubated 5 min on ice and spun at 15,000×g for 8 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific) and adjusted to 1 mg/mL using lysis buffer. Samples (300–500 μL) were precleared by rotating with 20 μL protein G agarose for 30 min at 4°C. Supernatants were then incubated with 1–2 μg of the appropriate antibody overnight. In the morning, 30 μL of protein G agarose was added and samples were returned to rotation for 1 h. Beads were washed with lysis buffer and centrifuged at 5000×g 3 times, and then aspirated with a 27G needle before resuspending in 1x PAGE sample buffer (3% β-mercaptoethanol, final) and heating at 80°C for 10 min. Figures are representative for at least 3 experimental replicates.
Immunoblotting
Cell lysates were made using RIPA lysis buffer (1% NP-40 Tergitol, 0.5% deoxycholate, 0.1% SDS, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 20 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 μM leupeptin, and 1 mM benzamidine). Mouse adrenal protein extracts were made by homogenizing whole glands in RIPA buffer. For experiments to detect S/T phosphoproteins, 10 mM beta glycerophosphate was added. Samples were incubated 5 min on ice and spun at 15,000×g for 8 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific). Gels were loaded with 10–30 μg protein after heating for 10 min at 80°C with PAGE sample buffer containing 3% (final) β-mercaptoethanol. Proteins were transferred to nitrocellulose or PDVF, incubated with ponceau S to measure total protein loading, blocked in 5% milk TBST for 30 min at RT, and probed with antibodies in 5% BSA TBST overnight at 4°C. Membranes were washed 3 times in TBST and then incubated with secondary antibodies conjugated to HRP diluted in 5% milk TBST for 1 h at RT. After washing again 3x in TBST, signals were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) on an Invitrogen iBright Imaging System. Quantification was performed with ImageJ analysis software by measuring signal minus background for each band and dividing by the appropriate control signal, as indicated in each figure.
Hormone measurements
Cells were washed once in PBS and media was changed prior to harvest (1–2 h, as indicated for each experiment). Harvested media samples were either used immediately or snap frozen until assayed. Mouse serum was extracted by centrifugation (2,000×g) of clotted blood samples. To assay, samples were diluted and subjected to measurement using Enzo Cortisol or Corticosterone ELISA kit, or an Abcam ACTH ELISA kit. Values were determined using an absorbance plate-reader and subsequent curve fit analysis. All experiments were normalized to control levels within replicates or same-sex mouse litters.
Photoactivation microscopy
NCI-H295R cells were grown in glass bottom dishes and transfected using Lipofectamine 3000 48 h before imaging. Mammalian expression plasmids with CMV promoters and encoding AKAP79-YFP, smAKAP-GFP, RIIα-iRFP, RIα-iRFP, and either WT, L205R, or W196R tagged with photoactivatable mCherry were used. Imaging was performed using a GE OMX SR system. Exposure and laser intensity were optimized for each experimental replicate and held constant among experimental conditions. Photoactivation laser duration was kept under 50 milliseconds to activate a discrete area with minimal spread in the first image collected after activation. Images were collected at 2 Hz in 3 channels. A baseline of 4 images was taken prior to activation of the PKAc fluorophore. Cells were selected for imaging only when R-iRFP signal was colocalized with the AKAP. Secondary screening for this was performed posthoc. Timecourses were measured using ImageJ analysis software (FIJI). A localization index (intensity of the activated region divided by intensity of cytosolic region 6–8 μm distal) was used to interrogate change in fluorescent signal localization over time (mobility). For representative images, deconvolution and alignment of green and far-red channels were performed using OMX software.
Proximity biotinylation and sample prep for MS
Stable NCI-H295R adrenal cell lines were made using lentivirus encoding a tetracycline-responsive promoter and variants of PKAc tagged with V5 and miniTurbo biotin ligase at the c-terminus. Induction with doxycycline (0.5–1 μg/mL) was optimized to yield a subtle overexpression of the bait constructs at 20% of endogenous PKAc levels, as determined by PKAc immunoblotting and quantification using ImageJ. miniTurbo-tagged variant expression was induced for 48 h before application of 50 μM biotin in DMSO. Cells were incubated for 1 h at 37°C, washed 2x for 1 min using 10 mL PBS to deplete excess biotin, and then lysed using RIPA buffer (as described above). Protein concentrations were measured by BCA and samples were diluted to 1 mL of 0.5 mg/mL in RIPA buffer and placed in low protein binding tubes (Thermo Fisher) containing 25 μL of Nanolink magnetic streptavidin beads. Tubes were rotated 1 h at RT and placed on a magnet. Supernatant was saved for diagnostics and samples were washed in RIPA 2x, 2 M urea in 20 mM Tris 2x, and 25 mM Tris 2x. For normal mass spec analysis, samples were resuspended in 8 M urea in 100 mM Tris pH 8.5 with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 10 mM chloroacetamide (CAM) and then incubated at 37°C for 1 h. For phosphopeptide mass spec analysis, samples were resuspended in 20% trifluoroethanol 25 mM Tris pH 7.8 with 5 mM TCEP and 10 mM CAM and incubated at 95°C for 5 min. For digestion, samples were diluted 2-fold with 100 mM TEAB and 1 μg LysC was added before incubation for 2 h shaking at 37°C. Samples were again diluted with 100 mM TEAB and 1 μg Trypsin was added before incubation overnight shaking at 37°C. In the morning, normal mass spec samples were acidified to 1% formic acid and loaded on C18 StageTips. Samples for phosphoproteomics were subjected to phosphopeptide enrichment using a Thermo Scientific High-Select Fe-NTA Phosphopeptide Enrichment Kit prior to StageTip loading.
LC-MS analysis
Peptides were eluted from StageTips using elution buffer (40% acetonitrile, 1% FA) and then loaded on a self-pulled 360 μm OD × 100 μm ID 20 cm column with a 7 μm tip packed with 3 μm Reprosil C18 resin (Dr. Maisch, Germany). For pull-down experiment, peptides were analyzed by nanoLC-MS in a 90 min gradient from 15% to 38% buffer B (for phosphopeptides 6%–35% buffer B) at 300 nL/min using a Thermo EASY nLC 1200 system (buffer A: 0.1% acetic acid; buffer B: 0.1% acetic acid, 80% acetonitrile). Mass spectra were collected from an Orbitrap Fusion Lumos Tribrid Mass Spectrometer using the following settings. For MS1, Orbitrap FTMS (R = 60 ,000 at 200 m/z; m/z 350–1600; 7e5 target; max 20 ms ion injection time); For MS2, Top Speed data-dependent acquisition with 3 s cycle time was used, HCD MS2 spectra were collected using the Orbitrap mass analyzer(R = 30,000 at 200 m/z; 31% CE; 5e4 target; max 100 ms injection time) an intensity filter was set at 2.5e4 and dynamic exclusion for 45 s.
Mass spectrometry data analysis
Mass spectra were searched against the UniProt human reference proteome downloaded on July 06th, 2016 using MaxQuant v1.6.2.6. Detailed MaxQuant settings: for phosphopeptide analysis, samples were set to fraction 1 and 5 for WT and mutant, respectively, to allow within-group “match between run”; for pull-down, “Label-free quantification” was turned on, but not “match between run”, no fractionation was set; Trypsin/P was selected in digestion setting. Other settings were kept as default. Protein network prediction and gene ontology analysis were performed using STRING database version 11.5 and gene ontology enrichment analysis was performed using The Gene Ontology Resource powered by PANTHER. For phosphoproteomic kinase predictions, NetworKIN analysis was performed on significantly enriched phosphosites for each mutant. Total probability was summed for each protein kinase and only the highest-ranking member of each kinase classification was used due to prediction overlap. For pie chart depiction, the top 15 kinase classification groups were pooled into categories based on recognition sequence characteristics.
Recombinant protein purification
WT, W196R and L205R PKA catalytic (C) subunit, RIIα regulatory subunit, AKAP79 (amino acids 297–427) and PKI were produced in BL21 (DE3) pLysS E. coli cells (Novagen) and expression induced with 0.5 mM IPTG for 18 h at 18°C and purified as N-terminal His6-tag or N-terminal His6-GST tag fusion proteins by affinity chromatography and size exclusion chromatography using a HiLoad 16/600 Superdex 200 column (GE Healthcare) equilibrated in 50 mM Tris/HCl, pH 7.4, 100 mM NaCl, 1 mM DTT and 10% (v/v) glycerol.
In vitro pull-down assays
Purified GST-AKAP79(297–427) fusion proteins containing 3C protease cleavage sites were incubated for 3 h with glutathione Sepharose beads at 4°C in the presence of 2 mM DTT, washed five times in binding buffer (50 mM Tris pH 7.4, 0.1 M NaCl) and resuspended at a final protein concentration of ~5μM. C-subunit (~16 μM final concentration) and RIIα-subunits (~4 μM final concentration) were incubated with AKAP79(297–427) beads in the presence or absence of 1 mM cAMP for 20 min at 30°C with constant agitation. The supernatant was then removed, and after three washes in binding buffer, complexes were eluted from the beads by incubation with 250 ng of 3C protease for 30 min at 30°C. Proteins were analyzed by SDS-PAGE on a 12% gel.
Protein kinase assays
PKA kinase assays were performed using a real-time mobility shift-based microfluidic system, as described previously (Byrne et al., 2016), in the presence of 2 μM of the fluorescent-tagged “Kemptide” substrate (LRRASLG) and 1 mM ATP (as standard). Pressure and voltage settings were −1.8 (PSI), 2250 V (upstream voltage), and −500 V (downstream voltage) respectively. All assays were performed in 50 mM HEPES (pH 7.4), 0.015% (v/v) Brij-35, 1 mM DTT and 5 mM MgCl2, and peptide phosphorylation was detected in real time as the ratio of phosphopeptide:peptide. Changes in PKA activity in the presence of regulatory proteins (PKI and RIIα, assayed at the indicated concentrations) was quantified as the rate of phosphate incorporation into the substrate peptide (pmol phosphate $ min-1 $ uM enzyme-1), and then normalized with respect to control assays. To prevent ATP depletion and consequential loss of assay linearity, phosphate incorporation into the peptide was generally limited to <20%. ATP KM values were determined by nonlinear regression analysis using GraphPad Prism software. Unless otherwise specified, PKA WT and W196R mutants were assayed at a final concentration of 0.3 nM, and PKA L205R was assayed at 6 μM to account for the lower rate of activity.
Differential scanning fluorimetry
Thermal-shift assays were performed using a StepOnePlus Real-Time PCR machine (Life Technologies) using Sypro-Orange dye (Invitrogen) and thermal ramping (0.3°C in step intervals between 25 and 94°C). PKA proteins were diluted to a final concentration of 5 μM in 50 mM Tris/HCl, pH 7.4 and 100 mM NaCl in the presence or absence of the indicated concentrations of ATP, PKI or staurosporine (final DMSO concentration no higher than 4% v/v) and were assayed as described previously (Byrne et al., 2016). Normalized data were processed using the Boltzmann equation to generate sigmoidal denaturation curves, and average Tm/ΔTm values calculated using GraphPad Prism software.
PKAc autophosphorylation mass spectrometry
Concentrated purified PKA (10 μg) were diluted to 400 μL in 100 mM ammonium bicarbonate pH 8.0 (~60-fold dilution) before being subject to reduction (with dithiothreitol) and alkylation (with iodoacetamide) as previously described (Ferries et al., 2017). The eluent was equally divided into two for digestion with either: 33:1 (w/w) trypsin gold (Promega) or 10:1 (w/w) elastase (Promega) at 37°C for 18 h with 600 rpm shaking. Digests were then subject to strong cation exchange using in-house packed stage tip clean-up, as previously described (Daly et al., 2021). Dried peptides were solubilized in 20 μL of 3% (v/v) acetonitrile and 0.1% (v/v) TFA in water, sonicated for 10 min, and centrifuged at 13,000 g for 15 min at 4°C before separation and analysis by LC-MS/MS using an Ultimate 3000 nano system (Dionex), over a 60-min gradient, as described in (Ferries et al., 2017). Briefly, samples were loaded at a rate of 12 μL/min onto a trapping column (PepMap100, C18, 300 μm × 5 mm) in loading buffer (3% (v/v) acetonitrile, 0.1% (v/v) TFA) before being resolved on an analytical column (Easy-Spray C18 75 μm × 500 mm, 2 μm bead diameter column) using a gradient of 97% A (0.1% (v/v) formic acid): 3% B (80% (v/v) acetonitrile, 0.1% (v/v) formic acid) to 80% B over 30 min at a flow rate of 300 nL/min. All data acquisition was performed using a Thermo Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific), with higher-energy C-trap dissociation (HCD) fragmentation set at 32% normalized collision energy for 2 + to 5 + charge states using a 3 s cycle time. MS1 spectra were acquired in the Orbitrap (120K resolution at 200 m/z) over a range of 350–1400 m/z, AGC target = standard, maximum injection time = auto, with an intensity threshold for fragmentation of 2e4. MS2 spectra were acquired in the Orbitrap (30K resolution at 200 m/z), AGC target = standard, maximum injection time = dynamic. A dynamic exclusion window of 20 s was applied at a 10 ppm mass tolerance. Data was analyzed by Proteome Discoverer 1.4 using a custom database of the Uniprot Escherichia coli proteome (updated January 2021) supplemented with the His-tagged mouse PKA protein and relevant mutants, searched with fixed modification = carbamidomethylation (C), variable modifications = oxidation (M) and phospho (S/T/Y), instrument type = electrospray ionization–Fourier-transform ion cyclotron resonance (ESI-FTICR), MS1 mass tolerance = 10 ppm, MS2 mass tolerance = 0.01 Da, and the ptmRS node on; set to a score >99.0. For label free relative quantification of phosphopeptide abundances in the different forms of PKA, the area under the curve of extracted ion chromatograms (in Xcalibur, Qual browser) of the phosphopeptide ions were normalized against 5 non-modified peptides to account for potential protein load variability during analysis.
Immunofluorescent staining
Cells were plated on acid-washed coverslips and either transfected with plasmids (using Lipofectamine 3000 or Mirus) or infected with PKAc variant lentivirus 48 h before harvest. Cells were fixed in 4% PFA for 15 min at RT and washed 3x in PBS. Coverslips were moved to humidity chamber and blocked for 1 h at RT in 3% BSA, 0.3% Triton X-100. Primary antibodies were diluted in blocking solution and applied to coverslips overnight at 4°C. Coverslips were washed 3x with PBS, incubated with fluorescent secondaries (1:1000) and DAPI (~1:10,000), and washed 3x in PBS again before mounting. Images were taken on either a Keyence BZ-X710 microscope or a GE Healthcare OMX and processed/analyzed using ImageJ analysis software (FIJI).
Pharmacology
For stress hormone measurements and acute immunoblots, drugs were applied to cells for a 10 min preincubation before washing and exchanging for fresh media and drugs for a 1 or 1.5 h incubation at 37°C. Media was then harvested and cells were lysed in RIPA buffer (see Immunoblotting above). For photoactivation experiments, drugs were applied to cells in glass bottom dishes ten minutes before the start of imaging. Sample use was limited to one hour after drug application to minimize variation among imaged cells. St-Ht31 experiments were performed similarly, but with a 2 h incubation and omission of the pre-incubation.
QUANTIFICATION AND STATISTICAL ANALYSES
Data quantification and statistical analyses were performed with GraphPad Prism 9 for Mac as indicated in each figure legend. All data are presented with individual values displayed when possible along with mean ± SEM unless otherwise noted in the figure legend. Individual figure legends contain specific information on statistical parameters. Experiments involving more than three conditions used 1-way ANOVA with subsequent t-tests corrected for multiple comparisons. Specific statistical approaches were determined based on the distributions and parameters for each dataset.
Supplementary Material
Highlights.
PKAc mutants found in adrenal Cushing’s syndrome are chronically mislocalized
Displacement from AKAPs is required for stress hormone overproduction by mutants
A PKAc-W196R knockin mouse recapitulates adrenal Cushing’s syndrome hallmarks
PKAc-L205R and PKAc-W196R drive distinct downstream signaling pathways
ACKNOWLEDGMENTS
The authors thank K. Rosenthal, L. Gabrovsek, J. Nelson, K. Suso, and H. Dahlin for technical assistance and helpful discussions. Work was supported by National Institutes of Health (NIH) Grants F32DK121415 (M.H.O.), R01DK119192 (J.D.S.), R01DK119186 (J.D.S.), and R01GM129090 (S.-E.O.), a grant from the Fibrolamellar Foundation (J.D.S.), funding from BBSRC BB/T018127/1 and BB/S018514/1 (C.E.E. and P.A.E.), and Cancer Research UK C1443/A22095 (C.E.E.). This work used an EASY-nLC1200 UHPLC and Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer purchased with funding from NIH SIG Grant S10OD021502.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111073.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as a member of the LGBTQ+community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Amunjela JN, Swan AH, and Brand T (2019). The role of the Popeye domain containing gene family in organ homeostasis. Cell 8. 10.3390/cells8121594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffi TR, Lordén G, Wozniak JM, Feichtner A, Yeung W, Kornev AP, King CC, Del Rio JC, Limaye AJ, Bogomolovas J, et al. (2021). mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci. Signal 14, eabe4509. 10.1126/scisignal.abe4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A, Fassnacht M, and Calebiro D (2019). Alterations in protein kinase A substrate specificity as a potential cause of cushing syndrome. Endocrinology 160, 447–459. 10.1210/en.2018-00775. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DMF, Dessauer CW, and Scott JD (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Molecular cell 23, 925–931. 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, et al. (2014). Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med 370, 1019–1028. 10.1056/NEJMoa1310359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M, and Lohse MJ (2021). Optical mapping of cAMP signaling at the nanometer scale. Cell 184, 2793. 10.1016/j.cell.2021.04.043. [DOI] [PubMed] [Google Scholar]
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling inliving cells and organisms with TurboID. Nat. Biotechnol 36, 880–887. 10.1038/nbt.4201. https://www.nature.com/articles/nbt.4201#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne DP, Vonderach M, Ferries S, Brownridge PJ, Eyers CE, and Eyers PA (2016). cAMP-dependent protein kinase (PKA) complexes probed by complementary differential scanning fluorimetry and ion mobility-mass spectrometry. Biochem. J 473, 3159–3175. 10.1042/bcj20160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, Zhou W, Li X, Zhong X, Lei Y, et al. (2014). Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 344, 913–917. 10.1126/science.1249480. [DOI] [PubMed] [Google Scholar]
- Daly LA, Brownridge PJ, Batie M, Rocha S, Sée V, and Eyers CE (2021). Oxygen-dependent changes in binding partners and post-translational modifications regulate the abundance and activity of HIF-1α/2α. Science signaling 14, eabf6685. 10.1126/scisignal.abf6685. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- De Meyts P (2000). The insulin receptor and its Signal Transduction Network, Endotext KR, Feingold B, Anawalt A, Boyce G, Chrousos WW, de Herder K, Dhatariya K, Dungan A, Grossman JM, and Hershman JH, et al. , eds. (MDText.com, Inc; ). [PubMed] [Google Scholar]
- Di Dalmazi G, Altieri B, Scholz C, Sbiera S, Luconi M, Waldman J, Kastelan D, Ceccato F, Chiodini I, Arnaldi G, et al. (2020). RNA sequencing and somatic mutation status of adrenocortical tumors: novel pathogenetic insights. J. Clin. Endocrinol. Metab 105, e4459–e4473. 10.1210/clinem/dgaa616. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullié M, et al. (2014). Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J. Clin. Endocrinol. Metab 99, E2093–E2100. 10.1210/jc.2014-2152. [DOI] [PubMed] [Google Scholar]
- Dostmann WR, Taylor SS, Genieser HG, Jastorff B, Døskeland SO, and Ogreid D (1990). Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3’, 5’-cyclic phosphorothioates. J. Biol. Chem 265, 10484–10491. 10.1016/S0021-9258(18)86973-3. [DOI] [PubMed] [Google Scholar]
- Dumontet T, Sahut-Barnola I, Septier A, Montanier N, Plotton I, Roucher-Boulez F, Ducros V, Lefrançois-Martinez AM, Pointud J-C, Zubair M, et al. (2018). PKA signaling drives reticularis differentiation and sexually dimorphic adrenal cortex renewal. JCI Insight 3, e98394. 10.1172/jci.insight.98394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espiard S, Knape MJ, Bathon K, Assié G, Rizk-Rabin M, Faillot S, Luscap-Rondof W, Abid D, Guignat L, Calebiro D, et al. (2018). Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 3, 98296. 10.1172/jci.insight.98296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferries S, Perkins S, Brownridge PJ, Campbell A, Eyers PA, Jones AR, and Eyers CE (2017). Evaluation of parameters for confident phosphorylation site localization using an Orbitrap fusion Tribrid mass spectrometer. J. Proteome Res 16, 3448–3459. 10.1021/acs.jproteome.7b00337. [DOI] [PubMed] [Google Scholar]
- Freedman BD, Kempna PB, Carlone DL, Shah MS, Shah M, Guagliardo NA, Barrett PQ, Gomez-Sanchez CE, Majzoub JA, and Breault DT (2013). Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev. Cell 26, 666–673. 10.1016/j.devcel.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Korah R, Suttorp AC, Dietrich D, et al. (2014). Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet 46, 613–617. 10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ramírez LC, and Stratakis CA (2018). Genetics of Cushing’s syndrome. Endocrinol. Metab. Clin. N. Am 47, 275–297. 10.1016/j.ecl.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Deuker MM, McCormick F, and McMahon M (2014). Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467. 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, and Linding R (2014). KinomeXplorer: an integrated platform for kinome biology studies. Nat. Methods 11, 603–604. 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- Isensee J, Kaufholz M, Knape MJ, Hasenauer J, Hammerich H, Gonc-zarowska-Jorge H, Zahedi RP, Schwede F, Herberg FW, and Hucho T (2018). PKA-RII subunit phosphorylation precedes activation by cAMP and regulates activity termination. J. Cell. Biol 217, 2167–2184. 10.1083/jcb.201708053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan J, Sczaniecka A, Heidary Arash E, Nguyen L, Chen CC, Ratkovic S, Klezovitch O, Attisano L, McNeill H, Emili A, and Vasioukhin V (2016). DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genesis 30, 2696–2709. 10.1101/gad.284539.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix A, Feelders RA, Stratakis CA, and Nieman LK (2015). Cushing’s syndrome. Lancet 386, 913–927. 10.1016/S0140-6736(14)61375-1. [DOI] [PubMed] [Google Scholar]
- Langeberg LK, and Scott JD (2015). Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell. Biol 16, 232–244. 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Ruth Vaseghi H, Qian L, and Liu J (2017). Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep 7, 2193. 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubner JM, Dodge-Kafka KL, Carlson CR, Church GM, Chou MF, and Schwartz D (2017). Cushing’s syndrome mutant PKA L 205R exhibits altered substrate specificity. FEBS. Lett 591, 459–467. 10.1002/1873-3468.12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Liu W, Zhang A, Pan L, Tang W, Jiang B, Wang F, Chen S, and Fu B (2019). Identification of a PRKAR1A mutation (c.491_492delTG) in familial cardiac myxoma: a case report. Medicine 98, e14866. 10.1097/md.0000000000014866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musheshe N, Schmidt M, and Zaccolo M (2018). cAMP: from long-range second messenger to nanodomain signalling. Trends. Pharmacol. Sci 39, 209–222. 10.1016/j.tips.2017.11.006. [DOI] [PubMed] [Google Scholar]
- Newton AC (2003). Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J 370, 361–371. 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Willis BS, Wallen A, Sweet IR, Jetton TL, Thompson BR, Wu C, Lange AJ, and McKnight GS (2005). Cre recombinase-dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase A. Genesis 43, 109–119. 10.1002/gene.20159. [DOI] [PubMed] [Google Scholar]
- Omar MH, and Scott JD (2020). AKAP signaling islands: venues for precision pharmacology. Trends. Pharmacol. Sci 41, 933–946. 10.1016/j.tips.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia-Campos A, Aoto PC, Machal EMF, Rivera-Barahona A, Soto-Bielicka P, Bertinetti D, Baker B, Vu L, Piceci-Sparascio F, Torrente I, et al. (2020). Germline and mosaic variants in PRKACA and PRKACB cause a multiple congenital malformation syndrome. Am. J. Hum. Genet 107, 977–988. 10.1016/j.ajhg.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SS, Monteiro MP, Costa MM, Ferreira J, Alves MG, Oliveira PF, Jarak I, and Pignatelli D (2019). MAPK/ERK pathway inhibition is a promising treatment target for adrenocortical tumors. J. Cell. Biochem 120, 894–906. 10.1002/jcb.27451. [DOI] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang X-B, Christensen AE, Schwede F, Genieser H-G, Bos JL, Doskeland SO, et al. (2008). Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278. 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- Ragazzon B, Lefrançois-Martinez AM, Val P, Sahut-Barnola I, Tournaire C, Chambon C, Gachancard-Bouya JL, Begue RJ, Veyssière G, and Martinez A (2006). Adrenocorticotropin-dependent changes in SF-1/DAX-1 ratio influence steroidogenic genes expression in a novel model of glucocorticoid-producing adrenocortical cell lines derived from targeted tumorigenesis. Endocrinology 147, 1805–1818. 10.1210/en.2005-1279. [DOI] [PubMed] [Google Scholar]
- Rinaldi L, Delle Donne R, Catalanotti B, Torres-Quesada O, Enzler F, Moraca F, Nisticò R, Chiuso F, Piccinin S, Bachmann V, et al. (2019). Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system. Nat. Commun 10, 2572. 10.1038/s41467-019-10037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I, Calebiro D, Schwarzmayr T, Appenzeller S, Rubin B, et al. (2016). Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J. Clin. Endocrinol. Metab 101, 3526–3538. 10.1210/jc.2016-1586. [DOI] [PubMed] [Google Scholar]
- Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X, and Bertherat J (2002). Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann. N. Y. Acad. Sci 968, 65–74. 10.1111/j.1749-6632.2002.tb04327.x. [DOI] [PubMed] [Google Scholar]
- Sandberg M, Butt E, Nolte C, Fischer L, Halbrügge M, Beltman J, Jahnsen T, Genieser HG, Jastorff B, Walter U, and Walter U (1991). Characterization of Sp-5, 6-dichloro-1-beta-D-ribofuranosylbenzimidazole- 3’, 5’-monophosphorothioate (Sp-5, 6-DCl-cBiMPS) as a potent and specific activator of cyclic-AMP-dependent protein kinase in cell extracts and intact cells. Biochem. J 279 (Pt 2), 521–527. 10.1042/bj2790521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, et al. (2014). Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344, 917–920. 10.1126/science.1252328. [DOI] [PubMed] [Google Scholar]
- Scott JD, and Pawson T (2009). Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224. 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj V, Stocco DM, and Clark BJ (2018). Current knowledge on the acute regulation of steroidogenesis. Biol. Reprod 99, 13–26. 10.1093/biolre/ioy102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabb JB (2001). Physiological substrates of cAMP-dependent protein kinase. Chem. Rev 101, 2381–2412. 10.1021/cr000236l. [DOI] [PubMed] [Google Scholar]
- Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, and Scott JD (2017). Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293. 10.1126/science.aaj1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Langeberg LK, Cellurale C, Pawson T, Morrison DK, Davis RJ, and Scott JD (2010). AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell. Biol 12, 1242–1249. 10.1038/ncb2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Omar MH, Nygren PJ, Soughayer J, Hoshi N, Lau H-T, Snyder CG, Branon TC, Ghosh D, Langeberg LK, et al. (2018). Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. U S A 115, E11465–E11474. 10.1073/pnas.1816614115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, and Gonen T (2013). Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2, e01319. 10.7554/eLife.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA (2018). Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): the expected, the unexpected, and what might be next. J. Pathol 244, 257–259. 10.1002/path.5014. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids. Res 47, D607–d613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskén K, and Aandahl EM (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev 84, 137–167. 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, and Kornev AP (2012). Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell. Biol 13, 646–658. 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnham RE, and Scott JD (2016). Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Genewatch 577, 101–108. 10.1016/j.gene.2015.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnham RE, Smith FD, Kenerson HL, Omar MH, Golkowski M, Garcia I, Bauer R, Lau H-T, Sullivan KM, Langeberg LK, et al. (2019). An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. Elife 8, e44187. 10.7554/eLife.44187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van AAN, Kunkel MT, Baffi TR, Lordén G, Antal CE, Banerjee S, and Newton AC (2021). Protein kinase C fusion proteins are paradoxically loss of function in cancer. J. Biol. Chem 296, 100445. 10.1016/j.jbc.2021.100445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viña D, Seoane N, Vasquez EC, and Campos-Toimil M (2021). cAMP compartmentalization in cerebrovascular endothelial cells: new therapeutic opportunities in alzheimer’s disease. Cells 10. 10.3390/cells10081951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C, Wang Y, Olivieri C, Karamafrooz A, Casby J, Bathon K, Calebiro D, Gao J, Bernlohr DA, Taylor SS, and Veglia G (2019). Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv 5, eaaw9298. 10.1126/sciadv.aaw9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, and Scott JD (2004). AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell. Biol 5, 959–970. 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Yarwood SJ (2020). Special issue on “new advances in cyclic AMP signalling”-an editorial overview. Cells 9. 10.3390/cells9102274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, and Guan KL (2013). The Hippo pathway: regulators and regulations. Genes & development 27, 355–371. 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhang M, Huang L, Ma Z, Gong X, Liu W, Zhang J, Chen L, Yu Z, Zhao W, and Liu Y (2019). ERK1 indicates good prognosis and inhibits breast cancer progression by suppressing YAP1 signaling. Aging 11, 12295–12314. 10.18632/aging.102572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, and Taylor SS (2012). Structure and allostery of the PKA RIIβ tetrameric holoenzyme. Science 335, 712–716. 10.1126/science.1213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The autophosphorylation mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030888 and https://doi.org/10.6019/PXD030888. The proximity biotinylation mass spectrometry proteomics data have been deposited to MassIVE with the identifier MSV000088654, ftp://massive.ucsd.edu/MSV000088654/. These mass spectrometry datasets and identifiers are also listed in the key resources table. All data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PKAc | BD Biosciences | 610981; RRID: AB_398294 |
| PKARIIα | BD Biosciences | 612243; RRID: AB_399566 |
| GFP | Rockland | 600-101-215; RRID: AB_218182 |
| AKAP79 | custom rabbit polyclonal, Scott lab | VO89 |
| PKARIβ | Santa Cruz | sc-907; RRID: AB_2237411 |
| V5-tag | Thermo Fisher | R96025; RRID: AB_2556564 |
| phospho-CREB/phospho-ATF1 | CST | 9198; RRID: AB_2561044 |
| CREB | CST | 9104; RRID: AB_490881 |
| phospho-PKA substrate | CST | 9624; RRID: AB_331817 |
| StAR | CST | 8449; RRID: AB_10889737 |
| phospho-RIIβ | Santa Cruz | sc-136460; RRID: AB_10655524 |
| RIIβ | BD Biosciences | 610626; RRID: AB_397958 |
| phospho-ERK1/2 | CST | 9101; RRID: AB_331646 |
| ERK1/2 | CST | 9102; RRID: AB_330744 |
| pan ERK | BD Biosciences | 610123; RRID: AB_397529 |
| YAP/TAZ | CST | 8418; RRID: AB_10950494 |
| Bacterial and virus strains | ||
| BL21 (DE3) Competent Cells | Novagen | 70235-M |
| GC10 Competent Cells | Genesee | 42–659 |
| One Shot Stbl3 Competent Cells | Invitrogen | C737303 |
| Stable Competent Cells | NEB | C3040I |
| Biological samples | ||
| Human adrenal gland tissue | NW BioSpecimen | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Adenosine-3′,5′-cyclic monophosphate (cAMP), free acid | Biolog | A 001–500 H |
| IPTG | Sigma | I5502 |
| Glutathione Sepharose | GE Healthcare | 17075605 |
| Biotin | Sigma | B4501 |
| NeutrAvidin-HRP | Pierce | 31030 |
| Nanolink magnetic streptavidin beads | Tri-link Biotechnologies, Vector Labs | M-1002–020 |
| Endoproteinase LysC | NEB | P8109S |
| Pierce Trypsin protease, MS grade | Thermo Fisher | 90057 |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | Thermo Scientific | A32992 |
| Reprosil-Pur 120 C18-AQ, 3 mm | Dr. Maisch | R13.AQ.0001 |
| H-89 | Enzo Life Sciences | BML-EI196–0005 |
| DAPI | Life Technologies | 62248 |
| Sp-5,6-DCl-cBIMPS [5,6-Dichlorobenzimidazole riboside-3′,5′-cyclic monophosphorothioate, Sp- isomer (Sp-5,6-DCl-cBIMPS), sodium salt] | Biolog | D 014–05 |
| 6-Bnz-cAMP [N6-Benzoyladenosine-3′,5′-cyclic monophosphate (6-Bnz-cAMP), sodium salt] | Biolog | B 009–10 |
| Sp-cAMPS [Adenosine-3′,5′-cyclic monophosphorothioate, Sp-isomer (Sp-cAMPS), sodium salt | Biolog | A 003–10 S |
| Forskolin | Santa Cruz | sc-3562 |
| IBMX [3-isobutyl-1-methylxanthine] | Sigma | I7018 |
| ACTH, human | Sigma | A0423 |
| St-Ht31 disruptor peptide | Tocris | 6286 |
| St-Ht31P control peptide | Tocris | 6287 |
| Qproteome FFPE Tissue | Qiagen | 37623 |
| Critical commercial assays | ||
| Mouse/Rat ACTH ELISA kit | Abcam | ab263880 |
| Cortisol ELISA kit | Enzo Life Sciences | ADI-900-071 |
| Corticosterone ELISA kit | Enzo Life Sciences | ADI-900-097 |
| Deposited data | ||
| Autophosphorylation mass spectrometry proteomics | ProteomeXchange Consortium via the PRIDE partner repository (deposited) | 10.6019/PXD030888 |
| Proximity biotinylation mass spectrometry proteomics | MassIVE (deposited) | MSV000088654 |
| Experimental models: Cell lines | ||
| HEK293T cell line | GE Lifesciences | HCL4517 |
| U2OS PKA KO cell line | Smith et al., (2017) | N/A |
| NCI-H295R cell line | ATCC | CRL-2128 |
| ATC7L cell line | Ragazzon et al., (2006) | N/A |
| H295R-PKAcWT-miniTurbo stable cell line | This paper | N/A |
| H295R-PKAcL205R-miniTurbo stable cell line | This paper | N/A |
| H295R-PKAcW196R-miniTurbo stable cell line | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: AS-Cre: Cyp11b2tm1.1(cre)Brlt/+ | Freedman et al., (2013) | N/A |
| Mouse: Floxed stop PRKACAW196R | Niswender et al., (2005) | MMRRC: 032825-MU |
| (CαR): B6.129X1-Prkacatm3Gsm/Mmmh | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain #:000664; RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| DNA primers | Table S1 | N/A |
| Recombinant DNA | ||
| pcDNA3.1_PKAcWT_v5 | This paper | N/A |
| pcDNA3.1_PKAcL205R_v5 | This paper | N/A |
| pcDNA3.1_PKAcW196R_V5 | This paper | N/A |
| pEYFP-N1_AKAP79 | This paper | N/A |
| pEGFP-N1_RIIα | This paper | N/A |
| pEGFP_smAKAP | This paper | N/A |
| piRFP-N1_RIα | This paper | N/A |
| piRFP-N1_RIIα | This paper | N/A |
| pPAmCherry-N1_PKAcWT | This paper | N/A |
| pPAmCherry-N1_PKAcL205R | This paper | N/A |
| pPAmCherry-N1_PKAcW196R | This paper | N/A |
| pCW57-PKAcWT-miniTurbo | This paper | N/A |
| pCW57-PKAcL205R-miniTurbo | This paper | N/A |
| pCW57-PKAcW196R-miniTurbo | This paper | N/A |
| pSMALB-PKAcWT-V5 | This paper | N/A |
| pSMALB-PKAcL205R-V5 | This paper | N/A |
| pSMALB-PKAcW196R-V5 | This paper | N/A |
| pSMALB-PKAcK72A-V5 | This paper | N/A |
| pSMALB-PKAcL205R; K72A-V5 | This paper | N/A |
| pSMALB-PKAcW196R; K72A-V5 | This paper | N/A |
| pSMALB-RIIα-PKAcWT-V5_fusion | This paper | N/A |
| pSMALB-RIIα-PKAcL205R-V5_fusion | This paper | N/A |
| pSMALB-RIIα-PKAcW196R-V5_fusion | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcWT-V5 | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcL205R-V5 | This paper | N/A |
| pSMALB-RIIα-P2A-PKAcW196R-V5 | This paper | N/A |
| pCW57-MCS1-P2A-MCS2 (Blast) | Adam Karpf; http://n2t.net/addgene:80921 | 80921; RRID: Addgene_80921 |
| psPAX2 | DidierTrono; http://n2t.net/addgene:12260 | 12260; RRID: Addgene_12260 |
| pMD2.G | DidierTrono; http://n2t.net/addgene:12259 | 12259; RRID: Addgene_12259 |
| pSMALB | John Dick & Peter van Galen; http://n2t.net/addgene:161786 | 161786; RRID: Addgene_161786 |
| Software and algorithms | ||
| ImageJ (FIJI) | https://imagej.net/downloads | N/A |
| GraphPad Prism 9 | www.graphpad.com | N/A |
| MaxQuant | https://www.maxquant.org/ | N/A |
| Perseus | https://www.maxquant.org/ | N/A |
| STRING database | https://string-db.org/ | N/A |
| The Gene Ontology Resource powered by PANTHER | http://geneontology.org/ | N/A |
| NetworKIN | https://kinomexplorer.info/ | N/A |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.