

Abstract
We review evidence challenging the hypothesis that memories are processed or consolidated in sleep. We argue that the brain is in an unconscious state in sleep, akin to general anesthesia, and hence is incapable of meaningful cognitive processing – the sole purview of waking consciousness. At minimum, the encoding of memories in sleep would require that waking events are faithfully transferred to and reproduced in sleep. Remarkably, however, this has never been demonstrated, as waking experiences are never truly replicated in sleep but rather appear in very altered or distorted forms. General anesthetics (GAs) exert their effects through endogenous sleep-wake control systems and accordingly GAs and sleep share several common features: sensory blockade, immobility, amnesia and lack of awareness (unconsciousness). The loss of consciousness in non-REM (NREM) sleep or to GAs is characterized by: (1) delta oscillations throughout the cortex; (2) marked reductions in neural activity (from waking) over widespread regions of the cortex, most pronounced in frontal and parietal cortices; and (3) a significant disruption of the functional connectivity of thalamocortical and corticocortical networks, particularly those involved in “higher order” cognitive functions. Several (experimental) reports in animals and humans have shown that disrupting the activity of the cortex, particularly the orbitofrontal cortex, severely impairs higher order cognitive and executive functions. The profound and widespread deactivation of the cortex in the unconscious states of NREM sleep or GA would be expected to produce an equivalent, or undoubtedly a much greater, disruptive effect on mnemonic and cognitive functions. In conclusion, we contend that the unconscious, severely altered state of the brain in NREM sleep would negate any possibility of cognitive processing in NREM sleep.
Keywords: Non-REM sleep (NREM), consciousness, general anesthetics, prefrontal cortex, functional connectivity, cognition, memory
Graphical Abstract
Locations of waking/arousal-related cell groups of the brainstem and caudal diencephalon at which general anesthetics exert anesthetic actions. They include the pontomesencephalic reticular formation (PRF), locus coeruleus (LC), parabrachial nucleus (PB), tuberomammillary nucleus (TMN) and orexin-containing cells of the lateral hypothalamus. General anesthetics have been shown to suppress the activity of each of these structures to induce anesthesia whereas their activation (e.g., by electrical stimulation) hastens the recovery from anesthesia.
Introduction
While several functions have been proposed for sleep including shaping brain development, energy conservation, removal of toxic wastes, modulation of immune responses, and restitution of energy stores, the current dominant theory in the field is that sleep serves a role in memory processing/consolidation (for review, Krueger et al., 2016; Zielinski et al., 2016; Frank and Heller, 2019).
The notion that memories are processed or consolidated in sleep is an attractive one, for among other reasons, it would suggest that time spent in sleep is not “wasted” but serves the useful purpose of organizing (or reorganizing) waking experiences to strengthen and/or integrate them into existing schema. In this sense, then, sleep would serve as an offline processor, continuing to perform in sleep the same or similar functions of waking consciousness. Under ideal conditions, the material acted on in sleep would be encoded and incorporated into existing memories to form new associations.
However, despite the attractiveness of this hypothesis, we believe that there is no compelling or convincing evidence to support a role for sleep in the processing or consolidation of memory – with memory defined as the ability to encode, store and retrieve information for immediate or future use. We will argue that the brain during sleep is in an unconscious state, akin to that under general anesthesia, and accordingly is incapable of meaningful cognitive processing – the sole purview of waking consciousness. As recognized, cognitive processing denotes the process of acquiring, storing, recalling, and utilizing information to direct behavior.
We (Vertes and Eastman, 2000; Vertes, 2004; Vertes and Siegel, 2005) and others (Siegel, 2001; Pan and Rickard, 2015; Dringenberg, 2019) have previously challenged the now seemingly entrenched view of sleep-dependent memory consolidation. The present treatment revisits this controversial issue.
Although there are several views on precisely how sleep serves to promote memory processing, it seems that at their core the various theories would maintain the following. The cognitive content of waking can be: (1) accessed in sleep; (2) repeated (or replayed) in sleep; (3) transformed in sleep; and (4) “improved” in sleep – rendering it more beneficial with, than without, intervening sleep. In the latter case, this could involve the strengthening of significant and/or eliminating insignificant material of waking. With the foregoing in mind, we examine and dispute the claim that cognitive processing is possible in the unconscious state of sleep – thereby challenging the sleep-memory consolidation (SMC) hypothesis (Stickgold, 2005; Stickgold and Walker, 2007; Rasch and Born, 2013; Poe, 2017; Sara, 2017).
Dreams – the sole window into the “cognitive” contents of sleep
If waking experiences are to be consolidated/enhanced in sleep, they would seemingly need to be faithfully, or nearly so, repeated/replicated in sleep. While it has been reported that patterns of neural activity of waking are reproduced in sleep, to possibly consolidate them (Wilson and McNaughton, 1994; O’Neill et al., 2010; Deuker et al., 2013; Foster, 2017), fortunately for humans, one is not limited to such indirect measures, but can directly compare the cognitive content of waking and sleep – through dreams. Dreams are the sole window into the cognitive contents of sleep, and accordingly any claims that waking material is transformed in sleep would reasonably look to dreams to understand how this material is represented in sleep. A priori, it would seem (at minimum) that any strengthening/consolidation of waking events in sleep (NREM/REM) would require their nearly precise replication in sleep – as revealed through dreams. Accordingly, dream mentation should closely resemble waking mentation – if waking events are to be accurately represented in sleep. However, as clearly recognized, there is no correspondence between waking experiences and the cognitive contents of sleep -- or dreams. This would indicate either that the faithful (or undistorted) reproduction of waking experiences occurs outside of dreams (which make up 30–50% of sleep) or that dream material becomes consolidated in sleep – in our view an unlikely possibility.
While the hypothesis that sleep serves a role in memory processing has received considerable recent attention, this is not a new (or even unreasonable) position. Freud (1900), in fact, grappled with this issue, entertaining the possibility that sleep serves to somehow enhance waking experiences. Freud, however, rejected this notion based on the lack of correspondence (or mismatch) between the cognitive content of waking and sleep. For instance, Freud (1900) stated that “It might perhaps occur to us that the phenomenon of dreaming could be reduced entirely to that of memory: dreams, it might be supposed, are a manifestation of a reproductive activity which is at work even in the night and which is an end in itself.” Continuing, “But views of this sort are inherently improbable owing to the manner in which dreams deal with the material to be remembered. As has been rightly pointed out, dreams do not reproduce experiences. They take one step forward, but the next step in the chain is omitted, or appear in altered form, or is replaced by something entirely extraneous. Dreams yield no more than fragments of reproductions; and this is so general a rule that theoretical conclusions may be based on it.”
Although admittedly much of an overstatement, the position that “theoretical conclusions” can be based on the observation that “dreams do not reproduce experiences” may have, in part, contributed to the development of psychoanalytic theory. Specifically, if dreams do not reflect waking experiences (manifest content), they can only be understood by reference to their latent (or hidden) content. Material hidden from consciousness (as in dreams) becomes the “unconscious” or the product of the unconscious mind -- a cornerstone of psychoanalytic theory. Unconscious material can be accessed through dreams, or as Freud famously stated: “dreams are the royal road to the unconscious”. Without embracing psychoanalytic theories of the unconscious, it is nonetheless evident that dreams have a “life of their own,” or are divorced from waking experiences. If waking events were, by some means, reproduced in whole (or in large part) in sleep, the case could legitimately be made that waking experiences are acted upon in sleep. However, waking experiences are not replicated in sleep (or dreams), hence the impossibility of processing these experiences in sleep.
In detailed analyses of dreams and their meaning, the late Ernest Hartmann (2010) concluded that dreams are constructions (or creations) and not replays of waking experiences. Hartmann acknowledged, as generally accepted, that dreams sometime contain elements (or fragments) of waking events (day residue) which can become incorporated into dream scenarios. However, these episodes are not replicas of waking experiences but are one of several elements woven into the dream narrative. Whereas Hartmann (2010) did not discount the possibility that dreams could “faithfully” reproduce daytime experiences, he indicated that he never observed this. Specifically, Hartmann noted that even the repetitive dreams (or nightmares) of veterans suffering from post-traumatic stress disorder (PTSD) were not precise replicas of waking experiences; that is, they “turn out on examination to be creations, not simple replays of waking events”.
In a striking example of the degree of disconnect between waking and sleep mentation, Hartmann and Brezler (2008) analyzed the dreams of 44 subjects immediately before and after the tragedy of 9/11. In all, 880 dreams were examined -- 440 before and 440 after 9/11. Remarkably, of the 440 dreams examined after 9/11, none of them contained instances of planes hitting buildings (or similar scenarios) even though as the authors indicated, all participants had repeatedly seen images of the 9/11 events and were emotionally moved by them. In fact, none of the post-9/11 dreams even contained elements resembling the actual events, leading the authors to conclude that “even something as striking as the 9/11 attacks do not appear in dreams as a replay”.
While almost everyone (of a certain age) has vivid memories of the events surrounding 9/11, it is evident from the foregoing, that the (relative) permanence of these memories does not depend on sleep – as the events are not rehearsed, and hence processed, in sleep. As the images of 9/11 were repeatedly shown on TV (waking replay), discussed, shared and often recalled, it begs the question as to what more sleep could contribute to their encoding – probably nothing. Finally, if such an emotionally charged event as 9/11 is not replayed in sleep (and thus committed to memory through sleep) it seems very unlikely that less emotionally charged, or relatively inconsequential, experiences are expressed and processed in sleep.
Supporting the foregoing, Stickgold and colleagues (Fosse et al., 2003) systematically examined the relationship between episodic events of waking and the cognitive content of sleep and found virtually no correspondence. Specifically, they compared detailed accounts of daytime activity to dream reports of 29 subjects over a 14-day period that included locations, actions, objects, characters, themes, and emotions. Only 5 of 299 (1.4%) dream reports of NREM/REM sleep were judged to be very similar to waking experiences and thus possibly qualified as strong candidates for the transfer of episodic memories from waking to sleep. Accordingly, Stickgold and co-workers concluded that the results “are consistent with evidence that sleep has no role in episodic memory consolidation”. Taking this further, they stated that the “reactivation of episodic memories appears to be actively blocked during sleep” (Fosse et al., 2003).
In summary, there is (virtually) no correspondence between the episodic experiences of waking and sleep – even for highly-charged emotional events of waking that might be expected to be represented in sleep. This lack of transfer of cognitive material of waking to sleep would indicate that there is no access to waking experiences in sleep and as such no ability to act on these experiences in sleep – to strengthen, weaken or integrate them into existing memories.
While adherents of the SMC hypothesis might agree that waking events are not replicated or processed in sleep through dreams, or that the contents of dreams become consolidated in sleep, they might nonetheless argue that experiences of waking can be accessed, and hence acted upon, in sleep outside of dreams. Although highly improbable, this would suggest that waking experiences, through some unknown process, can be faithfully transferred from waking to “dreamless” sleep -- even though these experiences manage to evade any probe of cognitive processing in sleep. Accordingly, this would lead to the conclusion that material which reaches consciousness in sleep (dreams) is not processed, while at the same time holding that material that never reaches consciousness in sleep (whatever its nature) is somehow processed and consolidated in sleep. The SMC position would seem to require two parallel systems (seemingly sharing the same neural substrates during sleep) involved in processing waking material in sleep: a “sleep conscious” system (dreams) which distorts and imprecisely codes waking events, and a “sleep unconscious” system that faithfully reproduces and consolidates waking experiences. If the latter system exists, surprisingly it has so far escaped detection.
The following topics will be addressed in comparing sleep and general anesthesia (GA).
General anesthetics (GAs) operate through endogenous sleep control networks.
Cortical EEG activity of NREM sleep and GAs is very similar (or identical) in animals and humans, dominated by delta activity in deep NREM sleep and GA.
Sleep and GA can partially substitute one for the other.
Widespread areas of the cortex are severely suppressed in NREM sleep and GA – likely resulting in the significant disruption of normal (or waking) cognitive functions in these states.
The functional connections (FC) of major forebrain networks are significantly altered in NREM sleep and GA, likely producing a loss of functions normally performed by these networks in the waking state.
NREM sleep and general anesthesia (GA): parallel states of unconsciousness
Sleep and GA are similar states, sharing common defining features: immobility, sensory blockade, amnesia and a lack of awareness (unconsciousness) (Table 1). The brain is an unconscious state in sleep – comparable to that under general anesthesia. General anesthetics (GAs) alter consciousness by acting on the same neural substrates that are affected in sleep.
Table 1.
Direct parallels between the unconscious states of NREM sleep and general anesthesia (GA)
| Sensory Blockade | ✓ | ✓ |
| Immobility | ✓ | ✓ |
| Amnesia | ✓ | ✓ |
| Loss of Consciousness | ✓ | ✓ |
| Common Neural Substrates See Fig 1 and 2 | ✓ | ✓ |
| Cortical EEG - Rodent | 1–4 Hz Delta | 1–4 Hz Delta |
| Cortical EEG - Human | 1–4 Hz Delta | 1–4 Hz Delta |
| Widespread Cortical Suppression | ✓ | ✓ |
| Disruption of Functional Connectivity of Higher Order Cortical Areas | ✓ | ✓ |
| Cognitive Processing | no | no |
It is beyond the present scope to discuss the nature of consciousness (per se), as this is a vast, controversial and currently unresolved issue (Crick and Koch, 2003; Lamme, 2006; Cohen and Dennett, 2011; Strauss et al., 2015; Sikkens et al., 2019). However, an attractive hypothesis for consciousness is the predictive coding (PC) model which holds that consciousness involves feedback (top-down) cortical influences which serve to detect mismatches (deviance detection) between perceived and expected outcomes. Mismatches gives rise to a characteristic P3B (of the P300) sensory evoked potential which reflects the detection of so-called “global deviants” – a proposed measure of consciousness. In this regard, Sikkens et al. (2019) recently stated “Upon loss of consciousness, detection of local deviants remains, at least partially, present in the human brain. Detection of global deviants is, on the other hand, absent during states of unconsciousness and deep sleep”. Strauss et al. (2015) similarly reported that global responses (or mismatches) to auditory stimuli, as reflected in the P300, vanished in NREM and REM sleep. According to this model, then, consciousness is lost in sleep – as in the anesthetic state.
Whereas it is well established that memory and cognitive processing are blocked in the unconscious state of GA (Alkire and Gorski, 2004; Franks, 2008; Wang and Orser, 2011; Gross et al., 2019; Hemmings et al., 2019), this seemingly would be no less the case for the unconsciousness of sleep. For instance, addressing the effects of GAs on memory, Hemmings et al. (2019) recently commented that “Amnesia is one of the most sensitive behavioral endpoints of anesthetic action, with memory blockade occurring at plasma concentrations that are considerably lower than those required for unconsciousness and immobility.” In effect, we contend that cognition is the sole domain of consciousness and without consciousness there can be no cognitive processing – in sleep, general anesthesia or coma.
In a recent review of neural substrates of anesthetized states, Lydic et al. (2018) described what they termed the “shared circuit hypothesis” wherein the systems which evolved to generate sleep/wake states became usurped to induce states of general anesthesia. Essentially, over the course of evolution, species adapted to changing light-dark conditions by developing rest-activity cycles and over time the circuity to generate sleep-wake states. There was no evolutionary pressure to develop circuitries responsive to anesthetics. Nonetheless, as was pointed out, anesthetics abolish wakefulness in fruit flies (which have existed for several million years) as well as in humans, relatively new to the evolutionary scene. This would indicate a conserved neural network, developed for controlling sleep/wake states, that becomes responsive to anesthetics. The “shared circuit hypothesis” is similar to the notion that anesthetics have “hijacked” sleep/wake control systems (see below).
General anesthetics act on sleep/wake control systems: the arousal/wakefulness network
As discussed below, GAs exert effects through endogenous sleep/wake control networks -- to inhibit waking systems and/or activate sleep “centers” to thereby suppress wakefulness (Scharf and Kelz, 2013; Lydic et al., 2018).
Several excellent reviews have been devoted to a description of the neural circuitry controlling sleep/waking states (Vertes, 1984, 1990; Datta and MacLean, 2007; Siegel, 2009; Datta, 2010; Jones, 2011; Brown et al., 2012; Saper and Fuller, 2017; Scammell et al., 2017). In brief, the circuitry consists of mutual interacting wake/arousal nuclei of the brainstem and caudal diencephalon and sleep “center(s)” of the ventral basal forebrain.
Of the “classic” sites contributing to wakefulness, we will focus on the pontomesencephalic reticular formation (RF), the locus coeruleus (LC), the parabrachial nucleus (PB), the tuberomammillary (TMN) nucleus and the orexinergic (ORX) cell group of the caudal diencephalon. These nuclei are depicted in Figure 1. As described below, GAs that suppress or enhance the activity of these structures prolong or hasten recovery, respectively, from anesthesia (for review, Lu et al., 2008; Leung et al., 2014).
Figure 1.
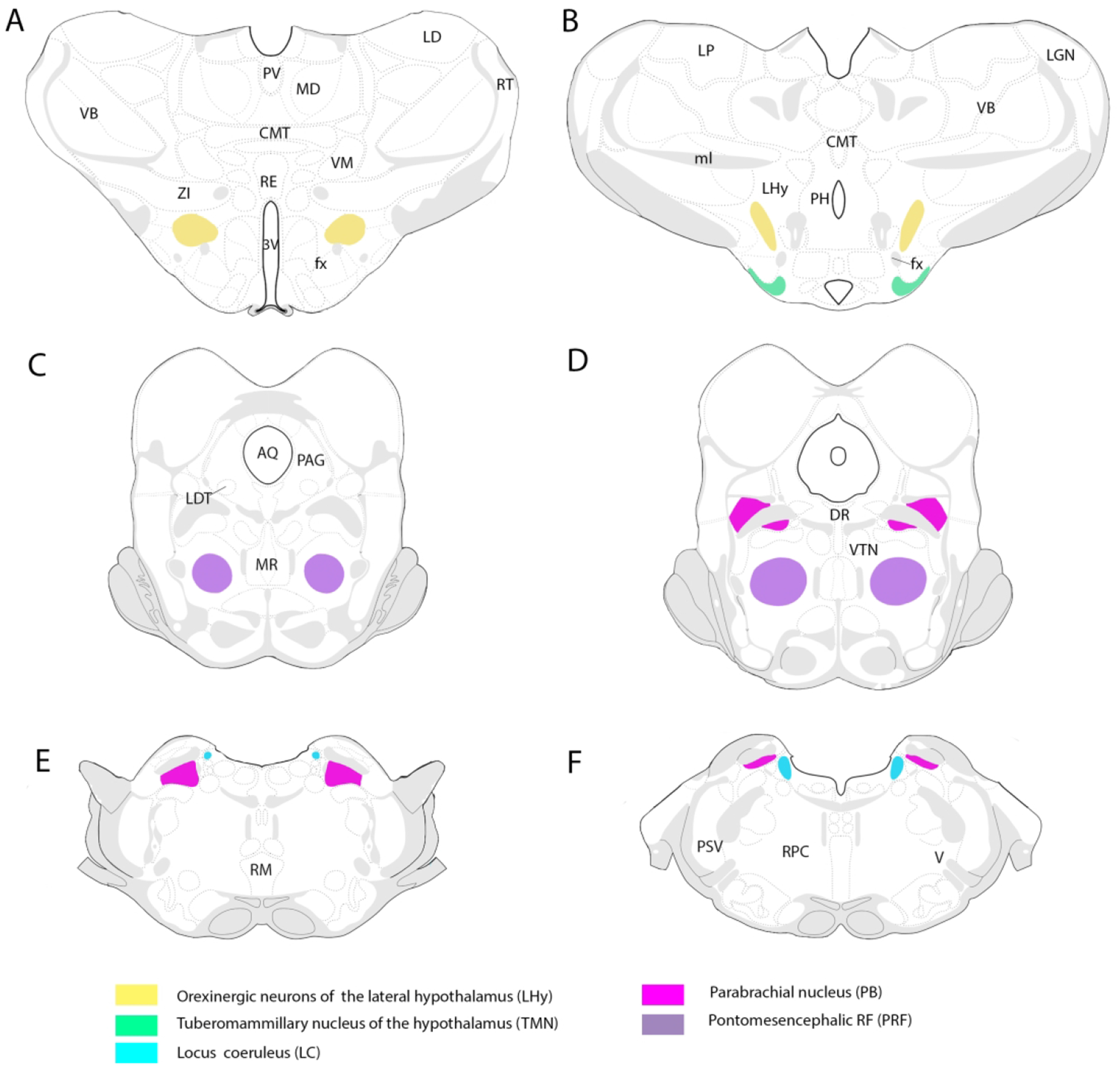
Locations of waking/arousal-related cell groups of the brainstem and caudal diencephalon at which general anesthetics (GAs) exert anesthetic actions. They include the pontomesencephalic reticular formation (PRF), locus coeruleus (LC), parabrachial nucleus (PB), tuberomammillary nucleus (TMN) and orexin-containing cells of the lateral hypothalamus (LHy). Abbreviations: 3V, third ventricle; AQ, cerebral aqueduct; CMT, central medial thalamic nucleus; DR, dorsal raphe nucleus; fx, fornix; LD, laterodorsal nucleus of the thalamus; LDT, laterodorsal tegmental nucleus; LGN, lateral geniculate nucleus of the thalamus; LP, lateral posterior nucleus of thalamus; MD, mediodorsal nucleus of thalamus; ml, medial lemniscus; MR, median raphe nucleus; PAG, periaqueductal gray; PH, posterior hypothalamus; PSV, principal sensory nucleus of trigeminal nerve; PV, paraventricular nucleus of thalamus; RE, nucleus reuniens of thalamus; RF, reticular formation; RM, raphe magnus, RPC, reticularis pontis caudalis; RT, reticular nucleus of thalamus; V, motor nucleus of trigeminal nerve; VB, ventrobasal complex of thalamus; VM, ventromedial nucleus of thalamus; VTN, ventral tegmental nucleus; ZI, zona incerta.
In a series of studies, Devor and colleagues (Sukhotinksy et al., 2016; Devor et al., 2016; Minert and Devor, 2016; Minert et al., 2017) identified a region of the pontomesencephalic RF, termed the mesopontine tegmental anesthesia area (MPTA), which when suppressed produces an anesthetic-like state. Specifically, microinjections of the GABAA agonist, muscimol, or the anesthetics, sodium pentobarbital or propofol, into the MPTA, but not into adjacent RF areas, rapidly induced an anesthetized state in rats (Devor et al., 2016; Minert et al., 2017). In addition, rats with MPTA lesions were resistant to the administration of these anesthetics (Minert and Devor, 2016).
The locus coeruleus (LC) serves a well-recognized role in arousal (Aston-Jones et al., 2001; Carter et al., 2010; Sara and Bouret, 2012), and hence is a likely target for anesthetics. In this regard, Vazey and Aston-Jones (2014) reported that the selective DREADD-induced activation of noradrenergic LC cells (NE-LC) produced a shift in cortical EEG activity from low (delta) to higher frequencies during maintained isoflurane anesthesia and accelerated emergence from this anesthetic. The effect was blocked by NE antagonists, and when given alone antagonists prolonged the duration of anesthesia. While the effects of GA at LC likely involve multiple LC targets, a critical one appears to be the central medial nucleus (CMT) of thalamus. Specifically, Fu et al. (2017) showed that microinjections of NE into the CMT significantly shortened the time of emergence from propofol anesthesia, and NE reversed a propofol-induced suppression of CMT activity in the slice.
The parabrachial nucleus (PB), of the dorsolateral pons, is an important site in the arousal circuitry (Fuller et al., 2011; Kaur et al., 2013; Qiu et al., 2016). Partial PB lesions markedly reduce wakefulness, whereas near-total parabrachial destruction produces a coma-like state (Fuller et al., 2011). Regarding PB’s role in anesthesia, Muindi et al. (2016) described enhanced levels of c-fos expression in PB during the (passive) emergence from isoflurane anesthesia, and further reported that PB stimulation produced arousal and return of the righting reflex during continuous isoflurane administration. Consistent with this, Luo et al. (2018) demonstrated that PB activity was suppressed during propofol or isoflurane anesthesia and strongly enhanced with emergence from anesthesia. Further, chemogenetic PB activation significantly shortened the recovery time from these GAs (Luo et al., 2018).
Two hypothalamic nuclei prominently contributing to arousal/wakefulness are the histaminergic tuberomammillary nucleus (TMN) and the ORX cell group of the lateral hypothalamus (Saper and Fuller, 2017; Scammell et al., 2017), With respect to TMN and GA, Nelson et al. (2002) described marked reductions in c-fos expression in the TMN to the anesthetics, pentobarbital or propofol (which act via GABAA receptors), and further showed that injections of GABAA antagonists into the TMN attenuated the hypnotic response to these anesthetics. In a similar manner, Luo and Leung (2011) reported that intracerebral injections of histaminergic antagonists or selective lesions of histaminergic TMN neurons prolonged the emergence from the GA, isoflurane.
Orexin serves a well-defined role in arousal (Brown et al., 2012; Jones, 2011; Saper and Fuller, 2017; Scammell et al., 2017) and accordingly several reports have described the effects of anesthetics on the orexin system (Kelz et al., 2008; Shirasaka et al., 2011; Zhang et al., 2012; 2016; Zhou et al., 2018). In an initial study, Kelz et al. (2008) found that isoflurane or sevoflurane suppressed c-fos expression of ORX cells, and that ORX-1 receptor antagonists, or the genetic ablation of ORX neurons, delayed the emergence from these anesthetics. More recently, Zhang et al. (2016) similarly showed that ORX-1 receptor antagonists prolonged the emergence from isoflurane anesthesia, and that administration of orexin-A (acting on ORX-1 receptors) facilitated recovery from anesthesia. Finally, Zhou et al. (2018) demonstrated that selective chemogenetic activation of ORX cells in orexin-Cre transgenic mice significantly shortened the time of emergence from isoflurane anesthesia.
In summary, GAs act at major hubs of the arousal/wakefulness system to dampen activity to induce/prolong anesthesia, or reversing GA effects hastens the emergence from anesthesia. This indicates that GAs act through an endogenous wakefulness system.
General anesthetics act on sleep/wake control systems: the NREM sleep network
As briefly discussed, switching between sleep and wake states involves reciprocal inhibitory actions of wake control systems of the brainstem/diencephalon and sleep control networks, mainly of the ventrolateral preoptic area (VLPO) and the median preoptic area (MnPO) (Fig. 2) of the ventrobasal forebrain (Saper et al., 2010; Brown et al., 2012; Saper and Fuller, 2017).
Figure 2.
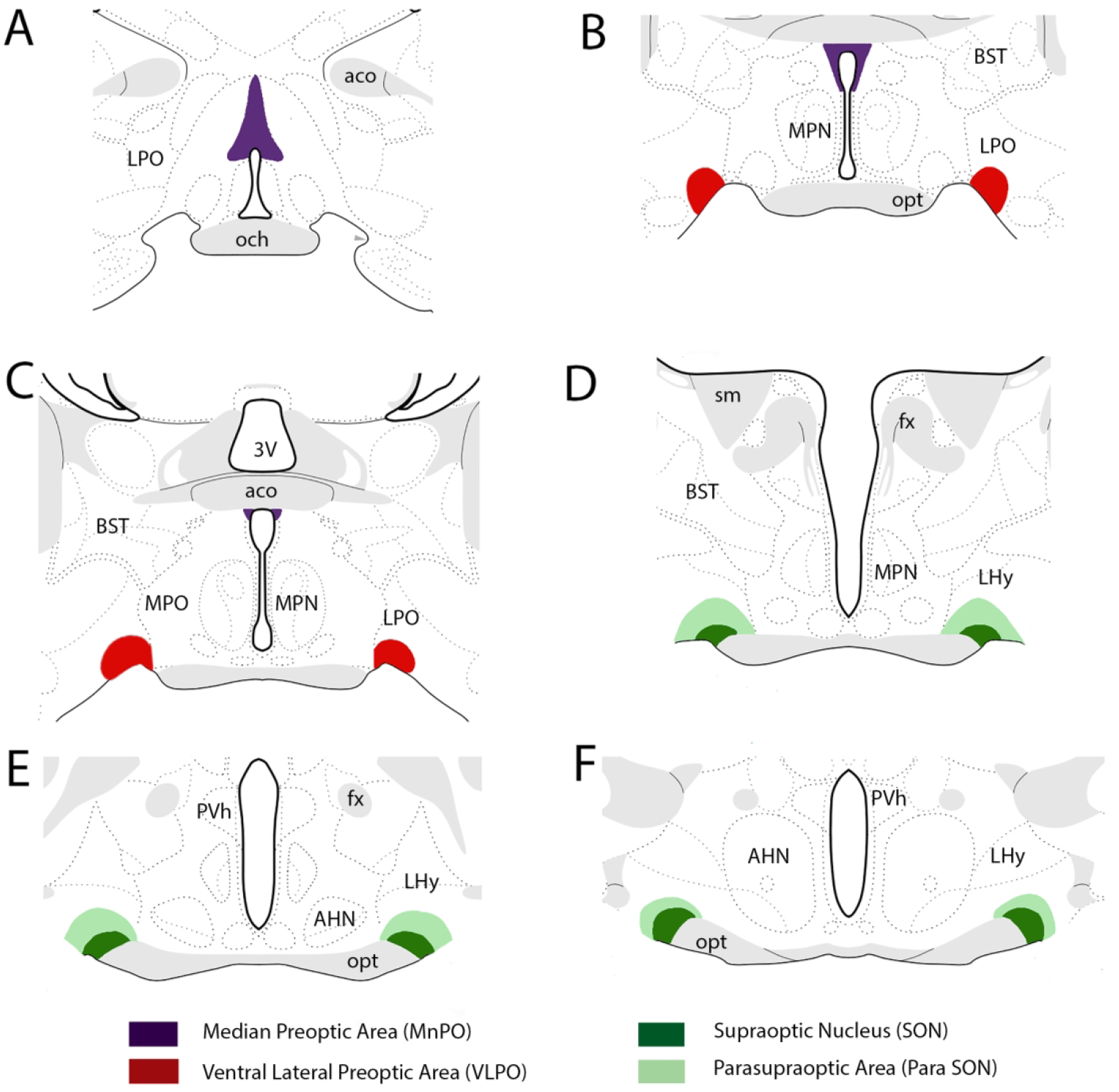
Locations of NREM sleep “centers” of the basal forebrain at which general anesthetics (GAs) exert anesthetic actions. They include the ventral lateral preoptic area (VLPO), the median preoptic area (MnPO), the supraoptic nucleus (SON) and the para-SON area. Abbreviations: 3V, third ventricle; aco, anterior commissure; AHN, anterior hypothalamus; BST, bed nucleus of stria terminalis; fx, fornix; LHy, lateral hypothalamus; LPO, lateral preoptic area; MPN, medial preoptic nucleus; MPO; medial preoptic area; och, optic chiasm; opt, optic tract; PVh, paraventricular nucleus of hypothalamus; sm, stria medullaris.
In an initial report, Tung et al. (2001) showed that injections of propofol into the medial basal forebrain of rats, shortened sleep onset latencies and significantly increased NREM and total sleep times. Supporting this, several studies have shown that various anesthetics activate sleep-inducing VLPO neurons. For instance, Moore et al. (2012) described enhanced levels of c-fos expression in VLPO of mice to the administration of the GAs, isoflurane or halothane, and further showed that isoflurane greatly increased the activity of sleep-active VLPO neurons, whereas non-sleep related VLPO cells were unaffected. In follow up study, Han et al. (2014) described increases in c-fos expression to isoflurane or halothane in VLPO, and in the MnPO, but no changes in either nucleus to the “nonimmobolizer”, 1.2, dichlorohexafluorocylobutane (F6), with anesthetic-like qualities. Additionally, as the sleep-active VLPO neurons were GABAergic cells, isoflurane, but not F6, enhanced c-fos expression of GABAergic VLPO neurons in the slice preparation.
Sleep-active VLPO cells contain various adrenergic receptors and are inhibited by NE (Matsuo et al., 2003; Kumar et al., 2006). Examining the role of NE on the GA modulation of VLPO neurons, McCarren et al. (2014) showed that microinjections of the α2-adrenergic agonist, dexmedetomidine, into VLPO aroused isoflurane-anesthetized mice and reversed isoflurane-elicited depolarization of VLPO neurons in the slice. As VLPO and NE-LC reciprocally interact, Zhang et al. (2015) reported that rats with VLPO lesions were much less sensitive to propofol anesthesia and that VLPO lesions reversed the inhibitory actions of propofol on LC activity.
Finally, a recent report by Jiang-Xie et al. (2019) described a common substrate for GAs and (NREM) sleep in the ventral preoptic area of the hypothalamus (Fig. 2). Specifically, they identified a unique population of cells of the preoptic area of behaving mice which showed enhanced c-fos expression to the GA isoflurane, and further demonstrated that a subset of these cells (termed anesthesia-activated neurons, AAN) discharged at significantly increased rates of activity to isoflurane – with the onset and offset of firing leading to the loss and return to consciousness. AANs were localized to a region in and around the supraoptic nucleus (SON), just caudal to VLPO, identified as the SON and para-SON region (Fig. 2). Interestingly, AAN cells were activated by all anesthetics examined including isoflurane, propofol, ketamine and dexmedetomidine. Regarding the effects of AANs on sleep, it was shown that: (1) the DREADD-induced activation of AAN cells strongly enhanced NREM sleep; (2) brief optogenetic stimulation of AANs similarly elicited sleep – with effects lasting 10 min post-stimulation; and (3) the selective ablation of AANs profoundly disrupted natural NREM sleep. The authors concluded that the findings “identify a common neural substrate at the intersection of sleep and GA”.
In summary, in addition to direct anesthetic actions on the arousal/wakefulness circuitry, GAs also act through a sleep promoting network of the preoptic area including VLPO, MnPO and the recently identified SON and para-SON regions to induce anesthesia.
Cortical EEG activity in NREM sleep
If, as indicated, GAs act through the sleep/wake circuitry, the effects of GAs on cortical EEG activity should be similar to that observed with natural sleep. As well recognized, the progression from waking to (deep) sleep (stages 3/4) in humans involves an orderly shift from low voltage, high frequency (LVHF) cortical EEG activity (beta/gamma) to low frequency, high amplitude activity (delta) (Pace-Schott, 2009; Mendelson, 2017). Specifically, the wake/NREM sleep cycle consists of the following: waking, LVHF activity in the beta/gamma range (15–100 Hz); stage 1 (NREM), low voltage activity, mixed with higher amplitude activity in the theta and alpha range; stage 2 (NREM), background of mixed frequencies (comparable to stage 1) with superimposed K complexes and sleep spindles (7 −14 Hz); stages 3 and 4 (slow wave sleep, SWS), low frequency, high amplitude activity in the delta range (1–4 Hz), with delta waves comprising approximately 20% of the record in stage 3 and greater than 50% in stage 4 sleep. Stages 3 and 4 are thus termed delta sleep (Pace-Schott, 2009; Mendelson, 2017). The progressive decreases in frequency and increases in amplitude of the cortical EEG from stages 1–4 of NREM sleep reflects the descent into deeper stages of sleep. Unlike for humans, the sleep of rodents (and most non-primate species) generally consists of a single NREM period (rather than discrete stages) characterized by continuous low frequency, high amplitude (largely delta) activity of the cortical EEG (Brown et al., 2012).
Cortical EEG activity in NREM sleep and GA are very similar, including progressive frequency/amplitude changes in the descent from lighter to deeper states of anesthesia. The popular notion that anesthetics “puts one to sleep” rings true.
Cortical EEG activity in general anesthesia in rodents
Guidera et al. (2017) recently examined the effects of the anesthetic, sevoflurane, on the cortical EEG and behavior in rats, and showed that the transition to the anesthetic state, determined by the loss of movement (LOM) followed by loss of the righting reflex (LORR), was accompanied by abrupt changes in cortical EEG activity from beta/low gamma oscillations (12–40 Hz) to slow delta activity (0.1–4.0 Hz). Specifically, they recorded EEG activity and local field potentials (LFPs) from the frontal (PFC) and parietal (PC) cortices and the central thalamus (CT) and demonstrated: (1) a sharp decrease in beta/low gamma coherence between the CT and the PFC with the LOM and (2) significant increases in delta power and delta coherence for all sites (CT, PFC, PC) with the LOM, which became more marked (delta power/coherence) with the LORR.
Flores et al. (2017) described a similar sequence of thalamocortical EEG changes with the induction and recovery from the GA, propofol, in rats. During the initial stages of induction of anesthesia, the cortical EEG was dominated by beta oscillations, which switched to alpha with LORR and then to delta frequencies with the loss of movement (LOM). Thalamic and cortical oscillations were highly coherent in the alpha and delta range with LORR and the LOM, respectively. The sequence of events differed with the emergence from GA. In contrast to the relatively slow stepwise progression from beta > alpha > delta oscillations with induction, there was a relatively sharp transition from delta/alpha activity to beta oscillations with emergence from GA -- with an associated loss of thalamic and cortical coherence at low (delta) frequencies.
In one of the few such reports, Baker et al. (2014) compared patterns of thalamocortical EEG activity under general anesthesia (with dexmedetomidine or propofol) and sleep in the same group of rats. Specifically, they recorded LFPs and EEG activity from the central medial (CMT) and ventrobasal (VB) nuclei of the thalamus and their cortical targets, the anterior cingulate (AC) and barrel cortices, respectively, and described very similar changes in pairs of the thalamocortical structures in the transition to GA or to NREM sleep. In brief, during the initial stages of GA or sleep, high frequency activity (of waking) switched to a pattern of low frequency/high amplitude oscillations in all structures, with CMT leading the other sites. When these states (GA or sleep) became fully established, marked by LORR for anesthesia, a strong phase coupling at delta frequencies developed between the two thalamic sites and their respective cortical regions, with the thalamic nuclei leading the cortical structures by 90° for both conditions.
In summary, the progression to GA or to NREM sleep in the rat, with an associated LOC, involves a shift from beta/low gamma activity in waking to progressively slower oscillations with deepening states of GA or NREM sleep: alpha > theta > delta. This indicates that GA and NREM sleep are similar states of unconsciousness.
Cortical EEG activity in general anesthesia in humans
In early reports, John and colleagues (Gugino et al., 2001; John et al., 2001) described changes in the cortical EEG following the induction and subsequent LOC to the GAs, sevoflurane or propofol. Specifically, with induction, there was a decrease in alpha activity in posterior (or occipital) regions of the cortex together with a gradual shift from alpha/beta to theta activity in frontal cortices. With the LOC, theta and particularly delta waves predominated in the frontal cortex with subsequent spread to posterior regions of the cortex.
In a series of studies, Emery Brown and colleagues comprehensively examined the effects of major classes of anesthetics on patterns of cortical EEG activity in humans (Purdon et al., 2013, 2015; Akeju et al., 2014a,b; Akeju et al., 2016). With respect to the GA, propofol, Purdon et al. (2013) reported that 10–30 sec following iv injections of propofol, the cortical EEG changed from a (waking) pattern of beta/low gamma activity to slow delta oscillations (0.1 – 5 Hz) which increased in power during approximately the first 5 min post-injection. The changes in patterns of cortical EEG activity to propofol (and associated LOC) were very striking, for they noted “the slow-delta oscillation amplitudes can be 5 to 20 times larger than the gamma and beta oscillations” of wakefulness. With the gradual recovery from propofol, the delta oscillations gave way to a mixed pattern of delta and alpha activity – comparable to stage 2 sleep.
The spectral EEG characteristics to anesthesia induced with the major inhalation GAs (sevoflurane, isoflurane, desflurane) are similar to those with propofol, with some notable differences (Akeju et al., 2014b; Purdon et al., 2015). Principally, alpha activity is present during the initial stages of induction with the volatile GAs, but as levels deepen, the EEG becomes a mixture of delta, theta (~ 5 Hz) and alpha oscillations of generally equal power, thus spanning an approximate 1–15 Hz frequency range. The presence of generally higher frequency oscillations with hypnotic levels of the volatile anesthetics, compared to delta with propofol, would suggest a lighter anesthetic state with these GAs – or comparable to (or bridging) stages 2 and 3 of NREM sleep.
In one of the few comparisons of the cortical EEG during anesthesia and sleep in humans, Murphy et al. (2011) reported that patterns of cortical EEG activity with the LOC were essentially the same in GA or NREM sleep. Specifically, propofol anesthesia produced a shift in the frontal cortical EEG from alpha/low beta (12–25 Hz) activity to delta oscillations – which were virtually indistinguishable from the delta oscillations of deep NREM sleep. Accordingly, the authors concluded: “we observed no clear differences between the slow waves observed during sleep and those recorded during propofol LOC.” In summary, the equivalent patterns of cortical EEG activity in humans in general anesthesia and NREM sleep further attests to the marked similarity of these unconscious states.
Interactions between sleep and anesthesia
If, as argued, anesthetics work through the sleep/wake network, GAs and sleep should mutually influence each other – or at least partially, substitute for each other. In fact, this was demonstrated by Tung, Mendelson and associates in a series of reports (for review, Tung and Mendelson, 2004). In the first of the series (Tung et al., 2001), propofol was continuously administered to rats for 12 hours during the light phase of the sleep/wake cycle (normal rest phase) and rats were then allowed to naturally sleep. Importantly, they reported decreases in NREM sleep (and the power of delta) for the 4-hour period following the termination of anesthesia – rather than an expected rebound increase in NREM sleep if rats were in fact sleep deprived during the 12-hour period of anesthesia. In effect, the anesthesia substituted for NREM sleep, or according to the authors, anesthesia “was a restorative process reversing the natural accumulation of sleep need that occurs during wakefulness” (Tung et al., 2001). As anesthesia can affect sleep, sleep can also influence anesthesia. Specifically, Tung et al. (2002) showed that 24 hours of sleep deprivation (SD) in rats significantly shortened the induction time to isoflurane and propofol anesthesia, indicating that sleep pressure enhances the effects of anesthetics. Finally, Tung et al. (2004) reported that the recovery period (or rebound) from 24 hours of sleep deprivation was the same for rats allowed to sleep or given propofol anesthesia (for 6 hours post SD), showing that anesthesia, like sleep, serves a restorative function -- or the two processes are interchangeable. In summary, the findings that sleep and GA can substitute for each other further attests to the general equivalency of these two states.
The cortex/brain is in a very depressed state in NREM sleep
Several human imaging studies have shown that the cortex/brain is “deactivated” in NREM sleep -- or activity is severely reduced from levels of wakefulness. For instance, a number of early reports, using PET, described global decreases in glucose metabolism or cerebral blood flow (CBF) ranging from 25–40% in NREM sleep compared to wakefulness (Buchsbaum et al., 1989; Maquet et al., 1990; Madsen et al., 1991a). In a comprehensive PET examination of regional differences in CBF in sleep, Braun et al. (1997) found major changes in CBF in NREM and REM sleep from waking in structures throughout the brain including the brainstem, basal forebrain, thalamus, amygdala, hippocampus/parahippocampus and neocortex. Activity (CBF) decreased in most structures in NREM sleep from waking, and with some notable exceptions (see below) increased in REM to equivalent or greater levels than in waking. Specifically, activity was depressed virtually throughout the cortex in NREM, most strongly in the parahippocampal, parietal and frontal cortices. CBF, however, was not reduced in NREM in primary and secondary sensory cortices – or unimodal (non-associative) sensory cortices. It was suggested that a minimal (“self-generating’) level of activity is maintained in these sensory areas in NREM to prepare (or prime) for sudden arousal in sleep, but these (unimodal) sensory regions remain disconnected from higher order association cortices in sleep to which they are functionally coupled in waking.
Braun et al. (1997) drew particular attention to their findings that regions of the cortex which are strongly deactivated in NREM perform the highest order cognitive functions – with the clear implication that these functions are lost in NREM sleep. They stated: “During normal awake consciousness, the prefrontal cortices perform the highest order processing of neural information, integrating sensory, cognitive and limbic information, organizing meaningful, temporally sequenced behavioral responses and subserving working memory. The inferior parietal cortices are involved in cross-modal association of perceptual material necessary for higher order cortical activities such as language. The onset of non-REM sleep was associated with dramatic and specific deactivation of these regions.”
Several subsequent imaging studies have similarly demonstrated a pronounced deactivation of the cortex, particularly frontal cortical areas, in NREM sleep. For example, in a H215O PET study, Hofle et al. (1997) found that activity was very depressed in the orbitofrontal and cingulate cortices in NREM sleep but increased in primary/secondary visual and auditory cortices – which they attributed to dream-like mentation in NREM sleep. Kajimura et al. (1999) compared CBF in light and deep NREM sleep and showed that CBF was reduced in frontal and parietal cortices in light NREM, and bilaterally throughout the cortex in deep NREM sleep – except for primary visual and somatosensory cortices. The decreased activity in light NREM sleep led the authors to conclude that: “deactivation of the high-order association cortices, and selective deactivation of the language areas may take place during the early stage of NREM sleep”.
Consistent with the foregoing, Maquet and colleagues (Maquet et al., 1997. Maquet, 2000) reported that CBF was strongly reduced in NREM in the orbitofrontal, anterior cingulate and precuneus regions of the cortex, with largest changes in the orbitofrontal cortex, but they (nonetheless) observed that NREM sleep “profoundly affects the activity of the whole cortex, not only in areas in which CBF is lowest” (Maquet, 2000).
A few recent reports have described changes in activity during sleep using fMRI -- blood oxygen level-dependent (BOLD) changes (Czisch et al., 2004; Kaufman et al., 2006; Tushaus et al., 2017). In accord with previous studies, Kaufmann et al. (2006) demonstrated a specific sequence of changes (BOLD) in transitions to stage 1 (S1), stage 2 (S2) and deep NREM sleep. In short, activity was significantly reduced in the posterior cingulate, right insular and occipital cortices in S1, additionally in the frontal, parietal and temporal cortices in S2, and finally, in the anterior cingulate, left insular and hippocampal regions in deep NREM sleep. Tushaus et al. (2017) similarly reported that as sleep deepened, progressively more structures showed reduced activity (BOLD) in NREM. The cortical regions exhibiting the largest decreases in activity in NREM were the prefrontal, cingulate and precuneus cortices.
In summary, the foregoing shows that: (1) the activity of virtually the entire cortex, including the hippocampus, is suppressed in NREM sleep; (2) the deactivation of the cortex begins in light NREM (stages 1 and 2) and becomes progressively stronger (more structures, greater suppression) with increasing depths of sleep; and (3) the cortical regions most profoundly deactivated in NREM are the prefrontal/frontal, parietal, cingulate and precuneus cortices – or those responsible for higher order cognitive processing.
A plethora of experimental studies in animals and humans have shown that disruption of activity of the cortex, particularly the orbitofrontal, parietal or cingulate cortices, severely impairs higher order cognitive and executive functions (for review, Goldman-Rakic, 1995; Dalley et al., 2004; Barbas and Garcia-Cabezas, 2017; Eichenbaum, 2017). The global deactivation of the cortex in NREM sleep would be expected to produce an equivalent, or undoubtedly a much greater, disruptive effect on cognitive, mnemonic and executive functions.
Higher order regions of the cortex are depressed in REM sleep
Fewer reports have examined metabolic activity/CBF in REM sleep than in NREM sleep (Maquet et al., 1990; Madsen et al., 1991a,b; Maquet et al., 1996; Braun et al., 1997; Maquet, 2000). Early reports described widespread increases in activity in REM from NREM sleep, generally equal to levels of wakefulness (Maquet et al., 1990; Madsen et al., 1991a,b). Examining regional differences in CBF across the sleep/wake cycle, Braun et al. (1997) similarly reported that most areas of the cortex showed “rebound” increases in activity in REM from depressed levels of NREM sleep (see above). Importantly, however, activity over a relatively large area of the frontal and parietal cortex remained depressed in REM sleep; that is, to levels of NREM sleep. With respect to this atypical pattern of cortical activity in REM, Braun (1997) commented that the cortex is generally “active” in REM sleep “with the specific exclusion of areas which normally participate in the highest order analysis and integration of neural information”.
In like manner, Maquet and colleagues (Maquet et al., 1996; Maquet, 2000) described increases in CBF in REM sleep from NREM in the pontine tegmentum, thalamus, the amygdala, and the anterior cingulate and right parietal cortices, but “striking” reductions in activity in REM over an extensive area of the cortex which included the right dorsolateral PFC (Brodmann’s areas, 8–11 and 48), the left dorsolateral PFC (Brodmann’s areas, 10, 11, 46, 47), the right and left parietal cortex, the precuneus and posterior cingulate cortex. Addressing the functional significance of this pattern of cortical deactivation in REM, Maquet et al. (1996) stated that: “Dorsolateral prefrontal areas and precuneus have been shown to participate in the encoding and retrieval of episodic memory, whereas the inferior parietal lobules (and prefrontal areas) participate in working memory. Our results suggest that these memory processes are not prominent during REM sleep”.
In summary, the pronounced suppression in REM sleep of several cortical regions serving higher order cognitive and executive functions would indicate that higher order functions are lost in REM sleep -- as they are in NREM sleep.
Disruption of functional connectivity (FC) of the brain/cortex with general anesthetics
General anesthetics have been shown to produce global reductions in activity of the brain ranging from 30 to 65%, with large decreases in CBF/glucose metabolism in frontal, parietal and cingulate cortices (Alkire et al., 1995, 1997; Vesalis et al., 1997; Fiset et al., 1999). Further, GAs also significantly disrupt communication between structures (or networks) of the brain – or their functional connectivity (FC) (for review, Heine et al., 2012; Hudetz, 2012).
Using fMRI, Boveroux et al. (2010) initially reported that propofol significantly reduced functional connectivity (FC) within and between two well-characterized frontoparietal resting state networks (RSNs): the default mode network (DMN) and the executive control network (ECN) (Greicius et al., 2003; Fox et al., 2005; Seeley et al., 2007). By contrast, connectivity was preserved in (lower order) visual and auditory systems. Schrouff et al. (2011) similarly demonstrated a propofol-induced disruption of the DMN as well as several additional networks including motor, attentional and salience networks, with connections between frontal-parietal and parietal-temporal areas most severely affected. In like manner, Schroter et al. (2012) described “brain wide” reductions in spatiotemporal (functional) interactions to propofol, with marked disruptions of long range corticocortical connections. Finally, Guldenmund et al. (2016) confirmed widespread decreases of FC with propofol, showing reductions in connectivity for eight RSNs – with each containing a frontal cortical component.
Similar to propofol, the volatile anesthetic, sevoflurane, also significantly alters cortical functional connections (Deshpande et al. 2010; Martuzzi et al. 2010; Ku et al., 2011; Huang et al. 2014; Palanca et al. 2015; Ranft et al., 2016). In initial fMRI studies, Deshpande et al. (2010) reported that sevoflurane disrupted the entire DMN network, while Martuzzi et al. (2010) showed that “higher order” systems were particularly vulnerable to sevoflurane, especially memory circuits. More recently, Palanca et al. (2015) described cortical-wide decreases in activity (BOLD) with sevoflurane, particularly for structures of the DMN and ventral attention (VAN) networks. For instance, they reported a breakdown in FC along the anterior/posterior axes of the DMN and VAN networks -- and between the thalamus and these networks. Regarding functional implications, Palanca et al. (2015) noted that disruptions of cortical FC involves (or underlies) all forms of altered consciousness stating that: “disorders of consciousness, absence epilepsy, slow wave sleep, and propofol and sevoflurane anesthesia are all associated with reduced correlation between the posterior (posterior cingulate/precuneus cortex, PCC) and anterior (medial prefrontal cortex) components of the DMN”.
In summary, GAs significantly disrupt functional connections of several cortical networks, particularly higher order resting state networks. Associated with the loss of FC, is a corresponding loss of functions normally performed by these higher order networks.
Disruption of functional connectivity (FC) of the cortex during non-REM sleep
Massimini et al. (2005) were among the first to describe a breakdown in cortical FC in sleep, showing that the transcranial magnetic stimulation (TMS)-induced propagation of activity from the premotor cortex to distant cortical sites was disrupted in NREM sleep but not in waking -- or effects remained local in NREM sleep. Using fMRI, Horovitz et al. (2009) reported that FC with the DMN was significantly altered in NREM; specifically, anterior (PFC) and posterior (posterior cingulate/precuneus) regions of the DMN were strongly decoupled in NREM sleep. Spoormaker and colleagues (Spoormaker et al. 2010, 2012; Samann et al., 2011) described changes in FC in light and deep NREM sleep, showing initial sharp reductions in thalamocortical FC at the onset and early stages of NREM sleep and a marked breakdown of frontoparietal connectivity in deep NREM sleep. The frontoparietal disconnection was depicted as a complete disintegration of frontoparietal networks in NREM which “are associated with some of the most complex cognitive functions in humans” (Spoormaker et al., 2012).
More recently, Picchioni et al. (2014) reported that the central median nucleus of thalamus becomes functionally disconnected from several higher order cortical regions in NREM sleep including the medial frontal gyrus, PCC/precuneus and parahippocampal gyrus. This was characterized as a functional thalamic deafferentation of the cortex in NREM, and likened to that occurring in general anesthesia and coma. Finally, Jobst et al. (2017) described a large-scale breakdown in the functional (or effective) connectivity of the brain in NREM sleep, showing, in effect, that communication between regions of the brain becomes “constrained” (or local) in NREM sleep – as opposed to cross-regional in waking states.
In summary, like GAs, the FC among major cortical networks, particularly higher order structures/networks, are significantly disrupted in NREM sleep. The loss of FC of higher order networks in NREM would indicate a loss of higher order functions in NREM sleep.
Comparisons of sleep and GA states in rodents and humans
As was pointed out, NREM sleep for rodents essentially consists of a single stage, whereas that for humans it consists of four stages (1–4) -- with stages 3/4 termed slow wave sleep (SWS). Despite this difference, the deep stages of NREM sleep for both rodents and humans are characterized by delta activity (1–4 Hz) of the cortical EEG. GAs similarly produce delta EEG activity EEG in both species. The virtually identical patterns of cortical EEG activity to GAs and NREM sleep for rodents and humans suggests that these unconscious states (with GAs or in NREM sleep) are essentially the same for both species.
We described the neural circuitry controlling sleep-wake states and showed that GAs exert their effects through modulating endogenous sleep-waking control systems (Figs. 1,2). Whereas understandably research on the neural substrates for sleep-wake control has predominantly been done on lower animals (mainly rodents), it was clear from even the early (and oft cited) work of von Economo (1930) with encephalitis patients that the human brain contains wake and sleep “centers”, located in the caudal hypothalamus and basal forebrain, respectively. It has further been reported that GAs act on sleep-wake “centers” to induce anesthesia in humans. For instance, Zhang et al. (2010), using fMRI in humans, described marked decreases in activity in the posterior hypothalamus to the GA propofol.
We reviewed several reports in humans, using imaging techniques, that described: (1) general reductions in activity in higher order cortical structures of the (human) brain in NREM sleep and to GAs; and (2) the loss of functional connectivity (FC) among forebrain networks in NREM sleep and GA. Considerably fewer reports have examined the functional connectivity of forebrain networks in rodents and exceedingly few have compared the effects of GAs on these networks in rodents with reference to sleep/wake states. However, in one such study. Paasonen et al. (2018) showed that several GAs disrupted the FC of frontal regions of the DMN network and commented that “as non-rapid-eye-movement sleep and anesthesia share many similarities in neurophysiology and brain activity, our observations suggest similarities in DMN modulation”.
Conclusion and implications
The present report addresses fundamental issues which cast doubt on the involvement of sleep in cognitive processing, mainly: (1) the lack of transfer of waking experiences to sleep in humans; and (2) the suppression of cognitive processing in the parallel unconscious states of general anesthesia (GA) and NREM sleep.
Point 1:
If waking experiences were to be processed and consolidated in sleep it would be expected that waking experiences/events would be faithfully represented in sleep – but they are not. As well recognized, the verbal reports of the cognitive contents of consciousness of sleep (dreams), with spontaneous or forced awakening from sleep, bear no resemblance to waking experiences. This lack of transfer of waking experiences to sleep would seem to negate any possibility of waking experiences being consolidated in sleep, since these experiences are not represented/reproduced in sleep – to be acted upon. It could be argued that waking events could be transferred unconsciously from waking to sleep and processed unconsciously in sleep -- or without surfacing to consciousness, as in dreams. While this is conceivable, it seems highly unlikely that waking experiences which never reach consciousness in sleep would be faithfully (or exactly) reproduced in sleep when material that reaches consciousness in sleep (and can be verbalized) is completely unrepresentative of waking experiences.
Point 2:
Aside from the fact that sleep is more readily reversible than general anesthetics, the unconscious states of sleep and GA are very similar. As depicted in Table 1, NREM sleep and general anesthesia (GA) exhibit the following common characteristics: sensory blockade, immobility, amnesia, and lack of awareness (unconsciousness). In addition, GAs exert their effects by suppressing “wake control” systems of the brainstem/caudal diencephalon (Fig. 1) and by activating “sleep control” systems of the medial basal forebrain (Fig. 2). In effect, NREM sleep and GA share common neural substrates. Further, it has been shown that: (1) widespread areas of the cortex, primarily the prefrontal/frontal, parietal, cingulate and precuneus cortices, are severely depressed in NREM sleep and GA; and (2) the functional connectivity (FC) within and across major cortical networks, including the default mode network (DMN), executive control network (ECN) and ventral attention network (VAN), are profoundly disrupted in NREM sleep and GA. This would indicate that these “higher order” cortical regions are non-functional (or offline) in both NREM sleep and GA. As has been thoroughly demonstrated in experimental studies in animal and humans, the disruption of activity of these same cortical regions severely impairs mnemonic, cognitive and executive functions. Whereas few would argue that memory and cognitive functions are blocked in the unconscious state of general anesthesia, we would argue that the same is true for the unconscious state of NREM sleep. To conclude, we contend that there is no cognitive processing in the unconscious, anesthetic-like, state of NREM sleep.
Acknowledgements:
This was supported by NIH grant: NS108259.
References
- Akeju O, Pavone KJ, Westover MB, Vazquez R, Prerau MJ, Harrell PG, Hartnack KE, Rhee J, Sampson AL, Habeeb K, Gao L, Pierce ET, Walsh JL, Brown EN, & Purdon PL (2014a). A comparison of propofol- and dexmedetomidine-induced electroencephalogram dynamics using spectral and coherence analysis. Anesthesiology, 121, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeju O, Westover MB, Pavone KJ, Sampson AL, Hartnack KE, Brown EN, & Purdon PL (2014b). Effects of sevoflurane and propofol on frontal electroencephalogram power and coherence. Anesthesiology, 121, 990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeju O, Kim SE, Vazquez R, Rhee J, Pavone KJ, Hobbs LE, Purdon PL, & Brown EN (2016). Spatiotemporal Dynamics of Dexmedetomidine-Induced Electroencephalogram Oscillations. Public Library of Science One 11, e0163431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkire MT, & Gorski LA (2004). Relative amnesic potency of five inhalational anesthetics follows the Meyer-Overton rule. Anesthesiology, 101, 417–429. [DOI] [PubMed] [Google Scholar]
- Alkire MT, Haier RJ, Barker SJ, Shah NK, Wu JC, & Kao YJ (1995). Cerebral metabolism during propofol anesthesia in humans studied with positron emission tomography. Anesthesiology, 82, 393–403. [DOI] [PubMed] [Google Scholar]
- Alkire MT, Haier RJ, Shah NK, & Anderson CT (1997). Positron emission tomography study of regional cerebral metabolism in humans during isoflurane anesthesia. Anesthesiology, 86, 549–557. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chen S, Zhu Y, & Oshinsky ML (2001). A neural circuit for circadian regulation of arousal. Nature Neuroscience, 4, 732–738. [DOI] [PubMed] [Google Scholar]
- Baker R, Gent TC, Yang Q, Parker S, Vyssotski AL, Wisden W, Brickley SG, & Franks NP (2014). Altered activity in the central medial thalamus precedes changes in the neocortex during transitions into both sleep and propofol anesthesia. Journal of Neuroscience, 34, 13326–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbas H, & García-Cabezas MÁ (2017). Prefrontal Cortex Integration of Emotion and Cognition. In Watanabe M (Ed.) The Prefrontal Cortex as an Executive, Emotional, and Social Brain (pp. 51–76). Tokyo, JP: Springer Press. [Google Scholar]
- Braun AR, Balkin TJ, Wesenten NJ, Carson RE, Varga M, Baldwin P, Selbie S, Belenky G, & Herscovitch P (1997). Regional cerebral blood flow throughout the sleep-wake cycle. An H215O PET study. Brain, 120, 1173–1197. [DOI] [PubMed] [Google Scholar]
- Boveroux P, Vanhaudenhuyse A, Bruno MA, Noirhomme Q, Lauwick S, Luxen A, Degueldre C, Plenevaux A, Schnakers C, Phillips C, Brichant JF, Bonhomme V, Maquet P, Greicius MD, Laureys S, & Boly M (2010). Breakdown of within- and between-network resting state functional magnetic resonance imaging connectivity during propofol-induced loss of consciousness. Anesthesiology, 113, 1038–1053. [DOI] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, & McCarley RW (2012). Control of sleep and wakefulness. Physiological Reviews, 92, 1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum MS, Gillin JC, Wu J, Hazlett E, Sicotte N, Dupont RM, & Bunney WE Jr. (1989). Regional cerebral glucose metabolic rate in human sleep assessed by positron emission tomography. Life Sciences, 45, 1349–1356. [DOI] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, & de Lecea L (2010). Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nature Neuroscience, 13, 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MA, & Dennett DC (2011). Consciousness cannot be separated from function. Trends in Cognitive Science, 15, 358–364. [DOI] [PubMed] [Google Scholar]
- Crick F, & Koch C (2003). A framework for consciousness. Nature Neuroscience, 6, 119–126. [DOI] [PubMed] [Google Scholar]
- Czisch M, Wehrle R, Kaufmann C, Wetter TC, Holsboer F, Pollmächer T, & Auer DP (2004), Functional MRI during sleep: BOLD signal decreases and their electrophysiological correlates. European Journal of Neuroscience, 20, 566–574. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, & Robbins TW (2004). Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neuroscience and Biobehavioral Reviews, 28, 771–784. [DOI] [PubMed] [Google Scholar]
- Datta S (2010). Cellular and chemical neuroscience of mammalian sleep. Sleep Medicine, 11, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, & Maclean RR (2007). Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neuroscience and Biobehavioral Reviews, 31, 775–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, Kerssens C, Sebel PS, & Hu X (2010). Altered local coherence in the default mode network due to sevoflurane anesthesia. Brain Research, 1318, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuker L, Olligs J, Fell J, Kranz TA, Mormann F, Montag C, Reuter M, Elger CE, & Axmacher N (2013). Memory consolidation by replay of stimulus-specific neural activity. Journal of Neuroscience, 33, 19373–19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M, Zalkind V, Fishman Y, & Minert A (2016). Model of anaesthetic induction by unilateral intracerebral microinjection of GABAergic agonists. European Journal of Neuroscience, 43, 846–858. [DOI] [PubMed] [Google Scholar]
- Dringenberg HC (2019). Sleep and memory consolidation: Conceptual and methodological challenges. In Dringenberg HC, (Ed.) Handbook of Sleep Research (pp. 489–501). New York, NY: Elsevier. [Google Scholar]
- Eichenbaum H (2017). Prefrontal–hippocampal interactions in episodic memory. Nature Reviews Neuroscience, 18, 547–558. [DOI] [PubMed] [Google Scholar]
- Fiset P, Paus T, Daloze T, Plourde G, Meuret P, Bonhomme V, Hajj-Ali N, Backman SB, & Evans AC (1999). Brain mechanisms of propofol-induced loss of consciousness in humans: a positron emission tomographic study. Journal of Neuroscience, 19, 5506–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores FJ, Hartnack KE, Fath AB, Kim SE, Wilson MA, Brown EN, & Purdon PL (2017). Thalamocortical synchronization during induction and emergence from propofol-induced unconsciousness. Proceedings of the National Academy of Sciences, USA, 114, E6660–E6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosse MJ, Fosse R, Hobson JA, & Stickgold RJ (2003). Dreaming and episodic memory: A functional dissociation? Journal of Cognitive Neuroscience, 15, 1–9. [DOI] [PubMed] [Google Scholar]
- Foster DJ (2017). Replay Comes of Age. Annual Review of Neuroscience, 40, 581–602. [DOI] [PubMed] [Google Scholar]
- Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, & Raichle ME (2005). The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proceedings of the National Academy of Sciences, USA, 102, 9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, & Heller HC (2019). The function(s) of sleep. Handbook of Experimental Pharmacology, 253, 3–34. [DOI] [PubMed] [Google Scholar]
- Franks NP (2008). General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nature Reviews Neuroscience, 9, 370–386. [DOI] [PubMed] [Google Scholar]
- Freud S (1900). In Strachey J, (Trans. & Ed.) The Interpretation of Dreams. New York, NY: Basic Books. [Google Scholar]
- Fu B, Yu T, Yuan J, Gong X, & Zhang M (2017). Noradrenergic transmission in the central medial thalamic nucleus modulates the electroencephalographic activity and emergence from propofol anesthesia in rats. Journal of Neurochemistry, 140, 862–873. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Sherman D, Pedersen NP, Saper CB, & Lu J (2011). Reassessment of the structural basis of the ascending arousal system. Journal of Comparative Neurology, 519, 933–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS (1995). Cellular basis of working memory. Neuron, 14, 477–485. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, & Menon V (2003). Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proceedings of the National Academy of Sciences, USA,100, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross WL, Lauer KK, Liu X, Roberts CJ, Liu S, Gollapudy S, Binder JR, Li SJ, & Hudetz AG (2019). Propofol sedation alters perceptual and cognitive functions in healthy volunteers as revealed by functional magnetic resonance imaging. Anesthesiology, 131, 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugino LD, Chabot RJ, Prichep LS, John ER, Formanek V, & Aglio LS (2001). Quantitative EEG changes associated with loss and return of consciousness in healthy adult volunteers anaesthetized with propofol or sevoflurane. British Journal of Anaesthesia, 87, 421–428. [DOI] [PubMed] [Google Scholar]
- Guidera JA, Taylor NE, Lee JT, Vlasov KY, Pei J, Stephen EP, Mayo JP, Brown EN, & Solt K (2017). Sevoflurane induces coherent slow-delta oscillations in rats. Frontiers in Neural Circuits, 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldenmund P, Demertzi A, Boveroux P, Boly M, Vanhaudenhuyse A, Bruno MA, Gosseries O, Noirhomme Q, Brichant JF, Bonhomme V, Laureys S, & Soddu A (2013). Thalamus, brainstem and salience network connectivity changes during propofol-induced sedation and unconsciousness. Brain Connectivity, 3, 273–285. [DOI] [PubMed] [Google Scholar]
- Guldenmund P, Gantner IS, Baquero K, Das T, Demertzi A, Boveroux P, Bonhomme V, Vanhaudenhuyse A, Bruno MA, Gosseries O, Noirhomme Q, Kirsch M, Boly M, Owen AM, Laureys S, Gómez F, & Soddu A (2016). Propofol-induced frontal cortex disconnection: a study of resting-state networks, total brain connectivity, and mean BOLD signal oscillation frequencies. Brain Connectivity, 6, 225–237. [DOI] [PubMed] [Google Scholar]
- Han B, McCarren HS, O’Neill D, & Kelz MB (2014). Distinctive recruitment of endogenous sleep-promoting neurons by volatile anesthetics and a nonimmobilizer. Anesthesiology 121, 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E (2010). The dream always makes new connections: the dream is a creation, not a replay. Sleep Medicine Clinics, 5, 241–248. [Google Scholar]
- Hartmann E, & Brezler T (2008). A systematic change in dreams after 9/11/01. Sleep 31, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine L, Soddu A, Gómez F, Vanhaudenhuyse A, Tshibanda L, Thonnard M, Charland-Verville V, Kirsch M, Laureys S, & Demertzi A (2012). Resting state networks and consciousness: alterations of multiple resting state network connectivity in physiological, pharmacological, and pathological consciousness states. Frontiers in Psychology, 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings HC, Riegelhaupt PM, Kelz MB, Solt K, Eckenhoff RG, Orser BA, & Goldstein PA (2019). Towards a comprehensive understanding of anesthetic mechanisms of action: A decade of discovery. Trends in Pharmacological Sciences, 40, 464–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofle N, Paus T, Reutens D, Fiset P, Gotman J, Evans AC, & Jones BE (1997). Regional cerebral blood flow changes as a function of delta and spindle activity during slow wave sleep in humans. Journal of Neuroscience, 17, 4800–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz SG, Braun AR, Carr WS, Picchioni D, Balkin TJ, Fukunaga M, & Duyn JH (2009). Decoupling of the brain’s default mode network during deep sleep. Proceedings of the National Academy of Sciences, USA, 106, 11376–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wang Z, Zhang J, Dai R, Wu J, Li Y, Liang W, Mao Y, Yang Z, Holland G, Zhang J, & Northoff G (2014). Altered temporal variance and neural synchronization of spontaneous brain activity in anesthesia. Human Brain Mapping, 35, 5368–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudetz AG (2012). General anesthesia and human brain connectivity. Brain Connectivity, 2, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang-Xie L-F, Yin L, Zhao S, Prevosto V, Han B-X, Dzirasa K, & Wang F (2019). A common neuroendocrine substrate for diverse general anesthetics and sleep. Neuron, 102, 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John ER, Prichep LS, Kox W, Valdés-Sosa P, Bosch-Bayard J, Aubert E, Tom M, di Michele F, & Gugino LD (2001). Invariant reversible QEEG effects of anesthetics. Consciousness and Cognition, 10, 165–183. [DOI] [PubMed] [Google Scholar]
- Jobst BM, Hindriks R, Laufs H, Tagliazucchi E, Hahn G, Ponce-Alvarez A, Stevner ABA, Kringelbach ML, & Deco G (2017). Increased stability and breakdown of brain effective connectivity during slow-wave sleep: mechanistic insights from whole-brain computational modelling. Science Reports, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE (2011). Neurobiology of waking and sleeping. Handbook of Clinical Neurology, 98, 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura N, Uchiyama M, Takayama Y, Uchida S, Uema T, Kato M, Sekimoto M, Watanabe T, Nakajima T, Horikoshi S, Ogawa K, Nishikawa M, Hiroki M, Kudo Y, Matsuda H, Okawa M, &Takahashi K (1999). Activity of midbrain reticular formation and neocortex during the progression of human non-rapid eye movement sleep. Journal of Neuroscience, 19, 10065–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann C, Wehrle R, Wetter TC, Holsboer F, Auer DP, Pollmächer T, & Czisch M (2006). Brain activation and hypothalamic functional connectivity during human non-rapid eye movement sleep: an EEG/fMRI study. Brain, 129, 655–667. [DOI] [PubMed] [Google Scholar]
- Kaur S, Pedersen NP, Yokota S, Hur EE, Fuller PM, Lazarus M, Chamberlin NL, & Saper CB (2013). Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. Journal of Neuroscience, 33, 7627–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelz MB, Sun Y, Chen J, Cheng Meng Q, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, & Yanagisawa M (2008). An essential role for orexins in emergence from general anesthesia. Proceedings of the National Academy of Sciences, USA, 105, 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Frank MG, Wisor JP, & Roy S (2016). Sleep function: Toward elucidating an enigma. Sleep Medicine Reviews, 28, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SW, Lee U, Noh GJ, Jun IG, & Mashour GA (2011). Preferential inhibition of frontal-to-parietal feedback connectivity is a neurophysiologic correlate of general anesthesia in surgical patients. Public Library of Science One, 6, e25155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar VM, Vetrivelan R, & Mallick HN (2006). Alpha-1 adrenergic receptors in the medial preoptic area are involved in the induction of sleep. Neurochemical Research, 31, 1095–1102. [DOI] [PubMed] [Google Scholar]
- Lamme VA (2006). Towards a true neural stance on consciousness. Trends in Cognitive Science, 10, 494–501. [DOI] [PubMed] [Google Scholar]
- Leung LS, Luo T, Ma J, & Herrick I (2014). Brain areas that influence general anesthesia. Progress in Neurobiology, 122, 24–44. [DOI] [PubMed] [Google Scholar]
- Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, & Saper CB (2008). Role of endogenous sleep-wake and analgesic systems in anesthesia. Journal of Comparative Neurology, 508, 648–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Yu S, Cai S, Zhang Y, Jiao Y, Yu T, & Yu W (2018). Parabrachial neurons promote behavior and electroencephalographic arousal from general anesthesia. Frontiers in Molecular Neuroscience, 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, and Leung LS (2011). Involvement of tuberomamillary histaminergic neurons in isoflurane anesthesia. Anesthesiology, 115, 36–43. [DOI] [PubMed] [Google Scholar]
- Lydic R, Baghdoyan HA, & May AL (2018). Neurochemistry of Anesthetic States. Methods in Enzymology, 603, 237–255. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Holm S, Vorstrup S, Friberg L, Lassen NA, & Wildschiødtz G (1991b). Human regional cerebral blood flow during rapid-eye-movement sleep. Journal of Cerebral Blood Flow & Metabolism, 11, 502–507. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Schmidt JF, Wildschiødtz G, Friberg L, Holm S, Vorstrup S, & Lassen NA (1991a). Cerebral O2 metabolism and cerebral blood flow in humans during deep and rapid-eye-movement sleep. Journal of Applied Physiology, 70, 2597–2601. [DOI] [PubMed] [Google Scholar]
- Maquet P (2000). Functional neuroimaging of normal human sleep by positron emission tomography. Journal of Sleep Research, 9, 207–231. [DOI] [PubMed] [Google Scholar]
- Maquet P, Dive D, Salmon E, Sadzot B, Franco G, Poirrier R, von Frenckell R, & Franck G (1990). Cerebral glucose utilization during sleep-wake cycle in man determined by positron emission tomography and [18F]2-fluoro-2-deoxy-D-glucose method. Brain Research, 513, 136–143. [DOI] [PubMed] [Google Scholar]
- Maquet P, Péters J, Aerts J, Delfiore G, Degueldre C, Luxen A, & Franck G (1996). Functional neuroanatomy of human rapid-eye-movement sleep and dreaming. Nature, 383, 163–166. [DOI] [PubMed] [Google Scholar]
- Maquet P, Degueldre C, Delfiore G, Aerts J, Péters JM, Luxen A, & Franck G (1997). Functional neuroanatomy of human slow wave sleep. Journal of Neuroscience, 17, 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuzzi R, Ramani R, Qiu M, Rajeevan N, & Constable RT (2010). Functional connectivity and alterations in baseline brain state in humans. Neuroimage, 49, 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimini M, Ferrarelli F, Huber R, Esser SK, Singh H, & Tononi G (2005). Breakdown of cortical effective connectivity during sleep. Science, 309, 2228–2232. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Jang IS, Nabekura J, & Akaike N (2003). Alpha 2-Adrenoceptor-mediated presynaptic modulation of GABAergic transmission in mechanically dissociated rat ventrolateral preoptic neurons. Journal of Neurophysiology, 89, 1640–1648. [DOI] [PubMed] [Google Scholar]
- McCarren HS, Chalifoux MR, Han B, Moore JT, Meng QC, Baron-Hionis N, Sedigh-Sarvestani M, Contreras D, Beck SG, & Kelz MB (2014). α2-Adrenergic stimulation of the ventrolateral preoptic nucleus destabilizes the anesthetic state. Journal of Neuroscience, 34, 16385–16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson WB (2017). The Science of Sleep. Chicago, IL: The University of Chicago Press. [Google Scholar]
- Minert A, & Devor M (2016). Brainstem node for loss of consciousness due to GABA(A) receptor-active anesthetics. Experimental Neurology, 275, 38–45. [DOI] [PubMed] [Google Scholar]
- Minert A, Yatziv SL, & Devor M (2017). Location of the mesopontine neurons responsible for maintenance of anesthetic loss of consciousness. Journal of Neuroscience, 37, 9320–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JT, Chen J, Han B, Meng QC, Veasey SC, Beck SG, & Kelz MB (2012). Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Current Biology, 22, 2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muindi F, Kenny JD, Taylor NE, Solt K, Wilson MA, Brown EN, & Van Dort CJ (2016). Electrical stimulation of the parabrachial nucleus induces reanimation from isoflurane general anesthesia. Behavioural Brain Research, 306, 20–25. [DOI] [PubMed] [Google Scholar]
- Murphy M, Bruno MA, Riedner BA, Boveroux P, Noirhomme Q, Landsness EC, Brichant JF, Phillips C, Massimini M, Laureys S, Tononi G, & Boly M (2011). Propofol anesthesia and sleep: a high-density EEG study. Sleep, 34, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, & Maze M (2002). The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nature Neuroscience, 5, 979–984. [DOI] [PubMed] [Google Scholar]
- O’Neill J, Pleydell-Bouverie B, Dupret D, & Csicsvari J (2010). Play it again: Reactivation of waking experience and memory. Trends in Neurosciences, 33, 220–229. [DOI] [PubMed] [Google Scholar]
- Paasonen J, Stenroos P, Salo RA, Kiviniemi V, & Gröhn O (2018) Functional connectivity under six anesthesia protocols and the awake condition in rat brain. Neuroimage, 172, 9–20. [DOI] [PubMed] [Google Scholar]
- Pace-Shott EF (2009). Sleep architecture. In Stickgold R and Walker M (Eds.) The Neuroscience of Sleep (pp. 11–17). San Diego, CA: Academic Press. [Google Scholar]
- Palanca BJ, Mitra A, Larson-Prior L, Snyder AZ, Avidan MS, & Raichle ME (2015). Resting-state functional magnetic resonance imaging correlates of sevoflurane-induced unconsciousness. Anesthesiology, 123, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan SC, & Rickard TC (2015). Sleep and motor learning: Is there room for consolidation? Psychological Bulletin, 141, 812–834. [DOI] [PubMed] [Google Scholar]
- Picchioni D, Pixa ML, Fukunaga M, Carr WS, Horovitz SG, Braun AR, & Duyn JH (2014). Decreased connectivity between the thalamus and the neocortex during human nonrapid eye movement sleep. Sleep, 37, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe GR (2017). Sleep is for forgetting. Journal of Neuroscience, 37, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdon PL, Pierce ET, Mukamel EA, Prerau MJ, Walsh JL, Wong KF, Salazar-Gomez AF, Harrell PG, Sampson AL, Cimenser A, Ching S, Kopell NJ, Tavares-Stoeckel C, Habeeb K, Merhar R, & Brown EN (2013). Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proceedings of the National Academy of Sciences, USA, 110, E1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdon PL, Sampson A, Pavone KJ, & Brown EN (2015). Clinical Electroencephalography for Anesthesiologists: Part I: Background and Basic Signatures. Anesthesiology, 123, 937–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu MH, Chen MC, Fuller PM, & Lu J (2016). Stimulation of the pontine parabrachial nucleus promotes wakefulness via extra-thalamic forebrain circuit nodes. Current Biology, 262, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranft A, Golkowski D, Kiel T, Riedl V, Kohl P, Rohrer G, Pientka J, Berger S, Thul A, Maurer M, Preibisch C, Zimmer C, Mashour GA, Kochs EF, Jordan D, & Ilg R (2016). Neural correlates of sevoflurane-induced unconsciousness identified by simultaneous functional magnetic resonance imaging and electroencephalography. Anesthesiology, 125, 861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch B, & Born J (2013). About sleep’s role in memory. Physiological Reviews, 93, 681–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sämann PG, Wehrle R, Hoehn D, Spoormaker VI, Peters H, Tully C, Holsboer F, & Czisch M (2011). Development of the brain’s default mode network from wakefulness to slow wave sleep. Cerebral Cortex, 21, 2082–2093. [DOI] [PubMed] [Google Scholar]
- Saper CB, & Fuller PM (2017). Wake-sleep circuitry: an overview. Current Opinion in Neurobiology, 44, 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, & Scammell TE (2010). Sleep state switching. Neuron, 68, 1023–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ (2017). Sleep to remember. Journal of Neuroscience, 37, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ, & Bouret S (2012). Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron, 76, 130–141. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, & Lipton JO (2017). Neural circuitry of wakefulness and sleep. Neuron, 93, 747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, & Kelz MB (2013). Sleep and anesthesia interactions: a pharmacological appraisal. Current Anesthesiology Reports, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröter MS, Spoormaker VI, Schorer A, Wohlschläger A, Czisch M, Kochs EF, Zimmer C, Hemmer B, Schneider G, Jordan D, & Ilg R (2012). Spatiotemporal reconfiguration of large-scale brain functional networks during propofol-induced loss of consciousness. Journal of Neuroscience, 32, 12832–12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrouff J, Perlbarg V, Boly M, Marrelec G, Boveroux P, Vanhaudenhuyse A, Bruno MA, Laureys S, Phillips C, Pélégrini-Issac M, Maquet P, & Benali H (2011). Brain functional integration decreases during propofol-induced loss of consciousness. Neuroimage, 57, 198–205. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, Reiss AL, & Greicius MD (2007). Dissociable intrinsic connectivity networks for salience processing and executive control. Journal of Neuroscience, 27, 2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaka T, Yonaha T, Onizuka S, & Tsuneyoshi I (2011). Effects of orexin-A on propofol anesthesia in rats. Journal of Anesthesia, 25, 65–71. [DOI] [PubMed] [Google Scholar]
- Siegel JM (2001). The REM sleep-memory consolidation hypothesis. Science, 294, 1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2009). The neurobiology of sleep. Seminars in Neurology, 29, 277–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkens T, Bosman CA, & Olcese U (2019). The role of top-down modulation in shaping sensory processing across brain states: implications for consciousness. Frontiers in Systems Neuroscience, 24, 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoormaker VI, Schröter MS, Gleiser PM, Andrade KC, Dresler M, Wehrle R, Sämann PG, & Czisch M (2010). Development of a large-scale functional brain network during human non-rapid eye movement sleep. Journal of Neuroscience, 30, 11379–11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoormaker VI, Gleiser PM, & Czisch M (2012). Frontoparietal connectivity and hierarchical structure of the brain’s functional network during sleep. Frontiers in Neurology, 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickgold R (2005). Sleep-dependent memory consolidation. Nature, 437, 1272–1278. [DOI] [PubMed] [Google Scholar]
- Stickgold R, & Walker MP (2007). Sleep-dependent memory consolidation and reconsolidation. Sleep Medicine, 8, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss M, Sitt JD, King JR, Elbaz M, Azizi L, Buiatti M, Naccache L, van Wassenhove V, & Dehaene S (2015). Disruption of hierarchical predictive coding during sleep. Proceedings of the National Academy of Sciences, USA, 112, E1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhotinsky I, Minert A, Soja P, & Devor M (2016). Mesopontine switch for the induction of general anesthesia by dedicated neural pathways. Anesthesia & Analgesia, 123, 1274–1285. [DOI] [PubMed] [Google Scholar]
- Tüshaus L, Omlin X, Tuura RO, Federspiel A, Luechinger R, Staempfli P, Koenig T, & Achermann P (2017). In human non-REM sleep, more slow-wave activity leads to less blood flow in the prefrontal cortex. Science Reports, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung A, & Mendelson WB (2004). Anesthesia and sleep. Sleep Medicine Reviews, 8, 213–225. [DOI] [PubMed] [Google Scholar]
- Tung A, Bluhm B, & Mendelson WB (2001). The hypnotic effect of propofol in the medial preoptic area of the rat. Life Sciences, 69, 855–862. [DOI] [PubMed] [Google Scholar]
- Tung A, Lynch JP, & Mendelson WB (2001). Prolonged sedation with propofol in the rat does not result in sleep deprivation. Anesthesia & Analgesia, 92, 1232–1236. [DOI] [PubMed] [Google Scholar]
- Tung A, Szafran MJ, Bluhm B, & Mendelson WB (2002). Sleep deprivation potentiates the onset and duration of loss of righting reflex induced by propofol and isoflurane. Anesthesiology, 97, 906–911. [DOI] [PubMed] [Google Scholar]
- Tung A, Bergmann BM, Herrera S, Cao D, & Mendelson WB (2004). Recovery from sleep deprivation occurs during propofol anesthesia. Anesthesiology, 100, 1419–1426. [DOI] [PubMed] [Google Scholar]
- Vazey EM, & Aston-Jones G (2014). Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proceedings of the National Academy of Sciences, USA, 111, 3859–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP (1984). Brainstem control of the events of REM sleep. Progress in Neurobiology, 22, 241–288. [DOI] [PubMed] [Google Scholar]
- Vertes RP (1990). Brainstem mechanisms of slow-wave sleep and REM sleep. In R Klemm W and Vertes RP, (Eds). Brainstem mechanisms of behavior, (pp. 535–583). New York, NY: John Wiley & Sons. [Google Scholar]
- Vertes RP (2004). Memory consolidation in sleep: Dream or reality. Neuron, 44, 135–148. [DOI] [PubMed] [Google Scholar]
- Vertes RP, & Eastman KE (2000). The case against memory consolidation in REM sleep. Behavioral and Brain Sciences, 23, 867–876. [DOI] [PubMed] [Google Scholar]
- Vertes RP, & Siegel JM (2005). Time for the sleep community to take a critical look at the purported role of sleep in memory processing. Sleep, 28, 1228–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselis RA, Reinsel RA, Beattie BJ, Mawlawi OR, Feshchenko VA, DiResta GR, Larson SM, & Blasberg RG (1997). Midazolam changes cerebral blood flow in discrete brain regions: an H2(15)O positron emission tomography study. Anesthesiology, 87, 1106–1117. [DOI] [PubMed] [Google Scholar]
- von Economo C (1930). Sleep as a problem of localization. Journal of Nervous and Mental Disease, 71, 249–259. [Google Scholar]
- Wang DS, & Orser BA (2011). Inhibition of learning and memory by general anesthetics. Canadian Journal of Anaesthesia, 58, 167–177. [DOI] [PubMed] [Google Scholar]
- Wilson MA, & McNaughton BL (1994). Reactivation of hippocampal ensemble memories during sleep. Science, 265, 676–679. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wang W, Zhao Z, Ge Y, Zhang J, Yu D, Chai W, Wu S, & Xu L (2010). The action sites of propofol in the normal human brain revealed by functional magnetic resonance imaging. The Anatomical Record, 293, 1985–1990. [DOI] [PubMed] [Google Scholar]
- Zhang LN, Li ZJ, Tong L, Guo C, Niu JY, Hou WG, & Dong HL (2012). Orexin-A facilitates emergence from propofol anesthesia in the rat. Anesthesia & Analgesia, 115, 789–796. [DOI] [PubMed] [Google Scholar]
- Zhang LN, Yang C, Ouyang PR, Zhang ZC, Ran MZ, Tong L, Dong HL, & Liu Y (2016). Orexin-A facilitates emergence of the rat from isoflurane anesthesia via mediation of the basal forebrain. Neuropeptides, 58, 7–14. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu T, Yuan J, & Yu BW (2015). The ventrolateral preoptic nucleus is required for propofol-induced inhibition of locus coeruleus neuronal activity. Neurological Sciences, 36, 2177–2184. [DOI] [PubMed] [Google Scholar]
- Zhou W, Cheung K, Kyu S, Wang L, Guan Z, Kurien PA, Bickler PE, & Jan LY (2018). Activation of orexin system facilitates anesthesia emergence and pain control. Proceedings of the National Academy of Sciences, USA, 115, E10740–E10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, McKenna JT, & McCarley RW (2016). Functions and mechanisms of sleep. AIMS Neuroscience, 3, 67–104. [DOI] [PMC free article] [PubMed] [Google Scholar]