

ABSTRACT
Dendritic cells (DCs) have received considerable attention as potential targets for the development of novel cancer immunotherapies. However, the clinical efficacy of DC-based vaccines remains suboptimal, largely reflecting local and systemic immunosuppression at baseline. An autologous DC-based vaccine (DCVAC) has recently been shown to improve progression-free survival and overall survival in randomized clinical trials enrolling patients with lung cancer (SLU01, NCT02470468) or ovarian carcinoma (SOV01, NCT02107937), but not metastatic castration-resistant prostate cancer (SP005, NCT02111577), despite a good safety profile across all cohorts. We performed biomolecular and cytofluorometric analyses on peripheral blood samples collected prior to immunotherapy from 1000 patients enrolled in these trials, with the objective of identifying immunological biomarkers that may improve the clinical management of DCVAC-treated patients. Gene signatures reflecting adaptive immunity and T cell activation were associated with favorable disease outcomes and responses to DCVAC in patients with prostate and lung cancer, but not ovarian carcinoma. By contrast, the clinical benefits of DCVAC were more pronounced among patients with ovarian carcinoma exhibiting reduced expression of T cell-associated genes, especially those linked to TH2-like signature and immunosuppressive regulatory T (TREG) cells. Clinical responses to DCVAC were accompanied by signs of antitumor immunity in the peripheral blood. Our findings suggest that circulating signatures of antitumor immunity may provide a useful tool for monitoring the potency of autologous DC-based immunotherapy.
KEYWORDS: Cancer immunotherapy, metastatic castrate-resistant prostate cancer, non-small cell lung carcinoma, epithelial ovarian carcinoma, circulating biomarkers, anti-PD-1
Introduction
Immunotherapy is currently the most rapidly advancing area of clinical oncology and has markedly improved the clinical management of multiple types of cancer.1 Although, immune checkpoint inhibitors (ICIs) have revolutionized the clinical management of various solid tumors, only about 20% of patients with the most common solid tumors respond to ICIs as standalone therapies, although the proportion varies greatly among different indications.2,3 Thus, novel strategies are needed alongside the identification of biomarkers that can prospectively identify patients who may benefit from specific immunotherapeutic regimens.4–6
Dendritic cells (DCs) have received considerable attention as potential targets for the development of cancer immunotherapies in recent decades.7 Notably, the activity of DCs is associated with or underlies the efficacy of currently approved cancer therapies, such as ICIs.8 Therefore, combining DC vaccination with different therapeutic approaches has been proposed. Nonetheless, the clinical efficacy of DC-based vaccines used as monotherapy remains suboptimal, which reflects the baseline level of circulating and/or intratumoral immune responses and the extent of immunosuppression.9,10
Although tumor sampling is widely implemented for biomarker identification and analysis, there are several challenges including limited accessibility, heterogeneity of the biopsy site, and the patient’s condition.11 Therefore, identification of potential predictive biomarkers using more accessible peripheral blood is critical for the development and clinical utility of biomarkers.12,13 Recent technological, analytical, and mechanistic advances in immunology have enabled the identification of several circulating cancer biomarkers including, but not limited to: circulating tumor cells in breast and prostate cancer, tumor genomic alterations such as discrete oncogenic variants (e.g. EGFR, PBRM1 and JAK1/2), microsatellite instability, tumor mutational burden-related metrics, peripheral immune-cell function, and analyses of immune-related cytokines and plasma proteins.14–20 Because personalized DC-based cancer immunotherapy is largely dependent on preexisting circulating immunity, the identification of immune signatures associated with the response to therapy might provide a useful stratification tool.
We recently published the results of three independent open-label, randomized Phase I/II, II and III clinical studies that compared the efficacy of an autologous DC-based vaccine (DCVAC) delivered in the context of standard of care chemotherapy (SOC) versus SOC alone in patients with advanced non-small cell lung carcinoma (NSCLC; SLU01, NCT02470468),21 epithelial ovarian cancer (EOC; SOV01, NCT02107937),22,23 or metastatic castration-resistant prostate cancer (mCRPC; SP005, NCT02111577).24 In these settings, DCVAC was well tolerated and significantly extended the progression-free survival (PFS) and overall survival (OS) of EOC or NSCLC patients.21,22 In mCRPC patients, DCVAC combined with SOC and continued as maintenance treatment showed a favorable safety profile but did not extend OS.24
Here, we performed biomolecular and cytofluorometric analyses using peripheral blood samples collected prior to immunotherapy for 1000 patients enrolled in these trials of DCVAC. We found that a circulating immune-related gene signature associated with adaptive immunity and T cell activation was associated with an improved response to DC-based immunotherapy in mCRPC and NSCLC patients enrolled in SP005 and SLU01, although not in EOC patients enrolled in SOV01. Conversely, the clinical benefit of DCVAC was more pronounced in EOC patients with gene expression levels below median for TH2-like and immunosuppressive gene signatures associated with a low frequency of circulating CD4+CD25+FoxP3+ T cells, as determined by molecular and flow cytometry analyses. Pending validation in independent studies, our findings suggest that the circulating immune signature is a potential tool for stratification of patients prior to cellular immunotherapy, largely reflecting the oncologic indication.
Materials and methods
Patient characteristics
In SP005 (NCT02111577), 1182 mCRPC patients were randomized between June 2014 and November 2017 across 177 hospital clinics in Europe and the United States (US). Of these, 787 were assigned to DCVAC and 395 to placebo.24 Patients in both arms received SOC, and DCVAC was continued as maintenance therapy. In SLU01 (NCT02470468), 112 patients with advanced NSCLC were randomized to one of three arms between January 2015 and November 2016. Patients in arm A received DCVAC/LuCa and chemotherapy (n = 45), patients in arm B received DCVAC/LuCa, chemotherapy and immune enhancers (n = 29), and patients in arm C received chemotherapy alone (arm C, n = 38).21 In SOV01 (NCT02107937), 99 EOC patients were randomized to one of three arms between November 2013 and May 2015. All patients underwent debulking surgery followed by adjuvant SOC combined with DCVAC administered in parallel with SOC (arm A, n = 34) or sequential to SOC (arm B, n = 34). Patients in arm C (n = 31) received SOC alone.22 The designs of these studies are briefly described in the Supplemental Materials and Methods. In SP005 and SLU01, the primary endpoint was overall survival (OS) defined as the time from randomization until death due to any cause. In SOV01, the primary efficacy endpoint was PFS. Of 1182 mCRPC patients, 112 NSCLC patients, and 99 EOC patients randomized to treatment, peripheral blood samples and data were available for 804 (68%), 103 (92%), and 93 (96%), respectively. Written informed consent was obtained according to the Declaration of Helsinki, and the study was approved by appropriate Ethical Committees. The results of all three clinical trials have been reported.21,22,24 The baseline characteristics for patients included in this study were similar across the relevant treatment groups (Supplemental Table 1).
Preparation of DCVAC
Each DCVAC dose comprises DCs loaded with antigens derived from the EOC cell lines (OV-90 and SK-OV-3) in SOV01, NSCLC cell lines (H522 and H520) in SLU01, and a human prostate adenocarcinoma cell line (LNCaP) in SP005. To prepare DCVAC, the peripheral blood mononuclear cells, obtained via leukapheresis and gradient centrifugation, are first cultured in a medium containing interleukin-4 and granulocyte-macrophage colony-stimulating factor. Immature DCs are separated, co-cultured (pulsed) with high hydrostatic pressure-treated tumor cell lines, and matured using polyinosinic:polycyticylic acid.25,26 The resulting product is cryopreserved at a concentration of approximately 107 DCs in 1 mL of CryoStor CS10 (StemCell) per vial.
Isolation of RNA from peripheral blood mononuclear cells (PBMCs) and reverse transcription
Total RNA was isolated with RNeasy Mini Kits (Qiagen). Cell lysates in RLT buffer enriched with 1% 2-mercaptoethanol were quickly thawed and processed according to the manufacturer’s instructions, including DNase I digestion. The RNA concentration and purity were determined using a NanoDrop 2000c (Thermo Scientific). Purified RNA samples were stored at −80°C until further use. cDNA for the detection of 93 selected genes associated with the immune system (Supplemental Table 2) was synthesized from 100 ng of total RNA using the TATAA GrandScript cDNA Synthesis Kits (TATAA Biocenter).
cDNA preamplification
Ten microliters of cDNA samples diluted 1:2 was used in a 50 μL preamplification reaction with TATAA PreAmp GrandMaster® Mix and the relevant primers at a final concentration of 40 nM per primer. Targeted pre-amplification was implemented on a T100 Thermal Cycler (Bio-Rad) with the following conditions: 95°C for 3 min, followed by 14 cycles of amplification (95°C for 20s, 55°C for 3 min and 72°C for 20s). After a final extension step (10 min), the samples were immediately frozen and stored at −20°C until analysis.
High-throughput quantitative real-time polymerase chain reaction (qPCR)
High-throughput qPCR was performed on the Biomark HD system (Fluidigm) using the 48.48 Dynamic Array Chip for Gene Expression and probe-based detection. Each reaction sample (5 µL) contained 1 µL of the pre-amplification products (diluted 1:10), 2.74 µL of Probe GrandMaster Mix (TATAA Biocenter), 0.25 µL of 20× GE Sample Loading Reagent (Fluidigm), 0.01 µL of ROX (Life Technologies; final concentration: 50 nM), and DNA/DNAse-free water. The assay reaction mix (5 µL) contained 2.5 µL of Assay Loading Reagent (Fluidigm) and 2.5 µL of a 5 µM mix of the reverse and forward primers plus 2.5 µM probes. Priming and loading of the dynamic array were performed according to the manufacturer’s instructions using the IFC controller HX (Fluidigm). The thermal conditions comprised thermal mixing at 50°C for 2 min followed by 70°C for 40 min and 25°C for 10 min, hot-start activation at 95°C for 30s and 40 cycles of amplification (95°C for 10s and 60°C for 60s). Melting curve analysis was performed in the range of 60°C to 95°C with increments of 0.5°C/s. The amplification data were analyzed with Fluidigm Real-Time PCR Analysis software, applying the linear derivative baseline subtraction method and a user-defined global threshold to obtain Cq values.
Flow cytometry
The frequency of CD4+CD25+FoxP3+ regulatory T cells was assessed by flow cytometry using standard procedures. Briefly, peripheral blood mononuclear cells (PBMCs) were stained with CD45-HV500 (BD Biosciences) CD3-A700 (Exbio), CD4-ECD (Beckman Coulter), and CD25-PE (Exbio) conjugates plus Aqua Blue Live/Dead cell viability dye (Life Technologies) (Supplemental Table 3). Thereafter, cells were fixed with fixation/permeabilization buffer (BD Bioscience), permeabilized with permeabilization buffer (BD Bioscience), and incubated with FoxP3-A488 (Thermo Fisher Scientific). Flow cytometry was performed on an LSRFortessa Analyzer (BD), and data were analyzed using FlowJo software (Tree Star, Inc). After excluding dead cells, regulatory T cells were determined as CD45+CD4+CD25+FoxP3+ cells (Supplementary Figure 1).
Multiplex assay
The serum levels of IL6, IL10 and IL13 in SOV01 patients were measured using a MAGPIX system (Luminex) with Magnetic Bead Panel HCYTOMAG-60 K, 3-plex (Merck). Samples were stored at −80°C until analyzed.
Statistical analysis
These analyses were conducted in a prospective exploratory manner using data collected from prospective clinical trials. PFS was defined as the time from randomization to the date of the first radiological progression or death, whichever came first. OS was calculated as the time from randomization to death from any cause. Survival analyses were estimated by Cox proportional hazard regression and the Kaplan–Meier method using R survival package, and differences between the groups of patients were calculated using the log-rank test. For log-rank tests, the prognostic value of continuous variables was assessed using cluster stratification or median cutoff for each gene or the frequency of circulating CD4+CD25+FOXP3+ regulatory T cells. PCR data were analyzed using GenEx software (MultiD Analyses). The relative gene expression levels were calculated using the ΔΔCt method and were normalized to the expression levels of reference genes selected by Normfinder. Genes for which the expression was below the assay’s detection limit were excluded from further analyses (SLU01: IL4, IL13, NIS2, NCR2, MPPED1, NPR1; SOV01: NOS2, NCR2, MPPED1, CCL17, NPR1; SP005: IL4, NOS2, NCR2, CCL17, NPR1). Heatmaps were prepared using ComplexHeatmap R package.27 The EnrichGo function in ClusterProfiles R package was used to identify enriched GO terms based on hypergeometric distribution.28 p values were adjusted for multiple comparisons using the Benjamini–Hochberg method. Wilcoxon’s test was used to compare the frequency of immune markers before and after therapy. Fisher’s exact test was used to compare patient distribution across subgroups. All analyses were performed with Prism 8.4.2 (GraphPad), SAS software V.9.4, and R (http://www.r-project. org/). p values <0.05 were considered statistically significant.
Results
The immune-related gene signature in peripheral blood predicted survival and the response to DCVAC in mCRPC patients
We first performed biomolecular analyses to compare the gene expression profile associated with the immune system in pre-treatment peripheral blood samples collected from 804 mCRPC patients enrolled in SP005 (Supplemental Table 1A). We focused on the detection of 93 genes classified into 9 clusters reflecting various immune subsets and functions, including (but not limited to): B cells, cytotoxicity, DCs, immune populations, immunosuppression, natural killer (NK) cell function, T cell activation, and TH1 vs TH2 polarization (Supplementary Table 2). Unsupervised hierarchical clustering identified two main patient clusters, which were well balanced across the study arms (Figure 1a). Cluster 1, a high inflammatory cluster, was significantly enriched with 68 genes compared with cluster 2, a low inflammatory cluster (Supplementary Table 4). Functional studies revealed a significant association between the differentially expressed genes (DEGs), particularly positive regulation of adaptive immune responses, and cytotoxic T cell- and NK cell-mediated immunity (Supplemental Figure 2A).
Figure 1.
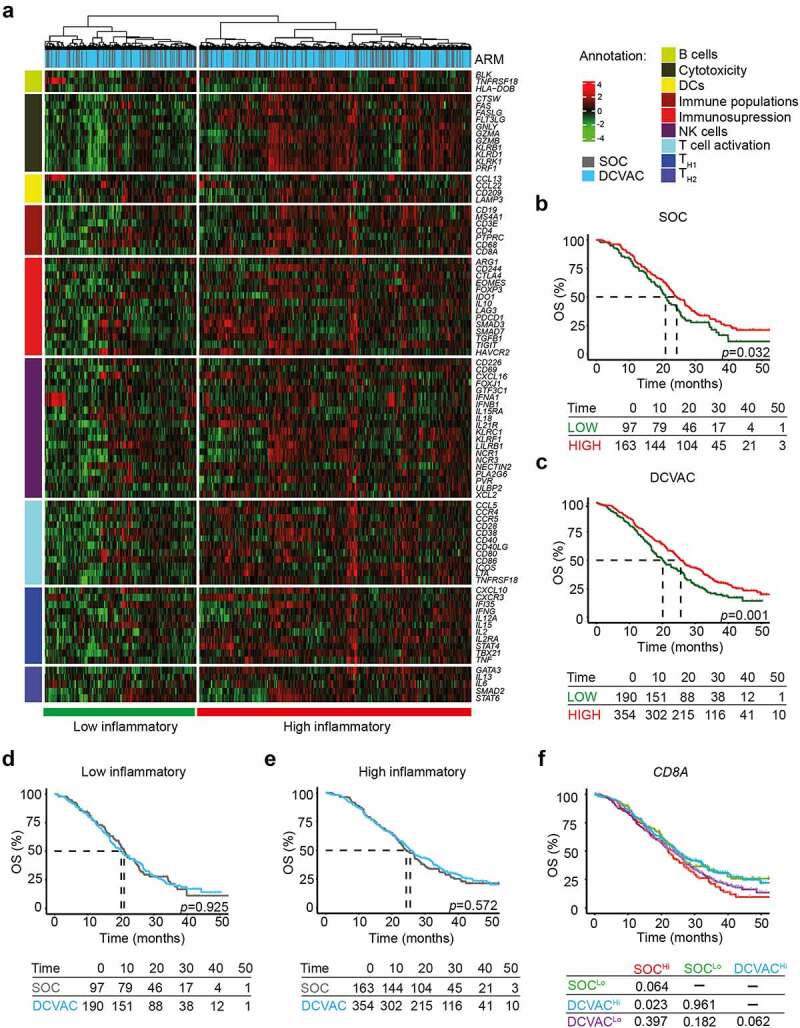
High expression of CD8A in peripheral blood is correlated with favorable prognosis and response to DCVAC in mCRPC patients in SP005. (a) Unsupervised hierarchical clustering of 804 mCRPC patients in SP005 based on the expression of 93 genes classified into clusters related to B cells, cytotoxicity, DCs, immune populations, immunosuppression, NK cells, T cell activation, and TH1 and TH2 signatures. (b, c) OS of 260 patients from the SOC arm (b) and 544 patients from the DCVAC arm (c) following stratification by unsupervised hierarchical clustering into low and high inflammatory clusters. (d, e) Direct comparison of OS of SOC and DCVAC patients following stratification by unsupervised hierarchical clustering into low (d) and high inflammatory clusters. (f) OS of 804 mCRPC patients stratified by the median CD8A expression and study arm. Survival curves were estimated using the Kaplan–Meier method and differences between groups were evaluated using the log-rank test. The numbers of patients at risk and p values are reported.
To assess the prognostic value of the immune-related gene signatures in peripheral blood, we compared OS between the distinct clusters of patients. In both study arms, the high inflammatory cluster was associated with longer OS (p <0.001) compared with the low inflammatory cluster (Figure 1b,) (SOC: p =0.032; DCVAC: p =0.001). In line with these findings, univariate Cox regression analyses revealed a strong prognostic value of 9 and 35 genes that were mainly associated with adaptive immunity and T cell activation. These genes were significantly overrepresented in the high inflammatory cluster in the SOC and DCVAC arms (Table 1).
Table 1.
Univariate Cox proportional hazard analyses for OS in mCRPC patients from the SOC and DCVAC arms in SP005.
| SOC arm |
|
DCVAC arm |
|||||
|---|---|---|---|---|---|---|---|
| Variable | HR (95% CI) | p-value | Variable | HR (95% CI) | p-value | ||
| ARG1 | 1.2 (1.1–1.3) | <0.001 | IL12A | 0.7 (0.61–0.8) | <0.001 | ||
| IL6 | 0.8 (0.7–0.9) | <0.001 | MS4A1 | 0.79 (0.72–0.87) | <0.001 | ||
| CD69 | 0.7 (0.5–0.09) | <0.001 | CCR5 | 1.3 (1.2–1.5) | <0.001 | ||
| GATA3 | 0.7 (0.6–0.8) | 0.001 | CD69 | 0.74 (0.65–0.85) | <0.001 | ||
| CCR5 | 1.3 (1.1–1.5) | 0.009 | CD19 | 0.82 (0.75–0.9) | <0.001 | ||
| KLRB1 | 0.7 (0.6–0.9) | 0.016 | IL6 | 0.82 (0.74–0.9) | <0.001 | ||
| CD209 | 0.83 (0.7–0.9) | 0.016 | CD68 | 1.4 (1.2–1.7) | <0.001 | ||
| CD4 | 0.7 (0.6–0.9) | 0.029 | BLK | 0.84 (0.76–0.92) | <0.001 | ||
| IL2 | 0.9 (0.8–1) | 0.042 | IL18 | 1.4 (1.2–1.7) | <0.001 | ||
| IL15 | 1.4 (1.1-1.7) | <0.001 | |||||
| IFI35 | 1.2 (1.1–1.4) | <0.001 | |||||
| CD226 | 1.3 (1.1–1.4) | <0.001 | |||||
| CD86 | 1.3 (1.1–1.6) | <0.001 | |||||
| TGFB1 | 1.3 (1.1–1.5) | 0.002 | |||||
| HAVCR | 1.3 (1.1–1.5) | 0.002 | |||||
| GNLY | 0.83 (0.74–0.93) | 0.002 | |||||
| SMAD2 | 1.4 (1.1–1.8) | 0.002 | |||||
| LTA | 0.8 (0.7–0.92) | 0.002 | |||||
| KLRB1 | 0.81 (0.7–0.92) | 0.002 | |||||
| ARG1 | 1.1 (1–1.1) | 0.002 | |||||
| HLA-DOB | 0.87 (0.8–0.95) | 0.002 | |||||
| LILRB1 | 1.3 (1.1–1.5) | 0.002 | |||||
| CD3E | 0.83 (0.74–0.94) | 0.004 | |||||
| GATA3 | 0.83 (0.72–0.95) | 0.005 | |||||
| TBX21 | 0.85 (0.75–0.96) | 0.009 | |||||
| NECTIN2 | 1.2 (1–1.3) | 0.011 | |||||
| IL2 | 0.91 (0.84–0.98) | 0.011 | |||||
| IL15RA | 1.2 (1–1.4) | 0.014 | |||||
| IFNG | 0.9 (0.83–0.98) | 0.016 | |||||
| PLA2G6 | 0.79 (0.65–0.96) | 0.017 | |||||
| CXCL16 | 1.2 (1–1.4) | 0.017 | |||||
| NCR3 | 0.87 (0.78–0.98) | 0.021 | |||||
| IL10 | 1.1 (1–1.1) | 0.032 | |||||
| CCL22 | 1.1 (1–1.2) | 0.032 | |||||
| STAT4 | 0.88 (0.78–1) | 0.043 | |||||
OS = overall survival; mCRPC = metastatic castration-resistant prostate cancer; SOC = standard of care chemotherapy; DCVAC, dendritic cell-based vaccination; HR = hazard ratio; CI = confidence interval
To determine the predictive value of the immune-related gene signature in peripheral blood of mCRPC patients, we also compared the OS between the two study arms for the low and high inflammatory cluster separately. However, DCVAC did not show a distinct OS advantage in either cluster (Figure 1d,e). To obtain additional insights into the predictive value of gene-signatures associated with B cells, cytotoxicity, DCs, immune population, immunosuppression, NK cell function, T cell activation, and TH1 and TH2 on DCVAC efficacy, we directly compared OS among patients stratified by median gene expression levels and study arms. We found, that DCVAC treatment conferred a significant OS advantage to mCRPC patients with high expression of CD8A (CD8A: p =0.023), but not to their low counterparts (Figure 1f). Conversely, we failed to identify a predictive impact of gene signatures associated with B cells, DCs, NK cells, or individual T cell subsets and their functional capacity (Supplemental Figure 3A).
Taken together, these findings indicate that high expression of immune-related genes, especially those related to adaptive immunity, T cells and NK cells, was associated with improved OS in a large cohort of mCRPC patients. However, only high CD8A expression in peripheral blood was associated with a significantly improved response to DC-based immunotherapy in mCRPC patients.
The immune-related gene signature in peripheral blood predicted survival and the response to DC-based immunotherapy in NSCLC patients
Inspired by our observation in mCRPC, we then compared the expression profile of the same panel of 93 genes using pre-treatment peripheral blood samples from 103 NSCLC patients enrolled in SLU01 (Supplemental Table 1B). Similar to the results for mCRPC, unsupervised hierarchical clustering identified two main clusters of NSCLC patients that were well balanced across the study arms (Figure 2a). Cluster 1, the high inflammatory cluster, was significantly enriched for 61 genes compared with cluster 2, the low inflammatory cluster (Supplementary Table 4). Similarly, to the findings for mCRPC, functional studies revealed significant associations between the DEGs with positive regulation of the adaptive immune response, and cytotoxic T cell- and NK cell-mediated immunity (Supplemental Figure 2B). To assess the prognostic value of the immune-related gene signatures in peripheral blood, we assess OS in each cluster. In both SOC and DCVAC arms, the low inflammatory cluster was associated with shorter OS compared to high inflammatory cluster (Figure 2b,). Consistent with these findings, univariate Cox regression analyses confirmed a strong prognostic value of 14 and 30 genes, mainly associated with T cell activation, that were significantly overrepresented in the high inflammatory cluster in the SOC and DCVAC arms, respectively (Table 2).
Figure 2.
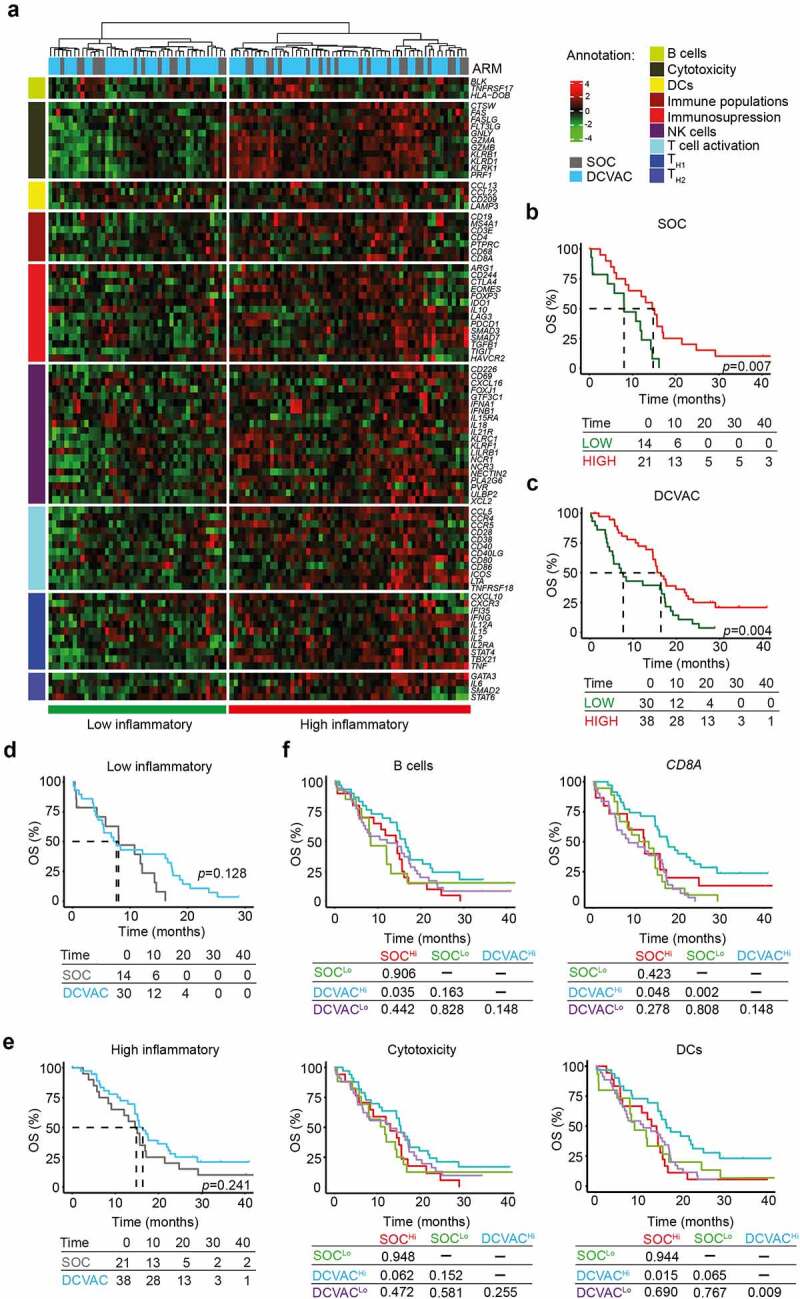
High expression gene signatures associated with B cells, CD8A, cytotoxicity, and DCs is correlated with favorable prognosis and response to DCVAC in NSCLC patients in SLU01. (a) Unsupervised hierarchical clustering of 103 NSCLC patients in SLU01 based on the expression of 93 genes classified into clusters related to B cells, cytotoxicity, DCs, immune populations, immunosuppression, NK cells, T cell activation, and TH1 and TH2 signatures. (b, c) OS of 35 patients from the SOC arm (b) and 68 patients from the DCVAC arm (c) following stratification by unsupervised hierarchical clustering into low and high inflammatory clusters. (d, e) Direct comparison of OS of SOC and DCVAC patients following stratification by unsupervised hierarchical clustering into low (d) and high inflammatory clusters (e). (f) OS of 103 NSCLC patients stratified by the median expression of genes associated with B cell signature, CD8A expression, cytotoxicity, and DCs, and study arm. Survival curves were estimated using the Kaplan–Meier method, and differences between groups were evaluated using the log-rank test. The numbers of patients at risk and p values are reported.
Table 2.
Univariate Cox proportional hazard analyses for OS in NSCLC patients from the SOC and DCVAC arms in SLU01.
| SOC arm |
DCVAC arm |
|||||
|---|---|---|---|---|---|---|
| Variable | HR (95% CI) | p-value | Variable | HR (95% CI) | p-value | |
| PLA2G6 | 0.1 (0.01–0.5) | 0.002 | CD28 | 0.4 (0.2–0.6) | <0.001 | |
| GATA3 | 0.45 (0.3–0.8) | 0.007 | CD3E | 0.4 (0.2–0.6) | <0.001 | |
| LILRB1 | 2.6 (1.3–5.4) | 0.008 | LILRB1 | 3.1 (1.7–5.8) | <0.001 | |
| LTA | 0.5 (0.3–0.9) | 0.011 | STAT4 | 0.4 (0.2–0.7) | <0.001 | |
| IFI35 | 2 (1.2–3.5) | 0.011 | CD8A | 0.6 (0.4–0.8) | <0.001 | |
| HAVCR | 2.6 (1.2–5.7) | 0.019 | FLT3LG | 0.4 (0.2–0.7) | <0.001 | |
| CD86 | 2.8 (1.2–6.7) | 0.021 | CD40LG | 0.5 (0.3–0.7) | 0.001 | |
| RORC | 0.7 (0.5–0.9) | 0.026 | FOXP3 | 0.6 (0.4–0.8) | 0.001 | |
| KLRB1 | 0.6 (0.3–0.9) | 0.036 | KLRB1 | 0.5 (0.3–0.8) | 0.002 | |
| CD28 | 0.45 (0.21–0.95) | 0.037 | IL15 | 3.1 (1.5–6.6) | 0.002 | |
| CD269 | 1.4 (1–2) | 0.038 | CD68 | 1.9 (1.3–3) | 0.003 | |
| CXCR3 | 0.6 (0.3–0.9) | 0.043 | TIGIT | 0.6 (0.4–0.8) | 0.003 | |
| CXCL16 | 2 (1–4) | 0.041 | CCL5 | 0.6 (0.4–0.8) | 0.003 | |
| IL2 | 0.8 (0.6–1) | 0.045 | GATA3 | 0.5 (0.3–0.8) | 0.004 | |
| KLRF1 | 0.5 (0.3–0.8) | 0.006 | ||||
| PLA2G6 | 0.4 (0.2–0.8) | 0.008 | ||||
| CTSW | 0.6 (0.4–0.9) | 0.011 | ||||
| NCR3 | 0.6 (0.4–0.9) | 0.012 | ||||
| IL21R | 0.5 (0.3–0.9) | 0.014 | ||||
| CTLA4 | 0.6 (0.4–0.9) | 0.014 | ||||
| IL12A | 0.6 (0.5–0.9) | 0.014 | ||||
| STAT6 | 2.3 (1.2–4.6) | 0.017 | ||||
| SMAD3 | 0.4 (0.2–0.9) | 0.018 | ||||
| TBX21 | 0.6 (0.4–0.9) | 0.023 | ||||
| IL10 | 1.3 (1–1.5) | 0.024 | ||||
| CD86 | 1.8 (1.1–3.2) | 0.027 | ||||
| CCR4 | 0.7 (0.4–0.9) | 0.028 | ||||
| IFI35 | 1.6 (1–2.4) | 0.032 | ||||
| TNFRSF18 | 0.6 (0.4–0.9) | 0.033 | ||||
| XCL2 | 0.7 (0.5–1) | 0.039 | ||||
OS = overall survival; NSCLC = non-small cell lung cancer; SOC = standard of care chemotherapy; DCVAC, dendritic cell-based vaccination; HR = hazard ratio; CI = confidence interval
To assess the predictive value of immune-related gene signature in peripheral blood of SLU01 patients, we compared OS between the study arms for the low and high inflammatory clusters separately (Figure 2d, e). Although, there was no advantage of DCVAC in either cluster, we found that DCVAC conferred an OS advantage to patients with high expression levels of gene signatures associated with B cells (p =0.035), CD8A (p =0.048), and DCs (p =0.015) (Figure 2f). A similar non-significant trend was also observed for gene signature associated with cytotoxicity (p =0.062) (Figure 2f). Importantly, we failed to observe a negative impact of TH2 and FoxP3 gene signature on the final response to DCVAC in NSCLC patients (Supplemental Figure 3B).
Taken together, these findings indicate that, similar to mCRPC, high expression of immune-related genes, particularly those related to adaptive immunity and T cells activation, are associated with improved disease outcome in NSCLC patients. However, a greater clinical benefit of DCVAC was observed in NSCLC patients with high expression levels of genes associated with B cells, effector CD8+ T cells, and DCs.
A low inflammatory gene signature in peripheral blood was correlated with improved PFS in EOC patients treated with DCVAC
Driven by our observations in mCRPC and NSCLC, we also compared the gene expression profile for the same panel of 93 genes in pre-treatment peripheral blood samples of 93 EOC patients enrolled in SOV01 (Supplemental Table 1C). Again, unsupervised hierarchical clustering identified two main patient clusters associated with low and high expression of immune-related genes that were well balanced across the study arms (Figure 3a). The high inflammatory cluster was significantly enriched for 68 genes compare with the low inflammatory cluster (Supplementary Table 4C). Functional studies revealed significant associations between the DEGs, especially positive regulation of adaptive immune responses, as well as cytotoxic T cell- and NK cell-mediated immunity (Supplemental Figure 2C).
Figure 3.
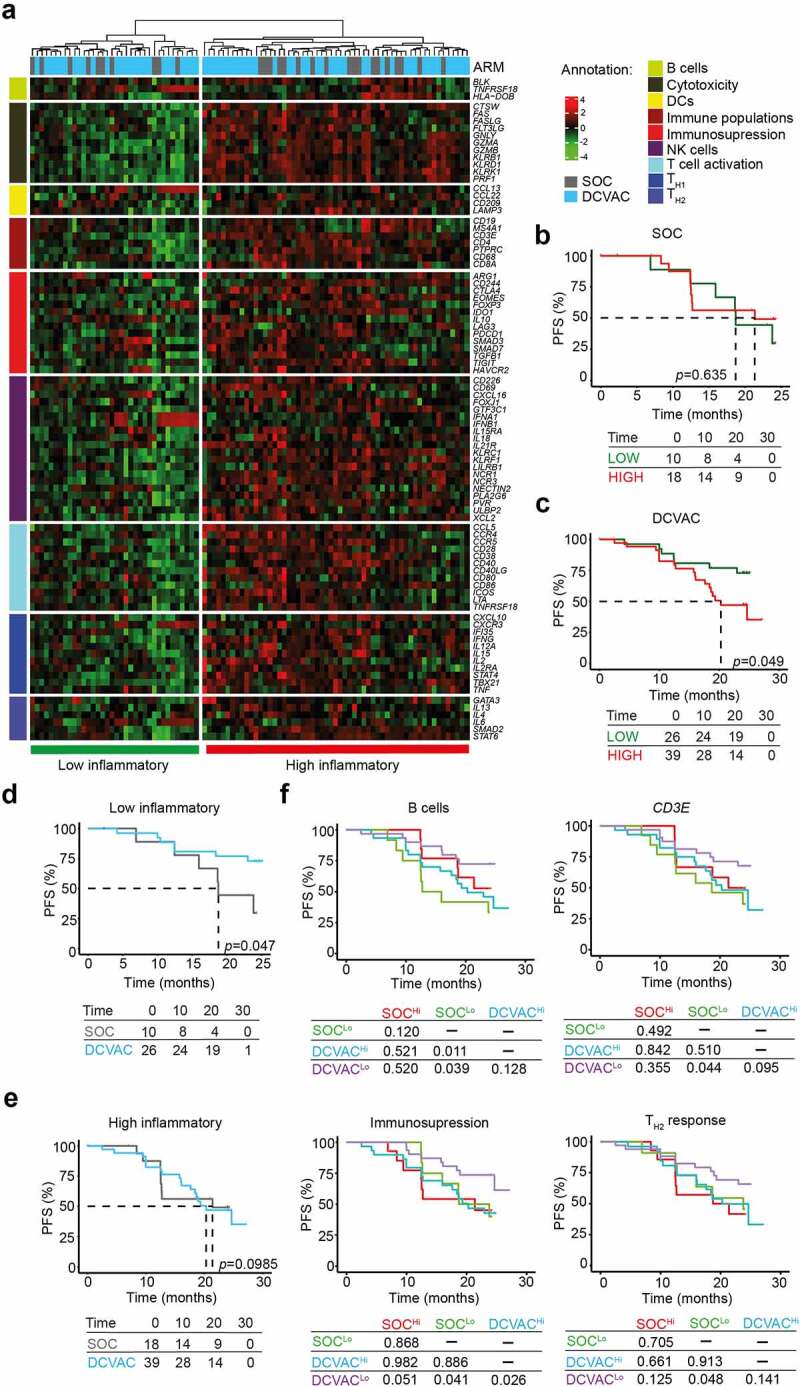
Low expression of genes associated with immunosuppression and TH2 signature is correlated with an improved response to DCVAC in EOC patients in SOV01. (a) Unsupervised hierarchical clustering of 93 EOC patients in SOV01 based on the expression of 93 genes classified into clusters related to B cells, cytotoxicity, DCs, immune populations, immunosuppression, NK cells, T cell activation, and TH1 and TH2 signatures. (b, c) PFS of 28 patients from the SOC arm (b) and 65 patients from the DCVAC arm (c) following stratification by unsupervised hierarchical clustering into low and high inflammatory clusters. (d, e) Direct comparison of PFS of SOC and DCVAC patients following stratification by unsupervised hierarchical clustering into low (d) and high inflammatory clusters (e). (f) PFS of 93 EOC patients upon stratification by the median expression of genes associated with B cell signature, CD3E, immunosuppression, and TH2 signature, and study arm. Survival curves were estimated using the Kaplan–Meier method, and differences between groups were evaluated using the log-rank test. The numbers of patients at risk and p values are reported.
To assess the prognostic value of immune gene signatures in peripheral blood, we evaluated PFS in distinct clusters of patients. Importantly, among patients treated with DCVAC, we observed worse PFS in the “high” inflammatory cluster than in the low inflammatory cluster (p =0.049). However, we failed to observe a similar trend in SOC patients (Figure 3b, c). In line with these findings, univariate COX regression analyses confirmed negative prognostic role of 5 genes namely CD3E, CD4, forkhead box P3 (FOXP3), granzyme A (GZMA), granzyme B (GZMB), HLA-DOB, and interleukin 4 (IL4), which were associated with poor disease outcomes in DCVAC-treated patients (Table 3).
Table 3.
Univariate Cox proportional hazard analyses for OS in EOC patients from the SOC and DCVAC arms in SOV01.
| SOC arm |
DCVAC arm |
|||||
|---|---|---|---|---|---|---|
| Variable | HR (95% CI) | p-value | Variable | HR (95% CI) | p-value | |
| IL10 | 2.1 (1.3–3.5) | 0.005 | NCR1 | 1.9 (1.2–3) | 0.007 | |
| IL15RA | 5.4 (1.6–1.8) | 0.005 | GZMA | 1.8 (1.1–2.9) | 0.012 | |
| CD8A | 1.6 (1.1–2.2) | 0.009 | IL4 | 2.4 (1.2–4.7) | 0.015 | |
| SMAD3 | 5.1 (1.3–2) | 0.019 | CD4 | 2.4 (1.2–5) | 0.017 | |
| TGFB1 | 3.6 (1.2–11) | 0.023 | CD3E | 2.1 (1.1–4.2) | 0.028 | |
| HLA-DOB | 0.6 (0.3–0.1) | 0.033 | HLA-DOB | 1.6 (1–2.6) | 0.037 | |
| NECTIN2 | 2.6 (1.1–6.4) | 0.033 | GZMB | 1.7 (1–2.9) | 0.044 | |
| IL6 | 0.6 (0.4–0.1) | 0.046 | FOXP3 | 2 (1–3.9) | 0.047 | |
PFS = progression-free survival; EOC = epithelial ovarian cancer; SOC = standard of care chemotherapy; DCVAC, dendritic cell-based vaccination; HR = hazard ratio; CI = confidence interval
To assess the predictive value of the immune gene signature in peripheral blood of EOC patients in SOV01, we directly compared PFS between the high and low inflammatory clusters of patients in both study arms (Figure 3d, e). Importantly, in the low inflammatory cluster, we found that DCVAC conferred a significant PFS advantage compared with their counterparts in the SOC arm (Figure 3d). By contrast, among patients included in the high inflammatory cluster, PFS was not significantly different between patients treated with SOC and DCVAC (Figure 3e). Consistent with this notion, DCVAC was associated with improved PFS compared with SOC among patients with expression levels below the median for gene signatures associated with B (p =0.039) and CD3E (p =0.044), immunosuppression (p =0.041), and TH2 response (p =0.048) in peripheral blood (Figure 3f).
Taken together, these findings indicate that low expression levels of T cells-like genes were associated with improved prognosis in EOC patients who received DC-based immunotherapy, opposite to the findings in mCRPC and NSCLC, where high expression levels were associated with improved OS.
High frequency of regulatory T cells in peripheral blood of EOC patients is associated with a poor response to DCVAC
Considering our findings for the individual cancer types, we next compared the expression levels of all 93 genes among mCRPC, NSCLC and EOC patients to examine whether there are differences in the baseline circulating immunity in distinct malignancies. Notably, we found that the expression levels of 8 genes were significantly higher in EOC patients than in mCRPC and NSCLC patients: arginase 1 (ARG1), FOXP3, interleukin 6 (IL6), interleukin 13 (IL13), programmed cell death 1 (PDCD1; best known as PD-1), transforming growth factor beta 1 (TGFB1), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and tumor necrosis factor A (TNFA) (Figure 4a,b). These findings indicate higher levels of cellular and humoral immunosuppression in peripheral blood of EOC patients compared with NSCLC and mCRPC patients (Figure 4a,b). Consistent with this notion, we observed increased expression of an immunosuppressive-like gene signature (FOXP3, HAVCR2, IDO1, IL10, LAG3, PDCD1, TGFB1, TIGIT) and decreased expression of an immunostimulatory-like gene signature (GNLY, GZMA, GZMB, IFNG, IL12A, PRF1, TBX21, CD8A) in EOC patients in SOV01 than in mCRPC and NSCLC patients in SP005 and SLU01 (Figure 4c). Additionally, mCRPC and NSCLC patients with immunostimulatory gene signatures above median levels showed improved responses to DCVAC (mCRPC: p =0.032; NSCLC: p =0.045) (Figure 4d,). However, the gene expression profile of immunosuppressive signature failed to impact disease outcomes (Supplemental Figure 4A, B). By contrast, DCVAC provided a significant benefit to EOC patients with expression levels of the immunosuppressive gene signature below the median (p =0.025) (Figure 4f), but the immunostimulatory gene signature did not have a significant impact on clinical outcomes (Supplemental Figure 4C).
Figure 4.
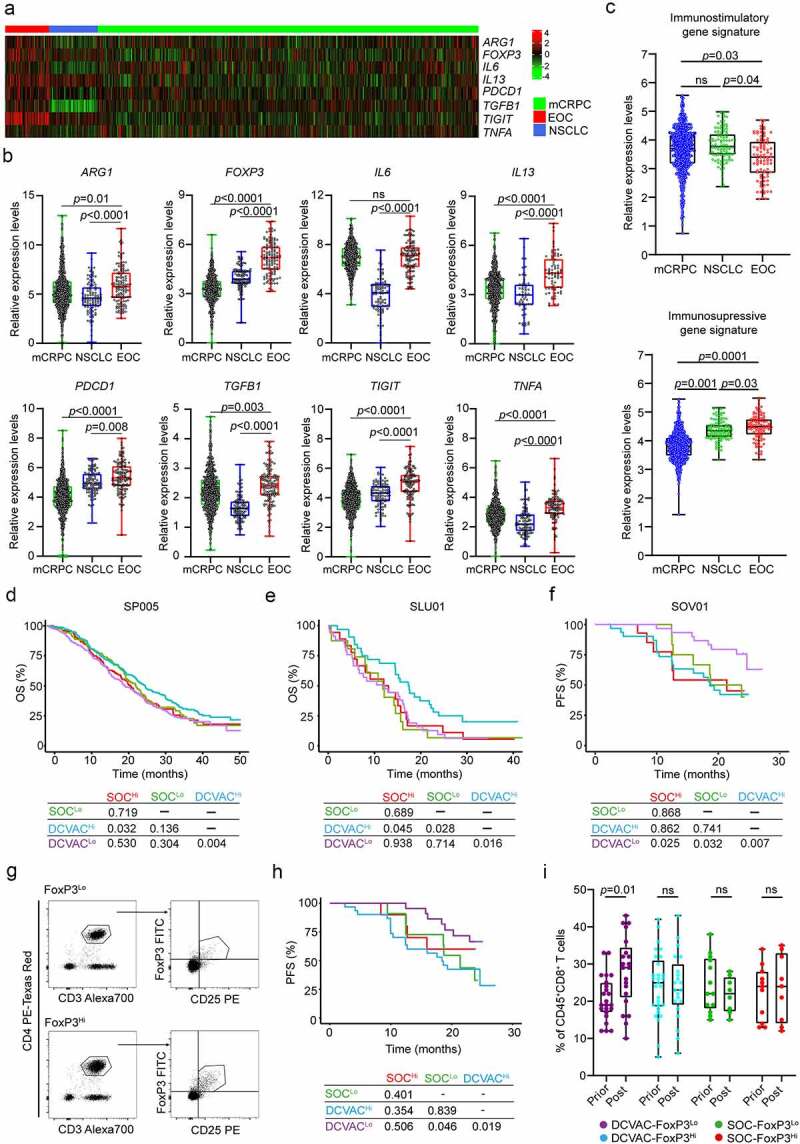
High frequency of regulatory T cells in peripheral blood of EOC patients is associated with poor response to DCVAC therapy. (a) Heat map and (b) relative expression levels of the differentially expressed genes (DEGs) ARG1, FOXP3, IL6, IL13, PDCD1, TGFB1, TIGIT and TNFA in pre-treatment peripheral blood samples among mCRPC, NSCLC, and EOC patients in SP005, SLU01, and SOV01. (c) Relative expression levels of immunostimulatory (CD8A, GNLY, GZMA, GZMB, IFNG, IL12A, PRF1, TBX21) and immunosuppressive (FOXP3, HAVCR2, IDO1, IL10, LAG3, PDCD1, TGFB1, TIGIT) gene signatures in mCRPC, NSCLC and EOC patients in SP005, SLU01, and SOV01. (d, e) OS of 804 mCRPC (d) and 103 NSCLC (e) patients following stratification by the median expression of the immunostimulatory-like gene signature and study arm. (f) PFS of 93 EOC patients following stratification by the median expression of the immunosuppressive-like gene signature and study arm. Survival curves were estimated using the Kaplan–Meier method, and differences between groups were evaluated using the log-rank test. The numbers of patients at risk and p values are reported. (g) Representative dot plots for CD4+CD25+FoxP3+ regulatory T cells in low and high EOC patients in SOV01. (h) PFS of EOC patients treated with SOC or DCVAC stratified by the median percentage of CD4+CD25+FoxP3+ regulatory T cells in peripheral blood. Survival curves were estimated using the Kaplan–Meier method, and differences between groups were evaluated using the log-rank test. (i) Percentage of CD8+ T cells in peripheral blood of SOC FoxP3Lo, SOC FoxP3Hi, DCVAC FoxP3Lo and DCVAC FoxP3Hi patients prior and post DCVAC therapy. Statistical significance was calculated by the Wilcoxon test. p values are indicated.
To investigate the potential impact of immunosuppressive soluble factors on DCVAC activity in EOC patients, we measured the serum levels of IL6, IL10 and IL13. Although high levels of IL6 and IL10 were associated with worse PFS in the SOC arm (IL6: p =0.007; IL10: p =0.021), the serum levels of IL6, IL10, and IL13 were not prognostic and predictive factors in the DCVAC arm (Supplemental Figure 5A-C). These findings suggest that humoral immunosuppression is not associated with the response to DCVAC therapy in EOC patients.
In terms of cellular immunosuppression, we found that DCVAC-treated FOXP3Hi patients did not show a favorable PFS as compared to FOXP3Lo counterparts, indicating a negative impact of immunosuppressive circulating regulatory T cells (Supplemental Figure 4D). To validate these findings using an independent approach, we performed flow cytometry to quantify the frequency of circulating CD4+CD25+FoxP3+ regulatory T cells in pretreatment peripheral blood samples from EOC patients in SOV01 (Figure 4g). The frequency of circulating CD4+CD25+FoxP3+ regulatory T cells was comparable between the DCVAC and SOC arms (Supplemental Figure 4E). To assess the prognostic value of CD4+CD25+FoxP3+ cells in EOC patients, we evaluated PFS after stratifying patients based on the median frequency. FoxP3Lo status was associated with improved PFS, but only in DCVAC-treated patients. These findings may indicate that DCVAC provides a significant PFS benefit in EOC patients with a low frequency of CD4+CD25+FoxP3+ regulatory T cells compared with patients with a high frequency of these cells (Figure 4h). By comparison, we did not observe a prognostic role of FOXP3 expression in PBMCs obtained from mCRPC and NSCLC patients (Supplemental Figure 5 F, G).
To confirm and extend these findings using another technological approach, we analyzed the circulating biomarkers of immune responses mediated by DCVAC therapy in EOC patients after treatment termination by performing flow cytometry (Supplemental Figure 1A). Confirming our transcriptional findings, we found that, although the frequency of circulating CD3+ T cells remained unchanged before and after DCVAC therapy (Supplemental Figure 5 H), there was a significant increase in the frequency of circulating CD8+ T cells in FoxP3Lo patients following DCVAC therapy (Figure 4i). Overall, these findings indicate that DCVAC improved effector functions in the peripheral blood of EOC patients with a low frequency of regulatory T cells that was associated with a significant PFS benefit compared with patients with a high frequency of these cells.
Although these data need to be confirmed in a larger cohort of DCVAC-treated EOC patients, our findings indicate that DCVAC boosts clinically relevant cytotoxic T lymphocyte (CTL) responses, especially in EOC patients with a low frequency of circulating FoxP3+ cells, which is the patient subset that obtained the greatest clinical benefit of DC-based immunotherapy in SOV01.
Discussion
Over the past decade, several immunotherapies have become available for the routine clinical management of cancer.1,29 These include (but are not limited to) ICIs targeting cytotoxic T lymphocyte-associated protein 4 (CTLA4), or PD-1 or its ligand PD-L1 in distinct solid cancer malignancies, including melanoma, NSCLC and urothelial carcinoma.2,30,31 Only about 20% of patients with the most common solid tumors respond to ICIs as standalone therapies.1,2,32 Moreover, some malignancies, particularly prostate and ovarian cancer are insensitive to ICIs as standalone immunotherapies or combined upfront with SOC.33,34 Thus, strategies to induce anticancer immune responses in patients with limited responses to ICIs as well as biomarkers that improve the decision making with respect to the (immuno)therapeutic approach in solid malignancies are eagerly awaited.7,35
DCs are a diverse group of specialized antigen-presenting cells with key roles in the initiation and regulation of innate and adaptive immunity.7,36 The use of DC vaccines for cancer has been extensively investigated, with more than 200 clinical trials completed to date.37–39 Many strategies have been developed to target DCs in cancer, including in situ vaccination approaches, in which DC antigen uptake and immune recognition of tumors is promoted by immunomodulators, as well as the generation of DC-based vaccines.7,40,41 The second approach largely depends on loading DCs with tumor antigens in vitro followed by administration of those DCs to patients, predominantly with melanoma, prostate cancer, glioblastoma, or renal carcinoma.24,42–44 Various types of canonical DC-based cancer vaccines have been explored but with limited clinical benefit, with overall response rates of just 8–15%.37 Thus, strategies to improve the development of anticancer immune responses, implementation of combinatorial immunotherapeutic strategies, and the identification of novel biomarkers for DC-based immunotherapy are needed.7
In line with this notion, we recently reported the results of four randomized clinical trials (SOV01, NCT02107937; SLU01, NCT02470468; SOV02, NCT02107950; SP005, NCT02111577) involving more than 1400 cancer patients demonstrating that DC-based immunotherapy DCVAC is well tolerated and significantly extends PFS and OS over SOC in EOC and NSCLC patients.21,22,45 Despite the favorable safety profile, DCVAC combined with SOC and continued as maintenance treatment did not extend OS in mCRPC patients.24 Here, using peripheral blood samples from 1000 patients enrolled these DCVAC studies, we have demonstrated that a circulating immune-related gene signature associated with adaptive immunity and T cell activation is associated with good prognosis and improved response to DC-based immunotherapy in mCRPC and NSCLC patients in SP005 and SLU01 (Figures 1 and 2). Although the same was not true for EOC patients in SOV01 (Figure 3), we unexpectedly found that DCVAC provided a significant benefit to the low inflammatory cluster of EOC patients. These unexpected findings might be explained by the fact that EOC, as compared with mCRPC and NSCLC, was associated with the lowest expression of the immunostimulatory-like gene signature. Conversely, the immunosuppressive-like gene signature associated with circulating soluble (ARG1, IL6, IL13, TGFB1 and TNFA) and cellular markers (FOXP3, PDCD1 and TIGIT) is over-represented in EOC patients compared with mCRPC and NSCLC patients, as shown by us and others (Figure 4).11,46,47 Supporting this perspective, circulating regulatory T cells, in particular, were shown to abolish the potential of DCs and CTLs for mediating anticancer effects through various mechanisms that included but not were limited to immunosuppressive cytokines, adenosine signaling, CTLA-4-dependent downregulation of CD80 and CD86 expression by a process termed trans-endocytosis, LAG-3 engagement of MHC-II molecules, and direct cytolytic effects mediated by GZMB and PRF1 on CTLs and antigen presenting cells.48–52 Supporting this notion, patients with a low inflammatory immune signature associated with low expression of the immunosuppressive regulatory T cells and TH2-like gene signatures in peripheral blood were shown to be permissive for the effector functions of DCVAC-driven CTLs because systemic immunosuppression has not been established (Figure 4).
These findings demonstrate robust systematic and intratumoral immunosuppression, particularly in EOC, and call for the development of combinatorial treatment strategies.8,53,54 Overcoming the immunosuppression is crucial for improving the response to immunotherapies, including DC-based immunotherapies. Accumulating preclinical and clinical evidence indicates that chemotherapy regimens and targeted anticancer agents used in the management of various malignancies, including EOC, can induce anticancer immunity by various mechanisms, including (1) selective depletion of immunosuppressive cells; (2) lymphodepletion associated with the renovation of the patient’s immunological repertoire; and (3) activation of immune effector cells.54,55 Therefore, chemotherapy and targeted anticancer agents appear to represent promising partners for combination with immunotherapies, and might improve the clinical benefit of DC-based therapies, particularly in combination with ICIs.56–58 However, compared to ICIs where several phase III clinical studies are currently evaluated the synergy with SOC, no advanced studies have focused on their potential synergy with DC-based immunotherapies in EOC patients.54
Our study has various limitations. First, it was an explorative retrospective study focusing on 93 pre-selected genes related to the circulating immune responses to prior therapy, with no preplanned statistical analysis, which limits the statistical power. Second, post-treatment blood samples were not analyzed in the study, which prevented us from investigating the alterations in the anti-tumor immune response elicited by DCVAC.
Because DC-based immunotherapies are promising candidates for management of immunoresistant solid cancers with minimal side effects, additional clinical trials are needed to address the potential value of the immune-related gene signature at baseline to identify biomarkers reflecting the disease origin and potential value of combinatorial approaches that respect the clinical management of individual cancers.22,45 In particular, DC-based immunotherapies combined with ICIs appear to represent an promising strategy because the transferred DCs may encourage initial antigen-specific effector T cell activation, which is eventually curtailed by the coinhibitory activity that is controlled by ICIs.8,59 Thus, clinical studies investigating this synergistic approach are urgently needed.
Supplementary Material
Acknowledgments
This study was sponsored by Sotio Biotech (Czech Republic). JL and AR were supported by the project BBMRI-CZ LM2018125, the program PROGRES Q40/11, the Cooperatio Program (research area DIAG), and the European Regional Development Fund-Project BBMRI-CT.: Biobank network – a versatile platform for the research of the etiopathogenesis of diseases, No: EF16_013/0001674. LR, MHa and JD were supported by the program Cooperatio program 207035, Maternal and Childhood Care, 3rd Faculty Medicine, Charles University. Research in ADG’s lab is supported by Research Foundation Flanders (FWO) (Fundamental Research Grant, G0B4620N to ADG; Excellence of Science/EOS grant, 30837538, for ‘DECODE’ consortium, for ADG), KU Leuven (C1 grant, C14/19/098, C3 grant, C3/21/037, and POR award funds, POR/16/040 to ADG), Kom op Tegen Kanker (Stand Up To Cancer, the Flemish Cancer Society) (KOTK/2018/11509/1, KOTK/2019/11955/1), and VLIR-UoS (iBOF grant, iBOF/21/048, for ‘MIMICRY’ consortium).
Funding Statement
This study was sponsored by Sotio Biotech (Czech Republic).
Contributions
Concept and design: MH, RS, JF; development of methodology: MH, JR, LK, JP, acquisition of data: MH, JR, LK, TL, JP, PH, MH, TH, PK, KS, DR, LS, JD, JL, RH, GH, TB, MH, LR, AL, DC; analysis and interpretation of data: MH, JR, LK, TLS, JP, PH, MH, TH, PK, KS, DR, LS, JD, JL, RH, GH, TB, MH, LR, AL, AC, IV, AG, AL, DC; writing, review, and/or revision of the manuscript: MH, AC, IV, AG, DC, JB, RS, JF; study supervision: MH, JB, RS, JF.
Data availability statement
The data generated in this study are available upon request to the corresponding author.
Disclosure statement
IV declares consulting for AstraZeneca, Clovis Oncology Inc., Carrick Therapeutics, Deciphera Pharmaceuticals, Elevar Therapeutics, F. Hoffmann-La Roche Ltd, Genmab, GSK, Immunogen Inc., Jazzpharma, Mersana, Millennium Pharmaceuticals, MSD, Novocure, Octimet Oncology NV, Oncoinvent AS, Sotio a.s., Verastem Oncology, Zentalis; contracted research for: Oncoinvent AS, Genmab; and research funding from Amgen and Roche. RS and JB are minority shareholders of Sotio. ADG received fees for consultancy, lectures or services from Boehringer Ingelheim (Germany), Miltenyi Biotec (Germany), Isoplexis (USA) and Novigenix (Switzerland). AR declares advisory services and invited lectures for Amgen, AstraZeneca, BMS, Eli-Lilly, Janssen-Cilag, MSD, and Roche. AC is a contracted researcher for Oncoinvent AS and Novocure and a consultant for Sotio Biotech a.s. MH, JR, LK, TL, JF, PH, MH, TH, PK, KS, DR, LS, JB, RS, and JF are employees of Sotio a.s. The other authors declare no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2101596.
References
- 1.Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A.. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10(459). doi: 10.1126/scitranslmed.aat7807. [DOI] [PubMed] [Google Scholar]
- 2.Garon EB, Rizvi NA, Hui R, Leighl N, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–14. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 3.Ansell SM, Lesokhin AM, Borrello I, Halwani A, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang AC, Postow MA, Orlowski RJ, Mick R, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hellmann MD, Callahan MK, Awad MM, Calvo E, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell. 2018;33(5):853–61 e4. doi: 10.1016/j.ccell.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassez A, Vos H, Van Dyck L, Floris G, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med. 2021;27(5):820–832. doi: 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- 7.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 8.Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017;38(8):577–593. doi: 10.1016/j.it.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 10.Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27(2):212–224. doi: 10.1038/s41591-021-01233-9. [DOI] [PubMed] [Google Scholar]
- 11.Sprooten J, Vankerckhoven A, Vanmeerbeek I, Borras DM, et al. Peripherally-driven myeloid NFkB and IFN/ISG responses predict malignancy risk, survival, and immunotherapy regime in ovarian cancer. J Immunother Cancer. 2021;9(11):e003609. doi: 10.1136/jitc-2021-003609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nixon AB, Schalper KA, Jacobs I, Potluri S, Wang IM, Fleener C. Peripheral immune-based biomarkers in cancer immunotherapy: can we realize their predictive potential? J Immunother Cancer. 2019;7(1):325. doi: 10.1186/s40425-019-0799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sprooten J, Coosemans A, Garg AD. A first-in-class, non-invasive, immunodynamic biomarker approach for precision immuno-oncology. Oncoimmunology. 2022;11(1):2024692. doi: 10.1080/2162402X.2021.2024692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scher HI, Graf RP, Schreiber NA, Jayaram A, et al. Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncol. 2018;4(9):1179–1186. doi: 10.1001/jamaoncol.2018.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Duan J, Cai S, Han M, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. 2019;5(5):696–702. doi: 10.1001/jamaoncol.2018.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss GJ, Beck J, Braun DP, Bornemann-Kolatzki K, et al. Tumor cell-free DNA copy number instability predicts therapeutic response to immunotherapy. Clin Cancer Res. 2017;23(17):5074–5081. doi: 10.1158/1078-0432.CCR-17-0231. [DOI] [PubMed] [Google Scholar]
- 18.Hong X, Sullivan RJ, Kalinich M, Kwan TT, et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc Natl Acad Sci U S A. 2018;115(10):2467–2472. doi: 10.1073/pnas.1719264115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee EY, Kulkarni RP. Circulating biomarkers predictive of tumor response to cancer immunotherapy. Expert Rev Mol Diagn. 2019;19(10):895–904. doi: 10.1080/14737159.2019.1659728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Indini A, Rijavec E, Grossi F. Circulating biomarkers of response and toxicity of immunotherapy in advanced non-small cell lung cancer (NSCLC): a comprehensive review. Cancers (Basel). 2021;13(8):1794. doi: 10.3390/cancers13081794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zemanova M, Cernovska M, Havel L, Bartek T, et al. Autologous dendritic cell-based immunotherapy (DCVAC/LuCa) and carboplatin/paclitaxel in advanced non-small cell lung cancer: a randomized, open-label, phase I/II trial. Cancer Treat Res Commun. 2021;28:100427. doi: 10.1016/j.ctarc.2021.100427. [DOI] [PubMed] [Google Scholar]
- 22.Rob L, Cibula D, Knapp P, Mallmann P, et al. Safety and efficacy of dendritic cell-based immunotherapy DCVAC/OvCa added to first-line chemotherapy (carboplatin plus paclitaxel) for epithelial ovarian cancer: a phase 2, open-label, multicenter, randomized trial. J Immunother Cancer. 2022;10(1):e003190. doi: 10.1136/jitc-2021-003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fucikova J, Hensler M, Kasikova L, Lanickova T, et al. An autologous dendritic cell vaccine promotes anticancer immunity in ovarian cancer patients with low mutational burden and cold tumors. Clin Cancer Res. pp.OF1–OF13. 2022. doi: 10.1158/1078-0432.CCR-21-4413 [DOI] [PubMed] [Google Scholar]
- 24.Vogelzang NJ, Beer TM, Gerritsen W, Oudard S, et al. Efficacy and safety of autologous dendritic cell-based immunotherapy, docetaxel, and prednisone vs placebo in patients with metastatic castration-resistant prostate cancer: the VIABLE Phase 3 randomized clinical trial. JAMA Oncol. 2022;8:546. doi: 10.1001/jamaoncol.2021.7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fucikova J, Moserova I, Truxova I, Hermanova I, et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer. 2014;135(5):1165–1177. doi: 10.1002/ijc.28766. [DOI] [PubMed] [Google Scholar]
- 26.Fucikova J, Rozkova D, Ulcova H, Budinsky V, et al. Poly I: c-activated dendritic cells that were generated in CellGro for use in cancer immunotherapy trials. J Transl Med. 2011;9:223. doi: 10.1186/1479-5876-9-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 28.Wu T, Hu E, Xu S, Chen M, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (N Y). 2021;2(3):100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dall’Olio FG, Marabelle A, Caramella C, Garcia C, et al. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2022;19(2):75–90. doi: 10.1038/s41571-021-00564-3. [DOI] [PubMed] [Google Scholar]
- 31.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monk BJ, Colombo N, Oza AM, Fujiwara K, et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22(9):1275–1289. doi: 10.1016/S1470-2045(21)00342-9. [DOI] [PubMed] [Google Scholar]
- 34.Beer TM, Kwon ED, Drake CG, Fizazi K, et al. Randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. 2017;35(1):40–47. doi: 10.1200/JCO.2016.69.1584. [DOI] [PubMed] [Google Scholar]
- 35.Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. doi: 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fucikova J, Palova-Jelinkova L, Bartunkova J, Spisek R. Induction of tolerance and immunity by dendritic cells: mechanisms and clinical applications. Front Immunol. 2019;10:2393. doi: 10.3389/fimmu.2019.02393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sprooten J, Ceusters J, Coosemans A, Agostinis P, et al. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology. 2019;8(11):e1638212. doi: 10.1080/2162402X.2019.1638212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vacchelli E, Vitale I, Eggermont A, Fridman WH, et al. Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology. 2013;2(10):e25771. doi: 10.4161/onci.25771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pol J, Bloy N, Buque A, Eggermont A, et al. Trial watch: peptide-based anticancer vaccines. Oncoimmunology. 2015;4(4):e974411. doi: 10.4161/2162402X.2014.974411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salmon H, Idoyaga J, Rahman A, Leboeuf M, et al. Expansion and activation of CD103(+) Dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44(4):924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garg AD, Vandenberk L, Koks C, Verschuere T, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8(328):328ra27. doi: 10.1126/scitranslmed.aae0105. [DOI] [PubMed] [Google Scholar]
- 43.Sarivalasis A, Boudousquie C, Balint K, Stevenson BJ, et al. A Phase I/II trial comparing autologous dendritic cell vaccine pulsed either with personalized peptides (PEP-DC) or with tumor lysate (OC-DC) in patients with advanced high-grade ovarian serous carcinoma. J Transl Med. 2019;17(1):391. doi: 10.1186/s12967-019-02133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charles J, Chaperot L, Hannani D, Bruder Costa J, et al. An innovative plasmacytoid dendritic cell line-based cancer vaccine primes and expands antitumor T-cells in melanoma patients in a first-in-human trial. Oncoimmunology. 2020;9(1):1738812. doi: 10.1080/2162402X.2020.1738812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cibula D, Rob L, Mallmann P, Knapp P, et al. Dendritic cell-based immunotherapy (DCVAC/OvCa) combined with second-line chemotherapy in platinum-sensitive ovarian cancer (SOV02): a randomized, open-label, phase 2 trial. Gynecol Oncol. 2021;162:652–660. doi: 10.1016/j.ygyno.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Coosemans A, Decoene J, Baert T, Laenen A, et al. Immunosuppressive parameters in serum of ovarian cancer patients change during the disease course. Oncoimmunology. 2016;5(4):e1111505. doi: 10.1080/2162402X.2015.1111505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Bruyn C, Ceusters J, Landolfo C, Baert T, et al. Neo-adjuvant chemotherapy reduces, and surgery increases immunosuppression in first-line treatment for ovarian cancer. Cancers (Basel). 2021;13(23):5899. doi: 10.3390/cancers13235899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang B, Workman C, Lee J, Chew C, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. 2008;180(9):5916–5926. doi: 10.4049/jimmunol.180.9.5916. [DOI] [PubMed] [Google Scholar]
- 49.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 50.Deaglio S, Dwyer KM, Gao W, Friedman D, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11(2):119–130. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kandalaft LE, Odunsi K, Coukos G. Immunotherapy in ovarian cancer: are we there yet? J Clin Oncol. 2019;37(27):2460–2471. doi: 10.1200/JCO.19.00508. [DOI] [PubMed] [Google Scholar]
- 54.Fucikova J, Palova-Jelinkova L, Klapp V, Holicek P, et al. Immunological control of ovarian carcinoma by chemotherapy and targeted anticancer agents. Trends Cancer. 2022;8:426–444. doi: 10.1016/j.trecan.2022.01.010. [DOI] [PubMed] [Google Scholar]
- 55.Petroni G, Buque A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39(3):310–345. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Stewart RA, Pilie PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78(24):6717–6725. doi: 10.1158/0008-5472.CAN-18-2652. [DOI] [PubMed] [Google Scholar]
- 57.Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22(8):1897–1906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 58.Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23(4):487–500. doi: 10.1038/s41590-022-01132-2. [DOI] [PubMed] [Google Scholar]
- 59.Coukos G, Tanyi J, Kandalaft LE. Opportunities in immunotherapy of ovarian cancer. Ann Oncol. 2016;27 Suppl 1:i11–i5. doi: 10.1093/annonc/mdw084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request to the corresponding author.