

ABSTRACT
In Eukarya, immature mRNA transcripts (pre-mRNA) often contain coding sequences, or exons, interleaved by non-coding sequences, or introns. Introns are removed upon splicing, and further regulation of the retained exons leads to alternatively spliced mRNA. The splicing reaction requires the stepwise assembly of the spliceosome, a macromolecular machine composed of small nuclear ribonucleoproteins (snRNPs). This review focuses on the early stage of spliceosome assembly, when U1 snRNP defines each intron 5’-splice site (5ʹss) in the pre-mRNA. We first introduce the splicing reaction and the impact of alternative splicing on gene expression regulation. Thereafter, we extensively discuss splicing descriptors that influence the 5ʹss selection by U1 snRNP, such as sequence determinants, and interactions mediated by U1-specific proteins or U1 small nuclear RNA (U1 snRNA). We also include examples of diseases that affect the 5ʹss selection by U1 snRNP, and discuss recent therapeutic advances that manipulate U1 snRNP 5ʹss selectivity with antisense oligonucleotides and small-molecule splicing switches.
KEYWORDS: RNA splicing, 5’-splice site, U1 snRNP, antisense oligonucleotides, splicing modifiers
Introduction
In the three kingdoms of life, information is encoded in DNA molecules that form the genomes of membrane-based organisms [1]. Archaea, Bacteria and Eukarya share the use of RNA molecules as obligatory mediators of DNA gene expression [1]. RNA molecules are transcribed from genomic DNA and are usually further processed into various categories of mature RNA molecules. RNA classification distinguishes between messenger RNAs (mRNAs) that code for proteins, and noncoding RNAs (ncRNAs) that do not encode proteins [2,3]. Each mRNA molecule is transcribed from a genomic DNA gene, and this is true for Prokarya (i.e. Archaea, Bacteria) and Eukarya. In the early days of RNA biology, a major finding was that mRNA precursors from eukaryotic organisms and viruses may contain intervening sequences that are removed from the immature mRNA transcript (pre-mRNA) to yield a mature mRNA [4–6]. Terminological rules emerged and intervening sequences were termed as introns, while their removal from the pre-mRNA produced a functional mRNA consisting of joined exons. This maturation process occurs within the nucleus and is defined as RNA splicing [7]. This discovery was therefore the basis for investigation of the discontinuous nature of eukaryotic DNA genes, which contrasted with the continuous organization of RNA-coding information in prokaryotic organisms [8]. It is now clear that eukaryotic protein coding genes are generally organized as a succession of exons that code for the sequence of amino acids, and are spaced by introns that do not code for amino acids. Nevertheless, long-standing debates remain on the origin of the eukaryotic gene structure [9,10]. Overall, the splicing reaction is a crucial maturation step by which non-coding sequences are spliced out from the pre-mRNA to yield a functional mRNA [11].
In this review, we briefly introduce the splicing reaction and machinery, and describe how it is important for gene expression and regulation. On this basis, we focus on the molecular details by which the U1 small nuclear ribonucleoprotein (snRNP) defines the intron 5’-splice site (5ʹss) at one end of the intron during pre-mRNA processing. We describe examples in which this 5ʹss selection is perturbed in a variety of diseases, leading to aberrant splicing and the production of a suboptimal to non-functional mRNA. Finally, we discuss recent therapeutic advances to manipulate U1 snRNP 5ʹss selectivity by using synthetic splicing switches.
Splicing description
The overall splicing reaction relies on a highly dynamic macromolecular machine called the spliceosome (Fig. 1) [11]. More than 150–300 proteins enter and exit the spliceosome with each step of the splicing cycle [12,13]. The major spliceosome requires five functional subunits known as small nuclear ribonucleoproteins (snRNPs): each is defined by a snRNP-specific small nuclear RNA (snRNA) to which common and snRNP-specific proteins are bound [14]. Given a prototypical pre-mRNA segment, represented here as exonn–intron–exonn+1, the splicing reaction is defined by the presence of a 5’-splice site (5ʹss) – or donor splice site – at the exonn–intron junction, and by a 3’-splice site (3ʹss) – or acceptor splice site – at the intron–exonn+1 junction. In addition, the branch point (BP) is an intron motif upstream of the 3ʹss that includes an essential adenosine required for the splicing reaction (Fig. 1).
Figure 1.
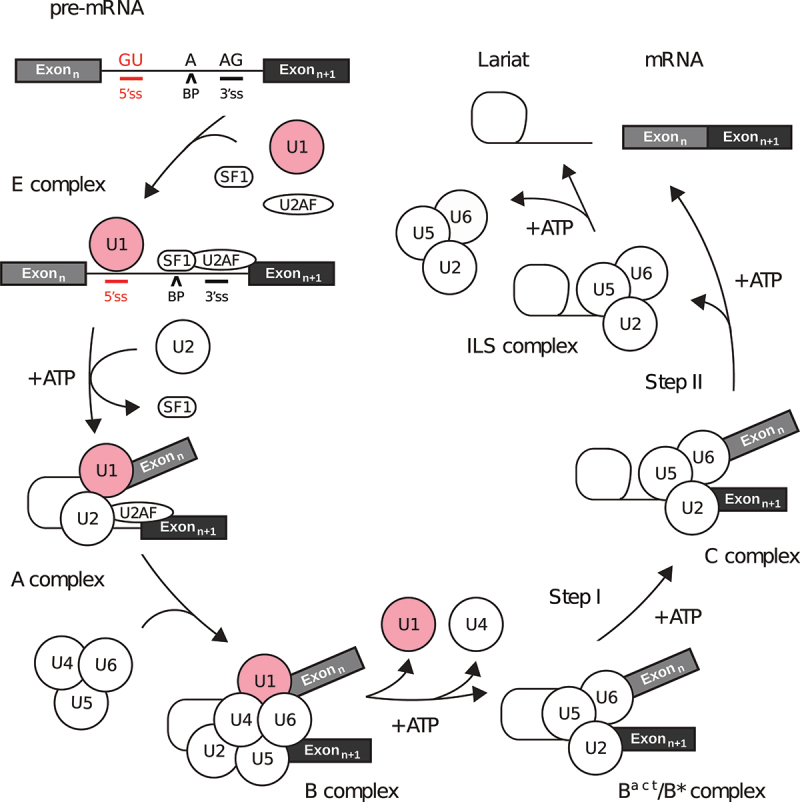
Overview of the splicing reaction. The pre-mRNA must contain at least two exons (boxes) interleaved by one intron (black line) to be relevant for splicing. A single intron is defined by a pair of 5’- and 3’-splice sites (5ʹss, 3ʹss), as well as the branch point (BP) adenosine. Five small nuclear ribonucleoproteins (snRNPs) are the core components of the spliceosome machinery (U1/2/4/5/6) which requires auxiliary factors such as SF1 and U2AF. For each step in the diagram, the name of the corresponding spliceosomal complex is given.
Spliceosome assembly is a stepwise process that begins with the binding of U1 snRNP, SF1 and U2AF proteins to the 5ʹss, branch point and 3ʹss, respectively. The binding of these essential splicing factors results in the formation of the E complex [15]. Next, U2 snRNP is recruited to the branch point by exchanging with SF1, where it interacts with U1 snRNP in an ATP-dependent manner to form the A complex. This leads to the pairing between the splice sites [16]. The tri-snRNP (U4/U6.U5) composed of U4, U5 and U6 snRNP is next recruited by the A complex, displacing U1 snRNP downstream of the 5ʹss, and leading to formation of the pre-B complex [17]. RNP remodelling of the pre-B complex results in the dissociation of U1 snRNP and the pre-catalytic B complex [18]. Further remodelling events induce the release of U4 snRNP, leading to the activated B complex (Bact) [19]. The resulting activated complex is converted into a catalytic pre-branching spliceosome (B*) that performs the first step of splicing. The essential adenine base within the branch point acts as a nucleophile, and attacks a guanine within the 5ʹss to form the spliceosomal C complex [20]. This reaction is followed by numerous RNP rearrangements that include a large-scale movement of U2 snRNP to result in the step II catalytically activated C* complex [21]. Splicing is completed upon ligation between the 3’-end of the cleaved 5ʹss and the 3ʹss, ultimately leading to exon–exon junction and release during the post-catalytic P complex, while the intron lariat to which U2, U5 and U6 remain bound is later released from the ILS complex [22]. In addition, research in multiple model organisms using various approaches have found that splicing is predominantly co-transcriptional, meaning that the spliceosome assembles on the nascent pre-mRNA during transcription [23,24]. Thanks to the cryo-EM revolution, most of the different spliceosome intermediates have now been studied at the atomic level, thus revealing the molecular mechanisms of the splicing reaction [25].
The nuclear spliceosome was also proposed to play a role in the splicing of mitochondrial RNA (mtRNA). The mitochondrial genome contains 13 protein-encoding genes that may contain group I/II introns. They harbour 5’ and 3’ boundaries similar to nuclei-like canonical splice sites and were proposed to be spliced out by the nuclear spliceosome [26]. Furthermore, the loss of U1 snRNP subunits shifts energy metabolism from glycolysis to OXPHOS in a cell-specific manner, in line with the implication of the nuclear spliceosome in the processing of mtRNAs [27].
Alternative splicing
An intron is any segment within a gene that is removed upon splicing, and the 5’- and 3’-splice sites (5ʹss, 3ʹss) define the intron/exon boundaries. Conversely, an exon is any part of a gene that is a part of the mature mRNA. Due to the regulation of splice site recognition, the splicing reaction may create dynamic patterns through alternative splicing, and the underlying molecular regulatory mechanisms are still under investigation. At the start of the Human Genome Project in the 1990s, ~100,000 protein-coding genes were expected to be found [28,29]. In the following decade, estimations based on the consensus human genome revised this number to ~26,000 protein-coding genes, which has now shrunk to ~20000 upon refined analysis of the complete genome [30,31]. From an anterior perspective, this low number of human genes was unexpected when compared with the ~6000 genes that had been predicted from the genome of Saccharomyces cerevisiae (yeast) [32]. The question at that time was how this relatively small difference in the number of protein-coding genes may account for the incredible gap of complexity between yeast and human. This discrepancy was partially answered based on the observation that only ~3% of yeast genes contain introns, whereas more than 97% of human genes have introns [33]. In terms of intron frequency per gene, the average is less than 0.1 in yeast, while it is ~8 in humans, which is the highest value across eukaryotic species [34]. These important differences are correlated with the effective size of the reference proteome for each species: 6050 proteins for yeast (UniProtKB UP000002311) and 79038 proteins for human (UniProtKB UP000005640) [35]. In terms of gene:protein ratio, the average is 1:1 in yeast and 1:4 in humans, although this number does not account for the protein isoforms that remain to be experimentally detected.
The link between intron frequency and the number of proteins associated with one gene is explained at the transcript level by this idea of alternative splicing [36–39]. That is, a single pre-mRNA may produce several mature mRNA transcripts due to a variable exon composition and with striking consequences for the proteome diversity [38,39]. Consistently, the proportion of genes subject to alternative splicing is correlated with the level of complexity of the organism, reaching higher levels in primates [40]. From the perspective of the interactome, interaction profiling experiments have shown that the majority of isoform pairs share less than 50% of their interactions. Alternative splicing thus produces isoforms that behave more like distinct proteins rather than minor variants, which again expands the functional potential of a single gene [41]. Because alternative splicing is achieved by the selection of subsets of 5ʹss and/or 3ʹss in a competitive manner, a small number of mutations within these motifs can dramatically change exon-selection patterns. This property also suggests that alternative splicing is an important contributor to the appearance of novel phenotypes [42,43], and several studies suggest that alternative splicing contributes to accelerated evolutionary changes because it can create evolutionary hotspots within a protein while retaining the original protein sequence [44,45]. In this context, the U1 snRNP has a crucial role because it initiates the splicing reaction by defining the 5ʹss, hence the selection of alternative 5ʹss is directly linked with changes in U1 snRNP recruitment.
Regulation of gene expression
Alternative splicing is an essential regulatory process that uses the intron 5’- and 3’-splice sites (5ʹss, 3ʹss), but with variation arising from distinct mechanisms: alternative first exon (AFE), alternative last exon (ALE), cassette exon, mutually exclusive exons, intron retention and alternative 5ʹss and/or 3ʹss selection (Fig. 2) [46–50]. Alternative splicing relies on alternative splice site selection by components of the spliceosome (i.e. U1 snRNP, U2AF) often based on both the pre-mRNA sequence as well as auxiliary splicing factors. The regulation of splicing is a major pathway in the regulation of gene expression [51], with two distinct but cooperative classes of regulatory elements. The first class is defined by RNA cis-acting regulatory sequences, such as 5ʹss and 3ʹss, branch point (BP), intronic and exonic splicing enhancers (ISE, ESE) and silencers (ISS, ESS) [52]. Proteins define the second class, with trans-acting splicing factors such as SR (Ser-Arg rich) and hnRNP proteins, which bind to cis-acting regulatory sequences in the pre-mRNA [53]. Depending on the position of cis-acting regulatory elements with respect to the 5ʹss, trans-acting splicing factors can act as activators or repressors [54]. Many trans-acting splicing factors are not ubiquitous but are expressed in specific tissues, during development or upon external triggers. Regulation of alternative splicing through tissue-specific trans-acting splicing factors plays an essential role in rewiring downstream protein interaction networks, which is crucial for cell differentiation and organ development [55].
Figure 2.
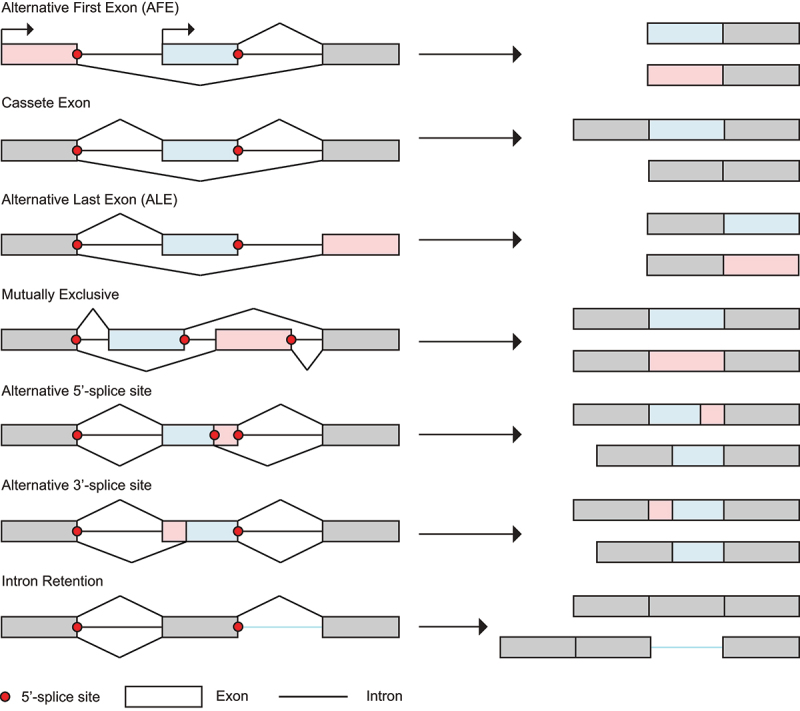
Alternative splicing events. Alternative splicing uses the intron 5’- and 3’-splice sites (3ʹss, 5ʹss), but with variations arising from different mechanisms: alternative first exon (AFE), alternative last exon (ALE), cassette exon, mutually exclusive exons, alternative 5ʹss and/or 3ʹss selection and intron retention.
Regulation of alternative splicing is widespread, and numerous examples exist in the literature [56]. We illustrate this diversity with a few selected cases. As a first example, temperature-dependent effects in gene expression can be linked to alternative splicing, since temperature-sensitive CDC-like kinases (CLKs) phosphorylate SR-proteins at lower temperatures. This post-translational modification of the SR-protein splicing factors in turn triggers changes in splicing patterns, with wide implications in circadian, tissue-specific and disease-associated settings [57]. Physiological splicing patterns may also be hijacked by viruses, such as in the suppression of interferon response upon viral infection [58]. Recent insights also highlight the significance of splicing and isoform-level regulatory mechanisms in promoting an effective immune response to vaccines [59]. In addition to the interplay between cis-acting sequences and trans-acting splicing factors, alternative splicing also integrates genetic and epigenetic factors such as transcriptional elongation, DNA methylation, chromatin architecture and histone modifications to finely tune gene expression patterns in a co-transcriptional context [56]. Finally, pre-mRNA modifications including but not limited to N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), pseudouridine (Ψ) and ribose-methylation (2’-O-Me) can contribute to the regulation of gene expression [60]. For instance, a recent report indicates that m6A methylation of a 3ʹss can block recognition by the essential splicing factors, hence inhibiting splicing [61].
Intrinsic features of the 5’-splice site that modulate U1 snRNP binding
U1 snRNP initiates the splicing reaction upon recognition and binding to the 5’-splice site (5ʹss) to define each exon–intron junction within the pre-mRNA. U1 snRNP consists of the U1 snRNA (164 nt), the seven Sm proteins (Sm-B/Sm-B’, Sm-D1, Sm-D2, Sm-D3, Sm-E, Sm-F, and Sm-G), and three U1-specific proteins (U1-A, U1-C, U1-70 K) [62] (Fig. 3a). The U1 snRNA topology consists of an unpaired 5’-end, a four-way junction of three stem-loops (SL1-3) in a trefoil fold, a Sm site, and a fourth stem-loop at the 3’-end (SL4) (Fig. 3b). With respect to U1 snRNA, U1-C, U1-70 K and U1-A are bound to the 5’-end, SL1 and SL2, respectively. The Sm proteins are arranged in a heptameric ring structure around the Sm site [62] (Fig. 3c). In this section, we describe how the unpaired 5’-end of U1 snRNA can recognize a variety of 5ʹss through specific base-pairing registers, prior to discuss to which extent the intrinsic features of the 5ʹss can drive the recognition by U1 snRNP.
Figure 3.
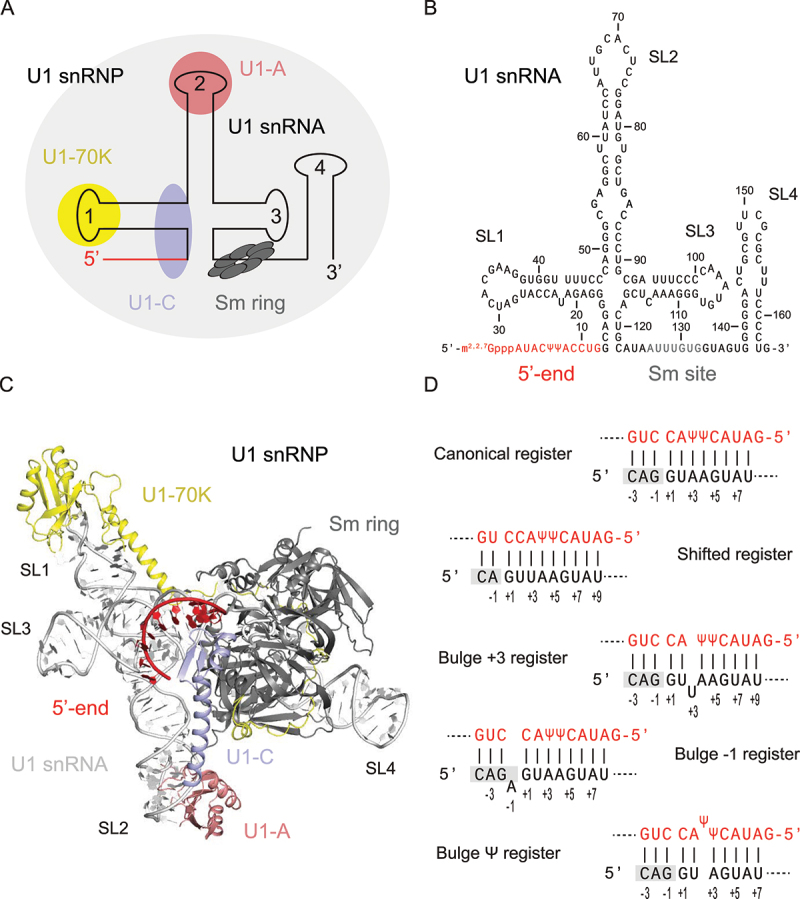
U1 snRNP structure and 5’-splice site (5ʹss) recognition. (a) Summary of U1 snRNP topology. U1 snRNP (light grey) is composed of the U1 snRNA (line), seven Sm proteins (dark grey) and three U1 snRNP specific proteins. (b) U1 snRNA sequence and topology. In U1 snRNA (164 nt), the 5’-end (red) binds to the 5ʹss, the downstream region contains SL1-3, followed by the Sm-site (grey), and SL4 in 3’-end. (c) Structural model of the complete U1 snRNP based on the crystal structure of human spliceosomal U1 snRNP [62]. U1 snRNA (light grey) and its 5’-end (red), the Sm-ring (grey) and U1-specific proteins U1-70 K (yellow), U1-A (pink) and U1-C (purple) are highlighted. (d) Base-pairing registers. U1 snRNA (red) and the 5ʹss (black) can adapt canonical and alternative registers.
U1 snRNA/5’-splice site base-pairing registers
In mammals, the canonical mechanism of 5ʹss recognition by U1 snRNP relies on base-pairing between the 5’-end of U1 snRNA (5’-m2,2,7GpppAUACΨΨACCUG, with Ψ standing for pseudouridine) and the consensus 5ʹss sequence CAG|GUAAGU defined over 9 nucleotides from position −3 to +6 with respect to the exon–intron junction [15] (Fig. 3d). However, the 5ʹss in pre-mRNA only rarely represents an exact matching sequence, but is instead highly degenerate with strict sequence conservation limited to the ubiquitous G+1 and U+2 nucleotides [63]. Within the 5’ end of U1 snRNA, it is suggested that the presence of pseudouridine (Ψ) bases provides an advantage in 5ʹss discrimination [64]. This RNA modification may be required for the stability of the U1 snRNA interaction with 5ʹss, such as in the case of HIV-1 SD4 RNA with Ψ-G base pairing [65]. The general helical U1 snRNA:5ʹss duplex can accommodate alternative registers that include shifted basepairing and non-canonical base-pairing with bulged nucleotides on either the 5ʹss or U1 snRNA strand (i.e. bulge registers) [63,66,67]. Notably, statistical analysis demonstrated that bulge registers are associated with increased alternative 5ʹss selection, yet with a mechanistic basis that remains to be investigated [63]. Furthermore, base pairing between the U1 snRNA and the 5ʹss is sometimes limited by the accessibility of the 5ʹss itself that can be sequestered into inhibitory secondary structure. This strategy is commonly used to regulate alternative splicing of cassette exons such as SMN2 exon 7 [68] or Map/Tau exon 10 [69]. The sequence of the 5ʹss as well as its accessibility are therefore two major components that influence the 5ʹss strength and usage. In the context of this review, the 5ʹss strength relates to the Gibbs free energy change (ΔG) upon formation of the RNA duplex between the 5’-end of U1 snRNA and the 5ʹss, while the 5ʹss usage relates to a splicing event actually occurring at the splice site.
Relevance of the 5’-splice site consensus
The diversity of base-pairing registers explains why the conservation of GU-flanking sequences with respect to 5ʹss consensus is not a reliable predictor of splice site strength or usage, thus complicating de novo predictions of alternative 5ʹss selection. Instead, empirical evidence suggests that 5ʹss selection is correlated with the binding free energy of U1 snRNA:5ʹss base-pairing for strong sites only, in sharp contrast with intermediate and weak splice sites that cannot be distinguished based on this single descriptor [64]. In terms of a general trend, 5ʹss recognition tends to be positively affected by increased base-pair complementarity with the 5’-end of U1 snRNA [70]. It is unclear how an extended complementarity between the segments flanking the 5’-end of U1 snRNA and the 5ʹss could impact splice site recognition and usage. It is suggested that extensive complementarity promotes 5ʹss recognition but leads to an excessively stable U1 snRNA:5ʹss complex, which would be inhibitory to downstream splicing steps [71]. However, it was also reported that an extended complementarity does not decrease splice site usage, but rather increases 5ʹss recognition and exon inclusion [70]. Despite opposite observations on 5ʹss usage, the positive impact of an extended complementarity on 5ʹss recognition is acknowledged on both sides, showing that increased recognition is not necessarily correlated with increased splice site usage.
The highly degenerate nature of 5ʹss sequences, in combination with the modifying influence of nearby cis-acting regulatory motifs, makes it difficult to model relationships between splice site sequence, recognition by U1 snRNA, and splicing efficiency. To understand the relevant numbers involved in these predictions, we can use an example analysis of a 9-nucleotide template of 5ʹss. In such a model system, with nucleotides constrained to NNN|GYNNNN with N ∈ {A,U,G,C}, the set of all unique 5ʹss would contain 74 or 32768 sequences. In a fascinating work, Wong and co-workers released the quantitative activity profile of all unique 5ʹss measured in human cells by using the method of Massively Parallel Splicing Assay (MPSA) with three distinct gene contexts (BRCA2 intron 17, SMN1 intron 7, IKBKAP intron 20) [72]. They found that the splice site sequence is a major determinant of 5ʹss recognition and usage within a given gene context. However, differences are substantial across gene contexts with the same sequence, indicating that the splice site sequence alone is indeed not a reliable descriptor of 5ʹss recognition and usage across multiple gene contexts. Nonetheless, subsets of splice-site sequences or patterns were found to correlate with splicing efficiency across different contexts, hence supporting the existence of context-independent models. For instance, only a minor subset of all GC-based 5ʹss sequences were recognized as functional, supporting that U > C substitution at position +2 may lead to suboptimal sequence for base-pairing with U1 snRNA [72]. Based on BRCA2 and SMN1 studies, the pattern G−1 … G+5 was highly preferential, with a single substitution at either position resulting in lower usage, and non-G substitutions at both positions being highly unfavourable. From this exhaustive search, it is speculated that G-C base-pairing at the −1 and +5 positions may contribute to the strong dependency observed [72].
Computational 5’-splice site prediction
Splice sites define exon/intron boundaries. Therefore, the accurate de novo prediction of splice sites is useful for the annotation of genes, and also to find alternative splice sites associated with diseases. In de novo prediction tasks, consensus sequences based on Position-Weight Matrix (PWM) are not appropriate because they assume statistical independence between positions, which is inaccurate. In the 1980ʹs, computational approaches based on Artificial Neural Networks (ANNs) first appeared in the RNA field to distinguish translational initiation sites, and outperformed consensus-based methods [73]. Among other computational objects that aim to predict splice sites, many Deep Neural Networks (DNNs) with similar architecture were recently released [74–80]. In these tools, the internal representation of the nucleotide sequence is a chain of characters. Instead, the recent use of evolutionary related sequences and multiple sequence alignment (MSA) was crucial to solve other sequence-related problems in biology [81]. Thus, further development in pre-mRNA sequence representation will undoubtedly propel DNN-based prediction of splice sites to the next level, as seen for other sequence-related problems [81].
Stabilization of U1 snRNP on weak 5’-splice sites via protein–protein interactions
While the 5’-end of U1 snRNA recognizes and base-pairs with the 5’-splice site (5ʹss), the recruitment of U1 snRNP to the 5ʹss is also regulated through direct interactions between U1-specific proteins (U1-70 K, U1-A, U1-C) and splicing factors. This mechanism is particularly common when the 5ʹss sequence is considered to be weak in strength. The auxiliary trans-acting splicing factors bind cis-regulatory elements near to the splice site using RNA binding domain(s) and contact the U1 snRNP though protein–protein interactions. The additional interactions provide a secondary link to the pre-mRNA that can result in increased recruitment of the 5ʹss recognition machinery onto otherwise weak splice sites. As detailed below, examples of trans-acting splicing factors have been found to participate in protein–protein interactions with each of the three essential U1-specific proteins.
Via U1-70 K
Serine/arginine-rich splicing factors (SR-proteins) are trans-acting splicing enhancers with a shared topology that includes an N-terminal region with one or two RNA binding domains and a C-terminal RS domain. In general, SR-proteins bind to exonic splicing enhancer (ESE) sequences on the pre-mRNA through their RNA binding domains that belong to the RNA recognition motif (RRM) or zinc finger (Znf) families. A secondary interaction is then primarily within U1 snRNP to aid in its recruitment to the 5ʹss. As a specific example, the SR-protein SRSF1, which contains two RRM domains, can recruit U1 snRNP at the 5ʹss through its interaction with U1-70 K [82] (Fig. 4a). The U1 small nuclear ribonucleoprotein 70 kDa (U1-70 K) contains an N-terminal disordered region, followed by an RRM domain that binds U1 snRNA SL1, and a C-terminal RS domain [62]. It was initially thought that the interaction between SRSF1 and U1-70 K was solely mediated by the C-terminal RS domains found in both proteins, and the phosphorylation of SRSF1 RS domain was shown to be crucial for U1 snRNP recruitment to the 5ʹss [83–86]. Recent reports indicate that the RRM domains of SRSF1 bound to the pre-mRNA could also interact with the RRM domain of U1-70 K, hence bridging the whole U1 snRNP to the pre-mRNA, near to the 5ʹss. This interaction is dependent on the phosphorylation state of SRSF1, because its RRM domains can only participate in intermolecular interactions after hyper-phosphorylation of the RS domain [82]. The U1-70 K mediated recruitment of U1 snRNP to the pre-mRNA by SRSF1 is proposed to seed spliceosome assembly.
Figure 4.
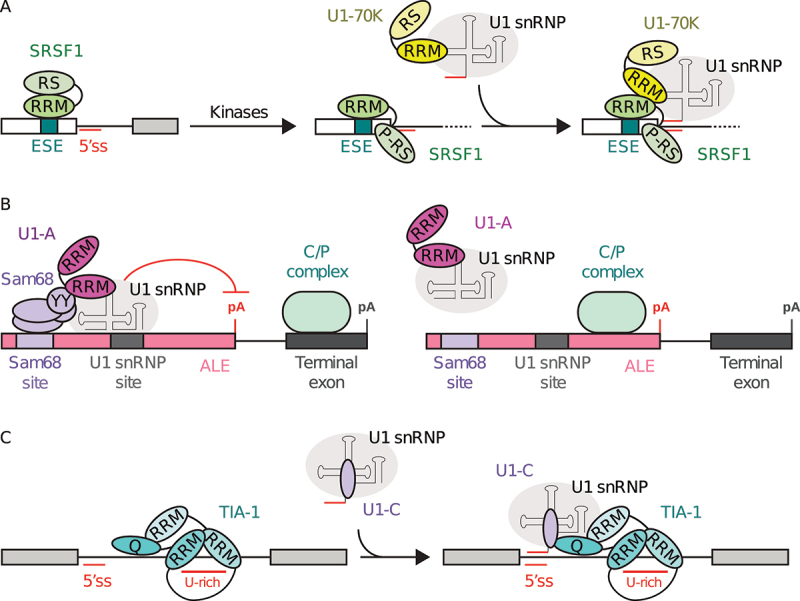
U1 snRNP-specific proteins interact with splicing factors to modulate 5’-splice site (5ʹss) recognition and spliceosome assembly. (a) Recruitment of U1 snRNP at the 5ʹss by SRSF1 upon interaction with U1-70 K [82]. The phosphorylation of ESE-bound SRSF1 in the RS domain makes its RRMs available for interaction with U1-70 K RRM, which contributes to recruiting U1 snRNP to the 5ʹss. (b) Sam68 interaction with U1 snRNP is mediated by U1-A to affect the definition of the alternative last exon [87,89]. The interaction between U1-A RRM1 and Sam68 YY-domain stabilizes the binding of U1 snRNP to the pre-mRNA, which in turn represses the polyadenylation signal and leads to inclusion of the terminal exon. In contrast, the absence of Sam68 does not allow U1 snRNP binding, which results in the inclusion of alternative last exon. (c) Recruitment of U1 snRNP at the 5ʹss by TIA-1 upon interaction with U1-C [94,95]. The binding of TIA-1 RRM1-2 to U-rich sequences downstream of the 5ʹss facilitates the recruitment of U1 snRNP through interactions between TIA-1 Q-rich domain and the U1-C protein.
Via U1-A
Modulation of U1 snRNP 5ʹss selectivity can also rely on the U1 small nuclear ribonucleoprotein A (U1-A) protein, which contains an N-terminal RRM domain (RRM1) bound to U1 snRNA SL2, followed by a long disordered segment and a C-terminal RRM domain (RRM2). U1 snRNP can be recruited to the 5ʹss by the Src associated in mitosis 68 kDa (Sam68) protein, which contains a STAR domain that binds to U(U/A)AA direct repeats in the pre-mRNA, and a C-terminal YY domain shown to interact with the RRM1 domain of the U1-A protein [87]. Sam68 regulates the alternative splicing of the mammalian target of rapamycin (mTor), and the interaction between Sam68 and U1-A promotes U1 snRNP recruitment to the 5ʹss in intron 5 of mTor pre-mRNA [88]. Disruption of the U1-A/Sam68 interaction through mutation of Sam68 or the cis-regulatory element abrogates U1-A mediated recruitment of U1 snRNP at the 5ʹss and splicing [87]. In meiotic cells, Sam68 is highly expressed and regulates alternative last exon (ALE) splicing events in genes required for spermatogenesis [89]. Sam68-regulated ALEs are characterized by the proximity between U1 snRNP and Sam68 binding motifs, and the recruitment of U1 snRNP to Sam68-regulated ALEs is impaired in Sam68−/− germ cells [89]. Upon Sam68 interaction with U1-A, the recruitment of the whole U1 snRNP near to internal polyadenylation sites prevents their recognition by the cleavage and polyadenylation (C/P) complex, abolishing premature transcript termination [89] (Fig. 4b). Overall, Sam68 modulates U1 snRNP recruitement at the 5ʹss through U1-A in a wide range of contexts [87,89].
Via U1-C
While the topology of U1 snRNP is widely conserved in Eukarya, notable variations of U1 snRNP composition can be observed between phyla. The U1 small nuclear ribonucleoprotein C (U1-C) contains an N-terminal zinc finger (Znf) domain and a long C-terminal tail. U1-C binds to the U1 core domain in an U1-70 K-dependent fashion, while the helix A of U1-C binds in the minor groove of the U1 snRNA/5ʹss RNA duplex [15]. In yeast, Nam8 is a constitutive U1-specific protein, and is composed of three RRM domains (RRM1-3) and a C-terminal Q-rich domain [90,91]. TIA-1 is the human homolog of yeast Nam8, although TIA-1 is not part of human U1 snRNP. In the yeast U1 snRNP, the C-terminal moiety of Nam8 interacts with the C-terminal domain of U1-C, while the N-terminal RRM domains of Nam8 binds to U-rich sequences on the pre-mRNA, which helps recruit yeast U1 snRNP to weak splicing sites [92,93]. In humans, TIA-1 enhances splicing of the K-SAM alternative exon that depends on U-rich intronic splicing enhancer sequences (IAS1) immediately downstream the 5ʹss, and in a U1 snRNP-dependent manner [94]. While not part of human U1 snRNP, TIA-1 still interacts with U1 snRNP through the U1-C protein. The Q-rich domain of TIA-1 makes a direct contact with the N-terminal region of U1-C, enhanced by contacts with the RRM1 domain, while the RRM2/3 domains bind the pre-mRNA [95]. The RRM1 and Q-rich domain of TIA-1 mediates the association with U1 snRNP, and both are required to facilitate its recruitment to the 5ʹss [95]. Consistently, TIA-1 is proposed to bind U-rich sequences downstream the 5ʹss of target exons and to recruit U1 snRNP by contacting U1-C [94,96,97] (Fig. 4c).
Stabilization of U1 snRNP on weak 5’-splice sites via protein–RNA interactions
Although the 5’-end of U1 snRNA is the structural segment that actually recognizes and base-pairs with the 5’-splice site (5ʹss), other U1 snRNA segments contribute to splicing regulation as sensors and interaction platforms for signalling. Within U1 snRNP, the stem-loops 3 (SL3) and 4 (SL4) are not bound to proteins and remain exposed to solvent, hence they are available for interactions with splicing modulators (Fig. 3c). The structure of U1 snRNP bound to a short mRNA fragment suggests that SL3 is oriented towards the exon while SL4 faces the downstream intron [17,62]. The analysis of the massive amount of cross-linking and immunoprecipitation (CLIP) data generated by the ENCODE project suggests that U1 snRNA SL3 and SL4 are targets for a number of RNA-binding proteins in vivo, and that competitive binding for these two stem loops would be a major determinant of splicing outcomes in mammalian cells [98,99] (Fig. 5a).
Figure 5.
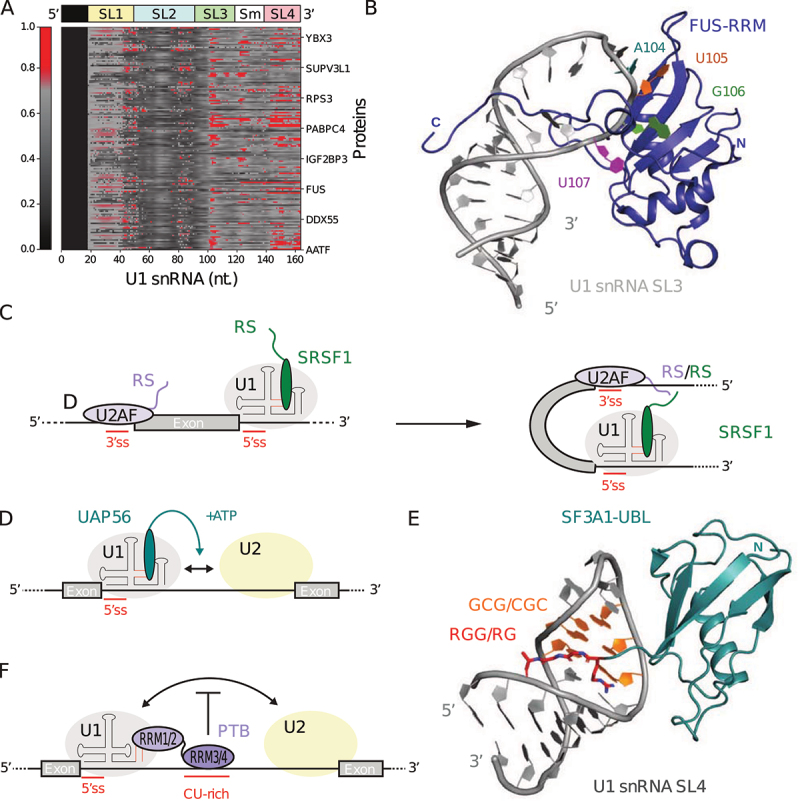
U1 snRNA SL3 and SL4 are targeted by splicing factors to modulate 5’-splice site (5ʹss) recognition and spliceosome assembly. (a) Protein cross-links to U1 snRNA in vivo [98,99]. The heat map shows the distribution of cross-links to U1 snRNA for 147 RNA-binding proteins. (b) Solution structure of FUS-RRM (blue) in complex with U1 snRNA SL3 (grey) (pdb code: 6SNJ) [101]. The structure corresponds to the lowest energy model from the NMR ensemble. (c) Model for exon independent recruitment of SRSF1 by U1 snRNP [98]. (d) Model for UAP56 mediated splicing enhancement [100]. (e) Crystal structure of SF3A1-UBL (cyan) in complex with U1 snRNA SL4 (grey) (pdb code: 7P0V) [108]. (f) Model for PTB mediated splicing repression [110,111].
Role of U1 snRNA SL3
The flexible U1 snRNA SL3 includes a 9 base-pair long stem with a single cytosine bulge and a 7 nt loop (Fig. 3b). Mutations in U1 snRNA SL3 are known to disrupt splicing events, highlighting the functional significance of SL3 in the context of U1 snRNP [100]. In the next paragraphs, we provide examples of interactions between the U1 snRNA SL3 and protein partners and discuss how they contribute to 5ʹss selection or spliceosome assembly.
FUS binds U1 snRNP to modulate 5ʹss selection via U1 snRNA SL3
The interaction between U1 snRNA SL3 and the RNA-binding protein Fused in Sarcoma (FUS) is particularly relevant for disease. Mutations causing Amyotrophic Lateral Sclerosis (ALS) in FUS are reported to result in aberrant contacts with cytoplasmic U1 snRNA at the Sm site, causing disruption of snRNP biogenesis [101]. FUS is a trans-acting splicing factor with a versatile and context-dependent impact on splicing regulation. Physiological and pathological RNA targets of FUS were recently identified using CLIP experiments. FUS strongly associates with pre-mRNAs, and its major nuclear RNA target is the U1 snRNA that is bound by FUS on SL3 [101]. In the solution structure of FUS in complex with a segment of U1 snRNA SL3, the RRM domain interacts with the apical region of U1 snRNA SL3 [101] (Fig. 5b). In this context, the RRM and zinc finger domain of FUS could recognize RNA elements separated by up to 80 Å using a bipartite RNA binding mode [102]. These results suggest that FUS could help position U1 snRNP on weak 5ʹss to modulate RNA splicing and repress premature polyadenylation [101].
Exon-independent recruitment of SRSF1 and exon definition
In the early stages of spliceosome assembly, U1 snRNA SL3 interacts with the SR-rich Splicing Factor 1 (SRSF1), a global trans-acting splicing enhancer [82]. Classically, SRSF1 is recruited by cis-acting exonic splicing enhancer (ESE) sequences, hence contributing to splice site selection by promoting U1 snRNP recruitment. While the interaction of SRSF1 with U1 snRNP has been known for decades, it has previously been attributed to protein–protein interaction between SRSF1 and U1-70 K [82]. Recently, it was demonstrated that SRSF1 binds to U1 snRNP in vitro and that SRSF1ΔSR retains interaction capabilities with U1 snRNP [98]. Using U1 snRNA SL3 alone, binding was still observed, and interaction surface mapping on SRSF1 showed that the RRM1 domain is bound to the CA motif at the 5’ side of the loop, while the RRM2 domain binds the 3’ side of the stem [98,103,104]. Consistently, an original and ESE-independent mechanism for SRSF1 recruitment was proposed, in which a single molecule of SRSF1 can be recruited by U1 snRNP at the 5ʹss through contact mediated by U1 snRNA SL3 [98]. From this perspective, SRSF1 binding to U1 snRNA SL3 enables RS domain interaction between SRSF1 and U2AF complex, facilitating the transition from the spliceosomal E complex to the A complex (Fig. 5c). Finally, the analysis of protein cross-links to U1 snRNA in vivo revealed that SRSF7 and SRSF9 show similar crosslink distribution as compared to SRSF1, suggesting that the interplay with U1 snRNA SL3 may be shared among a subset of SR-proteins [98].
UAP56 facilitates the transition from spliceosomal E to A complex
During spliceosome assembly, the transition from the spliceosomal E complex to the A complex can also be promoted by U1 snRNA SL3 interaction with the 56 kDa U2AF65-Associated Protein (UAP56), a DExD/H-box family RNA helicase involved in mRNA nuclear export and pre-spliceosome assembly. U1 snRNA SL3 interacts with UAP56 in an ATP-dependent manner, facilitating contact between U1 and U2 snRNP and the conversion to the spliceosomal A complex [100] (Fig. 5d). Addition of excess free SL3 in trans enhances splicing upon binding to endogenous UAP56 [100]. UAP56 knockdown or U1 snRNA SL3 mutations are phenotypically equivalent, which results in reduction of exon inclusion and lowered splicing efficiency [100]. The helicase activity of UAP56 may also facilitate the melting of SL3 and the binding of SRSF1. However, sequential binding of proteins or competition for binding on SL3 has not yet been explored experimentally.
Role of U1 snRNA SL4
The rigid stem-loop 4 (SL4) at the 3’-end of U1 snRNA is required for splicing to occur [105]. U1 snRNA SL4 includes a 5 base-pair stem followed by a 2 nt internal loop, a 3 base-pair GCG stem, and an apical UUCG structured tetraloop [106, 107] (Fig. 3b). Herein, we discuss how the interactions between splicing factors and the U1 snRNA SL4 can promote or inhibit the transition from spliceosomal E to A complex.
SF3A1 promotes spliceosomal E to A complex transition
U1 snRNA SL4 is a target of the splicing factor 3 subunit 1 (SF3A1) protein. SF3A1 is a constitutive component of U2 snRNP and interacts with U1 snRNA SL4 within the pre-spliceosomal complex A by mediating contact between the 5ʹss and 3ʹss [109]. The N-terminal RRM domains (RRM1/2) of PTBP1 interact with U1 snRNA SL4, while the C-terminal RRM domains (RRM3/4) bind downstream of the 5ʹss [110,111]. The direct interaction between U1 snRNA SL4 and PTBP1 was recently confirmed in vitro between intact U1 snRNP and either full-length PTBP1 or just the N-terminal RRM domains (RRM1/2) [111]. In the context of the c-src N1 exon, U1 snRNP is known to recognize the 5ʹss, but the presence of PTBP1 results in exon skipping [112]. The binding of PTBP1 to CU-rich motifs downstream of the 5ʹss, and the formation of a ternary complex with nearby U1 snRNP, is thought to prevent contact between U1 and U2 snRNP to inhibit spliceosomal complex A formation [110] (Fig. 5 F). The recent structure of SF3A1-SL4 suggests that PTBP1 and other RNA binding proteins may compete with the ubiquitin-like domain of SF3A1 for the binding to SL4. Thus, U1 snRNA SL4 represents a hotspot for splicing decisions.
Synergistic role of U1 snRNA SL3/4
Together, U1 snRNA SL3 and SL4 also have synergistic roles in maintaining U1 snRNP function, in regards to cross-intron contact with U2 snRNP to drive exon definition [98,100]. As a consequence, the interaction between U1 snRNA SL4 and the U2 snRNP protein SF3A1 is sensitive to events occurring on SL3, such as mutations or knockdown of the SL3 partner UAP56, which abrogates SL4 interaction with SF3A1 [100]. The analysis of protein cross-links to U1 snRNA in vivo also suggests that U1 snRNA SL3 and SL4 are preferential targets for a large number of RNA-binding proteins [98,99] (Fig. 5a). Overall, U1 snRNA SL3 and SL4 are major hubs for splicing regulation through their role in modulating 5ʹss selection, which contributes to exon definition and the transition from the pre-spliceosomal E complex to the A complex in the early stage of the spliceosome assembly.
Altered 5’-splice site selection and diseases
Alternative splicing needs to be tightly regulated to produce isoforms at the correct time and place, and any deviation can thus cause disease. As discussed in the introduction, alternative splicing is a key modulator of gene expression and can occur through distinct mechanisms including alternative 5ʹss and/or 3ʹss selection [50]. Due to the versatility and intricacy of splicing, patterns of mature mRNA transcripts are highly sensitive to single nucleotide polymorphisms (SNP) when they occur within cis-acting regulatory sequences, which explains why defects in RNA splicing have been associated with ~35% of diseases caused by inherited or somatic mutations [113]. It is estimated that ~10% of all disease-causing mutations impact either a 5ʹss or 3ʹss and consistently result in exon skipping, intron retention, or alternative splice site activation [113]. The online database DBASS documents new exon boundaries induced by pathogenic mutations in human disease genes [114]. In the following paragraph, we discuss how inherited diseases and cancers are linked with altered 5ʹss selection by U1 snRNP.
Spinal muscular atrophy (SMA)
Polymorphism at a cis-acting regulatory sequence can modify U1 snRNP 5ʹss selectivity. One of the most studied inherited diseases that falls into this category is spinal muscular atrophy (SMA), the leading genetic cause of infantile death [115]. SMA is an autosomal recessive neuromuscular disease characterized by a progressive degeneration of motor neurons in the spinal cord, leading to muscle weakness and atrophy [115]. More than 95% of SMA cases are caused by a homozygous inactivation of the SMN1 gene, which encodes for the Survival Motor Neuron (SMN) protein [116]. However, a paralogous SMN2 gene is also present in the human genome, and a positive correlation is found with a high copy number of the paralogous SMN2 gene accompanying milder SMA phenotypes [117]. Unfortunately, within the SMN2 gene a C > T mutation at position 6 of exon 7 weakens the 5ʹss, which results in exon 7 skipping for ~85% of spliced transcripts and the production of a truncated, non-functional SMN protein [118]. Because SMN2 is the sole source for SMN protein expression in SMA patients, the small fraction of functional SMN that is still produced is not sufficient to ensure physiological function, but it does explain the relationship between SMN2 copy number and the severity of the disease [117]. Consistently, increasing the amount of functional SMN protein produced by the SMN2 gene is a therapeutical strategy that was proven to be helpful to reduce the severity of the disease, as discussed later in the review.
Huntington disease (HD)
Also linked with U1 snRNP activity, Huntington disease (HD) is a rare and inherited condition that causes degeneration of nerve cells in the brain [119]. This autosomal dominant and progressive neurodegenerative disorder is caused by cytosine–adenine–guanine (CAG) repeat expansions in the exon 1 of the huntingtin gene (HTT), resulting in the production of a mutant huntingtin (mHTT) protein prone to form aggregates [119]. It was proposed that the trans-acting factor SRSF6 binds the CAG expansion in HTT exon 1, which could either interfere with U1 snRNP protection of polyA signals or negatively regulate the 5ʹss of intron 1, resulting in a short transcript that includes intron 1 and leads to pathological mHTT [120]. In this context, lowering the levels of mHTT by promoting the inclusion of a poison exon is a therapeutical strategy currently being investigated.
Familial dysautonomia (FD)
Related to the impairment of 5ʹss recognition, familial dysautonomia (FD) is an inherited autosomal recessive disorder that affects the nerve fibres, causing difficulties to feel pain, temperature, skin pressure, among other related issues [121]. FD is caused by the intervening sequence 20 (IVS20) +6 T > C splicing mutation of the inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein (IKBKAP) gene, causing abnormal exon 20 skipping in the mature mRNA due to weakened recognition of the mutant 5ʹss by U1 snRNP [122]. The loss of exon 20 leads to a frame shift and reduced expression of IKAP protein, compromising tRNA modification and neuronal cell survival [121]. Herein, restoring exon 20 inclusion with splicing modifiers that promotes 5ʹss recognition is a therapeutical strategy under investigations [123].
Hutchinson–Gilford Progeria Syndrome (HGPS)
Point mutations can sometimes create a stronger 5ʹss, like in the rare Hutchinson–Gilford Progeria Syndrome (HGPS). Occurring mainly in patients without familial history, HGPS is an autosomal dominant condition resulting from mutations in the lamin A gene (LMNA), which causes premature ageing and early death from related complications [124]. In ~90% of cases, HGPS is caused by a de novo point mutation C > T in position 1824 of the LMNA gene, within the exon 11, which creates an alternative 5ʹss and results in the production of progerin, a partially deleted form of nuclear lamin A responsible for dysfunctional nuclear membrane and premature senescence [125]. In preclinical studies, it was shown that the pharmacological blockade of the LMNA splice site leading to progerin production was a promising treatment approach for patients affected with HGPS [126].
Emerging role in cancer and global relevance
Finally, deregulation in alternative splicing is increasingly reported to play an important role in tumorigenesis [127]. Aberrant splicing can lead to loss of function in tumour suppressors or activation of oncogenes, which explains how alternative splicing deregulation may promote hallmarks of cancer such as increased cell proliferation, metabolism, genomic instability, and more [128]. For instance, deregulation of alternative 5ʹss selection in intron 2 of the Bcl2L1 gene can increase the ratio between large (Bcl-xL) and small (Bcl-xS) mRNAs that encode for the anti- and pro-apoptotic isoforms, hence contributing to apoptosis inhibition [128]. Many tumour-associated splicing changes arise due to alterations in particular components of the splicing machinery [129,130]. Regarding cis-regulatory sequences, the systematic analysis of multiple cancer types revealed that somatic mutations do not only disrupt canonical splice sites, but can also lead to de novo splice sites in cancer-related genes such as TP53, ATRX, BAP1 CTNNB1, RB1, and more [131]. In addition, recurrent mutation at 5’-end of U1 snRNA were recently correlated with specific tumour types such as Sonic hedgehog (SHH) medulloblastomas, where snRNA-mutant tumours have significantly disrupted RNA splicing patterns [132]. Overall, the relationship between splicing patterns and cancer explains why tumour-associated splicing variants such as Bcl-xL (e.g. lymphoma), Δ4-EGFR (e.g. glioma), Cyclin D1b (e.g. breast cancer), and more, are increasingly proposed as biomarkers in oncology [133].
In summary, mutations in the 5ʹss and other cis-acting regulatory sequences have a critical impact in the early stages of spliceosome assembly and associated clinical outcomes. Accordingly, the scientific community has developed synthetic tools to rationally manipulate 5ʹss selection for therapeutic purposes. In the following part, we discuss recent developments of these technologies that have already delivered innovative therapeutic solutions.
Correction of 5’-splice site selection with antisense oligonucleotides
Antisense therapy is a form of treatment relying on antisense oligonucleotides (ASOs) to target highly specific regions either in the pre-mRNA or mature mRNA. In the context of splicing, ASOs can be directed to mask cis-acting regulatory elements in the pre-mRNA, in order to induce appropriate changes in splicing pattern depending on the disease and specific gene context (Fig. 6 (a,b)). ASOs are typically designed to mask enhancer or silencer sequences near a specific 5ʹss, instead of targeting the splice site itself. By masking the regulatory sequence, the ASO prevents splicing factors from being associated with the pre-mRNA, thereby modulating the recruitment of the U1 snRNP to correct the pathological splicing pattern. The next paragraphs provide examples of ASO-mediated exon inclusion for the treatment of spinal muscular atrophy (SMA) or exon exclusion in the case of Duchene Muscular Dystrophy (DMD). We also describe how ASO could eventually be used for patient-customized therapy.
Figure 6.
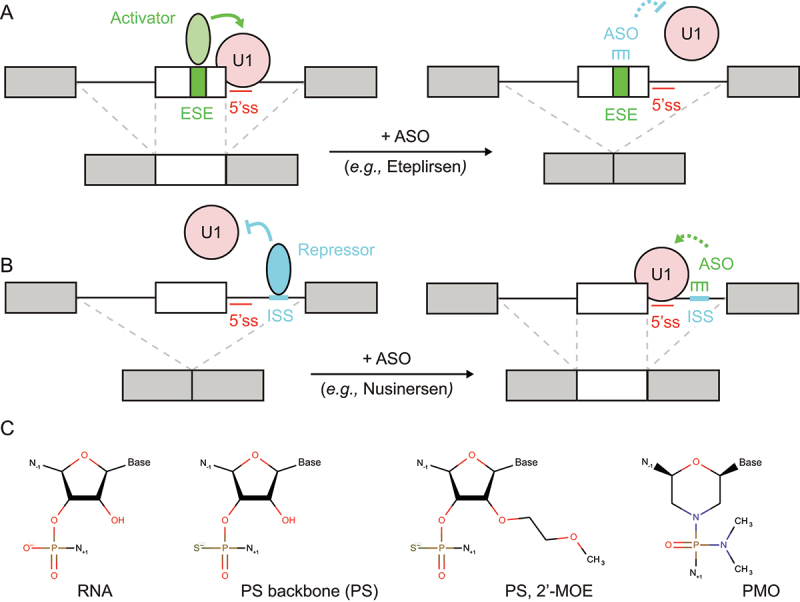
Antisense oligonucleotide (ASO) can modulate 5’-splice site (5ʹss) strength by masking nearby cis-regulatory sequences. (a) Exon skipping. ASO can mask enhancer sequences, promoting exon skipping. (b) Exon inclusion. ASO can mask silencer sequences, promoting exon inclusion. (c) RNA modification. Phosphorothioate (PS) backbone, 2’-O-methoxyethyl (2’-MOE), and phosphorodiamidate morpholino oligomer (PMO).
ASO-mediated exon inclusion
As described previously, in SMA patients the SMN2 gene produces only residual amount of functional Survival Motor Neuron (SMN) protein that is insufficient to compensate for the loss of SMN1 [116,117]. Compared to SMN1, the SMN2 gene has a C > T substitution at position 6 of exon 7 which weakens the 5’-splice site (5ʹss), resulting mostly in exon 7 exclusion and therefore a non-functional protein [118]. Based on this observation, splice-switching ASOs (SSOs) were developed in order to shift splicing towards exon 7 inclusion to recover physiological amount of functional SMN protein [134]. The intronic splicing silencer N1 (ISS-N1) was chosen as the target, since it is located immediately downstream of the weak 5ʹss of SMN2 exon 7 and is the binding site for the trans-acting splicing repressor hnRNPA1 [68,135] (Fig. 6a). The abrogation of hnRNPA1 binding to ISS-N1 by SSOs alleviates repression and mechanically promotes 5ʹss recognition and exon 7 inclusion in vivo [136–141]. On this premise, the SSO drug Nusinersen (SPINRAZA) was developed and thus became the first treatment approved for SMA [142]. Nusinersen is a 18 nt 2’-O-(2-methoxyethyl)-oligoribonucleotide (2’-O-MOE) SSO with a fully modified phosphorothioate (PS) backbone (Fig. 6c). The use of 2’-O-MOE bases and PS backbone confers resistance to nuclease degradation in the body [143]. Nusinersen is administered into the cerebrospinal fluid (CSF) via intrathecal injection with a prolonged half-life of 135–177 days in the CSF. Nusinersen improved neuromuscular functions in infants with SMA and is now used to treat infantile-onset SMA [142].
ASO-mediated exon skipping
In addition to exon inclusion, SSOs can be directed to promote exon skipping. On the one hand, Duchene Muscular Dystrophy (DMD) is caused by mutations in the DMD gene encoding for the dystrophin protein, inducing progressive muscle degeneration and weakness. DMD mutations often result in exon deletion, leading to the creation of premature stop codon and the rapid degradation of dystrophin mRNA by nonsense-mediated decay (NMD) [144]. On the other hand, Becker Muscular Dystrophy (BMD) is a less severe form of dystrophin-related disease. Mutations in the DMD gene are still present but do not disrupt the mRNA open reading frame, which results in a non-physiological dystrophin isoform, but partially retains its function [145]. Therefore, SSOs were designed in order to induce exon skipping in DMD genotypes, to prevent the creation of a premature stop codon and to mimic BMD genotypes. The SSO drug Eteplirsen (EXONDYS 51) was developed on this basis and is approved in the USA [146]. Eteplirsen targets dystrophin pre-mRNA and induces exon 51 skipping, which restores the mRNA open-reading frame and results in a shortened, but functional dystrophin. Eteplirsen binds to cis-acting exonic splicing enhancer (ESE) sequences in exon 51, which presumably prevents the binding of trans-acting splicing activators and subsequent recruitment of U1 snRNP [147] (Fig. 6b). Eteplirsen is a 30 nt SSO with phosphorodiamidate morpholino- (PMO-) based chemical modifications and is indicated for ~14% of all DMD patients. The PMO modification confers resistance to a variety of enzymes, and the drug is administered intravenously on a weekly basis, with a half-life of 3–4 h in the serum [146] (Fig. 6c).
Patient-customized therapy
Notably, SSOs represent a great opportunity for personalized medicine, with a recent report of patient-customized SSO therapy against ceroid lipofuscinosis 7 (CLN7) [148]. CLN7 is a form of Batten’s disease, a rare and fatal neurodegenerative disease, and the case was a six-year-old girl with symptoms that included blindness, ataxia, seizures, and developmental regression. Genetic profiling revealed a relevant mutation in MFSD8 gene with G > C substitution at position 1102, while RNA sequencing revealed mis-splicing of exon 6 into a cryptic splice-acceptor site in MFSD8 intron 6, overall predicting premature translational termination of MFSD8 protein. Based on experience gained from the development of Nusinersen for SMA, SSOs were designed to target enhancer sequences near the cryptic splice-acceptor site. This approach led to the creation of Milasen, a 22 nt SSO with 2’-O-(2-methoxyethyl)-oligoribonucleotide (2’-O-MOE bases) and a fully modified phosphorothioate (PS) backbone [148] (Fig. 6c). After completion of toxicity studies in animals, Milasen was administered to the patient by intrathecal injection over a year on a monthly basis. Milasen treatment tripled the ratio of normal:mutant MFSD8 protein in the patient’s fibroblasts, which alleviated lysosomal dysfunction. This patient-customized SSO therapy objectively reduced the frequency and duration of seizures in the patient, while demonstrating that ASOs can be used for the future development of personalized medicines.
Correction of splicing with small-molecule splicing modifiers
While ASO are powerful tools to correct splicing in disease, this class of drugs cannot cross the blood–brain barrier and therefore may require frequent intrathecal injections, such as for treatment of spinal muscular atrophy (SMA). Orally administered, small-molecule splicing modifiers are being actively developed to make 5’-splice site (5ʹss) correcting therapies more accessible to the patient. As detailed below, there are now examples of small drugs created to promote exon inclusion in the treatment of SMA and Huntington disease (HD). General details on how drugs can achieve splicing correction at the molecular level is also discussed.
Exon inclusion in spinal muscular atrophy (SMA)
For SMA, several orally administered small molecules that modify the splicing of SMN2 gene to promote exon 7 inclusion have been designed [149–151]. Despite their success in restoring effective SMN levels in vitro and in vivo, the initial molecular scaffolds raised safety concerns as assayed by in vitro phototoxicity and mutagenicity (due to coumarins, iso-coumarins) and in vivo toxicity upon long-term exposure at high concentration (from pyrido-pyrimidinones) [151]. In response, a pyridazine scaffold was developed to reduce toxicity. Using this scaffold, branaplam was developed to stabilize the interaction between the spliceosome and SMN2 pre-mRNA to promote exon 7 inclusion [152]. Branaplam showed efficacy in a mouse model of severe SMA with an increase of full-length RNA and protein levels for SMN, and extended survival [153]. Branaplam was the first oral small-molecule splicing modulator tested in SMA Type I patients, but was nevertheless discontinued in phase II/III trials due to approval of similar SMA therapies [154] (Fig. 7a). Among those therapies, drug design efforts led to the discovery of a novel series of SMN-C having a benzamide as a core, which shows an excellent in vitro and in vivo safety and efficacy profiles on two models of SMA mice (adult C/C-allele and neonatal Δ7) [155]. This approach has led to the newest SMA drug, risdiplam, a selective SMN2 splicing modifier [156] (Fig. 7a). The compound has gone through clinical trials for the treatment of SMA in patients of all ages and stages, and is now approved for therapy [157].
Figure 7.
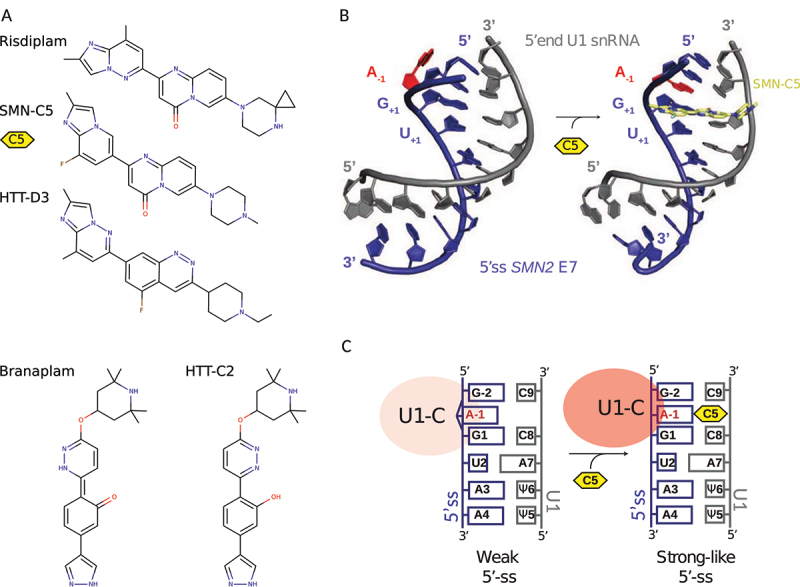
Small-molecule splicing modifiers can strengthen 5’-splice site (5ʹss) interaction with U1 snRNP through the mechanism of bulge-repair. (a) Chemical structure of Risdiplam, Branaplam, and analogues for the treatment of spinal muscular atrophy (SMA) and Huntington disease. (b) Solution structures of U1 snRNA 5’-end (grey) in complex with the 5ʹss (blue) of SMN exon 7, in absence (left, pdb code: 6HMI) and in presence (right, pdb code: 6HMO) of SMNC5 (yellow) [101]. Within the 5ʹss, the A-1 (red) bulging out in absence of small molecule (left) turns inward when SMN-C5 is present (right). (c) The bulge-repair concept. A weak 5ʹss may be strengthened by small-molecule splicing modifiers acting as a glue between U1 snRNP and the 5ʹss.
Pseudoexon inclusion in Huntington disease (HD)
In Huntington disease (HD), one of the key challenges is to ensure optimal delivery and distribution throughout the Central Nervous System (CNS). Therefore, compounds that cross the blood–brain barrier and can be administered orally are a priority for therapy development. As described previously, HD is caused by cytosine–adenine–guanine (CAG) repeat expansions in the huntingtin (HTT) gene, which produces a pathogenic mutant HTT (mHTT) protein [119,158]. In order to lower levels of mHTT, a strategy consists of promoting nonsense-mediated mRNA decay (NMD), a surveillance pathway that eliminates mRNA transcripts that contain premature termination codons. In this context, small-molecule splicing modifiers were designed to promote selective inclusion of a pseudoexon containing a premature termination codon [159]. The selected pseudoexon is located within intron 49 and contains a weak 5ʹss with non-canonical GA dinucleotide at positions −2 and −1; a pattern known to cause inefficient splicing, such as seen in the case of SMN2 exon 7. The initial lead compound HTT-C2 was selected because it strengthens the non-canonical 5ʹss of the selected pseudoexon, hence introducing a premature stop codon that prevents full-length protein production and promotes mRNA degradation via NMD [159] (Fig. 7a). Subsequently, the lead molecule HTT-D3 was developed with improved distribution in the body and was found to result in correlative and equal reduction of mHTT protein levels in plasma and cerebral spinal fluid of Hu97/18 mice [159] (Fig. 7a). Mechanistically, HTT-D3 has a strong preference for the non-canonical AGA|GUAAG 5ʹss, which is similar to the motif recognized by risdiplam. Consistently, branaplam was shown to be effective in mouse models of HD [160]. Overall, this shows that HTT-C2 and analogues strengthen the interaction between U1 snRNP and the 5ʹss, thus enabling exon definition by the spliceosome [159,161].
Molecular basis for splicing correction
How splicing modifiers may stabilize the interaction between the pre-mRNA and the spliceosome was further investigated in the context of spinal muscular atrophy (SMA). Recent efforts have determined the mode of action of the highly selective SMN2 splicing modifier SMN-C5, a chemical analogue of risdiplam with comparable efficacy in vitro and in vivo [161,162]. Combining RNA splicing assays, chemical proteomics, and nuclear magnetic resonance (NMR) spectroscopy, it was found that the drug functions at the molecular level by interacting with a tertiary RNA structure that includes the exonic splicing enhancer (ESE) sequence of SMN2 pre-mRNA exon 7, with the RNA helix formed by the 5ʹss and the 5’-end of U1 snRNA [162]. The solution structures of the intermolecular RNA helix (5ʹss/U1 snRNA) without and with SMN-C5 were solved by NMR spectroscopy, showing how the splicing modifier stabilizes an unpaired adenine in position −1 at the exon–intron junction in the RNA helix base stack through the bulge-repair mechanism [161] (Fig. 7b). An allosteric change caused by SMN-C5 promotes the binding of U1-C zinc finger, and thus whole U1 snRNP, to the 5ʹss of SMN2 pre-mRNA exon 7 (Fig. 7c). This conformational change thereby converts the otherwise weak 5ʹss to a stronger 5ʹss (Fig. 7c). Altogether, the risdiplam analogue SMN-C5 acts as a glue that strengthens the interaction between U1 snRNP and the 5ʹss of SM2 pre-mRNA, thus promoting exon 7 inclusion [161,162].
Conclusion
In recent years, ASOs and small-molecule splicing modifiers that target 5ʹss selection by U1 snRNP were approved for therapy [142,146,148,154,157]. Further research in splicing regulation at the 5ʹss will be key to develop innovative drugs against genetic diseases and cancer. Along these lines, there is a strong need for more systematic research to probe the interplay between 5ʹss and other cis-acting regulatory elements such as enhancer and silencer sequences within introns and exons. In addition, there is a need to investigate the interactome of U1 snRNP, which includes RNA-binding proteins that are associated with U1-specific proteins and U1 snRNA (e.g., SL3-4), all playing a role in 5ʹss selection and spliceosome assembly. Historically, splicing events were believed to be under the sole dependence of cis-acting regulatory motifs on the pre-mRNA, to which trans-acting splicing factors would bind and tune splice site recognition and spliceosome assembly. Therefore, the discovery of U1 snRNP as a binding platform that tunes 5ʹss definition is a conceptual breakthrough, since it shows that splicing can be modulated through U1-mediated protein–protein or protein–RNA interactions, sometimes independently from cis-acting regulatory sequences [82,87,94,98,100,101,105].
Given the variability of splicing patterns across tissues, between individuals, and along time, the pursuit of a set of formal rules that could predict splicing events from RNA features (i.e. a splicing code) remains largely unsolved. An explicit definition of such a code is made even more difficult by the new role of U1 snRNP as a binding platform for splicing factors. Indeed, the U1 snRNP interactome is now considered to be as essential as RNA features and splicing factors, which complicates the determination of the splicing rules. Nevertheless, the accumulation of experimental data sampling the interplay between gene context and splicing outcomes may eventually provide the basis for an implicit definition of the splicing code. Such analysis could use regressions based on Artificial Intelligence (AI) with supervised learning approaches such as Artificial Neural Networks (ANNs) [74–80]. While the latter do not generally provide a meaningful representation of the relationship between parameters and outcome, they may constitute an implicit definition of the splicing code to more reliably predict splicing events based on RNA features, tissue-specific splicing factors, and proteins that interfere with U1 snRNP. This type of modelling would be beneficial to the development of new ASO drugs and small-molecule splicing modifiers. Indeed, ASO development would take great advantage of reliable prediction tools, based on large ensemble of experimental data, in order to know in advance which RNA region to target in the pre-mRNA that will produce the desired splicing outcome. Similarly, small-molecule splicing modifiers that act as a glue between the 5’-end of U1 snRNA and the 5ʹss, in a fashion termed as bulge-repair, may benefit from modelling efforts to identify which particular 5ʹss may be targeted given the molecule [161].
Overall, understanding and correcting pre-mRNA splicing regulation at the 5ʹss provides a fundamental basis for therapy development. ASOs and small-molecule splicing modifiers are effective to treat a growing number of inherited diseases, and they are increasingly considered in oncology where many mutations relevant to splicing were correlated to various types of cancers [127,131]. In this context, the importance of studying U1 snRNP is even raised, because it is the central player to define the 5ʹss.
Funding Statement
This work was funded by the federal council of La Ligue contre le Cancer through the grant ARN 2021.LCC/SeC [to S.C.] and INSERM [to C.D.M and S.C].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Alberts B, Johnson A, and Lewis J.. Molecular biology of the cell. 4th ed. New York: Garland Science. 2003. [Google Scholar]
- [2].Brosius J, Raabe CA. What is an RNA? A top layer for RNA classification. RNA Biol. 2016;13(2):140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hombach S, Kretz M. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3–17. [DOI] [PubMed] [Google Scholar]
- [4].Berget SM, Moore C, Sharp PA. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci USA. 1977;74(8):3171–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Aloni Y, Dhar R, Laub O. Novel mechanism for RNA maturation: the leader sequences of simian virus 40 mRNA are not transcribed adjacent to the coding sequences. Proc Natl Acad Sci USA. 1977;74(9):3686–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Breathnach R, Mandel JL, Chambon P. Ovalbumin gene is split in chicken DNA. Nature. 1977;270(5635):314–319. [DOI] [PubMed] [Google Scholar]
- [7].Crick F. Split genes and RNA splicing. Science. 1979;204(4390):264–271. [DOI] [PubMed] [Google Scholar]
- [8].Sharp PA. On the origin of RNA splicing and introns. Cell. 1985;42(2):397–400. [DOI] [PubMed] [Google Scholar]
- [9].Rogozin IB, Carmel L, Csuros M. Origin and evolution of spliceosomal introns. Biol Direct. 2012;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Poverennaya IV, Roytberg MA. Spliceosomal Introns: features, functions, and evolution. Biochemistry. Biokhimiia. 2020;85(7):725–734. [DOI] [PubMed] [Google Scholar]
- [11].Wilkinson ME, Charenton C, Nagai K. RNA splicing by the spliceosome. Annu Rev Biochem. 2020;89(1):359–388. [DOI] [PubMed] [Google Scholar]
- [12].Rappsilber J, Ryder U, Lamond AI. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12(8):1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hegele A, Kamburov A, Grossmann A. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45(4):567–580. [DOI] [PubMed] [Google Scholar]
- [14].Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3(7):a003707–a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kondo Y, Oubridge C, van Roon AM. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5’ splice site recognition. eLife. 2015;4. DOI: 10.7554/eLife.04986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Plaschka C, Lin PC, Charenton C. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559(7714):419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Charenton C, Wilkinson ME, Nagai K. Mechanism of 5’ splice site transfer for human spliceosome activation. Science. 2019;364(6438):362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bertram K, Agafonov DE, Dybkov O. Cryo-EMStructureofaPre-catalyticHumanSpliceosomePrimedforActivation. Cell. 2017;170(4):701–713.e11. [DOI] [PubMed] [Google Scholar]
- [19].Zhang X, Yan C, Zhan X. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018;28(3):307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhan X, Yan C, Zhang X. Structure of a human catalytic step I spliceosome. Science. 2018;359(6375):537–545. [DOI] [PubMed] [Google Scholar]
- [21].Bertram K, Agafonov DE, Liu WT. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature. 2017;542(7641):318–323. [DOI] [PubMed] [Google Scholar]
- [22].Zhang X, Zhan X, Yan C. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res. 2019;29(4):274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brugiolo M, Herzel L, Neugebauer KM. Counting on co-transcriptional splicing. F1000Prime Rep. 2013;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang S, Aibara S, Vos SM. Structure of a transcribing RNA polymerase II-U1 snRNP complex. Science. 2021;371(6526):305–309. [DOI] [PubMed] [Google Scholar]
- [25].Fica SM, Nagai K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol. 2017;24(10):791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herai RH, Negraes PD, Muotri AR. Evidence of nuclei-encoded spliceosome mediating splicing of mitochondrial RNA. Hum Mol Genet. 2017;26(13):2472–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jourdain AA, Begg BE, Mick E. Loss of LUC7L2 and U1 snRNP subunits shifts energy metabolism from glycolysis to OXPHOS. Mol Cell. 2021;81(9):1905–1919.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].USDepartmentofHealthandHumanService . UnderstandingOurGeneticInheritance:TheUSHumanGenome project: the first five years FY 1991-1995. New York: National Institute of Health; 1990. [Google Scholar]
- [29].Liang F, Holt I, Pertea G. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25(2):239–240. [DOI] [PubMed] [Google Scholar]
- [30].ENCODE project consortium . An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Willyard C. New human gene tally reignites debate. Nature. 2018;558(7710):354–355. [DOI] [PubMed] [Google Scholar]
- [32].Goffeau A, Barrell BG, Bussey H. Life with 6000 genes. Science. 1996;274(5287):546, 563–7. [DOI] [PubMed] [Google Scholar]
- [33].Louhichi A, Fourati A, Rebaï A. IGD: a resource for intronless genes in the human genome. Gene. 2011;488(1– 2):35–40. [DOI] [PubMed] [Google Scholar]
- [34].Roy SW, Gilbert W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet. 2006;7(3):211–221. [DOI] [PubMed] [Google Scholar]
- [35].The UniProt Consortium . UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100–107. [DOI] [PubMed] [Google Scholar]
- [37].Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Severing EI, van Dijk AD, van Ham RC. Assessing the contribution of alternative splicing to proteome diversity in Arabidopsis thaliana using proteomics data. BMC Plant Biology. 2011;11(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu Y, Gonzàlez-Porta M, Santos S. Impact of alternative splicing on the human proteome. Cell Rep. 2017;20(5):1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen L, Bush SJ, Tovar-Corona JM. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol Biol Evol. 2014;31(6):1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang X, Coulombe-Huntington J, Kang S. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016;164(4):805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim E, Magen A, Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35(1):125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen L, Tovar-Corona JM, Urrutia AO. Alternative splicing: a potential source of functional innovation in the eukaryotic genome. Int J Evol Biol. 2012;2012:596274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cusack BP, Wolfe KH. Changes in alternative splicing of human and mouse genes are accompanied by faster evolution of constitutive exons. Mol Biol Evol. 2005;22(11):2198–2208. [DOI] [PubMed] [Google Scholar]
- [45].Xing Y, Lee C. Alternative splicing and RNA selection pressure–evolutionary consequences for eukaryotic genomes. Nat Rev Genet. 2006;7(7):499–509. [DOI] [PubMed] [Google Scholar]
- [46].Chen WH, Lv G, Lv C. Systematic analysis of alternative first exons in plant genomes. BMC Plant Biol. 2007;7(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee S, Wei L, Zhang B. ELAV/Hu RNA binding proteins determine multiple programs of neural alternative splicing. PLoS Genet. 2021;17(4):e1009439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cui Y, Cai M, Stanley HE. Comparative analysis and classification of cassette exons and constitutive exons. Biomed Res Int. 2017;2017:7323508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jin Y, Dong H, Shi Y. Mutually exclusive alternative splicing of pre-mRNAs. Wiley Interdiscip Rev RNA. 2018;9(3):e1468. [DOI] [PubMed] [Google Scholar]
- [50].Monteuuis G, Wong JJL, Bailey CG. The changing paradigm of intron retention: regulation, ramifications and recipes. Nucleic Acids Res. 2019;47(22):11497–11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gehring NH, Roignant JY. Anything but ordinary - emerging splicing mechanisms in eukaryotic gene regulation. Trends Genet. 2021;37(4):355–372. [DOI] [PubMed] [Google Scholar]
- [52].Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14(5):802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012. Jan-Feb;3(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Erkelenz S, Mueller WF, Evans MS. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA. 2013;19(1):96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ellis JD, Barrios-Rodiles M, Colak R. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 2012;46(6):884–892. [DOI] [PubMed] [Google Scholar]
- [56].Dvinge H. Regulation of alternative mRNA splicing: old players and new perspectives. FEBS Lett. 2018;592(17):2987–3006. [DOI] [PubMed] [Google Scholar]
- [57].Haltenhof T, Kotte A, De Bortoli F. A conserved kinase-based body-temperature sensor globally controls alternative splicing and gene expression. Mol Cell. 2020;78(1):57–69.e4. [DOI] [PubMed] [Google Scholar]
- [58].Banerjee AK, Blanco MR, Bruce EA. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183(5):1325–1339.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim EY, Che Y, Dean HJ. Transcriptome-wide changes in gene expression, splicing, and lncRNAs in response to a live attenuated dengue virus vaccine. Cell Rep. 2022;38(6):110341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shi H, Chai P, Jia R. Novel insight into the regulatory roles of diverse RNA modifications: redefining the bridge between transcription and translation. Mol Cancer. 2020;19(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mendel M, Delaney K, Pandey RR. Splice site m6A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell. 2021;184(12):3125–3142.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pomeranz Krummel DA, Oubridge C, Leung AK. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature. 2009;458(7237):475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Roca X, Akerman M, Gaus H. Widespread recognition of 5’ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012;26(10):109–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Roca X, Sachidanandam R, Krainer AR. Determinants of the inherent strength of human 5’ splice sites. RNA. 2005;11(5):683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Freund M, Asang C, Kammler S. A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res. 2003;31(23):6963–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Roca X, Krainer AR. Recognition of atypical 5’ splice sites by shifted base-pairing to U1 snRNA. Nat Struct Mol Biol. 2009;16(2):176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tan J, Ho JX, Zhong Z. Noncanonical registers and base pairs in human 5’ splice-site sel ection. Nucleic Acids Res. 2016;44(8):3908–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Singh NK, Singh NN, Androphy EJ. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chen JL, Zhang P, Abe M. Design, optimization, and study of small molecules that target tau Pre-mRNA and affect splicing. J Am Chem Soc. 2020;142(19):8706–8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Freund M, Hicks MJ, Konermann C. Extended base pair complementarity between U1 snRNA and the 5’ splice site does not inhibit splicing in higher eukaryotes, but rather increases 5’ splice site recognition. Nucleic Acids Res. 2005;33(16):5112–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lund M, Kjems J. Defining a 5’ splice site by functional selection in the presence and absence of U1 snRNA 5’ end. RNA. 2002;8(2):166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wong MS, Kinney JB, Krainer AR. Quantitative activity profile and context dependence of all human 5’ splice sites. Mol Cell. 2018;71(6):1012–1026.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stormo GD, Schneider TD, Gold L. Use of the ’Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 1982;10(9):2997–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bretschneider H, Gandhi S, Deshwar AG. COSSMO: predicting competitive alternative splice site selection using deep learning. Bioinformatics. 2018;34(13):i429–i437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Naito T. Human splice-site prediction with deep neural networks. J Comput Biol. 2018;25(8):954–961. [DOI] [PubMed] [Google Scholar]
- [76].Zuallaert J, Godin F, Kim M. SpliceRover: interpretable convolutional neural networks for improved splice site prediction. Bioinformatics. 2018;34(24):4180–4188. [DOI] [PubMed] [Google Scholar]
- [77].Wang R, Wang Z, Wang J. SpliceFinder: ab initio prediction of splice sites using convolutional neural network. BMC Bioinformatics. 2019;20(Suppl S23):652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535–548.e24. [DOI] [PubMed] [Google Scholar]
- [79].Tayara H, Tahir M, Chong KT. iSS-CNN: identifying splicing sites using convolution neural network. Chemometr Intell Lab Syst. 2019;188:63–69. [Google Scholar]
- [80].Albaradei S, Magana-Mora A, Thafar M. Splice2Deep: an ensemble of deep convolutional neural networks for improved splice site prediction in genomic DNA. Gene: X. 2020;5:100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Jumper J, Evans R, Pritzel A. Highly accurate protein structure prediction with alphafold. Nature. 2021;596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cho S, Hoang A, Sinha R. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci USA. 2011;108(20):8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kohtz JD, Jamison SF, Will CL. Protein-protein interactions and 5’-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368(6467):119–124. [DOI] [PubMed] [Google Scholar]
- [84].Cao W, Garcia-Blanco MA. A serine/arginine-rich domain in the human U1 70k protein is necessary and sufficient for ASF/SF2 binding. J Biol Chem. 1998;273(32):35–40. [DOI] [PubMed] [Google Scholar]
- [85].Yeakley JM, Tronchère H, Olesen J. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J Cell Biol. 1999;145(3):447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein- RNA interactions and is necessary for splicing. Genes Dev. 1997;11(3):499–509. [DOI] [PubMed] [Google Scholar]
- [87].Subramania S, Gagné LM, Campagne S. SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing. Nucleic Acids Res. 2019;47(8):4181–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Huot MÉ, Vogel G, Zabarauskas A. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol Cell. 2012;46(2):187–199. [DOI] [PubMed] [Google Scholar]
- [89].Naro C, Pellegrini L, Jolly A. Functional interaction between U1snRNP and Sam68 insures proper 3’ end Pre-mRNA processing during germ cell differentiation. Cell Rep. 2019;26(11):2929–2941.e5. [DOI] [PubMed] [Google Scholar]
- [90].Dember LM, Kim ND, Liu KQ. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J Biol Chem. 1996;271(5):2783–2788. [DOI] [PubMed] [Google Scholar]
- [91].Wang I, Hennig J, Jagtap PK. Structure, dynamics and RNA binding of the multi-domain splicing factor TIA-1. Nucleic Acids Res. 2014;42(9):5949–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li X, Liu S, Jiang J. CryoEM structure of SaccharomycescerevisiaeU1snRNPoffersinsightintoalternativesplicing. Naturecommunications. 2017;8(1):1035. [Google Scholar]
- [93].Qiu ZR, Schwer B, Shuman S. Determinants of Nam8-dependent splicing of meiotic pre-mRNAs. Nucleic Acids Res. 2011;39(8):3427–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Le Guiner C, Lejeune F, Galiana D. TIA-1 and TIAR activate splicing of alternative exons with weak 5’ splice sites followed by a U-rich stretch on their own pre-mRNAs. J Biol Chem. 2001;276(44):40638–40646. [DOI] [PubMed] [Google Scholar]
- [95].Förch P. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5’ splice sites. EMBO J. 2002;21(24):6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shukla S, Dirksen WP, Joyce KM. TIA proteins are necessary but not sufficient for the tissue-specific splicing of the myosin phosphatase targeting subunit 1. J Biol Chem. 2004;279(14):13668–13676. [DOI] [PubMed] [Google Scholar]
- [97].Förch P, Puig O, Kedersha N. The apoptosis promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol Cell. 2000;6(5):1089–1098. [DOI] [PubMed] [Google Scholar]
- [98].Jobbins AM, Campagne S, Weinmeister R. Exon-independent recruitment of SRSF1 is mediated by U1 snRNP stem-loop 3. EMBO J. 2022;41(1):e107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Van Nostrand EL, Freese P, Pratt GA. A large-scale binding and functional map of human RNA-binding proteins. Nature. 2020;583(7818):711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Martelly W, Fellows B, Kang P. Synergistic roles for human U1 snRNA stem-loops in pre-mRNA splicing. RNA Biol. 2021;18(12):2576–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Jutzi D, Campagne S, Schmidt R. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat Commun. 2020;11(1):6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Loughlin FE, Lukavsky PJ, Kazeeva T. The solution structure of FUS bound to RNA reveals a bipartite mode of RNA recognition with Both sequence and shape specificity. Mol Cell. 2019;73(3):490–504.e6. [DOI] [PubMed] [Google Scholar]
- [103].Cléry A, Sinha R, Anczuków O. Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc Natl Acad Sci USA. 2013;110(30):E2802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Cléry A, Krepl M, Nguyen CKX. Structure of SRSF1 RRM1 bound to RNA reveals an unexpected bimodal mode of interaction and explains its involvement in SMN1 exon7 splicing. Nat Commun. 2021;12(1):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sharma S, Wongpalee SP, Vashisht A. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev. 2014;28(22):2518–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ennifar E, Nikulin A, Tishchenko S. The crystal structure of UUCG tetraloop. J Mol Biol. 2000;304(1):35–42. [DOI] [PubMed] [Google Scholar]
- [107].Martelly W, Fellows B, Senior K. Identification of a noncanonical RNA binding domain in the U2 snRNP protein SF3A1. RNA. 2019;25(11):1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].de Vries T, Martelly W, and Campagne S. Sequence-specific RNA recognition by an RGG motif connects U1 and U2 snRNP for spliceosome assembly. Proc Natl Acad Sci U S A. 2022;119(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Oberstrass FC, Auweter SD, Erat M. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science. 2005;309(5743):2054–2057. [DOI] [PubMed] [Google Scholar]
- [110].Sharma S, Maris C, Allain FH. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell. 2011;41(5):579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Campagne S, de Vries T, Malard F. An in vitro reconstituted U1 snRNP allows the study of the disordered regions of the particle and the interactions with proteins and ligands. Nucleic Acids Res. 2021;49(11):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sharma S, Kohlstaedt LA, Damianov A. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol. 2008;15(2):183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Buratti E, Chivers M, Hwang G. DBASS3 and DBASS5: databases of aberrant 3’- and 5’-splice sites. Nucleic Acids Res. 2011;39(Database issue):D86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–2133. [DOI] [PubMed] [Google Scholar]
- [116].Lefebvre S, Bürglen L, Reboullet S. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):65–155. [DOI] [PubMed] [Google Scholar]
- [117].Mailman MD, Heinz JW, Papp AC. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002. Jan-Feb;4(1):20–26. [DOI] [PubMed] [Google Scholar]
- [118].Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010;24(5):438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Macdonald M, The Huntington’s Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell. 1993;72(6):971–983. [DOI] [PubMed] [Google Scholar]
- [120].Sathasivam K, Neueder A, Gipson TA. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA. 2013;110(6):2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Dietrich P, Dragatsis I. Familial dysautonomia: mechanisms and models. Genet Mol Biol. 2016. Oct-Dec;39(4):497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Ibrahim EC, Hims MM, Shomron N. Weak definition of IKBKAP exon 20 leads to aberrant splicing in familial dysautonomia. Hum Mutat. 2007;28(1):41–53. [DOI] [PubMed] [Google Scholar]
- [123].Ajiro M, Awaya T, Kim YJ. Therapeutic manipulation of IKBKAP mis-splicing with a small molecule to cure familial dysautonomia. Nat Commun. 2021;12(1):4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ahmed MS, Ikram S, Bibi N. Hutchinson-Gilford progeria syndrome: a premature aging disease. Molecular Neurobiology. 2018;55(5):4417–4427. Molecular neurobiology [DOI] [PubMed] [Google Scholar]
- [125].Noda A, Mishima S, Hirai Y. Progerin the protein responsible for the Hutchinson-Gilford progeria syndrome, increases the unrepaired DNA damages following exposure to ionizing radiation. Genes and Environment: the Official Journal of the Japanese Environmental Mutagen Society. 2015;37(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Osorio FG, Navarro CL, Cadiñanos J. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3(106):106ra107. [DOI] [PubMed] [Google Scholar]
- [127].Zhang Y, Qian J, Gu C. Alternative splicing and cancer: a systematic review. Signal Transduct Target Ther. 2021;6(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bonnal SC, López-Oreja I, Valcárcel J. Roles and mechanisms of alternative splicing in cancer -implications for care. Nat Rev Clin Oncol. 2020;17(8):457–474. [DOI] [PubMed] [Google Scholar]
- [129].Dvinge H, Kim E, Abdel-Wahab O. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16(7):413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Rahman MA, Krainer AR, Abdel-Wahab O. SnapShot: splicing alterations in cancer. Cell. 2020;180(1):208–208.e1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Jayasinghe RG, Cao S, Gao Q. Systematic analysis of splice site-creating mutations in cancer. Cell Rep. 2018;23(1):270–281.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Suzuki H, Kumar SA, Shuai S. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. 2019;574(7780):707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bessa C, Matos P, and Jordan P. Alternative splicing: expanding the landscape of cancer biomarkers and therapeutics. Int J Mol Sci. 2020;21(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Meijboom KE, Wood MJA, and McClorey G. Splice-switching therapy for spinal muscular atrophy. Genes (Basel). 2017;8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Beusch I, Barraud P, Moursy A. Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7. eLife. 2017;6. DOI: 10.7554/eLife.25736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hua Y,Vickers TA, Okunola HL. AntisensemaskingofanhnRNPA1/A2intronicsplicingsilencercorrectsSMN2splicingintransgenicmice. Americanjournalofhumangenetics. 2008;82(4):834–848. [Google Scholar]
- [137].Hua Y, Sahashi K, Rigo F. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Porensky PN, Mitrpant C, McGovern VL. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21(7):1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhou H, Janghra N, Mitrpant C. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther. 2013;24(3):331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Rigo F, Chun SJ, Norris DA. Pharmacology of a central nervous system delivered 2’-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hammond SM, Hazell G, Shabanpoor F. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci USA. 2016;113(39):10962–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Singh NN, Howell MD, Androphy EJ. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017;24(9):520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides: a primer. Neurol Genet. 2019;5(2):e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Bushby K, Finkel R, Birnkrant DJ. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. [DOI] [PubMed] [Google Scholar]
- [145].Thada PK, Bhandari J, Umapathi KK. Becker muscular dystrophy, 2022. [PubMed]
- [146].Syed YY. Eteplirsen: first global approval. Drugs. 2016;76(17):1699–1704. [DOI] [PubMed] [Google Scholar]
- [147].McNally EM, Wyatt EJ. Mutation-based therapy for Duchenne muscular dystrophy: antisense treatment arrives in the clinic. Circulation. 2017;136(11):979–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Kim J, Hu C, Moufawad El Achkar C. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381(17):1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Naryshkin NA, Weetall M, Dakka A. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–693. [DOI] [PubMed] [Google Scholar]
- [150].Woll MG, Qi H, Turpoff A. Discovery and optimization of small molecule splicing modifiers of survival motor neuron 2 as a treatment for spinal muscular atrophy. J Med Chem. 2016;59(13):6070–6085. [DOI] [PubMed] [Google Scholar]
- [151].Ratni H, Karp GM, Weetall M. Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. J Med Chem. 2016;59(13):6086–6100. [DOI] [PubMed] [Google Scholar]
- [152].Palacino J, Swalley SE, Song C. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11(7):511–517. [DOI] [PubMed] [Google Scholar]
- [153].Cheung AK, Hurley B, Kerrigan R. Discovery of small molecule splicing modulators of survival motor neuron-2 (SMN2) for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(24):11021–11036. [DOI] [PubMed] [Google Scholar]
- [154].Charnas L, Voltz E, Pfister C. Safety and efficacy findings in the first-in-human trial (FIH) of the oral splice modulator branaplam in type 1 spinal muscular atrophy (SMA): interim results. Neuromuscul Disord. 2017;27:S207–S208. [Google Scholar]
- [155].Pinard E, Green L, Reutlinger M. Discovery of a novel class of survival motor neuron 2 splicing modifiers for the treatment of spinal muscular atrophy. J Med Chem. 2017;60(10):4444–4457. [DOI] [PubMed] [Google Scholar]
- [156].Ratni H, Ebeling M, Baird J. Discovery of Risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15):6501–6517. [DOI] [PubMed] [Google Scholar]
- [157].Dhillon S. Risdiplam: first Approval. Drugs. 2020;80(17):1853–1858. [DOI] [PubMed] [Google Scholar]
- [158].Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci. 2016;18(1):91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Bhattacharyya A, Trotta CR, Narasimhan J. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun. 2021;12(1):7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Keller CG, Shin Y, Monteys AM. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun. 2022;13(1):1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Campagne S, Boigner S, Rüdisser S. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol. 2019;15(12):1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Sivaramakrishnan M, McCarthy KD, Campagne S. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017;8(1):1476. [DOI] [PMC free article] [PubMed] [Google Scholar]