

ABSTRACT
Aggressive triple-negative breast cancer (TNBC) is classically treated with chemotherapy. Besides direct tumor cell killing, some chemotherapeutics such as cisplatin provide additional disease reduction through stimulation of anti-tumor immunity. The cisplatin-induced immunomodulation in TNBC was here investigated in-depth using immunocompetent intraductal mouse models. Upon primary tumor transition to invasive carcinoma, cisplatin was injected systemically and significantly reduced tumor progression. Flow cytometric immunophenotyping was corroborated by immunohistochemical analyses and revealed both differential immune cell compositions and positivity for their programmed death (PD)-1 and PD-ligand (L)1 markers across body compartments, including the primary tumor, axillary lymph nodes and spleen. As key findings, a significant decrease in immunosuppressive and a concomitant increase in anti-tumor lymphocytic cell numbers were observed in the axillary lymph nodes and spleen, highlighting their importance in cisplatin-stimulated anti-tumor immunity. These immunomodulatory effects were already established following the first cisplatin dose, indicating that early cisplatin-mediated events may determine (immuno)therapeutic outcome. Furthermore, a single cisplatin dose sufficed to alleviate anti-PD-1 resistance in a 4T1-based model, providing add-on disease reduction without toxic side effects as seen upon multiple cisplatin dosing. Overall, these results highlight cisplatin as immunotherapeutic ally in TNBC, providing durable immunostimulation, even after a single dose.
KEYWORDS: triple-negative breast cancer, intraductal model, cisplatin, immunomodulation, immunotherapy, tumor immunology
Introduction
Breast cancer is the most diagnosed and deadliest cancer in women worldwide.1 Chemotherapy remains the standard of care treatment for these patients, especially for aggressive subtypes such as triple-negative breast cancer (TNBC).2,3 Chemotherapy efficacy in TNBC is mediated primarily by killing tumor cells, but some chemotherapeutics such as the platinum-based agent cisplatin were also reported to have immunomodulatory properties that create a permissive environment for anti-tumorigenic immune responses.4–6 The current understanding of cisplatin-mediated immunomodulation proposes four key mechanisms:5 (1) the direct reduction of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), (2) the increase of antigen presenting major histocompatibility complex class I (MHC I) expression on tumor cells for recognition by T-cells, (3) the direct recruitment and proliferation of anti-tumorigenic effector cells such as cytotoxic T-lymphocytes (CTLs), and (4) the increased lytic activity of CTLs, e.g. through increased tumor cell sensitivity to perforin/granzyme-mediated killing7 or increased immunogenic cell death stimulating the acquired anti-tumor immunity through the release of immunostimulatory molecules and antigen presentation by, e.g. dendritic cells (DCs).8 Moreover, cisplatin has been reported to influence anti-tumor immunity in different cancer types by modulating both the tumor and immune cell expression of immune checkpoint proteins programmed death (PD)-1 and its ligand (L) PD-L1.9–11 Overall, cisplatin chemotherapy is synergistic with immune checkpoint blockade (ICB, i.e. anti-PD-(L)1 treatment).11–15 The majority of these studies focus on the immunomodulation by frequently dosed cisplatin at the primary tumor level, while its immunomodulation at lower/less toxic dosing frequency and at the systemic level (i.e. in lymphoid and metastasis-bearing tissues), also with regard to the regulation of the PD-(L)1 axis, remains to be explored.
In order to gain a more comprehensive understanding of cisplatin-mediated immunomodulation in TNBC upon different dosing regimens, this study aimed to investigate the effects of cisplatin on immune components at both the local (primary tumor) and the systemic (including axillary lymph nodes and spleen) level using an in-house characterized immunocompetent 4T1-based intraductal model for TNBC.16–18 This model is based on the inoculation of 4T1 mammary tumor cells in the mammary ducts of lactating syngeneic BALB/c mice and ensures that tumor cells initially grow in the mammary ducts, being representative for the ductal carcinoma in situ (DCIS) stage of TNBC in patients. In line with a previous report from our group,19 cisplatin significantly decreased disease progression upon transition from the DCIS to the invasive carcinoma (IC) stage. This decrease was confirmed in an alternative, non-metastatic but highly proliferative Py230-based intraductal model as also characterized by our group.18 In the current study, the immunomodulatory effects of cisplatin were demonstrated in the 4T1-based model through differential changes in immunophenotype and PD-(L)1 immune cell positivity across the investigated body compartments. The lymphoid tissues, more specifically the axillary lymph nodes and spleen, were the most positively impacted sites following cisplatin treatment, with reduced numbers of immunosuppressive and increased numbers of anti-tumorigenic immune cell subsets, thus creating a favorable environment to relieve ICB resistance. Moreover, even a single cisplatin dose induced significant tumor and metastatic reduction as well as add-on disease reduction in combination with anti-PD-1 ICB in the 4T1-based model through early stimulation of anti-tumor immunity, again most significantly in both lymphoid tissues. Collectively, our innovative data in the context of TNBC underline the importance of durable cisplatin-mediated immunomodulation, adding crucial information on the efficacy of chemotherapy in combination with and without anti-PD-1 ICB.
Materials and methods
Cell culture
Firefly luciferase-expressing 4T1 mammary tumor cells that resemble metastatic human TNBC were provided as a kind gift from Prof. Clare Isacke (Breakthrough Breast Cancer Research Center, London, UK) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Thermo Fisher Scientific), 100 U/ml penicillin, and 100 μg/ml streptomycin (both from Sigma-Aldrich, Overijse, Belgium) at 37°C and 5% CO2. Py230 mammary tumor cells, derived from the American Type Culture Collection (ATCC) and reported to model TNBC,18,20 were cultured in Ham’s F-12 K Medium (Thermo Fisher Scientific) supplemented with 5% heat-inactivated FBS (Thermo Fisher Scientific), 0.1% MITO+ Serum Extender (Corning, New York, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich) at 37°C and 5% CO2. Checks for mycoplasma and other bacterial contamination were routinely performed using a PlasmoTestTM mycoplasma detection kit (Invivogen, San Diego, USA). Harvesting of the cells involved washing with phosphate buffered saline (PBS) and incubation with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich) for 5 minutes (min). The cells were subsequently centrifuged at 805 × g for 5 min, resuspended in PBS and counted with a Bürker chamber.
Animals
Eight-week (w) old wild-type BALB/c mice were purchased from Envigo (Horst, The Netherlands), provided with a health monitoring report, for inoculation of 4T1-luc cells. Albino Tyr−/− C57BL/6 mice, having a white coat due to lack of tyrosinase expression, were bred in-house for inoculation of Py230 cells. All animals were conventionally housed with ad libitum access to food and water. The animal research described in this study was conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Committee on the Ethics of Animal Experiments of The Faculty of Veterinary Medicine at Ghent University (approval numbers: EC2018-037, EC2020-014, EC2021-001, and EC2022-009).
Intraductal inoculation and treatment
Intraductal inoculations with 4T1-luc and Py230 cells were performed on lactating female mice, which were obtained by mating with male counterparts and weaning of the suckling pups 2 w after birth. More specifically, 1 hour (h) after weaning, a 32-gauge blunted needle was used to inoculate a 100 µl mixture containing either 5 × 104 4T1-luc cells or 1 × 105 Py230 cells resuspended in cold 1:10 PBS and Matrigel® (Corning) in the mammary ducts of the third mammary gland pair. The inoculations were conducted under inhalational anesthesia using a mixture of oxygen and 2–3% isoflurane and analgesia was provided through intraperitoneal (i.p.) administration of Vetergesic (10 µg/kg buprenorphine diluted in PBS, Val d’Hony-Verdifarm NV, Belgium). Following 3 w (for the 4T1-based model) or 5 w (for the Py230-based model) of tumor growth, cisplatin (6 mg/kg dissolved in 0.9% sodium chloride solution, Sigma-Aldrich) was i.p. administered every 5 d as also performed in a previous study from our group.19 A recombinant mouse monoclonal antibody against PD-1 (Anti-mPD-1-mIgG1e3 InvivoFit™ from Invivogen and derived from clone RMP1-14), specifically designed for in vivo mouse studies, was i.p. administered every w at 200 µg/mouse.
Analysis of disease progression
Body weight and temperature were weekly monitored to determine the health status of the animals during the study. Primary tumor growth was monitored through tumor volume measurements (length x width x height) on the one hand, and in vivo bioluminescence imaging (only in the 4T1-based model using the IVIS lumina II system, PerkinElmer, Zaventem, Belgium) on the other hand. In vivo bioluminescence imaging was performed under inhalation anesthesia and required i.p. injection with D-luciferin (4 mg/200 µl suspended in PBS; PerkinElmer). Bioluminescent signals in primary tumor regions were quantified by placing a region of interest (ROI) circle over the primary tumor area using the accompanying living image analysis software 3.2, and subsequently measuring the total flux density within that ROI. Bioluminescence imaging was also performed on isolated axillary lymph nodes, lungs and spleens to detect and quantify 4T1 metastases. Mice were therefore first sedated with a mixture of ketamine (100 mg/kg, Ketamidor, Ecuphar nv/sa, Oostkamp, Belgium) and xylazine (10 mg/kg, xylazini hydrochloridum, Val d’Hony-Verdifarm, Beringen, Belgium) and subsequently sacrificed through cervical dislocation. Spleen weight was also determined to investigate splenomegaly.
Analysis of protein levels
Proteins were extracted from primary tumor, axillary lymph node and spleen homogenates using lysis buffer as previously described.18 Serum was prepared from cardiac puncture-derived blood that was allowed to clot and centrifuged at 17,000 × g for 1 h. Chitinase 3-like 1 (CHI3L1) and lipocalin 2 (LCN2) levels were measured in primary tumor lysates, sera and spleen lysates using ELISA (Mouse Quantikine ELISA Kit, Bio-Techne, Minneapolis, MN, USA). PD-1 and PD-L1 levels were measured in primary tumor and spleen lysates using Luminex ProcartaPlex Assays (Thermo Fisher Scientific), interferon (IFN)-γ levels were measured in axillary lymph node lysates using a Luminex Simplex Assay and analyzed using the Bio-plex 200 system (Bio-Rad, CA, USA). Protein array analysis (Proteome Profiler Mouse XL Cytokine Array from Bio-Techne) was performed on 200 μg spleen lysates and analyte concentrations were determined using ImageJ. Subtraction of background signals and a normalization to positive controls on the membrane was performed to allow comparison of the results between blots. All assays were performed according to the manufacturer’s instructions.
Reverse transcription-quantitative (RT-q)PCR
RNA was extracted from primary tumors using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands) according to previously developed protocols18,21 to obtain a final elution volume of 70 µl with RNA sample. The RNA quantity and integrity was subsequently determined using the Agilent Bioanalyzer RNA 6000 Nano and cDNA was prepared from 2 µl (i.e. 230–420 ng) RNA sample using the High Capacity cDNA RT kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Nanodrop was used to determine cDNA concentration for subsequent RT-qPCR with 1045–2013 ng cDNA per 10 µl reaction. Mastermixes for the RT-qPCR reactions included Sybr green Perfecta Quanta Supermix (VWR, Leuven, Belgium) and the specific forward/reverse primers. The RT-qPCR was run on the qTower 3 g Real-Time PCR Instrument (Analytik Jena, Jena, Germany) with following cycling conditions: 3 min at 95°C for hot-start and 40 cycli with 15 sec at 95°C for denaturation, 30 sec at 60°C for annealing and 30 sec at 95°C for extension. Finally, a melting curve analysis was performed to control for specificity of the primers. Five candidate reference genes (PRDX1, CTBP, RPL13, USP7, GAPDH with primer sequences described in Table 1) were selected from a previous publication.22 The most stable reference genes were selected as previously described23 based on geNorm24 and NormFinder,25 after which we decided to normalize using CTBP, PRDX1, and RPL13. Hydrolysis probes were used to measure the quantification cycle Cq for PDCD1 (Assay ID Mm00435532_m1, Thermo Fisher Scientific) and CD274 (Assay ID Mm00452054_m1, Thermo Fisher Scientific), which was based on the cycle threshold (Ct) calculated by the qTower software qPCRsoft 4.0 (Analytik Jena). Normalized relative expression for PDCD1 and CD274 was calculated using the SLqPCR version 1.56.0 (Functions for analysis of real-time quantitative PCR data at SIRS-Lab GmbH).
Table 1.
Primer sequences used for RT-qPCR.
| Gene |
Forward primer sequence 5’-3’ |
Reverse primer sequence 5’-3’ |
| PRDX1 | AATGCAAAAATTGGGTATCCTGC | CGTGGGACACACAAAAGTAAAGT |
| CTBP1 | GTGCCCTGATGTACCATACCA | GCCAATTCGGACGATGATTCTA |
| RPL13A | CCCTCCACCCTATGACAAGA | GCCCCAGGTAAGCAAACTT |
| USP7 | CCACAAGGAAAACGACTGGG | GTAACACGTTGCTCCCTGATT |
| GAPDH | CTGGTGCTGCCAAGGCT | CTGCTTCACCACCTTCTTGATGTC ATCATA |
Flow cytometric immunophenotyping
Primary tumors, axillary lymph nodes, lungs, spleen, and blood were isolated from untreated and treated mice upon sacrification. Primary tumors and lungs were digested into single cells using a commercially available Tumor dissociation kit and gentleMACS Dissociator with accompanying C tubes (Miltenyi Biotec, Leiden, The Netherlands) as previously described.18
Axillary lymph nodes and spleens were processed into single cells by pushing the tissue fragments through a 22 G needle on a 1 ml syringe and subsequent filtering through a 70 µm strainer. The cellular solution was centrifuged at 400 × g for 5 min and resuspended in red blood cell lysis buffer (containing 168 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA, all from Sigma-Aldrich). Similarly, blood that was collected through cardiac puncture in heparin-coated tubes, was also centrifuged at 400 × g for 5 min and resuspended in red blood cell lysis buffer. Following a 2 min incubation at room temperature (RT) for cellular solutions derived from axillary lymph nodes and spleens, and a 5 min incubation on ice for cellular solutions derived from blood, DMEM was added and a final centrifugation step of 400 × g for 5 min was performed to pellet the cells. Blood cells were immediately resuspended in FACS buffer (PBS with 1% bovine serum albumin (BSA), 2.5 mM EDTA, and 0.01% sodium azide), whereas axillary lymph node and splenic cells were subjected to a second filtration through a 70 µm strainer to remove cellular clusters that may obstruct the flow cytometer.
In order to count the resulting number of cells, 20 μl of Trucount beads (BD Biosciences) was added to 80 µl of diluted cell suspension in a well of a 96 well plate for analysis on a Cytoflex flow cytometer (Analis, Ghent, Belgium). Based on Trucount technology and by taking the dilution factors into account, the absolute number of cells could be calculated. Cellular viability was also evaluated by adding propidium iodide (PI, 50 μg/ml) to the suspension and by staining with a viobilityTM 488/520 fixable dye (from Miltenyi Biotec) according to the manufacturer’s instructions. The remaining cell solutions were pelleted through centrifugation, resuspended in FACS buffer and subsequently divided across a 96 well plate by bringing 2 × 105–1 × 106 cells in each well. Cellular stainings for identification of immune cell subtypes in each well required a 30 min incubation at 4°C with prepared antibody cocktails supplemented in FACS buffer. Used antibodies and dilutions are described in Table 2. Staining with anti-F4/80-APC, anti-CD206-PE, anti-PD-1-PerCP-eFluor 710, anti-PD-L1-PE-Cy7 and Lineage Cell Detection Cocktail-Biotin (using anti-biotin-APC as secondary antibody) required treatment with FcR blocking reagent (from Miltenyi Biotec, diluted 1:10 in FACS buffer) to reduce aspecific FcR binding. After staining the surface markers and before staining nuclear FoxP3 and cytoplasmic granzyme B, respectively a FoxP3 staining kit and an Inside Stain Kit (both from Miltenyi Biotec) was used to fix and permeabilize the cells according to the manufacturer’s instructions. The Inside Stain Kit was used for additional cytoplasmic pan-cytokeratin staining. Inclusion of isotype-matched controls allowed the detection of positive staining upon flow cytometric analysis. After these stainings, cells were pelleted again at 800 x g for 5 min, washed twice with FACS buffer and resuspended in 100 µl final volume prior to flow cytometric analysis. The event recording threshold was set at 30,000 events in the viable CD45+ gate for all samples except for intracellular markers, which was set either at 60 000 events, because of the expected low positive percentage, or in a measuring time of 85 sec (acquisition velocity 60 µl/min). The collected data were processed using CytExpert v2.0.0.153 software (Beckman Coulter, Inc., California, USA).
Table 2.
Fluorescent antibodies used for flow cytometric immunophenotyping.
| Target |
Fluorophore |
Clone |
Dilution |
Supplier |
| CD45 | VioBlue | REA737 | 1:50 | Miltenyi Biotec |
| CD11b | APC-Vio770 | REA592 | 1:50 | Miltenyi Biotec |
| CD14 | PE-Vio770 | REA934 | 1:50 | Miltenyi Biotec |
| Ly6C | PE | REA796 | 1:50 | Miltenyi Biotec |
| Ly6G | APC | REA526 | 1:50 | Miltenyi Biotec |
| F4/80 | APC | Cl:A3-1 | 1:20 | Bio-Rad |
| CD206 | PE | C068C2 | 1:40 | BioLegend |
| MHC II | PE | REA813 | 1:50 | Miltenyi Biotec |
| CD80 | PerCP-Vio700 | REA983 | 1:50 | Miltenyi Biotec |
| CD11c | PE | REA754 | 1:50 | Miltenyi Biotec |
| CD3ε | APC-Vio770 | REA606 | 1:50 | Miltenyi Biotec |
| CD4 | PE or PE-Vio770 | REA604 | 1:50 | Miltenyi Biotec |
| FoxP3 | PE | REA788 | 1:50 | Miltenyi Biotec |
| CD8α | APC | REA601 | 1:50 | Miltenyi Biotec |
| Granzyme B | PE | REA226 | 1:50 | Miltenyi Biotec |
| CD19 | PE | REA749 | 1:50 | Miltenyi Biotec |
| CD45R (B220) | PE-Vio770 | REA755 | 1:50 | Miltenyi Biotec |
| CD335 (NKp46) | APC | REA815 | 1:50 | Miltenyi Biotec |
| Biotin | APC | REA746 | 1:50 | Miltenyi Biotec |
| Pan-cytokeratin | APC | C-11 | 1:50 | Abcam |
| PD-1 | PerCP-eFluor 710 | J43 | 1:160 | Thermo Fisher Scientific |
| PD-L1 | PE-Cy7 | 10F.9G2 | 1:200 | BioLegend |
Histology and immunohistochemistry
Primary tumor, axillary lymph node, lung, and spleen tissue were fixed for 24 h in 3.5% buffered formaldehyde for subsequent embedding in paraffin. Histology required deparaffinization and rehydration of 5 µm sections followed by short (5 min) incubation in hematoxylin and eosin (H&E) staining buffers. Microscopic analysis subsequently required dehydration and mounting with a cover glass.
Immunohistochemistry required pressurized antigen retrieval of 2–3 µm sections at 95°C for 30 min using either 10 mM tri-sodium citrate buffer (Santa Cruz Biotechnology, Heidelberg, Germany) at pH 6 or 10 mM Tris-1 mM EDTA buffer (Thermo Fisher Scientific) at pH 9, both containing 0.05% Tween-20 (Sigma-Aldrich) as previously described.18,19 Following a 10 min blocking step for endogenous peroxidase (using 3% H2O2) and aspecific binding (using serum-free protein block from Dako, Heverlee, Belgium), stainings with primary antibodies diluted in Antibody diluent (Dako) were performed for 1 h. Primary antibodies, clones and dilutions used in the study are described in Table 3. Secondary antibodies were incubated for 30 min and included either Rat-on-Mouse HRP-Polymer (Biocare Medical, CA, USA) or Dako EnVision+ Rabbit (Dako). Visualization of positive staining required treatment with 3,3′-diaminobenzidine (DAB)-containing buffer (Dako) for 10 min, counterstaining with hematoxylin for 1–5 min and dehydration of the tissue slides for mounting. Throughout the staining, slides were kept in a humidified box and were washed between each incubation step with tris-buffered saline (TBS, Biocare Medical). ImageJ was applied to quantify positive staining (color deconvolution and automatic counting of % area), ImageJS was applied to determine Ki67 proliferation indices as previously described.18,19,26
Table 3.
Primary antibodies used for immunohistochemistry.
| Target |
Host species |
Clone |
Dilution |
Antigen retrieval |
Supplier |
| Ki67 | Rabbit | SP6 | 1:50 | Citrate pH 6 | Thermo Fisher Scientific |
| CD31 | Rabbit | EPR17259 | 1:2000 | Citrate pH6 | Abcam |
| CAIX | Rabbit | NB100-417 | 1:1000 | Citrate pH6 | Novus Biologicals |
| CD45 | Rabbit | polyclonal | 1:1000 | Citrate pH6 | Abcam |
| CD11b | Rabbit | EPR1344 | 1:2000 or 1:4000 | Citrate pH6 | Abcam |
| Ly6G | Rat | 1A8 | 1:1000 | Citrate pH6 | BioLegend |
| CD163 | Rabbit | EPR19518 | 1:500 | Citrate pH6 | Abcam |
| CD11c | Rabbit | D1V9Y | 1:100 | Citrate pH6 | Cell Signaling Technology |
| α-SMA | Rabbit | EPR5368 | 1:2000 | Citrate pH6 | Abcam |
| FAP-α | Rabbit | polyclonal | 1:500 | Tris/EDTA pH9 | Abcam |
| CD3ε | Rabbit | EPR20752 | 1:1000 | Citrate pH6 | Abcam |
| FoxP3 | Rat | FJK-16s | 1:100 | Citrate pH6 | Thermo Fisher Scientific |
| CD19 | Rat | 6OMP31 | 1:1000 | Citrate pH6 | Thermo Fisher Scientific |
| NCR-1 | Rabbit | EPR23097-35 | 1:500 | Tris/EDTA pH9 | Abcam |
| granzyme B | Rabbit | polyclonal | 1:1000 | Citrate pH6 | Abcam |
Statistical analysis
Student’s t-tests and analysis of variance (ANOVA) testing with Newman–Keuls post hoc test for multiple comparison were performed in Prism GraphPad. Whenever necessary, data were normalized through log10 transformation.
Results
Multiple cisplatin dosing reduces progression, proliferation, and vascularity in the primary tumor, but has general cytotoxic effects
Upon their inoculation through the teat canal, 4T1 mammary tumor cells progressively grow from within the mammary ducts to break through the epithelial barrier and invade the mammary fat pad over a period of 3 w. As this transition to IC is a prognostic indicator for chemotherapy in the clinic, cisplatin treatment was started at 3 w p.i. and doses were provided every 5 d up to 6 w p.i. (Figure 1a).
Figure 1.

Multiple cisplatin dosing reduces disease progression in a 4T1-based intraductal model. (a) Experimental timeline with H&E histology of primary tumors showing initial growth of intraductally inoculated tumor cells in the mammary ducts (indicated by the arrow) at 1 w p.i. and transition to invasive carcinoma at 3 w p.i. Scale bars = 50 µm. (b) Primary tumor volume measurements as indication for primary tumor growth (n = 18 for both groups). (c) In vivo imaging of bioluminescence signals in the primary tumor (indicated as total flux density in p/s/cm2) for verification of primary tumor growth (n = 6 for the untreated group, n = 8 for the 5x cisplatin-treated group). (d) Representative images of the bioluminescent signals in primary tumors from untreated and 5x cisplatin-treated mice at 6 w p.i. (e, f) Quantification based on total flux density (in p/s/cm2) in axillary lymph nodes (e) and lungs (f) of untreated (n = 9 ax. lymph nodes and n = 3 lungs) and 5x cisplatin-treated mice (n = 12 ax. lymph nodes and n = 4 lungs) at 6 w p.i. Representative images of bioluminescence signals derived from 4T1 metastases in axillary lymph nodes and lungs of untreated and 5x cisplatin-treated mice at 6 w p.i. are also shown. (g, h) H&E histology of axillary lymph node (g) and lung (h) metastases in untreated and 5x cisplatin-treated mice at 6 w p.i. Scale bars = 200 µm. (i) Representative images of the spleen from untreated and 5x cisplatin-treated mice at 6 w p.i., with the spleen of a healthy mouse also highlighted for comparison. (j) Weight measurements of the spleen from untreated (n = 3) and 5x cisplatin-treated mice (n = 4) at 6 w p.i. The dotted line represents the mean spleen weight of 4 healthy mice. (k, l) CHI3L1 (k) and LCN2 (l) biomarker levels in primary tumor, serum and spleen from untreated (n = 4) and 5x cisplatin-treated mice (n = 6) at 6 w p.i. Data in panels b, c, e, f and j-l are presented as the means ± standard error of the mean (SEM). *: P < .05, **: P < .01, ***: P < .001.
Caliper-based tumor volume measurements showed that cisplatin treatment for 3 w significantly reduced tumor growth (Figure 1b). More specifically, a significant reduction in tumor growth was already detected after 1 w of cisplatin treatment. This decrease was corroborated by in vivo imaging of 4T1 bioluminescence signals in the primary tumor area (Figure 1 c and d). Metastases in axillary lymph nodes and lungs were also significantly decreased by cisplatin treatment based on reduced 4T1 bioluminescence signals in the tissue (Figure 1 e and f) and H&E histology (Figure 1 g and h). Splenomegaly is a characteristic of progressive disease in the 4T1 model,16–19 and cisplatin treatment was able to restore the splenic size and weight back to near normality (Figure 1 i and j). As an additional confirmation of reduced disease progression, the levels of two selected immune-related disease biomarkers, CHI3L1 and LCN2,16–19 were significantly reduced in primary tumor lysates, sera, and spleen lysates of cisplatin-treated mice at 6 w p.i. (Figure 1 k and l).
To investigate tumor-dependency, the disease reductive capacity of cisplatin was also evaluated in an alternative Py230-based intraductal model for TNBC. In contrast to the 4T1-based model, we previously reported these Py230 tumors to be less metastatic but highly proliferative with a more immunosuppressed tumor microenvironment (TME).18 Therefore, they were allowed to grow for 5 w, reaching progression of DCIS to IC and a tumor volume of ± 60 mm3, before starting the treatment with cisplatin for 2 w (Supplementary Fig. 1A). Cisplatin treatment also significantly reduced Py230 tumor volume compared to untreated mice (Supplementary Fig. 1B).
H&E histology of primary tumor tissue sections from both the 4T1- and Py230-based model revealed cisplatin-mediated attenuation of tumor aggressiveness as reduced areas of cellular necrosis and spindle-shaped tumor cells undergoing epithelial-to-mesenchymal transition (EMT) for subsequent metastasis were observed (Supplementary Fig. 1C and 2A). In the Py230-based model, necrotic and EMT areas were even completely abolished upon cisplatin treatment (Supplementary Fig. 1C). In line with this reduced aggressiveness, tumor cell proliferation significantly decreased upon cisplatin treatment in both the 4T1 and Py230 tumors based on stainings with Ki67 and its proliferation index (Supplementary Fig. 1D and E, 2B). A less aggressive growth results into reduced oxygen consumption, hence hypoxia significantly decreased in cisplatin-treated 4T1 primary tumor tissue based on stainings for the hypoxia marker carbonic anhydrase 9 (CAIX)27 (Supplementary Fig. 2B). Significantly decreased CD31 stainings for vascular endothelial cells indicated an inhibitory effect of cisplatin on angiogenesis in the 4T1 primary tumor tissue (Supplementary Fig. 2B).
Despite these beneficial effects, the frequent chemotherapeutic dosing also induced substantial cytotoxicity as shown through a significantly decreased body temperature at 6 w p.i. and body weight by 5 w p.i. in cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Supplementary Fig. 3A and B). Toxic effects of multiple cisplatin doses were also seen in the cisplatin-treated Py230-based model with a significantly decreased body temperature and weight at 7 w p.i. (Supplementary Fig. 3C and D). Cessation of milk production following pup weaning induced a cisplatin-independent drop in body weight during the first w p.i. in all 4T1- and Py230 tumor-bearing mice (Supplementary Fig. 3B and D).
Multiple cisplatin dosing reduces the number of TAMs and DCs in the primary tumor, and enhances local PD-(L)1 immune cell positivity
To in-depth investigate the cisplatin-mediated changes in immune cells at different body locations (including most importantly primary tumor, axillary lymph nodes and spleen), flow cytometric immunophenotyping was performed on single-cell preparations at 6 w p.i. in the 4T1-based model and at 7 w p.i. in the Py230-based model (Supplementary Fig. 4 and 5). The antibody panels used were simplified to obtain an initial perspective of 9 general immune cell subtypes in the 4T1 and Py230 TME as partly adopted from Mosely et al.28 with myeloid cells subdivided into CD11b+ Ly6Cint Ly6G+ polymorphonuclear (PMN)-MDSCs, CD11b+ Ly6Chi Ly6G− monocytic (M)-MDSCs, CD11b+ F4/80+ macrophages/TAMs and CD11c+ DCs, T-cell subsets identified as CD3ε+ CD4+ CD8α− and CD3ε+ CD4− CD8α+, CD19+ cells identified as B-cells, and CD3ε− NKp46+ and CD3ε+ NKp46+ cells respectively identified as natural killer (NK) cells and NK-T cells.
Although cisplatin treatment had an overall toxic effect on the primary tumors, it did not significantly change the local percentage and absolute number of CD45+ leukocytes in the 4T1-based model (Figure 2 a and b). However, cisplatin significantly reduced myeloid cell populations in 4T1 primary tumors at the level of the TAMs and DCs (Figure 2c). This significant reduction was also seen at the level of CD206+ M2 pro-tumorigenic TAMs (Figure 2 d and e). In contrast, cisplatin treatment did not affect 2 other major myeloid cell types in these tumors, i.e. PMN- and M-MDSCs (Figure 2c). Cisplatin treatment also did not affect lymphocytic cell populations in the 4T1 primary tumors, and maintained similar numbers of CD4+ and CD8α+ T-cells as well as B-cells, NK and NK-T cells compared to untreated mice (figure 2f).
Figure 2.

Multiple cisplatin dosing induces distinct immunophenotypic changes in the primary tumors of a 4T1-based intraductal model. (a-i) Primary tumors were isolated from untreated and 5x cisplatin-treated mice at 6 w p.i. and digested into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (a) Percentage of CD45+ leukocytes within the total single cell suspension. (b) CD45+ leukocyte cell number per gram of primary tumor. (c) Number of myeloid cell subtypes (including PMN-MDSCs, M-MDSCs, macrophages/TAMs and DCs) per gram of primary tumor. (d) Percentage of M2 TAM subtypes within the CD45+ CD11b+ F4/80+ TAM population. (e) Number of M2 TAM subtypes per gram of primary tumor. (f) Number of lymphocytic cell subtypes (including CD4+ and CD8α+ T-cells, B-cells, NK cells and NK-T cells) per gram of primary tumor (g) Percentage of PD-1+ and PD-L1+ cells within the CD45+ leukocyte population. (h, i) Percentage of PD-1+ (h) and PD-L1+ cells (i) within each of the immune cell subtype populations. Data are presented as the means ± SEM. *: P < .05, **: P < .01, ***: P < .001.
In Py230 tumors, cisplatin treatment significantly decreased both the percentage and absolute number of CD45+ leukocytes (Supplementary Fig. 6A and B). This overall leukotoxic effect was displayed by a significant decrease in both myeloid and lymphocytic cell types (Supplementary Fig. 6C-E), in marked contrast to the effects on the immune landscape in 4T1 primary tumors.
In a follow-up analysis, it was evaluated whether cisplatin modulates the immune checkpoint proteins PD-1 and PD-L1 at the immune cell surface, focusing on the 4T1-based model (Supplementary Fig. 7A). Cisplatin treatment significantly increased the percentage of PD-1+ and PD-L1+ CD45+ leukocytes (Figure 2g). When investigating this positivity on each immune cell type (Supplementary Fig. 7B), 5 of the 9 subpopulations showed significantly enhanced PD-(L)1 positivity with cisplatin treatment, including PMN-MDSCs, CD4+ and CD8α+ T-cells (Figure 2 h and i). A cisplatin-mediated significant increase in PD-1, but not PD-L1 was also seen for M-MDSCs. NK cells, on the other hand, showed a significant increase in PD-L1, but not in PD-1 positivity upon cisplatin treatment. Corroborating the immunophenotyping, PD-1 and PD-L1 levels also significantly increased in primary tumor lysates of cisplatin-treated compared to untreated 4T1 tumor-bearing mice at 6 w p.i. (Supplementary Fig. 7C). This local increase in PD-1 and PD-L1 expression upon cisplatin treatment was confirmed at the RNA level based on RT-qPCR results for PDCD1 and CD274, albeit not significantly (Supplementary Fig. 7D).
Semi-quantitative immunohistochemistry for CD11b, CD163, and CD11c on 4T1 primary tumor tissue sections confirmed both the significant overall reduction in myeloid cells, and the specific reductions in M2 TAMs and DCs in cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 3). CD11b stained abundantly in both the tumor core and margin, indicative for a wide range of different myeloid cell types (Figure 3). CD163 stainings were typically localized at the tumor margin/stroma, while CD11c stainings were primarily located between the tumor cells in the tumor core (Figure 3). Immunohistochemistry for additional immune cell markers corroborated the comparable numbers of the other investigated immune cell types in the primary tumor tissue of cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Supplementary Fig. 8). Ly6G stainings for PMN-MDSCs and/or neutrophils was observed between tumor cells and at vascular sites. Characteristic for an inflamed TME, CD3ε stainings showed infiltration of T-cells in the tumor core. Forkhead box P3 (FoxP3) as a specific marker for the immunosuppressive regulatory T-cell (T-reg) subset was localized at the tumor margin and also showed a significant decreased staining in cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Supplementary Fig. 8). In line with the inflammatory nature of 4T1 tumors, CD19 stainings for B-cells and NCR-1 stainings for NK and NK-T cells were also found in the tumor core, although less abundantly than T-cells. Granzyme B stainings for CTLs were similar in the tumor core of cisplatin-treated compared to untreated 4T1 tumor-bearing mice.
Figure 3.

Immunohistochemical stainings confirm immunophenotypic changes and CAF reduction by multiple cisplatin dosing in the primary tumors of a 4T1-based intraductal model. Immunohistochemistry for the myeloid cell marker CD11b, the M2 TAM marker CD163, the DC marker CD11c and fibroblast/CAF markers α-SMA and FAP-α on primary tumor sections from untreated and 5x cisplatin-treated mice at 6 w p.i. (n = 8; 2 tissue slides from each group with 4 images per slide). Dashed inserts highlight stained tissue at a larger magnification. Black scale bars = 200 µm, red scale bars = 50 µm. Data are presented as the means ± SEM. *: P < .05, **: P < .01, ***: P < .001.
Additional immunohistochemistry for two fibroblast markers α-smooth muscle actin (α-SMA) and fibroblast activating protein-α (FAP-α) on 4T1 primary tumor sections indicated a significant decrease in the number of cancer-associated fibroblasts (CAFs) upon cisplatin treatment (Figure 3). CAFs were mainly detected in the tumor core, but also at the tumor margins with α-SMA staining the cell surface and FAP-α showing cytoplasmic staining (Figure 3).
Multiple cisplatin dosing increases the number of T-cells and reduces the number of MDSCs as well as PD-(L)1 immune cell positivity in the axillary lymph nodes, but does not affect blood leukocytes
Upon immunophenotyping, metastases-bearing axillary lymph nodes in the 4T1-based model were observed to consist almost exclusively of CD45+ leukocytes, representing 88% of the tissue suspension in untreated mice (Figure 4a). Upon cisplatin treatment, the CD45+ leukocyte fraction of total single cells in these axillary lymph nodes significantly increased to 93% (Figure 4a), although the number of CD45+ leukocytes remained unchanged (Figure 4b). The increase in the CD45+ leukocyte fraction was subsequently identified to be the result of a significant decrease in the percentage and absolute number of 4T1 metastatic tumor cells based on pan-cytokeratin stainings (Figure 4 c and d). Immunophenotyping with the same panel of markers used for the primary tumor revealed that cisplatin treatment in the 4T1-based model induced major changes in the axillary lymph node immune cell composition. It significantly reduced the number of myeloid cells, and more specifically the PMN- and M-MDSCs, and also the number of macrophages and DCs, albeit not significantly (Figure 4e). On the other hand, cisplatin treatment mediated a trend towards increased numbers of CD4+ and CD8α+ T-cells (figure 4f).
Figure 4.

Multiple cisplatin dosing provides local immunomodulation in support of anti-tumor immunity in the axillary lymph nodes of a 4T1-based intraductal model. (a-j) Axillary lymph nodes were isolated from untreated and 5x cisplatin-treated mice at 6 w p.i. and processed into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (a) Percentage of CD45+ leukocytes within the total single cell suspension. (b) CD45+ leukocyte cell number per gram of ax. lymph node. (c) Percentage of Pan-cytokeratin+ tumor cells within the total single cell suspension. (d) Pan-cytokeratin+ tumor cell number per gram of ax. lymph node. (e) Number of myeloid cell subtypes (including PMN-MDSCs, M-MDSCs, macrophages and DCs) per gram of ax. lymph node. (f) Number of lymphocytic cell subtypes (including CD4+ and CD8α+ T-cells, B-cells, NK cells and NK-T cells) per gram of ax. lymph node. (g) Percentage of PD-1+ and PD-L1+ cells within the CD45+ leukocyte population. (h) IFN-γ levels in lysates of ax. lymph nodes from untreated and 5x cisplatin-treated mice (n = 3 for both groups) at 6 w p.i. (i, j) Percentage of PD-1+ (i) and PD-L1+ cells (j) within each of the immune cell subtype populations. (k) Quantification and representative images of immunohistochemical stainings for the myeloid cell marker CD11b and the PMN-MDSC/neutrophil marker Ly6G on axillary lymph node sections from untreated and 5x cisplatin-treated mice at 6 w p.i. (n = 9; 3 tissue slides from each group with 3 images per slide). Dashed inserts highlight stained tissue at a larger magnification. Black scale bars = 200 µm, red scale bars = 50 µm. Data in panels a-k are presented as the means ± SEM. ND: not detected. *: P < .05, **: P < .01, ***: P < .001.
Axillary lymph nodes derived from the Py230-based model did not show significant changes in numbers of both myeloid and lymphocytic subsets upon cisplatin treatment (Supplementary Fig. 9A and B), indicating the absence of systemic disease and concomitant systemic cisplatin-mediated effects in this alternative TNBC model.
In analogy with the primary tumors from the 4T1-based model, cisplatin-mediated immunomodulation also occurred at the level of both PD-1 and PD-L1 in axillary lymph nodes, with a significant decrease in PD-(L)1 positivity on CD45+ leukocytes (Figure 4g). This decrease was not linked to a decrease in the production of IFN-γ, the major inducer of PD-L1 expression,29 as these levels even significantly increased in lysates of axillary lymph nodes derived from cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 4h). Alternatively, a significant reduction in PD-(L)1-positive B-cells, representing the largest immune cell subpopulation in axillary lymph nodes of the 4T1-based model, was identified as potential reason for the PD-(L)1 decrease (Figure 4 i and j). The decrease in PD-(L)1 positivity was not applicable for all lymphocytic cell types as both CD4+ and CD8α+ T-cells significantly increased in PD-(L)1 positivity in axillary lymph nodes from cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 4 i and j).
Immunohistochemical stainings for the immune cell markers CD11b and Ly6G on axillary lymph node tissue sections derived from the 4T1-based model further confirmed the significantly decreased numbers of overall myeloid cells and more specifically PMN-MDSCs upon cisplatin treatment in 4T1 metastases-bearing axillary lymph nodes (Figure 4k). CD11b primarily stained the surroundings of areas with metastasized tumor cells, whereas Ly6G stained both within and outside of these metastatic zones (Figure 4k).
Besides metastasizing via the lymphatic vasculature and lymph nodes, 4T1 tumor cells also spread hematogeneously to distant organs. Blood from 4T1 tumor-bearing mice that was denuded of red blood cells consisted of about 87% CD45+ leukocytes (Supplementary Fig. 10A). The blood leukocyte population consisted primarily of myeloid cells, with more specifically PMN-MDSCs as the biggest myeloid cell subtype, comprising about 90% of total blood leukocytes (Supplementary Fig. 10B). Cisplatin treatment did not affect either PMN-MDSC or other blood immune cells in the 4T1-based model, including macrophages, T- and B-cells (Supplementary Fig. 10B). It also did not affect PD-(L)1 positivity in circulating leukocytes (Supplementary Fig. 10C-E).
Multiple cisplatin dosing induces overall leukotoxicity in the lungs, reducing the relative fraction of DCs and B-cells and increasing the relative T-cell fraction and PD-(L)1 immune cell positivity
In line with the lower metastatic burden in the lung, cisplatin treatment significantly decreased the percentage of CD45+ pulmonary leukocytes in 4T1 tumor-bearing mice (Supplementary Fig. 11A). In lungs of untreated 4T1 tumor-bearing mice, myeloid cells comprised the largest immune cell fraction, dominated by PMN-MDSCs and smaller fractions of M-MDSCs, macrophages and DCs (Supplementary Fig. 11B). The relative fractions of PMN- and M-MDSC, as well as macrophage subpopulations within the CD45+ leukocyte population remained unchanged, whereas the DC fraction in the lungs from 4T1 tumor-bearing mice was significantly depleted by cisplatin treatment. Fractions of specific T-cell subsets in these lungs, and more specifically the CD4+ and the CD8α+ T-cell fraction, significantly increased upon cisplatin treatment (Supplementary Fig. 11B). In contrast, the relative B-cell fraction significantly decreased in lungs of cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Supplementary Fig. 11B). NK(-T) cell fractions remained similar in lungs upon cisplatin treatment (Supplementary Fig. 11B).
In analogy with the primary tumor and the axillary lymph nodes in the 4T1-based model, cisplatin increased PD-(L)1 positivity of CD45+ leukocytes in the lungs (Supplementary Fig. 11C). More specifically, PMN-MDSCs, M-MDSCs, CD4+ and CD8α+ T-cells, B-cells, and NK-T cells showed significantly increased positivity for PD-1, whereas increase in PD-L1 positivity was restricted to CD4+ and CD8α+ T-cells, B-cells and NK-T cells (Supplementary Fig. 11D and E).
Immunohistochemical stainings for the leukocyte marker CD45 on lung tissue sections of cisplatin-treated 4T1 tumor-bearing mice was mainly detected surrounding metastatic tumor cell areas and confirmed the significant decrease in the pulmonary leukocyte fraction upon cisplatin treatment (Supplementary Fig. 12). Moreover, cisplatin affected various immune cell types in 4T1 metastases-bearing lungs as CD11b stainings for overall myeloid cells, CD163 stainings for M2 macrophages, CD11c stainings for DCs and CD3ε stainings for T-cells significantly decreased in lung tissue sections upon cisplatin treatment (Supplementary Fig. 12). In analogy with the CD45 stainings, stainings for these four immune cell markers were observed at the border of 4T1 lung metastases (Supplementary Fig. 12).
Multiple cisplatin dosing reduces the number of myeloid cells and increases the number of T- and B-cells as well as PD-(L)1 immune cell positivity in the spleen
The spleen plays a crucial role in cancer-associated immunosuppression, yet its involvement in tumor progression is often neglected. Serving as a homing location for hematopoietic stem progenitor cells (HSPCs) and active site for extramedullary hematopoiesis (EMH), it produces and releases a large number of immunosuppressive cell types such as macrophages and MDSCs stimulated by tumor-secreted factors.30–32 This massive production of immunosuppressive cells has been reported, including by our group, to cause splenomegaly in the 4T1-based model, distorting the normal splenic architecture.16–19,32,33 Cisplatin treatment was able to restore the normal architecture of the splenic tissue in 4T1 tumor-bearing mice as shown by H&E staining (Figure 5a). More specifically, spleens of cisplatin-treated 4T1 tumor-bearing mice had distinguishable white pulp regions, which were distorted in untreated mice due to EMH-mediated expansion of the splenic red pulp regions with proliferating cells, including HSPCs, as shown by Ki67 stainings and its proliferation index (Figure 5b).
Figure 5.

Multiple cisplatin dosing induces an anti-tumorigenic environment in the spleen of a 4T1-based intraductal model. (a) H&E histology and immunohistochemistry for the cell proliferation marker Ki67 on spleen sections from untreated and 5x cisplatin-treated mice at 6 w p.i. Spleen sections from healthy mice were stained for comparison. White pulp areas (indicated with W) appear as hematoxylin-rich regions that do not stain for Ki67. Scale bars = 200 µm. (b) The Ki67 proliferation index in the spleen calculated as the proportion of Ki67+ nuclei relative to all identified nuclei on Ki67-stained tissue sections (n = 9; 3 tissue slides from each group with 3 images per slide). The dotted line represents the mean Ki67 proliferation index in spleens from healthy mice. (c-k) Spleens were isolated from untreated and 5x cisplatin-treated mice at 6 w p.i. and processed into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (c) Percentage of CD45+ leukocytes within the total single cell suspension. (d) CD45+ leukocyte cell number per gram of spleen. (e) Number of lineage-negative cells per gram of spleen. (f) Number of myeloid cell subtypes (including PMN-MDSCs, M-MDSCs, macrophages and DCs) per gram of spleen. (g) Number of M2 macrophage subtypes per gram of spleen. (h) Number of lymphocytic cell subtypes (including CD4+ and CD8α+ T-cells, B-cells, NK cells and NK-T cells) per gram of spleen. (i) Percentage of PD-1+ and PD-L1+ cells within the CD45+ leukocyte population. (j, k) Percentage of PD-1+ (j) and PD-L1+ cells (k) within each of the immune cell subtype populations. (l) Levels of PD-1 and PD-L1 in spleen lysates from untreated and 5x cisplatin-treated mice at 6 w p.i. (n = 4 for both groups). Data in panels b-l are presented as the means ± SEM. *: P < .05, **: P < .01, ***: P < .001.
Flow cytometric immunophenotyping demonstrated that cisplatin treatment in the 4T1-based model significantly increased the percentage of CD45+ leukocytes in the spleen compared to untreated mice (Figure 5c), whereas the absolute numbers of CD45+ leukocytes remained unchanged (Figure 5d). This relative increase was found to be the result of a significant decrease in HSPCs, characterized through splenic lineage-negative cells (Figure 5e). Cisplatin treatment also significantly decreased the number of splenic myeloid cells, i.e. PMN- and M-MDSCs, as well as macrophages (figure 5f). Moreover, the significant decrease in macrophage numbers was also detectable at the level of CD206+ M2 pro-tumorigenic macrophages (Figure 5g). However, cisplatin treatment did not significantly change the number of splenic DCs (figure 5f). In marked contrast to the myeloid cells, cisplatin treatment significantly increased the number of splenic CD4+ and CD8α+ T-cells compared to untreated 4T1 tumor-bearing mice (Figure 5h). Similarly, cisplatin treatment also significantly increased splenic B-cells, whereas NK(-T) cell numbers were not significantly affected (Figure 5h).
In analogy with the axillary lymph nodes and the absence of systemic cisplatin-mediated effects, cisplatin treatment did not induce significant changes in specific myeloid and lymphocytic splenic cell numbers in the Py230-based model (Supplementary Fig. 13A-C).
In line with the modulation of the splenic immunophenotype in the 4T1-based model, PD-1 and PD-L1 immune cell positivity was significantly increased in spleens of cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 5i). CD4+ and CD8α+ T-cells as well as B-cells all increased in both PD-1 and PD-L1 positivity, whereas M-MDSCs only increased in PD-1 positivity and macrophages only in PD-L1 positivity (Figure 5 j and k). Significantly enhanced PD-1 and PD-L1 levels were also detected in splenic lysates of cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 5l).
Immunohistochemistry for CD11b, Ly6G, and CD163 on splenic tissue sections derived from the 4T1-based model corroborated the reduction in overall myeloid cells and in the myeloid subpopulations, including PMN-MDSCs and M2 macrophages upon cisplatin treatment (Figure 6). Stainings for myeloid cell types were restricted to the red pulp area, where EMH and myeloid cell production typically takes place. In contrast, lymphocytic cell type CD3ε and CD19 stainings were restricted to the white pulp area and confirmed the increase of T- and B-cells following cisplatin treatment (Figure 6). Stainings for granzyme B as a marker for anti-tumorigenic CTLs were spread across the splenic tissue, but significantly increased in cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 6).
Figure 6.
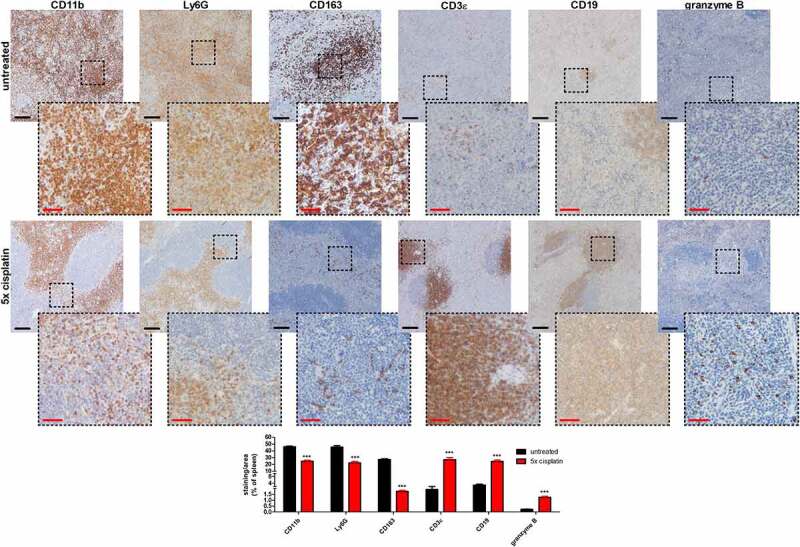
Immunohistochemical stainings confirm anti-tumorigenic immunomodulation by multiple cisplatin dosing in the spleen of a 4T1-based intraductal model. Immunohistochemistry for the myeloid cell marker CD11b, the PMN-MDSC/neutrophil marker Ly6G, the M2 macrophage marker CD163, the T-cell marker CD3ε, the B-cell marker CD19 and the CTL marker granzyme B on spleen sections from untreated and 5x cisplatin-treated mice at 6 w p.i. (n = 9; 3 slides with 3 images per slide). Black scale bars = 200 µm, red scale bars = 50 µm. Data are presented as the means ± SEM. ***: P < .001.
Cisplatin-mediated immunomodulation in the spleen of the 4T1-based model was also analyzed through protein profiling, with significantly increased levels of chemokines CCL5, CCL6, and CCL12 as well as the immune-related protein CD40 in splenic lysates of cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Supplementary Fig. 14A and B). Endoglin, which has been associated with immune responses in splenic macrophages,34 as well as coagulation factor III and FGF acidic as prothrombotic risk factors,35,36 were significantly increased, while MMP-9 as pro-tumorigenic factor37 was decreased by cisplatin in the splenic lysates (Supplementary Fig. 14A and B).
Cisplatin immunomodulation occurs early and principally affects lymphoid tissues
Although 5 cisplatin doses induced anti-tumorigenic immune responses, it remains unclear whether cisplatin also significantly modulates anti-tumor immunity at lower dosing frequency and if it occurs as an early effect after the first cisplatin dose, even before the reduction of tumor progression through direct tumor cell killing by cisplatin. In a subsequent experiment, 4T1 tumor-bearing mice were treated with one dose of cisplatin, followed by scarification 2 d later, i.e. 23 d p.i. (Figure 7a). At this early timepoint, both primary tumor growth and induced systemic disease, as shown by an increased spleen size, were not yet affected by cisplatin chemotherapy (Figure 7 b and c). Flow cytometric immunophenotyping of primary tumor tissue at 23 d p.i. showed a significant decrease in TAMs and DCs in cisplatin-treated compared to untreated 4T1 tumor-bearing mice (Figure 7d), similarly as detected following multiple-cisplatin doses. Additional markers MHC II and CD80 were now included to differentiate anti-tumorigenic M1 from pro-tumorigenic CD206+ M2 TAM subtypes in the CD11b+ F4/80+ TAM population (Supplementary Fig. 15A) and identified a significant decrease in both TAM subtypes in 4T1 primary tumors following cisplatin treatment (Figure 7e). As the numbers of M1 and M2 TAMs almost equally decreased upon cisplatin treatment, the primary tumor M1/M2 ratio did not significantly alter between cisplatin-treated and untreated 4T1 tumor-bearing mice (figure 7f). Moreover, M2 TAMs were more abundant than M1 TAMs at this early timepoint, providing an M1/M2 ratio <1.
Figure 7.

Single cisplatin dosing induces early immunomodulation in the primary tumors of a 4T1-based intraductal model. (a) Experimental timeline with intraductal inoculation of 4T1 mammary tumor cells, start of systemic treatment with cisplatin at 3 w p.i. and euthanasia 2 d later indicated. (b) Primary tumor weight measurements as indication for primary tumor growth at 23 d p.i. in untreated and 1x cisplatin-treated mice (n = 3 for both groups). (c) Weight measurements of the spleen from untreated and 1x cisplatin-treated mice (n = 3 for both groups) at 23 d p.i. (d-h) Primary tumors were isolated from untreated and 1x cisplatin-treated mice at 23 d p.i. and digested into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (d) Number of myeloid cell subtypes (including PMN-MDSCs, M-MDSCs, macrophages/TAMs and DCs) per gram of primary tumor. (e) Number of M1 and M2 TAM subtypes per gram of primary tumor. (f) M1/M2 ratio in primary tumors calculated based on M1 and M2 TAM subtype cell numbers. (g) Number of lymphocytic cell subtypes (including CD4+ and CD8α+ T-cells, B-cells, NK cells and NK-T cells) per gram of primary tumor. (h) Number of T-regs and CTLs per gram of primary tumor. Data in panels b-h are presented as the means ± SEM. *: P < .05.
In contrast to myeloid cells, lymphocytic cell types remained unchanged in cisplatin-treated 4T1 primary tumors at 23 d p.i. (Figure 7g), again similarly as following multiple cisplatin doses. CD4+ and CD8α+ T-cells, subdivided into immunosuppressive T-regs and anti-tumorigenic CTLs using FoxP3 and granzyme B as markers, respectively (Supplementary Fig. 15B), were also unaffected by cisplatin treatment at 23 d p.i. (Figure 7h).
However, immunophenotyping of axillary lymph node and splenic tissue showed more notable changes in both myeloid and lymphocytic cell types at 2 d following one dose of cisplatin. More specifically, the number of PMN- and M-MDSCs significantly decreased in both tissues (Figure 8 a and b) and the number of macrophages also significantly reduced in splenic tissue (Figure 8b). In accordance with the primary tumors, both splenic M1 and M2 macrophage subtypes significantly and equally decreased following cisplatin treatment (Figure 8c), providing a similar splenic M1/M2 ratio compared to untreated mice (Figure 8d). Moreover, as M1 and M2 were present in almost equal numbers, the splenic M1/M2 ratio approximated 1. Lymphocytic cell types including CD4+ and CD8α+ T-cells in both lymphoid tissues, as well as B-cells and NK-(T) cells in splenic tissue remained unchanged (Figure 8 e and f). In marked contrast, a significant increase in CTLs in both axillary lymph nodes and spleen, and a significant decrease of T-regs in splenic tissue was established upon cisplatin treatment at 23 d p.i. (Figure 8 e and f).
Figure 8.

Single cisplatin dosing induces early systemic immunomodulation in the axillary lymph nodes and spleen of a 4T1-based intraductal model. (a-f) Axillary lymph nodes and spleens were isolated from untreated and 1x cisplatin-treated mice at 23 d p.i. and digested into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (a, b) Number of myeloid cell subtypes (including PMN- and M-MDSCs in both ax. lymph nodes and spleen, and also macrophages and DCs in the spleen only) per gram of ax. lymph node (a) or spleen (b). (c) Number of M1 and M2 macrophage subtypes per gram of spleen. (d) M1/M2 ratio in the spleen calculated based on M1 and M2 macrophage subtype cell numbers. (e, f) Number of lymphocytic cell subtypes (including CD4+ T-cells, T-regs, CD8α+ T-cells and CTLs in both ax. lymph nodes and spleen, and also B-cells, NK cells and NK-T cells in the spleen only) per gram of ax. lymph node (e) or spleen (f). Data are presented as the means ± SEM. *: P < .05.
Cisplatin alleviates anti-PD-1 resistance in the 4T1-based model by stimulating anti-tumor immunity, already after a single dose
Given the importance of CTLs in the success and immunosuppressive myeloid cells as well as T-regs in the failure of and resistance to ICB in TNBC patients, it is of interest to further investigate whether these early, principally systemic, immunological changes following a single cisplatin dose are sufficient or whether multiple cisplatin doses are needed to increase ICB efficacy in the 4T1-based model. In a subsequent experiment, intraductally inoculated 4T1 tumor-bearing mice were treated with either single or frequent doses of cisplatin, now with and without anti-PD-1, to check for synergistic effects of chemotherapy in combination with ICB (Figure 9a). The combination treatment with anti-PD-1 precluded longer treatment than 2 w (i.e. until 5 w p.i.) as in a preliminary experiment we observed that anti-PD-1 ICB becomes toxic to 4T1 tumor-bearing mice upon 3 doses (data not shown). The number of cisplatin doses in the frequent dosing regimen was therefore also reduced from 5 to 3 (i.e. 6 mg/kg every 5 d from 3 to 5 w p.i.).
Figure 9.

Single cisplatin dosing alleviates anti-PD-1 resistance and reduces disease progression to the same extent as multiple cisplatin dosing in a 4T1-based intraductal model. (a) Experimental timeline with intraductal inoculation of 4T1 mammary tumor cells, start of systemic treatment with cisplatin (1 or 3 doses) and anti-PD-1 at 3 w p.i., and euthanasia at 5 w p.i. indicated. (b) Primary tumor volume measurements as indication for primary tumor growth (n = 8 for all groups). (c) In vivo imaging of bioluminescence signals in the primary tumor (indicated as total flux density in p/s/cm2) for verification of primary tumor growth (n = 8 for all groups). (d) Representative images of the bioluminescent signals in primary tumors from every treatment group at 5 w p.i. (e) Quantification based on total flux density (in p/s/cm2) in axillary lymph nodes (n = 6 for the untreated, anti-PD-1- and 3x cisplatin-treated group; n = 8 for all other groups) and lungs (n = 3 for the untreated, anti-PD-1- and 3x cisplatin-treated group; n = 4 for all other groups) at 5 w p.i. (f, g) Representative images of bioluminescence signals derived from 4T1 metastases in axillary lymph nodes (f) and lungs (g) at 5 w p.i. (h) Weight measurements of the spleen from all treatment groups (n = 4) at 5 w p.i. The dotted line represents the mean spleen weight of 4 healthy mice. Data in panels b, c, e and h are presented as the means ± SEM. *: P < .05, **: P < .01, ***: P < .001.
The 4T1-based model showed resistance to anti-PD-1 ICB based on similar primary tumor volume measurements compared to untreated mice (Figure 9b). This was corroborated by in vivo imaging, where similar 4T1 bioluminescence signals were observed in the primary tumors of anti-PD-1-treated compared to untreated mice (Figure 9 c and d). However, treating the 4T1 tumor-bearing mice with a single dose of cisplatin induced a similar tumor growth reduction compared to treatment with 3 doses of cisplatin based on both tumor volume measurements and in vivo bioluminescence imaging (Figure 9 b-d). Moreover, as displayed by normal body temperature and weight, a single cisplatin dosing regimen did not induce toxic effects, in marked contrast to multiple cisplatin dosing (Supplementary Fig. 16A and B). In combination with anti-PD-1, both 1 and 3 doses of cisplatin provided similar add-on tumor growth reduction compared to both these cisplatin treatments alone (Figure 9b-d). These growth reductive effects were also detectable at the metastatic level, with significantly decreased 4T1 metastases-derived signals in axillary lymph nodes and lungs upon treatment with 1 and 3 cisplatin doses compared to either untreated or anti-PD-1-treated controls, and an additional decrease upon combination with anti-PD-1 (Figure 9 e-g). Spleen sizes were also similarly significantly decreased following 1 and 3 doses of cisplatin, with an add-on decrease in combination with anti-PD-1 (Figure 9h), concomitant with a significantly decreased systemic disease progression.
Flow cytometric immunophenotyping identified comparable immune cell changes induced by 1 and 3 cisplatin doses in primary tumors at 5 w p.i. More specifically, the number of TAMs decreased similarly significant following 1 and 3 doses of cisplatin compared to untreated and anti-PD-1-treated mice, in line with our previous results both at 23 d (single dose) and 6 w p.i. (frequent dosing), and this decrease was maintained upon combination with anti-PD-1 (Figure 10a). In accordance with total TAM numbers, anti-tumorigenic M1 and pro-tumorigenic M2 TAM subtypes also decreased similarly significant by 1 and 3 cisplatin doses as well as the cisplatin + anti-PD-1 combination treatments (Figure 10b), providing comparable M1/M2 ratios of approximately 1 in all groups (Figure 10c). Although DC numbers were decreased by both cisplatin dosing regimens, concomitant with our primary tumor data both at 23 d and 6 w p.i., the combination treatments with anti-PD-1 induced a significant increase in their numbers, even to a higher extent than in primary tumors of untreated and anti-PD-1-treated mice (Figure 10a). Changes in lymphocytic cell types compared to untreated and anti-PD-1-treated controls included a similarly significant decrease in T-reg numbers following 1 and 3 doses of cisplatin in combination with and without anti-PD-1 (Figure 10d). In contrast, the number of CTLs significantly increased, albeit only upon combination of cisplatin with anti-PD-1 (Figure 10d).
Figure 10.

Single and multiple cisplatin dosing comparably mediates distinct immunophenotypic changes in primary tumors of a 4T1-based intraductal model. (a-d) Primary tumors were isolated from every treatment group at 5 w p.i. and digested into a single cell suspension for flow cytometric immunophenotyping (n = 3 for all groups). (a) Number of myeloid cell subtypes (including PMN-MDSCs, M-MDSCs, macrophages/TAMs and DCs) per gram of primary tumor. (b) Number of M1 and M2 TAM subtypes per gram of primary tumor. (c) M1/M2 ratio in primary tumors calculated based on M1 and M2 TAM subtype cell numbers. (d) Number of lymphocytic cell subtypes (including CD4+ T-cells, T-regs, CD8α+ T-cells, CTLs, B-cells, NK cells and NK-T cells) per gram of primary tumor. Data are presented as the means ± SEM. *: P < .05, **: P < .01.
Again in accordance with our previous data at earlier (23 d p.i.) and later (6 w p.i.) timepoints, flow cytometric immunophenotyping of myeloid cells in the axillary lymph node and spleen identified a similarly significant decrease in the number of PMN- and M-MDSCs in both lymphoid tissues (Figure 11 a and b), and an additional similarly significant decrease in the number of macrophages in spleen (Figure 11b) upon both 1 and 3 doses of cisplatin compared to untreated and anti-PD-1-treated controls. Combination of cisplatin with anti-PD-1 did not further significantly decrease these myeloid cell types. Similarly to the primary tumor, both pro-tumorigenic M1 and anti-tumorigenic M2 macrophage subtypes in the spleen decreased similarly significant following 1 and 3 cisplatin doses in combination with and without anti-PD-1 (Figure 11c), providing a splenic M1/M2 ratio of approximately 1 (Figure 11d). Both 1 and 3 cisplatin doses similarly increased the numbers of lymphocytic cell types, and more specifically CD4+ and CD8α+ T-cells as well as CTLs, in both lymphoid tissues (Figure 11 e and f). Moreover, these lymphocytic subset numbers even further increased significantly compared to untreated and anti-PD-1-treated controls, upon both cisplatin combinations with anti-PD-1 (Figure 11 e and f). Again only additionally in the spleen alone, the number of T-regs also significantly decreased and the number of B-cells significantly increased upon 1 and 3 cisplatin doses compared to untreated and anti-PD-1-treated mice (Figure 11f). Combination of cisplatin with anti-PD-1 maintained these changes.
Figure 11.

Single and multiple cisplatin dosing comparably mediates similar anti-tumor stimulation in lymphoid tissues of a 4T1-based intraductal model. (a-f) Axillary lymph nodes and spleens were isolated from every treatment group at 5 w p.i. and digested into a single cell suspension for flow cytometric immunophenotyping (n = 3 for both groups). (a, b) Number of myeloid cell subtypes (including PMN- and M-MDSCs in both ax. lymph nodes and spleen, and also macrophages and DCs in the spleen only) per gram of ax. lymph node (a) or spleen (b). (c) Number of M1 and M2 macrophage subtypes per gram of spleen. (d) M1/M2 ratio in the spleen calculated based on M1 and M2 macrophage subtype cell numbers. (e, f) Number of lymphocytic cell subtypes (including CD4+ T-cells, T-regs, CD8α+ T-cells and CTLs in both ax. lymph nodes and spleen, and also B-cells, NK cells and NK-T cells in the spleen only) per gram of ax. lymph node (e) or spleen (f). Data are presented as the means ± SEM. *: P < .05, **: P < .01.
Superior gating strategy for in-depth myeloid and lymphocytic immune cell subset characterization confirms the comparable immunomodulation upon single and multiple cisplatin dosing
As the gating strategy could be improved for in-depth characterization of myeloid and lymphocytic immune cell types, flow cytometric immunophenotyping was reperformed including additional markers. Immune cell changes in primary tumors, axillary lymph nodes and spleen upon 1 and 3 cisplatin doses in combination with and without anti-PD-1 were evaluated for their reproducibility and these data were extended. More specifically, applying CD14 allowed to better characterize the myeloid compartment (Supplementary Fig. 17A) resulting in a CD45+ CD11b+ CD14+ Ly6Cint Ly6G+ PMN-MDSC (Figure 12a), a CD45+ CD11b+ CD14+ Ly6Chi Ly6G− M-MDSC (Figure 12b) and a CD45+ CD11b+ CD14− Ly6Cint Ly6G+ neutrophil/tumor-associated neutrophil (TAN) subset (Figure 12c) in the primary tumor and both lymphoid tissues of the 4T1-based model. Still, a significant decrease of either myeloid subset in lymphoid tissues upon cisplatin dosing in combination with and without anti-PD-1 was herewith confirmed. Applying CD14 to the existing CD45+ CD11b+ F4/80+ antibody panel for macrophages/TAMs, and more specifically to the M1 and M2 macrophage subsets, did not alter the initial primary tumor and splenic numbers nor their significant decrease upon 1 or 3 cisplatin doses in combination with and without anti-PD-1 (Figure 12d-f). Specifying CD11c+ DCs as CD11b+ also did not alter DC numbers in either primary tumor or spleen and verified an increase in primary tumor DCs upon treatment with cisplatin + anti-PD-1 combination (Figure 12g). Conventional CD4+ T-cells were now specified as FoxP3− and their numbers again similarly significantly increased in axillary lymph nodes and spleen upon both 1 and 3 cisplatin doses (Figure 13a). Moreover, their numbers even further increased upon combination with anti-PD-1 (Figure 13a). Subdividing B-cells into 3 populations using CD19 and B220 as specific markers (Supplementary Fig. 17B), again identified a similarly significant increase in splenic CD19 single-positive (Figure 13b) and CD19/B220 double-positive (Figure 13d), but not in splenic B220 single-positive B-cell numbers (Figure 13c) upon a 1 and 3 cisplatin dose treatment with and without anti-PD-1.
Figure 12.

Addition of cellular markers increases myeloid cell type specification and confirms comparable immunomodulation by single and multiple cisplatin dosing. (a) Number of CD45+ CD11b+ CD14+ Ly6Cint Ly6G+ PMN-MDSCs per gram of primary tumor, ax. lymph node or spleen. (b) Number of CD45+ CD11b+ CD14+ Ly6Chi Ly6G− M-MDSCs per gram of primary tumor, ax. lymph node or spleen. (c) Number of CD45+ CD11b+ CD14− Ly6Cint Ly6G+ neutrophils or TANs per gram of primary tumor, ax. lymph node or spleen. (d) Number of CD45+ CD11b+ CD14+ F4/80+ macrophages or TAMs per gram of primary tumor or spleen. (e) Number of CD45+ CD11b+ CD14+ F4/80+ MHC II+ CD80+ M1 macrophage or TAM subtypes per gram of primary tumor or spleen. (f) Number of CD45+ CD11b+ CD14+ F4/80+ CD206+ M2 macrophage or TAM subtypes per gram of primary tumor or spleen. (g) Number of CD45+ CD11b+ CD11c+ DCs per gram of primary tumor or spleen. Data are presented as the means ± SEM. *: P < .05, **: P < .01.
Figure 13.

Addition of cellular markers increases lymphocytic cell type specification and confirms comparable immunomodulation by single and multiple cisplatin dosing. (a) Number of CD45+ CD3ε+ CD4+ CD8α− FoxP3− T-cells per gram of primary tumor, ax. lymph node or spleen. (b) Number of CD45+ CD3ε− NKp46− CD19+ B220− B-cells per gram of primary tumor or spleen. (c) Number of CD45+ CD3ε− NKp46− CD19− B220+ B-cells per gram of primary tumor or spleen. (d) Number of CD45+ CD3ε− NKp46− CD19+ B220+ B-cells per gram of primary tumor or spleen. Data are presented as the means ± SEM. *: P < .05, **: P < .01.
Discussion
Cisplatin is a platinum-based chemotherapeutic that relies on the alkylation of DNA for its anti-tumoral activity. More specifically, cisplatin intercalates with purine bases forming DNA-adducts, which interfere with DNA repair and eventually induce cellular apoptosis.38 Severe toxic effects such as kidney injury and gastro-intestinal problems that lead to a decrease in body weight and temperature are well known following repetitive dosing, and are also observed in preclinical models.39,40 However, cisplatin is still widely used in the clinic, especially in the context of TNBC where few alternative treatments are currently available.41 Lowering the cisplatin dosing frequency could reduce and even exclude toxicity, but it is currently unknown whether such dosing regimen also has similar anti-tumor efficacy in TNBC. Moreover, systemic effects induced by cisplatin remain understudied, but could be of key importance for chemotherapeutic efficacy.
Using fully immunocompetent complementary 4T1- and Py230-based intraductal mouse models for TNBC, multiple cisplatin dosing was here shown to attack primary tumor cells and reduce their proliferation, but also to influence their EMT and subsequent metastasis. Corroborating our observations, a recent clinical study by Wang et al. reported that cisplatin treatment prevents breast cancer metastasis by inducing expression of activating transcription factor 3 (ATF3) and antagonizing transforming growth factor (TGF)-β-mediated early EMT.42 Both our current and previous preclinical data19 describe that frequent cisplatin treatment also reduces the hypoxic conditions in the 4T1 primary tumor, which may reduce its aggressive progression given the growth-stimulating properties of tumor hypoxia. Stromal components of the TME, including the CD31+ vascular endothelial cells are likely an additional target for cisplatin, which may also explain the increased risk for hypertension and thromboembolic complications in patients treated with cisplatin.43 In accordance, a prothrombotic effect of cisplatin was here detected in the intraductal 4T1-based TNBC model, as spleens from cisplatin-treated 4T1 tumor-bearing mice showed enhanced expression of coagulation factor III and FGF acidic. More importantly, a key finding was that a single dose of cisplatin could prevent the problematic toxicity of frequent cisplatin dosing in this model, shown by significant decreases in body temperature and weight, while the desired beneficial growth reductive effect on primary tumors as well as metastases in axillary lymph nodes and lungs was retained.
Cisplatin is also a promising candidate chemotherapeutic agent for clinical use in combination with immunotherapeutic strategies such as anti-PD-1 ICB. More specifically, in metastatic TNBC patients, it was reported to enhance the sensitivity to PD-1 blockade, providing an objective response rate of >20%.15 Simulating a potential clinically relevant cisplatin and anti-PD-1 combination, we here chose to compare a single cisplatin with a multiple cisplatin dosing regimen, both with and without ICB combination in the intraductal 4T1-based model. A single dose of cisplatin already alleviated anti-PD-1 resistance and this combination even provided an add-on disease reduction to the same extent as anti-PD-1 combined with multiple cisplatin doses. These data show that one dose of cisplatin suffices to mediate a durable anti-tumor effect.
An important part of cisplatin efficacy lies in its immunomodulation of the TME.4–6 Currently, this process remains not well characterized. Therefore, it was additionally investigated here whether the observed durable treatment effect with a single cisplatin dose is related to a change in both immunosuppressive and anti-tumor immune cells. In extension of previous preclinical data,19 our current study shows that in 4T1 primary tumors, following multiple cisplatin doses, significantly decreased numbers of immunosuppressive CAFs, myeloid cell types such as TAMs and DCs, and immunosuppressive lymphocytic T-regs are found.
In contrast to 4T1 primary tumors, multiple cisplatin doses had an overall leukotoxic effect in Py230 primary tumors, which could be explained by the fact that Py230 tumors are more susceptible to cisplatin-mediated apoptosis due to their enhanced proliferation index.
In line with a similar disease reduction, a single cisplatin dose mediated a similar decrease in TAM, DC and T-reg numbers compared to multiple cisplatin doses in 4T1 primary tumors, with M1 and M2 TAM numbers also being equally decreased. Moreover, these reductions were already detectable at 2 d after the single cisplatin dose (i.e. before disease reduction), highlighting that cisplatin-mediated immunomodulation in primary tumors is an early effect. Despite the overall decrease in TAMs as a major immunosuppressive population, additional myeloid cell subsets such as PMN-MDSCs remained unaffected by cisplatin in 4T1 primary tumors. This is regarded as a major hurdle for effective immunotherapeutic responses in TNBC patients. Indeed, PMN-MDSCs are produced and accumulate in different body compartments as a result of tumor-secreted factors such as G-CSF that bring hematopoiesis into overdrive as a compensatory reaction to restore normal blood cell production (so-called emergency myelopoiesis).44–46 In line with the concern regarding myeloid cell-mediated immunosuppression in the primary tumors, the De Visser group reported that combining anti-Ly6G with cisplatin is required to increase anti-tumor immunity in a highly immunosuppressive breast cancer model.47 Nevertheless, our current study shows that combination with single or multiple cisplatin doses alleviates anti-PD-1 resistance in the aggressive 4T1-based intraductal model and also triggers significant increase in tumor cell-killing CTLs and DCs, the latter most likely destined for enhanced tumor antigen presentation.
Cisplatin-mediated immunomodulation at metastatic sites and other lymphoid organs such as the axillary lymph nodes and spleen, may also play a major role in enhancing immunotherapeutic efficacy. We here at first investigated these body compartments in the 4T1-based intraductal model and observed that cisplatin significantly reduced immunosuppressive myeloid cell types in axillary lymph nodes and spleen. This key finding correlates with the substantially reduced splenomegaly, upon both dosing regimens. Again highlighting the differential immunomodulation, cisplatin showed a general leukotoxic effect in the lungs, which can be regarded as a normalization effect and is in line with an almost complete abrogation of metastatic 4T1 tumor growth. Last but not least, the failure of cisplatin to modulate the immune cell composition in peripheral blood, even upon multiple dosing, could again be regarded as potentially problematic for its immunotherapeutic efficacy. Yet, Balog et al. elegantly demonstrated in the 4T1 model after fat pad inoculation, that rather than affecting cellular numbers, cisplatin immunomodulates blood leukocytes at the phenotypic level, and measured enhanced anti-tumorigenic cytokine IFN-γ levels in cisplatin-treated blood PMN-MDSCs.48 These and other authors also reported that cisplatin treatment changes the splenic immunophenotype by increasing T- and B-cell numbers, improving systemic anti-tumor immunity.48,49 Our current splenic immunophenotyping confirmed this key observation and further extended it to the axillary lymph nodes, upon single and multiple cisplatin doses. More specifically, we observed that single and multiple cisplatin dosing regimens induced an increase in CD4+ and CD8α+ T-cell subsets as well as CTLs in both lymphoid tissues, but did so most significantly upon combination with anti-PD-1 ICB. Similarly to primary tumor immunomodulation, the reduction in immunosuppressive cell types and increase in anti-tumorigenic lymphocytes was already detectable at 2 d following a single cisplatin dose in both lymphoid tissues. This major finding is again in line with our statement above on the early immunomodulatory effect by cisplatin that occurs most notably systemically and is durably maintained in the 4T1-based intraductal model.
The increase in PD-1 and PD-L1 immune cell positivity may further lie at the base for a successful cisplatin + anti-PD-1 combination therapy.9–11 Indeed, enhanced immune checkpoint expression provides more ICB targets and may therefore enhance therapeutic responses. Our innovative data demonstrate increase of PD-(L)1 positivity on the immune cell surface in all investigated tissues, except for the axillary lymph nodes. The latter observation could be explained by the significant decrease in PD-(L)1 on B-cells (i.e. representing the largest proportion of non-myeloid cells in this lymphoid organ). T-cells, as major anti-tumorigenic immune cell types, are important leukocyte targets for anti-PD-(L)1 immunotherapy and showed enhanced PD-1 and PD-L1 positivity in all tissues. Yet, PD-L1 expression on T-cells may rather be detrimental as these have been reported to restrain effector T-cells and stimulate tumorigenesis.50 Enhanced PD-(L)1 positivity on myeloid cells has also widely been shown to negatively impact immunotherapeutic success. More specifically, deletion of PD-1 positivity on MDSCs and macrophages is known to mediate metabolic reprogramming that stimulates anti-tumor immunity and reduces myelopoiesis, necessary for ICB efficacy.51 Accordingly, another study showed that PD-1 expression by TAMs negatively correlates with anti-tumorigenic phagocytic potency.52 Hedgehog-stimulated PD-L1 positivity on TAMs is also critical for suppressing anti-tumorigenic CD8+ T-cell functions in tumor-bearing mice53 and associates with a negative outcome in ICB therapy.54 PD-L1 positivity on PMN-MDSCs and TANs also drives their immunosuppressive activity, which can be abrogated by inhibiting a fatty acid transport protein (FATP2) and stimulates the efficacy of several immune checkpoint blockers.55 Yet, with regards to TANs, another study has shown that PD-L1 positivity can be additionally found on anti-tumorigenic neutrophils,56 highlighting that anti-PD-(L)1 ICB efficacy relies on a delicate balance between tumor-promoting and -antagonizing PD-(L)1-expressing leukocytes.
In conclusion and based on the reduction of immunosuppressive and increase of anti-tumorigenic cells observed in the current study, we here postulate that axillary lymph nodes and spleen as lymphoid tissues are the most positively affected body locations following cisplatin treatment and may thus be the main drivers of ICB efficacy, potentially even after a single cisplatin dose. In line with our postulation, Fransen et al. reported that tumor-draining lymph nodes (TDLNs) are necessary for CTL accumulation in tumors following ICB, as therapeutic efficacy was mitigated by locking T-cells in the TDLNs or TDLN resection.57 Axillary lymph nodes can therefore be regarded as main contributor to the CTL number increase in cisplatin + anti-PD-1-treated primary tumors seen in our current study. Recent insights, reviewed by our group32 and others,30,31 also increasingly highlight the spleen as a driver of tumor progression through massive production and systemic release of immunosuppressive cell types. The spleen has even been opted as an immune barometer that potentially prognosticates disease outcome and instructs on the presence of an anti-tumorigenic immune response in cancer patients.32,58,59 These recent studies and our current findings on splenic immunomodulation warrant further research to unravel the role of the spleen in immunotherapeutic efficacy of cisplatin in preclinical models, e.g. by studying its impact through splenectomy60 or through use of nanoparticle therapeutics.61 Moreover, analyzing TNBC patient data, especially with regard to systemic immunomodulatory effects of cisplatin by checking e.g. immune responses in blood, is also warranted to translate the current mouse data to a clinical setting.
Supplementary Material
Acknowledgments
The authors kindly acknowledge Lobke De Bels (Department of Morphology, Faculty of Veterinary Medicine, Ghent University, Merelbeke, Belgium) for assistance with histology and tissue slide preparation, Sophie Godefroidt and Michelle Wouters (Department of Veterinary and Biosciences, Faculty of Veterinary Medicine, Ghent University, Merelbeke, Belgium) for assistance with immunohistochemical stainings, Jurgen De Craene for assistance with RT-qPCR set-up, and Alice De Vliegher for help with the artwork. This work was in part supported by a junior postdoctoral fellowship from the Research Foundation Flanders (FWO, 12Y3122N) awarded to JS, a grant from the Fund Suzanne Duchesne (managed by the King Baudouin Foundation) awarded to JS and a grant from the Research Foundation Flanders (FWO, G.0621.10) awarded to NNS.
Funding Statement
This work was supported by the Koning Boudewijnstichting [Fund Suzanne Duchesne]; Research Foundation Flanders [G.0621.10]; Research Foundation Flanders [12Y3122N].
Data availability statement
The data that support the findings of this study are available from the corresponding author, JS, upon reasonable request.
Disclosure statement
TP discloses interest through co-foundership, shareholdership, and a management consultancy agreement with OCTIMET Oncology NV, Beerse, Belgium. EM received support through a research contract from OCTIMET Oncology NV, Beerse, Belgium. All other authors report no conflict of interest.
Author contributions
JS and EM concepted and designed the study. JS, JB, NVE, KD and JDV acquired the data. JS, KD and JDV analyzed and interpreted the data. JS drafted the manuscript. TP, ODW, WVDB, WDS, NNS and EM revised the manuscript. All authors approved the final version of the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2103277
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–24. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat. 2011;125(3):627–636. doi: 10.1007/s10549-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta GK, Collier AL, Lee D, Hoefer RA, Zheleva V, Siewertsz van Reesema LL, Tang-Tan AM, Guye ML, Chang DZ, Winston JS, et al. Perspectives on triple-negative breast cancer: current treatment strategies, unmet needs, and potential targets for future therapies. Cancers. 2020;12(9):2392. doi: 10.3390/cancers12092392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hato SV, Khong A, de Vries Ij, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res. 2014;20(11):2831–2837. doi: 10.1158/1078-0432.CCR-13-3141. [DOI] [PubMed] [Google Scholar]
- 5.de Biasi Ar, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20(21):5384–5391. doi: 10.1158/1078-0432.CCR-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rébé C, Demontoux L, Pilot T, Ghiringhelli F. Platinum derivatives effects on anticancer immune response. Biomolecules. 2019;10(1):13. doi: 10.3390/biom10010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markasz L, Skribek H, Uhlin M, Otvos R, Flaberg E, Eksborg S, Olah E, Stuber G, Szekely L. Effect of frequently used chemotherapeutic drugs on cytotoxic activity of human cytotoxic T-lymphocytes. J Immunother. 2008;31(3):283–293. doi: 10.1097/CJI.0b013e3181628b76. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 9.Fournel L, Wu Z, Stadler N, Damotte D, Lococo F, Boulle G, Ségal-Bendirdjian E, Bobbio A, Icard P, Trédaniel J, et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019;464:5–14. doi: 10.1016/j.canlet.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Ock CY, Kim S, Keam B, Kim S, Ahn YO, Chung EJ, Kim JH, Kim TM, Kwon SK, Jeon YK, et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget. 2017;8(58):97920–97927. doi: 10.18632/oncotarget.18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakita D, Iwai T, Harada S, Suzuki M, Yamamoto K, Sugimoto M. Cisplatin augments antitumor T-cell responses leading to a potent therapeutic effect in combination with PD-L1 blockade. Anticancer Res. 2019;39(4):1749–1760. doi: 10.21873/anticanres.13281. [DOI] [PubMed] [Google Scholar]
- 12.Tran L, Allen CT, Xiao R, Moore E, Davis R, Park SJ, Spielbauer K, Van Waes C, Schmitt NC. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol Res. 2017;5(12):1141–1151. doi: 10.1158/2326-6066.CIR-17-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cubas R, Moskalenko M, Cheung J, Yang M, McNamara E, Xiong H, Hoves S, Ries CH, Kim J, Gould S. Chemotherapy combines effectively with Anti-PD-L1 treatment and can augment antitumor responses. J Immunol. 2018;201(8):2273–2286. doi: 10.4049/jimmunol.1800275. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, He S, Wu H, Xie H, Zhang T, Deng Z. Blocking the PD-1/PD-L1 axis enhanced cisplatin chemotherapy in osteosarcoma in vitro and in vivo. Environ Health Prev Med. 2019;24(1):79. doi: 10.1186/s12199-019-0835-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver Kk, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–928. doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 16.Steenbrugge J, Breyne K, Denies S, Dekimpe M, Demeyere K, De Wever O, Vermeulen P, Van Laere S, Sanders NN, Meyer E. Comparison of the adipose and luminal mammary gland compartment as orthotopic inoculation sites in a 4T1-based immunocompetent preclinical model for triple-negative breast cancer. J Mammary Gland Biol Neoplasia. 2016;21(3–4):113–122. doi: 10.1007/s10911-016-9362-7. [DOI] [PubMed] [Google Scholar]
- 17.Steenbrugge J, Breyne K, Demeyere K, De Wever O, Sanders NN, Van Den Broeck W, Colpaert C, Vermeulen P, Van Laere S, Meyer E. Anti-inflammatory signaling by mammary tumor cells mediates prometastatic macrophage polarization in an innovative intraductal mouse model for triple-negative breast cancer. J Exp Clin Cancer Res. 2018;37(1):191. doi: 10.1186/s13046-018-0860-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steenbrugge J, Vander Elst N, Demeyere K, De Wever O, Sanders NN, Van Den Broeck W, Dirix L, Van Laere S, Meyer E. Comparative profiling of metastatic 4T1- vs. non-metastatic Py230-Based mammary tumors in an intraductal model for triple-negative breast cancer. Front Immunol. 2019;10:2928. doi: 10.3389/fimmu.2019.02928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steenbrugge J, Vander Elst N, Demeyere K, De Wever O, Sanders NN, Van Den Broeck W, Ciamporcero E, Perera T, Meyer E. OMO-1 reduces progression and enhances cisplatin efficacy in a 4T1-based non-c-MET addicted intraductal mouse model for triple-negative breast cancer. NPJ Breast Cancer. 2021;7(1):27. doi: 10.1038/s41523-021-00234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao L, Cardiff RD, Steinbach P, Messer KS, Ellies LG. Multipotent luminal mammary cancer stem cells model tumor heterogeneity. Breast Cancer Res. 2015;17(1):137. doi: 10.1186/s13058-015-0615-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Laere S, Van der Auwera I, Van den Eynden GG, Fox SB, Bianchi F, Harris AL, van Dam P, Van Marck EA, Vermeulen PB, Dirix LY. Distinct molecular signature of inflammatory breast cancer by cDNA microarray analysis. Breast Cancer Res Treat. 2005;93(3):237–246. doi: 10.1007/s10549-005-5157-z. [DOI] [PubMed] [Google Scholar]
- 22.van de Moosdijk AA, van Amerongen R. Identification of reliable reference genes for qRT-PCR studies of the developing mouse mammary gland. Sci Rep. 2016;6(1):35595. doi: 10.1038/srep35595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Spiegelaere W, Dern-Wieloch J, Weigel R, Schumacher V, Schorle H, Nettersheim D, Bergmann M, Brehm R, Kliesch S, Vandekerckhove L, et al. Reference gene validation for RT-qPCR, a note on different available software packages. PLoS One. 2015;10:e0122515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 26.Almeida JS, Iriabho EE, Gorrepati VL, Wilkinson SR, Grüneberg A, Robbins DE, Hackney JR. ImageJS: personalized, participated, pervasive, and reproducible image bioinformatics in the web browser. J Pathol Inform. 2012;3(1):25. doi: 10.4103/2153-3539.98813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf Dem Keller U, Leung S, Huntsman D, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71(9):3364–3376. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- 28.Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5(1):29–41. doi: 10.1158/2326-6066.CIR-16-0114. [DOI] [PubMed] [Google Scholar]
- 29.Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of IFNγ in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. 2016;22(10):2329–2334. doi: 10.1158/1078-0432.CCR-16-0224. [DOI] [PubMed] [Google Scholar]
- 30.Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39(5):806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu C, Hua Q, Zheng L. Generation of myeloid cells in cancer: the spleen matters. Front Immunol. 2020;11:1126. doi: 10.3389/fimmu.2020.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steenbrugge J, De Jaeghere EA, Meyer E, Denys H, De Wever O. Splenic hematopoietic and stromal cells in cancer progression. Cancer Res. 2021;81(1):27–34. doi: 10.1158/0008-5472.CAN-20-2339. [DOI] [PubMed] [Google Scholar]
- 33.DuPre’ SA, Hunter KW Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol. 2007;82(1):12–24. doi: 10.1016/j.yexmp.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Ojeda-Fernández L, Recio-Poveda L, Aristorena M, Lastres P, Blanco FJ, Sanz-Rodríguez F, Gallardo-Vara E, de Las Casas-Engel M, Corbí Á, Arthur HM, et al. Mice lacking endoglin in macrophages show an impaired immune response. PLoS Genet. 2016;12(3):e1005935. doi: 10.1371/journal.pgen.1005935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lechner D, Kollars M, Gleiss A, Kyrle PA, Weltermann A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J Thromb Haemost. 2007;5(12):2445–2452. doi: 10.1111/j.1538-7836.2007.02788.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhao M, Ross JT, Itkin T, Perry JM, Venkatraman A, Haug JS, Hembree MJ, Deng CX, Lapidot T, He XC, et al. FGF signaling facilitates postinjury recovery of mouse hematopoietic system. Blood. 2012;120(9):1831–1842. doi: 10.1182/blood-2011-11-393991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joseph C, Alsaleem M, Orah N, Narasimha PL, Miligy IM, Kurozumi S, Ellis IO, Mongan NP, Green AR, Rakha EA. Elevated MMP9 expression in breast cancer is a predictor of shorter patient survival. Breast Cancer Res Treat. 2020;182(2):267–282. doi: 10.1007/s10549-020-05670-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McSweeney KR, Gadanec LK, Qaradakhi T, Ali BA, Zulli A, Apostolopoulos V. Mechanisms of cisplatin-induced acute kidney injury: pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers. 2021;13(7):1572. doi: 10.3390/cancers13071572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delgado ME, Grabinger T, Brunner T. Cell death at the intestinal epithelial front line. FEBS J. 2016;283(14):2701–2719. doi: 10.1111/febs.13575. [DOI] [PubMed] [Google Scholar]
- 41.Guan X, Ma F, Fan Y, Zhu W, Hong R, Xu B. Platinum-based chemotherapy in triple-negative breast cancer: a systematic review and meta-analysis of randomized-controlled trials. Anticancer Drugs. 2015;26(8):894–901. doi: 10.1097/CAD.0000000000000260. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Guo S, Kim SJ, Shao F, Ho JWK, Wong KU, Miao Z, Hao D, Zhao M, Xu J, et al. Cisplatin prevents breast cancer metastasis through blocking early EMT and retards cancer growth together with paclitaxel. Theranostics. 2021;11(5):2442–2459. doi: 10.7150/thno.46460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cameron AC, Touyz RM, Lang NN. Vascular complications of cancer chemotherapy. Can J Cardiol. 2016;32(7):852–862. doi: 10.1016/j.cjca.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schultze JL, Mass E, Schlitzer A. Emerging principles in myelopoiesis at homeostasis and during infection and inflammation. Immunity. 2019;50(2):288–301. doi: 10.1016/j.immuni.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 45.Yvan-Charvet L, Ng LG. Granulopoiesis and neutrophil homeostasis: a metabolic, daily balancing act. Trends Immunol. 2019;40(7):598–612. doi: 10.1016/j.it.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Wildes TJ, DiVita Dean B, Flores CT. Myelopoiesis during solid cancers and strategies for immunotherapy. Cells. 2021;10(5):968. doi: 10.3390/cells10050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salvagno C, Ciampricotti M, Tuit S, Hau CS, van Weverwijk A, Coffelt SB, Kersten K, Vrijland K, Kos K, Ulas T, et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol. 2019;21(4):511–521. doi: 10.1038/s41556-019-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balog JÁ, Hackler L Jr, Kovács AK, Neuperger P, Alföldi R, Nagy LI, Puskás LG, Szebeni GJ. Single cell mass cytometry revealed the immunomodulatory effect of cisplatin via downregulation of splenic CD44+, IL-17A+ MDSCs and promotion of circulating IFN-γ+ myeloid cells in the 4T1 metastatic breast cancer model. Int J Mol Sci. 2019;21(1):170. doi: 10.3390/ijms21010170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye J, Zou MM, Li P, Lin XJ, Jiang QW, Yang Y, Huang JR, Yuan ML, Xing ZH, Wei MN, et al. Oxymatrine and cisplatin synergistically enhance anti-tumor immunity of CD8+ T cells in non-small cell lung cancer. Front Oncol. 2018;8:631. doi: 10.3389/fonc.2018.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. 2020;21(4):442–454. doi: 10.1038/s41590-020-0620-x. [DOI] [PubMed] [Google Scholar]
- 51.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. 2020;5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petty AJ, Dai R, Lapalombella R, Baiocchi RA, Benson DM, Li Z, Huang X, Yang Y. Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight. 2021;6(6):e146707. doi: 10.1172/jci.insight.146707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, Schalper KA, Rimm DL. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26(4):970–977. doi: 10.1158/1078-0432.CCR-19-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–78. doi: 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yajuk O, Baron M, Toker S, Zelter T, Fainsod-Levi T, Granot Z. The PD-L1/PD-1 axis blocks neutrophil cytotoxicity in cancer. Cells. 2021;10(6):1510. doi: 10.3390/cells10061510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, van Hall T, Ossendorp F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight. 2018;3(23):e124507. doi: 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Markel JE, Noore J, Emery EJ, Bobnar HJ, Kleinerman ES, Lindsey BA. Using the spleen as an in vivo systemic immune barometer alongside osteosarcoma disease progression and immunotherapy with α-PD-L1. Sarcoma. 2018;2018:8694397. doi: 10.1155/2018/8694397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Jaeghere EA, Laloo F, Lippens L, Van Bockstal M, De Man K, Naert E, Van Dorpe J, Van de Vijver K, Tummers P, Makar A, et al. Splenic 18F-FDG uptake on baseline PET/CT is associated with oncological outcomes and tumor immune state in uterine cervical cancer. Gynecol Oncol. 2020;159(2):335–343. doi: 10.1016/j.ygyno.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 60.Stöth M, Freire Valls A, Chen M, Hidding S, Knipper K, Shen Y, Klose J, Ulrich A, Ruiz de Almodovar C, Schneider M, et al. Splenectomy reduces lung metastases and tumoral and metastatic niche inflammation. Int J Cancer. 2019;145(9):2509–2520. doi: 10.1002/ijc.32378. [DOI] [PubMed] [Google Scholar]
- 61.Duan X, He C, Kron SJ, Lin W. Nanoparticle formulations of cisplatin for cancer therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8(5):776–791. doi: 10.1002/wnan.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, JS, upon reasonable request.