

Abstract
The amount of data available from genomic medicine has revolutionized the approach to identify the determinants underlying many rare diseases. The task of confirming a genotype–phenotype causality for a patient affected with a rare genetic disease is often challenging. In this context, the establishment of the Matchmaker Exchange (MME) network has assumed a pivotal role in bridging heterogeneous patient information stored on different medical and research servers. MME has made it possible to solve rare disease cases by “matching” the genotypic and phenotypic characteristics of a patient of interest with patient data available at other clinical facilities participating in the network. Here, we present PatientMatcher (https://github.com/Clinical-Genomics/patientMatcher), an open‐source Python and MongoDB‐based software solution developed by Clinical Genomics facility at the Science for Life Laboratory in Stockholm. PatientMatcher is designed as a standalone MME server, but can easily communicate via REST API with external applications managing genetic analyses and patient data. The MME node is being implemented in clinical routine in collaboration with the Genomic Medicine Center Karolinska at the Karolinska University Hospital. PatientMatcher is written to implement the MME API and provides several customizable settings, including a custom‐fit similarity score algorithm and adjustable matching results notifications.
Keywords: gene discovery, genomic API, Matchmaker Exchange, matchmaking, rare disease
1. INTRODUCTION
The increasing accessibility of accurate genomic data via next‐generation sequencing (NGS) (Metzker, 2010; Shendure & Ji, 2008) has opened new avenues to a cost‐effective diagnosis of the genetic determinants underlying many rare diseases (RDs) (Boycott et al., 2013). Obtaining a molecular diagnosis for a patient with a rare disease often constitutes a challenging task; currently typically less than 50% of patients receive a molecular diagnosis despite a strong suspicion of an underlying genetic determinant (Lee et al., 2014; Soden et al., 2014; Stranneheim et al., 2021; Yang et al., 2014). However, a powerful approach for disease gene discovery is through identification of other patients with a similar phenotype. By establishing a cohort of similar patients, the likelihood of identifying the shared genomic determinant is strongly increased. The establishment of the Matchmaker Exchange (MME) (Boycott et al., 2015) federated network has dramatically improved the process of “matchmaking” patients across clinical laboratories’ and research centers’ databases (Azzariti & Hamosh, 2020). MME APIs (Buske et al., 2015) simplify sharing of selected patient data with the purpose of identifying cases with shared phenotypes and genotypic profiles. An MME matching event results in a notification sent to the patients’ data submitters, each of which belong to separate participating centers. The centers can evaluate the matching features and eventually establish a causative gene or variant for the given patient features. The obvious advantage of this tool is that single users utilizing the service do not need to worry about different database standards and data formats, as MME nodes communicate via standardized protocols and return results in a common and language‐independent data format (JSON).
The Clinical Genomics facility at Science for Life Laboratory (SciLifeLab) Stockholm has been collaborating with the regional healthcare at Karolinska University Hospital to provide whole genome sequencing (WGS)‐based rare disease (RD) diagnostics since 2015. Through this collaboration, termed Genomic Medicine Center Karolinska (Stranneheim et al., 2021), more than 6000 RD patients corresponding to more than 10,000 samples (6000 at the time PatientMatcher was launched) have been analyzed to date making this the largest clinical WGS effort in Sweden. This collaboration is responsible for the genetic testing of the vast majority of RD cases in the Stockholm region, accounting for ~2500 samples sequenced annually. Additionally, Clinical Genomics is a founding member of Genomic Medicine Sweden (https://www.genomicmedicine.se) and the Nordic Alliance for Clinical Genomics (http://www.nordicclinicalgenomics.org). In this framework, PatientMatcher was developed by Clinical Genomics as a clinical diagnostic decision support tool to aid clinicians and researchers at partner institutes solving RD cases. PatientMatcher is now being implemented at the Genomic Medicine Center Karolinska to establish a controlled, fully integrated data sharing possibility as part of the diagnostic workflow.
2. DESIGN AND IMPLEMENTATION
At the time this software was developed, there already existed four open‐source solutions, which could be adopted by a patient database owner to connect to the MME network as an independent node (https://github.com/ga4gh/mme-apis/wiki/Implementations). After an analysis of the existing implementations, we came to the conclusion that none of them addressed our needs. The only software that was written in Python (the language of choice for most of the applications developed at our facility, and for this reason ensuring better maintenance for the project over time) was in fact the Matchmaker Reference Server (https://github.com/MatchmakerExchange/reference-server), a very helpful yet simple implementation to illustrate the setup of a Matchmaker server. The authors describe this software as an example only, not intended to be used in production settings.
As previously mentioned, the first technical reason that prompted us to launch PatientMatcher was the need to develop an application written in Python (https://www.python.org/). Another obvious advantage is that developing the solution in a very popular programming language, will likely increase the chances that PatientMatcher or some of its modules will be used by other research centers or diagnostic laboratories willing to connect to MME as distinct nodes. The second technical challenge that led us to develop a custom solution, was the necessity of storing data in a document‐oriented database such as MongoDB (https://www.mongodb.com/), where patient data documents are very similar to data objects used in Scout (https://github.com/Clinical-Genomics/scout), the application used by our clinical laboratories for handling results from NGS analyses. Additionally, MongoDB saves documents in JSON, the same format used by MME nodes for exchanging patient data via HTTP requests. Technical considerations aside, our primary reason to develop the software from scratch was the opportunity to introduce a highly customizable patient similarity scoring algorithm, to help data contributors to fine‐tune the parameters of interest to be used in the patient similarity computation. PatientMatcher consists of a Python (3.6+) backend connected to a web app built in Flask 2.0+ (https://flask.palletsprojects.com/en/2.0.x/). The application data is stored in a MongoDB database.
See the README provided in PatientMatcher's GitHub repository (https://github.com/Clinical-Genomics/patientMatcher) for a quick introduction to the software.
The program backend contains the command to update database resources: HPO and disease term ontologies, respectively, downloaded from the OBO Foundry (https://github.com/OBOFoundry) and the Jenkins automation server from the Monarch Institute (https://ci.monarchinitiative.org/). These resources are the core of the software's phenotype similarity score algorithm. In addition, the command line is a used to add or remove MME clients (connected nodes allowed to run queries on PatientMatcher by exhibiting a security token that is unique for each node) and MME nodes (external nodes queried by PatientMatcher using a token assigned in turn by these servers). A recent addition to the command line options allows software admins to reassign patients, present in the database with a given user contact, to another user contact.
PatientMatcher is basically a Representation State Transfer (REST) API tool that enables submission of data, downloading of results and performing exhaustive comparison against the internal database data set or submission of queries to external nodes. The application implements the MME API specifications (Buske et al., 2015). The available server endpoints are illustrated in Table 1.
Table 1.
PatientMatcher server HTTP API endpoints
| Endpoint | Method | Rule | Purpose |
|---|---|---|---|
| indexa | GET | / | Landing page, showing statistics and MME node disclaimer. |
| add | POST | /patient/add | Adds or updates one patient by submitting a json payload structured as described in the MME API. |
| delete | DELETE | patient/delete/<patient_id> | Deletes the patient with the given ID and all its matching results from the database. |
| heartbeata | GET | /heartbeat | Returns a heartbeat response as defined in the MME API. |
| match_external | POST | /match/external/<patient_id> | Matches data from a patient already stored in PatientMatcher with a given ID against connected MME nodes. |
| match_internal | POST | /match | Matches json data received from a request sent from a connected node against the patients stored in PatientMatcher's database. Returns a response to the requester with eventual matches. |
| matches | GET | /matches/<patient_id> | Returns all positive matches stored in the database for the patient with a given ID. |
| metricsa | GET | /metrics | Returns a json object with server statistics described in the MME API. |
| nodes | GET | /nodes | Returns a response describing all external nodes connected to the server. |
Note: The PatientMatcher endpoint named in column Endpoint can be accessed by HTTP(S) scheme requests, with HTTP Method as in column Method, with URL path formed as in Rule. The endpoint usage is further described in the column Purpose. The API conforms to MatchMaker API (Buske et al., 2015) to allow exchange also with Match maker Exchange nodes implementing other server software.
Endpoints not exposing sensitive information and therefore not requiring a security token to be accessed.
2.1. Matching algorithm
When the server receives a matching request from an external node (external matching) or from a user wishing to match a specific patient against all other patients on the server (internal matching), the query triggers a matching algorithm, which computes the similarity between the query patient and all patients stored in the database. Characteristics associated with a patient that are taken into consideration when matching the patient across nodes, are generally defined as “features”. Patients’ features can describe either genetic components, such as genes or gene variants (genotype features) or a phenotype (phenotype features). As for other MME implementations and per MME API specifications, patient similarity is measured by a similarity score between 0 (no matching features) and 1 (exact matching of all patient's features). Given the different number and heterogeneous nature of features that can be provided to describe a patient, we consider it unlikely that two patients in PatientMatcher would be identical, and when compared with an external sample, return exactly the same matching score. For this reason, and because we believe that accuracy over number is to be preferred when presenting results to a clinician, we have defined a “MAX_RESULTS” key in the software settings. This number corresponds to the maximum number of patients returned by the server and its default value is 5. This number is obviously arbitrary and can be increased to yield more conservative results. Patient matches are returned in order of descending similarity with the query patient, that is, high similarity matches are presented first in the list of results.
Similarity score computation in PatientMatcher is taking into account genotype and phenotype similarity across patients. The weight of these factors is numerically evaluated into a “GTScore” and a “PhenoScore”, where the sum of these two contributes to the total similarity score (result score) between query and matched patient. The relative importance of GTScore and PhenoScore in the computation can be customized by the server administrator by modifying the values of the parameters named “MAX_GT_SCORE” and “MAX_PHENO_SCORE” in the app configuration settings. The default value for both these parameters is 0.5, meaning an equivalent impact of phenotype and genotype similarity on the result score. This design was made to address diverse requirements from different data contributors. For example, a clinical laboratory might be storing patient genetic information with little availability to diagnoses or phenotype terms. In that case it makes sense to set the weight of the phenotype matching to zero and rely on genotype matching only. On the other hand, country regulations might not allow sharing of accurate genetic information, for instance variant details, but only gene symbols. If detailed patient diagnoses are also available for these patients, using both GTScore and PhenoScore when running the similarity algorithm will increase the chances of producing meaningful matches.
2.1.1. Genotype matching algorithm
When the parameter MAX_GT_SCORE is set to a valuer higher than zero and the query data contains genotype features (gene or variant information), a genotype similarity score will be evaluated between query patient and every patient (matched patient) contained in the database. All patients matching at least one of the candidate genes present in the query will be initially selected as matches. As specified in the MME API, candidate genes should preferably be described by an Ensembl ID (i.e., “ENSG00000101680”), but it is possible to search the database using patients with genes represented by HGNC symbols (i.e., “LAMA1”) and Entrez IDs (i.e., “6481”). The algorithm is designed to assign higher matching scores to patients with fewer genotype features. For instance, a query patient connected with a unique gene (A) that matches a database patient connected with the same gene (A) will produce a higher genotype score than a query patient connected with two genes (A and B). Genotype score (GT_SCORE) is quantified by the formula:
This number is calculated by dividing the MAX_GT_SCORE by the sum of the feature scores (fs) measured from the match of each genotype feature of a query patient against a matching patient. For example, according to this definition, assuming a MAX_GT_SCORE of 0.5, each gene from a patient connected with three genes will have a fs of a third of 0.5 (0.1666). If a gene from the query patient does not match any gene of the matched patient, then the fs for that feature would have a value of 0. In the eventuality of exact matching of a specific gene and a specific gene variant, the fs would be assigned with the highest possible value for the feature (0.1666). Incomplete gene matches (gene matching and no variant matching or no variant metadata available for the provided genes) are assigned with an arbitrary value or a quarter of the fs for the feature (0.1666/4). By calculating the GT_SCORE in this manner, the algorithm produces an accurate numerical estimate of the similarity between all genotype features of matching patients. This, in turn, allows the server to return patient hits sorted by descending genetic similarity with the query patient and not simply all patients that match any of the associated genes.
PatientMatcher provides also the possibility to evaluate and assign scores to matching variants that are not contained within genes. Feature scores from such variant matches are assigned with the same fs as exact (gene + variant) matchings. It is worth mentioning that the genotype matching algorithm contains a liftover functionality that allows quantification of the similarity between patients containing genomic features described in different genome builds.
2.1.2. Phenotype matching algorithm
PatientMatcher is calculating phenotype matching scores based on both patient features and disorders. Patient features are described by HPO terms (Köhler et al., 2014) provided for query and matched patients, while disorders are represented by Decipher (Firth et al., 2009), OMIM (Amberger et al., 2015) or Orphanet (Pavan et al., 2017) entries. When comparing patients using both features and disorders, these descriptors will both be accounted for and each of them will contribute to half of the resulting phenotype score (PhenoScore). Similarity between HPO features will solely be considered in the computation when disorders are not provided for one or both patients. Whereas comparison of the disease terms in the algorithm is still relatively unpolished (it consists of a pairwise comparison of diagnoses between the patients), semantic similarity metrics between HPO terms and their ancestor terms are calculated as simGIC measures (Pesquita et al., 2008). The original algorithm used for creating the phenotype ontology and comparing the patients in PatientMatcher is available in the Patient‐Similarity package (https://github.com/buske/patient-similarity). Since the HPO is curating resources bridging disease terms with their associated HPO entries, we envision that in future software releases, disease similarity comparisons will also be calculated as semantic relationships between terms.
2.2. Email notifications
Email notifications can be enabled by administrators via specific parameters present in the software configuration file. To modulate the amount of information included in the email notification body and thereby limit the extent of potentially sensitive information distributed via email, there exist two notification options: (1) complete notifications containing the entire description of matching patients (including gene names, variants and phenotypes), and (2) partial notification reports with only the patient ID and the patient's clinician's contact information. Email notifications are sent to the patient contact only in case of positive matches from requests triggered by the same user, by another user within PatientMatcher (internal matches) or an external user from an MME connected node (external matches).
3. INTEGRATION WITH SCOUT DATA AT GENOMIC MEDICINE CENTER KAROLINSKA, STOCKHOLM
PatientMatcher was developed as a standalone software with the aim of providing an easy‐to‐administer MME server for any research institute or clinical laboratory wanting to pursue a connection to the MME network as an independent node. Except for a basic landing page showing general information and database statistics, the application does not have a graphical user interface (GUI) and patient data entry is achieved by handling incoming HTTP POST requests containing authentication tokens as well as patient data information. The instance of PatientMatcher hosted at GMCK contains an integration with Scout, the browser‐based decision support software platform used to display and analyze WGS analyses from RD cases. These cases include mostly patients from the three collaborating clinical diagnostic laboratories at the Karolinska University Hospital: The Center for Inherited Metabolic Diseases, the Department of Clinical Genetics and the Department of Immunology and Transfusion Medicine. These patients, analyzed either as singletons, trios or larger family groups, present symptoms such as intellectual disabilities, inborn errors of metabolism, mitochondrial and neuromuscular diseases, primary immune deficiencies as well as connective tissues and skeletal diseases, among other disorders (Stranneheim et al., 2021). In the current setup, Scout and PatientMatcher are distinct software instances residing on a single server, but depending on local IT infrastructure they can, if needed, be installed on different servers as they communicate via REST APIs. On the Scout portal, the MME integration feature is visible by all users. Access to the functionality is, however, granted to designated users authorized to submit cases to the MME network. A typical interpretation of a clinical case using Scout involves reviewing variants available for the affected individual(s) of a case with the goal of identifying one or a few candidates responsible for a specific phenotype. Phenotypes in Scout can be assigned at the case and the individual level as a list of HPO terms and/or OMIM diagnoses (https://omim.org/). The requirements to submit a case to PatientMatcher is that the Scout case should have one to three variants “pinned” as possible causatives and/or its phenotype should be described (by HPO and/or OMIM terms). It is noteworthy that since diagnoses in Scout are currently only represented by OMIM terms, it is not possible at the moment to submit from this platform patients described by the Orphanet (https://www.orpha.net/consor/cgi-bin/index.php) or Decipher (https://www.deciphergenomics.org/) ontologies, even though PatientMatcher supports all three ontologies. Hence, all phenotype features from Scout patients present on the Swedish PatientMatcher node will only be described by HPO terms and OMIM diagnoses. This limitation of the Scout software will also have an impact on the matching of patients: in fact, any patient described by Decipher or Orphanet terms from an external node will be matched against the Swedish node only based on the genotype features.
As shown in Figure 1, gender might be optionally assigned to a patient to be submitted to the MME.
Figure 1.
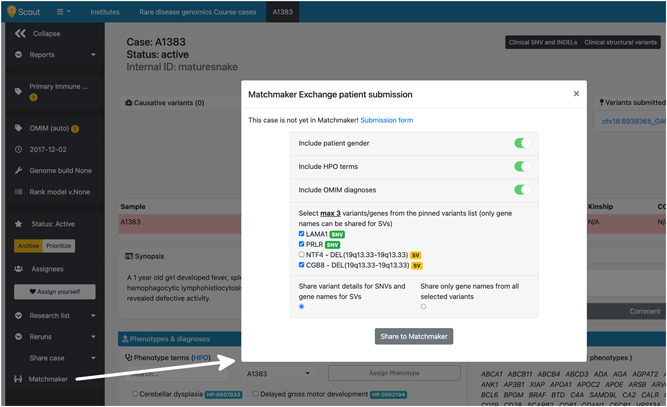
Matchmaker Exchange (MME) patient submission form in Scout. The submission of a Scout case to MME is initiated by clicking on a link present on the case page. The Scout user chooses which type of information (gender, HPO terms, OMIM terms, specific variant information or gene symbols only) will be submitted for the affected individuals of the case. While it is possible to share details at the specific variant level for any single‐nucleotide variant or short insertion/deletion (SNVs, marked with a green “SNV” badge in the figure), only general gene information can be shared for structural variants (SVs, denoted by a yellow “SV” badge). This figure shows how it is possible to submit one or more genes from the same structural variant (NTF4 and CGB8 in the example). Regardless of their nature (SNV or SVs), it is only possible to share a maximum of three candidate genotype features for each patient using Scout. Demo data was used to generate this figure
As regulations concerning genomic data sharing diverge depending on national legislation (Phillips, 2018), and even though initiatives like the EU's General Data Protection Regulation (GDPR) exist to harmonize the rules for data processing and sharing across borders in Europe and internationally (Bonomi et al., 2020), data controllers might not feel at ease disclosing the specific candidate variant(s) for a certain case (Molnár‐Gábor & Korbel, 2020). For this reason, we have included the option in Scout to describe MME patient's genotype features at the variant level (at the specific variant genotype level) or at the more generic gene level (only at the candidate gene level). These two options are illustrated in the bottom section of the patient's submission form of Figure 1.
The Scout user submitting a patient to the MME network automatically becomes its contact person and will be notified if the submitted case is positively matched.
The case data is stored in the database indefinitely and is subjected to internal and external queries, but can be reviewed (Figure 2), modified and eventually removed at any moment by other authorized users from the same institution. In the eventuality that another user modifies a patient previously submitted to the PatientMatcher, then the patient contact information will be updated with the email of the second user, which will become its new contact person. To ensure that all Scout cases submitted to the MME node are actively followed over time, we enforced a routine that makes it impossible to remove users from Scout before all their assigned patients are re‐assigned to another user.
Figure 2.
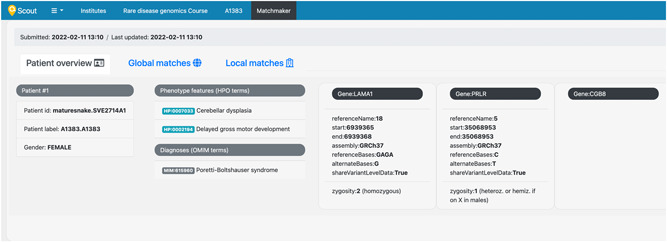
Overview of an Matchmaker Exchange (MME) patient in Scout. A dedicated page in Scout summarizes the information associated to the patient submitted to MME. The patient submitted in the example contains three candidate genotype features, two of them with variant‐level data (LAMA1 and PRLR gene variants) and a third with general gene information (CGB8 gene). Demo data was used to generate this figure
MME nodes connected to PatientMatcher are displayed and can independently be searched for patients similar to the query case (external matching). Alternatively, similar patients can also be retrieved from the list of other Scout patients in PatientMatcher (internal matching) (Figure 3).
Figure 3.
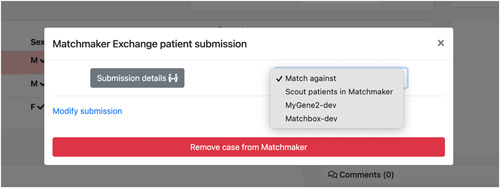
Matching options selection. Matchmaker Exchange (MME) nodes connected to PatientMatcher are displayed and can be independently searched for patients similar to the query case (external matching). Alternatively, similar patients can be also retrieved from the list of other Scout patients in PatientMatcher (internal matching). The example illustrated in this figure shows the real settings of the staging server of Scout, which is connected to the development instances of two other MME nodes: MyGene2 (https://www.mygene2.org/MyGene2/) and Matchbox (https://seqr.broadinstitute.org/matchmaker/matchbox), developed and maintained respectively by the Center for Mendelian Genomics, University of Washington (https://uwcmg.org/) and the Center for Mendelian Genomics at the Broad Institute (https://cmg.broadinstitute.org/)
Both “active matches” (Scout patient has initiated the search and a matching patient has been found in a connected node or in PatientMatcher) and “passive matches” (an external party has initiated the matching and the Scout patient is among the results in PatientMatcher) are displayed in dedicated tabs named “Global matches” and “Local matches,” respectively, displaying results for a patient matched against other nodes or other Scout patients (Figure 4).
Figure 4.
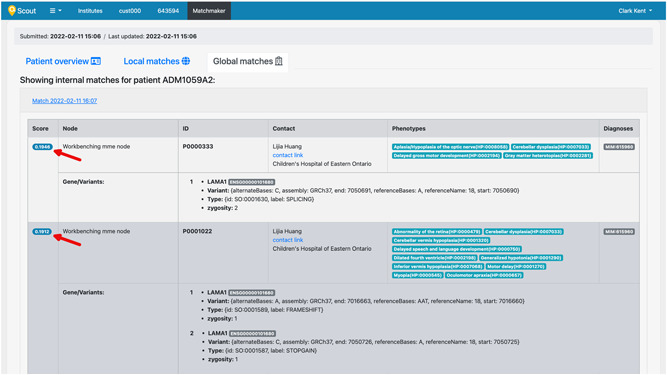
Patient matching results page in Scout. “Global Matches” and “Local Matches” tabs, respectively, display matching results for a patient against other nodes or other Scout patients. Red arrows designate the similarity score computed between query sample and matched sample. Demo data was used to generate this figure
4. SOFTWARE AVAILABILITY AND INSTALLATION
PatientMatcher is open‐source and available on GitHub (https://github.com/Clinical-Genomics/patientMatcher). The software is distributed under the MIT license (https://github.com/Clinical-Genomics/patientMatcher/blob/master/LICENSE) and we encourage all interested parties to use and modify its code according to their needs. The main GitHub repository is curated by Clinical Genomics, but we look forward to establishing a collaborative environment where other users could help improving the code, adding or simply requesting new useful features.
The simplest way to run and test the server is to use the up‐to‐date container image with a basic software installation that can be pulled from Docker Hub (https://hub.docker.com/r/clinicalgenomics/patientmatcher). On the GitHub pages of the repository, we also provide instructions and support files to test PatientMatcher with real data without needing to install any software, except Docker. For this purpose, we compiled a multi‐container Docker Compose file that, when launched by a single command from the terminal, provides a complete setup of the server, including a running instance of MongoDB containing the 50 benchmarking patients spanning 22 disorders reported in the original paper describing the MME API (Buske et al., 2015). This server represents a standalone MME instance, ready to accept HTTP requests on localhost and port 8000. For development and testing reasons we have also created a more sophisticated Docker Compose setup, with an MME server connected to another two MME nodes (other instances of PatientMatcher), both containing demo patient data. This file is available under the “/containers” folder in the GitHub page of the PatientMatcher software. Deploying the software in a production environment using the official Docker image file could be achieved using Kubernetes (https://kubernetes.io/) or via Podman (https://podman.io/) system services. Another tested way to deploy the software is installing it from the Python Package Index (PyPI) using the Python installer Pip. In this case it is recommended to operate in a virtual environment, such as Conda (https://docs.conda.io/), after installing Python 3.6+. All these options, together with other server settings, are extensively described on the software's GitHub pages.
5. CONCLUSIONS
PatientMatcher is curated and maintained by Clinical Genomics, SciLifeLab in collaboration with the Genomic Medicine Center Karolinska at the Karolinska University Hospital, Stockholm. It is an open‐source solution for clinical laboratories and research facilities who wish to join the federated MME network as independent nodes. Among the advantages of administering an independent node is the control over the data submitted to the server. National legislation, for instance, might hinder storing sensitive data on cloud solutions or on servers located in other geographical areas. In a time of rapidly increasing genetic data generation, this MME implementation is meant to provide an easy‐to administer tool to collect patient information and perform extensive comparisons between patients within the internal database or from external nodes. To reach as many users as possible, we have designed a standalone application with customizable settings to harmonize the matching algorithm and notifications with data structures and routines present in different host centers. At the same time, we have established a pipeline where candidate variants or genes with linked patient phenotypes analyzed using the Scout decision support solution can easily be shared to the MME. To improve the code and better meet user expectations we look forward to collaborating with interested third parties to further develop the tool and its underlying matching algorithms. In conclusion, we look forward to teaming up with other clinical laboratories to share candidate gene‐to‐phenotype associations to contribute to the accelerating disease gene discovery.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
WEB RESOURCES
https://github.com/Clinical-Genomics/patientMatcher
https://www.genomicmedicine.se
http://www.nordicclinicalgenomics.org
https://github.com/ga4gh/mme-apis/wiki/Implementations
https://github.com/MatchmakerExchange/reference-server
https://github.com/Clinical-Genomics/scout
https://flask.palletsprojects.com/en/2.0.x/
https://ci.monarchinitiative.org/
https://github.com/buske/patient-similarity
https://www.orpha.net/consor/cgi-bin/index.php
https://www.deciphergenomics.org/
ACKNOWLEDGMENTS
We acknowledge the support of the BigMed project funded by the Norwegian Research Council and of the Genomic Medicine Sweden initiative funded by the Swedish Innovation Agency (Vinnova) both of which have financially contributed to the development of PatientMatcher. This study was supported by the BIGMED grant from the Norwegian Research Council.
Rasi, C. , Nilsson, D. , Magnusson, M. , Lesko, N. , Lagerstedt‐Robinson, K. , Wedell, A. , Lindstrand, A. , Wirta, V. , & Stranneheim, H. (2022). PatientMatcher: A customizable Python‐based open‐source tool for matching undiagnosed rare disease patients via the Matchmaker Exchange network. Human Mutation, 43, 708–716. 10.1002/humu.24358
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no data sets were generated or analyzed during the current study.
REFERENCES
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzariti, D. R. , & Hamosh, A. (2020). Genomic data sharing for novel Mendelian disease gene discovery: The Matchmaker Exchange. Annual Review of Genomics and Human Genetics, 21, 305–326. [DOI] [PubMed] [Google Scholar]
- Bonomi, L. , Huang, Y. , & Ohno‐Machado, L. (2020). Privacy challenges and research opportunities for genomic data sharing. Nature Genetics, 52, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. , Hamosh, A. , Rehm, H. , Philippakis, A. A. , Azzariti, D. R. , Beltran, S. , Brookes, A. J. , Brownstein, C. A. , Brudno, M. , Brunner, H. G. , Buske, O. J. , Carey, K. , Doll, C. , Dumitriu, S. , Dyke, S. O. M. , den Dunnen, J. T. , Firth, H. V. , Gibbs, R. A. , Girdea, M. , … Rehm, H. L. (2015). The Matchmaker Exchange: A platform for rare disease gene discovery. Human Mutation, 36, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Vanstone, M. R. , Bulman, D. E. , & MacKenzie, A. E. (2013). Rare‐disease genetics in the era of next‐generation sequencing: Discovery to translation. Nature Reviews Genetics, 14, 681–691. [DOI] [PubMed] [Google Scholar]
- Buske, O. J. , Schiettecatte, F. , Hutton, B. , Dumitriu, S. , Misyura, A. , Huang, L. , Hartley, T. , Girdea, M. , Sobreira, N. , Mungall, C. , & Brudno, M. (2015). The Matchmaker Exchange API: Automating patient matching through the exchange of structured phenotypic and genotypic profiles. Human Mutation, 36, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth, H. V. , Richards, S. M. , Bevan, A. P. , Clayton, S. , Corpas, M. , Rajan, D. , Vooren, S. V. , Moreau, Y. , Pettett, R. M. , & Carter, N. P. (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. American Journal of Human Genetics, 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, S. , Doelken, S. C. , Mungall, C. J. , Bauer, S. , Firth, H. V. , Bailleul‐Forestier, I. , Black, G. C. , Brown, D. L. , Brudno, M. , Campbell, J. , FitzPatrick, D. R. , Eppig, J. T. , Jackson, A. P. , Freson, K. , Girdea, M. , Helbig, I. , Hurst, J. A. , Jähn, J. , Jackson, L. G. , … Robinson, P. N. (2014). The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Research, 42, D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. , Deignan, J. L. , Dorrani, N. , Strom, S. P. , Kantarci, S. , Quintero‐Rivera, F. , Das, K. , Toy, T. , Harry, B. , Yourshaw, M. , Fox, M. , Fogel, B. L. , Martinez‐Agosto, J. A. , Wong, D. A. , Chang, V. Y. , Shieh, P. B. , Palmer, C. G. S. , Dipple, K. M. , Grody, W. W. , … Nelson, S. F. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. Journal of the American Medical Association, 312, 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker, M. L. (2010). Sequencing technologies—The next generation. Nature Reviews Genetics, 11, 31–46. [DOI] [PubMed] [Google Scholar]
- Molnár‐Gábor, F. , & Korbel, J. O. (2020). Genomic data sharing in Europe is stumbling—Could a code of conduct prevent its fall? EMBO Molecular Medicine, 12, e11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavan, S. , Rommel, K. , Mateo Marquina, M. E. , Höhn, S. , Lanneau, V. , & Rath, A. (2017). Clinical practice guidelines for rare diseases: The Orphanet database. PLoS One, 12, e0170365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesquita, C. , Faria, D. , Bastos, H. , Ferreira, A. E. , Falcão, A. O. , & Couto, F. M. (2008). Metrics for GO based protein semantic similarity: A systematic evaluation. BMC Bioinformatics, 9, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, M. (2018). International data‐sharing norms: From the OECD to the General Data Protection Regulation (GDPR). Human Genetics, 137, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure, J. , & Ji, H. (2008). Next‐generation DNA sequencing. Nature Biotechnology. 26, 1135–1145. [DOI] [PubMed] [Google Scholar]
- Soden, S. E. , Saunders, C. J. , Willig, L. K. , Farrow, E. G. , Smith, L. D. , Petrikin, J. E. , LePichon, J.‐B. , Miller, N. A. , Thiffault, I. , Dinwiddie, D. L. , Twist, G. , Noll, A. , Heese, B. A. , Zellmer, L. , Atherton, A. M. , Abdelmoity, A. T. , Safina, N. , Nyp, S. S. , Zuccarelli, B. , … Kingsmore, S. F. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Science Translational Medicine, 6, 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranneheim, H. , Lagerstedt‐Robinson, K. , Magnusson, M. , Kvarnung, M. , Nilsson, D. , Lesko, N. , Engvall, M. , Anderlid, B.‐M. , Arnell, H. , Johansson, C. B. , Barbaro, M. , Björck, E. , Bruhn, H. , Eisfeldt, J. , Freyer, C. , Grigelioniene, G. , Gustavsson, P. , Hammarsjö, A. , Hellström‐Pigg, M. , … Wedell, A. (2021). Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Medicine, 13, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Muzny, D. M. , Xia, F. , Niu, Z. , Person, R. , Ding, Y. , Ward, P. , Braxton, A. , Wang, M. , Buhay, C. , Veeraraghavan, N. , Hawes, A. , Chiang, T. , Leduc, M. , Beuten, J. , Zhang, J. , He, W. , Scull, J. , Willis, A. , … Eng, C. M. (2014). Molecular findings among patients referred for clinical whole‐exome sequencing. Journal of the American Medical Association, 312, 1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no data sets were generated or analyzed during the current study.