

Abstract
A total synthesis of the cyclic lipodepsipeptide natural product orfamide A was achieved. By developing a synthesis format using an aminoacid ester building block and SPPS protocol adaptation, a focused library of target compounds was obtained, in high yield and purity. Spectral and LC‐HRMS data of all library members with the isolated natural product identified the 5Leu residue to be d‐ and the 3’‐OH group to be R‐configured. The structural correction of orfamide A by chemical synthesis and analysis was confirmed by biological activity comparison in Chlamydomonas reinhardtii, which indicated compound configuration to be important for bioactivity. Acute toxicity was also found against Trypanosoma brucei, the parasite causing African sleeping sickness.
Keywords: algae, natural products, peptides, structure-activity relationship, total synthesis
For the total synthesis of orfamide A an efficient synthesis format for cyclic lipodepsipeptide natural products on solid phase was developed. A strategy using side chain anchoring, an ester building block, and O→N acyl migration suppression enabled high‐yielding synthesis on solid support and facilitated compound library synthesis. The configuration of the proposed structure of orfamide A was corrected at two positions. Studies in Chlamydomonas algae uncovered the impact of orfamide A stereochemistry on compound bioactivity.

Introduction
Pseudomonads are ubiquitous, Gram‐negative gammaproteobacteria that produce a range of biologically active natural products.[ 1 , 2 ] Among them, cyclic lipodepsipeptides (CLPs) act as biosurfactants, as antimicrobials, or as biocontrol agents. [3] Typical CLPs biosynthesized by pseudomonads such as viscosin (1), orfamide A (2,3), or anikasin (4) contain 8–25 amino acids of which 4–10 residues form a macrocyle (Figure 1). [2] Moreover, all feature a lipidated N‐terminus. Orfamide A from Pseudomonas protegens Pf‐5 or CHA0[ 4 , 5 ] has been characterized as an insecticidal,[ 6 , 7 ] an antifungal, [8] and as an algicidal agent. [9] Intriguingly, orfamide A triggers an increase in cytosolic Ca2+ in the green alga Chlamydomonas reinhardtii, causing its deflagellation, and thus leads to its immobilization (IC50 =4.1 μM).[ 9 , 10 ] Orfamide A immobilizes several flagellate algae from the class of Chlorophyceae but not select algae from other classes. [9] A total synthesis of orfamide A has not been reported to date, except for a patent claim. [11] We report here on the total synthesis and the resulting revision of the chemical structure of orfamide A (2→3), based on an improved, generally reliable synthetic route to CLP natural products.
Figure 1.

Cyclic lipodepsipeptides (CLPs) viscosin, [28] orfamide A, [4] and anikasin [29] from Pseudomonas spp. Amino acid (A.A.) residues above are color‐coded to indicate preserved (red), similar (blue), or stereoisomeric residues (green).
Results and Discussion
Orfamide A features ten amino acids and a β‐hydroxytetradecanoic acid (β‐HTDA). The 3’‐hydroxy group had been allocated S‐configuration (→2), [4] in contrast to many β‐hydroxy acids found in other CLPs from Pseudomonas, [2] which are R‐configured. In order to clarify this issue, we synthesized both enantiomers of β‐HTDA in high e.e. by using Noyori's hydrogenation chemistry[ 12 , 13 , 14 ] (see Supporting Information for details). Then, a method was adapted that had originally been reported for the synthesis of pseudodesmin A. [12] By applying solid‐phase peptide synthesis (SPPS) with side‐chain anchored serine, on‐resin esterification, and subsequent cyclization, the orfamide stereoisomers 2 and 5 (Table 1) were initially obtained in approx. 2 % yield (see Supporting Information, Scheme S3).
Table 1.
Orfamide A isomers prepared in this study. The configuration that was finally confirmed is highlighted.
|
Compound |
A.A. residue 1 |
A.A. residue 5 |
3’‐OH |
Yield[a] |
|---|---|---|---|---|
|
2 |
l‐Leu |
l‐Leu |
S |
2 %, 36 %[b] |
|
5 |
l‐Leu |
l‐Leu |
R |
2 %, 38 %[b] |
|
6 |
d‐Leu |
l‐Leu |
S |
18 %[b] |
|
7 |
d‐Leu |
l‐Leu |
R |
6 %, 40 %[b] |
|
8 |
l‐Leu |
d‐Leu |
S |
36 %[b] |
|
3 |
l‐Leu |
d‐Leu |
R |
7 %, 35 %[b] |
|
9 |
d‐Leu |
d‐Leu |
S |
44 %[b] |
|
10 |
d‐Leu |
d‐Leu |
R |
12 %, 44 %[b] |
[a] Yield calculated from resin loading; [b] yield obtained by using optimized procedures (Scheme 2).
Compounds 2 and 5 were compared with an authentic sample by using LC/HRMS analysis (Figure 2) and 1H NMR spectroscopy. Both synthetic CLPs were surprisingly distinct from the natural product, indicating the originally proposed structure 2 to deserve a more detailed reinvestigation beyond side chain hydroxylation stereochemistry.
Figure 2.
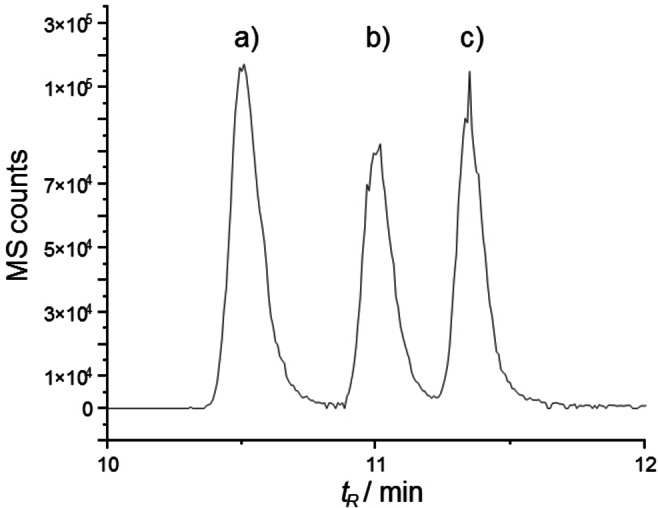
Extracted ion chromatogram (MS range: 1295.8±0.5) of a mixed sample containing synthesized compounds 2 and 5 and isolated orfamide A (NP). a) 2, b) 5, c) NP. See Supporting Information for experimental details.
NMR‐ and MS data indicated the compound in question to be an isomer of the proposed structure, likely a stereoisomer. The NRPS (non‐ribosomal peptide synthetase) biosynthesis genes of orfamide A in P. protegens Pf‐5 were hence analyzed using antiSMASH version 6.0. [15] The NRPS genes orfA, orfB, and orfC, which code for the biosynthetic machinery of the orfamides, were further examined using NRPSPredictor2 to determine the amino acid specificity of the adenylation (A) domains. In addition, a phylogenetic algorithm was employed to assign the condensation (C) domain functionalities (Figure 3). [16] The A‐domain analysis was in good agreement with the structure proposed earlier. [4] All but two A‐domain specificities showed a 100 % match with the code introduced by Stachelhaus et al. [17] and were in accordance with the originally assigned structure of the orfamides. The only exceptions were the A domains 2 and 10, which were predicted to incorporate Asp (80 % match) and a hydrophobic amino acid (not further specified), respectively. Such ambiguous or deviating classifications are regularly encountered in bioinformatics analyses. Importantly, the predicted character of the two amino acids (acidic and hydrophobic) matched the expected structure. However, analysis of the C domains revealed stereochemical inconsistencies for the residues 1Leu and 5Leu. These amino acids had originally been assigned with l‐configuration, but condensation‐epimerization (C/E) domains subsequent to modules 1 and 5 should produce d‐configured residues. Nonetheless, non‐functional epimerization domains have been observed in Pseudomonas,[ 2 , 18 ] precluding an assignment from NRPS gene sequencing data a priori in these organisms today. Therefore, all possible stereoisomers at the ambiguous positions were selected as candidates for comparison by synthesis (3, 6–10, Table 1).
Figure 3.

Module structure of the three NRPS genes that code for the biosynthesis of the orfamides in P. protegens Pf‐5 (Gene Bank accession number CP000076), as predicted by antiSMASH. Expected A‐domain specificity (NRPSPredictor2) and C‐domain subtype (NaPDoS) are indicated (see Supporting Information for methods). The residue loaded by module 10 is predicted to be hydrophobic. Abbreviations: orf=Orfamide synthase; C/E=Condensation‐epimerization domain; CStart=Starter condensation domain; LCL=Condensation domain for linking two L−A.A.s; TE=Thioesterase domain; A=Adenylation domain; T=Thiolation domain.
In our early synthesis experiments (Supporting Information, Scheme S3), we had found the on‐resin esterification procedure to be slow and prone to epimerization, in line with other reports.[ 19 , 20 ] Synthesis reorganization only led to modest improvements (Supporting Information, Schemes S2 and S3). To establish a more reliable method, we envisioned using a building block with a preformed ester bond.[ 20 , 21 ] Such building blocks were previously employed for the synthesis of A54145B [22] and ophiotine. [23] Notably, O→N acyl migrations from β‐oxygen atoms to proximal α‐amino groups are facile under basic conditions and have indeed been applied to dedicated peptide bond syntheses.[ 20 , 21 , 24 ] To study if regular Fmoc protection could be kept for SPPS and the migration still be prohibited, Fmoc‐d‐allo‐Thr (11) was protected with a diphenylmethyl (Dpm) group[ 25 , 26 ] and esterified with Alloc‐l‐Val to yield ester 13 in excellent stereoisomeric purity (>97 %, Scheme 1 and Supporting Information). TFA‐mediated cleavage of the Dpm group produced the ester building block 14 in 81 % overall yield for direct use. Isomer 14’ was prepared for comparison (d.e. determination).
Scheme 1.

Synthesis of dipeptide building blocks 14 and 14′. Reagents and conditions: a) Benzophenone hydrazone, PIDA, cat. I2, CH2Cl2, 0 °C, 93 %;[ 25 , 26 ] b) Alloc‐l‐Val or Alloc‐d‐Val, EDCI, DMAP, CH2Cl2, 0 °C to r.t.; c) TFA, TIS, CH2Cl2, 0 °C to r.t.
Automated SPPS using Fmoc/tBu strategy and side‐chain anchored serine on trityl resin at moderate loading (0.5 mmol/g) cleanly provided the resin‐bound peptides 16 and 17 (Scheme 2). Coupling with dimer 14 gave the linear octadepsipeptides 18 and 19. During the coupling of ester 14, elimination of Alloc‐l‐Valine to give a dehydrothreonine residue was observed as a side reaction, [20] which was suppressed after suitable optimization (see Supporting Information for details). Simultaneous allyl deprotection and on‐resin macrolactamization efficiently led to the cyclic octadepsipeptides 20 and 21. Fmoc deprotection tests with linear peptide 19 and cyclic peptide 21 indicated the O→N acyl migration rate of cyclic peptide 21 (23→24) to be significantly lower than that of linear peptide 19, probably owing to a more rigid structure (see Supporting Information for details). After optimization, the O→N acyl migration could be suppressed for the cyclic peptides 20 and 21 by applying rapid Fmoc deprotection by brief (30s, 2x) exposure to 2 % DBU/2 % piperidine/DMF solution and rapid washing with DMF to minimize the peptide exposure period to basic conditions, immediately followed by subsequent amino acid couplings. Finally, acylation with β‐HTDA completed the syntheses of the target compounds 2, 3, and 5–10 in very appreciable yields (Table 1), likely the result of significantly facilitated compound purification.
Scheme 2.

Total synthesis of orfamide A and its stereoisomers (see Table 1). Reagents and Conditions: a) piperidine/DMF (20 vol %); 14, HATU, HOAt, 2,4,6‐collidine, DMF, r.t.; b) Pd(PPh3)4, PhSiH3, CH2Cl2, r.t.; c) HATU, HOAt, 2,4,6‐collidine, DMF, r.t.; d) DBU/piperidine/DMF (2 vol %, 2 vol %), 2×30 s; e) Fmoc‐A.A., HBTU, HOBt, DIEA, DMF, r.t.; piperidine/DMF (20 vol %); f) (R)‐ or (S)‐3‐TBSoxy tetradecanoic acid, HBTU, HOBt, DIEA, DMF, r.t.; g) 0.1 N HCl/HFIP, TIS (1 vol %), r.t.[ 27 , 30 ]
All synthesized candidates were subsequently characterized by using RP‐HPLC. Candidates with retention times on RP‐HPLC similar to authentic material were further compared by using LC/HRMS (Figure 4). Only the characteristics of compound 3 (l‐1Leu, d‐5Leu, 3’‐R‐HTDA) agreed with the isolated natural product. Unambiguous verification of the structure of orfamide A was then achieved by 1H NMR through comparison of synthetic 3, the natural product, and their mixture, all showing identical 1H NMR spectra in acidic buffer (see Supporting Information, Figures S5–S10).
Figure 4.
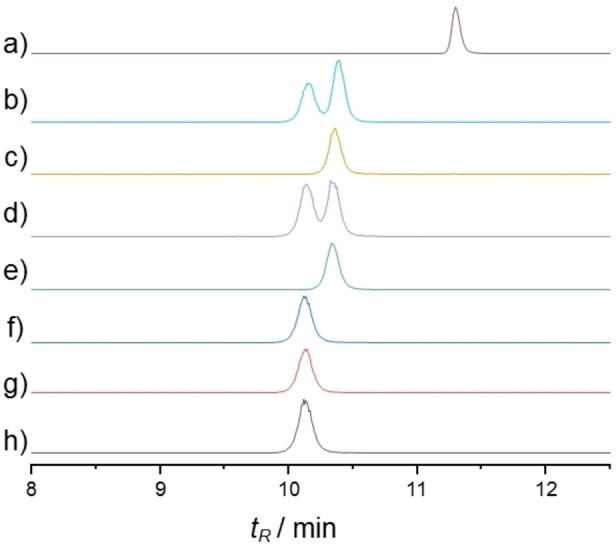
Extracted ion chromatogram (m/z=1295.8±0.5 Da) of the natural product (NP) and selected orfamide A derivatives (see Table 1): a) 10; b) mixture of 9 and NP; c) 9; d) mixture of 7 and NP; e) 7; f) 3; g) mixture of 3 and NP; h) NP. Peak heights normalized for comparison, for individual full traces see Supporting Information (Figure S12).
To compare the biological response of synthetic CLPs 2 and 3 to isolated orfamide A in C. reinhardtii, we used the transgenic AEQ34 cell line that expresses apo‐aequorin for measuring cytosolic Ca2+ levels (for details see Supporting Information).[ 9 , 10 ] CLP 3 showed identical characteristics to the natural product, with a rapidly occurring initial Ca2+ release signal and a secondary Ca2+‐release that persisted over several minutes. These data additionally confirmed that compound 3 has the correct structure of orfamide A. CLP 2, which differs from CLP 3 only in the configuration of two residues, showed a distinct Ca2+ signature, where the initial signal was strongly reduced and the secondary release was absent (Figure 5). Beyond biological identity, these data indicate that its stereochemistry profoundly affects the biological properties of orfamide A. This is corroborated by other biological activities and the activity of further orfamide A variants. [31]
Figure 5.
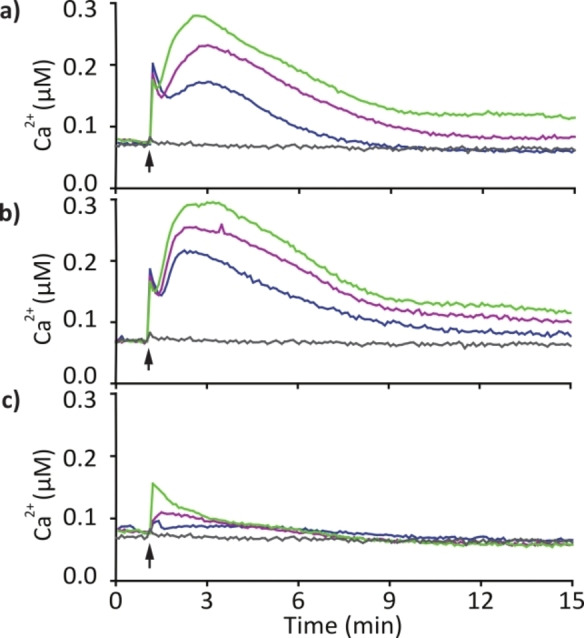
Time course of cytosolic Ca2+ concentrations in C. reinhardtii upon compound treatment. Compounds were dissolved as a stock solution in MeOH and further diluted in TAP medium. Each line represents the mean of three independent biological replicates, and each biological replicate includes three technical replicates. For details see Supporting Information. Color code: MeOH control (gray), 2 μM compound (blue), 5 μM (purple), 10 μM (green). a) Synthetic orfamide A (3), b) isolated orfamide A (NP), c) compound 2.
Since orfamide A causes immobilization in several flagellated chlorophyte algae, [9] we also studied the medically relevant Trypanosoma brucei, a flagellated parasitic microorganism that causes African sleeping sickness. While orfamide A (3) did not cause deflagellation of T. brucei, it nonetheless inhibited its growth (IC50 =6 μM, see Supporting Information for details), as it did in C. reinhardtii. [9] It was furthermore found to be acutely toxic to trypanosoma cells immediately upon exposure.
Conclusion
In summary, we have completed the first total synthesis of orfamide A and determined its correct structure (3) by independently applying synthetic chemistry, bioinformatics analysis, and bioassays. These results emphasize once again the importance of total synthesis in natural products chemistry, especially when stereochemistry is concerned. [32] We have established a novel, robust, versatile, and high yielding synthetic route to CLPs by using a preformed ester bond in building block 14 and by suppressing O→N acyl migration during Fmoc deprotection. This methodology was implemented on solid support and is compatible with automated SPPS. Thereby, orfamide A analogs and other related CLPs will now be easily accessible for detailed biological or structural studies.
Conflict of interest
The authors declare no conflicts of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
M. M., P. S. and H.‐D. A. received funding from the Deutsche Forschungsgemeinschaft (DFG) within the collaborative research center SFB1127/2 ChemBioSys‐Project ID 239748522 (subprojects A02, A04, and C03). Support from the DFG Cluster of Excellence EXC2051 Balance of the Microverse‐Project‐ID 390713860 (to U. A. H., P. S., H.D.A., and M.M.) is kindly acknowledged. P. S. is grateful for financial support from the Werner Siemens‐Stiftung. Y. B. received a predoctoral fellowship grant of the YOSHIDA foundation (JPN). This work benefitted from an equipment grant of the DFG (INST 275/442‐1 FUGG). Open Access funding enabled and organized by Projekt DEAL.
Y. Bando, Y. Hou, L. Seyfarth, J. Probst, S. Götze, M. Bogacz, U. A. Hellmich, P. Stallforth, M. Mittag, H.-D. Arndt, Chem. Eur. J. 2022, 28, e202104417.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Sampedro I., Parales R. E., Krell T., Hill J. E., FEMS Microbiol. Rev. 2015, 39, 17–46. [DOI] [PubMed] [Google Scholar]
- 2. Götze S., Stallforth P., Nat. Prod. Rep. 2020, 37, 29–54. [DOI] [PubMed] [Google Scholar]
- 3. Geudens N., Martins J. C., Front. Microbiol. 2018, 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gross H., Stockwell V. O., Henkels M. D., Nowak-Thompson B., Loper J. E., Gerwick W. H., Chem. Biol. 2007, 14, 53–63. [DOI] [PubMed] [Google Scholar]
- 5. Ma Z., Geudens N., Kieu N. P., Sinnaeve D., Ongena M., Martins J. C., Höfte M., Front. Microbiol. 2016, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jang J. Y., Yang S. Y., Kim Y. C., Lee C. W., Park M. S., Kim J. C., Kim I. S., J. Agric. Food Chem. 2013, 61, 6786–6791. [DOI] [PubMed] [Google Scholar]
- 7. Loper J. E., Henkels M. D., Rangel L. I., Olcott M. H., Walker F. L., Bond K. L., Kidarsa T. A., Hesse C. N., Sneh B., Stockwell V. O., Taylor B. J., Environ. Microbiol. 2016, 18, 3509–3521. [DOI] [PubMed] [Google Scholar]
- 8. Ma Z., Ongena M., Höfte M., Plant Cell Rep. 2017, 36, 1731–1746. [DOI] [PubMed] [Google Scholar]
- 9. Aiyar P., Schaeme D., García-Altares M., Carrasco-Flores D., Dathe H., Hertweck C., Sasso S., Mittag M., Nat. Commun. 2017, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rose M. M., Scheer D., Hou Y., Hotter V. S., Komor A. J., Aiyar P., Scherlach K., Vergara F., Yan Q., Loper J. E., Jakob T., van Dam N. M., Hertweck C., Mittag M., Sasso S., Environ. Microbiol. 2021, 23, 5525–5540. [DOI] [PubMed] [Google Scholar]
- 11.Hybio Pharmaceutical Co Ltd. The solid phase synthesis process of a kind of cyclic ester peptide Orfamide A. Chinese Patent CN 103626848 B, May 4, 2016.
- 12. de Vleeschouwer M., Sinnaeve D., van den Begin J., Coenye T., Martins J. C., Madder A., Chem. Eur. J. 2014, 20, 7766–7775. [DOI] [PubMed] [Google Scholar]
- 13. Noyori R., Ohkuma T., Kitamura M., Takaya H., Sayo N., Kumobayashi H., Akutagawa S., J. Am. Chem. Soc. 1987, 109, 5856–5858. [Google Scholar]
- 14. Ratovelomanana-Vidal V., Girard C., Touati R., Tranchier J. P., Ben Hassine B., Genêt J. P., Adv. Synth. Catal. 2013, 345, 261–274. [Google Scholar]
- 15. Blin K., Shaw S., Kloosterman A. M., Charlop-Powers Z., van Wezel G. P., Medema M. H., Weber T., Nucleic Acids Res. 2021, 49, W29-W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rausch C., Hoof I., Weber T., Wohlleben W., Huson D. H., BMC Evol. Biol. 2007, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stachelhaus T., Mootz H. D., Marahiel M. A., Chem. Biol. 1999, 6, 493–505. [DOI] [PubMed] [Google Scholar]
- 18. Arp J., Götze S., Mukherji R., Mattern D. J., García-Altares M., Klapper M., Brock D. A., Brakhage A., Strassmann J. E., Queller D. C., Bardl B., Willing K., Peschel G., Stallforth P., Proc. Natl. Acad. Sci. USA 2018, 115, 3758–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Vleeschouwer M., Martins J. C., Madder A., J. Pept. Sci. 2016, 22, 149–155. [DOI] [PubMed] [Google Scholar]
- 20. Coin I., Dölling R., Krause E., Bienert M., Beyermann M., Sferdean C. D., Carpino L. A., J. Org. Chem. 2006, 71, 6171–6177. [DOI] [PubMed] [Google Scholar]
- 21. Coin I., Schmieder P., Bienert M., Beyermann M., J. Pept. Sci. 2008, 14, 299–306. [DOI] [PubMed] [Google Scholar]
- 22. Chow H. Y., Chen D., Li X., Org. Biomol. Chem. 2020, 18, 4401–4405. [DOI] [PubMed] [Google Scholar]
- 23. Wang J., Liu C., Hu H. G., Zou Y., Zhao Q. J., Ye G. M., Chem. Nat. Compd. 2020, 56, 883–887. [Google Scholar]
- 24. Sohma Y., Hayashi Y., Skwarczynski M., Hamada Y., Sasaki M., Kimura T., Kiso Y., Pept. Sci. 2004, 76, 344–356. [DOI] [PubMed] [Google Scholar]
- 25. Lapatsanis L., Milias G., Paraskewas S., Synthesis 1985, 513–515. [Google Scholar]
- 26. Zhu Y., Gieselman M. D., Zhou H., Averin O., van der Donk W. A., Org. Biomol. Chem. 2003, 1, 3304–3315. [DOI] [PubMed] [Google Scholar]
- 27. Palladino P., Stetsenko D. A., Org. Lett. 2012, 14, 6346–6349. [DOI] [PubMed] [Google Scholar]
- 28. Burke T. R., Knight M., Chandrasekhar B., Ferretti J. A., Tetrahedron Lett. 1989, 30, 519–522. [Google Scholar]
- 29. Götze S., Herbst-Irmer R., Klapper M., Görls H., Schneider K. R. A., Barnett R., Burks T., Neu U., Stallforth P., ACS Chem. Biol. 2017, 12, 2498–2502. [DOI] [PubMed] [Google Scholar]
- 30. de Vleeschouwer M., van Kersavond T., Verleysen Y., Sinnaeve D., Coenye T., Martins J. C., Madder A., Front. Microbiol. 2020, 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hou Y., Bando, Flores D. C., Hotter V. S., Schiweck B., Arndt H.-D., Mittag M., bioRxiv 2021.07.24.453618, 10.1101/2021.07.24.453618. [DOI] [Google Scholar]
- 32. de Roo V., Verleysen Y., Kovács B., Matthias D. V., Girard L., Höfte M., De Mot R., Madder A., Geudens N., Martins J. C., bioRxiv 2022.1.7.475420, 10.1101/2022.01.07.475420. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.