

Abstract
Individuals with Down syndrome show cellular and clinical features of dysregulated aging of the immune system, including a shift from naïve to memory T cells and increased incidence of autoimmunity. However, a quantitative understanding of how various immune compartments change with age in Down syndrome remains lacking. Here we performed deep immunophenotyping of a cohort of individuals with Down syndrome across the lifespan, selecting for autoimmunity-free individuals. We simultaneously interrogated age- and sex-matched healthy controls and people with type 1 diabetes as a representative autoimmune disease. We built an analytical software, IMPACD (Iterative Machine-assisted Permutational Analysis of Cytometry Data), that enabled us to rapidly identify many features of immune dysregulation in Down syndrome shared with other autoimmune diseases. We found quantitative and qualitative dysregulation of naïve CD4+ and CD8+ T cells in individuals with Down syndrome and identified interleukin (IL)-6 as a candidate driver of some of these changes, thus extending the consideration of immunopathologic cytokines in Down syndrome beyond interferons. We used immune cellular composition to generate three linear models of aging (immune clocks) trained on control participants. All three immune clocks demonstrated advanced immune aging in individuals with Down syndrome. One of these clocks, informed by Down syndrome-relevant biology, also showed advanced immune aging in individuals with type 1 diabetes. Orthologous RNA sequencing-derived immune clocks also demonstrated advanced immune aging in individuals with Down syndrome. Together, our findings demonstrate an approach to studying immune aging in Down syndrome which may have implications in the context of other autoimmune diseases.
One Sentence Summary:
Permutational analysis of the immune system reveals advanced immune aging in individuals with Down syndrome and in individuals with type 1 diabetes.
INTRODUCTION
Down syndrome (DS, trisomy 21) is the most common congenital chromosomal abnormality, affecting about 1 in 800 births (1). Immune dysregulation is a central feature of DS, manifested clinically by a greatly increased predisposition to many autoimmune diseases including autoimmune thyroid disease, type 1 diabetes (T1D), and celiac disease (2, 3). Autoimmunity is an important and growing burden on quality of life in people with DS due in part to the increasing lifespan of people with DS increasing the duration of this chronic comorbidity (2). Understanding how triploidy of the 233 genes on chromosome 21 remodels the immune landscape is essential to enable rational selection and development of therapies to specifically mitigate DS-associated autoimmunity (2). Furthermore, this knowledge can advance our understanding of how related mechanisms promote autoimmunity in the broader population.
Previous studies have identified several features of immune remodeling in DS associated with autoimmunity, including altered immune cellular subsets, such as increased memory:naïve T cell ratios, elevated circulating concentrations of pro-inflammatory cytokines, and transcriptional evidence of a hyper-response to type 1 interferons (IFN) (4–9). Interestingly, many of these features have also been described in the context of inflammaging, an inflammation-driven remodeling of the immune landscape during aging associated with increased risk of autoimmunity (10–12). The role of accelerated immune aging has not been well-studied in the context of DS. It remains to be clarified (i) how DS impacts immune remodeling across the lifespan, (ii) what features of immune remodeling are DS-specific, (iii) the role of type 1 IFN and other cytokines in DS-specific immune remodeling, and (iv) how immune remodeling in the context of DS resembles that in autoimmunity. Prior immunologic studies frequently focus on either pediatric or adult populations with DS, hampering a comprehensive study of immune changes across the lifespan. Additionally, many DS cohorts are enriched for autoimmunity, which may influence immune age (13). Therefore, we currently lack a quantitative understanding of how immune age is altered in DS, when during the lifespan immune age differences begin and whether these differences contribute to the development of or result from comorbidities associated with DS.
In this study, we addressed these questions by evaluating the immune landscape in a cohort of individuals with DS, ages 2 to 55 years old, with a low incidence of autoimmunity. We included age- and sex-matched (i) healthy typical control (TC) participants and (ii) typical individuals with T1D to identify changes related to one DS-relevant autoimmune disease. Pertinent to this study and unlike other autoimmune diseases, individuals with T1D span a wide range of ages and do not receive immunomodulatory therapy that might alter the immune landscape. To enable exhaustive analysis of our mass cytometry data, we built a software tool, IMPACD (Iterative Machine-assisted Permutational Analysis of Cytometry Data). This tool improves analytic rigor of manual gating analyses and performs exhaustive permutational analysis of mass cytometry data with zero down-sampling. Using IMPACD, we found extensive dysregulation of naïve T cells in DS and identified interleukin (IL)-6 as a potential driver of several of these qualitative changes. We also found evidence of co-regulation of CD4+ T cells and natural killer T (NKT) cells in individuals with DS. Importantly, IMPACD empowered us to use immune subsets to build linear models that quantitate immune age (here termed immune clocks). These cellular immune clocks quantitatively demonstrate advanced immune aging in DS. This finding was independently corroborated by RNA sequencing (RNAseq)-based immune clocks, which pointed to mechanistic hypotheses. Furthermore, a DS-informed cytometry-based immune clock also demonstrated advanced immune aging in people with T1D. These findings advance our understanding of altered immune architecture in people with and without DS.
RESULTS
The immune landscape is broadly altered in individuals with DS.
To investigate features of immune dysregulation in DS that may underlie predisposition to autoimmunity, we used mass cytometry (CyTOF) to immunophenotype peripheral blood mononuclear cells (PBMCs) from 28 individuals with DS (ages 2 to 55 years), 28 age- and sex-matched healthy typical controls (TCs) and 25 age- and sex-matched typical individuals with T1D (Fig. 1A and B). Autoimmunity in our DS cohort was limited to Hashimoto’s disease, a common cause of hypothyroidism, in 2 of 28 participants. As cytomegalovirus (CMV) infection can substantially alter the immune landscape, we measured anti-CMV antibodies as evidence of prior infection and found similar proportions of seropositive participants in all cohorts (fig. S1A) (14).
Figure 1. Broad changes in immune architecture in Down syndrome are revealed by mass cytometry.
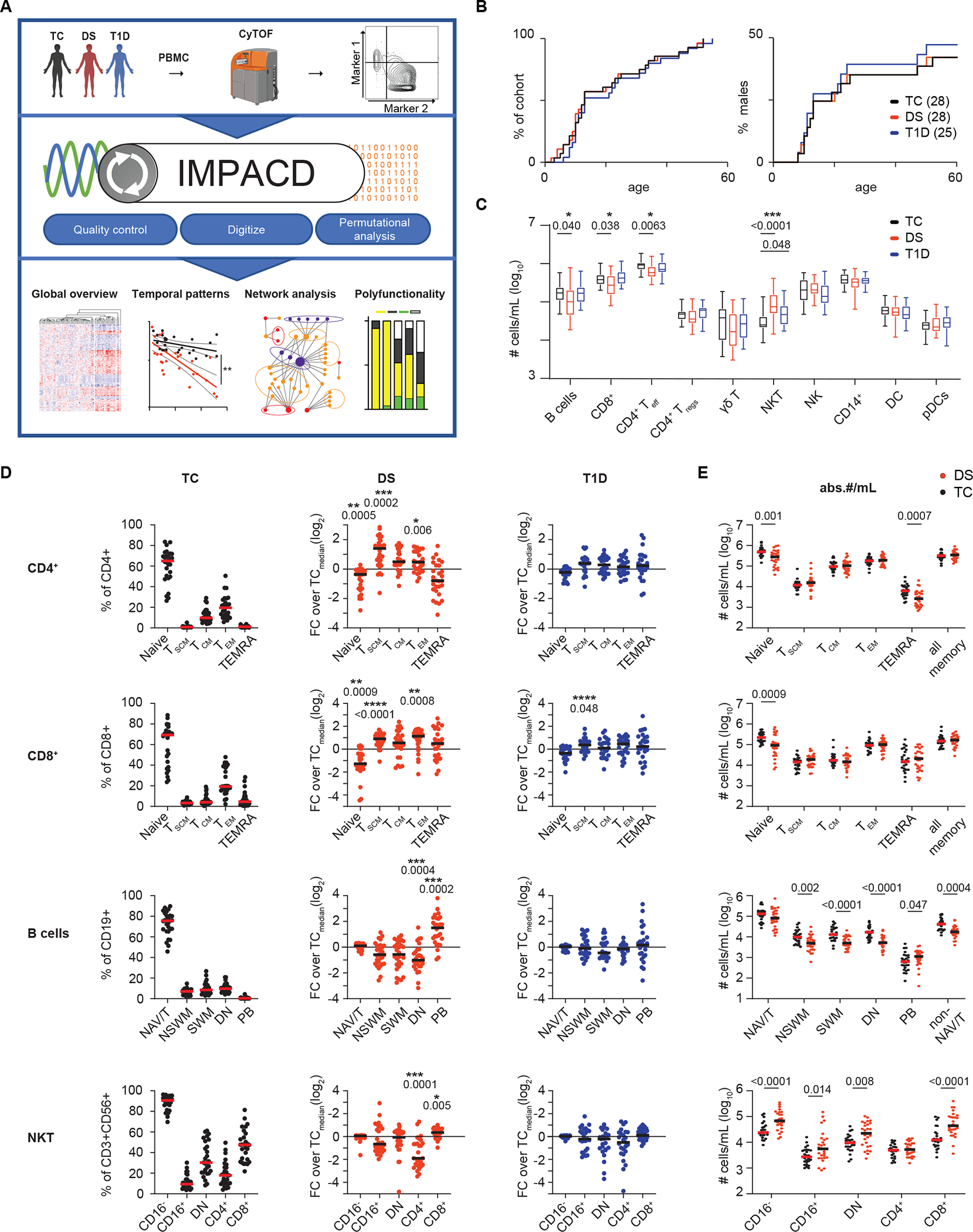
(A) Schematic of study design. PBMCs from age and sex-matched participants with Down syndrome (DS, n=28), typical controls (TC, n=28) or typical participants with type 1 diabetes (T1D, n=25) were immunophenotyped by mass cytometry. A software tool, IMPACD, was developed to perform rigorous and exhaustive permutational analysis. IMPACD’s data output is readily analyzed using omics-relevant approaches. (B) Age and sex distribution of cohorts are shown. (C) Absolute numbers of major immune cell types are shown by cohort. (D) Frequencies of CD4+ T cell, CD8+ T cell, B cell and NKT cell subsets in TC, DS, and T1D participants are shown. Data from participants with DS or T1D are scaled (FC, fold change) to the corresponding subset’s median value in TC participants (TCmedian) and statistically compared to TCs. T cell (stem cell memory (TSCM), central memory (TCM), effector memory (TEM) and terminally differentiated effector memory T cells (TEMRA)), B cell (naïve/transitional (NAV/T), non-switched memory (NSWM), switched memory (SWM), double negative (DN) and plasmablast (PB)) and NKT (CD4−CD8− double negative (DN)) subsets are shown. (E) Absolute numbers per mL of blood (abs.#/mL) of CD4+ T, CD8+ T, B cell and NKT subsets in TC and DS participants are shown. (B to E) n = 28 (B and D) or n = 26 (C and E) TC, n = 28 DS, n = 25 T1D, across 5 batches. (C and D) Asterisks indicate statistical significance by Kruskal-Wallis test (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). Posthoc Mann-Whitney tests were used; p-values are shown. (E) Mann-Whitney tests were used; p-values are shown.
We first quantitated proportions of major innate and adaptive cell types by manual gating and absolute numbers using contemporaneous complete blood count (CBC) data. We found decreased absolute numbers of B cells, CD4+ effector T (Teff) cells and CD8+ T cells, consistent with previous studies showing varying degrees of B- and T-lymphopenia in DS (Fig. 1C and fig. S1B) (6, 9, 15). Numbers of regulatory T (Treg) cells were comparable between cohorts (Fig. 1C). CD3+CD56+ NKT cells were increased in DS and, to a lesser extent, T1D (Fig. 1C). Analyses of cellular frequencies among total PBMCs showed similar results (fig. S1C).
As T and B cells were the most quantitatively altered subsets in DS, we next assessed qualitative remodeling in these cells, first enumerating naïve and memory subsets as previously defined (16). Given the wide range of frequencies between these subsets, we show the raw percentage in TC and normalize the percentage in people with DS or T1D to the corresponding median value in TC to effectively display all data simultaneously (Fig. 1D). Consistent with previous studies, we found decreased frequencies of naïve CD4+ Teff and CD8+ T cells in DS (Fig. 1D and fig. S1B) (6, 9). The frequency of memory T cell subsets in DS was increased, including stem cell memory T (TSCM) and effector memory T (TEM) cells (Fig. 1D and fig. S1B). We did not observe any differences in frequencies of naïve or memory cells amongst Tregs (fig. S1D). We calculated the absolute number of cells in each subset and found that naïve CD4+ and CD8+ T cells were significantly decreased (P=0.001 and 0.0009 respectively) without any increase in any memory T cell subset; in fact, CD4+ terminally differentiated effector memory T (TEMRA) cells were decreased in DS (Fig. 1E). These results show that the increased memory:naïve T cell ratio is driven by decreased absolute numbers of naïve T cells in DS, consistent with prior studies suggesting decreased thymic output of naïve CD4+ T cells in individuals with DS (17).
In contrast, the B cell compartment in PBMCs from individuals with DS showed decreased absolute numbers of most memory B cell subsets, except plasmablasts (Fig. 1E). The frequency of double negative (DN) B cells was decreased in DS (Fig. 1D). Frequency and absolute numbers of plasmablasts were increased in DS (Fig. 1D and E). These results demonstrate that decreased numbers of memory B cells drive a memory to naïve B cell shift in DS. Previous studies showing intact proliferation and somatic hypermutation in memory B cells in DS suggest that these differences reflect impaired T cell help, although B cell-intrinsic dysregulation cannot be excluded (18, 19).
We further examined NKT cells using two well-established classification schemes of CD16 or CD4 and CD8 expression (20, 21). We found a decreased proportion of CD4+ NKT cells that was driven by increased absolute numbers of CD8+ and CD4−CD8− (DN) NKT cells (Fig. 1D and E). Previous studies demonstrating increased cytotoxic capacity of CD8+ and DN NKT cells suggests a potential immunomodulatory role (22, 23).
Dysregulation of T, B, and NKT compartments in PBMCs from individuals with T1D was less profound than from PBMCs from individuals with DS and included increased frequency of CD8+ TSCM cells (Fig. 1D). Together, these results extend prior studies showing CD8+ TSCM cells are important to T1D pathobiology and provide a unified view of major lymphocyte subset alterations in people with DS and a direct comparison to a DS-relevant autoimmune disease, T1D (24, 25).
High-rigor manual gating with exhaustive permutational analysis using IMPACD enhances deep immune subset profiling.
We hypothesized that extending our analysis to encompass all markers would reveal important differences related to DS. However, the large number of markers (greater than 40 in CyTOF) challenges the consistency of manual gating analysis of high-dimensional cytometry data (26). One challenge is threshold variability, where graphical thresholding of one parameter in multiple plots leads to inadvertent, if slight, variation (fig. S2A). Another challenge is “versioning”, which arises from the need to adjust and reapply gates to multiple subsets. To overcome these challenges, we developed a software tool, IMPACD, that reads and displays gate values from standard flow cytometry workspace (WSP) files. This allowed us to easily identify and harmonize discrepant threshold values within and between batches (fig. S2A). Infrequent use of batch-specific threshold values was guided by an internal control. Thus, IMPACD maximizes the consistency and rigor of manual gating analyses. Next, we used IMPACD to read standard flow cytometry (FCS) files, using the harmonized thresholds to binarize each cell as positive or negative for each marker (fig. S2A). IMPACD and FlowJo software counted similar absolute numbers of cells as positive for each of 29 markers in all 81 individuals over a wide dynamic range, validating IMPACD’s performance (fig. S2B).
We then used IMPACD to define differences in the immune landscape of DS, T1D, and TC. Focusing on “root nodes” of biologically important T, B, and NKT subsets described above, we exhaustively interrogated all permutations of up to three additional markers (Fig. 2A). We found diminishing yield of examining more markers, which mostly generated rare subsets (fig. S2C). This approach allows us to map differences onto a scaffold of known biology, such as cell type or developmental stage. Each subset is described as a “path” of serially queried markers comprising the “root node”, 0 to 2 “modifier nodes” and the “terminal node” (Fig. 2A). Key features of our analysis include (i) zero down-sampling, (ii) rigorous multiple testing correction, (iii) exclusion of small (less than 10 cells in both cohorts) subsets and (iv) exclusion of uninformative “modifier nodes”, which curated the output (Fig. 2B). Interpretation of orthologous (FlowSOM) clustering analyses (27) was hampered by (i) substantial over- or under-binning (where clusters were typically either large or very small, fig. S2D), reflecting limited control over the analytical granularity, which can often be driven by non-biologically meaningful differences in marker expression (28) and (ii) down-sampling impairing statistically robust consideration of smaller subsets. IMPACD directly addresses these limitations.
Fig. 2. IMPACD performs deep immune subset profiling by permutational analysis.
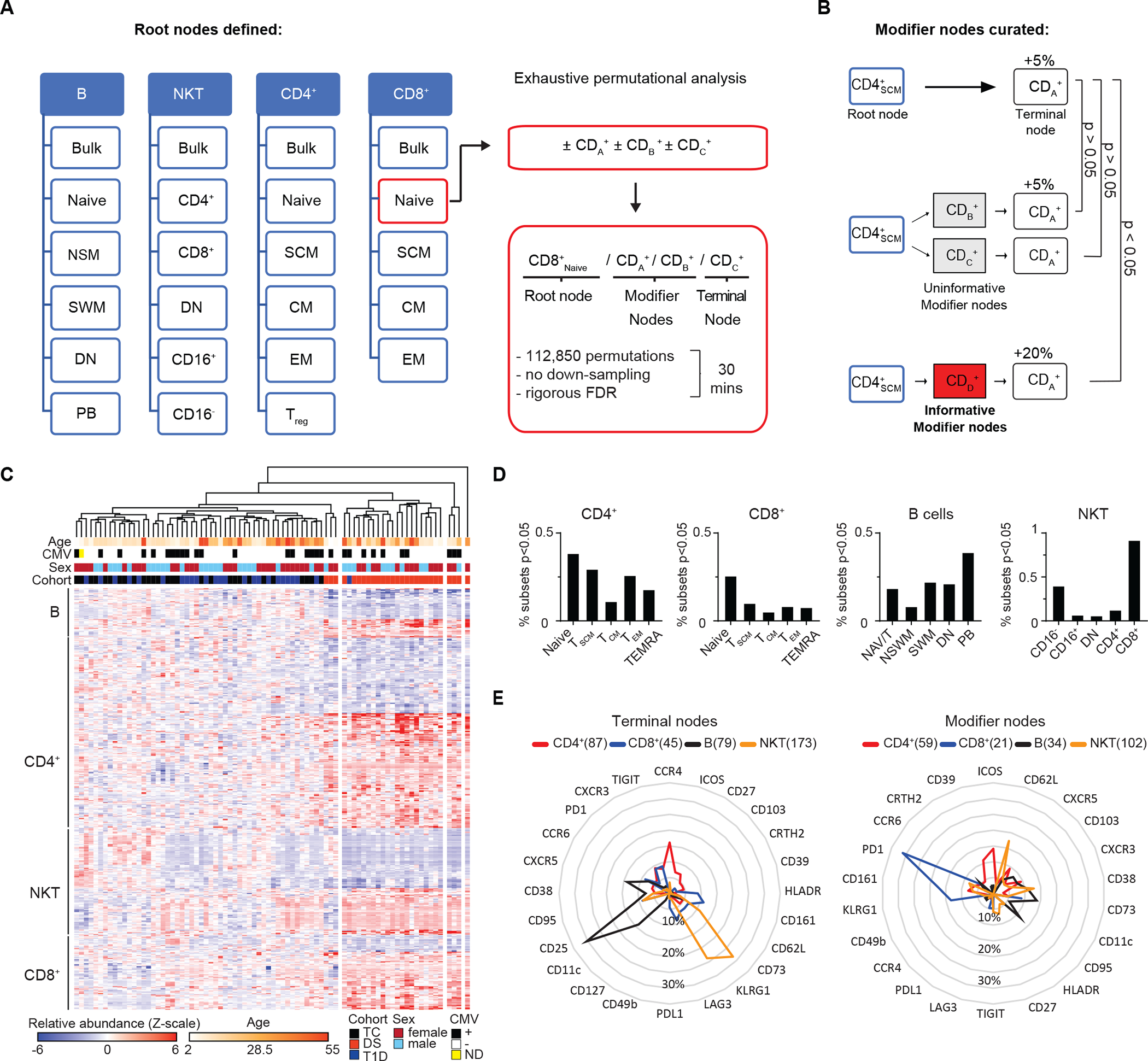
(A) IMPACD uses thresholds from digital gating to identify pre-defined subsets and perform permutational analysis using all remaining markers, illustrated here by the hypothetical markers cluster of differentiation (CD) A, B and C. Subsets are described by a path including root, modifier, and terminal nodes as shown. IMPACD analysis can be accomplished in 30 minutes. (B) An outline of modifier analysis is shown, which curates differentially represented subsets for maximal informativeness. In this example examining the hypothetical markers CDA, CDB, CDC, and CDD, expression of CDA is altered in CD4SCM cells. Many descendant subsets (CD4SCMCDB+, CD4SCMCDC+) would be expected to reflect this same difference and are curated out (gray boxes). IMPACD’s modifier analysis includes only subsets that show statistically significant (Wilcoxon ranksum), additional differences (CD4SCMCDD+, red box). (C) A heatmap shows B, T, and NKT cell subsets identified by IMPACD as differentially abundant between control (TC) and DS or T1D. Unsupervised hierarchical clustering shows distinct clustering of individuals with DS. (D) The percentage of subsets, relative to the total number of subsets in each cell type or memory subset, that were differentially abundant in DS versus TC are shown (Wilcoxon ranksum test, nominal p<0.05). (E) Analysis of subsets differentially abundant in DS versus TC are shown, comparing frequency of each modifier and terminal node in each cell type, as a proportion of all modifier or terminal nodes in that cell type.
Qualitative immune landscape remodeling is observed in PBMCs from individuals with DS.
IMPACD analysis of B, T, and NKT cells identified 651 subsets, or combinations of expressed markers, that were differentially abundant in DS compared to TC (Fig. 2C). Using these subsets readily separated DS from admixed TC and T1D participants with either principal component analysis (PCA, fig. S2E) or unsupervised hierarchical clustering (Fig. 2C). Hierarchical clustering further showed (i) differences in these subsets were broadly coordinated in each participant with DS and (ii) TC and T1D participants broadly clustered by age with three of the youngest individuals with DS clustered closer to (older) TC and T1D participants (Fig. 2C). These findings support the notion that age-related differences contribute to DS-related differences in a spectrum that overlaps with TC or T1D participants. To reveal organizing principles, we categorized these 651 subsets according to cell type and differentiation stage. We found that dysregulated subsets in DS were relatively concentrated in naïve CD4+, naïve CD8+, memory B cells, and CD8+ NKT cells (Fig. 2D). From this global viewpoint, qualitative changes tended to be concentrated in subsets that were also quantitatively altered in DS.
Next, we looked for common themes underlying these dysregulated subsets. First, we asked if similar markers were being dysregulated across different cell types by enumerating terminal nodes in each cell type. This showed that dysregulated terminal nodes exhibited highly cell-type-specific patterns with some limited overlap (Fig. 2E). Next, we assessed for commonalities between the subsets that exhibited dysregulated marker expression by enumerating modifier nodes in each cell type. We found that modifier nodes also exhibit cell-type-specific patterns with some limited overlap (Fig. 2E). The observation that each cell type exhibits dominant terminal and modifier nodes supports the notion that immune dysregulation in the context of DS engages common mechanistic programs shared between subsets of each major cell type. The observation that different cell types exhibit different dominant terminal and modifier nodes could reflect regulation by different effector mechanisms in each cell type or by common effectors modulated by cell type-specific epigenetic landscapes.
Qualitative remodeling of B and T cells isolated from individuals with DS shows autoimmunity-related features.
Dysregulated B and T cell homeostasis is a key feature of many autoimmune responses. To better understand similarities between B cell remodeling in DS and autoimmunity, we used IMPACD to build a temporal map of how B cells are dysregulated in DS at sequential maturation stages including naïve/transitional (NAV/T), non-switched memory (NSM), switched memory (SWM), double negative (DN) and plasmablasts (PB) (fig. S1B and Fig. 3A). Qualitative remodeling of the B cell compartment in DS shared many features with other autoimmune diseases, including (i) increased expression of CD11c in non-PB B cell subsets, as seen in rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and multiple sclerosis (MS) (29–31), (ii) decreased expression of CXCR5 and CD25 in non-PB B cell subsets, as in SLE (32), (iii) increased expression of programmed cell death protein 1 (PD-1) in non-PB B cell subsets, as in RA (33), (iv) increased CXCR3 and decreased CCR6 expression in SWM B cells, as in RA (34) and SLE (35), (v) increased CD11c+CXCR5− DN2 and CD11c+CXCR5− activated naïve (aNAV) B cells, as in SLE (32) and (vi) increased expression of CD95 in DN B cells, as in SLE (36) (Fig. 3A and B). In contrast, the B cell compartment of individuals with T1D showed less profound remodeling (Fig. 3A). Together, these results highlight how the B cell compartment is broadly remodeled in DS and exhibits features consistent with several autoimmune diseases and impaired activation or function.
Fig. 3. Remodeling of B and T cell compartments in individuals with DS shows autoimmunity-related features.
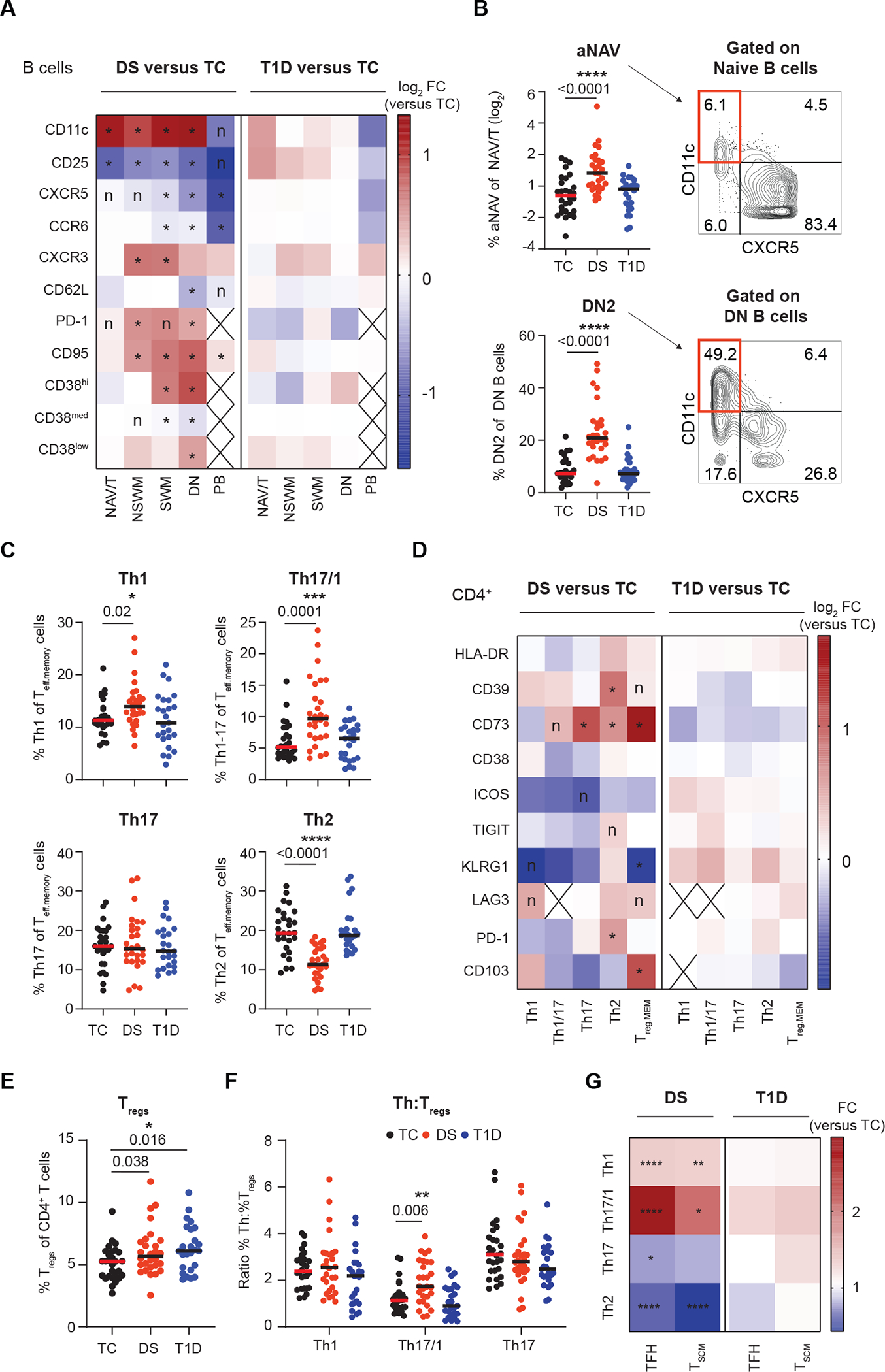
(A) IMPACD compares expression of selected markers in each B cell memory subset in samples from individuals with either DS or T1D versus controls (TC). NAV/T, naïve/transitional; NSWM, non-switched memory; SWM, switched memory; DN, double negative; PB, plasmablast. (B) Frequency of autoimmunity-related activated naïve (aNAV) and DN2 B cells (amongst naïve and DN B cells respectively) in each cohort are shown. The gating strategy is shown on the right. (C) Frequencies of Th subsets are shown among non-TFH (CXCR5−) CD45RO+ Teffs (Teff.memory) in each cohort. (D) Expression of selected markers in Th subsets is shown in either DS or T1D versus TC. Treg.mem indicates memory Tregs. (E) Frequency of Tregs (CD127lowCD25hi) amongst CD4+ T cells are shown in each cohort. (F) Ratio of Th1, Th17/1 and Th17 cell frequencies to Treg frequencies are shown for each cohort. (G) Frequency of Th subsets within total TFH (T follicular helper, CXCR5+CD45RO+ CD4+ Teff) or TSCM cells from people with DS or T1D are shown as fold change (FC) relative to TCs. (A to G) n = 28 TC, n = 28 DS, n = 25 T1D, across 5 batches. (A, D, and G) Heatmap shows the ratio of median percentage of marker-expressing cells. (A and D) Wilcoxon ranksum test with Benjamini-Hochberg correction was used; *, FDR-adjusted p<0.05; n, nominal p<0.05; cross (X), the median number of cells in that group was < 10. (B, C, E, and F) Asterisks indicate statistical significance by Kruskal-Wallis test (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). Posthoc Mann-Whitney tests were used; p-values are shown. (G) Mann-Whitney test, * p<0.05, **p<0.01,*** p<0.001, **** p<0.0001.
Similarly, we found that remodeling of T cells in DS also shared many features with other autoimmune diseases. Increased pro-inflammatory Th1, Th17 and Th17/1 subsets have been observed in MS (37), Crohn’s disease (38, 39), and psoriasis (40). Anti-inflammatory Tregs have been observed to be either increased or decreased in autoimmune diseases such as RA and SLE, suggesting complex regulation (41–43). In comparison to TC, we found increased Th1 and decreased Th2 frequency in samples from individuals with DS, based on surface expression of CXCR3, CCR6, and CCR4 in CXCR5− non-T follicular helper (TFH) cells (Fig. 3C). Although frequencies of Th17 cells were similar, the increase in Th17/1 cells, which are IFN-γ+IL-17+ cells that arise from Th17 precursors, points to quantitative and qualitative remodeling of the Th17 compartment in the context of DS (Fig. 3C) (44). These results are consistent with previous reports of pro-inflammatory Th cell bias in DS (5, 6). Next, we found that qualitative remodeling of surface markers was focused in Th2 and Th17 cells (Fig. 3D). Intracellular staining correspondingly showed disproportionately increased expression of activation-associated cytokines such as tumor necrosis factor (TNF)-α and IL-2 in IL-17+ Th17 cells (fig. S3A). Frequencies of Tregs were similarly increased in both DS and T1D (Fig. 3E). Memory Tregs were also qualitatively remodeled specifically in DS, including increased expression of CD39 and CD73, resembling changes seen in the joints of patients with RA (Fig. 3D) (45). The ratio of pro-inflammatory Th17/1 to anti-inflammatory Tregs was specifically increased in DS, highlighting Th17/1 cells as putative drivers of pathologic inflammation in DS (Fig. 3F). Finally, we examined two T cell subpopulations of particular interest in autoimmunity. TFH cells help drive the B cell response and TSCM cells can regenerate effector T cells, which may empower long-lived autoimmune responses (46, 47). TFH frequency was not altered, but the frequency of both CD4+ and CD8+ TSCM cells was increased in DS, as has been observed in RA (48) and aplastic anemia (49) (Fig. 1D and fig. S3B). Both TSCM and TFH compartments are qualitatively remodeled in DS, showing similar skewing towards Th1- and Th17-like cells and away from Th2-like cells (Fig. 3G) (50). Taken together, these results provide a detailed map of how Th cell subsets are dysregulated in a pro-inflammatory autoimmunity-relevant manner in DS.
Naïve CD4+ T cells from individuals with DS exhibit a poised state driven in part by IL-6.
IMPACD also readily highlighted immune differences in the context of DS in high resolution agnostic of previously described autoimmunity-relevant differences. Focusing on T cells, we found dysregulation at all stages of maturation, particularly in naïve CD4+ Teff and naive CD8+ T cells, again showing qualitative remodeling focused on quantitatively altered subsets (Fig. 2D and Fig. 4A). Interestingly, naïve T cell changes involved many markers associated with T cell activation, including CD62L and CD38 (downregulated with T cell maturation) as well as CXCR3, T cell immunoglobulin and ITIM domain (TIGIT), Killer cell lectin-like receptor subfamily G member 1 (KLRG1) and Human Leukocyte Antigen – DR isotype (HLA-DR) (upregulated with T cell activation) (6, 51). Some, but not all, markers were similarly dysregulated in naïve CD4+ Teff and CD8+ T cells from participants with DS as compared to TCs. Dysregulation of CD73, CD38 and Inducible Co-Stimulator (ICOS) was specific to naïve CD4+ Teff cells, upregulation of KLRG1 and HLA-DR was specific to naïve CD8+ T cells, and decreased CD62L and increased CXCR3, CD39 and TIGIT was common to both naïve CD4+ Teff and CD8+ T cells from individuals with DS (Fig. 4A). Changes in NKT cells broadly resembled the corresponding CD4+ or CD8+ T cell subset (fig. S3C). Remodeling in individuals with T1D was less profound (Fig. 4A). These findings suggest that naïve CD4+ and CD8+ T cells in individuals with DS exist in a hyperactivated state, poised for activation. This is further supported by our CyTOF findings of increased expression of intracellular activation-related markers including Ki-67, IL-2, and TNF-α in naïve CD4+ Teff and CD8+ T cells from individuals with DS, as well as previous studies showing enhanced effector differentiation potential of and cytokine expression by CXCR3+ naïve cells (fig. S3D) (6, 51, 52).
Fig. 4. Remodeling of naïve T cells in individuals with DS suggests a state of poised activation driven in part by IL-6.
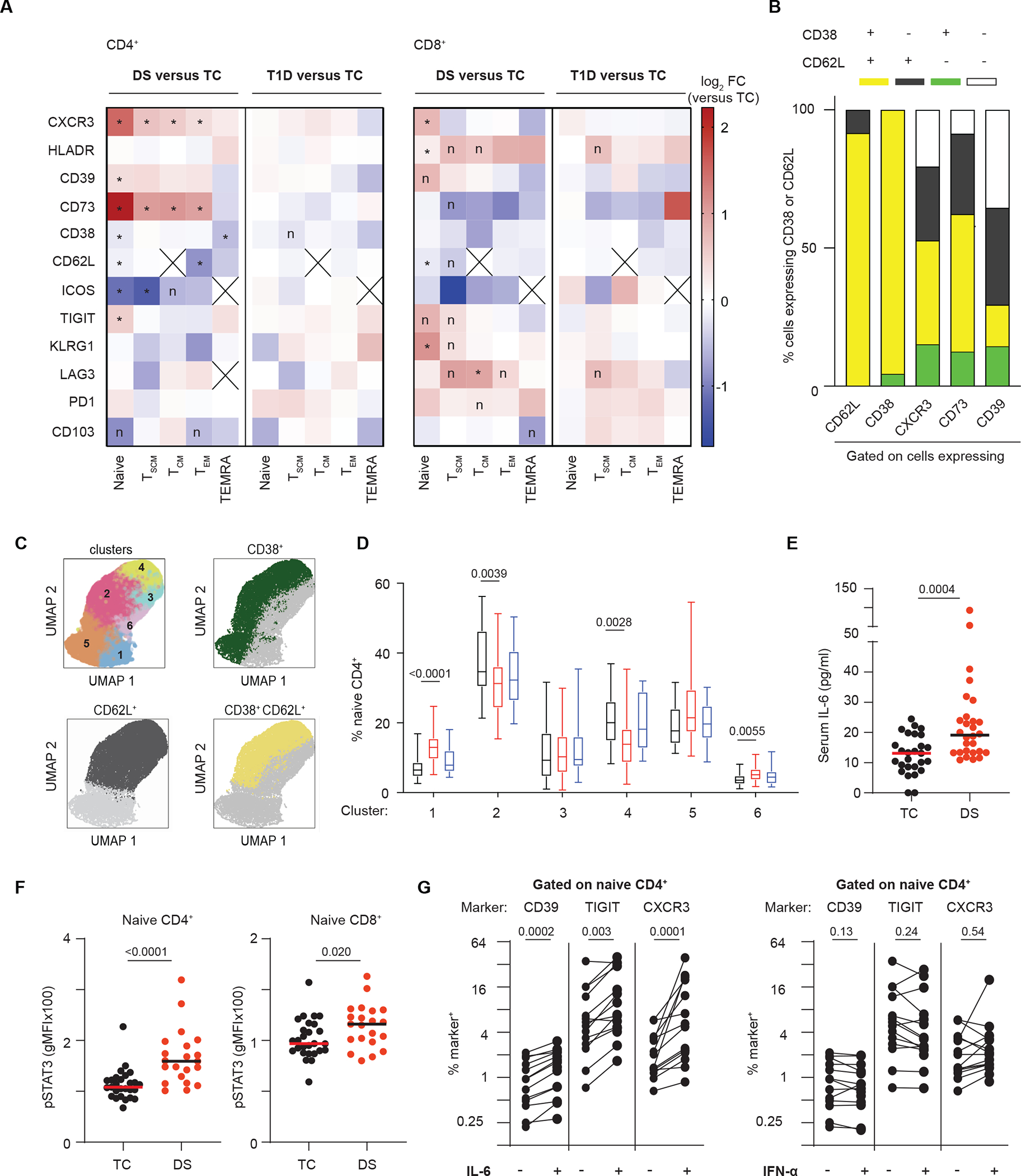
(A) Expression of selected markers in T cell memory subsets are shown in either DS or T1D versus TC. Heatmap shows the ratio of median percentage of marker-expressing cells. (B) Polyexpression analysis of selected markers dysregulated in naïve CD4+ T cells from people with DS shows CD38 and CD62L co-expression (yellow). (C) FlowSOM analysis of naïve CD4+ T cells isolated from TCs or individuals with DS or T1D identifies 6 clusters (upper left); overlays show CD38 and CD62L expression (manual gating thresholds). (D) Percent naïve CD4+ T cells in each FlowSOM cluster were quantified. (E) Serum IL-6 is increased in individuals with DS relative to TCs. (F) Baseline pSTAT3 is increased in naïve CD4+ and CD8+ T cells from individuals with DS. gMFI, geometric mean fluorescence intensity. (G) Expression of CD39, TIGIT, and CXCR3 in naïve CD4+ T cells from controls is increased by IL-6 but not IFN-α stimulation. Lines connect samples from the same donor. (A and B) n = 28 TC, n = 28 DS, n = 25 T1D, across 5 batches. (C and D) n = 22 TC, n = 22 DS, n = 19 T1D, across 4 batches. (E) n = 27 TC, n = 28 DS across one batch. (F) n = 27 TC, n = 21 DS, across 5 batches. (A) Wilcoxon ranksum test was used with Benjamini-Hochberg correction; *, FDR-adjusted p<0.05; n, nominal p<0.05; cross (X), either median number of cells in that group was < 10 or part of subset definition (TCM=CD62L+). (D) Quasibinomial logistic models with age as a covariate and Holm correction was used with p-values shown. See data file S1. (E and F) Mann-Whitney test was used; p-values are shown. (G) n = 13 to 14 in 3 independent experiments analyzed by Wilcoxon matched-pairs signed rank test; p-values are shown. See data file S1.
To better understand whether any of these markers were coordinately dysregulated, we performed a polyexpression (analogous to polyfunctionality) analysis of these markers in naïve CD4+ T cells. We found a striking decrease in CD62L+CD38+ double-positive cells in samples from individuals with DS, highlighting that CD62L+ and CD38+ are coordinately decreased in naïve CD4+ T cells (Fig. 4B). This was supported by independent FlowSOM analysis, which grouped naïve CD4+ T cells into 6 major clusters with CD62L+CD38+ cells predominating in clusters 2 and 4 (Fig. 4C). Both of these clusters were decreased in samples from individuals with DS (Fig. 4D). FlowSOM analysis also showed some increase in a CD62L+ and a CD38−CD62L− cluster, although no other definitive markers were identified (Fig. 4C and D). Demonstrating the power of IMPACD’s down-sampling-free and controlled-granularity approach, polyexpression analysis of CXCR3+, CD39+, and CD73+ cells revealed that the increased representation of these cells was associated with CD62L and CD38 co-expression (Fig. 4B). These results point to extensive remodeling of a CD62L+CD38+ compartment in individuals with DS, with a dramatic quantitative decrease accompanied by qualitative remodeling exemplified by upregulation of CXCR3, CD39, or CD73 expression.
To explore the role of cytokine signaling on the naïve T cell dysregulation in DS, we examined the response of TC T cells to IFN-α and IL-6 in the absence of TCR stimulation. We focused on these cytokines for two reasons. First, IFN signaling is enhanced in DS due to overexpression of chromosome 21-encoded IFN receptor subunits and supported by our findings of elevated phospho (p) Signal transducer and activator of transcription 1 (STAT1) in DS T cells (fig. S3E). Second, IL-6 is associated with autoimmunity, increased in serum from participants with DS, and its functional relevance is supported by increased pSTAT3 in CD4+ T cells from individuals with DS (Fig. 4E and F) (7, 8, 53, 54). Cytokine treatment did not deplete naïve T cells, supporting feasibility of our approach (fig. S3F). Unexpectedly, we found that treating TC T cells with IL-6, but not IFN-α, recapitulated many differences found in naïve CD4+ T cells from people with DS, including increased expression of CD39, TIGIT and CXCR3 (Fig. 4G). We additionally performed whole blood RNAseq to identify genes differentially expressed in DS; gene set enrichment (GSEA) analysis against 50 hallmark pathways revealed enrichment of IL-6–JAK–STAT3 signaling, further supporting a mechanistic role of IL-6 in DS-associated immunodysregulation (fig. S3G) (55, 56).
Defining a CD4-NKT module that is differentially coregulated in individuals with DS.
To illuminate organizing principles underlying immune dysregulation in DS, we analyzed the 651 subsets differentially abundant in DS for modules that were coordinately regulated in DS differently from TC. We identified 145 pairs that were co-regulated in DS and showed either non-significant (P>0.05) coregulation or opposite correlation in TC. We categorized pairs according to the cell types involved and found overrepresentation in the CD4+-NKT group (Fig. 5A). To validate these findings, we performed a simulation experiment where we randomly re-assorted individuals to DS or TC cohorts (10,000 iterations) before assessing differentially correlated subsets, generating a false discovery rate (FDR)-adjusted p-value that was statistically significant in 7 of 10 groups of interactions (P<0.05, Fig. 5A).
Fig. 5. Identifying differentially co-regulated subsets in DS.
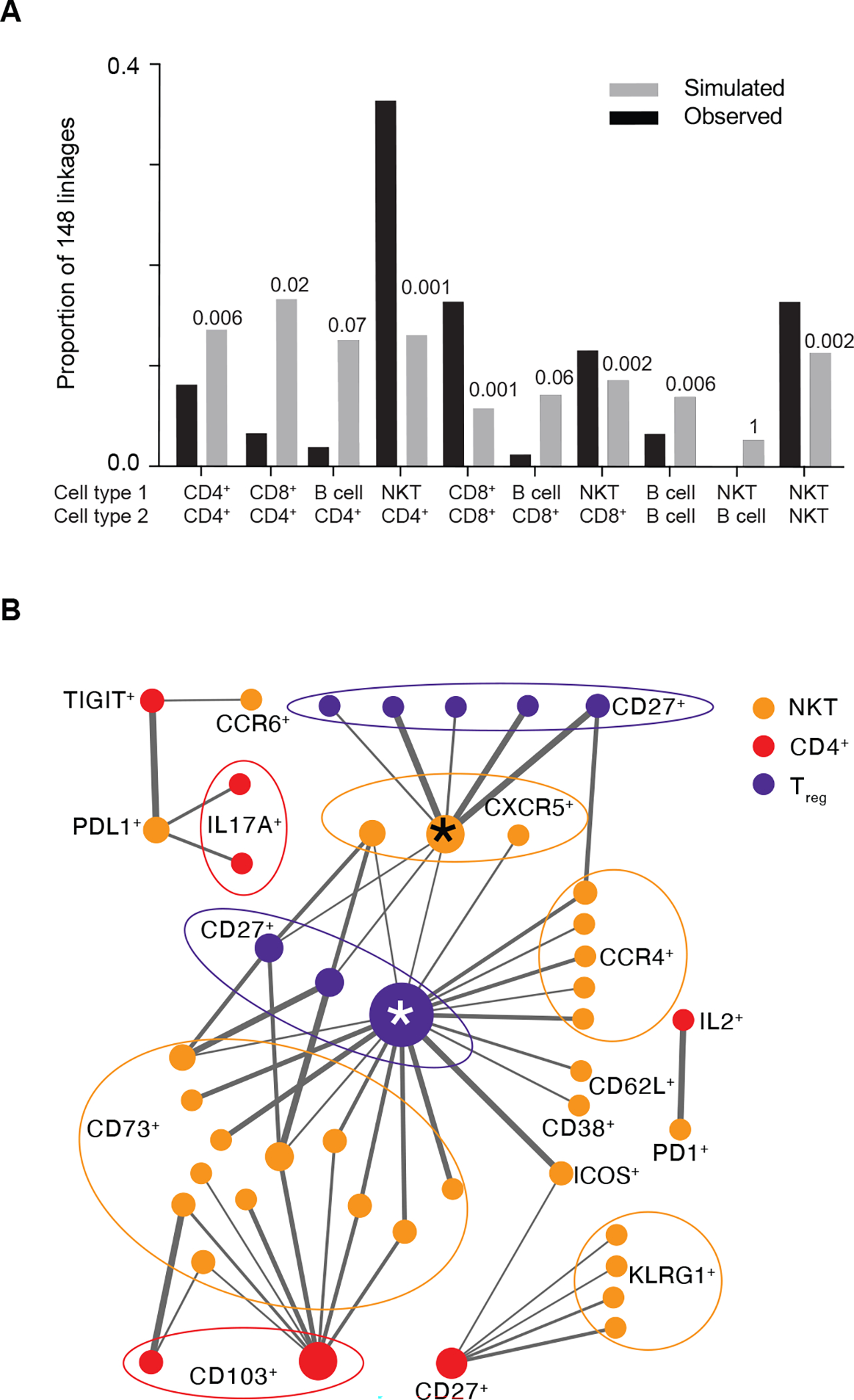
(A) Diffcoex analysis of IMPACD data identifies subsets differentially co-regulated in DS versus TC, here organized by interacting cell types. For comparison, results from random choice simulations (x10,000) are shown in gray. Numbers above each column show the fraction of simulations where the number of differential correlations in each group reached or exceeded the same number as were actually observed. (B) Network map of CD4+ T cell-NKT cell interactions in DS reveals distinct organizational logic, including hubs. Two major inter-connected hubs are highlighted (*). Nodes are colored according to root node; size is proportional to number of connections. Terminal nodes are indicated, using boundary ellipses as appropriate. Edge thickness reflects significance of differential correlation in DS versus TC. (A and B) n = 28 TC, n = 28 DS, n = 25 T1D, across 5 batches.
We focused on the CD4+-NKT group, which had the lowest p-value and the largest number of co-regulated subsets (P=0.001, Fig. 5A and data file S2). We generated a network graph of the CD4+-NKT linkages and found distinct organizational logic. We found that TIGIT+ naïve CD4+ Teff cells were co-regulated with two NKT cell subsets specifically in the context of DS (Fig. 5B and data file S2). We also uncovered larger patterns including two inter-connected hubs, CD45RO+CD103+HLADR−CD27+ Treg cells and CD95+CCR6−CXCR5+CD16− NKTs (marked *), that were coordinately regulated with several (CD8+CD73+ and CD16−CCR4+) NKT and (CD27+) Treg cell subsets (Fig. 5B). This suggests that related mechanisms regulate these subsets in DS and highlights another way that IMPACD’s high-dimensional output can identify organizational principles.
Advanced immune aging is associated with DS and T1D.
Inflammaging describes how immune system remodeling during aging exhibits pro-inflammatory features (10, 11). Although people with DS show clinical features of inflammaging, a quantitative understanding of inflammaging in DS remains lacking but is critical to inform mechanistic and translational studies. Our results identified qualitative differences in people with DS suggestive of inflammaging, including decreased naïve T cells, increased CD11c+ B cells and increased IL-6 (Fig. 1D to E, Fig. 3A and Fig. 4E). We sought to use the wide age range of our cohort to investigate how these features change with age in people with and without DS. First, we found that naïve T cells (particularly CD8+) decreased faster with age in DS versus TC, whereas memory CD4+ and CD8+ T cells increased with age indistinguishably in both groups (fig. S4A). Second, CD11c+ B cells, which are increased in murine aging and in people with rheumatoid arthritis, were present at higher abundance in individuals with DS than TCs, but did not increase with age in either population (fig. S4B) (29, 57, 58). Third, pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-22 were elevated in serum from people with DS versus TCs as previously reported, but did not increase linearly with age in either cohort (Fig. 4E and fig. S4C and D) (7). These results highlight how individual features of inflammaging can change with age at different rates, which critically informs modeling efforts.
Based on these findings, we built three linear models of inflammaging, which we term immune clocks, all trained using only CyTOF data from TC. In our first “Unfiltered” model, we leveraged IMPACD’s high-dimensionality output to evaluate each of the 294,061 subsets identified in TC and found 61 informationally non-redundant immune subsets that correlated most linearly with age in TC (p<0.001). We used similarity clustering (cutoff p<0.75) to identify 19 representative subsets to prevent overfitting and principal component analysis (PCA) to generate a linear model using these 19 subsets. This “Unfiltered” model showed excellent correlation with age (r2=0.92), demonstrating the utility of our approach (Fig. 6A). We validated this approach in two ways. First, we generated training and validation datasets from our TC data by withholding data from 5 randomly selected individuals in a validation dataset. We used an identical approach to build a linear model of age with the remaining 23 individuals and interrogated the quality of age prediction in the validation dataset. Over 5 iterations, using different individuals in each validation dataset, we found reproducibly consistent predictions, supporting our approach (fig. S4E). Second, we used PCA to build a model based on the 27 subsets in our study that were also found to change with age in a previous publication by Alpert et al. (59). This “Alpert-filtered” model, generated using an independent set of markers not enriched for linear correlation with age, predicted age with reasonable if lower accuracy (r2 = 0.46, Fig. 6A). Lastly, we focused on the 651 subsets identified by IMPACD as differently abundant in DS versus TC. From these, we identified 41 subsets that correlated linearly with age in TC (Fig. 6A). These included subsets of all major cell types with a slight over-representation of CD4+ subsets (fig. S4F). To prevent overfitting, we used similarity clustering (cutoff p<0.85) to extract a representative set of 24 subsets (fig. S4F and G). We used PCA to generate a linear model with these 24 subsets that correlated with age extremely well (“DS-filtered” model, r2 = 0.77, Fig. 6A). Therefore, we built three immune clocks that accurately describe how the immune landscape of TC changes with age.
Fig. 6. Advanced immune aging is observed in peripheral blood from individuals with DS.
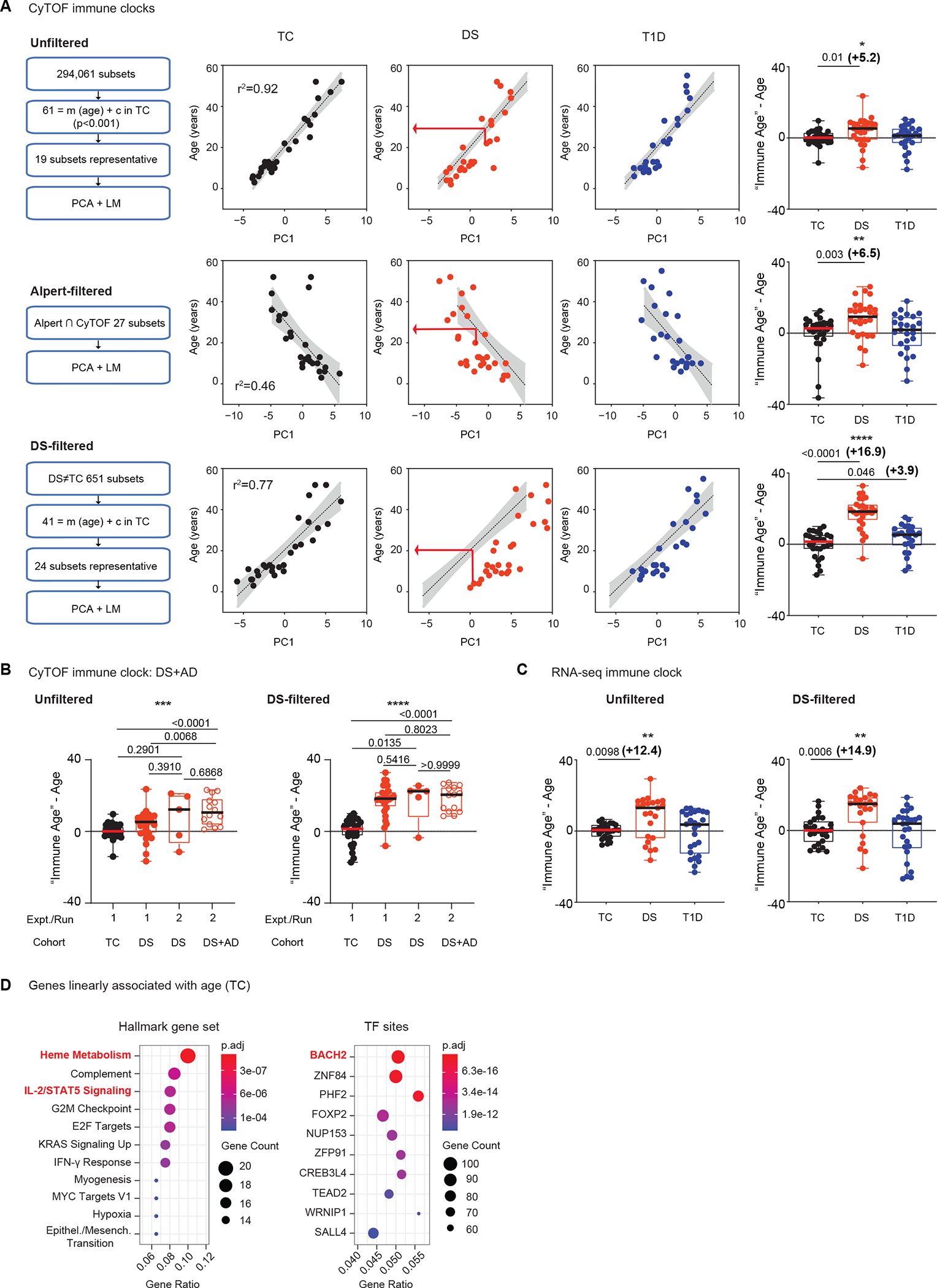
(A) The approach to build (CyTOF-based) control- or TC-trained linear models of age is shown on the left, including identifying subsets that vary linearly with age (= m(age) + c), identifying representative subsets (similarity clustering) to prevent overfitting, principal component analysis (PCA) and linear modeling (LM). The TC-derived algorithm was used to calculate immune age in DS and T1D cohorts (middle, red arrow). The difference (“immune age” – age) is summarized per cohort (right). (B) An identical approach was used in a separate experiment (run 2) to assess immune age of individuals with DS and autoimmunity (DS+AD) and five additional autoimmunity-free individuals with DS. Results from run 1 TC and DS cohorts (also in Fig. 6A) are included for reference. (C) Whole blood RNA-seq-based TC-trained linear models of age were used to calculate immune age of DS and T1D individuals. The difference (“immune age”– age) is summarized per cohort. (D) Gene set enrichment analyses of the 747 genes that vary linearly with age in TC are presented, showing enriched hallmark gene sets (FDR p<5×10−4) and transcription factor (TF) binding sites (FDR p<5×10−11). (A ) n = 28 TC, n = 28 DS, n = 25 T1D, across 5 batches. (B) n = 5 DS, n = 14 DS+AD, across 2 batches. (C and D) n = 26 TC, n = 23 DS, n = 26 T1D, across 1 batch. (A to C) Asterisks indicate statistical significance by Kruskal-Wallis test (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). Posthoc Mann-Whitney tests were used; p-values are shown.
We next used these immune clocks to compare the “immune age” of participants with DS, calculated using their individual immune subset values (red arrow, Fig. 6A), to their actual age. All three immune clocks showed advanced immune aging in DS (Fig. 6A). The “Unfiltered” and “Alpert-filtered” models predicted comparable magnitude of advanced immune aging beginning in childhood (+5.2 and +6.5 years, respectively). This suggests that these models quantitate similar aspects of the immune landscape. Notably, the “DS-filtered” model predicted that the immune system of a person with DS resembles that of a person 16.9 years older on average (Fig. 6A, right). This difference was observed beginning at the earliest age of our cohort, suggesting again that advanced immune aging in DS begins in childhood (Fig. 6A). This model also predicted linear immune system change with age in DS (r2 = 0.88, Fig. 6A). Decreasing difference between DS and TC at older ages may be a result of the upper age limit of our training cohort (55 years old, Fig. 1B) constraining linear range of our models. Samples taken from a subset of individuals with DS at separate time points showed comparable advanced immune aging, further supporting the robustness of this finding (fig. S4H). Models conversely first trained on participants with DS also showed advanced immune aging in DS (versus TC), demonstrating that this observation is not due to unique features of immune aging in TC or DS. (fig. S4I). Taken together, these findings exemplify our ability to generate quantitative linear models (immune clocks) that unanimously point to inflammaging (advanced immune aging) in people with DS. Importantly, even though all immune clocks were built on data from the same TCs, comparison of predictions from all three models suggests that the “DS-filtered” model can provide unique DS-relevant biological insight.
To further compare the “Unfiltered” and “DS-filtered” models, we assessed PC1 loading coefficients in both models and found that all components contributed equitably, arguing against the dominant effect of few components (fig. S4J). We next compared features of the subsets that correlated linearly with age in TCs in both models, prior to similarity clustering to ensure an accurate and comprehensive view. We found that root nodes were utilized at similar frequencies in both models (fig. S4K). Differences were observed in the most used modifier nodes, which were CD161+ and Helios+ in the “Unfiltered” model and PD-1+ and CD73+ in the “DS-filtered” model. Terminal nodes used in both models showed some similarities, such as high representation of CD38+, CD95+ and naïve+; the “DS-filtered” model showed relative over-representation of naïve+, Helios+ and CD25+ terminal nodes (fig. S4K). These findings show how selection of different subsets, even within the same major cell types, that vary linearly with age in TC can reveal distinct features of aging-related biology.
To better understand how co-existing autoimmunity impacts DS-immune aging, we interrogated samples from 14 individuals with DS and autoimmunity (DS+AD) using an identical CyTOF approach. Autoimmune diseases in this cohort included Hashimoto’s thyroiditis, Crohn’s disease, psoriasis, rheumatoid arthritis, and T1D (fig. S5A). We calculated the immune age of these individuals using the same “Unfiltered” and “DS-filtered” models from our first cohort. Compared to TC, individuals with DS+AD showed significantly advanced immune age in both models, further supporting our observation of advanced immune age in DS (P<0.0001, Fig. 6B). Immune age of individuals with DS+AD was more advanced than individuals with DS (from the first cohort) specifically in the “Unfiltered” model, raising the possibility that co-existing autoimmunity may further alter immune architectural differences associated with DS (Fig. 6B). As a control, we simultaneously assessed a separate cohort of 5 people with DS (DS run 2). Compared to these 5 individuals, participants with DS+AD did not exhibit advanced immune age in either model (Fig. 6B); whether this is due to small cohort size remains to be clarified. As another control, we ran aliquots from 4 previously assessed controls spanning a wide range of ages in each batch. Immune age of each control, in both “Unfiltered” and “DS-filtered” models, was comparable in all batches, supporting reasonable inter-batch variation (fig. S5B). Taken together, these findings enhance our understanding of advanced immune aging in people with DS and autoimmunity.
To further support our findings, we applied an analogous analytic approach to whole blood RNAseq data from the same participants in our first cohort. Briefly, we identified genes whose expression varied linearly with age in TCs, used similarity clustering to identify <28 representative genes (to prevent overfitting) and performed PCA and linear modeling. This allowed us to build RNAseq-based “Unfiltered” and “DS-filtered” immune clocks that showed advanced immune aging in individuals with DS of +12.4 and +14.9 years respectively, beginning in childhood (Fig. 6C and fig. S5C and D). To highlight organizing principles, we performed GSEA analysis of all 747 genes that change linearly with age in TC (nominal p<0.05, data file S3) (55, 56). Of 50 hallmark pathways, the most enriched was heme metabolism, which recent GWAS/eQTL meta-analyses found to be the pathway most strongly associated with aging (Fig. 6D) (60). Heme metabolism was also the most enriched pathway when considering the subset of 99 genes that were additionally differentially expressed in samples from individuals with DS (fig. S5E). Data from TrisomExplorer (8) show increased expression of the gene encoding 5’-Aminolevulinate Synthase 1 (ALAS1), the rate-limiting enzyme in heme biosynthesis, in monocytes and bulk T cells from individuals with DS but not in whole blood, suggesting cell-specific links between heme metabolism and immune aging in DS (fig. S5F). Similar to the GWAS/eQTL meta-analyses (60), we also found the IL-2–STAT5 signaling pathway to be enriched in the 747 TC-age-associated genes, suggesting broader links with heme metabolism and immune aging (Fig. 6D). Using the chromatin immunoprecipitation (ChIP)-seq-based Gene Transcription Regulation Database (GTRD), we found the 747 TC-age-associated genes to be most enriched for binding sites to BTB Domain And CNC Homolog 2 (BACH2), a transcriptional repressor strongly linked to inflammation and aging (Fig. 6D) (61). Data from TrisomExplorer (8) and our RNAseq results show decreased BACH2 expression in T cells and whole blood from individuals with DS, consistent with a pro-inflammatory phenotype (fig. S5F) (61). GSEA analysis of the 747 TC-age-associated genes did not find enrichment of genes on chromosome 21, supporting that our RNAseq-immune clock findings are not a simple consequence of trisomy-driven overexpression of component genes. However, 3 of the 7 age-associated genes on chromosome 21 (ZBTB21, HMGN1 and SETD4) are transcription factors or epigenetic remodelers, overexpressed in various immune cell types in people with DS, that could broadly impact the expression of many genes (fig. S5F) (62). Together, these analyses provide independent validation of advanced immune age in DS, highlight candidate key pathways and transcription factors, and point to putative driver genes on chromosome 21.
Finally, we compared the CyTOF-based “immune age” of participants with T1D to their actual age. The “DS-filtered” model showed that the immune systems of participants with T1D resemble those of TC individuals who are 3.9 years older on average (Fig. 6A). This difference is evident even in the youngest participants (Fig. 6A). Neither the “Unfiltered” nor the “Alpert-filtered” model showed evidence of advanced immune aging in participants with T1D, although the relative differences in predicted immune age correlated well with the “DS-filtered” model (Fig. 6A and fig. S5G). Together, these findings point to an association between advanced immune age and T1D and highlight how studying immune dysregulation in DS can highlight specific aspects of biology in other autoimmune diseases.
DISCUSSION
Aging is associated with increased autoimmunity, increased immune exhaustion, loss of protective immunity and increased pro-inflammatory cytokines, especially IL-6 (13). These features are comprised by the term inflammaging (11). Associated changes in epigenetics, signaling and immune subsets may contribute to autoimmunity risk, although mechanistic details remain incompletely elucidated (59, 63). Drawing causal links to autoimmunity in the general population is further challenged by diverse pathogenic subtypes (64). In this context, DS represents a unique need and opportunity. Individuals with DS exhibit clinical, cellular, and molecular features of inflammaging, including increased autoimmunity, early-onset Alzheimer’s disease, decreased naïve T cells, altered epigenetics or glycomics, and increased serum IL-6 (2, 5–7, 63, 65).Thus, trisomy 21 is a genetic driver of inflammaging in DS. Specific therapeutic strategies may best mitigate inflammaging-associated diseases in individuals with DS and in pathophysiologically-related subsets of the general population.
The high-granularity output of our software, IMPACD, enabled us to build three linear models of immune age (immune clocks) using PBMC-CyTOF data from only TCs. All three immune clocks quantitatively demonstrate advanced immune aging in people with DS beginning from childhood. Independent RNAseq-based models showed similar results. These findings exemplify how quantitative and integrative analysis of immune age might inform mechanistic investigations of healthy aging and translational efforts to identify specific pathways of dysregulated aging, as we observe in DS and other groups. Our cohort clarifies that DS-associated inflammaging is independent of autoimmunity and may help explain why autoimmunity develops more frequently and at younger ages in people with DS (3). This may also help explain other clinical differences in DS, including decreased threshold age for poor outcome from SARS-CoV-2 infection (40 in DS versus 60 in the typical population) (66). These findings provide a framework to mechanistically investigate how DS-associated inflammaging impacts other aspects of health in DS. Responses to vaccination and infection are of particular interest, given that inflammaging was observed even in our youngest participants with DS, previous studies suggest impaired vaccine response in DS, and pulmonary infections represent the leading cause of death in individuals with DS (67, 68). Mechanistic studies are needed to better understand the key forces driving DS-associated inflammaging, which may in turn guide DS-specific therapeutic selection and development. Longitudinal studies in larger cohorts will help establish the predictive value of immune age. Further studies are needed to understand how DS-specific immune features that do not vary linearly with age contribute to DS-associated conditions. Observations that autoimmunity shares features of immune dysregulation with DS and may exacerbate immune age advancement in DS highlight the need to consider immune comorbidities in future studies of DS.
Independent and orthologous whole blood RNAseq analyses similarly show advanced aging in DS, further supporting our CyTOF findings and enabling integrative and mechanistic analyses. Deeper interrogation of the genes that comprise the RNAseq-aging models, particularly in individual cell types, may help define precise hypotheses. Our findings share striking concordance with recent GWAS meta-analyses, supporting the importance of heme metabolism and IL-2–STAT5 signaling in immune aging (60). Our data suggesting a role for BACH2, which is known to regulate peripheral tolerance (61), in regulating immune aging cannot exclude a possible role for BACH1 (not assessed in GTRD), whose DNA-binding bZIP domain is homologous to BACH2’s (69). BACH1 activity is regulated by heme, and BACH1 deficiency is protective in several models of inflammation. Further, BACH1 expression is increased in T cells and whole blood from individuals with DS (61). The absence of enrichment of chromosome 21-encoded genes suggests that advanced immune aging in DS is driven by trisomy of specific genetic elements (such as transcription factors) on chromosome 21; dysregulation of these elements may also impact immune aging in people without DS.
Our “DS-filtered” CyTOF-model demonstrates that T1D is associated with inflammaging and highlights how studying immune dysregulation in DS can advance our understanding of other immune-mediated diseases. Dysregulation of B, CD4+ T, CD8+ T, innate immune, or islet b cells can drive type 1 diabetes risk and progression (64). Our findings highlight an immunologic commonality between T1D and DS, providing a basis for mechanistic and translational inquiry. Qualitative concordance between all 3 models supports that studying immune aging in T1D is likely to be broadly important; how this might stratify prediabetic individuals for risk of progression, prioritize individuals for specific disease-delaying therapy, or predict response to different therapies is of interest. The similar results of the “Unfiltered” and the “Alpert-filtered” models suggests that they reflect similar aspects of aging-related biology. The “DS-filtered” model’s quantitatively distinct results exemplify how DS-immunodysregulation can inform our understanding of other immune diseases. This use of a signature defined in one context to highlight overlapping biology in a second context is analogous to GSEA analysis of RNAseq data and highlights how IMPACD allows application of this concept to flow cytometry data. Our findings demonstrate how additional reference cytometric signatures, generated by future experiments, could more broadly highlight shared immune features of different diseases, while incorporating clinical metadata like age.
We found additional interesting features of DS-immunodysregulation. First, our findings suggest that naïve CD4+ T cells in DS are hyperactivated, due in part to elevated IL-6 (7). Our findings extend the consideration of pathogenic cytokines in DS beyond type 1 IFN, the best-known drivers of DS-immunopathology (8). Future work will evaluate how these cytokines coordinately impact naïve CD4+ T cell response to activation and Th cell differentiation. Although the cellular source of increased IL-6 in the context of DS remains undetermined, IL-6 is linked to autoimmunity in the general population and possibly to early-onset Alzheimer’s disease in DS, suggesting an immune contribution to DS-Alzheimer’s (70, 71). Second, DS-immunodysregulation centrally impacts NKT cells, including quantitative changes, qualitative remodeling, and differential co-regulation with CD4+ T cells, suggest overlapping DS-specific mechanistic pathways. NKT cells are autoimmunity-relevant; further study of NKT dysregulation in DS may inform mechanistic studies in the typical population and clarify their role (72). Third, B cell compartment remodeling in DS recapitulates several features of other autoimmune diseases, including increased CD11c+ aNAV cells, a major source of antibody secreting cells and serum autoantibodies in SLE (30), increased CXCR3 and decreased CCR6 in SWM B cells, suggestive of altered migratory potential to inflammation sites in RA (34) and SLE (35) and increased PD-1 expression, reminiscent of hyperactive B cells in the joints of patients with RA (33). These may point to how B cell dysregulation promotes autoimmunity in DS; causal links to quantitative and qualitative defects in T cell help or B cell-intrinsic dysregulation remain to be clarified. Prior studies show that immune traits can be associated with specific genetic loci, including autoimmunity-associated SNPs. These traits include immune subsets dysregulated in DS (such as decreased CXCR5+ B cells with Sjögren’s syndrome-associated ds4938573 and increased Th1 cells with PTPN22 C1858T) (73–75) and immune features associated with increased risk of autoimmunity in TCs (76, 77). These findings support the notion that trisomy 21 alters immune architecture in ways that are associated with, and might drive, increased risk of autoimmunity. Taken together, our findings expand our understanding of how immune architecture is altered by trisomy 21 and enhance our framework to define different contributing mechanisms. Future studies to clarify specific causal immune features will drive therapeutic efforts.
Our analysis was facilitated by IMPACD, which advances analysis of high-dimensional cytometry data by using digital (manual or automated) gating strategies, improving the rigor of manual gating analyses, and performing exhaustive permutational analysis with zero down-sampling robust multiple testing correction. Continued interest in innovating digital and manual gating methods is exemplified by approaches including SYLARAS and FAUST (28, 78). The advantages of digital gating methods include longstanding understanding of many markers, ability to adjust for batch effects, and direct translation of results into sorting or analytic strategies for mechanistic studies. IMPACD may be synergistically used with clustering-based algorithms, particularly when digital gating thresholds are less self-evident. Key distinguishing features between IMPACD and clustering approaches include control over granularity of the analysis by sequentially evaluating individual markers, unambiguous definition of cellular subsets for future studies and zero down-sampling allows robust interrogation of smaller subsets. Here, we showed how IMPACD can direct analysis in the context of known biologic frameworks and generate high-density data that can be mined using omics-related methods to highlight distinct mechanistic hypotheses. We envision that broader usage will generate more context- and perturbation-specific cellular signatures to enhance functionality and that integration with other data modalities will advance hypothesis generation efforts.
Our study has several limitations. Our sample size limits our ability to capture inter-individual heterogeneity in immune age within each cohort, and smaller differences between cohorts. Larger studies will help identify which subsets correlate most robustly with immune age. Our CyTOF models are defined by our choice of markers, and we expect to reveal additional biology by using expanded sets of markers informed by these and other studies.
In conclusion, we have broadly defined immune alterations in individuals with DS, using a custom-built analytic pipeline (IMPACD). These studies quantitatively demonstrate advanced immune aging and autoimmunity-relevant changes in individuals with DS, which in turn informs our understanding of immune aging in individuals with T1D.
MATERIALS AND METHODS
Study design
The aim of this study was to identify how immune alterations in individuals with DS recapitulate those associated with either autoimmunity or aging. We used CyTOF to interrogate PBMCs from individuals with DS across a wide age range (ages 2–55), and age- and sex-matched TCs and individuals with T1D. We used cytokine stimulation of PBMCs and whole blood RNAseq to validate key findings. Research protocols were approved by the Benaroya Research Institute Institutional Review Board. All participants with DS provided assent. Participants or their parents provided written informed consent before participation in the study. The initial CyTOF study comprised n=28 individuals with DS with no history of autoimmune disease (except 2 individuals with Hashimoto’s disease), n=28 TCs with no history of autoimmune disease and n=25 individuals with T1D. Sex and age-matched samples from each cohort (TC, DS, T1D) were randomly assigned to one of five batches for CyTOF and phospho-flow (n= 27 TC, n=21 DS) experiments to control for batch effects. The follow-up CyTOF study included n=14 individuals with DS and autoimmunity, n=5 additional individuals with DS (collected at 2 time points) and n=4 TCs from the first CyTOF study. Sex and age-matched samples from each cohort were randomly assigned to one of two batches. For time-point comparisons all samples from an individual were analyzed in the same batch. Cytokine stimulation experiments included n=14 distinct TCs. Whole blood RNAseq studies included n=23 individuals with DS, n=26 TCs and n=26 individuals with T1D. No power calculations were performed. All experiments and data analysis were performed by investigators blinded to sample demographics. Sampling and experimental replicates are specified in the figure legends.
Statistical analysis
Unless otherwise described, statistical testing (Mann-Whitney, Wilcoxon matched-pairs signed rank test, Kruskal-Wallis, Wilcoxon ranksum testing, linear regression) was performed using Prism (GraphPad). Statistical details and significance levels are specified in the figure legends. Central horizontal lines in column dot plots display median values. Boxplots display the median with a central line, the 25th and 75th percentiles with the bottom and top edges of the boxes respectively, and the minimum and maximum values with the whiskers.
Supplementary Material
Materials and Methods
Fig. S1. Characterization of the cohorts.
Fig. S2. IMPACD enables analysis of manually gated cytometry data.
Fig S3. Dysregulation of cellular subsets and signaling are observed in samples from individuals with T1D and DS.
Fig. S4. Interrogating immune aging in samples from individuals with DS using CyTOF.
Fig. S5. Interrogating immune aging in samples from individuals with DS using RNAseq.
Table S1. Source of Reagents.
Data file S2. CD4-NKT subsets differentially co-regulated in DS.
Date file S3. Genes that vary linearly with age in TC.
Data file S1.
Acknowledgments:
We gratefully acknowledge members of the BRI Translational and Diabetes Clinical Research Programs, including Gina Marchesini, Kassidy Benoscek, Marli McCulloch-Olson, Clementine Chalal, Kaytlyn Ly, Laynee Laube, Rebecca Rawlings, Claire Mangan, Catie Wandell, Jenna Snavely, Ezra Graziano, Lisa Miller, McKenzie Lettau, and Jani Klein for participant recruitment and the BRI Clinical core, including Thien-Son Nguyen and Nicole Gilbert for assistance with sample processing and handling. We thank Hamid Bolouri, Kirsten Diggins and Tee Bahnson for helpful discussion. We thank Anne Hocking for editing and proofing the manuscript.
Funding:
This work was supported by National Institutes of Health grants R01AI132774 (to J.H.B.) and UL1TR002319 (to B.K.) and a Heidner Foundation grant (to B.K.).
Footnotes
Competing interests: BK consults for TeraImmune and BrickellBio outside the submitted work. JHB reported serving as scientific co-founder and on boards for GentiBio, consulted for Bristol-Myers Squibb, Hotspot Therapeutics, Janssen, La Jolla Institute for Immunology, Oklahoma Medical Research Foundation and UpToDate, and owning stock in Omeros and Gentibio outside the submitted work. GG serves as founder of Gestalt Strategies outside the submitted work. CJG has served on boards for Altheia Science, Enthera Pharma, Veilo Bio and Merck outside the submitted work. All other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials. RNAseq data were deposited in the GEO repository under accession number GSE183701.
References and Notes
- 1.de Graaf G, Buckley F, Skotko BG, Estimation of the number of people with Down syndrome in the United States., Genetics in medicine : official journal of the American College of Medical Genetics 19, 439–447 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH, Down syndrome, Nature reviews. Disease primers, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aitken RJ, Mehers KL, Williams AJ, Brown J, Bingley PJ, Holl RW, Rohrer TR, Schober E, Abdul-Rasoul MM, Shield JPH, Gillespie KM, Early-Onset, Coexisting Autoimmunity and Decreased HLA-Mediated Susceptibility Are the Characteristics of Diabetes in Down Syndrome, Diabetes care 36, 1181–1185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinowe SL, Rubin IL, George KL, Adri MN, Eisenbarth GS, Trisomy 21 (Down’s syndrome): autoimmunity, aging and monoclonal antibody-defined T-cell abnormalities., Journal of autoimmunity 2, 25–30 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Schoch J, Rohrer TR, Kaestner M, Abdul-Khaliq H, Gortner L, Sester U, Sester M, Schmidt T, Quantitative, Phenotypical, and Functional Characterization of Cellular Immunity in Children and Adolescents With Down Syndrome., The Journal of infectious diseases 215, 1619–1628 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Araya P, Waugh KA, Sullivan KD, Núñez NG, Roselli E, Smith KP, Granrath RE, Rachubinski AL, Estrada BE, Butcher ET, Minter R, Tuttle KD, Bruno TC, Maccioni M, Espinosa JM, Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity., Proceedings of the National Academy of Sciences of the United States of America 116, 24231–24241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, Rachubinski AL, Wolter-Warmerdam K, Hickey F, Espinosa JM, Blumenthal T, Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation., Scientific reports 7, 14818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, DeGregori J, Espinosa JM, Trisomy 21 consistently activates the interferon response., eLife 5, 1709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waugh KA, Araya P, Pandey A, Jordan KR, Smith KP, Granrath RE, Khanal S, Butcher ET, Estrada BE, Rachubinski AL, McWilliams JA, Minter R, Dimasi T, Colvin KL, Baturin D, Pham AT, Galbraith MD, Bartsch KW, Yeager ME, Porter CC, Sullivan KD, Hsieh EW, Espinosa JM, Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome., CellReports 29, 1893–1908.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cossarizza A, Monti D, Montagnani G, Ortolani C, Masi M, Zannotti M, Franceschi C, Precocious aging of the immune system in Down syndrome: alteration of B lymphocytes, T-lymphocyte subsets, and cells with natural killer markers., American journal of medical genetics. Supplement 7, 213–218 (1990). [DOI] [PubMed] [Google Scholar]
- 11.Fülöp T, Larbi A, Dupuis G, Page AL, Frost EH, Cohen AA, Witkowski JM, Franceschi C, Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes?, Frontiers in immunology 8, 1960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gensous N, Bacalini MG, Franceschi C, Garagnani P, Down syndrome, accelerated aging and immunosenescence., Seminars in immunopathology 42, 635–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goronzy JJ, Li G, Yang Z, Weyand CM, The janus head of T cell aging - autoimmunity and immunodeficiency., Frontiers in immunology 4, 131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJL, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, Maecker HT, Davis MM, Variation in the human immune system is largely driven by non-heritable influences., Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusters MA, Verstegen RH, de Vries E, Down syndrome: is it really characterized by precocious immunosenescence?, Aging and disease 2, 538–545 (2011). [PMC free article] [PubMed] [Google Scholar]
- 16.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E, The who’s who of T-cell differentiation: human memory T-cell subsets., European journal of immunology 43, 2797–2809 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Bloemers BLP, Bont L, de Weger RA, Otto SA, Borghans JA, Tesselaar K, Decreased thymic output accounts for decreased naive T cell numbers in children with Down syndrome., The Journal of Immunology 186, 4500–4507 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Carsetti R, Valentini D, Marcellini V, Scarsella M, Marasco E, Giustini F, Bartuli A, Villani A, Ugazio AG, Reduced numbers of switched memory B cells with high terminal differentiation potential in Down syndrome, European journal of immunology 45, 903–914 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verstegen RHJ, Driessen GJ, Bartol SJW, van Noesel CJM, Boon L, van der Burg M, van Dongen JJM, de Vries E, van Zelm MC, Defective B-cell memory in patients with Down syndrome., The Journal of allergy and clinical immunology 134, 1346–1353.e9 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Norris S, Doherty DG, Collins C, McEntee G, Traynor O, Hegarty JE, O’Farrelly C, Natural T cells in the human liver: cytotoxic lymphocytes with dual T cell and natural killer cell phenotype and function are phenotypically heterogenous and include Valpha24-JalphaQ and gammadelta T cell receptor bearing cells., Human immunology 60, 20–31 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Kim HY, FcgRIII engagement provides activating signals to NKT cells in antibody-induced joint inflammation, Journal of Clinical Investigation, 1–10 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Hill BJ, Darko S, Song K, Quigley MF, Asher TE, Morita Y, Greenaway HY, Venturi V, Douek DC, Davenport MP, Price DA, Roederer M, The peripheral differentiation of human natural killer T cells, Immunology and cell biology 97, 586–596 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, CD8+NKT-like cells regulate the immune response by killing antigen-bearing DCs, Nature Publishing Group, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vignali D, Cantarelli E, Bordignon C, Canu A, Citro A, Annoni A, Piemonti L, Monti P, Detection and Characterization of CD8+ Autoreactive Memory Stem T Cells in Patients With Type 1 Diabetes., Diabetes 67, 936–945 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Skowera A, Ladell K, McLaren JE, Dolton G, Matthews KK, Gostick E, Kronenberg-Versteeg D, Eichmann M, Knight RR, Heck S, Powrie J, Bingley PJ, Dayan CM, Miles JJ, Sewell AK, Price DA, Peakman M, β-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure., Diabetes 64, 916–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newell EW, Cheng Y, Mass cytometry: blessed with the curse of dimensionality., Nature immunology 17, 890–895 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Gassen SV, Callebaut B, Helden MJV, Lambrecht BN, Demeester P, Dhaene T, Saeys Y, FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data., Cytometry. Part A : the journal of the International Society for Analytical Cytology 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Baker GJ, Muhlich JL, Palaniappan SK, Moore JK, Davis SH, Santagata S, Sorger PK, SYLARAS: A Platform for the Statistical Analysis and Visual Display of Systemic Immunoprofiling Data and Its Application to Glioblastoma, Cell Systems, 1–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P, Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity, Blood 118, 1305–1315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, Schieferl S, Hom J, Jenks S, Feldman RJ, Mehr R, Wei C, Lee FE-H, Cheung WC, Rosenberg AF, Sanz I, Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus., Nature immunology 16, 755–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claes N, Fraussen J, Vanheusden M, Hellings N, Stinissen P, Wijmeersch BV, Hupperts R, Somers V, Age-Associated B Cells with Proinflammatory Characteristics Are Expanded in a Proportion of Multiple Sclerosis Patients., The Journal of Immunology 197, 4576–4583 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z, Bugrovsky R, Blalock EL, Scharer CD, Tipton CM, Wei C, Lim SS, Petri M, Niewold TB, Anolik JH, Gibson G, Lee FE-H, Boss JM, Lund FE, Sanz I, Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus, Immunity 49, 725–739.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Floudas A, Neto N, Marzaioli V, Murray K, Moran B, Monaghan MG, Low C, Mullan RH, Rao N, Krishna V, Nagpal S, Veale DJ, Fearon U, Pathogenic, glycolytic PD-1+ B cells accumulate in the hypoxic RA joint, JCI insight 5, 1894–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berek C, [Do B cells play an important role in the pathogenesis of rheumatoid arthritis?]., Zeitschrift fur Rheumatologie 64, 383–388 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Nicholas MW, Dooley MA, Hogan SL, Anolik J, Looney J, Sanz I, Clarke SH, A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE., Clinical immunology (Orlando, Fla) 126, 189–201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobi AM, Reiter K, Mackay M, Aranow C, Hiepe F, Radbruch A, Hansen A, Burmester GR, Diamond B, Lipsky PE, Dörner T, Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: delineation by expression of CD27, IgD, and CD95., Arthritis Rheum 58, 1762–1773 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, Ferrero B, Eid P, Novelli F, T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta., Annals of neurology 65, 499–509 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Li J, Ueno A, Gasia MF, Luider J, Wang T, Hirota C, Jijon HB, Deane M, Tom M, Chan R, Barkema HW, Beck PL, Kaplan GG, Panaccione R, Qian J, Iacucci M, Gui X, Ghosh S, Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease., Inflammatory bowel diseases 22, 1779–1792 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, McCauley JL, Abreu MT, Unutmaz D, Sundrud MS, Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids., The Journal of experimental medicine 211, 89–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, Szeliga W, Wang Y, Liu Y, Welling TH, Elder JT, Zou W, Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis., The Journal of Immunology 181, 4733–4741 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han GM, O’Neil-Andersen NJ, Zurier RB, Lawrence DA, CD4+CD25high T cell numbers are enriched in the peripheral blood of patients with rheumatoid arthritis., Cellular Immunology 253, 92–101 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellor-Pita S, Citores MJ, Castejon R, Tutor-Ureta P, Yebra-Bango M, Andreu JL, Vargas JA, Decrease of regulatory T cells in patients with systemic lupus erythematosus., Annals of the rheumatic diseases 65, 553–554 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crispin JC, Martínez A, Alcocer-Varela J, Quantification of regulatory T cells in patients with systemic lupus erythematosus., Journal of autoimmunity 21, 273–276 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT, Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis., Proceedings of the National Academy of Sciences of the United States of America 112, 7061–7066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrath J, Chemin K, Albrecht I, Catrina AI, Malmström V, Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion., European journal of immunology 44, 2979–2989 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Förster R, Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production., The Journal of experimental medicine 192, 1545–1552 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP, A human memory T cell subset with stem cell-like properties., Nature Medicine 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeshita M, Suzuki K, Kondo Y, Morita R, Okuzono Y, Koga K, Kassai Y, Gamo K, Takiguchi M, Kurisu R, Mototani H, Ebisuno Y, Yoshimura A, Takeuchi T, Multi-dimensional analysis identified rheumatoid arthritis-driving pathway in human T cell., Annals of the rheumatic diseases 78, 1346–1356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hosokawa K, Muranski P, Feng X, Townsley DM, Liu B, Knickelbein J, Keyvanfar K, Dumitriu B, Ito S, Kajigaya S, VI JGT, Kaplan MJ, Nussenblatt RB, Barrett AJ, O’Shea J, Young NS, Memory Stem T Cells in Autoimmune Disease: High Frequency of Circulating CD8 +Memory Stem Cells in Acquired Aplastic Anemia, Journal of immunology (Baltimore, Md : 1950) 196, 1568–1578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ottaviano G, Gerosa J, Santini M, Leo PD, Vecchione A, Jofra T, Trimarchi C, Pellegrin MD, Agosti M, Aiuti A, Marinoni M, Cicalese MP, Fousteri G, A Prevalent CXCR3+ Phenotype of Circulating Follicular Helper T Cells Indicates Humoral Dysregulation in Children with Down Syndrome., Journal of clinical immunology, 1–9 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Simone GD, Mazza EMC, Cassotta A, Davydov AN, Kuka M, Zanon V, Paoli FD, Scamardella E, Metsger M, Roberto A, Pilipow K, Colombo FS, Tenedini E, Tagliafico E, Gattinoni L, Mavilio D, Peano C, Price DA, Singh SP, Farber JM, Serra V, Cucca F, Ferrari F, Orrù V, Fiorillo E, Iannacone M, Chudakov DM, Sallusto F, Lugli E, CXCR3 Identifies Human Naive CD8+ T Cells with Enhanced Effector Differentiation Potential., The Journal of Immunology 203, 3179–3189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, Knox K, Bush EC, Sims PA, Sinari S, Billheimer D, Haddad EK, Murray KO, Wertheimer AM, Nikolich-Žugich J, Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses., Nature immunology 17, 966–975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M, Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis., European journal of immunology 18, 1797–1801 (1988). [DOI] [PubMed] [Google Scholar]
- 54.Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR, Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis., Journal of immunology (Baltimore, Md : 1950) 147, 117–123 (1991). [PubMed] [Google Scholar]
- 55.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles., Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection., Cell Systems 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP, A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice, Blood 118, 1294–1304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, Samuels J, Berman J, Buckner JH, Cunningham-Rundles C, Meffre E, Complement receptor 2/CD21− human naive B cells contain mostly autoreactive unresponsive clones, Blood 115, 5026–5036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alpert A, Pickman Y, Leipold M, Rosenberg-Hasson Y, Ji X, Gaujoux R, Rabani H, Starosvetsky E, Kveler K, Schaffert S, Furman D, Caspi O, Rosenschein U, Khatri P, Dekker CL, Maecker HT, Davis MM, Shen-Orr SS, A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring., Nature Medicine 25, 487–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Timmers PRHJ, Wilson JF, Joshi PK, Deelen J, Multivariate genomic scan implicates novel loci and haem metabolism in human ageing, Nat Commun 11, 3570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Igarashi K, Kurosaki T, Roychoudhuri R, BACH transcription factors in innate and adaptive immunity, Nat Rev Immunol 17, 437–450 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Mowery CT, Reyes JM, Cabal-Hierro L, Higby KJ, Karlin KL, Wang JH, Kimmerling RJ, Cejas P, Lim K, Li H, Furusawa T, Long HW, Pellman D, Chapuy B, Bustin M, Manalis SR, Westbrook TF, Lin CY, Lane AA, Trisomy of a Down Syndrome Critical Region Globally Amplifies Transcription via HMGN1 Overexpression, Cell Reports 25, 1898–1911.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang W, Qu J, Liu G-H, Belmonte JCI, The ageing epigenome and its rejuvenation, Nature reviews Molecular cell biology 21, 1–14 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, Bosi E, Brusko TM, DiMeglio LA, Evans-Molina C, Gitelman SE, Greenbaum CJ, Gottlieb PA, Herold KC, Hessner MJ, Knip M, Jacobsen L, Krischer JP, Long SA, Lundgren M, McKinney EF, Morgan NG, Oram RA, Pastinen T, Peters MC, Petrelli A, Qian X, Redondo MJ, Roep BO, Schatz D, Skibinski D, Peakman M, Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes., Diabetes care 43, 5–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borelli V, Vanhooren V, Lonardi E, Reiding KR, Capri M, Libert C, Garagnani P, Salvioli S, Franceschi C, Wuhrer M, Plasma N-Glycome Signature of Down Syndrome., Journal of proteome research 14, 4232–4245 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Hüls A, Costa ACS, Dierssen M, Baksh RA, Bargagna S, Baumer NT, Brandão AC, Carfi A, Carmona-Iragui M, Chicoine BA, Ghosh S, Lakhanpaul M, Manso C, Mayer M-A, Ortega M. del C., de Asua DR, Rebillat A-S, Russell LA, Sgandurra G, Valentini D, Sherman SL, Strydom A, T. C.−19 Initiative, Medical vulnerability of individuals with Down syndrome to severe COVID-19–data from the Trisomy 21 Research Society and the UK ISARIC4C survey, Eclinicalmedicine 33, 100769 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valentini D, Marcellini V, Bianchi S, Villani A, Facchini M, Donatelli I, Castrucci MR, Marasco E, Farroni C, Carsetti R, Generation of switched memory B cells in response to vaccination in Down syndrome children and their siblings., Vaccine 33, 6689–6696 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Colvin KL, Yeager ME, What people with Down Syndrome can teach us about cardiopulmonary disease, 1–16 (2017). [DOI] [PMC free article] [PubMed]
- 69.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, Igarashi K, Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site., Mol Cell Biol 16, 6083–6095 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iulita MF, Ower A, Barone C, Pentz R, Gubert P, Romano C, Cantarella RA, Elia F, Buono S, Recupero M, Romano C, Castellano S, Bosco P, Nuovo SD, Drago F, Caraci F, Cuello AC, An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: Relation to cognitive decline and longitudinal evaluation, Alzheimer’s Dementia 12, 1132–1148 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Raha-Chowdhury R, Raha AA, Henderson J, Ghaffari SD, Grigorova M, Beresford-Webb J, Allinson K, Chakraborty S, Holland A, Zaman SH, Impaired Iron Homeostasis and Haematopoiesis Impacts Inflammation in the Ageing Process in Down Syndrome Dementia, J Clin Medicine 10, 2909 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaer LV, Wu L, Therapeutic Potential of Invariant Natural Killer T Cells in Autoimmunity, Frontiers in immunology 9, 297–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orrù V, Steri M, Sidore C, Marongiu M, Serra V, Olla S, Sole G, Lai S, Dei M, Mulas A, Virdis F, Piras MG, Lobina M, Marongiu M, Pitzalis M, Deidda F, Loizedda A, Onano S, Zoledziewska M, Sawcer S, Devoto M, Gorospe M, Abecasis GR, Floris M, Pala M, Schlessinger D, Fiorillo E, Cucca F, Complex genetic signatures in immune cells underlie autoimmunity and inform therapy, Nature Genetics, 1–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aqrawi LA, Ivanchenko M, Björk A, Sepúlveda JIR, Imgenberg-Kreuz J, Kvarnström M, Haselmayer P, Jensen JL, Nordmark G, Chemin K, Skarstein K, Wahren-Herlenius M, Diminished CXCR5 expression in peripheral blood of patients with Sjögren’s syndrome may relate to both genotype and salivary gland homing., Clinical and experimental immunology 192, 259–270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vang T, Landskron J, Viken MK, Oberprieler N, Torgersen KM, Mustelin T, Tasken K, Tautz L, Rickert RC, Lie BA, The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses, Human immunology 74, 574–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Habib T, Long SA, Samuels PL, Brahmandam A, Tatum M, Funk A, Hocking AM, Cerosaletti K, Mason MT, Whalen E, Rawlings DJ, Greenbaum C, Buckner JH, T. 1 D. T. S. Group, Dynamic Immune Phenotypes of B and T Helper Cells Mark Distinct Stages of T1D Progression., Diabetes 68, 1240–1250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vecchione A, Fonte RD, Gerosa J, Jofra T, Cicalese MP, Napoleone V, Ippolito E, Galvani G, Ragogna F, Stabilini A, Bianconi E, Grogan P, Bonura C, Bonfanti R, Frontino G, Nano R, Melzi R, Pellegrin MD, Laurenzi A, Meschi F, Barera G, Rigamonti A, Indirli R, Bosi E, Piemonti L, Aiuti A, Battaglia M, Fousteri G, Reduced PD-1 expression on circulating follicular and conventional FOXP3+ Treg cells in children with new onset type 1 diabetes and autoantibody-positive at-risk children, Clinical immunology (Orlando, Fla) 211, 108319 (2020). [DOI] [PubMed] [Google Scholar]
- 78.Greene E, Finak G, D’Amico LA, Bhardwaj N, Church CD, Morishima C, Ramchurren N, Taube JM, Nghiem PT, Cheever MA, Fling SP, Gottardo R, New interpretable machine learning method for single-cell data reveals correlates of clinical response to cancer immunotherapy, 81, 35–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, Chen J, Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline., PLoS computational biology 12, e1005112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tesson BM, Breitling R, Jansen RC, DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules., BMC bioinformatics 11, 497–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for integrated models of biomolecular interaction networks., Genome research 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner., Bioinformatics (Oxford, England) 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data., Bioinformatics (Oxford, England) 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies., Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Characterization of the cohorts.
Fig. S2. IMPACD enables analysis of manually gated cytometry data.
Fig S3. Dysregulation of cellular subsets and signaling are observed in samples from individuals with T1D and DS.
Fig. S4. Interrogating immune aging in samples from individuals with DS using CyTOF.
Fig. S5. Interrogating immune aging in samples from individuals with DS using RNAseq.
Table S1. Source of Reagents.
Data file S2. CD4-NKT subsets differentially co-regulated in DS.
Date file S3. Genes that vary linearly with age in TC.
Data file S1.
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. RNAseq data were deposited in the GEO repository under accession number GSE183701.