

Abstract
Dedicated to Professor Manfred Scheer on the occasion of his 65th birthday
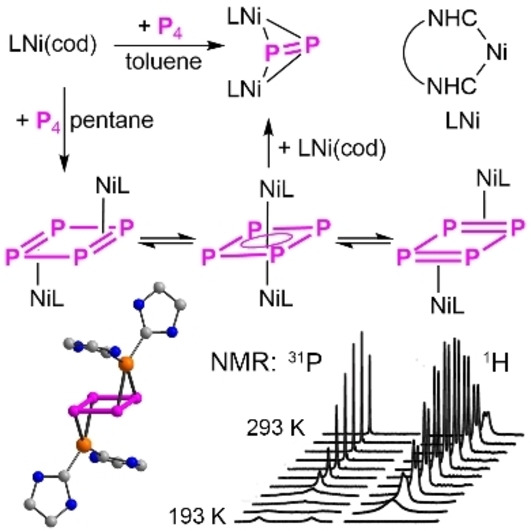
The reaction of (1)Ni(η2‐cod), 2, incorporating a chelating bis(N‐heterocyclic carbene) 1, with P4 in pentane yielded the dinuclear complex [(2)Ni]2(μ2,η2 : η2‐P4), 3, formally featuring a cyclobutadiene‐like, neutral, rectangular, π‐bridging P4‐ring. In toluene, the butterfly‐shaped complex [(1)Ni]2(μ2,η2 : η2‐P2), 4, with a formally neutral P2‐unit was obtained from 2 and either P4 or 3. Computational studies showed that a haptotropic rearrangement involving two isomers of the μ2,η2 : η2‐P4 coordination mode and a low‐energy μ2,η4 : η4‐P4 coordination mode, as previously predicted for related nickel cyclobutadiene complexes, could explain the coalescence observed in the low‐temperature NMR spectra of 3. The insertion of the (1)Ni fragment into a P−P bond of P7(SiMe3)3, forming complex 5 with a norbornane‐like P7 ligand, was also observed.
Keywords: Haptotropism, N-Heterocyclic Carbene, Nickel, Phosphorus, π-Ligands
The solvent‐dependent reaction of white phosphorus with a Ni0 reagent incorporating a chelating bis(N‐heterocyclic carbene) leads to complexes with neutral P2 and P4 ligands. The latter mirrors the coordination of cyclobutadiene to Ni0, displaying olefin‐like bonding and η2‐η4‐haptotropism, and can be converted to the former. These findings emphasize the fluidity of cyclo‐P4 coordination modes.
Transition metal complexes incorporating P x ligands have been extensively investigated due to their appealing structural variety and intriguing bonding. [1] White phosphorus is the entryway to the production of most phosphorus compounds, [2] and more recently the interest toward P x metal complexes has expanded to include the metal‐mediated activation and further transformation of this molecule. [3] Complexes incorporating P4 ligands are of particular interest because they are hypothesized to constitute the first stage in the activation of P4. [4] Hydrocarbon‐based π‐ligands such as cyclobutadiene, [5] cyclopentadienyl, and benzene have been used as a guideline for systematizing the chemistry of substituent‐free, or “naked”, phosphorus ligands because the CH and P fragments are isolobal. [6]
Mirroring cyclobutadiene complexes A (Figure 1), the planar cyclo‐P4 ligand forms mononuclear, 18‐valence‐electron sandwich and half sandwich complexes B (M=V, [7] Nb, [8] Ta, [9] L n =Cpx(CO)2; M=Mo, [10] L n =(CO)2(CNR)2; (CO)I2(CNR)2; M=Fe, [11] L n =(Cy2PCH2CH2)2PPh; M=Co, [12] L n =Cpx), B − (M=Mo, [13] L n =(CO)I(CNR)2; M=Fe, [14] L n =Cpx; M=Co, [15] L =bis(2,6‐Dipp)phenanthrene‐9,10‐diimine), and B 2− (M=Mo, [13] L n =(CO)(CNR)2, (CO)2(CNR)). The phosphorus ring in B can σ‐donate up to four lone pairs to additional metals, generating multinuclear complexes. [1] By analogy to cyclobutadiene, the P4 ligand in B is considered to be dianionic, and this view is supported by Mössbauer measurements [11] and computational studies.[ 13 , 14 ] cyclo‐P4 2− has been described as antiaromatic as a free ligand, [16] lone pair aromatic in alkali metal salts, [17] and aromatic in transition metal complexes.[ 11 , 13 ]
Figure 1.
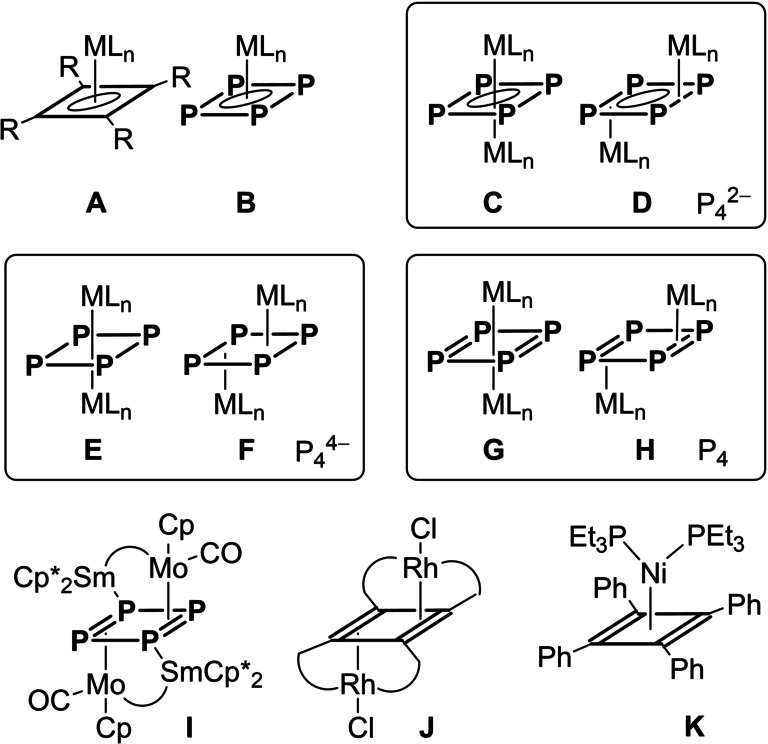
Reported coordination modes of the planar cyclo‐P4 ligand (B–I) and relevant cyclobutadiene analogues (A, J, and K).
In contrast to cyclobutadiene, which rarely π‐bridges transition metals, [18] cyclo‐P4 forms a variety of bridged complexes where the planar ligand can be formally considered P4 2− (C and D), P4 4− (E and F), or P4 (G and H), based on structural, spectroscopic, and computational data. For example, the μ2 : η4,η4‐bridging mode has been observed in complexes of type C (M=Fe, [19] L n =β‐diketiminato; M=Sm, [20] L n ={[DippN]2CH}2), C − (M=Co, L n =β‐diketiminato),[ 21 , 22 ] E (M=Zr, L n =PhP(CH2SiMe2NSiMe2CH2)2PPh), [23] E − (M=Co, L n =BIAN) [24] and G (M=Co, L n =β‐diketiminato),[ 21 , 22 ] while complexes D (M=U, L n =Cp*(COT)) [25] and F (M=Nb, [26] L n =(DippO)3; M=Nb, Ta, [27] L=(β‐diketiminato)(tBuN)) display μ2 : η2,η2‐ and μ2 : η3,η3‐bridging modes, respectively. Complex I is the only known example featuring a neutral μ2 : η2,η2‐P4 ligand, presumably because the constrained geometry does not allow for η4‐coordination. [28] This mirrors the behaviour of cyclobutadiene, which is known to coordinate in μ2 : η2,η2‐fashion only in J. [29]
Although the chemistry of nickel with P4 has been extensively investigated, leading to the isolation of a variety of (L n Ni)yP x derivatives (x=2–5, 8),[ 1 , 3 , 30 ] a planar, π‐coordinating cyclo‐P4 nickel complex has remained elusive. [31] The excellent π‐donating properties of the metal in [(1)Ni(η2‐cod)], [32a] 2 (Scheme 1; 1=bis(NHC)), [32b] recommend it as a promising synthon for the stabilization of π‐bonded P x ‐ligands. In a similar manner, the (Et3P)2Ni fragment has allowed the isolation of complex K, having structural features consistent with Ni0 and a neutral, η4‐cyclobutadiene ligand, as opposite to NiII and cyclobutadienyl. [33] Furthermore, a computational study on the model system (cyclobutadiene) Ni(PH3)2 suggested that the η4‐ and η2‐coordination modes had similar energies and a tiny interconversion barrier. [34] Higher‐level calculations for the analogous (cyclobutadiene)Pt(diphosphinylethane) gave a slightly greater energy difference. [35] These findings led us to investigate the reactivity of 2 with P4 in pursuit of the elusive μ2 : η2,η2‐P4 bridging mode H. This chemistry, as well as the reactivity of 2 with P7(SiMe3)3, will be reported herein.
Scheme 1.
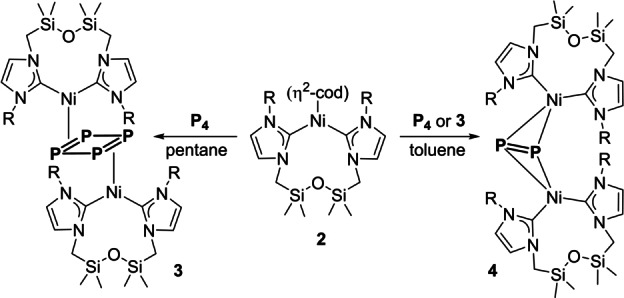
Synthesis of 3 and 4.
Reaction of 2 with P4 in pentane afforded diamagnetic, crystalline complex [(1)Ni]2P4, 3, irrespective of stoichiometry (Scheme 1). An X‐ray diffraction experiment revealed a dinuclear, C i‐symmetric structure with a planar, rectangular μ2 : η2,η2‐P4 core (Figure 2). The cyclo‐P4 ligand features alternating long (2.242(8) Å) and short (2.149(3) Å) P−P bonds (cf. P−P 2.1994(3) Å in P4 [36] and P=P 2.140(1) in PhP=PPh coordinated to Ni0), [37] suggestive of a neutral P4 ligand bound by two (1)Ni0 fragments, i.e., H. For comparison, the P4 ligand adopts a very similar geometry in G, M=CoI (av. P−P 2.29 Å and P=P 2.13 Å).[ 21 , 22 ]
Figure 2.
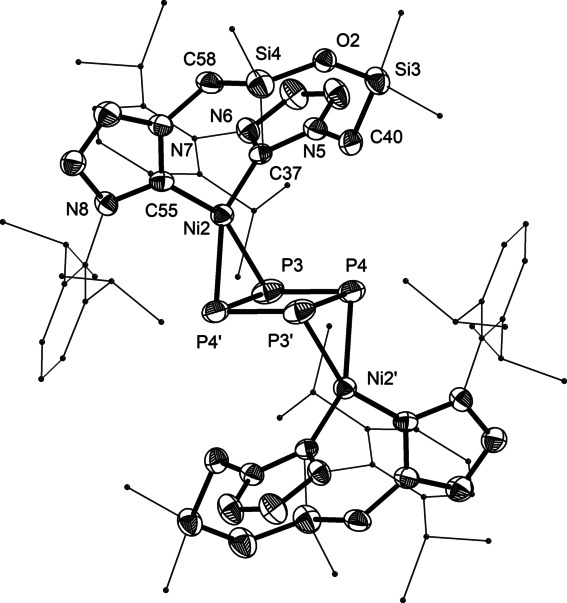
Solid‐state structure of one of the two independent molecules of 3, with 50 % thermal ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: P3–P4′ 2.145(2), 2.149(2), P3–P4 2.237(2), 2.242(2), Ni–P 2.2436(15)–2.2822(14), Ni–C 1.911(5)–1.933(5); P‐P‐P 89.58(7)–90.42(7), C‐Ni‐C 109.9(2), 110.8(2). [38]
A room‐temperature 31P NMR spectrum of 3 in toluene‐d 8 revealed a single, broad resonance at 45 ppm (cf. 128 and 169 ppm for G, M=CoI, in the solid state), [21] indicative of a dynamic process. The variable‐temperature study revealed two coalescence temperatures (Figure 3). At low temperature, the presence of two 31P resonances (21 and 63 ppm) is consistent with a centrosymmetric C i‐structure, as observed in the solid state. Upon heating above 210 K, all phosphorus atoms become equivalent on the NMR time scale. Resonance broadening suggestive of a second coalescence is apparent slightly above room temperature, but incipient decomposition precluded the investigation of this process. The 1H NMR spectrum features four AB doublets for the methylene protons (3.3, 4.3, 5.0, and 8.2 ppm, Figure S1), and two resonances for the carbene carbons are observed in 13C NMR (192.8 and 202.7 ppm, Figure S2). This indicates that the solid‐state structure of the (1)Ni fragment is retained in solution between 210–293 K.
Figure 3.
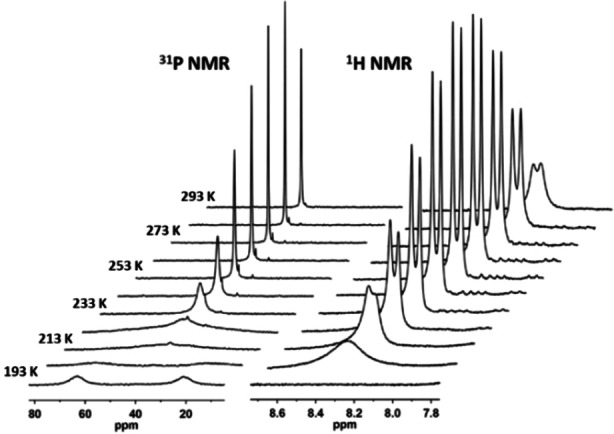
Stack plots of variable‐temperature (193–293 K) 31P (left) and 1H (right) NMR spectra of 3 in toluene‐d 8.
DFT calculations on 3 yielded a closed‐shell singlet ground state free of instabilities, supporting the proposed coordination mode H. An ETS‐NOCV analysis of bonding in 3 using closed‐ shell fragments ((1)Ni)2 and P4 showed significant interaction of nickel d‐orbitals with the π‐type orbitals of P4, giving two strongly stabilizing contributions (Figure S24). Population analyses suggested a net flow of 0.7–0.8 e− from the two (1)Ni0 fragments to P4, underlining the fact that the use of integer oxidation states and charges is inevitably an oversimplification.
Further inspection of the potential energy surface of 3 with DFT revealed that a μ2 : η4,η4‐bound C i‐isomer (3′′) with a square‐like cyclo‐P4 ring is only 14 kJ mol−1 higher in energy than 3, while a second C i‐symmetric μ2 : η2,η2‐bound isomer (3′) has a relative energy of only 8 kJ mol−1 (Scheme 2, Figure S23). Both 3 and 3′ connect to 3′′ with barriers <50 kJ mol−1, in agreement with the value of 44 kJ mol−1 calculated from the coalescence temperature. These results are in accordance with a haptotropic rearrangement (P4‐ring whizzing) accounting for the low temperature coalescence in the 31P NMR data of 3. Fluxional P4‐ring behaviour has been proposed for cyclo‐P4 complexes.[ 11 , 23 , 26 ] The high‐temperature coalescence is tentatively assigned to ligand symmetrisation via dynamic motion that was shown to involve an activation energy of 53 kJ mol−1 in (1)NiGeCl2; [32b] the barrier for 3 is expected to be higher due to increased steric strain.
Scheme 2.
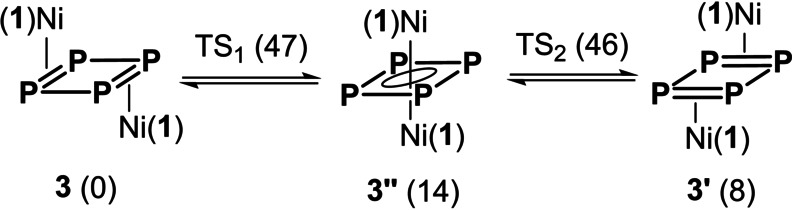
Calculated haptotropic rearrangement between 3 and 3′ (which differ in the relative orientation of ligand 1 with respect to cyclo‐P4), via 3′′ over transition states TS1 and TS2. Relative Gibbs free energies (in kJ mol−1 at 298 K) in parenthesis.
The singlet ground state of 3′′ shows an instability, leading to a broken‐symmetry singlet solution in which approximately 0.6 α spin becomes localized on one Ni centre and 0.6 β spin on the other. This suggests that coordination mode C, involving two (1)NiI fragments bridged by a dianionic P4 2− ring, is a reasonable first‐order approximation of bonding in the intermediate 3′′. This description is well in line with the square‐shaped structure calculated for the cyclo‐P4 ring in 3′′ having two almost equal bond lengths (2.163 and 2.172 Å) and bond angles (88 and 92°).
In toluene, the reaction of 2 with either a stoichiometric or an excess amount of P4 yields exclusively 4 (Scheme 1), which can also be prepared from 3 and 2 in toluene; notably, 3 does not convert to 4 upon dissolution in toluene. Under an inert atmosphere at −40 °C, solids 3 and 4 are stable for months but at room temperature in solution decomposition of both compounds leads within hours to the disappearance of all 31P resonances and formation of unidentified products. The solid state structure of 4 (Figure S21) features a butterfly‐shaped Ni2P2 core with a very short (2.0784(16) Å) P−P bond and a dihedral angle of 120.87(3)°. In similar complexes [{(NHC)2Ni}2(μ,η2 : η2‐P2)], the P2 unit (P−P 2.0906(8) Å) was described as P2 4− based on the visual inspection of frontier orbitals and their localization on P2. [39] Quantitative bonding analyses performed for 4 show that, while its frontier Kohn–Sham orbitals are similar to those of [{(NHC)2Ni}2(μ,η2 : η2‐P2)] (Figure S25), the contribution from P2 is much less than 50 % in all cases. Thus, complex 4 can be formally described as a neutral P2 ligand bound by two (1)Ni(0) fragments, akin to the reported dinuclear Ni0‐alkyne complexes. [40] An ETS‐NOCV analysis of 4 using this fragmentation scheme shows charge flow between the d‐orbitals on the metals and the π‐type orbitals on P2, resulting in both metal‐to‐ligand and ligand‐to‐metal bonding contributions (Figure S26). Population analyses indicate a net flow of 0.5–0.6 e− from ((1)Ni)2 to P2, illustrating, again, that the discussed promolecular fragments should only be treated as good first‐order approximations of bonding in 3 and 4.
The most accessible phosphorus clusters other than P4 are P7R3, which were used extensively as a ligands; [41] P−P bond activation has only been reported for the parent Zintl ion P7 3−. [42] Reaction of 2 with P7(SiMe3)3 [43] resulted into the formation of 5 (Scheme 3). At room temperature, short reaction times were needed to limit thermal decomposition. Crystallographic analysis revealed that the (1)Ni fragment inserted into the P3 ring of P7(SiMe3)3, generating a complex with a norbornane‐like P7 ligand (Figure 4). This mirrors the transformations observed for the activation of P7 3−, [44] except in the latter case η4‐P7 3− complexes were usually obtained. The insertion was accompanied by an inversion at phosphorus, leading to a change of the P7(SiMe3)3 conformation from syn, which is typical to all P7R3 analogues, to anti. The Ni−P bonds in 5 measure 2.2253(16) and 2.2950(16) Å, while the P−P bonds change little in comparison to P7(SiMe3)3. [45] The 31P NMR spectrum of 5 in toluene‐d 8 features seven multiplet signals that were assigned using 2D NMR.
Scheme 3.
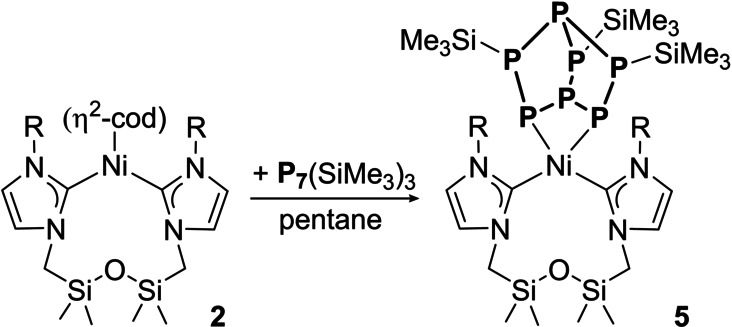
Synthesis of 5.
Figure 4.
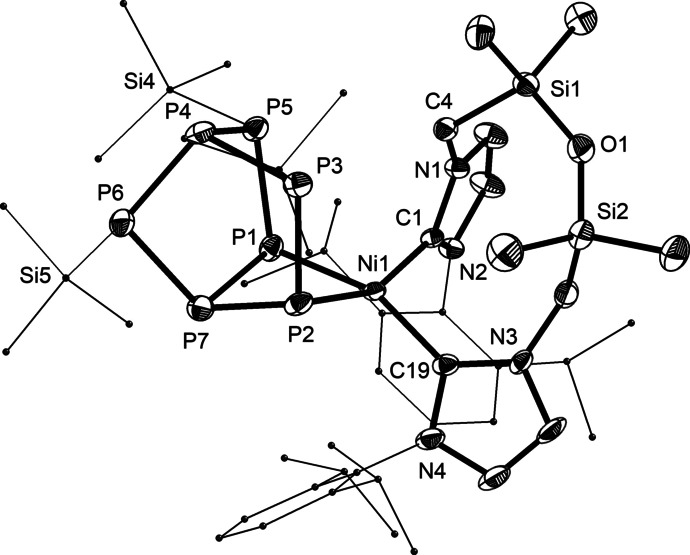
Solid‐state structure of 5 with 50 % thermal ellipsoids and hydrogen atoms omitted for clarity. Selected bond lengths [Å]: P4–P3 2.184(2), P4–P5 2.186(2), P4–P6 2.201(2), P1–P5 2.227(2), P2–P3 2.233(2), P7–P6 2.160(2), P1–P7 2.205(2), P2–P7 2.220(2), P1⋅⋅⋅P2 3.007(2), Ni–C 1.954(5), 1.992(5), Ni1–P1 2.2253(16), Ni1–P2 2.2950(16). [38]
In conclusion, complex 2 displays solvent‐dependent reactivity with P4, leading to 3 in pentane and 4 in toluene. Moreover, in reaction with 2, 3 generates 4, supporting the postulate that cyclo‐P4 complexes represent the first step in the activation of P4 by transition metals. Compound 3 features the elusive μ2 : η2,η2‐P4 bridging mode H, previously only characterized in the geometry‐constrained system I. The low‐temperature coalescence observed in the NMR spectra of 3 can be explained with a haptotropic rearrangement involving two isomers of the crystallographically characterized μ2 : η2,η2 bridging mode, as well as a slightly less stable isomer with a μ2 : η4,η4‐bound ligand. This leads to the equivalence of all phosphorus atoms of 3 on the NMR timescale and mirrors the ring‐whizzing mechanism proposed for the analogous nickel‐cyclobutadiene complex K. Employing 2, the first P−P bond activation in a P7R3 cluster was also achieved, leading to complex 5. The low energy barrier haptotropism of 3 and the formalism that best describes the structures 3, 3′, and 3′′ suggest that the classification of cyclo‐P4 complexes in categories C–H (Figure 1) is somewhat fluid.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
Financial support was provided by the Universities of Calgary and Jyväskylä, the Academy of Finland (Grant #340060 to C.G.), as well as the NSERC of Canada in the form of Discovery Grant #2019‐07195 to R.R. The project received funding from the European Research Council under the EU's Horizon 2020 programme (Grant #772510 to H.M.T.). Computational resources were provided by the Finnish Grid and Cloud Infrastructure (persistent identifier urn : nbn : fi : research‐infras‐2016072533).
C. Gendy, J. Valjus, H. M. Tuononen, R. Roesler, Angew. Chem. Int. Ed. 2022, 61, e202115692; Angew. Chem. 2022, 134, e202115692.
Contributor Information
Prof. Heikki M. Tuononen, Email: heikki.m.tuononen@jyu.fi.
Prof. Roland Roesler, Email: roesler@ucalgary.ca.
References
- 1.
- 1a. Giusti L., Landaeta V. R., Vanni M., Kelly J. A., Wolf R., Caporali M., Coord. Chem. Rev. 2021, 441, 213927. [Google Scholar]
- 2.
- 2a. Geeson M. B., Cummins C. C., Science 2018, 359, 1383–1385; [DOI] [PubMed] [Google Scholar]
- 2b. Scott D. J., Cammarata J., Schimpf M., Wolf R., Nat. Chem. 2021, 13, 458–464. [DOI] [PubMed] [Google Scholar]
- 3. Hoidn C. M., Scott D. J., Wolf R., Chem. Eur. J. 2021, 27, 1886–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo G., Du S., Wang P., Liu F., Zhang W.-X., Luo Y., Chem. Eur. J. 2020, 26, 13282–13287. [DOI] [PubMed] [Google Scholar]
- 5. Shoji Y., Ikabata Y., Ryzhii I., Ayub R., El Bakouri O., Sato T., Wang Q., Miura T., Karunathilaka B. S. B., Tsuchiya Y., Adachi C., Ottosson H., Nakai H., Ikoma T., Fukushima T., Angew. Chem. Int. Ed. 2021, 60, 21817–21823; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21988–21994. [Google Scholar]
- 6. Hoffmann R., Angew. Chem. Int. Ed. Engl. 1982, 21, 711–724; [Google Scholar]; Angew. Chem. 1982, 94, 725–739. [Google Scholar]
- 7. Herberhold M., Frohmader G., Milius W., J. Organomet. Chem. 1996, 522, 185–196. [Google Scholar]
- 8. Scherer O. J., Vondung J., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1989, 28, 1355–1357; [Google Scholar]; Angew. Chem. 1989, 101, 1395–1397. [Google Scholar]
- 9. Scherer O. J., Winter G., Wolmershäuser G., Z. Anorg. Allg. Chem. 1993, 619, 827–835. [Google Scholar]
- 10. Mandla K. A., Moore C. E., Rheingold A. L., Figueroa J. S., Angew. Chem. Int. Ed. 2019, 58, 1779–1783; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1793–1797. [Google Scholar]
- 11. Cavaillé A., Saffon-Merceron N., Nebra N., Fustier-Boutignon M., Mézailles N., Angew. Chem. Int. Ed. 2018, 57, 1874–1878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1892–1896. [Google Scholar]
- 12. Dielmann F., Timoshkin A., Piesch M., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 1671–1675; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1693–1698. [Google Scholar]
- 13. Mandla K. A., Neville M. L., Moore C. E., Rheingold A. L., Figueroa J. S., Angew. Chem. Int. Ed. 2019, 58, 15329–15333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15473–15477. [Google Scholar]
- 14. Chakraborty U., Leitl J., Mühldorf B., Bodensteiner M., Pelties S., Wolf R., Dalton Trans. 2018, 47, 3693–3697. [DOI] [PubMed] [Google Scholar]
- 15. Hoidn C. M., Maier T. M., Trabitsch K., Weigand J. J., Wolf R., Angew. Chem. Int. Ed. 2019, 58, 18931–18936; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19107–19112. [Google Scholar]
- 16. Jiménez-Halla J. O. C., Matito E., Robles J., Solà M., J. Organomet. Chem. 2006, 691, 4359–4366. [Google Scholar]
- 17.
- 17a. Kraus F., Aschenbrenner J. C., Korber N., Angew. Chem. Int. Ed. 2003, 42, 4030–4033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4162–4165; [Google Scholar]
- 17b. Kraus F., Korber N., Chem. Eur. J. 2005, 11, 5945–5959; [DOI] [PubMed] [Google Scholar]
- 17c. Kraus F., Hanauer T., Korber N., Inorg. Chem. 2006, 45, 1117–1123. [DOI] [PubMed] [Google Scholar]
- 18. Patel D., McMaster J., Lewis W., Blake A. J., Liddle S. T., Nat. Commun. 2013, 4, 2323. [DOI] [PubMed] [Google Scholar]
- 19. Spitzer F., Graßl C., Balázs G., Zolnhofer E. M., Meyer K., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 4340–4344; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4412–4416. [Google Scholar]
- 20. Schoo C., Bestgen S., Köppe R., Konchenko S. N., Roesky P. W., Chem. Commun. 2018, 54, 4770–4773. [DOI] [PubMed] [Google Scholar]
- 21. Yao S., Lindenmaier N., Xiong Y., Inoue S., Szilvási T., Adelhardt M., Sutter J., Meyer K., Driess M., Angew. Chem. Int. Ed. 2015, 54, 1250–1254; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1266–1270. [Google Scholar]
- 22. Spitzer F., Graßl C., Balázs G., Mädl E., Keilwerth M., Zolnhofer E. M., Meyer K., Scheer M., Chem. Eur. J. 2017, 23, 2716–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seidel W. W., Summerscales O. T., Patrick B. U., Fryzuk M. D., Angew. Chem. Int. Ed. 2009, 48, 115–117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 121–123. [Google Scholar]
- 24. Pelties S., Maier T., Herrmann D., de Bruin B., Rebreyend C., Gärtner S., Shenderovich I. G., Wolf R., Chem. Eur. J. 2017, 23, 6094–6102. [DOI] [PubMed] [Google Scholar]
- 25. Frey A. S. P., Cloke F. G. N., Hitchcock P. B., Green J. C., New J. Chem. 2011, 35, 2022–2026. [Google Scholar]
- 26. Velian A., Cummins C. C., Chem. Sci. 2012, 3, 1003–1006. [Google Scholar]
- 27. Camp C., Maron L., Bergman R. G., Arnold J., J. Am. Chem. Soc. 2014, 136, 17652–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arleth N., Gamer M. T., Köppe R., Pushkarevsky N. A., Konchenko S. N., Fleischmann M., Bodensteiner M., Scheer M., Roesky P. W., Chem. Sci. 2015, 6, 7179–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okamoto K., Omoto Y., Sano H., Ohe K., Dalton Trans. 2012, 41, 10926–10929. [DOI] [PubMed] [Google Scholar]
- 30. Carenco S., Resa I., Le Goff X., Le Floch P., Mézailles N., Chem. Commun. 2008, 2568–2570. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Yao S., Xiong Y., Milsmann C., Bill E., Pfirrmann S., Limberg C., Driess M., Chem. Eur. J. 2010, 16, 436–439; [DOI] [PubMed] [Google Scholar]
- 31b. Pelties S., Herrmann D., de Bruin B., Hartl F., Wolf R., Chem. Commun. 2014, 50, 7014–7016. For a wider selection of L n Ni–P x complexes, see Ref #2 in the Supporting Information. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Puerta Lombardi B. M., Gendy C., Gelfand B. S., Bernard G. M., Wasylishen R. E., Tuononen H. M., Roesler R., Angew. Chem. Int. Ed. 2021, 60, 7077–7081; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 7153–7157; [Google Scholar]
- 32b. Gendy C., Mansikkamäki A., Valjus J., Heidebrecht J., Hui P. C.-Y., Bernard G. M., Tuononen H. M., Wasylishen R. E., Michaelis V. K., Roesler R., Angew. Chem. Int. Ed. 2019, 58, 154–158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 160–164. [Google Scholar]
- 33. Eisch J. J., Piotrowski A. M., Aradi A. A., Krüger C., Romão M. J., Z. Naturforsch. B 1985, 40, 624–635. [Google Scholar]
- 34. Silvestre J., Albright T. A., Nouv. J. Chim. 1985, 9, 659–668. [Google Scholar]
- 35. Oloba-Whenu O. A., Albright T. A., Soubra-Ghaoui C., Beilstein J. Org. Chem. 2016, 12, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cossairt B. M., Cummins C. C., Head A. R., Lichtenberger D. L., Berger R. J. F., Hayes S. A., Mitzel N. W., Wu G., J. Am. Chem. Soc. 2010, 132, 8459–8465. [DOI] [PubMed] [Google Scholar]
- 37. Fenske D., Merzweiler K., Angew. Chem. Int. Ed. Engl. 1984, 23, 635–637; [Google Scholar]; Angew. Chem. 1984, 96, 600–602. [Google Scholar]
- 38.Deposition Numbers 2115737, 2115738, and 2115739 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 39. Zarzycki B., Zell T., Schmidt D., Radius U., Eur. J. Inorg. Chem. 2013, 2051–2058. [Google Scholar]
- 40. Barrios-Francisco R., Benítez-Páez T., Flores-Alamo M., Arévalo A., García J. J., Chem. Asian J. 2011, 6, 842–849. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Baudler M., Glinka K., Chem. Rev. 1993, 93, 1623–1667; [Google Scholar]
- 41b. Fritz G., Scheer P., Chem. Rev. 2000, 100, 3341–3401. [DOI] [PubMed] [Google Scholar]
- 42. Turbervill R. S. P., Goicoechea J. M., Chem. Rev. 2014, 114, 10807–10828. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Fritz G., Holderich W., Naturwissenschaften 1975, 62, 573–575. [Google Scholar]
- 44.
- 44a. Charles S., Bott S. G., Reinghold A. L., Eichhorn B. W., J. Am. Chem. Soc. 1994, 116, 8077–8086; [Google Scholar]
- 44b. Charles S., Fettinger J. C., Bott S. G., Eichhorn B. W., J. Am. Chem. Soc. 1996, 118, 4713–4714; [Google Scholar]
- 44c. Kesanli B., Charles S., Lam Y.-F., Bott S. G., Fettinger J., Eichhorn B., J. Am. Chem. Soc. 2000, 122, 11101–11107; [Google Scholar]
- 44d. Knapp C. M., Large J. S., Rees N. H., Goicoechea J. M., Chem. Commun. 2011, 47, 4111–4113. [DOI] [PubMed] [Google Scholar]
- 45. Hönle W., von Schnering H. G., Z. Anorg. Allg. Chem. 1978, 440, 171–182. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information