

Abstract
Although the rates of disease gene discovery have steadily increased with the expanding use of genome and exome sequencing by clinical and research laboratories, only ~16% of genes in the genome have confirmed disease associations. Here we describe our clinical laboratory's experience utilizing GeneMatcher, an online portal designed to promote disease gene discovery and data sharing. Since 2016, we submitted 246 candidates from 243 unique genes to GeneMatcher, of which 111 (45%) are now clinically characterized. Submissions meeting our candidate gene‐reporting criteria based on a scoring system using patient and molecular‐weighted evidence were significantly more likely to be characterized as of October 2021 versus genes that did not meet our clinical‐reporting criteria (p = 0.025). We reported relevant findings related to these newly characterized gene–disease associations in 477 probands. In 218 (46%) instances, we issued reclassifications after an initial negative or candidate gene (uncertain) report. We coauthored 104 publications delineating gene–disease relationships, including descriptions of new associations (60%), additional supportive evidence (13%), subsequent descriptive cohorts (23%), and phenotypic expansions (4%). Clinical laboratories are pivotal for disease gene discovery efforts and can screen phenotypes based on genotype matches, contact clinicians of relevant cases, and issue proactive reclassification reports.
Keywords: clinical validity, data sharing, disease gene discovery, exome sequencing, gene–disease validity, GeneMatcher, Matchmaker Exchange
Selective sharing of candidates with strong evidence aids efficient gene characterization.
1. INTRODUCTION
The past decade has been a time of rapid disease gene discovery, driven by the rise in popularity of next‐generation sequencing (NGS) technologies and the increasing use of web‐based collaborative data‐sharing initiatives, such as the Matchmaker Exchange (https://www.matchmakerexchange.org/; Bamshad et al., 2019; Boycott et al., 2019; Chong et al., 2015; Sobreira et al., 2015, 2017). Matchmaker Exchange enhances data sharing and characterization of novel gene–disease associations by connecting multiple genomic and phenotypic databases through a shared application programming interface (Sobreira et al., 2017). One component of Matchmaker Exchange is GeneMatcher (http://www.genematcher.org), which connects scientists and clinicians to share standardized data on candidate genes and the associated phenotypes of individuals with presumed Mendelian disorders (Sobreira et al., 2015). By sharing candidate gene information through GeneMatcher, researchers can assemble a critical mass of probands to support the characterization of new gene–disease associations, thus increasing the overall diagnostic yield of testing and the potential to identify new therapeutic targets (Bamshad et al., 2019; Sobreira et al., 2015). Ultimately, disease gene discovery impacts patients by ending the notorious “diagnostic odyssey,” providing more tailored clinical care and informing reproductive risks. However, most of the data generated by clinical laboratories around the globe are not adequately available for data sharing and matchmaking due to a lack of platforms to systematically upload raw sequencing data and data‐sharing policies (Boycott et al., 2019).
A spike in disease gene discovery rates occurred as the adoption of NGS technologies became more prominent (Bamshad et al., 2019; Boycott et al., 2017). However, the rates of publications reporting these discoveries are not keeping up (Bamshad et al., 2019). The elusive gene–disease relationships that remain to be described may be due to several factors, including complex inheritance or the difficulty in ascertaining probands with extremely rare disorders. Publications may be delayed until the collection of a large enough cohort with robust clinical data curation and paired functional studies. This can hinder the characterization of gene–disease associations for extremely rare disorders that are less conducive to cohort studies. Moving forward, participation in data‐sharing initiatives is even more imperative to help identify the elusive, ultrarare diagnoses.
Therefore, clinical laboratories that offer diagnostic exome sequencing (DES) are valuable partners for disease gene discovery, especially if variants in uncharacterized genes are evaluated as part of the DES reporting workflow (Bamshad et al., 2019; Farwell Hagman et al., 2017; Retterer et al., 2016). However, the reporting criteria for uncharacterized candidate genes vary between clinical laboratories. Published reports cite 6%–24% of DES cases include a reported candidate gene (Farwell Hagman et al., 2017; Retterer et al., 2016). Our laboratory developed a standardized and validated scoring metric for evaluating gene–disease validity (GDV) (Smith et al., 2017). Each gene–disease relationship receives a GDV score and genes may have multiple curated disease associations. Genes with limited evidence for a specific disease association are considered uncharacterized and those with a GDV score of moderate or higher are considered characterized for that phenotype. Variants identified in characterized and uncharacterized gene associations may be reported, and GDV scores are continuously updated as new evidence becomes available (Farwell et al., 2015; Farwell Hagman et al., 2017). We submit variants in uncharacterized genes that meet or nearly fulfill reporting criteria to GeneMatcher. A systematic approach to which variants are the most likely to be disease‐causing in a proband before submitting to data‐sharing initiatives is critical to ensure positive outcomes to matches.
Our laboratory submits to GeneMatcher if the existing knowledge of the gene function is consistent with the proband's reported clinical presentation and there is compelling evidence for the variant pathogenicity. This process selects for high‐confidence, potentially disease‐causing variants representing the strongest candidates and is consistent with the “gene‐to‐patient” model proposed by Seaby et al. (2021), to reduce the burden of sifting through large volumes of unvetted variants (“analytical noise”). The resulting connections ideally lead to peer‐reviewed publications and sufficient evidence to confirm new gene–disease associations (Figure 1).
Figure 1.
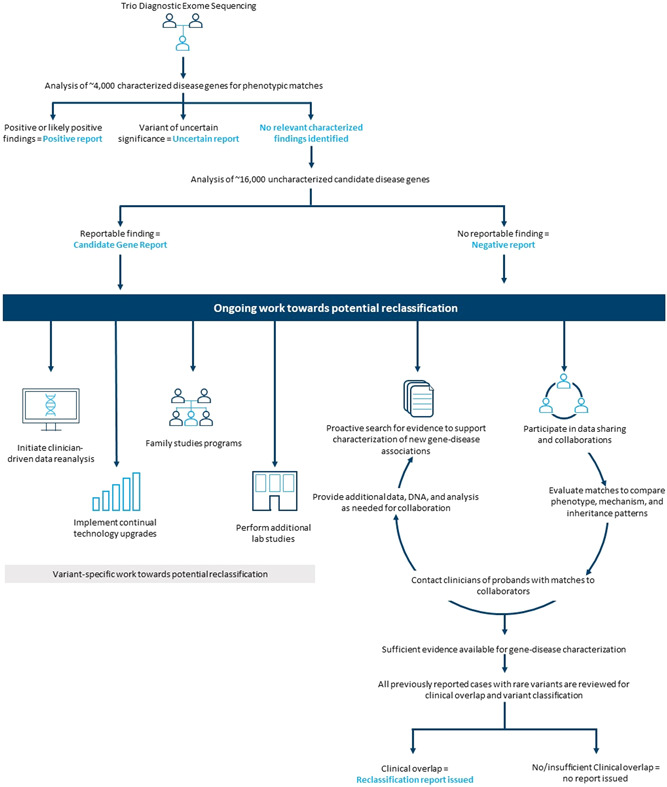
Uncharacterized gene reporting and reclassification workflow Figure 1: Up until 2020, uncharacterized gene analysis was performed for informative trios with an uncertain or negative result following characterized gene analysis. Currently, uncharacterized genes are analyzed for negative cases or for probands where the identified characterized gene finding only accounts for part their phenotype (“partial overlap”). Genes are continually evaluated based on the latest literature to determine whether the published evidence is sufficient to characterize the gene–disease association. Once characterized, all previously reported diagnostic exome sequencing (DES) cases with rare variants in a gene are assessed for clinical overlap and proactive reclassification reports are issued to appropriate cases. Our laboratory has several workflows addressing uncertain results for reclassification. New gene characterizations result in our highest rate of reclassifications. Participation in data‐sharing initiatives include GeneMatcher, ClinVar, the Undiagnosed Disease Network, and collaborations established directly with investigators. Only gene and variant information are shared on these platforms with follow‐up clinical details provided on a case‐by‐case basis. Ordering clinicians are contacted for a promising lead and connected to collaborators for additional data sharing and patient consent to participate in publications or additional studies. Other efforts for reclassification include performing reanalysis by clinician request and implementing bioinformatic pipeline upgrades and other technology upgrades, both of which are largely time dependent. Family studies and additional laboratory studies (i.e., RNA analysis and structural modeling) are variant‐dependent and, to date, not widely available for most alterations
Comprehensive phenotypic data, ideally in the form of clinical notes summarizing the salient medical history, are crucial for accurately identifying relevant variants in uncharacterized genes for probands undergoing DES (Seaby & Ennis, 2020). This is especially true when evaluating clinical overlap for candidate genes with limited or no patient reports in the literature. Robust clinical data are beneficial to identify links between a proband's features and a gene with limited evidence based on animal models and other functional data. In addition, detailed and specific clinical information is valuable when evaluating matches with other GeneMatcher users.
Here we report our laboratory's experience with GeneMatcher, how it has impacted the characterization of gene–disease associations, and the resulting publications on gene–disease associations.
2. MATERIALS AND METHODS
2.1. GDV assessment and gene characterization
Our clinical laboratory has established standardized guidelines for curating and scoring GDV that were independently validated (Smith et al., 2017). This tiered system is similar to the ClinGen model and scores evidence of gene–disease relationships as definitive, strong, moderate, limited, no reported evidence, or conflicting evidence reported (Strande et al., 2017). A team of scientists curate peer‐reviewed literature on an ongoing basis and determine the GDV score for each gene–disease relationship. Scores of “moderate” or higher are considered characterized, and those with a score of “limited” or lower are uncharacterized. Some genes have evidence to support associations with more than one phenotype, inheritance pattern, or disease mechanism. This could include genes with multiple characterized associations or a combination of characterized and uncharacterized associations (Smith et al., 2017). Gene–disease relationships classified as “limited” by GDV assessment may be included as an uncharacterized candidate for clinical‐reporting purposes. Genes with supporting evidence, but which do not meet the reporting criteria, are not included on reports but may be reported as “notable” findings and are available to clinicians by way of filtered variant lists (Farwell Hagman et al., 2017).
2.2. Exome sequencing
DES was performed as previously described (Farwell et al., 2015; Farwell Hagman et al., 2017). In brief, samples were prepared using either the SureSelect Target Enrichment System (Agilent Technologies), SeqCap EZ VCRome 2.0 (Roche NimbleGen), or the IDT xGen Exome Research Panel V1.0 (Integrated DNA Technologies). Sequencing was performed using paired‐end, 100 or 150‐cycle chemistry on the Illumina HiSeq, NovaSeq, or NextSeq (Illumina). Bioinformatics filtering removed common benign variants, intergenic and 3′/5′‐untranslated region variants, intronic variants outside ±6, and nonsplice‐related synonymous variants. Family history‐based filtering and inheritance models were applied to the data. Alterations that have clinical overlap with the patient's reported phenotype and are classified as variants of uncertain significance or higher are reported (Farwell et al., 2015).
2.3. Candidate gene analysis for diagnostic exome cases
All probands with informative trios, defined as having samples from family members representing both the maternal and paternal lineages, and negative results after characterized gene review undergo additional candidate gene analysis at our laboratory as previously described (Farwell Hagman et al., 2017). If an identified characterized gene finding only accounts for part of a patient's phenotype, uncharacterized genes can be analyzed for causes of the unaccounted‐for clinical features. Uncharacterized genes that meet reporting criteria are considered candidate genes with uncertain clinical significance (Richards et al., 2015). Alterations in uncharacterized genes identified during analysis as having potential relevance undergo dual scientist and director review to determine if there is adequate evidence to report as a candidate finding (Farwell Hagman et al., 2017). These reporting criteria are based on a scoring system using weighted evidence including previous patients reports, animal models, microdeletion/duplication syndromes encompassing the gene of interest, and functional evidence including expression profiles and interaction with genes associated with similar phenotypes (Farwell Hagman et al., 2017).
2.4. Data sharing to GeneMatcher
Our laboratory began submitting to GeneMatcher in March 2016. Alterations reported as a candidate gene finding or classified as “insufficient evidence” but with substantiation for disease relevance are entered on an ongoing basis. Separate entries are submitted for cases with novel mechanisms or inheritance models associated with a gene.
2.5. Follow‐up to matches
Matches go to a designated inbox (genematcher@ambrygen.com) monitored by research staff. Upon receiving a match notification, all cases with rare variants and consistent genotypes in a matched gene of interest are reviewed for clinical overlap. High‐level phenotypic data and variant information are shared in a Health Insurance Portability and Accountability Act (HIPAA)‐compliant manner with GeneMatcher collaborators to assess the integrity of the match. Ordering clinicians for matched cases are contacted by email with a general description of the collaboration, the investigator's contact information, and the gene of interest. A second follow‐up message to the ordering provider is sent by encrypted email and contains the proband's identifiable information and variant of interest. Other relevant information may also be included, for example, if an alteration did not meet our reporting criteria at the time of the initial report, and therefore there is no documentation of the alteration or if it was not Sanger confirmed. We do not share clinician or patient contact information with the collaborating investigator without consent. Once a connection is established, the clinician works with the family to obtain appropriate consent for inclusion in any resulting work. We continue as a partner to share additional data as requested (Sanger or integrative genomics viewer imaging, testing methodology details), DNA (if available), and additional family testing for variant cosegregation. For some cases, our laboratory issues research‐grade single‐site analysis reports to document alterations not previously reported to aid in the clinician's discussion with the family.
2.6. Data analysis
To assess the overall impact of our laboratory's involvement with GeneMatcher, we analyzed the outcomes of our submissions to date. For purposes of this analysis, duplicate entries for a candidate gene–disease relationship with the same inheritance, zygosity, and alteration type were consolidated and counted as a single entry. We retrospectively reviewed all our DES cases from 2011 through January 2021 and tabulated the number of probands with reported alterations in genes with a GeneMatcher submission. We only counted cases associated with the phenotype related to the GeneMatcher entry for genes with more than one disease association. We assessed the classification of the gene at the time of the initial report (uncharacterized or characterized), the current GDV score (uncharacterized, moderate, strong, or definitive), and the number of probands who received a reclassification report due to gene characterization. We used Fisher's exact test for statistical analysis.
To quantify our laboratory's contribution to disease gene discovery efforts, we queried PubMed (https://pubmed.ncbi.nlm.nih.gov/) for peer‐reviewed publications focused on gene characterization efforts, which listed authors affiliated with Ambry Genetics. These articles were reviewed for content and categorized as (1) description of new gene–disease association, (2) follow‐up case report or supportive evidence (i.e., functional or animal model evidence), (3) follow‐up descriptive cohort, and (4) follow‐up expansion of the phenotype. Publications that included a broadening and further defining of the phenotypic spectrum but not a major expansion in the phenotype were counted as a follow‐up descriptive cohort. We cross‐referenced this published gene list with our GeneMatcher submissions.
3. RESULTS
In total, we submitted 246 candidate entries representing 243 unique genes to GeneMatcher from our laboratory between March 2016 and September 2021 (Table S1). Four gene entries (EIF2C, ITGA11, LRFN2, and UNC45B) were subsequently marked as “suspended” because they no longer met candidate gene criteria per our laboratory's GDV scoring metric. All four of these genes were in cases reported before the establishment of our current candidate gene criteria (Farwell Hagman et al., 2017). Upon reanalysis of these cases, the evidence for the variant and gene's relevance to the case was determined to be insufficient and the entries were suspended. By practice, we remove cases from GeneMatcher if the initial evidence used to score it is no longer valid. However, cases are not removed solely, because a match was not made. Of note, one of these suspended genes, UNC45B, was later characterized for an autosomal dominant congenital myopathy in December 2020; however, we had originally submitted it as an autosomal recessive cause of Joubert syndrome with skeletal and ocular anomalies. Three genes (ANK3, RYR3, and SPTBN4) each had two entries representing distinct phenotypes of interest including differences in inheritance models or suspected mechanism of disease.
As of October 2021, 45% of entries have been characterized (Table 1). Most candidate gene entries were for autosomal dominant de novo occurrences; however, there are no significant differences for characterization based on zygosity, inheritance pattern, or alterations type. Of the 246 entries, 153 (62%) had at least one case with a reported uncharacterized gene finding, meaning the gene, variant, proband combination met candidate gene‐reporting criteria described in Farwell Hagman et al. (2017). Submissions that had at least one case meeting our candidate criteria were significantly more likely to be characterized (78/153; 51%) as of October 2021 compared with genes with no candidates meeting reporting criteria (33/93; 35%; p = 0.025). There were 32 genes with uncharacterized reports issued to multiple probands (range 2–4). These gene candidates were significantly more likely to be characterized compared with genes with only one candidate report (p = 0.003) and no candidate reports (p < 0.001).
Table 1.
Characteristics of GeneMatcher entries
| Entries (n = 246) | Entries (%) | Characterized (n = 111) | Characterized (%) | |
|---|---|---|---|---|
| Inheritance | ||||
| Autosomal dominant—de novo | 140 | 57% | 65 | 46% |
| Autosomal recessive | 83 | 34% | 37 | 45% |
| Autosomal dominant—inherited mutation | 10 | 4% | 3 | 30% |
| X‐linked | 10 | 4% | 5 | 50% |
| Autosomal dominant—unknown inheritance | 3 | 1% | 1 | 33% |
| Zygosity | ||||
| Heterozygous | 153 | 62% | 69 | 45% |
| Compound heterozygous | 44 | 18% | 20 | 45% |
| Homozygous | 40 | 16% | 18 | 45% |
| Hemizygous | 9 | 4% | 4 | 44% |
| Alteration type | ||||
| Missense | 192 | 58% | 82 | 43% |
| Predicted LOF | 103 | 31% | 52 | 50% |
| Predicted abnormal splicing | 29 | 9% | 12 | 41% |
| Nonframeshift deletion/duplication | 7 | 2% | 3 | 43% |
| Candidate reporting status | ||||
| Met candidate reporting criteria (all cases) | 153 | 62% | 78 | 51%* |
| Met candidate reporting criteria (2+ reports) | 32 | 13% | 24 | 75% ** |
| Did not meet candidate reporting criteria | 93 | 38% | 33 | 35% |
| Current clinical validity score | ||||
| Characterized | 111 | 45% | ||
| Uncharacterized | 135 | 55% | ||
Note: Inheritance, zygosity, and alteration types for GeneMatcher entries are listed. Entries with homozygous or compound heterozygous alterations reported were counted for each alteration type to accurately record the percentage of each alteration type. There were no statistical differences within the inheritance, zygosity, or alteration type categories. Entries that met our candidate gene‐reporting criteria for one (p = 0.025) or multiple probands (p < 0.001) were significantly more likely to be characterized as of October 2021 compared with entries that we did not report as candidate findings. Genes with multiple candidate reports were also significantly more likely to be characterized versus genes with only one candidate report issued (p = 0.003).
p = 0.025
p < 0.001.
In total, 196 probands received a candidate gene report involving one of the genes entered in GeneMatcher. Subsequently, 477 probands received a characterized report, which accounts for 12% (477/4012) of all characterized gene reports from our laboratory (data not shown). Of these 477 cases, 218 (46%) were reclassifications of an original result (Figure 2). Reclassifications include upgrades from candidate gene results (uncertain) or the addition of the finding to a previous report. Seventy‐five genes had more than one reported characterized case (range of 2–31 patients).
Figure 2.
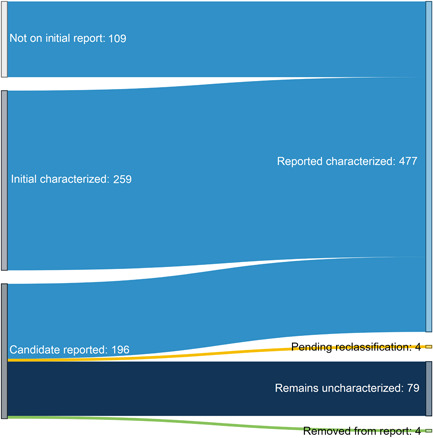
Overall impact of gene characterization following GeneMatcher entry Figure 2: 477 characterized gene reports were issued as of October 2021 for gene–disease associations entered in GeneMatcher. In total, 218 (45%) of these were reclassifications proactively issued by the laboratory. One hundred and nine of these reports were upgraded reclassifications from candidate gene reports and 109 were reclassifications issued to individuals who did not initially meet our candidate gene reporting criteria. After establishing a gene–disease association, 259 additional probands received a relevant finding in one of these genes, illustrating the impact disease gene discoveries have on the overall diagnostic exome sequencing (DES) diagnostic rate. For 79 cases for which we issued a candidate gene report, the gene–disease association remains uncharacterized. In four cases, further review of a reported candidate gene no longer met our criteria. These findings were removed from the proband's report and reclassifications were issued. In addition, these matches were marked as “suspended” in GeneMatcher. Currently, we have four cases pending review for reclassification based on recent literature
Our laboratory coauthored 104 peer‐reviewed papers describing gene–disease associations for 105 unique genes between 2013 and 2021 (Table S2). Thirty‐nine percent (41/104) of these publications were for genes we submitted to GeneMatcher. Other publications were the result of collaborations made through other sources including with ordering clinicians, investigators recruiting through ClinVar, and researchers who contacted our laboratory directly for recruitment purposes. Most publications were the initial description of a gene–disease association (60%). These studies also include follow‐up publications that further defined rare Mendelian disorders through additional case reports, subsequent descriptive cohorts, and phenotypic expansions (Figure 3).
Figure 3.
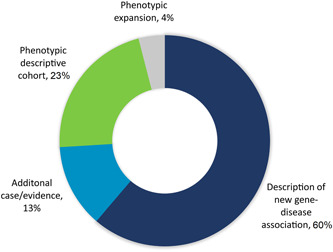
Coauthored peer‐reviewed publications Figure 3: Since 2013, we have co‐authored 104 peer‐reviewed papers describing gene–disease associations. These included descriptions of 105 unique genes. Four genes (BPTF, FDXR, IQSEC. 2, and SON) were the topic of more than one study. A comprehensive list of these publications can be found in Table S2
4. DISCUSSION
Patient identification is a critical but limiting step in the disease gene discovery process for rare disorders. Clinical laboratories can aid these efforts by screening large testing cohorts for relevant phenotypes based on genotype matches, contacting clinicians of appropriate cases, and issuing proactive reclassification reports. Here we show that selective submission of cases with considerable evidence for a new disease association creates meaningful contributions to disease gene discoveries.
Over the last 5 years, our laboratory has entered 246 candidate entries into GeneMatcher of which 45% are now characterized. Entries that met our candidate gene reporting criteria were significantly more likely to be characterized. This was especially true for genes with multiple candidate reports issued, which highlights that more prevalent disorders are easier to ascertain and therefore characterized. Although de novo and biallelic alterations represent a large percentage of our GeneMatcher entries (57% and 34%, respectively), there was no significant difference for the likelihood to be characterized based on inheritance, zygosity, or alteration type. This demonstrates how our process of evaluating and reporting existing evidence for a suspected gene–disease association is a strong predictor regardless of the mechanism. We maximize positive match outcomes by limiting submissions to those with substantial evidence. Follow‐up communications to screen matches and recontact patients are laborious. Therefore, contributors should integrate standardized methods to vet candidates before submitting to GeneMatcher to ensure the highest‐quality data are available and superfluous communications can be avoided.
Identifying these strong candidates is contingent on having access to quality clinical data submitted at the time of testing. Comprehensive clinic notes that summarize the salient points of a proband's phenotype should be submitted with all DES testing. These records are helpful not only for accurate variant assessment but also for potential gene characterization work. Robust clinical data can help identify links between a proband's features and a gene with limited published evidence. In our laboratory's experience with reviewing cases, those with more specific and complete clinical information are more likely to result in positive matches compared to probands with only vague phenotypic descriptions. In some instances, we amass sufficient clinical data and evidence to characterize a gene–disease association based on internal data. For example, we identified six internal probands with de novo rare ZNF292 alterations and similar neurodevelopmental phenotypes more than one year before the manuscript describing the association was published (Mirzaa et al., 2020).
Once a match is made based on our phenotypic review, we connect collaborators to the clinicians to share more in‐depth medical histories. Detailed and sizable case series are often needed to garnish enough evidence to meet the criteria for gene–disease characterization. For example, CHD3 was reported as a gene of interest in patients with neurodevelopmental disorders in several large cohort studies (Deciphering Development Disorders Study, 2017; Iossifov et al., 2014; O'Roak et al., 2014), but did not meet our criteria for characterization until a case series with detailed phenotypic data years after the first published report (Snijders Blok et al., 2018). Large cohort studies typically take longer to publish, delaying the necessary evidence for characterization to be available. This delay is more profound for gene–disease associations in extremely rare conditions that are less conducive to cohort studies and disorders with clinical heterogeneity, which require large cohorts to identify the phenotypic commonalities.
Significantly, these delays can stall delivery of diagnostic findings to patients and providers. The characterization of new gene–disease relationships have a cascading effect with the potential for multiple patients to receive a reclassified diagnostic finding. Here we found genes with GeneMatcher entries ultimately resulted in 477 probands receiving a characterized reportable finding. This accounts for a substantial portion (12%) of our total DES cohort with reported characterized findings. Importantly, nearly half of the probands with reported variants in these genes received a reclassification report. Laboratories should have processes to curate and score new GDV evidence in real time and retrospectively evaluate cases for proactive reclassification.
In addition to issuing reclassification following a new gene–disease association, clinical laboratories can serve as a recruitment hub to quickly identify additional probands for follow‐up reports. Additional patients can further power studies to clarify genotype–phenotype correlations and phenotypic spectrums. Publications describing larger cohorts often follow an initial report of a new gene–disease relationship. For example, we coauthored Peng et al. (2017), which described the association between biallelic FDXR alterations and auditory neuropathy and optic atrophy. The following year, we collaborated with Slone et al. (2018) on a follow‐up study to include additional patient reports and functional evidence. We collaborated on 25 subsequent cohort studies and 14 publications describing a single case report or other data in support of a newly described association. Five of our publications describe phenotypic expansions to include major features not previously reported (citations listed in Table S2).
There are several other ways clinical laboratories can further assist the research process. In cases where a candidate gene may not have met reporting criteria, we can provide reports documenting the presence of an alteration, but not commenting on the clinical relevance until sufficient information is amassed and reported that meets our characterization status. Clinicians request these reports to facilitate discussion with families about an alteration that was not originally included on a DES report and the option to enroll in a study further investigating a disease association. We also share remaining banked DNA (if available and with the appropriate permissions) with researchers and provide cascade testing to other family members for purposes of risk prediction, recurrence risk estimates, and variant segregation within a family. In addition, involving clinical laboratories in collaboration shortens the time from publication to reclassification report issuance. We previously found that clinically relevant reclassification results were issued faster for publications on which we were collaborators compared with genes for which we were not included in the publication (presented previously at Towne et al., 2019). The involvement of clinical laboratories in data‐sharing platforms such as the Matchmaker Exchange expedites the delivery of diagnostic findings to patients.
4.1. Next steps
Most of our GeneMatcher entries (91%) were for autosomal dominant de novo occurrences or autosomal recessive reports. Further work is needed to identify disease genes with lower penetrance or complex inheritance patterns and will require extensive involvement in data‐sharing initiatives. As DES use extends into new clinical settings, there is potential to discover disease genes associated with additional phenotypes. For example, there is the opportunity to identify severe disorders that are typically lethal in the prenatal setting. Ongoing participation in GeneMatcher and other Matchmaker Exchange platforms will heighten the diagnostic and disease gene discovery power of DES.
5. CONCLUSION
Clinical laboratories are valuable partners for research initiatives, especially those involving disease gene discovery efforts. We continually curate evidence for gene–disease associations by incorporating standardized GDV scoring and uncharacterized gene analysis into our laboratory's DES workflow. By prioritizing the cases with substantial evidence through GeneMatcher, we streamline the matching process and optimize outcomes. In turn, this has a meaningful impact on DES diagnostic rates and allows for efficient reclassification of variants for previously tested individuals.
CONFLICT OF INTERESTS
All authors are salaried employees of Ambry Genetics.
ETHICS STATEMENT
All HIPAA‐covered patient identifiers were removed. Western Institutional Review Board determined the study to be exempt from the Office for Human Research Protections Regulations for the Protection of Human Subjects (45 CFR 46) under Category 4. Retrospective data analysis of deidentified data exempted the study from the requirement to receive consent from patients.
Supporting information
Supporting Information.
Supporting Information.
ACKNOWLEDGMENTS
We are thankful for all the families for their participation, and for the clinicians for sharing the clinical histories and playing a critical role in recruitment, and researchers for their ongoing collaboration. We thank Sha Tang, Zoe Powis, and Taylor Cain for their work with GeneMatcher entry evaluations and submissions. No grant funding was used for this study.
Towne, M. C. , Rossi, M. , Wayburn, B. , Huang, J. M. , Radtke, K. , Alcaraz, W. , Farwell Hagman, K. D. , & Shinde, D. N. (2022). Diagnostic testing laboratories are valuable partners for disease gene discovery: 5‐year experience with GeneMatcher. Human Mutation, 43, 772–781. 10.1002/humu.24342
DATA AVAILABILITY SATEMENT
Ambry Genetic supports open data sharing. Data supporting the work presented here can be found at www.genematcher.org or in the supporting files attached. Further, classified alterations identified during diagnostic testing are deposited into ClinVar.
REFERENCES
- Bamshad, M. J. , Nickerson, D. A. , & Chong, J. X. (2019). Mendelian gene discovery: fast and furious with no end in sight. American Journal of Human Genetics, 105, 448–455. 10.1016/j.ajhg.2019.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Hartley, T. , Biesecker, L. G. , Gibbs, R. A. , Innes, A. M. , Riess, O. , Belmont, J. , Dunwoodie, S. L. , Jojic, N. , Lassmann, T. , Mackay, D. , Temple, I. K. , Visel, A. , & Baynam, G. (2019). A diagnosis for all rare genetic diseases: The horizon and the next frontiers. Cell, 177, 32–37. 10.1016/j.cell.2019.02.040 [DOI] [PubMed] [Google Scholar]
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , Brookes, A. J. , Brudno, M. , Carracedo, A. , den Dunnen, J. T. , Dyke, S. , Estivill, X. , Goldblatt, J. , Gonthier, C. , Groft, S. C. , Gut, I. , Hamosh, A. , Hieter, P. , Höhn, S. , … Lochmüller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. American Journal of Human Genetics, 100(5), 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. X. , Buckingham, K. J. , Jhangiani, S. N. , Boehm, C. , Sobreira, N. , Smith, J. D. , Harrell, T. M. , McMillin, M. J. , Wiszniewski, W. , Gambin, T. , Coban Akdemir, Z. H. , Doheny, K. , Scott, A. F. , Avramopoulos, D. , Chakravarti, A. , Hoover‐Fong, J. , Mathews, D. , Witmer, P. D. , Ling, H. , … Bamshad, M. J. (2015). The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. American Journal of Human Genetics, 97, 199–215. 10.1016/j.ajhg.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study . (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell Hagman, K. D. , Shinde, D. N. , Mroske, C. , Smith, E. , Radtke, K. , Shahmirzadi, L. , El‐Khechen, D. , Powis, Z. , Chao, E. C. , Alcaraz, W. A. , Helbig, K. L. , Sajan, S. A. , Rossi, M. , Lu, H. M. , Huether, R. , Li, S. , Wu, S. , Nuñes, M. E. , & Tang, S. (2017). Candidate‐gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19, 224–235. 10.1038/gim.2016.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell, K. D. , Shahmirzadi, L. , El‐Khechen, D. , Powis, Z. , Chao, E. C. , Tippin Davis, B. , Baxter, R. M. , Zeng, W. , Mroske, C. , Parra, M. C. , Gandomi, S. K. , Lu, I. , Li, X. , Lu, H. , Lu, H. M. , Salvador, D. , Ruble, D. , Lao, M. , Fischbach, S. , … Tang, S. (2015). Enhanced utility of family‐centered diagnostic exome sequencing with inheritance model‐based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17, 578–586. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- Iossifov, I. , O'Roak, B. J. , Sanders, S. J. , Ronemus, M. , Krumm, N. , Levy, D. , Stessman, H. A. , Witherspoon, K. T. , Vives, L. , Patterson, K. E. , Smith, J. D. , Paeper, B. , Nickerson, D. A. , Dea, J. , Dong, S. , Gonzalez, L. E. , Mandell, J. D. , Mane, S. M. , Murtha, M. T. , … Wigler, M. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa, G. M. , Chong, J. X. , Piton, A. , Popp, B. , Foss, K. , Guo, H. , Harripaul, R. , Xia, K. , Scheck, J. , Aldinger, K. A. , Sajan, S. A. , Tang, S. , Bonneau, D. , Beck, A. , White, J. , Mahida, S. , Harris, J. , Smith‐Hicks, C. , Hoyer, J. , … Bamshad, M. J. (2020). De novo and inherited variants in ZNF292 underlie a neurodevelopmental disorder with features of autism spectrum disorder. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 22, 538–546. 10.1038/s41436-019-0693-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak, B. J. , Stessman, H. A. , Boyle, E. A. , Witherspoon, K. T. , Martin, B. , Lee, C. , Vives, L. , Baker, C. , Hiatt, J. B. , Nickerson, D. A. , Bernier, R. , Shendure, J. , & Eichler, E. E. (2014). Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nature Communications, 5, 5595. 10.1038/ncomms6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y. , Shinde, D. N. , Valencia, C. A. , Mo, J. S. , Rosenfeld, J. , Truitt Cho, M. , Chamberlin, A. , Li, Z. , Liu, J. , Gui, B. , Brockhage, R. , Basinger, A. , Alvarez‐Leon, B. , Heydemann, P. , Magoulas, P. L. , Lewis, A. M. , Scaglia, F. , Gril, S. , Chong, S. C. , … Huang, T. (2017). Biallelic mutations in the ferredoxin reductase gene cause novel mitochondriopathy with optic atrophy. Human Molecular Genetics, 26, 4937–4950. 10.1093/hmg/ddx377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer, K. , Juusola, J. , Cho, M. T. , Vitazka, P. , Millan, F. , Gibellini, F. , Vertino‐Bell, A. , Smaoui, N. , Neidich, J. , Monaghan, K. G. , McKnight, D. , Bai, R. , Suchy, S. , Friedman, B. , Tahiliani, J. , Pineda‐Alvarez, D. , Richard, G. , Brandt, T. , Haverfield, E. , … Bale, S. (2016). Clinical application of whole‐exome sequencing across clinical indications. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 18, 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. , ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaby, E. G. , & Ennis, S. (2020). Challenges in the diagnosis and discovery of rare genetic disorders using contemporary sequencing technologies. Briefings in Functional Genomics, 19, 243–258. 10.1093/bfgp/elaa009 [DOI] [PubMed] [Google Scholar]
- Seaby, E. G. , Rehm, H. L. , & O'Donnell‐Luria, A. (2021). Strategies to uplift novel Mendelian Gene Discovery for Improved Clinical outcomes. Frontiers in Genetics, 12, 674295. 10.3389/fgene.2021.674295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slone, J. , Peng, Y. , Chamberlin, A. , Harris, B. , Kaylor, J. , McDonald, M. T. , Lemmon, M. , El‐Dairi, M. A. , Tchapyjnikov, D. , Gonzalez‐Krellwitz, L. A. , Sellars, E. A. , McConkie‐Rosell, A. , Reinholdt, L. G. , & Huang, T. (2018). Biallelic mutations in FDXR cause neurodegeneration associated with inflammation. Journal of Human Genetics, 63, 1211–1222. 10.1038/s10038-018-0515-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E. D. , Radtke, K. , Rossi, M. , Shinde, D. N. , Darabi, S. , El‐Khechen, D. , Powis, Z. , Helbig, K. , Waller, K. , Grange, D. K. , Tang, S. , & Hagman, K. D. F. (2017). Classification of genes: Standardized clinical validity assessment of gene‐disease associations aids diagnostic exome analysis and reclassifications. Human Mutation, 38, 600–608. 10.1002/humu.23183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok, L. , Rousseau, J. , Twist, J. , Ehresmann, S. , Takaku, M. , Venselaar, H. , Rodan, L. H. , Nowak, C. B. , Douglas, J. , Swoboda, K. J. , Steeves, M. A. , Sahai, I. , Stumpel, C. , Stegmann, A. , Wheeler, P. , Willing, M. , Fiala, E. , Kochhar, A. , Gibson, W. T. , … Campeau, P. M. (2018). CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nature Communications, 9, 4619. 10.1038/s41467-018-06014-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N. , Arachchi, H. , Buske, O. J. , Chong, J. X. , Hutton, B. , Foreman, J. , Schiettecatte, F. , Groza, T. , Jacobsen, J. , Haendel, M. A. , Boycott, K. M. , Hamosh, A. , & Rehm, H. L. , Matchmaker Exchange Consortium . (2017). Matchmaker exchange. Current Protocols in Human Genetics, 95, 9.31.1–9.31.15. 10.1002/cphg.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Boehm, C. , Valle, D. , & Hamosh, A. (2015). New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web‐based tool for linking investigators with an interest in the same gene. Human Mutation, 36, 425–431. 10.1002/humu.22769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strande, N. T. , Riggs, E. R. , Buchanan, A. H. , Ceyhan‐Birsoy, O. , DiStefano, M. , Dwight, S. S. , Goldstein, J. , Ghosh, R. , Seifert, B. A. , Sneddon, T. P. , Wright, M. W. , Milko, L. V. , Cherry, J. M. , Giovanni, M. A. , Murray, M. F. , O'Daniel, J. M. , Ramos, E. M. , Santani, A. B. , Scott, A. F. , … Berg, J. S. (2017). Evaluating the clinical validity of gene‐disease associations: An evidence‐based framework developed by the clinical genome resource. American Journal of Human Genetics, 100, 895–906. 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne, M.C. , Schoenfeld B., Blanco, K. , Radtke, K. , Powis Z., Tang S., Farwell Hagman K.D., & Shinde D. N. (2019, April 2‐6). “N of One” is the loneliest number: How a lack of corroborating case reports is hindering the classification of genes [Poster Presentation]. American College of Medical Genetics and Genomics Annual Clinical Genetics Meeting, Seattle, WA, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.
Supporting Information.
Data Availability Statement
Ambry Genetic supports open data sharing. Data supporting the work presented here can be found at www.genematcher.org or in the supporting files attached. Further, classified alterations identified during diagnostic testing are deposited into ClinVar.