

Abstract
Pancreatic ductal adenocarcinoma (PDAC) represents one of the most lethal malignancies with very high mortality and short survival time. About 5–10% of the PDAC patients have a familial predisposition to the disease designated as familial pancreatic cancer (FPC), suggesting genetic modulation of FPC pathogenesis. It is estimated that currently identified sequence variants account for less than 20% of the genetic basis of FPC leaving the majority of the genetic architecture unclarified. We performed whole genome sequencing (WGS) analysis on benign formalin‐fixed paraffin‐embedded (FFPE) tissues from 35 FPC patients focusing on genes enriched by rare and functional sequence variants. We identified 40 genes hosting at least 2 protein truncating variants (PTVs). Significant overlaps of the 40 genes were found (p < 1 × 10–22) with cancer genes, cancer driver genes and genes found in previous studies on cancer, including ATM, POLE, BRCA2, TYR03, PABPC1 and SSC5D. The PTV genes are significantly overrepresented in biological pathways in cancer development and progression including extracellular matrix organization, signaling by RHO GTPases and RHO GTPase cycle. Association analysis using external controls detected 6 genes with p < 0.05. The WGS analysis revealed high heterogeneity in the detected rare variants among FPC patients and provides novel genes harboring potential mutational hotspots for future validation and replication.
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and fatal malignancies worldwide with an estimated 5‐year survival of just 5% (Naghavi et al., 2019; McGuigan et al., 2018). Familial pancreatic cancer (FPC) is defined as having two or more first‐degree relatives (FDRs) with PDAC without known inherited cancer syndrome, and is responsible for up to 10% of all cases of PDAC (Diaz & Lucas, 2019). Families fulfilling the FPC criteria represent up to 80% of all families with PDAC aggregation (Llach et al., 2020). We have recently estimated the heritability of FPC as high as 0.51 (Tan et al., 2021a). The estimated relatively high genetic contribution to FPC calls for efforts to find the genetic variants underlying the genetic predisposition to FPC. Studies using next‐generation sequencing (NGS) technique have detected rare sequence variations in BRCA1, BRCA2, CDKN2A, PALB2, and ATM genes to be related to FPC (Roberts et al., 2016; Zhen et al., 2015). However, those variants are only observed in about 12% of all FPC cases. The suspected germline contribution to over 80% of all FPC cases still remains unknown (Chaffee et al., 2018).
Whole genome sequencing (WGS) can be used to explore genomic alterations in cancer and help us to better understand the whole landscape of mutational signatures in the cancer genomes and to elucidate their functional or clinical implications (Nakagawa & Fujita, 2018). Using WGS analysis, Roberts et al. (2016) demonstrated that the genetic architecture of FPC is highly heterogeneous. Genetic heterogeneity refers to: (1) allelic heterogeneity, where different variants at a single gene locus cause the same or similar phenotypic expressions of a disease and (2) locus heterogeneity, where variants at different gene loci cause the same or similar phenotypes of a disease (McClellan & King, 2010). The genetic heterogeneity of FPC means that susceptibility variants could be private to certain individuals or families. The situation renders the traditional association analysis for common variants underpowered. Both allelic and locus heterogeneity impose challenges in identifying the relevant genetic variants for FPC. More high‐coverage sequencing analyses are required to uncover the genetic diversity in FPC.
We have performed a WGS on PDAC patients from 27 FPC predisposed families from a recently established national cohort—with a focus on detecting rare genetic variants for the disease. We report findings from the WGS study and compare with published results from previous studies to validate and verify the detected genetic alterations as potential hotspots of functional variations for FPC.
2. MATERIALS AND METHODS
2.1. Familial pancreatic cancer cases and external controls
From the national cohort of 27 FPC families (Tan et al., 2021a, 2021b), a total of 83 PDAC cases (38 males, 45 females) were identified. Familial predisposition for FPC was defined as presence of either: (1) Two FDRs with PDAC, with at least one of the cases debuting at age <50 year; or (2) at least three FDRs with PDAC. Available benign tissue samples (i.e. noncancerous tissue) of formalin‐fixed paraffin‐embedded (FFPE) nonpancreatic tissues were retrieved for 35 FPC patients (Table S1) from the relevant departments of pathology from all five Danish regions (including: the Capital Region of Denmark, Region Zealand, the Region of Southern Denmark, the Central Denmark Region, and the Region of Northern Denmark).
This study made use of the WGS data of the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) as a large external control group for association analysis. Being the world's largest public collection of human genetic variations, it is developed for aggregating and harmonizing both exome and genome sequencing data and serves as a popular resource for basic research and clinical variant interpretation. The version 2.1.1 dataset (GRCh37/hg19) spans 15,708 WGSs from unrelated individuals sequenced by various genetic studies. The use of gnomAD as external controls is enabled by the proxy external controls association test (ProxECAT) (Hendricks et al., 2018).
2.2. Ethics
Data and sample collection from the included individuals were conducted with the approvals from the Danish National Committee on Health Research Ethics (NVK) (project number: 1604008) and the Danish Data Protection Agency (project number: 18/54160). A written informed consent was obtained from all alive subjects involved in the study. Patient consent was waived in case of deceased status of patients, with the approval of the Danish National Committee on Health Research Ethics (NVK) (project number: 1604008) citing the Danish Scientific Ethical Committees Act §10.
2.3. Sequencing analysis
WGS was conducted using DNA extracted from FFPE benign tissue samples from each of the 35 FPC patients. From each sample, ≥100 ng of genomic DNA was sequenced using the TruSeq DNA PCR free kit (Illumina, Inc). Sequencing was performed on a NovaSeq 6000 (Illumina, Inc). Sequence reads were analyzed and aligned to the human reference genome (hg19) using Illumina DRAGEN Bio‐IT software version 3.7. Variants were annotated using VarSeq version 2.2.1 (Golden Helix, Inc.) (Tan et al., 2021b) with (i) functional consequence in RefSeq gene transcripts, (ii) zygosity, (iii) minor allele frequency (MAF) determined using publicly available variant databases (gnomAD), and (iv) presence in ClinVar.
2.4. Filtering and characterizing variants
VarSeq software (version 2.2.1, Golden Helix, Inc.) was used for annotation and variant filtering. Considering the quality of DNA extracted from preserved tissue samples, we filtered variants according to the number of reads that support each of the reported alleles, the allele depth. The total allele depth for a variant is defined as the sum of number of reads aligned at the position, which includes allele depths of the reference (REF) and alternative (ALT) alleles. Variants with total allele depth < 4 or alternative allele depth < 2 were dropped.
We further removed sequence variants belonging to (1) pseudogenes using annotations provided by EnsDb.Hsapiens.v86 package in Bioconductor (DOI: 10.18129/B9.bioc.EnsDb.Hsapiens.v86); and (2) segmental duplication (https://humanparalogy.gs.washington.edu). Multimapped reads and artefacts were also filtered out from subsequent analysis.
For the purpose of association analysis, the remaining sequence variants were classified into (1) a group of functional variants including: frameshift variants, inframe deletion, inframe insertion, initiator codon variants, splice acceptor variants, splice donor variants, stop gained variants (nonsense variants), and missense variants; and (2) a group of synonymous (i.e., low impact) variants including: splice region variants, stop retained variants, 5′ UTR premature start codon gain variants.
VarSeq (https://www.goldenhelix.com/products/VarSeq/) was used for functional prediction of nonsynonymous variants. Clinical significance (benign, likely benign, pathogenic, likely pathogenic, uncertain significance, etc.) of variants was assessed based on ClinVar submitted records as recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP), and on evaluation by local clinicians and biologists using an in‐house assessment catalog. Variants assessed as benign or likely benign were filtered out from the nonsynonymous group (i.e., functional group). Similarly, variants assessed as pathogenic or likely pathogenic were removed from the synonymous group (i.e., nonfunctional group).
Functional interpretation of single nucleotide variants (SNVs) (a single‐base substitution, insertion, or deletion) was provided by dbNSFP (database for nonsynonymous single nucleotide polymorphisms’ functional predictions), a database developed for functional prediction and annotation of all potential nonsynonymous SNVs in the human genome (Liu et al., 2016). The dbNSFP in VarSeq contains variant effect classifications from six functional prediction algorithms. Pathogenicity prediction was provided by the PHRED‐like score, a scaled score based on CADD (combined annotation‐dependent depletion) scores 1.4 (Rentzsch et al., 2019).
2.5. Defining protein‐truncating variants
Protein‐truncating variants or premature‐truncating variants (PTVs) form a type of sequence variants within a gene that creates an early stop codon, leading to a shortened or truncated protein product with serious functional consequences. Following Roberts et al. (2016), we defined PTVs using the following criteria: (i) nonsense variants, splice‐site variants (splice donor variants, splice acceptor variants), and frameshift INDELs (frameshift variants, stop‐loss variants); (ii) heterozygous in the germline; (iii) minor allele frequency (MAF) < 0.01, and (iv) present in only one individual or one family, i.e., “private.”
2.6. Hypergeometric test
We applied a hypergeometric test for overrepresentation analysis (ORA) to assess, if the overlap of identified genes with genes from a functional cluster (e.g. biological pathway, a compiled list of cancer related genes) is significantly different from being random by calculating a probability from the hypergeometric distribution. ORA has also been implemented in a web tool for biological pathway analysis, the gene‐set enrichment analysis (GSEA), to test if genes in one biological pathway are over‐represented in a list of identified genes. GSEA was performed on canonical pathways at https://www.gsea‐msigdb.org/gsea/index.jsp. The R function phyper() was used for calculating the hypergeometric probability.
2.7. Association test
Statistical testing for gene‐based association analysis was performed using the Proxy External Controls Association Test (ProxECAT) (Hendricks et al., 2018), a statistical method specifically developed for analysis of sequencing data using existing large databases like gnomAD as external controls. ProxECAT makes use of nonfunctional variants as a proxy for how well variants within a genetic region are sequenced and called within a sample. It compares the ratio between variant and proxy frequencies in cases with that in the external controls to adjust for group differences in sequencing technology, in DNA sample processing, and in read depth for creating the internal and external datasets. Adjustment for multiple testing was done by calculating the false discovery rate (FDR) (Benjamini & Hochberg, 1995). Statistical significance for association analysis is defined as FDR < 0.05.
A flow‐chart displaying the steps of the study from sampling, sequencing, filtering to data analyses is showing by Figure 1.
FIGURE 1.
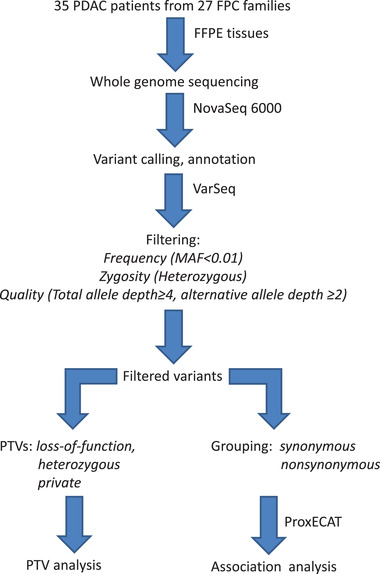
A flowchart describing the steps of WGS analysis in the study
3. RESULTS
In total, benign FFPE nonpancreatic tissue samples were available from 35 FPC patients (14 males, 21 females) for DNA extraction and sequencing analysis (Table 1, Table S1). The median age of FPC patients at diagnosis was 61.9 years (range: 33.5–86.9 years); the median age at death was 62.3 years (range: 35.4–87.2 years). The minimum and maximum survival times were 11 and 3696 days with a median of 241 days (Table 1). Two patients were censored at 2148 days (female) and 3696 days (male) respectively after diagnosis. No sex difference was found for age at death (p = 0.52), nor for time from diagnosis to death (p = 0.83) in the patient samples.
TABLE 1.
Sample description and basic statistics of 35 patients with familial pancreatic cancer
| Variables | Statistics |
|---|---|
| Sample size: | |
| Male | 14 |
| Female | 21 |
| Total | 35 |
| Year of birth: | |
| Range | 1912–1970 |
| Age at diagnosis (years): | |
| Median | 61.9 |
| Range | 33.5–86.9 |
| Age at death (years): | |
| Median | 62.3 |
| Range | 35.4–87.2 |
| Time from diagnosis to death (days): | |
| Median | 241 |
| Range | 11–3696 |
3.1. Sequencing outputs
All 35 FPC patients were sequenced on an Illumina NovaSeq 6000 platform. Sequence reads were filtered as described in the Methods section. After applying the filtering criteria, a total of 33,771 unique SNVs were detected and available for analysis. Among the 33,771 SNVs, 16,268 SNVs were detected in only once in 35 samples, accounting for nearly half of all the available SNVs.
3.2. Detection of PTVs
Following the criteria as described in Section 2, we detected a total of 865 PTVs harbored by 821 genes, including overlapping genes (see Table S2), with most of the genes hosting 1 PTV. There are 40 genes carrying more than one PTVs (Table 2), among them, five genes have three PTVs and 35 genes have two PTVs. The tables show that the detected PTVs exhibit high locus heterogeneity (multiple genes harboring PTVs) as well as high allelic heterogeneity (multiple PTVs within a host gene) (Scriver, 2001). Only one gene (RICTOR) has a variant shared by family members. The other 39 genes carry PTVs that were observed in only one patient. Annotations of the 865 PTVs are shown in Table S3. From the table, it can be seen that most of the variants within a gene were observed from different families indicating allelic heterogeneity.
TABLE 2.
A list of 40 detected genes harboring two or more protein truncating variants (PTVs) found in benign tissue samples from 35 patients with familial pancreatic cancer
| Genes | Chromosome | Number of PTVs | Gene Bank accession number |
|---|---|---|---|
| COL4A2,COL4A2‐AS2 | 13 | 3 | NM_001846.3,NM_001267044.1 |
| CRIPAK | 4 | 3 | NM_175918.3 |
| MYO15B | 17 | 3 | NM_001309242.1 |
| SYNE2 | 14 | 3 | NM_182914.2 |
| TYRO3 | 15 | 3 | NM_006293.3 |
| ABCA7 | 19 | 2 | NM_019112.3 |
| ABHD16B | 20 | 2 | NM_080622.3 |
| AGBL3 | 7 | 2 | NM_178563.3 |
| APLF | 2 | 2 | NM_173545.2 |
| ASGR1 | 17 | 2 | NM_001671.4 |
| ATM | 11 | 2 | NM_000051.3 |
| BRCA2 | 13 | 2 | NM_000059.3 |
| CNKSR3 | 6 | 2 | NM_173515.2 |
| CNOT2 | 12 | 2 | NM_014515.5 |
| EPPK1 | 8 | 2 | NM_031308.3 |
| FBRSL1 | 12 | 2 | NM_001142641.1 |
| GPSM3 | 6 | 2 | NM_022107.2 |
| GSX1 | 13 | 2 | NM_145657.2 |
| HABP2 | 10 | 2 | NM_004132.4 |
| HOOK3 | 8 | 2 | NM_032410.3 |
| KIAA0100 | 17 | 2 | NM_014680.3 |
| LOC101928841 | 13 | 2 | NM_001304433.1 |
| NEK8 | 17 | 2 | NM_178170.2 |
| OXER1 | 2 | 2 | NM_148962.4 |
| PABPC1 | 8 | 2 | NM_002568.3 |
| PDE3B | 11 | 2 | NM_000922.3 |
| PIH1D1 | 19 | 2 | NM_017916.2 |
| POLE | 12 | 2 | NM_006231.3 |
| PRKCD | 3 | 2 | NM_006254.3 |
| RICTOR | 5 | 2 | NM_001285439.1 |
| RP1L1 | 8 | 2 | NM_178857.5 |
| RPS6KA4 | 11 | 2 | NM_003942.2 |
| SENP7 | 3 | 2 | NM_020654.4 |
| SPG11 | 15 | 2 | NM_025137.3 |
| SSC5D | 19 | 2 | NM_001144950.1 |
| ST8SIA4 | 5 | 2 | NM_005668.5 |
| STAB1 | 3 | 2 | NM_015136.2 |
| TTC27 | 2 | 2 | NM_017735.4 |
| TTC6 | 14 | 2 | NM_001310135.1 |
| ZNF628 | 19 | 2 | NM_033113.2 |
We additionally observed genetic diversity at individual level or intraindividual genetic heterogeneity in our FPC patients. As shown in Table S3, the following genes display intraindividual allelic heterogeneity (multiple PTVs in the same gene in the same patient): CNOT2 (patient 24, two PTVs), EPPK1 (patient 8, two PTVs), KIAA0100 (patient 30, two PTVs), LOC101928841 (patient 2, two PTVs), MYO15B (patient 11, two PTVs), and TTC6 (patient 24, two PTVs). Meanwhile, we also observed intraindividual locus heterogeneity (multiple PTV genes in the same patient) exemplified by patient 24 with 126 PTVs in 124 genes and by patient 17 with 125 PTVs in 125 genes.
The 40 genes in Table 2 carry 85 PTVs among which 36 are frameshift variants, 29 are stop‐gain variants (i.e., nonsense variants), 10 splice acceptor variants, and 10 splice donor variants. The median PHRED score of the 85 PTVs is 35, similar to the median score for the rest of the 780 PTVs in Supplementary Table S3. As shown by Figure S1, most of the PTVs have high PHRED scores of over 20, and there is a highly significant positive correlation between PHRED score and the number of votes for being predicted as damaging using dbNSFP (p < 1.45e‐07). The Human Genome Variation Society (HGVS) nomenclature for each PTV gene is shown in Table S3.
3.3. Enrichment of functional pathways
The 821 genes were submitted to GSEA (https://www.gsea‐msigdb.org) for overrepresentation analysis of enriched functional pathways. We found 70 significant pathways showing corrected significance FDR < 0.01, with the top 20 pathways (p = 2.54e‐6, FDR = 3.71e‐4) shown in Table 3. The matrix of overlapping genes for the top 10 pathways is displayed in Figure S2. A high proportion of overlapping genes is observed in pathways of the extracellular matrix (ECM). Seven of the significant pathways (including collagen formation, collagen biosynthesis, and modifying enzymes) in Table 3 are related to organization, assembly, remodeling, and degradation of ECM involving ECM‐associated proteins (glycoproteins, collagens, and proteoglycans). Multiple pathways in Table 3 are related to cancer growth (cell cycle, RHO GTPase cycle, metabolism of lipids and RNA, collagen formation, etc.). Of note, the integrated breast cancer pathway is also highly significantly enriched (p = 4.15e‐8, FDR = 1.73e‐5).
TABLE 3.
Top 20 functional clusters overrepresented by protein truncating variant (PTV) genes found in 35 patients with familial pancreatic cancer
| Gene set name [number of genes (K)] | Description | Number of overlapping genes (k) | p‐Value | q‐Value |
|---|---|---|---|---|
| REACTOME EXTRACELLULAR MATRIX ORGANIZATION [301] | Extracellular matrix organization | 26 | 2.5 e−9 | 5.79 e−6 |
| REACTOME RHOA GTPASE CYCLE [149] | RHOA GTPase cycle | 18 | 3.96 e−9 | 5.79 e−6 |
| REACTOME RHO GTPASE CYCLE [444] | RHO GTPase cycle | 31 | 1.26 e−8 | 1.14 e−5 |
| REACTOME RNA POLYMERASE II TRANSCRIPTION [1374] | RNA Polymerase II Transcription | 63 | 1.83 e−8 | 1.14 e−5 |
| REACTOME SIGNALING BY RHO GTPASES MIRO GTPASES AND RHOBTB3 [717] | Signaling by Rho GTPases, Miro GTPases and RHOBTB3 | 41 | 1.94 e−8 | 1.14 e−5 |
| NABA MATRISOME [1026] | Ensemble of genes encoding extracellular matrix and extracellular matrix‐associated proteins | 51 | 3.63 e−8 | 1.73 e−5 |
| WP INTEGRATED BREAST CANCER PATHWAY [154] | Integrated Breast Cancer Pathway | 17 | 4.15 e−8 | 1.73 e−5 |
| REACTOME COLLAGEN FORMATION [90] | Collagen formation | 13 | 6.71 e−8 | 2.2 e−5 |
| REACTOME SIGNALING BY RECEPTOR TYROSINE KINASES [504] | Signaling by Receptor Tyrosine Kinases | 32 | 6.79 e−8 | 2.2 e−5 |
| REACTOME METABOLISM OF RNA [672] | Metabolism of RNA | 38 | 8.53 e−8 | 2.33 e−5 |
| REACTOME POST TRANSLATIONAL PROTEIN MODIFICATION [1435] | Post‐translational protein modification | 63 | 8.76 e−8 | 2.33 e−5 |
| REACTOME METABOLISM OF LIPIDS [741] | Metabolism of lipids | 40 | 1.33 e−7 | 3.25 e−5 |
| REACTOME CELL CYCLE [693] | Cell Cycle | 37 | 5.11 e−7 | 1.15 e−4 |
| NABA ECM REGULATORS [238] | Genes encoding enzymes and their regulators involved in the remodeling of the extracellular matrix | 19 | 1.14 e−6 | 2.37 e−4 |
| REACTOME TRANSCRIPTIONAL REGULATION BY TP53 [363] | Transcriptional Regulation by TP53 | 24 | 1.43 e−6 | 2.62 e−4 |
| REACTOME VESICLE MEDIATED TRANSPORT [724] | Vesicle‐mediated transport | 37 | 1.44 e−6 | 2.62 e−4 |
| REACTOME COLLAGEN BIOSYNTHESIS AND MODIFYING ENZYMES [67] | Collagen biosynthesis and modifying enzymes | 10 | 1.61 e−6 | 2.66 e−4 |
| KEGG FOCAL ADHESION [199] | Focal adhesion | 17 | 1.64 e−6 | 2.66 e−4 |
| REACTOME DEGRADATION OF THE EXTRACELLULAR MATRIX [140] | Degradation of the extracellular matrix | 14 | 2.12 e−6 | 3.26 e−4 |
| NABA CORE MATRISOME [275] | Ensemble of genes encoding core extracellular matrix including ECM glycoproteins, collagens, and proteoglycans | 20 | 2.54 e−6 | 3.71 e−4 |
3.4. Overlap with cancer‐related genes
Dietlein et al. (2020) compiled a list of 460 cancer driver genes, whose variations increase net cell growth under specific microenvironmental conditions. Among the 821 PTV genes, 32 were observed in the list of cancer driver genes. For the 40 multiple‐PTV genes in Table 2, six overlaps were found in the cancer driver gene list (ATM, POLE, BRCA2, TYRO3, PABPC1, SSC5D). Hypergeometric test showed that the overlaps of the 32 genes and the six genes, respectively, were both significantly different from being random (p < 1e‐22).
Next, we investigated the overlap of the 821 PTV genes with previously reported PTV genes in FPC studies. Roberts et al. (2016) identified 16 genes with more than two PTVs. Two out of their 16 genes (ATM, BRCA2) were found in our 821 PTV genes. The hypergeometric test showed very high significance (p < 1e‐22) for the observed overlaps. Finally, we tested overlaps between the 821 PTV genes with our recently reported 448 PTV genes detected in first‐degree relatives of FPC patients (Tan et al., 2021b). There were a total of 62 overlaps observed leading to an extremely high statistical significance (hypergeometric p < 1e‐22). Among the 40 genes carrying multiple PTVs (Table 2), six genes were found in our previously reported 448 PTV genes (COL4A2, CRIPAK, ABCA7, AGBL3, APLF, TTC27). Again, hypergeometric test showed that the overlap is highly significant (p = 4.32e‐17).
3.5. Association analysis
We performed a gene‐based association analysis by applying ProxECAT and using gnomAD as an external control group. Six genes showed nominal significance with p < 0.05, including: MORN1 (p = 6.1e‐03), MYO16 (p = 1.43e‐02), PIEZO1 (p = 1.87e‐02), KLHL5 (p = 2.56e‐02), PTS (p = 2.59e‐02), and CEP95 (p = 4.81e‐02). After correcting for multiple testing, none was significant (0.17 < FDR < 0.27). The rare variants carried by the six genes are all missense mutations except for CEP95 (Table S4). The missense variants all have relatively high PHRED scores of over 20 and high numbers of being predicted as damaging (above four out of six predictions) except MYO16. The missense variant in PTS (chr11:112101362, C‐>T) is pathogenic with a PHRED score of 28.8 and with all 6 predictions as damaging. The rare variant in CEP95 is a frameshift variant with a PHRED score of 26.
4. DISCUSSION
In contrast to common variants identified in genome‐wide association studies, rare variants revealed by sequencing analysis have played unique roles in the genetics of complex diseases in humans due to their distinctive features. Their unique roles are hypothesis‐free evidence for gene causality, serving as precise targets of functional analysis for understanding disease mechanisms as well as genetic markers for personalized medicine (Momozawa & Mizukami, 2021). By performing whole genome sequencing on benign, noncancerous tissues of FPC patients, we were able to focus on rare germline variants, aiming to characterize the hereditary basis of FPC. Our finding of a large number of genes hosting loss‐of‐function variants (PTVs, Table S2) and genes enriched by multiple PTVs (Table 2) revealed a high genetic heterogeneity in the form of both locus and allelic heterogeneity (16). The genetic diversity of FPC patients was observed not only across samples in our cohort, but also within individual FPC patients showing intraindividual allelic heterogeneity (multiple PTVs of the same gene in the same patient) and locus heterogeneity (multiple PTV genes in the same patient). High genetic heterogeneity of FPC patients has been reported by previous sequencing studies in cohorts from the United States and Germany (Roberts et al., 2016; Slater et al., 2021). Our results provide new evidence reconfirming FPC as a genetically heterogeneous disease associated with rare germline variants.
The high diversity in the genetic architecture of FPC imposes a challenge to current strategies for screening predisposed individuals. Traditional genetic screening is based on analyzing classical high penetrance genes that only explain the genetic predisposition in a reduced number of families. It is estimated that the currently identified variants in FPC susceptibility genes including BRCA1/2, ATM, CDKN2A, PALB2, etc. explain less than 20% of FPC cases leaving the genetic basis of more than 80% of FPC patients unknown (Roberts et al., 2016). A very recent WGS study on FPC patients failed to detect pathogenic variants in BRCA1/2, CDKN2A, or PALB2 (Slater et al., 2021). Likewise, association testing of our recent WGS on first‐degree relatives of FPC patients did not find rare variants in any of the reported FPC candidate genes (Tan et al., 2021b). Meanwhile, a comprehensive analysis of 35 candidate genes associated with hereditary cancer revealed that variants in previously described cancer‐predisposition genes including MLH1, CDKN2A, POLQ, TET2, and FANCM are found in 19% of FPC cases (Earl et al., 2020). In a recent genome‐wide meta‐analysis, NOC2L was also suggested as a pancreatic cancer susceptibility gene (Klein et al., 2018). The situation implies that traditional genetic testing based on classical high penetrance genes will most likely miss the majority of genetically predisposed individuals to FPC. More individualized testing strategies such as the NGS‐based panel testing (Nagahashi et al., 2019) that take genetic heterogeneity into account are called for.
The extracellular matrix (ECM) is a major structural component of the tumor microenvironment that provides both structural and biochemical support to regulate proliferation, self‐renewal, and differentiation of cancer stem cells (Nallanthighal et al., 2019). Among the 20 gene sets in Table 3, seven are related to ECM involving organization, assembly, remodeling, degradation, and coding of ECM‐associated proteins. PDAC has an extraordinarily dense fibrotic stroma primarily made of ECM whose stiffness confers mechanical properties of the tumor microenvironment and provides important biochemical and physical cues that promote survival, proliferation, and metastasis of cancer cells (Weniger et al., 2018). Nonsense and frameshift variants like PTVs and nonsynonymous variants that change the sequence and structure of coding proteins reduce the production of ECM proteins to impair matrix integrity, composition, and assembly due to quantitative ECM defects (Lamandé & Bateman, 2020). In a recent WGS study, we have observed a significant enrichment of the ECM pathway by genes carrying rare nonsynonymous variants in first‐degree relatives of FPC patients (Tan et al., 2021b). Moreover, a recent network‐based analysis of gene expression data on FPC and sporadic pancreatic cancer patients reported increased activity in extracellular structure and ECM organization (Tan et al., 2020). Our previous and current results from pathway analysis concerning ECM may help functionally characterizing the identified rare variants in ECM composition, assembly, and degradation as accomplices in the development and progression of FPC.
Guanosine triphosphate (GTP) is one of the building blocks needed for the synthesis of RNA during the transcription process. It is also used as a source of energy for protein synthesis and gluconeogenesis or has the role of an activator of substrates in metabolic reactions. Its binding proteins (Rho GTPases) play central roles in numerous cellular processes with dysregulation of Rho GTPase signaling observed in a broad range of human cancers (Jung et al., 2020). Although large scale sequencing efforts have revealed that variants in the Rho GTPase family are rare (Pajic et al., 2015), our results showed significant overrepresentation of PTV genes in the Rho GTPase pathways (Table 3) involving genes such as ROCK1 (Rho Associated Coiled‐Coil Containing Protein Kinase 1) (Figure S2). In a study by Nakashima et al. (2011), a suppressive role of ROCK in pancreatic cancer cell proliferation was characterized. The PTV we observed in ROCK1 is a frameshift variant (chr18:18566913, C:‐) that may result in a complete loss of protein structure and functionality, with the latter potentially beneficial for FPC development and progression. This observation serves as an example of support to Nakashima et al. (2011).
Extremely significant overlaps of our detected PTV genes have been found in cancer driver genes and previously reported cancer genes. Among the overlapping genes, the roles of ATM, BRCA2 in hereditary breast and ovarian cancers have been well characterized (Kobayashi et al., 2013). Variants in BRCA and ATM genes occur in both hereditary and sporadic PDAC causing deficiency in DNA repair pathways and provoke genomic instability (Perkhofer et al., 2021). In a recent study of 130 families with 2,227 family members with FPC predisposition, it was shown that individuals with an ATM variant had a cumulative risk for pancreatic cancer of 6.3% by age 70 and 9.5% by age 80 (Hsu et al., 2021). Both ATM and BRCA2 variants have previously been identified as inherited germline variants related to pancreatic cancer (Hu et al., 2018; Huang et al., 2018; Zhen et al., 2015).
Other overlapping genes that we found include POLE, TYRO3, PABPC1, and SSC5D. Variants in POLE have been found in individuals with early onset colorectal cancer, large numbers of adenomatous colorectal polyps and/or significant family history of colorectal cancer (Bellido et al., 2016). Some families with POLE variants include individuals with a wide range of cancers including pancreatic cancer (Hansen et al., 2015; Mur et al., 2020). TYRO3 is constitutively expressed in pancreatic cancer cells and is required for cell proliferation and invasion of pancreatic cancer (Morimoto et al., 2020). PABPC1 (Poly(A) Binding Protein Cytoplasmic 1) encodes PABP1 protein which binds mRNA and facilitates a variety of functions such as transport into and out of the nucleus, degradation, translation, and stability. A recent whole exome sequencing study reported that sequence variations in PABPC1 are associated with familial prostate cancer (Schaid et al., 2021). The SSC5D gene codes for soluble scavenger receptor cysteinerich domain‐containing protein (SSC5D), which binds to extracellular matrix proteins as a pattern recognition receptor and may play a role in the innate defense and homeostasis of certain epithelial surfaces. Al‐Sukhni et al. (2012) reported gain in DNA copy number in SSC5D gene region in familial pancreatic cancer. The RICTOR gene has recently been shown to be amplified in cancer, highlighting its role in cancer development and its potential as a therapeutic target (Jebali & Dumaz, 2018). Overall, previously published studies show that variations in the overlapping PTV genes have been associated with cancer development or directly with pancreatic cancer or FPC.
In addition to cancer related genes, our identified PTV genes also overlap significantly with genes reported by previously published WGS studies on FPC or FPC families. For example, enrichment of PTVs was also observed in BRCA2 and ATM in a WGS on FPC patients (Roberts et al., 2016). Such nonrandom overlaps could indicate that these genes are mutation hotspots serving as oncogenic drivers in FPC development. Moreover, it is interesting that the PTV genes found in FPC patients in this study also overlap significantly with the PTV genes detected in unaffected first‐degree relatives of FPC patients (Tan et al., 2021b). This is important as, with cancer events available from follow‐up, the burden of private variants in these genes can be calculated for each individual and used to build models for PDAC risk prediction and prognosis in the predisposed relatives of FPC patients.
Although with limited sample size of FPC patients, our gene‐based association test using ProxECAT was able to identify six genes (MORN1, MYO16, PIEZO1, KLHL5, PTS, and CEP95) with nominal significance of p < 0.05. Among the genes, KLHL5 (Kelch Like Family Member 5) was previously shown to represent an eligible prognostic predictor for gastric malignancy (Wu et al., 2020), and knockdown of the gene increases cellular sensitivity to anticancer drugs (Schleifer et al., 2018). PIEZO1 (Piezo Type Mechanosensitive Ion Channel Component 1) encodes a protein that induces mechanically activated currents in various cell types. The gene has been demonstrated to play oncogenic roles in gastric cancer cell proliferation, migration and invasion to promote gastric cancer progression (Zhang et al., 2018). Multiple studies have shown that the expression of PIEZO1 is related to the clinical characteristics of senescence and cancer, making the gene a new biomarker for diagnosis and prognosis of a variety of human cancers (Yu & Liao, 2021). Although some of the genes found by association test have been reported in cancer studies, their roles in pancreatic cancer need further verification and validation.
As mentioned, a big limitation of the study is the small sample size of FPC patients, which has limited the statistical power of our association test and the ability in detecting PTVs. Another limitation is the quality of DNA samples from formalin‐fixed paraffin‐embedded benign tissues of FPC patients. As full blood samples were not available for the FPC cases, FFPE samples of non‐pancreatic benign tissues were used for DNA extraction. The latter led to low sequencing coverage or depth due to an insufficient number of sequencing reads. As a result, the number of genes available for association testing was also limited, as ProxECAT requires both nonsynonymous and synonymous SNVs to conduct an association test. This is exacerbated by the sample size issue which resulted in a low number of genes found by association analysis. Moreover, ProxECAT assumes that the cases and the external controls match by ancestral population. The included FFPE samples were retrieved from cancer‐free, nonpancreatic sites and have been carefully examined by an experienced pathologist at the time of retrieval—eliminating the risk of potential contamination of samples from tumor cells. Although this ensures that the detected PTVs are germline variants, the big number of over 800 PTV genes should be treated with caution as the majority of them carry one PTV (only 40 genes carry multiple PTVs, see Table 2) from a small‐scale study.
With the establishment of a nation‐wide cohort of FPC families, high quality DNA samples from all families (including first‐degree relatives included in our nationwide FPC screening program) have been collected, sequenced, or stored to correlate with future cancer events (Tan et al., 2021a, 2021b)—which will eventually help validate our current findings.
5. CONCLUSIONS
Whole genome sequencing on FPC patients detected a multitude of rare variants displaying a high degree of allelic and locus heterogeneity in FPC. The hosting genes of detected variants significantly over‐represent cancer driver genes and/or cancer‐related genes that mediate cancer cell proliferation, migration, and invasion. The genetic heterogeneity of FPC is functionally characterized by significant enrichment of multiple biological pathways including the ECM and Rho GTPase pathways that jointly may contribute to the development and progression of FPC.
AUTHOR CONTRIBUTION
Conceptualization, M.T., M.T.J. and O.B.S.M.; methodology, M.T. and KB; software, M.T., K.B., and M.J.L.; validation, M.T., K.B. and S.D.; formal analysis, M.T.; investigation, M.T.; resources, M.T.J., O.B.S.M., K.B. and S.D.; data curation, M.T.; writing—original draft preparation, M.T.; writing—review and editing, M.T., M.T.J., O.B.S.M., K.B., S.D., M.B.M., A.‐M.G. and M.J.L.; visualization, M.T.; supervision, M.T.J., O.B.S.M., K.B. and S.D.; project administration, M.T., M.T.J., O.B.S.M.; funding acquisition, M.T., M.T.J., O.B.S.M. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supplementary Figure S1. Correlation between PHRED score and prediction as damaging.
Supplementary Figure S2. Overlap of genes among the top ten pathways enriched by PTV genes.
Table S1. Distribution of tissue samples by site of biopsy
Table S2. A list of PTV genes ordered by number of hosted PTVs
Table S3. Annotation of detected PTVs
Table S4. Annotation of functional variants detected by association tests
ACKNOWLEDGMENTS
The authors want to issue a special thanks to the following hospital departments for their contribution with relevant benign FFPE tissue samples for the FPC patients included in this study: the Department of Pathology, Aalborg University Hospital, Aalborg, Denmark; the Department of Pathology, Aarhus University Hospital, Aarhus, Denmark; the Department of Pathology, Herlev Hospital, Herlev, Denmark; the Department of Pathology, Hospital of South West Jutland (Sydvestjysk Sygehus)—Esbjerg, Esbjerg, Denmark; the Department of Pathology, Hospital Sønderjylland (Sygehus Sønderjylland)—Sønderborg, Sønderborg, Denmark; the Department of Pathology, Hvidovre Hospital, Hvidovre, Denmark; the Department of Pathology, Næstved Hospital (Næstved Sygehus), Næstved, Denmark; the Department of Pathology, Odense University Hospital, Odense, Denmark; the Department of Pathology, Randers Regional Hospital (Regionshospitalet Randers), Randers, Denmark; the Department of Pathology, Rigshospitalet, Copenhagen, Denmark; the Department of Pathology, Vejle Hospital—Sygehus Lillebaelt, Vejle, Denmark; the Department of Pathology, Viborg Regional Hospital (Regionshospitalet Viborg), Viborg, Denmark; the Department of Pathology, Zealand University Hospital—Roskilde, Roskilde, Denmark.
Tan, M. , Brusgaard, K. , Gerdes, A‐M. , Larsen, M. J. , Mortensen, M. B. , Detlefsen, S. , de Muckadell, O. B. S. , & Joergensen, M. T. (2022). Whole genome sequencing identifies rare genetic variants in familial pancreatic cancer patients. Annals of Human Genetics, 86, 195–206. 10.1111/ahg.12464
Funding Information
This project was jointly supported by: The Danish Cancer Society (ref.: R218‐A13150), NEYE Fonden, Odense Pancreas Center (OPAC), Odense University Hospital Research Fund, The Danish Cancer Research Fund (Dansk Kræftforskningsfond), Fonden til Lægevidenskabens Fremme (A.P. Møller Fonden), Fabrikant Einar Willumsens Mindelegat, the Rigshospitalet (RH)/Odense University Hospital (OUH) Research Fund (ref.: A3183), Aase og Ejnar Danielsens Fond (ref.: 18‐10‐0686).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Al‐Sukhni, W. , Joe, S. , Lionel, A. C. , Zwingerman, N. , Zogopoulos, G. , Marshall, C. R. , Borgida, A. , Holter, S. , Gropper, A. , Moore, S. , Bondy, M. , Klein, A. P. , Petersen, G. M. , Rabe, K. G. , Schwartz, A. G. , Syngal, S. , Scherer, S. W. , & Gallinger, S . (2012). Identification of germline genomic copy number variation in familial pancreatic cancer. Human Genetics, 131(9), 1481–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido, F. , Pineda, M. , Aiza, G. , Valdés‐Mas, R. , Navarro, M. , Puente, D. A. , Pons, T. , González, S. , Iglesias, S. , Darder, E. , Piñol, V. , Soto, J. L. , Valencia, A. , Blanco, I. , Urioste, M. , Brunet, J. , Lázaro, C. , Capellá, G. , Puente, X. S. , & Valle, L . (2016). POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genetics in Medicine, 18(4), 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y . (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. [Google Scholar]
- Chaffee, K. G. , Oberg, A. L. , McWilliams, R. R. , Majithia, N. , Allen, B. A. , Kidd, J. , Singh, N. , Hartman, A. R. , Wenstrup, R. J. , & Petersen, G. M. (2018). Prevalence of germ‐line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genetics in Medicine, 20(1), 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, K. E. , & Lucas, A. L . (2019). Familial pancreatic ductal adenocarcinoma. American Journal of Pathology, 189(1), 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietlein, F. , Weghorn, D. , Taylor‐Weiner, A. , Richters, A. , Reardon, B. , Liu, D. , Lander, E. S. , Van Allen, E. M. , & Sunyaev, S. R . (2020). Identification of cancer driver genes based on nucleotide context. Nature Genetics, 52(2), 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl, J. , Galindo‐Pumariño, C. , Encinas, J. , Barreto, E. , Castillo, M. E. , Pachón, V. , Ferreiro, R. , Rodríguez‐Garrote, M. , González‐Martínez, S. , Ramon Y Cajal, T. , Diaz, L. R. , Chirivella‐Gonzalez, I. , Rodriguez, M. , de Castro, E. M. , García‐Seisdedos, D. , Muñoz, G. , Rosa, J. , Marquez, M. , Malats, N. , & Carrato, A. (2020). A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine, 53, 102675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, M. F. , Johansen, J. , Bjørnevoll, I. , Sylvander, A. E. , Steinsbekk, K. S. , Sætrom, P. , Sandvik, A. K. , Drabløs, F. , & Sjursen, W . (2015). A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Familial Cancer, 14(3), 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks, A. E. , Billups, S. C. , Pike, H. N. C. , Farooqi, I. S. , Zeggini, E. , Santorico, S. A. , Barroso, I. , & Dupuis, J . (2018). ProxECAT: Proxy external controls association test. A new case‐control gene region association test using allele frequencies from public controls. Plos Genetics, 14(10), e1007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, F. C. , Roberts, N. J. , Childs, E. , Porter, N. , Rabe, K. G. , Borgida, A. , Ukaegbu, C. , Goggins, M. G. , Hruban, R. H. , Zogopoulos, G. , Syngal, S. , Gallinger, S. , Petersen, G. M. , & Klein, A. P . (2021). Risk of pancreatic cancer among individuals with pathogenic variants in the ATM Gene. JAMA Oncology, 7(11), 1664–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, C. , Hart, S. N. , Polley, E. C. , Gnanaolivu, R. , Shimelis, H. , Lee, K. Y. , Lilyquist, J. , Na, J. , Moore, R. , Antwi, S. O. , Bamlet, W. R. , Chaffee, K. G. , DiCarlo, J. , Wu, Z. , Samara, R. , Kasi, P. M. , McWilliams, R. R. , Petersen, G. M. , & Couch, F. J . (2018). Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA, 319(23), 2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, K. L. , Mashl, R. J. , Wu, Y. , Ritter, D. I. , Wang, J. , Oh, C. , Paczkowska, M. , Reynolds, S. , Wyczalkowski, M. A. , Oak, N. , Scott, A. D. , Krassowski, M. , Cherniack, A. D. , Houlahan, K. E. , Jayasinghe, R. , Wang, L. B. , Zhou, D. C. , Liu, D. , Cao, S. , Kim, Y. W. , … Ding, L . (2018). Pathogenic germline variants in 10,389 adult cancers. Cell, 173(2), 355–70.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebali, A. , & Dumaz, N . (2018). The role of RICTOR downstream of receptor tyrosine kinase in cancers. Molecular cancer, 17(1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, H. , Yoon, S. R. , Lim, J. , Cho, H. J. , & Lee, H. G . (2020). Dysregulation of Rho GTPases in human cancers. Cancers (Basel), 12(5), 1179. 10.3390/cancers12051179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, A. P. , Wolpin, B. M. , Risch, H. A. , Stolzenberg‐Solomon, R. Z. , Mocci, E. , Zhang, M. , Canzian, F. , Childs, E. J. , Hoskins, J. W. , Jermusyk, A. , Zhong, J. , Chen, F. , Albanes, D. , Andreotti, G. , Arslan, A. A. , Babic, A. , Bamlet, W. R. , Beane‐Freeman, L. , Berndt, S. I. , Blackford, A. , … Amundadottir, L. T . (2018). Genome‐wide meta‐analysis identifies five new susceptibility loci for pancreatic cancer. Nature Communication, 9(1), 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, H. , Ohno, S. , Sasaki, Y. , & Matsuura, M . (2013). Hereditary breast and ovarian cancer susceptibility genes (review). Oncology Reports, 30(3), 1019–1029. [DOI] [PubMed] [Google Scholar]
- Lamandé, S. R. , & Bateman, J. F . (2020). Genetic disorders of the extracellular matrix. Anatomical Record (Hoboken), 303(6), 1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E . (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llach, J. , Carballal, S. , & Moreira, L . (2020). Familial pancreatic cancer: Current perspectives. Cancer Management and Research, 12, 743–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan, J. , & King, M. C . (2010). Genetic heterogeneity in human disease. Cell, 141(2), 210–217. [DOI] [PubMed] [Google Scholar]
- McGuigan, A. , Kelly, P. , Turkington, R. C. , Jones, C. , Coleman, H. G. , & McCain, R. S . (2018). Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World Journal of Gastroenterology, 24(43), 4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momozawa, Y. , & Mizukami, K . (2021). Unique roles of rare variants in the genetics of complex diseases in humans. Journal of Human Genetics, 66(1), 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, M. , Horikoshi, Y. , Nakaso, K. , Kurashiki, T. , Kitagawa, Y. , Hanaki, T. , Sakamoto, T. , Honjo, S. , Umekita, Y. , Fujiwara, Y. , & Matsura, T . (2020). Oncogenic role of TYRO3 receptor tyrosine kinase in the progression of pancreatic cancer. Cancer Letters, 470, 149–160. [DOI] [PubMed] [Google Scholar]
- Mur, P. , García‐Mulero, S. , Del Valle, J. , Magraner‐Pardo, L. , Vidal, A. , & Pineda, M. , Cinnirella, G. , Martín‐Ramos, E. , Pons, T. , López‐Doriga, A. , Belhadj, S. , Feliubadaló, L. , Munoz‐Torres, P. M. , Navarro, M. , Grau, E. , Darder, E. , Llort, G. , Sanz, J. , Ramón Y Cajal, T. , Balmana, J. , … Valle, L . (2020). Role of POLE and POLD1 in familial cancer. Genetics in Medicine, 22(12), 2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi, M. , Shimada, Y. , Ichikawa, H. , Kameyama, H. , Takabe, K. , Okuda, S. , & Wakai, T . (2019). Next generation sequencing‐based gene panel tests for the management of solid tumors. Cancer Science, 110(1), 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghavi, M. on behalf of the GBD 2017 Pancreatic Cancer Collaborators . (2019). The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol, 4(12), 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, H. , & Fujita, M . (2018). Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Science, 109(3), 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, M. , Adachi, S. , Yasuda, I. , Yamauchi, T. , Kawaguchi, J. , Hanamatsu, T. , Yoshioka, T. , Okano, Y. , Hirose, Y. , Kozawa, O. , & Moriwaki, H . (2011). Inhibition of Rho‐associated coiled‐coil containing protein kinase enhances the activation of epidermal growth factor receptor in pancreatic cancer cells. Molecular Cancer, 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallanthighal, S. , Heiserman, J. P. , & Cheon, D.‐J . (2019). The role of the extracellular matrix in cancer stemness. Frontiers in Cell and Developmental Biology, 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic, M. , Herrmann, D. , Vennin, C. , Conway, J. R. , Chin, V. T. , Johnsson, A.‐K. E. , Welch, H. C. , & Timpson, P. (2015). The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases, 6(2), 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkhofer, L. , Gout, J. , Roger, E. , Kude de Almeida, F. , Baptista Simões, C. , Wiesmüller, L. , Seufferlein, T. , & Kleger, A . (2021). DNA damage repair as a target in pancreatic cancer: State‐of‐the‐art and future perspectives. Gut, 70(3), 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M . (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47(D1), D886–d94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, N. J. , Norris, A. L. , Petersen, G. M. , Bondy, M. L. , Brand, R. , Gallinger, S. , Kurtz, R. C. , Olson, S. H. , Rustgi, A. K. , Schwartz, A. G. , Stoffel, E. , Syngal, S. , Zogopoulos, G. , Ali, S. Z. , Axilbund, J. , Chaffee, K. G. , Chen, Y. C. , Cote, M. L. , Childs, E. J. , Douville, C. , … Klein, A. P . (2016). Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discovery, 6(2), 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid, D. J. , McDonnell, S. K. , FitzGerald, L. M. , DeRycke, L. , Fogarty, Z. , Giles, G. G. , Giles, G. G. , MacInnis, R. J. , Southey, M. C. , Nguyen‐Dumont, T. , Cancel‐Tassin, G. , Cussenot, O. , Whittemore, A. S. , Sieh, W. , Ioannidis, N. M. , Hsieh, C. L. , Stanford, J. L. , Schleutker, J. , Cropp, C. D. , Carpten, J. , Hoegel, J. , … Thibodeau, S. N . (2021). Two‐stage study of familial prostate cancer by whole‐exome sequencing and custom capture identifies 10 novel genes associated with the risk of prostate cancer. European Urology, 79(3), 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifer, R. J. , Li, S. , Nechtman, W. , Miller, E. , Bai, S. , Sharma, A. , She, J. X . (2018). KLHL5 knockdown increases cellular sensitivity to anticancer drugs. Oncotarget, 9(100), 37429–37438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriver, C. R . (2001). Allelic and locus heterogeneity. eLS. [Google Scholar]
- Slater, E. P. , Wilke, L. M. , Böhm, L. B. , Strauch, K. , Lutz, M. , Gercke, N. , Matthäi, E. , Hemminki, K. , Försti, A. , Schlesner, M. , Paramasivam, N. , & Bartsch, D. K . (2021). Combinations of low‐frequency genetic variants might predispose to familial pancreatic cancer. Journal of Personalized Medicine, 11(7), 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, M. , Brusgaard, K. , Gerdes, A.‐M. , Mortensen, M. B. , Detlefsen, S. , Schaffalitzky de Muckadell, O. B. , & Joergensen, M. T. (2021a). Cohort profile and heritability assessment of familial pancreatic cancer: A nation‐wide study. Scandinavian Journal of Gastroenterology, 56(8), 965–971. [DOI] [PubMed] [Google Scholar]
- Tan, M. , Brusgaard, K. , Gerdes, A.‐M. , Mortensen, M. B. , Detlefsen, S. , Schaffalitzky de Muckadell, O. B. , & Joergensen, M. T. (2021b). Whole genome sequencing identifies rare germline variants enriched in cancer related genes in first degree relatives of familial pancreatic cancer patients. Clinical Genetics, 100(5), 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, M. , Schaffalitzky de Muckadell, O. B. , & Jøergensen, M. T . (2020). Gene expression network analysis of precursor lesions in familial pancreatic cancer. Journal of Pancreatic Cancer, 6(1), 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weniger, M. , Honselmann, K. C. , & Liss, A. S . (2018). The extracellular matrix and pancreatic cancer: A complex relationship. Cancers (Basel), 10(9). 10.3390/cancers10090316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Q. , Yin, G. , Lei, J. , Tian, J. , Lan, A. , & Liu, S . (2020). KLHL5 is a prognostic‐related biomarker and correlated with immune infiltrates in gastric cancer. Frontiers in Molecular Biosciences, 7, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. L. , & Liao, H. Y . (2021). Piezo‐type mechanosensitive ion channel component 1 (Piezo1) in human cancer. Biomedicine & Pharmacotherapy, 140, 111692. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Zhou, Y. , Huang, T. , Wu, F. , Liu, L. , Kwan, J. S. H. , Cheng, A. , Yu, J. , To, K. F. , & Kang, W . (2018). PIEZO1 functions as a potential oncogene by promoting cell proliferation and migration in gastric carcinogenesis. Molecular Carcinogenesis, 57(9), 1144–1155. [DOI] [PubMed] [Google Scholar]
- Zhen, D. B. , Rabe, K. G. , Gallinger, S. , Syngal, S. , Schwartz, A. G. , Goggins, M. G. , Hruban, R. H. , Cote, M. L. , McWilliams, R. R. , Roberts, N. J. , Cannon‐Albright, L. A. , Li, D. , Moyes, K. , Wenstrup, R. J. , Hartman, A. R. , Seminara, D. , Klein, A. P. , & Petersen, G. M . (2015). BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genetics in Medicine, 17(7), 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Correlation between PHRED score and prediction as damaging.
Supplementary Figure S2. Overlap of genes among the top ten pathways enriched by PTV genes.
Table S1. Distribution of tissue samples by site of biopsy
Table S2. A list of PTV genes ordered by number of hosted PTVs
Table S3. Annotation of detected PTVs
Table S4. Annotation of functional variants detected by association tests
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.