

Abstract
Aim
To explore how bariatric surgery (BS) modified the obesity‐associated gut microbiome, the host metabolome, and their interactions in obese Korean patients.
Materials and Methods
Stool and fasting blood samples were obtained before and 1, 3, 6, and 12 months after BS from 52 patients enrolled in the Korean Obesity Surgical Treatment Study. We analysed the gut microbiome by 16S rRNA gene sequencing and the serum metabolome, including bile acids, by nuclear magnetic resonance spectroscopy and ultrahigh‐performance liquid chromatography/triple quadrupole mass spectrometry.
Results
Stool metagenomics showed that 27 microbiota were enriched and 14 microbiota were reduced after BS, whereas the abundances and diversity of observed features were increased. The levels of branched‐chain amino acids and metabolites of energy metabolism in serum were decreased after surgery, whereas the levels of metabolites related to microbial metabolism, including dimethyl sulphone, glycine, and secondary bile acids, were increased in the serum samples. In addition, we found notable mutual associations among metabolites and gut microbiome changes attributed to BS.
Conclusions
Changes in the gut microbiome community and systemic levels of amino acids and sugars were directly derived from anatomical changes in the gastrointestinal tract after BS. We hypothesized that the observed increases in microbiome‐related serum metabolites were a result of complex and indirect changes derived from BS. Ethnic‐specific environmental or genetic factors could affect Korean‐specific postmetabolic modification in obese patients who undergo BS.
Keywords: bariatric surgery, correlation analysis, gut microbiome, metabolomics, omics integration
1. INTRODUCTION
Obesity has been a worldwide problem for a long time and is treated as a pivotal risk factor for metabolic diseases such as diabetes, hypertension, dyslipidaemia, and cardiovascular diseases. 1 Several interventions, including diet control, exercise, and drug treatment, have been used to improve obesity, but these interventions are known to be of little help and not sustainable over the long term. 2 By contrast, bariatric surgery (BS) has been considered one of the most effective treatments to improve obesity and its complications. 3
BS has been reported to modulate the gut microbiota, which is a major environmental factor influencing host metabolism in obesity. For this reason, several studies have attempted to explore the impact of BS on alterations in the gut microbiome, 4 host metabolism, and their associations. 5 Ilhan et al. investigated the correlations between gut microbiota and stool metabolites, including amino acid fermentation and degradation products, branched‐chain fatty acids, and secondary bile acids (BAs), after gastric bypass surgery. 6
Although the stool metabolome is directly linked to the gut microbiome compared to the circulating metabolome, the circulating metabolome reflects systemic metabolism related to pathological metabolic disorders and improvements in host metabolism attributed to a differential microbiome after BS. 7 In recent years, there have been ongoing efforts to understand the correlations between host serum or plasma metabolites and the gut microbiota. For instance, associations between gut microbial species and serum metabolites in obese Han Chinese individuals have been reported to differ from those in their lean counterparts. 8 Additionally, a previous study reported that the gut microbiome partially mediated circulating metabolic improvement after BS in American and Spanish patients. 9
However, studies on the associations between the microbiome and various circulating metabolites through global profiling in BS are still lacking, especially for Korean obese patients. Although the traditional East Asian diet is largely composed of carbohydrate sources, relative fat consumption compared with total energy consumption rose rapidly in Korea from 7.2% in 1969 to 19.0% in 1998. 10 However, the low prevalence of obesity in Korea among Organisation for Economic Cooperation and Development countries could still be attributed to an ethnic‐specific lifestyle and diet pattern characterized by a carbohydrate‐rich and animal protein‐poor diet. 11 Furthermore, the Korean diet differs in its impact on the gut microbiome and host metabolism compared with Western or other Asian diets. 12 Thus, to appreciate BS‐associated Korean‐specific responses, it is necessary to evaluate metabolic and microbial changes in Korean individuals after BS.
Here, we aimed to identify alterations in the gut microbiome and a wide range of systemic metabolomes, including circulating metabolites and BAs, following BS in an obese Korean population; also, the notable associations between the altered gut microbiota and serum metabolites were identified through the integration of metagenomic and metabolomic approaches.
2. SUBJECTS AND METHODS
2.1. Study population
The population of the current study was a part of the BS group enrolled in the Korean Obesity Surgical Treatment Study (KOBESS), 13 a prospective, multicentre, single‐arm, observational cohort study of obese Korean patients. KOBESS was performed at five university hospitals from August 2016 to April 2019. Obese patients were eligible if they had a body mass index (BMI) of 35 kg/m2 or higher or a BMI of 30.0‐34.9 kg/m2 with obesity‐related co‐morbidities, such as diabetes, hypertension, or hyperlipidaemia. The patients underwent Roux‐en‐Y gastric bypass (RYGB) or sleeve gastrectomy (SG) based on their medical condition, including the risk of gastric cancer, reflux esophagitis, and mental health status, as determined by interviews with investigators. Twenty‐two patients underwent RYGB, and 30 patients underwent SG. The nutritional status of each patient was assessed by professional dieticians before the surgery and monitored on follow‐up visits after the surgery. The patients were asked to record their daily food intake, based upon which they were advised to adjust eating habits and diet to adapt to the new gastrointestinal physiology. Nutritional deficits, such as vitamin B12 deficiencies, were also monitored and managed by professional dieticians. Anthropometric data (height, sex, age, weight, and systolic and diastolic blood pressures) and stool and fasting blood samples were obtained before and 1, 3, 6, and 12 months after the surgery from the eligible patients (N = 52). The difference in clinical outcomes between individuals who underwent two distinct operations was not detected in the KOBESS research. 14 Interindividual variations in metabolomics data related to meals or physical health are generally acknowledged. 15 For this reason, we also collected serum samples from each patient after they had fasted.
Unfortunately, only a portion of the subjects had linked faeces and serum samples. Stool samples were collected from 31 patients, serum samples were collected from 38 patients, and both stool and serum samples were collected from 17 patients and were further used for the correlation analysis. This study was registered at ClinicalTrials.gov (NCT03100292) and approved by the Institutional Review Board of each centre (Approval number for the coordinating investigator: INHA 2016‐06‐015).
2.2. Metagenomic analysis
The quality of the raw sequence reads was analysed using FastQC. 16 Illumina adapter sequences of the paired‐end reads were removed using Cutadapt version 2.2. 17 Then the trimmed sequences were processed using the QIIME 2 202.8 release.
2.3. Global profiling analysis by nuclear magnetic resonance
For nuclear magnetic resonance (NMR) analysis, 130 μl of serum sample was diluted with 500 μl of saline solution (0.9% sodium chloride in deuterium oxide) and transferred to 5‐mm NMR tubes. 1H‐NMR spectra of sera were acquired by an Advance III HD 800 MHz NMR spectrometer (Bruker BioSpin, Ettlingen, Germany) with a Bruker 5‐mm CPTCI Z‐GRD probe. For all spectra of each serum, 256 transients were collected using a water‐suppressed Carr–Purcell–Meiboom–Gill spin‐echo pulse sequence. The internal reference was trimethylsilylpropanoic acid, and the experimental methodology followed that of earlier studies. 18
2.4. BA targeted analysis
For BA analysis using ultrahigh‐performance liquid chromatography/triple quadrupole mass spectrometry (UPLC/TQ‐MS), 20 μl of serum sample was extracted using 80 μl of methanol in a 96‐well plate, and 50 μl of supernatant was diluted with 50 μl of 20% methanol (v/v) containing 40 nM internal standard mixture. Targeted analysis of serum BAs was performed on a Waters ACQUITY UPLC I‐Class PLUS and a Xevo TQ‐XS Triple Quadrupole MS system (Waters) in multiple reaction monitoring modes.
2.5. Integration of microbiome and metabolomics data
Partial correlation analysis was carried out to evaluate the relationship between changes in metabolites and the microbiome using SPSS software, version 15.0. The measured values for each metabolite and the microbiota were adjusted by individual age and sex. We only evaluated invariant and intrinsic traits as possible confounding factors because some variables closely connected or associated with obesity could be the result of the surgical operation. Additionally, because correlation coefficient‐based network analysis of microbiome analysis frequently uses relative levels of species or optional taxonomic units, our delta changes in relative abundances of the microbiome could be useful in our study. 19 , 20 Although the delta change might be a result of an increase in taxa or the extinction of other microorganisms, this delta change in relative intensity was employed in another article, 21 and our results help to clarify our understanding of surgical microbiota influences. Network analysis of the data obtained from correlation analysis was performed by measuring stress centrality using Cytoscape v. 3.8.0. All data are expressed as the median and interquartile range and considered statistically significant at P less than .05.
3. RESULTS
3.1. General characteristics
Among the 52 patients in our study, metagenomics profiles from the stool samples of 31 participants (group 1), metabolomics profiles from the serum samples of 38 participants (group 2), and both profiles from the paired stool and serum samples of 17 participants, were obtained. The clinical characteristics of the participants before and 6 months after BS were also compared (Tables S1 and S2). We could not find heterogeneity in clinical characteristics between group 1 and group 2. BMIs in group 1 and group 2 significantly decreased after BS from 36.5 to 29.2 kg/m2 (P = .01) and from 37.8 to 27.0 kg/m2 (P < .01), respectively. We also observed notable improvements in lipid profiles (total cholesterol, triglyceride, HDL cholesterol, and LDL cholesterol levels) and markers of liver functions (aspartate transaminase, alanine aminotransferase, alkaline phosphatase, and γ‐glutamyl transpeptidase levels) from measurements at 6 months postsurgery. Additionally, blood glucose levels in group 1 and group 2 dropped to normal levels (< 100 mg/dl), and most of them became lower than the cut‐off criteria for impaired fasting glucose, a type of prediabetes defined as fasting glucose levels between 100 and 125 mg/dl according to the 2019 treatment guideline for diabetes from the Korean Diabetes Association. 22
3.2. Gut microbiome diversity
To confirm how BS converts the gut microbiome from a pro‐obesity environment to an antiobesity environment, we performed 16S RNA sequencing. First, we analysed the alpha diversity and beta diversity among the microbiome in the stool samples of obese patients collected before the surgery and the stool samples of obese patients 1, 3, 6 and 12 months after surgery. We identified the differential diversity among the groups (P = .054), and the abundances of the observed features tended to increase after BS compared with before surgery (Figure S1A), whereas evenness showed a jagged tendency after surgery (P = .69) (Figure S1B). The Shannon index was similar among all time points (P = .52) (Figure S1C). The intercomparison of microbial composition through beta diversity using two distance measures, Bray–Curtis and weighted Uni‐Frac, showed significantly diverse microbial compositions dependent on BS (P = .003 for both distances) (Figure S1D).
3.3. Changed microbial components after BS
We used MaAsLin2 to run a comparative study to determine which microbiomes were more numerous at different time intervals. Only two species were substantially more common at 1 month after surgery than at the other time points (3, 6, and 12 months after surgery), whereas additional multiple comparisons among all the time points revealed no significant differences in microbiome abundance (data not shown). This result suggests that in this research, surgery‐induced microbial changes were maintained within 1 year after the surgery. Additionally, in linear mixed model analysis to find time‐specific microbiota changes, when we assigned the PREOP (preoperation) as the reference of the factor variable ‘timepoint’, there was a similar microbiota profile after the surgery. However, if we assigned 12MO (12 months after surgery) as the reference for the analysis, then PREOP and 1MO (1 month after surgery) had distinct patterns when compared with the other time points (Figure S2). Because the recuperation time following surgery is typically 2 weeks to 1 month, 23 the results from the first month may be influenced by pathological surgical wounds. As a result of our mixed model results, we anticipated that the microbiome alteration observed 3 months after surgery would be maintained for 12 months. Additionally, we conducted linear discriminant analysis to identify differentially abundant bacterial taxa before and 1, 3, 6 and 12 months after BS (Extended data 1).
For this reason, the single time point we chose, 6 months after the surgery, could represent the microbiota changes attributed to BS. This result implies that the surgically induced microbial alterations in this study were maintained 1 year after the surgery. As a result, the single time point we chose, 6 months after surgery, may indicate microbiome changes related to BS. We performed a paired comparison of the microbiome obtained at different time points to further establish the differential abundance changes in our microbiome data (at 1, 3, and 12 months after BS). As a result, we discovered the following common list of differential microbiota between before surgery and 6 months after surgery versus the other three time points (1, 3, and 12 months after surgery): 1 month (23/27, 85%), 3 months (24/28, 85.7%), and 12 months (22/24, 91.7%), where (the number of simultaneously identified differential microbiomes from the analysis based on each time point were compared with the 6 months after the surgery/differentially abundant microbiome at each time point, percentage %) (Extended data 2).
Specifically, we investigated the microbiome in stool samples before surgery versus 6 months after surgery, the postoperative time point at which the serum samples were analysed for metabolic profiling and multiomics data integration. When we compared these two representative time points, the abundances of the observed features significantly increased after BS (P = .009) (Figure 1A, middle). Similarly, although not significant (P = .12), the Shannon index for patients 6 months after surgery tended to increase compared with that for patients before surgery (Figure 1A, right). Beta diversity between these two representative groups was again estimated by Bray–Curtis distance and weighted UniFrac distance. These analyses showed that the microbiome community between patients before surgery and patients 6 months after surgery was significantly different (P = .001 for both distances) (Figure 1B). Interestingly, the observed features were increased after surgery despite limited gut environments because of surgery, whereas evenness was decreased. This observation suggests that gastric acid, which was reduced because of BS, could orchestrate the balance of the microbiome of patients. Additionally, because an acidic pH prevents a specific microbiome from propagating in the gut, the microbiome community could lose its heterogeneity.
FIGURE 1.
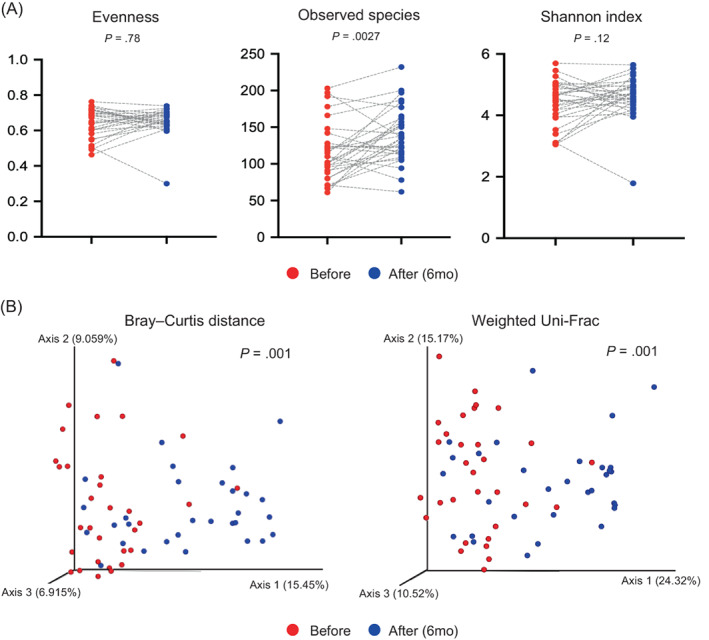
Changes in gut microbiome diversity. A, Boxplot of alpha diversity in patients before surgery and 6 months after surgery by evenness, observed features, and Shannon index. The grey dashed lines indicate the pairs connecting subjects before and after surgery. B, The beta diversity in patients before surgery and 6 months after surgery was compared by Bray–Curtis distance and weighted Uni‐Frac. The significances of alpha and beta diversity were calculated using the Wilcoxon signed‐rank test and PERMANOVA with 999 permutations, respectively
To investigate how the gut microbiome changed after the surgery, we observed the relative abundance of the gut microbiome at the phylum, genus, and species levels (Figure S3). MaAsLin2 analysis revealed that the gut microbiomes of patients before surgery differed from those after surgery (Figure 2; Figure S4). First, at the phylum level, Fusobacteria, Proteobacteria, and Verrucomicrobia were the most increased phyla in stools after surgery; Streptococcus, Oscillospira, and Akkermansia were increased, whereas Bifidobacterium, Turicibacter, and Prevotella were decreased after surgery at the genus level.
FIGURE 2.
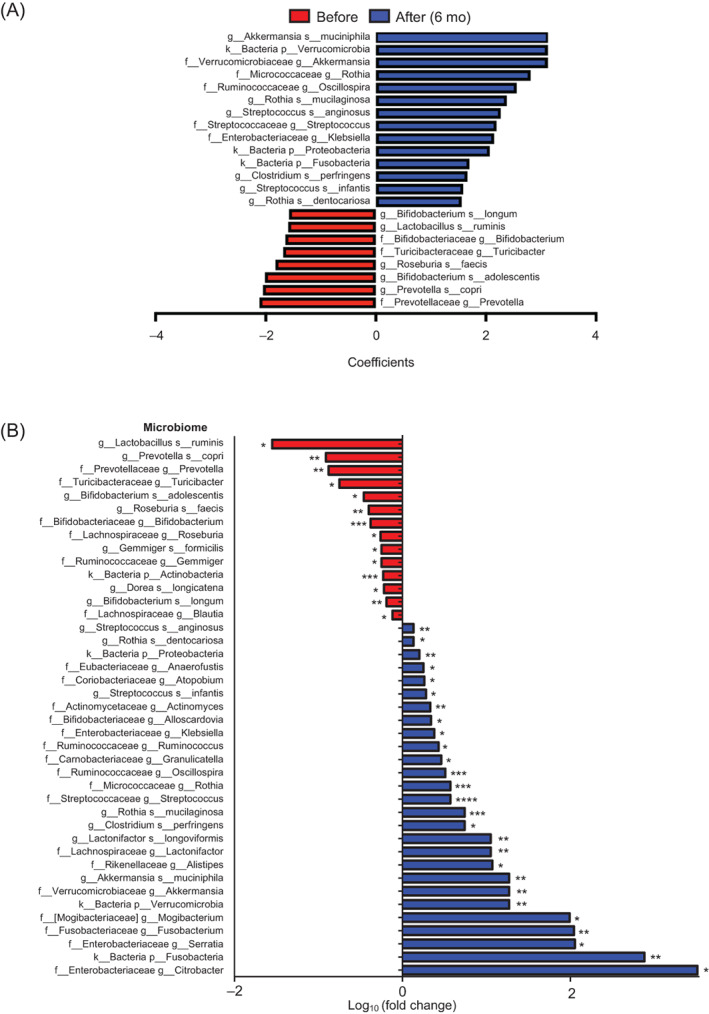
Change in abundances of the gut microbiota after bariatric surgery. A, Result of MaAsLin2 between before and after surgery (6 months) with confounding factors (age, sex, type of surgery, and participant's identifier) at the species level. The x‐axis indicates the coefficients of each time point with the microbiome (|coefficients| > 1.5). Microbiomes with false discovery rate (FDR) < 0.25 are shown. B, Fold changes in the relative abundances of the gut microbiota in patients 6 months after surgery compared with before surgery. Statistical significance levels were determined by the Wilcoxon signed‐rank test. *, **, ***, and **** indicate P < .05, P < .01, P < .001, and P < .0001, respectively. f, family; g, genus; k, kingdom; p, phylum; s, species
Collectively, these results indicated that the increased abundances of species, including Akkermansia muciniphila and Rothia mucilaginosa, after BS could have an antiobesity effect, whereas the decreased abundances of species, including Prevotella copri, Roseburia faecis, and Bifidobacterium longum, after BS could aggravate obesity.
3.4. NMR‐based global metabolomic profiling
Because the metagenomics analysis showed no significant differences among samples at any time point after surgery (Figure S1), we used 6 months as a representative postsurgery time point for our metabolomic profiling analysis using NMR spectroscopy. Forty‐three serum metabolites, including amino acids, organic acids, carbohydrates, and short‐chain fatty acids, were identified, and the concentrations of 38 metabolites were estimated (Table S4).
We employed multivariate statistical analysis to compare metabolites measured before and after BS. Significant metabolic changes after surgery were confirmed by principal component analysis (PCA) and partial least squares discriminant analysis (PLS‐DA) (Figure S5A; Figure 3A). Metabolites, including valine, glycine, tyrosine, leucine, isoleucine, dimethyl sulphone (DMSO2), O‐acetylcholine, glucose, lysine, glutamine, glutamate, mannose, choline, alanine, glycerol, and lactate, were significantly changed after surgery in the PLS‐DA model (variable importance in the projection score > 1) (Figure 3B); their residing pathways were identified as differential metabolic pathways using MetaboAnalyst (Figure S5B).
FIGURE 3.
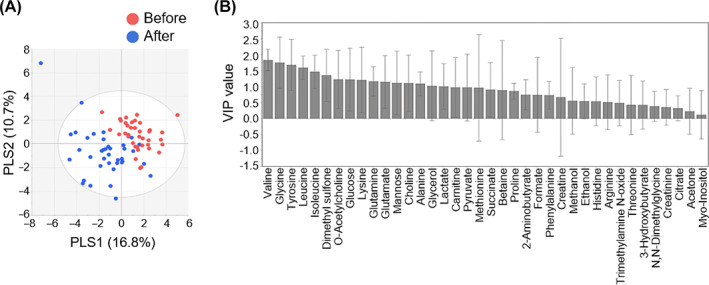
Multivariate statistical analyses of nuclear magnetic resonance (NMR) profiling data. A, Partial least squares discriminant analysis (PLS‐DA) score plot of serum metabolite concentrations obtained from 1H‐NMR spectra reveals a separation before and 6 months after surgery (R 2 X = 27.5%, R 2 Y = 60.3%, Q 2 = 40.9%). B, The variable importance in the projection (VIP) plot shows the metabolites influencing the classification
Among 38 quantified serum metabolites, the concentrations of 15 metabolites were significantly changed after surgery based on the Wilcoxon signed‐rank test (Figure 4; Table S4). After surgery, the concentrations of creatine and glutamine related to amino acid metabolism were significantly altered. The levels of metabolites classified as branched‐chain amino acids (BCAAs) or aromatic amino acids (AAAs), such as isoleucine, leucine, valine, and tyrosine, were significantly decreased after surgery compared with before surgery; similarly, the levels of metabolites related to energy metabolism, such as alanine, glucose, glutamate, lactate, and mannose, were significantly decreased. Interestingly, BCAAs, glucose, and mannose were positively correlated with total cholesterol, LDL cholesterol, and triglycerides, which are clinical indicators associated with lipid metabolism (Figure S6). Additionally, DMSO2 and glycine metabolites that are related to gut microbial metabolism were remarkably increased after surgery and indicated the changes in the gut microbiome community after surgery. In addition, we observed notable changes in lipid metabolites. For instance, the concentration of glycerol was significantly decreased, whereas that of O‐acetylcholine was increased after surgery (Figure 4). Collectively, our results showed that BS caused a significant decrease in the major metabolites involved in BCAA, AAA, and energy metabolism and an increase in microbial‐related metabolites such as DMSO2 and glycine.
FIGURE 4.
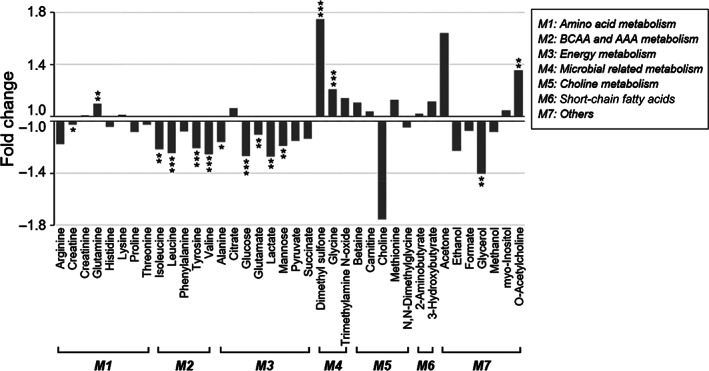
Relative fold changes in the concentrations of serum metabolites 6 months after surgery compared with before surgery. Significant alterations in branched‐chain amino acid (BCAA) and aromatic amino acid (AAA), energy, and microbial‐related metabolism were observed. The vertical axis represents the ratio of metabolic change; a fold change > 1 indicates that the metabolite is increased after surgery (C after/C before), and fold changes from the metabolite decreased after surgery are represented as –C before/C after. Statistical significance levels were determined by the Wilcoxon signed‐rank test. *, **, and *** indicate P < .05, P < .01, and P < .001, respectively
3.5. UPLC/TQ‐MS–based BA analysis
The levels of serum BAs were separately quantified via targeted analysis using UPLC/TQ‐MS. A total of 15 circulating BAs, including six primary and nine secondary BAs, were quantified (Table S5). Less than 30% of coefficient of variables (CVs) from the QC samples indicated the sufficient reproducibility of our experiment. Moreover, because the linear correlation coefficients (R 2) of the calibration curves for each BA were 0.99 or higher, we could obtain reliable quantities (Table S3). The total BA concentrations tended to increase after surgery compared with before surgery; however, they mainly came from increased concentrations of secondary BAs (P < .05) and not primary BAs (Figure 5A). In particular, secondary BAs such as glycodeoxycholic acid (GDCA), taurodeoxycholic acid (TDCA), lithocholic acid (LCA), glycolithocholic acid (GLCA), and taurolithocholic acid (TLCA) significantly increased after surgery, while primary BAs did not show any significant changes (Figure 5B).
FIGURE 5.
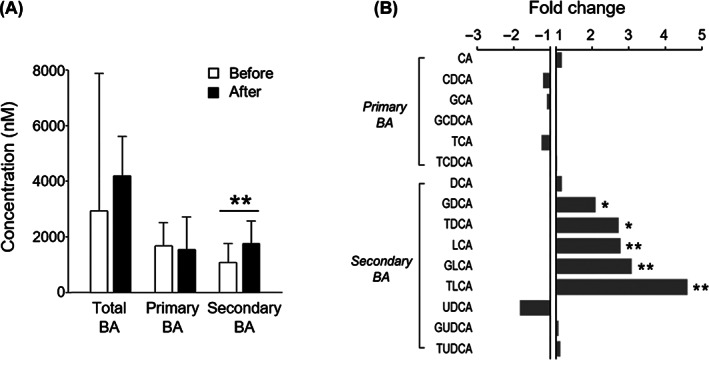
Quantification of serum bile acids (BAs) using ultrahigh‐performance liquid chromatography/triple quadrupole mass spectrometry. A, Total, primary, and secondary BA concentrations before and 6 months after surgery. B, Relative fold changes in BA concentrations 6 months after surgery compared with before surgery. A fold change > 1 indicates that the BA is increased after surgery (C after/C before), and fold changes from the metabolite decreased after surgery are represented as –C before/C after. Statistical significance levels were determined by the Wilcoxon signed‐rank test. *, and ** indicate P < .05, and P < .01, respectively. CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; GCA, glycocholic acid; GCDCA, glycochenodeoxycholic acid; GDCA, glycodeoxycholic acid; GLCA, glycolithocholic acid; GUDCA, glycoursodeoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid
3.6. Integration of the stool microbiome and serum metabolome
We performed partial correlation analysis while adjusting individual age and sex to evaluate the associations between the changes in gut microbial abundances and those in metabolite concentrations after BS (Figure S7A, Table S6). We identified many metabolites highly correlated with alterations in the gut microbiome, such as the Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Fusobacteria phyla.
For the BAs, each subtype was differentially correlated with the gut microbiota. There was a negative association between GDCA and Ros. faecis. TDCA was positively correlated with Gemmiger and Gemmiger formicili and negatively correlated with Mogibacterium, Fusobacterium, and Streptococcus anginosus. LCA was positively correlated with St. anginosus and negatively correlated with Granulicatella, Prevotella, and P. copri. GLCA had positive relationships with Alistipes and Turicibacter, but TLCA had strong negative relationships with Roseburia and Ros. faecis. Interestingly, BCAAs (isoleucine, leucine, and valine) and glucose had strong positive correlations in common with Roseburia and Ros. faecis. BCAAs also had positive correlations with Dorea longicatena. By contrast, isoleucine, leucine, and glucose were negatively correlated with Granulicatella.
Alanine and glutamate, which are related to energy metabolism, showed negative associations with Blautia and Lactobacillus ruminis, respectively. Lactate had positive linear relationships with Granulicatella, Rothia, and Rot. mucilaginosa and a negative relationship with L. ruminis. Mannose was positively correlated with St. anginosus but negatively correlated with Granulicatella and Turicibacter. The changes in glycine and DMSO2, metabolites related to microbial metabolism, were associated with various gut microbiota. Glycine had positive correlations with Actinobacteria and Bifidobacterium and negative correlations with Actinomyces, Rothia, Streptococcus, Atopobium, Blautia, Rothia dentocariosa, Salmonella infantis, and St. anginosus. DMSO2 showed strong positive correlations with Granulicatella, Actinomyces, Rothia, Rot. dentocariosa, Rot. mucilaginosa, and Sa. infantis. Among the metabolites pertinent to the amino acid metabolism pathway, creatine was negatively correlated with Clostridium perfringens and B. longum, and glutamine was negatively correlated with Blautia and St. anginosus. In addition, glycerol was negatively related to Blautia. O‐acetylcholine was positively related to Citrobacter, Serratia, Roseburia, D. longicatena, and Ros. faecis but negatively related to Proteobacteria.
Although we identified a large number of significant associations, we aimed to identify key factors among them. To address this issue, we visualized our data through a correlation coefficient network and quantified the stress centrality measures of each node, which represents either the metabolite or the microbiome. The number of shortest paths flowing through a node is known as stress centrality, and it can be used as a proxy indicator of importance. 24 In this network analysis, we identified several key factors, such as glycine, mannose, LCA, TDCA, glucose, Granulicatella, Fusobacterium, St. anginosus, and B. infantis (Figure S7B).
4. DISCUSSION
There is a scarcity of multiomics studies to observe perturbations caused by surgical intervention on the gut microbiome and the host metabolome for the same individuals. In this study, we explored the distinct features of the intestinal microbiome, host metabolism, and their association attributed to BS. The composition of the microbiome is highly affected by ethnic‐dependent factors, such as the environment, culture, and diet. 25 For this reason, because diverse alterations in the microbiome could be observed depending on the study model of different ethnicities, it is plausible to find differential changes in gut microbiota from Korean participants compared with previous studies recruiting participants of other ethnicities, even if the same BS was performed on patients. In fact, looking at the metabolite and microbiome data of Chinese patients who underwent the same surgery, the metabolites changed similarly to those of Korean patients, but the gut microbiota was significantly different, even although both populations were of Asian descent. 8 It is also known that an altered microbiome influences host metabolism through the regulation of protein degradation, amino acid fermentation, and BA composition. 26 , 27 , 28 , 29 Microbe–microbe interactions play a crucial role in secondary BA biosynthesis. 30 Therefore, studies on the mutual regulation of changes in the gut microbiome and host metabolism after BS enable us to improve obesity management. However, there have been few attempts to clarify the relationships between the changes in the intestinal bacterial community and host metabolome after BS.
Intriguingly, we found that only secondary BAs (GDCA, TDCA, LCA, GLCA, and TLCA), which are metabolized by gut microbiota, 31 were significantly increased in serum after BS. The concentration of total BA, including primary and secondary BAs together, was often increased after BS. 32 , 33 However, in our research, the primary BA concentration did not change. Considering that the gut microbiome can produce secondary BAs, this finding cautiously suggests that increased secondary BAs in blood could be affected by enriched gut microbiota after BS, such as Granulicatella, Alistipes, Mogibacterium, Fusobacterium, and St. anginosus. In colonic epithelial cells, high secondary BA concentrations cause the formation of reactive oxygen species, which cause cell membrane rupture. 34 DCA levels were found to be significantly greater in the serum of patients with colorectal adenomas, 35 while DCA levels were found to be higher in the unconjugated fraction, which was obtained directly from the colon, in the serum of colorectal adenoma patients. 36
Interestingly, the elevation of GLCA was positively correlated with Alistipes, which was enriched in lean individuals. 37 In addition, we found a correlation between the increase in LCA and Granulicatella, a microbiota known to be enriched in stool and mucosa after RYGB. 6 LCA is known to act as an anti‐inflammatory agent in the colon and is negatively correlated with Prevotella, and intriguingly, an included species, P. copri, was reported to increase inflammation. 38 , 39 Collectively, these results suggest how the microbiome altered by BS could affect the metabolism of the host, such as through changes in inflammation; our study shows that after BS in the gut, the changes in BAs and microbiota could mutually influence each other and further modulate the circulating BA profile in host blood.
After BS, we found a significant increase in serum DMSO2, which can be produced in part from methanethiol. 40 Colonic bacteria can convert methionine to methanethiol, a highly reactive molecule that is then transformed to dimethyl sulphoxide (DMSO) and then to DMSO2. Although there have been reports of dimethyl sulphoxide‐related adverse responses in humans, 41 the potential impact of DMSO2 on humans is mainly unknown. However, because the amount of DMSO is proportional to the amount of DMSO2, DMSO2 may have a beneficial or negative effect on humans. An increase in DMSO2 after BS was reported once in a previous study, but its relevance to the gut microbiota has not been reported. 42 In our results, a remarkably elevated DMSO2 concentration was accompanied by increased abundances of Granulicatella, Actinomyces, Rothia, Sa. infantis, Rot. dentocariosa, and Rot. mucilaginosa. Rothia mucilaginosa was observed to be enriched in lean African individuals, 43 while it was abundant in obese Japanese individuals, 44 which supports the idea that the same microbiota can show differential alterations according to host ethnicity. Interestingly, DMSO2 had strong positive correlations with the microbiota classified as Actinobacteria, including Actinomyces and Rothia spp. Thus, the increased DMSO2 concentration in this study may reflect authentic changes in the gut microbiome of Korean patients after BS. Although whether the gut microbiota can affect the systemic level of DMSO2 has not yet been identified, our findings suggest the possibility that the concentration of circulating DMSO2 might be affected by the gut microbiota. More advanced interpretations regarding the relationship between DMSO2 and gut microbiota could be provided from further research.
A remarkable increase in serum glycine concentration was also observed after BS, consistent with previous research. 45 An elevation in glycine concentration can have an antiobesity effect, 46 , 47 while systemic glycine deficiency has been consistently reported in obesity. 48 Alves et al. showed that host glycine metabolism was regulated by the gut microbiota and that gut microbiota dysbiosis may play an important role in reducing glycine availability in obesity‐related metabolism. 48 We found that glycine had correlations with microbiota, including Streptococcus spp., Blautia, Actinomyces, Rothia spp., Bifidobacterium, and Atopobium. In particular, increased glycine had a strong positive correlation with decreased Actinobacteria and Bifidobacterium, which are microbiota associated with obesity. 49 , 50 Interestingly, Blautia is a microbial genus that is altered in response to a Korean diet rather than a Western diet 12 ; therefore, the increase in Blautia after BS may be attributed to the diet behaviours of Korean patients. These observations potentially suggest that increased serum glycine concentration after BS may have been affected by obesity‐associated gut microbiota, and thus, the alterations in circulating glycine concentration may reflect the changes in the intestinal environment specifically in obese Korean patients.
In the current study, serum BCAA concentrations were significantly decreased after BS. BCAAs are known to be elevated in obese patients 42 , 51 and reduced after BS 42 , 45 in many previous studies. Notably, we observed that the reductions in BCAAs and glucose were accompanied by decreased abundances of Roseburia, Ros. faecis, and D. longicatena, which are all classified in the Firmicutes phylum reported to be present in high abundance in obese individuals. 49 , 52 Surprisingly, the identified Roseburia genus, including the species Ros. faecis, has genes related to BCAA biosynthesis in its genome, such as SerA and SerB, and for this reason these microbiota are positively involved in BCAA synthesis. 53 Additionally, Roseburia was associated with glucose metabolism and possesses genes residing in the carbohydrate metabolism pathway.
This study provides insights into the inter‐related changes in host metabolism and the intestinal environment following BS via a wide range of serum metabolome and gut microbiome profiles in obese individuals who underwent BS. There were some limitations as follows: because a comparatively small number of subjects (17 matched participants) were included in the correlation analysis, our results may not be universally reflective. Further experiments are needed to verify the correlation between serum metabolites and gut microbiota in our findings. Additionally, although Lactobacillus is one of the known microbes related to obesity, in our data, the abundance was decreased 6 months after surgery. However, its role in obesity is different by species, as Lactobacillus reuteri is associated with obesity, whereas Lactobacillus rhamnosus contributes to weight loss. Further study would clarify species‐specific effects on obesity. 54 , 55 In the future, whole‐genome shotgun analysis can be used to differentiate microbiota at the strain level. Nevertheless, notably, this study produced an integrated omics dataset from the same individuals to reveal metabolic changes attributed to BS. Furthermore, to the best of our knowledge, our research is the first multiomics study to investigate this subject in a Korean population. Our findings support previous reports on the relationship between the gut microbiota and host metabolome attributed to BS and simultaneously suggest new research hypotheses; additionally, this study extends our knowledge of the potential perspectives related to obesity interventions that target the gut microbiota.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
AUTHOR CONTRIBUTIONS
Yeyoung Han, Gihyeon Kim, Eunyong Ahn, Hansoo Park, and Geum‐Sook Hwang designed the study; Hansoo Park, and Geum‐Sook Hwang supervised the study; Yeyoung Han and Gihyeon Kimanalysed, integrated, and visualized the data and wrote the first version of the manuscript; Eunyong Ahn wrote the final version of the manuscript; Yeyoung Han recruited the subjects and performed the bariatric surgeries; Eunyoung Ha collected samples and anthropometric data; Yeyoung Han, Gihyeon Kim, and Yunjae Kim investigated the data; Yeyoung Han, Gihyeon Kim, Eunyong Ahn, Sunhee Jung, Youngae Jung, Hansoo Park, and Geum‐Sook Hwang interpreted the data; Youngae Jung, Do Hyun Ryu, Hansoo Park, and Geum‐Sook Hwang reviewed and edited the manuscript; and all the authors read and approved the final version of the manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14689.
Supporting information
DataS1
ACKNOWLEDGEMENTS
Korea Basic Science Institute; Grant/Award Number: C270200. National Research Foundation of Korea (NRF) grant funded by the Korean government; Grant/Award Numbers: 2019M3A9D5A01102796 and NRF‐2020R1A2C2007835. Korea Health Technology R&D Project, Ministry of Health & Welfare; Grant/Award Number: HC15C1322.
Han Y, Kim G, Ahn E, et al. Integrated metagenomics and metabolomics analysis illustrates the systemic impact of the gut microbiota on host metabolism after bariatric surgery. Diabetes Obes Metab. 2022;24(7):1224‐1234. doi: 10.1111/dom.14689
Yeyoung Han, Gihyeon Kim, and Eunyong Ahn contributed equally to this work.
Funding information Korea Basic Science Institute, Grant/Award Number: C270200; National Research Foundation of Korea, Grant/Award Numbers: 2019M3A9D5A01102796, NRF‐2020R1A2C200783; Ministry of HEalth & Welfare, Grant/Award Number: HC15C1322
Contributor Information
Gihyeon Kim, Email: kkra12@gm.gist.ac.kr.
Hansoo Park, Email: hspark27@gist.ac.kr.
Geum‐Sook Hwang, Email: gshwang@kbsi.re.kr.
DATA AVAILABILITY STATEMENT
The data described in the manuscript, codebook, and analytic code will be made available upon request pending application and approval by the corresponding author. The sequencing data analyzed in this paper have been deposited in the European Nucleotide Archive (accession no. PRJEB48942, status: confidential).
REFERENCES
- 1. Solomon CG, Manson JE. Obesity and mortality: a review of the epidemiologic data. Am J Clin Nutr. 1997;66(4 Suppl):1044s‐1050s. [DOI] [PubMed] [Google Scholar]
- 2. Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med. 2017;376(3):254‐266. [DOI] [PubMed] [Google Scholar]
- 3. Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta‐analysis. JAMA. 2004;292(14):1724‐1737. [DOI] [PubMed] [Google Scholar]
- 4. Ulker İ, Yildiran H. The effects of bariatric surgery on gut microbiota in patients with obesity: a review of the literature. Biosci Microbiota Food Health. 2019;38(1):3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samczuk P, Ciborowski M, Kretowski A. Application of metabolomics to study effects of bariatric surgery. J Diabetes Res. 2018;2018:6270875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ilhan ZE, DiBaise JK, Dautel SE, et al. Temporospatial shifts in the human gut microbiome and metabolome after gastric bypass surgery. NPJ Biofilms Microbiomes. 2020;6(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang A, Sun H, Wang X. Serum metabolomics as a novel diagnostic approach for disease: a systematic review. Anal Bioanal Chem. 2012;404(4):1239‐1245. [DOI] [PubMed] [Google Scholar]
- 8. Liu R, Hong J, Xu X, et al. Gut microbiome and serum metabolome alterations in obesity and after weight‐loss intervention. Nat Med. 2017;23(7):859‐868. [DOI] [PubMed] [Google Scholar]
- 9. Shen N, Caixàs A, Ahlers M, et al. Longitudinal changes of microbiome composition and microbial metabolomics after surgical weight loss in individuals with obesity. Surg Obes Relat Dis. 2019;15(8):1367‐1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kweon S, Kim Y, Jang M‐J, et al. Data resource profile: the Korea National Health and nutrition examination survey (KNHANES). Int J Epidemiol. 2014;43(1):69‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. OECD . Health at a Glance. New York, NY: Nature Publishing Company; 2017. [Google Scholar]
- 12. Shin J‐H, Jung S, Kim S‐A, et al. Differential effects of typical Korean versus American‐style diets on gut microbial composition and metabolic profile in healthy overweight Koreans: a randomized crossover trial. Nutrients. 2019;11(10):2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park YS, Park DJ, Lee JH, et al. Korean OBEsity surgical treatment study (KOBESS): protocol of a prospective multicentre cohort study on obese patients undergoing laparoscopic sleeve gastrectomy and Roux‐en‐Y gastric bypass. BMJ Open. 2017;7(10):e018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park DJ, An S, Park YS, et al. Bariatric surgery versus medical therapy in Korean obese patients: prospective multicenter nonrandomized controlled trial (KOBESS trial). Ann Surg Treat Res. 2021;101(4):197‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krug S, Kastenmüller G, Stückler F, et al. The dynamic range of the human metabolome revealed by challenges. FASEB J. 2012;26(6):2607‐2619. [DOI] [PubMed] [Google Scholar]
- 16. Davis MP, van Dongen S, Abreu‐Goodger C, Bartonicek N, Enright AJ. Kraken: a set of tools for quality control and analysis of high‐throughput sequence data. Methods. 2013;63(1):41‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin M, Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal. 2011;17(1):10. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 18. Jung J, Ha TK, Lee J, et al. Changes in one‐carbon metabolism after duodenal‐jejunal bypass surgery. Am J Physiol Endocrinol Metab. 2016;310(8):E624‐E632. [DOI] [PubMed] [Google Scholar]
- 19. Agler MT, Ruhe J, Kroll S, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14(1):e1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Faust K, Sathirapongsasuti JF, Izard J, et al. Microbial co‐occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8(7):e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mohammed Y, Kootte RS, Kopatz WF, et al. The intestinal microbiome potentially affects thrombin generation in human subjects. J Thromb Haemost. 2020;18(3):642‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim MK, Ko SH, Kim BY, et al. 2019 clinical practice guidelines for type 2 diabetes mellitus in Korea. Diabetes Metab J. 2019;43(4):398‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher BL. Comparison of recovery time after open and laparoscopic gastric bypass and laparoscopic adjustable banding. Obes Surg. 2004;14(1):67‐72. [DOI] [PubMed] [Google Scholar]
- 24. Ahn E, Lee J, Han J, Lee S‐M, Kwon K‐S, Hwang G‐S. Glutathione is an aging‐related metabolic signature in the mouse kidney. Aging. 2021;13(17):21009‐21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma N, Ma X. Dietary amino acids and the gut‐microbiome‐immune axis: physiological metabolism and therapeutic prospects. Compr Rev Food Sci Food Saf. 2019;18(1):221‐242. [DOI] [PubMed] [Google Scholar]
- 27. Mayneris‐Perxachs J, Fernández‐Real JM. Exploration of the microbiota and metabolites within body fluids could pinpoint novel disease mechanisms. FEBS J. 2020;287(5):856‐865. [DOI] [PubMed] [Google Scholar]
- 28. Martin AM, Sun EW, Rogers GB, Keating DJ. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front Physiol. 2019;10:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diether NE, Willing BP. Microbial fermentation of dietary protein: an important factor in diet–microbe–host interaction. Microorganisms. 2019;7(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. bioRxiv. 2017. https://www.biorxiv.org/content/10.1101/229138v1 [Google Scholar]
- 31. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30(3):332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pournaras DJ, Glicksman C, Vincent RP, et al. The role of bile after Roux‐en‐Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology. 2012;153(8):3613‐3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sayin SI, Wahlström A, Felin J, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(2):225‐235. [DOI] [PubMed] [Google Scholar]
- 34. Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bayerdörffer E, Mannes GA, Richter WO, et al. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology. 1993;104(1):145‐151. [DOI] [PubMed] [Google Scholar]
- 36. Bayerdörffer E, Mannes GA, Ochsenkühn T, Dirschedl P, Wiebecke B, Paumgartner G. Unconjugated secondary bile acids in the serum of patients with colorectal adenomas. Gut. 1995;36(2):268‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Verdam FJ, Fuentes S, de Jonge C, et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity. 2013;21(12):E607‐E615. [DOI] [PubMed] [Google Scholar]
- 38. Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151(4):363‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ward JBJ, Lajczak NK, Kelly OB, et al. Ursodeoxycholic acid and lithocholic acid exert anti‐inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol. 2017;312(6):G550‐G558. [DOI] [PubMed] [Google Scholar]
- 40. Engelke UF, Tangerman A, Willemsen MA, et al. Dimethyl sulfone in human cerebrospinal fluid and blood plasma confirmed by one‐dimensional (1)H and two‐dimensional (1)H‐(13)C NMR. NMR Biomed. 2005;18(5):331‐336. [DOI] [PubMed] [Google Scholar]
- 41. Kollerup Madsen B, Hilscher M, Zetner D, Rosenberg J. Adverse reactions of dimethyl sulfoxide in humans: a systematic review. F1000Research. 2018;7:1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gralka E, Luchinat C, Tenori L, Ernst B, Thurnheer M, Schultes B. Metabolomic fingerprint of severe obesity is dynamically affected by bariatric surgery in a procedure‐dependent manner. Am J Clin Nutr. 2015;102(6):1313‐1322. [DOI] [PubMed] [Google Scholar]
- 43. Eid HM, Wright ML, Anil Kumar NV, et al. Significance of microbiota in obesity and metabolic diseases and the modulatory potential by medicinal plant and food ingredients. Front Pharmacol. 2017;8:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andoh A, Nishida A, Takahashi K, et al. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J Clin Biochem Nutr. 2016;59(1):65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wijayatunga NN, Sams VG, Dawson JA, Mancini ML, Mancini GJ, Moustaid‐Moussa N. Roux‐en‐Y gastric bypass surgery alters serum metabolites and fatty acids in patients with morbid obesity. Diabetes Metab Res Rev. 2018;34(8):e3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pérez‐Torres I, Ibarra B, Soria‐Castro E, et al. Effect of glycine on the cyclooxygenase pathway of the kidney arachidonic acid metabolism in a rat model of metabolic syndrome. Can J Physiol Pharmacol. 2011;89(12):899‐910. [DOI] [PubMed] [Google Scholar]
- 47. Sekhar RV, McKay SV, Patel SG, et al. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care. 2011;34(1):162‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alves A, Bassot A, Bulteau AL, Pirola L, Morio B. Glycine metabolism and its alterations in obesity and metabolic diseases. Nutrients. 2019;11(6):1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koliada A, Syzenko G, Moseiko V, et al. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017;17(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abenavoli L, Scarpellini E, Colica C, et al. Gut microbiota and obesity: a role for probiotics. Nutrients. 2019;11(11):2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Newgard CB, An J, Bain JR, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, Lobley GE. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate‐producing bacteria in feces. Appl Environ Microbiol. 2007;73(4):1073‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hillman Ethan T, Kozik Ariangela J, Hooker Casey CA, et al. Comparative genomics of the genus Roseburia reveals divergent biosynthetic pathways that may influence colonic competition among species. Microb Genom. 2020;6(7):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Million M, Maraninchi M, Henry M, et al. Obesity‐associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii . Int J Obes. 2012;36(6):817‐825. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Wang T, Yan H, Lu Y, et al. Anti‐obesity effect of lactobacillus rhamnosus LS‐8 and lactobacillus crustorum MN047 on high‐fat and high‐fructose diet mice base on inflammatory response alleviation and gut microbiota regulation. Eur J Nutr. 2020;59(6):2709‐2728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DataS1
Data Availability Statement
The data described in the manuscript, codebook, and analytic code will be made available upon request pending application and approval by the corresponding author. The sequencing data analyzed in this paper have been deposited in the European Nucleotide Archive (accession no. PRJEB48942, status: confidential).