

Abstract
The liver carries out a wide range of functions ranging from the control of metabolites, nutrient storage, and detoxification to immunosurveillance. While inflammation is essential for the tissue remodeling and maintenance of homeostasis and normal liver physiology, constant exposure to dietary and microbial products creates a niche for potentially prolonged immune activation and unresolved inflammation in susceptible host. Failure to restrain inflammation can lead to development of chronic liver diseases characterized by fibrosis, cirrhosis and eventually liver failure. The liver maintains close interactions with numerous organs which can influence its metabolism and physiology. It is also known that oral cavity microenvironment can influence the physiological conditions of other organs and emerging evidence implicates that this could be true for the liver as well. Presence of chronic inflammation and dysbiotic microbiota is a common feature leading to clinical pathology both in periodontitis and chronic liver diseases (CLDs). In fact, known CLDs appear to have some relationship with periodontitis, which impacts the onset or progression of these conditions in a bidirectional crosstalk. In this review, we explore the emerging association between oral‐gut‐liver axis focusing on periodontitis and common CLDs including nonalcoholic fatty liver disease, chronic viral hepatitis, liver cirrhosis, and hepatocellular cancer. We highlight the immune pathways and oral microbiome interactions which can link oral cavity and liver health and offer perspectives for future research.
Keywords: chronic viral hepatitis, cirrhosis, fatty liver disease, hepatocellular cancer, liver, periodontal, periodontitis
1. INTRODUCTION
Periodontitis is an oral chronic inflammatory disease which has been associated with a wide range of systemic disorders some of which include diabetes, inflammatory bowel diseases, cardiovascular diseases, cancer, neurodegeneration, rheumatoid arthritis, and chronic liver diseases. 1 , 2 , 3 , 4 , 5 , 6 , 7 Periodontitis and these systemic conditions all share some degree of similarity in their pathophysiology which is characterized by a long‐lasting deregulated inflammatory response.
Today it is recognized that genetic predisposition, exposure to acquired risk factors and epigenetic changes throughout life play a decisive role for the development of immune‐ based disorders by modifying the host's susceptibility. 8 , 9 Given that time is required for their onset, the inflammatory imbalance resulting from one disease could directly alter the pathophysiology of the other or they could simply occur in a host sharing a similarly disturbed immunological background favorable for the development of general chronic disorders. Regardless of the perspective to be taken into account, periodontitis has an important place, whether as a warning time marker for the predisposition of other chronic diseases, or as an independent factor for their development or clinical worsening with direct effects on patients' uality of life, treatment outcome, and prognosis.
The increasing knowledge also suggests that imbalances in the microbiota‐immunity interactions may contribute to the onset of a multitude of immune‐mediated diseases. 10 While how and when this dysbiosis develops in different organs is still unknown, oral bacteria such as Porphyromonas gingivalis and Fusobacterium nucleatum have been associated with various disorders distant from the oral cavity. 11 , 12 , 13 In this context, interactions between oral and gut microbiomes are complex, dynamic, and context‐dependent since both sites are part of the multivariate environment of the gastrointestinal tract. Under physiological conditions they can keep a fine‐tuned balance across the lifespan, but failures in this crosstalk due to a host‐microbe disequilibrium have the potential to activate inflammatory networks in different organs. 14
The liver is among the organs that can be more affected by these biological networks and pathogenic disturbances since it maintains anatomical proximity and intense physiological interdependence with the intestine via metabolic exchange and translocation of bacteria. For this reason, interest in the so‐called oral‐gut‐liver axis has emerged, along with the idea that the oral dysbiosis caused by unrestrained inflammation could affect the pathophysiology of chronic liver diseases via blood circulation or enteral route. 1 , 15 , 16 , 17 The activation of a susceptible host immune system by pathogen‐ and damage‐ associated molecular patterns (PAMPs; DAMPs) in the periodontal milieu could induce the systemic overexpression of an array of pro‐inflammatory cytokines and chemokines hence potentially impacting the hepatic metabolism. 11 In parallel, oral bacteria in the context of periodontal dysbiosis could also invade the gut, spreading the adverse events to intestinal microbiome, and likely affecting liver functions. 18 Similarly, liver dysfunction could also exacerbate the on‐going pathology and clinical outcomes within the oral cavity. 15 Herein, we provide an overview of the current clinical and epidemiological evidence for the association between periodontitis and chronic liver diseases, whilst we track their related mechanisms and discuss gaps to be filled by future research.
2. LIVER: AN ORGAN WITH MULTIPLE FUNCTIONS
The liver is the largest solid organ in the body and through the uptake, metabolism, and secretion of solutes performs an important role in the metabolism of lipids (lipogenesis), carbohydrates (gluconeogenesis), proteins (synthesis of heptoglobin, albumin, and clotting factors), haemoglobin, bile salts, iron, vitamins, cupper, and drugs. 19 , 20 Most of the chemical levels in the blood leaving stomach and intestine are regulated by the liver exposing it to a variety of dietary products, pathogens, endotoxin and microbial components coming from the gastrointestinal tract by the portal vein. 20 , 21 Thus, liver serves as an important immunological site for cytokine signaling and acute‐phase protein production thereby participating in the maintenance of the equilibrium between immune surveillance against pathogens and tolerance to innocuous antigens and commensal bacteria. 22
Hepatocytes are the dominant parenchymal cells in the liver constituting about 70%‐ 85% of its volume and responsible to regulate part of its immune functions. Hepatocytes are also major producers of the liver extracelular matrix (ECM). In its physiological state, liver ECM is mainly composed of collagens, glycoproteins, proteoglycans, and hyaluronic acid. 20 , 23
Hepatic stellate cells (HSCs) are another major cell type in the liver. They are primarily located in the space of Dissé between the sinusoidal endothelial cells and hepatic epithelial cells, and account for about 5%‐8% of the liver volume. Under homeostatic conditions, HSCs are non‐parenchymal quiescent cells that contain numerous vitamin A lipid droplets, constituting the largest reservoir of this vitamin in the body. 19 , 24 , 25
On the immunological side, Kupffer cells are resident macrophages (Mφs) of the liver and form the main cell population. Their functions include the clearance of particles, immune complexes, senescent red blood cells, and endotoxins. 26
3. CHRONIC LIVER DISEASES
Chronic liver disease (CLD) is an umbrella term which is used for reference to a set of pathologies characterized by a progressive deterioration of liver functions over six months or more. Recent reports suggest that CLDs account for approximately 2 million deaths per year worldwide (3.5% of all deaths), although exact statistics are not always available due to lack of a clear, well‐established separation of CLD burden according to etiology and stage of disease across population‐based studies. 27 , 28 A broad spectrum of factors, such as viral infections, toxin exposure, alcohol abuse, autoimmune diseases, genetic and metabolic disorders, can trigger persistent inflammation with a consequent destruction of the liver parenchyma leading to the release of aspartate‐ and alanine‐ aminotransferases (AST; ALT), important markers of hepatic injuries. 27 , 28 , 29 The following events include gradual replacement of the liver structures by fibrous tissues, and presence of compensatory hepatocyte hyperplasia which can culminate in end‐stage liver diseases, namely cirrhosis and hepatocellular carcinoma. 30 , 31 The pathological fibrosis occurs mainly as a consequence of a continuous excessive deposition of the ECM by injured mesenchymal cells. Due to persistent inflammation, ECM is subjected to qualitative changes in its composition with an accumulation of proteins, especially collagen type I and III. 23 , 32 This process occurs because HSCs undergo transformation to become profibrogenic myofibroblasts, which can regulate matrix deposition and resorption whilst also have contractile properties. After activation, HSCs can enter in a perpetuation phase that comprises many functional properties including proliferation, fibrogenesis, chemotaxis, contractility, retinoid loss, and cytokine expression. 21 , 25 , 33 , 34 , 35 These subsequent phenotypic changes eventually determine the course of the disease. The Figure 1 summarizes the CLD stages.
FIGURE 1.
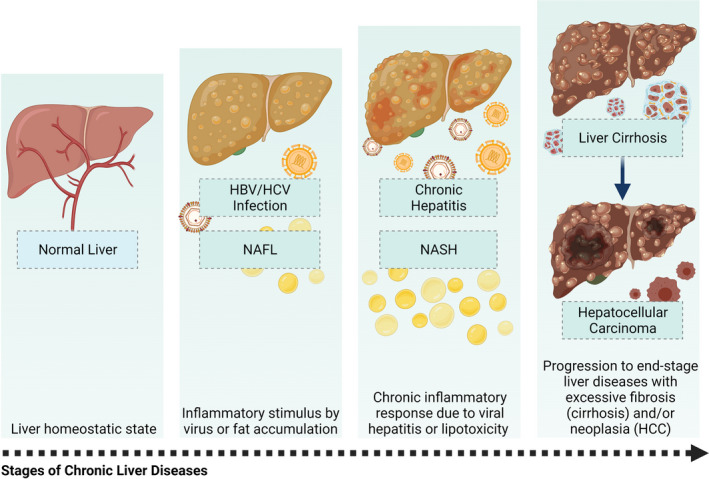
Clinicopathological stages of chronic liver diseases. The liver in its homeostatic state performs its normal physiological functions, which can be altered by viral infections with HBV or HCV and by the accumulation of fat (NAFL) due to systemic dysfunctions associated with different conditions such as diabetes and obesity. Non‐resolution of viral infection leads to chronic hepatitis, and the continuous accumulation of fat in the liver increases the chances of developing NASH via lipotoxicity events. These conditions are characterized by an underlying chronic inflammation that can lead to irreversible liver damage through an increase in the fibrotic response (cirrhosis) which can be accompanied by liver cell malignancy (HCC). This figure was created using BioRender
4. THE ASSOCIATION BETWEEN PERIODONTITIS AND CHRONIC LIVER DISEASES
The initial studies investigating the relationship between periodontitis and CLDs date back to 50s and 60s. 36 , 37 Today, the interest in evaluating the interaction between oral and hepatic diseases has grown even further as we develop a more comprehensive understanding of the onset and progression of chronic diseases. Although occurring in distant parts of the body, these pathologies share several etiological factors on their onset and portray an unbalanced inflammatory response in a susceptible host. Therefore, it is not surprising that recent epidemiological and clinical studies suggest an association between CLDs and periodontitis (Figure 2).
FIGURE 2.
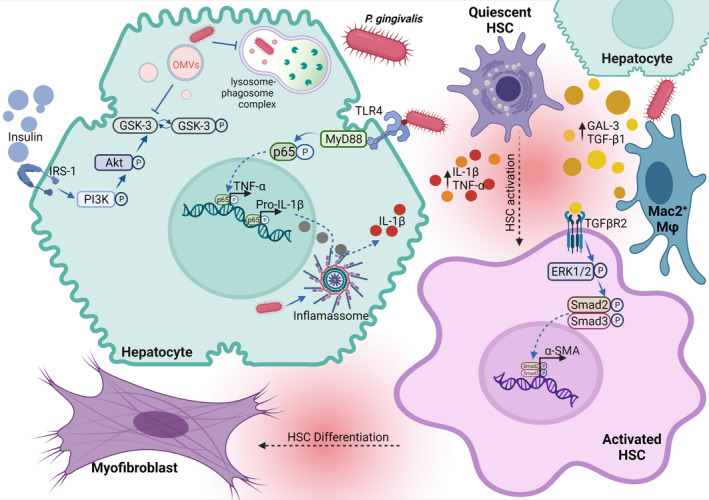
Proposed effects of P gingivalis on hepatic cells. Once the bacteria reache the liver, its invasion into hepatocytes is triggered by the P gingivalis induced‐impaired function of the lysosome‐autophagosome complex (LC3‐/LAMP2+). P gingivalis outer‐membrane vesicles can be released into the cytoplasm and reduce the conversion of phospho‐GSK3 (inactive form) into GSK3 (active form), subsequently inhibiting the glycogenesis mediated by the Insulin/IRS‐1 receptor signaling pathway. In parallel, P gingivalis‐derived LPS triggers TLR‐4 signaling cascade that increases transcription of pro‐inflammatory genes, such as TNF‐ α and IL‐1β. The released inactive pro‐IL‐1β can be converted into active IL‐1β, given that P gingivalis‐derived LPS also up‐regulates the inflammasome complex formed by NLRP3 and Caspase‐1. A boost in the concentration of pro‐inflammatory cytokines in the hepatic milieu removes HSCs from their quiescent state. Further, HSCs can also be activated by the release of galectin‐3 (Gal‐3) and TGF‐β1 by hepatocytes and Mac2+ Kupffer cells upon LPS exposure. Subsequently, the TGF‐β1/TGFβR2 downstream cascade in activated HSCs up‐regulates the transcription of α‐SMA which is responsible for HSC differentiation into myofibroblasts. This figure was created using BioRender
4.1. Non‐alcoholic fatty liver disease and Periodontitis
Non‐alcoholic fatty liver disease (NAFLD) is a condition characterized by the storage of excess fat in the liver in the absence of excessive alcohol consumption, a process that in most patients occur due to metabolic risk factors, such as obesity, diabetes mellitus, and dyslipidemia. 38 , 39 NAFLD encompasses a spectrum of liver diseases including simple fatty liver, also called nonalcoholic fatty liver (NAFL), and nonalcoholic steatohepatitis (NASH). NAFL is characterized by the excess accumulation of triglycerides in hepatocytes (>5% fat content in the liver; referred to as hepatic steatosis) with little or no inflammation or liver cell damage. By contrast, NASH is defined as the presence of hepatic steatosis along with lobular or portal inflammation, and ballooning degeneration of hepatocytes (a form of cell death) that entail liver injuries with or without fibrosis. 38 , 40 The rate of NAFLD is estimated to be 24% worldwide and the disease incidence continues to rise likely due to an increase in the prevalence of its risk factors such as obesity and diabetes. Based on the prevalence of biopsy‐ proven NASH in patients with NAFLD (6.7%–29.9%), it is assumed that approximately 1.5% to 6.5% of the general US population has NASH. 28 , 41 , 42
Lipotoxicity is one of the key pathophysiological processes in NAFLD and occurs when the liver is exposed to high levels of circulating free fatty acids, as well as high levels of insulin, which is produced to compensate for systemic insulin resistance in obesity and/or diabetes, as well as other highly toxic lipid metabolites such as ceramides, diacylglycerols, and oxidized cholesterol metabolites. 43 In this lipotoxic milieu, the liver itself also contributes to hepatic steatosis by synthesizing lipids from carbohydrates by de novo lipogenesis, which in a homeostatic liver is not a primary source of lipid. 38 , 44 The demand for metabolizing excess fatty acids places strain on hepatocyte mitochondria, which over time increases the production of reactive oxygen species (ROS). 45 Periodontitis and NAFLD share numerous risk factors and the progression of both diseases can be aggravated by insulin resistance and the increased systemic pro‐inflammatory profile found in diabetic and obese individuals. Additionally, both diseases also appear to affect the course of other systemic disorders such as cardiovascular diseases. 11 , 39 In fact, the overproduction of ROS is a common finding in periodontitis patients as well. 46
While recent epidemiological studies and systematic reviews investigating the relationship between periodontitis and NAFLD suggest possible associations, a causal link between the two conditions is still elusive. 16 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 Akinkugbe et al 48 investigated whether periodontitis could contribute to the onset of NAFLD through evaluating data from the Pomerania‐based cohort. The incidence of NAFLD and periodontal disease status was monitored in 2,623 participants for 7.7 years follow‐up. After adjusting for confounders such as excessive alcohol consumption, hepatitis diagnosis in past years, and use of hepatotoxic medications, the NAFLD incidence was moderately elevated with a ratio of 1.60 (1.05‐2.43) in subjects who at baseline had ≥30% of periodontal sites with CAL≥3mm. 48
In another cohort, Kuroe et al 55 investigated whether periodontitis could affect the progression of NAFLD towards fibrotic liver injuries in a Japanese population. 341 subjects with NAFLD diagnosis without liver fibrosis at baseline were followed‐up for 5‐years. The results indicated that having moderate to severe periodontitis increased the risk of developing liver fibrosis with an odds ratio of 2.06 (0.89‐4.76). Similarly, in a national Finnish cohort of 6,165 individuals who have been monitored for ~12‐years, Helenius‐Hietala and collaborators showed that the presence of advanced periodontitis and diagnosis of NAFLD at baseline increased the incidence of severe liver events including hospitalization due to liver disease, liver‐related death, or a diagnosis of primary liver cancer. 56
Most of these studies used hepatic ultrasound and/or measurement of serum levels of general liver injury markers, such as AST and ALT to diagnose and monitor NAFLD without making any distinction between NAFL and NASH. The inflammatory component of liver injury is present in NASH but not in NAFL and the actual diagnosis of NASH requires microscopic evaluation of liver to check for ballooning degeneration, which is an obstacle for populational‐based studies and identifying associations with periodontitis. 27 , 38 To overcome this, Alazawi and colleagues conducted a longitudinal study which monitored several parameters associated with NAFLD and periodontitis in a group of 45 NAFLD cases. 49 The investigators performed biopsies to differentiate those with NAFL and NASH and defined NASH with minimum or no fibrosis, or NASH with significant fibrosis. The results demonstrated increased periodontitis prevalence in patients with liver biopsy‐proven NASH fibrosis. Similarly, Iwasaki et al 51 reported increased prevalence of NAFLD which was accompanied by higher stages of periodontitis severity and elevated serum levels of CRP in a cross‐sectional study. Consistently, Akinkugbe and collaborators reported positive association between presence of periodontitis, prevalence of NAFLD and elevated serum CRP levels. 47 Together, these findings indicate an association between NAFLD and periodontitis.
Studies also assessed the association of periodontal pathogens and other oral bacteria with NAFLD/NASH. Alazawi et al 49 evaluated the link between periodontitis and liver fibrosis in a populational‐based study including 8,153 NHANES III participants and reported significant association between increased levels of Streptococcus noxia and S oralis serum antibodies and hepatic steatosis. Komazaki et al 57 investigated serum antibody response against periodontal bacteria, including Aggregatibacter actinomycetemcomitans, F nucleatum, and P gingivalis, in 52 individuals diagnosed with NAFLD and reported significant correlation between anti‐ A actinomycetemcomitans IgG antibody titers and the levels of AST. However, the results were not adjusted for confounding variables. Similarly, Nakahara et al 58 demonstrated a significant correlation between fibrosis progression and serum antibody titers against P gingivalis fimA type 4 in biopsy‐proven NAFLD patients and suggested that this pathogen could regulate the transition from NAFL to NASH. In another study, Furusho et al 59 detected P gingivalis in 21 of 40 liver biopsy specimens collected from patients with NASH, corresponding to 52.5%. Further, P gingivalis‐positive cases presented higher total fibrosis scores including perisinusoidal and periportal fibrosis. 59 Whether this periodontal pathogen could migrate from the oral cavity to the liver and contribute to the underlying intestinal dysbiosis found in NAFLD/NASH patients is still not known and need to be further investigated.
Few recent clinical studies investigated the impact of periodontal treatment on NAFLD/NASH outcome. Yoneda et al 60 performed an interventional study to assess whether periodontal treatment in NAFLD patients with periodontitis could regulate NAFLD biochemical parameters and reported a reduction in ALT and AST levels following periodontal therapy within 3 months. Recently, Kamata and colleagues published a protocol for a multicenter randomized clinical trial aiming to evaluate whether periodontal treatment decreases liver damage and endotoxemia in patients with NAFLD and the results of this study are expected to be available in the near future. 61
As the emerging evidence supports a possible link between NAFLD and periodontitis, future studies are needed to determine the power of these associations and the biological mechanisms connecting these conditions. It is also possible that the effect might be bidirectional and NAFLD can affect periodontitis outcomes as well, which again needs to be further investigated. In addition, so far populational‐based cohorts have only been carried out in Europeans and Japanese, which limits the external validity of these findings and warrants investigations in other cohorts.
4.2. Chronic hepatitis and Periodontitis
Chronic hepatitis B (CHB) infection is characterized by the detection of HBsAg antibodies and viral genomic materials in the serum, resulting from enduring active replicative and transcriptional activities of the human hepatitis B virus (HBV). The HBV is a member of the Hepadnaviridae Family, and its long‐term infection strongly depends on age at acquisition. 62 It is estimated that worldwide 257 million (3.5%) people have CHB, and the National Health and Nutrition Examination Survey (NHANES) estimates the US prevalence as 840,000 (0.35%). 27 , 63 This is the highest prevalence among hepatitis viruses despite existence of an effective vaccine to prevent this infection. From a public health perspective, large‐scale HBV vaccination would be expected to reduce the incidence of new infections and their complications.
Chronic hepatitis C (CHC) is defined by active long‐term carriage of the human hepatitis C virus (HCV), a hepatotrophic RNA flavivirus. Most of the individuals infected with HCV fail to clear the infection, which commonly leads to a stage of CHC. The global prevalence of HCV infection is estimated in 1%, with 71 million people living with CHC. 27 , 64 Despite the availability of testing and a new generation of highly effective specific direct‐acting antivirals, approximately 400,000 people still die each year due to CHC related complications. 27
The association between chronic hepatitis and periodontitis was initially discussed by the fact that viruses, such as HCV and HBV, can be found in saliva. Oral fluids are effectively used for large‐scale HBV detection and are known routes of transmission for this virus. 65 Oral transmission of HCV, on the other hand, is a matter of debate. This virus is transmitted mainly by the parenteral route but in 30%‐40% of CHC patients the source of infection is unidentifiable, and, besides its hepatotrophic nature, the HCV has a strong lymphotropism. 66 , 67 Not surprisingly, this virus can be traced in fluids that have mononuclear blood cells in their composition, including saliva and gingival crevicular fluid (GCF); some studies reported prevalence of HCV ranging from 31% to 100% in saliva and GCF samples with a great variability according to the study population and method of viral RNA extraction and detection. 68 , 69 , 70 Its potential sources could include transmigrated leukocytes through periodontal epithelium, salivary glands, and peripheral blood mononuclear cells from bleeding tissues in the oral mucosa. Interestingly, serum viral load and periodontal status do not appear to affect the detection of HCV RNA in the saliva of patients with CHC. 71 Given that these viruses can be traced in GCF and saliva, their potential effect on the development and/or progression of periodontal diseases in individuals affected by their respective chronic infections has also been investigated, 72 although no definitive conclusion has been reached.
Extrahepatic manifestations are common in chronic viral hepatitis, and it is thought that they can be related to complex immunological mechanisms (de) regulated by viruses. For example, in the oral cavity, lichen planus and xerostomia have been reported in both CHC and CHB patients. 73 A cross‐sectional study assessed possible impact of HCV on periodontal diseases where individuals with chronic infection by HCV were compared to the general population by using the Community Periodontal Index Treatment Needs (CPITN) and reported no differences in clinical parameters between groups. 74 Some of the limitations of the study included lack of calibration among clinical evaluators and collection of some of the data through a telephone interview survey. 75 Surlin et al 76 compared 13 systemically healthy periodontitis patients versus 11 asymptomatic CHC patients and reported similar degrees of periodontitis severity despite higher levels of IL‐1α and IL‐1β in the GCF of the CHC group. This study, however, did not report clinical attachment loss and relevant confounding demographic data. In another study, the degree of inflammation was reported to be higher in gingival biopsies from periodontitis patients with chronic HCV infection compared to those without HCV infection using optical coherence tomography. 78 Intriguingly, Marrone and collaborators followed a cohort of patients with implants of at least 5 years of function for the frequency of mucositis and peri‐implantitis and reported that those with a history of hepatitis were more susceptible to develop peri‐implantitis. 79
When compiling these data, it is difficult to conclude whether chronic liver infections by HBV or HCV could play a role in the etiopathogenesis or progression of periodontitis. The scientific evidence available on this theme is very scarce and only a few epidemiological studies have been published which did not address important factors. Notwithstanding, it is increasingly clear that the mammalian virome, which includes diverse commensal and pathogenic viruses, can evoke a broad range of immune responses. 80 As part of this notion, a two‐way interaction between chronic hepatitis and other inflammatory conditions like rheumatoid arthritis has been assessed. 81 , 82 These conditions have similarities with periodontitis, which push the periodontology to move forward in order to perform well‐designed epidemiological and mechanistic studies to investigate possible link.
4.3. Cirrhosis and Periodontitis
Liver cirrhosis is an end‐stage CLD that is characterized by the formation of regenerative nodules surrounded by fibrous bands in response to several years of inflammation and fibrosis. Clinically, it can be presented in compensated or decompensated stages. Decompensated stage coexists with life‐threatening complications, such as chickenpox hemorrhage, ascites, spontaneous bacterial peritonitis, and hepatic encephalopathy. 31 Globally, the number of patients with cirrhosis is increasing, and currently it accounts for 1.16 million deaths per year, which makes it the 11th most common cause of death. In Western and industrialised countries NAFLD/NASH is overtaking chronic hepatitis as the primary etiology for cirrhosis, whereas in China and other Asian countries CHB continues to be a leading underlying cause. 27 , 83
Cirrhosis develops following multiple lifelong injuries and can have several etiologies, including alcohol abuse, chronic hepatitis, and NAFLD/NASH, 31 thereby the assessment of another risk factor, such as periodontitis in this process is not an easy task. Earlier studies reported poor oral hygiene to explain the higher prevalence, extent, and severity of periodontitis in patients with cirrhosis. 84 , 85 However, the current evidence suggests this crosstalk can be far more complex, and periodontitis could impact the course of this liver disorder and vice versa.
In a Danish cohort study, Ladegaard Grønkjær et al 86 followed 184 patients diagnosed with cirrhosis (most of them alcohol‐ or cholestatic‐cirrhosis) for approximately 1 year and reported that the diagnosis of severe periodontitis at baseline predicted a higher cirrhosis‐related mortality with an adjusted odds ratio of 2.29 (1.04‐4.99). This study highlights the need for closer attention to oral health in the clinical care of patients with cirrhosis. The results of a recent systematic review showed increased association between cirrhosis without any distinguished underlying etiology and periodontitis, which could reach up to a OR of 2.28 (1.5‐3.48). 50 Notably, Costa et al 87 reported that individuals with cirrhosis (72% due to alcohol abuse) exhibited two times increased prevalence of periodontitis than healthy controls independent of alcohol use status. These findings suggest that there might be a bidirectional effect between these two conditions and that the pro‐inflammatory environment related to cirrhosis might impact periodontitis as well. In support of this, Jaiswal et al 88 reported a strong positive correlation between periodontal breakdown and serum alkaline phosphatase level in cirrhotic patients with or without periodontitis.
Studies that evaluated the benefits of periodontal treatment for patients with cirrhosis are still scarce. Lins et al 89 reported that subjects with cirrhosis (most of them due to CHC) that underwent periodontal treatment presented a lower mortality when compared to non‐treated patients. In this study, they did not mention which periodontal treatment was performed, as well as the follow‐up period, which limits a generalization of the findings. Notably, a recent clinical trial conducted by Bajaj and colleagues evaluated the impact of periodontal treatment on different hepatic parameters in cirrhotic patients with multiple underlying etiologies including CHC, alcohol abuse, and NASH. 1 Periodontal intervention reduced serum levels of endotoxin, LPS binding protein, and IL‐6, as well as salivary levels of IL‐1β, principally in cases of cirrhosis with hepatic encephalopathy one‐month post‐therapy. In addition to this systemic immunomodulatory effect, an improvement in the cognitive functions was also reported after the therapy. 1 The overall findings of this study underscore the periodontal treatment as a target to reduce inflammation and endotoxemia in these individuals, which needs to be further investigated in larger cohorts and long‐term trials.
Remarkably, there is also evidence of possible crosstalk between the oral microbiota and liver function in cirrhosis. Jensen et al 90 evaluated the subgingival microbiome of 21 patients suffering from cirrhosis and periodontitis and reported the abundance of Firmicutes and to a lesser extend Actinobacteria and Bacteroidetes. In the same study, bacteria considered as periodontal pathogens, like Porhyromonas gingivalis, T forsythia, and T denticola, were found in low abundancy. The authors suggested that a compromised immune system in cirrhosis could lead to dysbiosis and render commensal bacteria pathogenic. In another study, Ghapanchi et al 91 reported increased level of E coli in the saliva of cirrhotic patients when compared to age‐ and sex‐matched healthy controls suggesting that oral cavity could possibly serve as a reservoir and alter the intestinal microenvironment. Consistently, Qin et al 18 reported that major changes in the gut microbiota in individuals with cirrhosis could be associated with the invasion of the gut by oral bacterial species. The study showed a high proportion of denoted metagenomic species belonged to Veillonella and Streptococcus taxa in the gut microbiome of cirrhotic patients compared to healthy controls. 18 While the origin of these species can be both oral cavity and small intestine, an elegant genetic tracking revealed that most of them were of buccal origin. It was further hypothesized that an altered epiphenomenon allowed by an impaired gastric acid and bile secretion that is prevalent in cirrhosis could render the gut more permissible or accessible to oral bacteria. 18 , 92 Among the cirrhosis‐enriched species in the intestine were S anginosus, Veillonella atypica, Veillonella dispar, Veillonella sp oral taxon, and Campylobacter concisus. Indeed, other studies also showed a higher abundance of Streptococcaceae and Veillonellaceae in the intestinal microbiome of chronic hepatitis and cirrhotic patients albeit the findings for Veillonellaceae seem to be conflicting. 93 , 94 , 95 One of the major findings of these studies was to highlight the translocation of oral microbiota as a factor for the gut dysbiosis in cirrhosis. More recently, Dubinkina and colleagues reported increased oral microbes, S constellatus, S salivarius, V atypica, V dispar, and V parvula in the stool of cirrhotic patients. The authors also observed an increase of typical commensal species, namely Bifidobacterium (B longum, B dentium, and B breve), Streptococcus (S thermophilus and S mutans), and Lactobacillus (L salivarius and L crispatus). 96 Consistently, Bajaj et al 17 described that the salivary dysbiosis in cirrhosis is characterized by an increased abundance of Enterobacteriaceae, and reduction of autochthonous taxa (Lachnospiraceae, Ruminococcaceae). It was noted that this microbial composition could predict a higher rate of hospitalization regardless of disease severity, in a similar fashion to that reported for intestinal dysbiosis. Furthermore, in a proof‐of‐concept clinical trial, it was demonstrated that the periodontal treatment modulates the gut microbiome of patients with cirrhosis. 1 Specifically, the authors reported reduced levels of Streptococcaceae and Enterobacteriaceae and increased proportion of Ruminococcaceae in patients’ faeces when compared to non‐ treated cirrhotic control. Streptococcaceae cluster was also decreased in the salivary microbiome. 1 Increased levels of enterobacteria can weaken the gut barrier by reducing the expression of tight‐junction proteins, which increases the permeability and translocation of intestinal bacteria and their products to the liver. 97 Therefore, decreased levels of these bacteria as a result of periodontal treatment seem to provide beneficial effect. Intriguingly, the study also found higher levels of Veillonellaceae and Lactobacillaceae (commonly associated with a healthy periodontal status) and lower abundance of Porphyromonadaceae and Fusobacteria (commonly associated with a pathogenic periodontal environment) in the saliva of cirrhotic patients, which remained unaltered even after periodontal therapy. 1 It is also of note that higher salivary levels of IL‐6, IL‐1β, and secretory IgA along with lower levels of lysozyme and histatins in cirrhosis further indicate an overall mucosal‐immune imbalance and strengthen the notion that oral‐gut‐liver axis can be an alternative source of inflammatory load in these patients. 1
4.4. Hepatocellular carcinoma and Periodontitis
Hepatocellular carcinoma (HCC) is the sixth most frequent neoplasia and the fourth leading cause of cancer‐related deaths worldwide, causing over 800,000 deaths annually. About 80% of HCC cases is due to CHB and CHC infections and the rest involves alcohol abuse, NASH and NAFLD. 27 , 28 These factors can act independently or together to increase susceptibility for cirrhosis, which in turn creates an environment permissive for the development of HCC. Therefore, current screening recommendations focus primarily on patients with cirrhosis. 30 In fact, around 90% of HCC cases exhibit cirrhosis, yet less than 5% of patients with cirrhosis progress to HCC. Emerging paradigm proposes that these end‐ stage liver diseases could originate from independent pathological responses controlled by different molecular mechanisms rather than a sequential series of events. 98
Epidemiological studies suggest a positive correlation between periodontitis and overall cancer risk including HCC with systemic inflammation serving as the main focus for biological plausibility. 99 However, additional prospective studies are needed to better elucidate the strength of these relationships. A cohort based on NHANES III showed an association between severe periodontitis and mortality risk for oral digestive cancers including HCC, but the authors discussed that the study had limited power to evaluate mortality risks for specific cancers. 100 To address this gap, a recent prospective study reported sufficient power to examine an association between tooth loss and liver cancer incidence. In a Finnish cohort with 29,096 male smokers who were followed up for 17 years, Yang et al 101 reported that individuals presenting 11‐31 permanent teeth loss or edentulism at the baseline (surrogate markers of periodontitis) were more likely to develop HCC; HR of 1.42 (1.01‐1.98) and 1.45 (1.00‐2.10), after adjusting for confounding factors, respectively. Since this study only included smokers, its generalizability is limited, albeit it outlines the impact of a possible history of periodontitis on hepatocarcinogenesis. One of the plausible mechanisms for this association might be the increased levels of oxidizing compounds in the circulation due to the inflammatory destruction of periodontal tissues. 46 Consistently, a case‐control study reported 25.8% higher levels of reactive oxygen species (ROS) in HCC individuals with moderate/severe periodontitis compared to those without periodontitis. 102 Patients with periodontitis exhibited worse cancer staging and liver function when compared to non‐ periodontally affected controls, indicating poor prognosis. 102 Yet, further studies are warranted to fully establish this association.
4.5. Liver transplant recipients and Periodontitis
Liver transplantation (LT) is a lifesaving procedure for patients with chronic end‐stage liver disease and acute liver failure when there are no available medical and surgical treatment options. Most countries use the model for end‐stage liver disease (MELD) score for LT listing and recipient selection. Globally, LT is the second most common solid organ transplantation. 27 Cirrhosis accounts for most of the LTs performed in adults, although the underlying etiologies have changed over the past few years. According to the Organ Procurement and Transplantation Network/Scientific Registry of Transplant Recipients (OPTN/SRTR) 2018 annual data, cirrhosis secondary to alcohol abuse or other/unknown factors (often NASH) contributed for 27.4% and 33.3% of the LTs, respectively. HCC is other well recognized indication for LT, accounting for about 16.9% of these procedures. 103 In the past, chronic HCV infection was responsible for the largest number of LTs, but such cases have reduced dramatically due to the rapid advance in the treatment of HCV with highly effective and well tolerated drugs. 104 At a long‐term, 5 and 10‐year patient survival outcomes have been shown more favorable for those with non‐HCC diagnoses. 103 Post‐transplant long‐term complications are highly related to the immunosuppressive treatment. Increased risk for osteoporosis or altered bone metabolism in LT recipients result from a combination of low bone mineral density before transplantation due to hepatic osteodystrophy, malnutrition, and steroid use after transplantation. 105 The combination of tacrolimus, mycophenolate, and steroids is reported to be the predominant regimen, and must be optimized according to each underlying etiology, as well as the control of each associated risk factor, in order to achieve the long‐term success of the therapy. 103
Patients with end‐stage chronic liver disease candidates for LT in general have worse periodontal indexes and higher prevalence of severe periodontitis. 106 , 107 Thus, it is routine clinical practice to treat periodontitis prior to LT. 108 This is based on the premise that untreated dental disease may pose a risk for infection and sepsis, although there is no evidence that this has occurred in LT candidates or recipients. Much more common, changes in periodontal tissues ensue following LT likely because of immunosuppressive treatment. LT is associated with an overall medical and functional recovery for most patients but a longitudinal improvement in periodontal parameters is unclear. Thus, the question to be answered is whether there could be any changes in the status of periodontal diseases in post‐ transplanted (post‐LT) recipients. In this regard, Machtei et al 109 evaluated the periodontal status of 17 post‐LT patients in a non‐interventional prospective study of 10 years and showed that periodontal probing depth was significantly lower within the post‐LT group after the followed‐up period. A clinical trial assessed the impact of the nonsurgical periodontal therapy on cyclosporine‐ induced gingival overgrowth. After 12 months intervention, total inflammatory cells, gingival vessels, and fibroblast proliferation rate were reduced in gingival biopsy specimens, indicating a strict biofilm control program as critical to maintain periodontal health in post‐LT patients taking this immunosuppressant. 110 Further, a recent observational study reported that pre‐ and post‐LT patients have similar needs for periodontal treatment and comparable rates of moderate or severe periodontitis. 111 Given that there is interplay between periodontal tissues and hepatic pathophysiology, it still remains to be determined the outcome of an uncontrolled periodontitis on LT survival.
5. ORAL‐GUT‐LIVER AXIS
Considering the evidence from the epidemiological and clinical studies discussed above, it is plausible that an unbalanced oral‐gut‐liver axis could trigger further liver inflammation. Yet, while the precise source or direction of these pathological changes remains elusive, it is widely accepted that occurrence of dysbiotic microbiota coupled to dysregulated immune response drives the disease outcomes. 15 , 97 , 112 , 113 One of the most important links between gut microbiota and extra‐intestinal organs is the bidirectional gut‐liver axis. 114 This reciprocal interaction is established through the vascular route of the portal vein that carries gut‐derived products directly to the liver, and the liver feedbacks by the route of bile acids and antibody secretion to the intestine. 92 , 115 , 116 , 117 Thus, the liver shapes the intestinal microbiome which mutually impacts liver functions, especially in the context of dysbiosis. 117 A dysfunctional gut microbiota weakens intestinal tight junctions and accordingly increases mucosal permeability, which enables bacterial metabolites and MAMPs/DAMPs to reach the liver via portal circulation and in several cases promote endotoxemia. These molecules then act directly on hepatocytes, Kupffer cells, and HSCs through activation of pattern recognition receptors, including TLRs and NOD‐like receptors, hence triggering downstream pro‐inflammatory cascades. 97 , 113 , 118 Therefore, given the role of an intestinal dysfunction in the pathophysiology of CLDs and considering the aforementioned data reporting gut microbiome associated changes linked to oral bacteria in patients with CLDs, it is likely that oral‐gut‐liver axis could trigger or substantiate hepatic inflammation which warrants further investigations. In this regard, pre‐clinical assays using well established disease models are useful tools to dissect the possible biological mechanisms linking oral cavity to liver pathology which will be discussed in the following sections.
6. PLAUSIBLE MECHANISMS CONNECTING THE ORAL‐GUT‐LIVER AXIS
6.1. Effect of periodontal pathogens on liver pathophysiology
Several studies have used in vivo disease models in mice, rats, and rabbits to assess the effect of major periodontal bacteria, particularly P gingivalis, on liver tissues using various delivery routes such as oral gavage, or injection of live bacteria and/or bacterial components into dental pulp, intraperitoneal, intravascular, and intragingival spaces. The results of majority of these investigations support the hypothesis that keystone periodontal bacteria may in fact induce direct liver damage. 58 , 59 , 60 , 119 , 120 , 121 , 122 , 123 , 124 In a murine model of NAFLD induced by high‐fat diet, Yoneda et al 60 noted increased body and liver weight accompanied by exacerbated lipid accumulation in liver tissues, and elevated ALT and liver triglyceride levels following injection of P gingivalis Type II fimA via jugular vein. These hepatic changes were not observed with other non‐pathogenic periodontal bacteria such as S mutans, S sanguinis, and S salivalius, suggesting that a NAFLD progression could be a P gingivalis‐specific event. 60 In a similar model in rabbits, intragingival injection of LPS from P gingivalis promoted progression from NAFLD to NASH by boosting hepatic lobular inflammation and serum levels of gamma‐glutamyl transferase (GGT) which is a surrogate marker of liver injuries. 124 In another study, Fujita et al 125 tracked biodistribution of radiolabeled P gingivalis LPS from gingiva to the brain, liver, kidney, and spleen and noted increased predilection of this endotoxin for the liver. The study also reported elevated lipid deposition, presence of large fat droplets, and focal necrosis with inflammatory cells, and numerous ballooning degenerations around the central vein which resemble histopathological characteristics compatible with NASH following gingival application of LPS. 125 Corroborating the in vivo data, human HepG2 hepatocytes which were challenged with oleic acid to mimic NASH milieu and P gingivalis‐derived LPS exhibited increased accumulation of intracellular lipids, upregulation of MyD88 expression and phosphorylation of p65 and JNK, and increased release of pro‐inflammatory cytokines (eg IL‐1β, IL‐8, and TNF‐α) compared to those cells that were not treated with LPS. 126 Further, the lipid accumulation was reduced following suppression of this signaling pathway which suggests a possible role of MyD88‐NF‐kB cascade in the NASH pathology associated to this pathogen. 126
In an odontogenic infection model that used high‐fat diet fed mice and bacteria injection into the dental pulp, P gingivalis was able to exacerbate NASH lesions and increased the levels of MCP‐1, TNF‐α and IL‐17 in the liver. 59 There was also increased infiltration of liver paranchyma with Mac2+ (Galectin‐3) Mφs, a marker which is considered as one of the most important molecules to induce liver fibrosis towards cirrhosis. 127 Consistently, Nakahara et al 58 reported upregulation of NLRP3 and Casp‐1 in hepatocytes by P gingivalis‐LPS which could potentially contribute to the overproduction of pro‐inflammatory cytokines. It was noted that the disruption of liver metabolism by the bacterial infection and inflammatory microenvironment can lead to an increase in the proportion of oleic acid (C18:1) and palmitoleic acid (C16:1) which further contributes to the lipotoxicity associated to hepatic steatosis in NASH. 13 Consistently, by using the same odontogenic infection model, Nagasaki et al 121 reported presence of P gingivalis in hepatocytes and increased liver fibrosis. The authors further evaluated plausible mechanisms by which this pathogen could promote liver pathology through its effect on HSC and hepatocytes. The data showed upregulation of Smad2, Smad3, and ERK1/2 phosphorylations and TGF‐β1 production in P gingivalis infected HSCs. Following bacterial challenge, HSCs also produced increased Gal‐ 3, which activated downstream pathways such as Smad phosphorylation and α‐SMA expression, a key factor that leads to myofibroblastic differentiation. In a paracrine route, live P gingivalis‐infection and LPS could sustain this pathological microenvironment by inducing TGF‐β1 and Gal‐3 production from hepatocytes as well. 121
There is also evidence implicating the participation of lysosome‐autophagosome complex in P gingivalis mediated liver damage. In a model of hepatic steatosis using oleic acid, it has been noted that P gingivalis co‐localizes with LC3‐/LAMP2+ compartments in the hepatocytes. 128 LC3 is an autophagosome marker, while LAMP2 is representative for lysosomes. It is speculated that the lipid accumulation and subsequent inhibition of the lysosome‐autophagosome complex might provide a favorable niche for this periodontal pathogen to survive longer in the NAFLD hepatic environment. 128
Insulin resistance is pivotal for hepatic lipotoxicity in NAFLD/NASH. 43 Glycogenesis is one of the major activities performed by the liver which regulates glucose metabolism 20 and there is evidence that P gingivalis can interfere with this process likely through its gingipains/proteases which can reach liver via circulating bacterial outer membrane vesicles (OMVs). 123 In fact, Arg‐X (RGP, Arg‐gingipain) and Lys‐X (KGP, Lys‐gingipain) specific proteases have been shown in the liver sinusoids of mice following intraperitoneal injection of P gingivalis OMVs. 123 Using in vivo fluorescence monitoring of liver, heart, lung, spleen, and kidney, labeled P gingivalis OMVs demonstrated a long‐standing distribution only to the liver. 123 Once internalized into hepatocytes, P gingivalis decreases the rate of Insulin Receptor Substrate‐1 and Akt/GSK‐3β phosphorylation induced by insulin which drastically inhibits glycogen deposition. 129 It has been hypothesized that accumulation of these vesicles and proteases in the liver possibly attenuate insulin sensitivity and generate higher blood glucose levels. Consistently, in vitro stimulation of hepatocytes revealed that P gingivalis OMVs, but not LPS, regulated Akt/GSK‐3β signaling pathway further suggesting a critical role of gingipains on insulin sensitivity. 123 Similar to their effect on the liver and glucose metabolism, P gingivalis OMVs and gingipains have been shown to participate in various other pathologies distant from the oral cavity including Alzheimer's disease, rheumatoid arthritis, cardiovascular disease, diabetes, and even cancer. 130 To the best of our knowledge, no study has yet evaluated whether this pathogen would have any specific role in the viral hepatitis chronicity or in the etiopathogenesis of HCC, although its role in the carcinogenesis of other types of cancer has already been described. 131
The oral cavity is a complex microbial ecosystem; thus, from the perspective of species‐specific mechanisms, other bacteria hypothetically can have an important role in the regulation of liver functions. To assess this, germ‐free mice were orally administered with either oral pathobionts (P gingivalis, Filifactor alocis, and F nucleatum) or bacteria associated with periodontal health (Actinomyces naeslundii, S mitis, and V rogosae) and monitored for bacterial accumulation, antibody levels and inflammatory mediator expression. 132 The only bacteria isolated from liver specimens was F nucleatum whereas strong systemic antibody response was detected for P gingivalis and V rogosae. In addition, the endoplasmic reticulum stress‐related gene DNA Damage‐Inducible Transcript‐4 was upregulated in the liver samples from the group orally infected with the pathogenic consortium. 132 V rogosae was the only microorganism detected within the intestinal ecosystem. In fact, the oral commensal consortium increased the expression of TNF‐α in small intestine samples, whereas pathogenic species upregulated IL‐17a. 132 Collectively, this finding shows that a symbiotic relationship between these bacteria and the host can be reached in the oral environment, but not in the intestinal mucosa, where their presence can cause an immune imbalance and possible pathology.
While all of these investigations are valuable to understand the mechanistic link between periodontitis and systemic complications, the results should be interpreted with caution. One key limitation of these preclinical studies is that putative periodontal pathogens are not part of the murine microbiota and the parenteral administration, and the high microbial concentration used in several assays may not be a good representation of the normal human physiology. Additionally, for future studies, it will be important to include control infections with other microorganisms to understand whether this effect is bacterial‐specific or simply a widespread effect of bacterial inoculation.
6.2. Effect of periodontal pathogens on liver pathophysiology via gut induced dysbiosis
The oral cavity harbors the second‐largest and diverse microbiota after the gut and interacts with various microbial populations at different sites of the body. 77 Indeed, individuals with periodontitis may also present with intestinal dysbiosis, although the precise source or direction of this oral‐gut dysbiosis route remains elusive. 133 , 134 Studies with bacteria associated with periodontitis, especially strains of P gingivalis and A actinomycetemcomitans, have shown that these microorganisms can promote liver dysfunction by inducing gut dysbiosis with changes in the composition of the intestinal microbiota similar to those observed in patients with CLDs (Figure 3).
FIGURE 3.
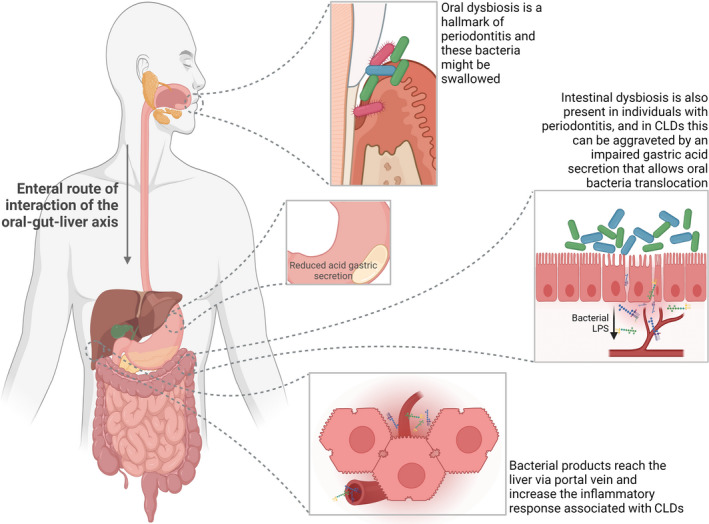
Proposed role of oral microbiome in oral‐gut‐liver axis. Individuals with periodontitis present microbial dysbiosis in different compartments of the oral cavity, especially in periodontal pockets, which is also accompanied by dysbiosis in the intestinal microbiota. In cases of chronic liver diseases, particularly cirrhosis, swallowed bacteria of the oral niche can reach the intestine since an underlying reduction of gastric acid secretion can occur in these conditions. An overgrowth of oral bacteria in the intestine triggers an inflammatory response that increases the permeability of the intestinal mucosa. Then, MAMPs, particularly LPS, can cross the damaged gut epithelial barrier and reach the liver via portal circulation where they trigger or exacerbate pre‐existing chronic liver inflammation. This figure was created using BioRender
In mice with a normal diet, the oral gavage administration of P gingivalis increased intestinal abundance of Bacteroidetes over Firmicutes which was then followed by down‐ regulation of the tight junction proteins in the small intestine and up‐regulation of the proinflammatory cytokines IL‐6, IL‐12β, IFN‐γ, and IL‐17c in the large intestine which collectively favor the breakdown of the gut barrier. 119 Tracking a flow of events along a biological axis, P gingivalis infection also increased the number of lipid containing hepatocytes. There was also elevated number of transcripts of TNF and IL‐6, as well as fat storage inducing transmembrane protein‐2 and perilipin‐2 in the liver, both of which strongly associated with lipid droplet formation. 119 Another study used intravenous injection of sonicated P gingivalis in high fat diet fed mice and noted that lysates of the pathogen reduced the presence of Ruminococcaceae in the gut, whereas L johnsonii and L reuteri became overrepresented. 122 Notably, these gut imbalances induced by P gingivalis match with the clinical findings for most of the CLDs. 93 , 135 , 136 , 137 , 138 Moreover, sonicated P gingivalis increased liver steatosis and upregulated the hepatic expression of acetyl‐CoA carboxylase, glucokinase, glucose transporter‐2, and glucose‐6‐phosphate, as well as of a set of genes related to fatty acid metabolism. 122 A shift in the gut microbiota composition and increased liver steatosis were also observed during oral infection with A actinomycetemcomitans. This periodontal pathogen impaired glucose tolerance and insulin resistance in mice feeding a normal or high fat diet. 57 In addition, marked lipid accumulation was found in the liver, which consistently exhibited higher gene transcription of Acetyl‐CoA carboxylase, an enzyme involved in hepatic lipid metabolism, and glucokinase, a master glucose sensor for regulation of insulin release. 57 The results of these investigations are similar to those reported in P gingivalis infection model, and, by examining the intestinal microbiome following A actinomycetemcomitans infection, there was significantly less abundance of Turicibacter, member from the phylum Firmicutes, in faeces. 57
Collectively, there is general agreement between clinical and experimental studies suggesting a specific role for some periodontal pathogens in the regulation of the oral‐gut‐liver axis, either through hematogenous or enteral route, especially in the context of NAFLD/NASH progression.
6.3. Host susceptibility in oral‐gut liver axis
As in any chronic inflammatory disease, it is the host response which dictates the clinical outcomes in the course of periodontitis and CLDs. 13 , 139 , 140 Therefore, it will likely be the host which sets the relationship between these conditions as well. Yet, the studies are still scarce characterizing specific immune pathways in relation to oral‐gut‐liver axis. A study in which a cohort was followed over 8 years revealed an association between elevated serum levels of ALT, a well‐known marker of liver injury, and higher periodontal probing depth. 141 Consistently, using murine ligature‐induced periodontitis model Mester et al 142 reported extensive microvesicular steatosis around the central liver vein as well as expansion of the portal spaces with deposition of mature fibrous tissue. The study also revealed elevated levels of malondialdehyde (a marker of lipid peroxidation), and MMP8 in serum, gingiva, and liver. 142 As these emerging investigations are starting to shed light on the possible immunological mechanisms which can favor the progression of liver diseases in patients with periodontitis, and vice versa, it will be important to carefully dissect the information from the existing literature for each individual condition and plan future studies to fully elucidate the interplay between oral and liver diseases (see Figure 4).
FIGURE 4.
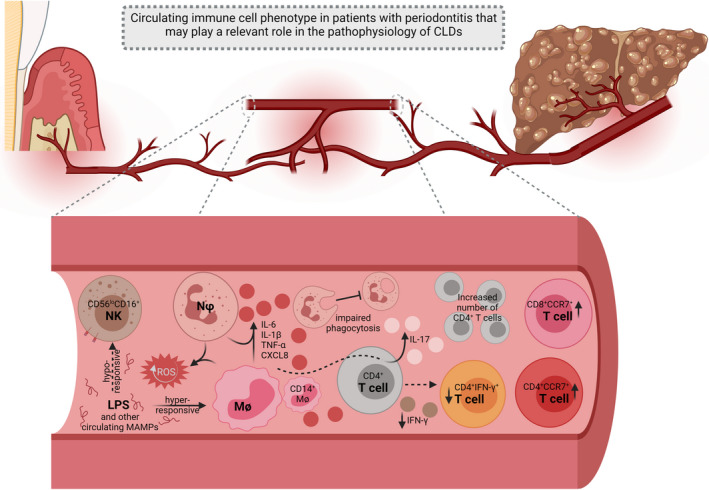
Proposed role of host response in oral‐gut‐liver axis. An altered systemic immune profile in periodontitis can exacerbate chronic liver inflammation and/or virus survival in hepatic cells. Circulating CD56loCD16+ Natural Killer (NK) cells are anergic in the peripheral blood of individuals with periodontitis, which can favor HBV and HCV survival. Neutrophils (Nφs) and monocytes (Mϕs) are hyper‐responsive and synthesize large concentrations of pro‐ inflammatory cytokines upon circulating LPS stimulus, hence potentiating this effect in distant organs, especially in cases underlied by systemic dysfunctions, such as NAFLD/NASH. These cells also exhibit a reduced phagocytic capacity, which can compromise the immune surveillance being carried out by the liver. Additionally, Nφs and Mϕs constituvely produce more reactive oxygen species (ROS) which can overlap the elevated concentration of these metabolites in HCC and overall CLDs. Higher concentrations of IL‐6 and IL‐1β released by peripheral CD14+ Mϕs prompt CD4+ T cells to produce IL‐17, a known pro‐fibrogenic mediator, which can trigger a pro‐cirrhotic milieu. This can be potentialized by the increased number of circulating CD4+ T. However, these cells have diminished ability to synthesize IFN‐y and accordingly reduced peripheral populations of CD4+IFN‐γ+ cells are found in periodontitis; this phenotypic change further compromises anti‐virus defense. The systemic increase in the number of CD4+CCR7+ and CD8+CCR7+ T cells may also contribute to an exacerbation of the inflammatory response in CLDs. This figure was created using BioRender
In the course of periodontal disease, circulating neutrophils (Nφs) and monocyte‐ derived Mφs isolated from the peripheral blood of patients with periodontitis elicit a hyperinflammatory phenotype characterized by elevated basal expression of proinflammatory cytokines (eg IL‐1, IL‐6, TNF, CXCL8) and ROS. 143 , 144 , 145 These responses are further aggravated upon priming of these cells with LPS. 146 , 147 In addition, these cells also exhibit altered phagocytic activity, 145 which is further decompensated by hyporesponsiveness to chemoattractants. 148 Similarly, Mφs isolated from patients suffering with NAFLD also exhibit a hyperinflammatory phenotype and reduced phagocytic function. 26 , 74 While the consequences of these cellular alterations on liver diseases are not yet known, these common pathways may likely drive the clinical pathology. Specifically, under the recent perspective an altered training of the innate immune response could mediate the association of periodontitis with other inflammatory comorbities but we do not know yet if this applies to oral‐gut‐liver axis. 13
Mφs are considered as the major cells mediating NAFLD where resident Kupffer Mφs play essential role during the early phases of liver injury through the production of TNF‐α and chemoattractants. During the later stages of the disease, CCR2+ monocyte‐derived Mφs are recruited from peripheral blood and acquire a pro‐inflammatory phenotype that increase the susceptibility to endotoxemia and amplify the development of NASH and fibrosis. 74 , 149 , 150 , 151 In addition, the overproduction of ROS metabolites is also a shared feature of the NAFLD lipotoxicity and hepatocarcinogenesis 38 , 45 which can likely be a mechanism to accelerate the progression of NASH and HCC. Consistently, a recent study using a mass cytometry immunoassay in the peripheral blood of patients with moderate to severe periodontitis and healthy controls revealed that monocytes from periodontitis patients display a hyperinflammatory phenotype through the canonical activation of the NF‐kB/ERK pathway, while CD56loCD16+ NK cells unexpectedly stay hypo‐responsive characterized by reduced response to GM‐CSF and TNF‐α challange. 152 Considering NK cells are effectors of virus clearance and cancer surveillance, 153 it can be speculated that their anergy could contribute to HBV/HCV survival and HCC development. In fact, this overlapping feature could be further aggravated by the presence of other dysfunctions in the NK phenotype in CHB and CHC. For example, during chronic hepatic infections, NK cells display reduced capacity to produce IFNγ, which is an important effector function for HCV control, and an upregulated expression of the apoptosis‐inducing ligand TRAIL that can induce virus‐specific CD8+ T cells depletion. 154 , 155 , 156
At the intersection of the innate and adaptive immune systems, it is worth noting that circulating CD14+ monocytes of periodontitis patients also induce CD4+ T cells to overproduce IL‐17, prompting them to a Th17 response. 157 Similarly, IL‐17 is a known pro‐fibrogenic interleukin in the pathophysiology of liver cirrhosis and its excess is also related to intestinal barrier dysfunctions associated with the disease. 158 In fact, both cytotoxic and helper T cells appear to be altered in the peripheral blood of individuals with severe periodontitis and a recent study noted an increase in the population of both CD8+CCR7+ and CD4+CCR7+ central memory T cells. 159 While the total number of CD4+ T cells is increased in individuals presenting with severe periodontitis, these cells exhibit reduced ability to produce IFN‐γ even after stimulation with pathogens. In addition, the number of circulating CD4+IFN‐γ+ is also reduced in this patient population. 160 , 161 IFNs are key for the immunity against viruses, and a deficiency in their synthesis could potentially compromise the antiviral immune defense in chronic hepatitis. HBV is termed as a “stealth virus” since its polymerase inhibits the transcription of Type I IFNs (IFNα and IFNβ), which together with type III IFNs (IFNλ) are major mediators of the antiviral immune system. 162 Therefore, HBV has very strong interference mechanisms for the IFN response 162 , 163 and CHB progression could be altered due to this unbalanced systemic environment existing in periodontitis patients.
Emerging evidence also implicates microRNAs as critical regulators of the immune response based on their ability to interfere with the post‐transcriptional expression of multiple target genes. Periodontitis patients seem to have an altered systemic profile of microRNAs, and their serum or peripheral blood mononuclear cells exhibit an up‐regulation of several of these small RNAs, including miR‐9, miR‐155, miR‐203a, miR‐147, miR‐182, miR‐183, miR‐ 664a, miR501, and miR‐21. 164 , 165 An array of microRNAs has also been reported in the pathophysiology of chronic hepatitis, progression from NAFLD to NASH, and hepatocarcinogenesis, 166 , 167 , 168 and a differential expression of miR155 seems to be overlapping periodontal and liver diseases. 164 , 167 , 168 Given that these small RNAs can elicit diverse effects by reaching distant organs, presumably they can be another source of interaction which deserves more attention in future research which evaluates oral and liver diseases.
7. CONCLUSIONS AND PERSPECTIVE
Collectively, the clinical evidence shows an association between periodontitis, NAFLD/NASH and cirrhosis, although a causal relationship is unclear. To fill this gap in the knowledge, more mechanistic and clinical studies are needed to assess whether the control of periodontitis in these patients could have any beneficial effect on the clinical outcomes of the liver diseases. To date, periodontal treatment seems to alter the composition of the intestinal microbiota of cirrhotic patients and modulate their systemic immune response. Evidence also suggests that the intestinal exposure to oral commensal microbiome has the potential to exacerbate the gut dysbiosis underlying NAFLD and cirrhosis. This exposure can lead to an unprecedent increase in the abundance of oral species in the intestine, hence triggering an overstimulation of the immune response with reflexes in liver physiology via portal circulation. The periodontitis‐associated dysbiosis seems to aggravate the liver pathology, but the mechanisms of ectopic colonization of the gut by oral bacteria are underexplored. Therefore, further studies are needed to clarify whether oral microorganisms can reach the liver via hematogenous route, or whether indirect modulation via oral‐gut‐liver axis prevails. In addition, it should be noted that similar to cirrhosis and NAFLD, periodontitis is also characterized by intestinal dysbiosis; thereby, it must be elucidated whether this microbial shift is a causal marker, or a consequence of a dysregulated inflammatory response shared by both diseases.
There is not enough evidence showing any relation between periodontitis and chronic viral hepatitis. It is therefore plausible that chronic hepatitis could offer another platform for exploring the role of viruses in the etiopathogenesis of periodontitis. Similarly, the link between periodontitis and HCC is also not defined due to lack of epidemiological evidence. Further studies are needed to understand the mechanisms by which periodontitis can alter the process of hepatocarcinogenesis. In this regard the systemic role of ROS metabolites produced in excess by immune cells during the course of periodontitis deserves attention.
From an immunological point of view, much has to be done to clarify the association between periodontitis and CLDs. Hypothetically, the pro‐inflammatory systemic profile found in periodontitis could promote catastrophic consequences on the liver pathophysiology under high oxidative stress or endotoxemia in cases of NAFLD/NASH and cirrhosis, or under chronic pathogenic challenge in cases of viral hepatitis. Recently, maladaptive bone marrow‐mediated trained immunity has been hypothesized to occur in periodontitis which could potentially link it to inflammatory comorbidities. 13 Whether and how this maladaptive immune training of bone marrow progenitors could alter the human hepatic environment is unclear and needs to be investigated.
ACKNOWLEDGEMENTS
This work was supported by US Public Health Service grants R01DE025037 and R01DE027374 to SE Sahingur from the National Institute of Dental and Craniofacial Research/National Institutes of Health.
Albuquerque‐Souza E, Sahingur SE. Periodontitis, chronic liver diseases, and the emerging oral‐gut‐liver axis. Periodontol 2000. 2022;98:125–141. doi: 10.1111/prd.12427
REFERENCES
- 1. Bajaj JS, Matin P, White MB, et al. Periodontal therapy favorably modulates the oral‐gut‐hepatic axis in cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2018;315:G824‐G837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartold PM, Lopez‐Oliva I. Periodontitis and rheumatoid arthritis: An update 2012–2017. Periodontol 2000. 2020;83(1):189‐212. [DOI] [PubMed] [Google Scholar]
- 3. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7:738‐748. [DOI] [PubMed] [Google Scholar]
- 4. Liccardo D, Marzano F, Carraturo F, et al. Potential bidirectional relationship between periodontitis and Alzheimer's disease. Front Physiol. 2020;11:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nwizu N, Wactawski‐Wende J, Genco RJ. Periodontal disease and cancer: Epidemiologic studies and possible mechanisms. Periodontol 2000. 2020;83(1):213‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papageorgiou SN, Hagner M, Nogueira AV, Franke A, Jäger A, Deschner J. Inflammatory bowel disease and oral health: systematic review and a meta‐analysis. J Clin Periodontol. 2017;44:382‐393. [DOI] [PubMed] [Google Scholar]
- 7. Van Dyke TE, Kholy KE, Ishai A, et al. Inflammation of the periodontium associates with risk of future cardiovascular events. J Periodontol. 2021;92:348‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stylianou E. Epigenetics of chronic inflammatory diseases. J Inflamm Res. 2018;12:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hajishengallis G, Darveau RP, Curtis MA. The keystone‐pathogen hypothesis. Nat Rev Microbiol. 2012;10:717‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21:426‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lira‐Junior R, Boström EA. Oral‐gut connection: one step closer to an integrated view of the gastrointestinal tract? Mucosal Immunol. 2018;11:316‐318. [DOI] [PubMed] [Google Scholar]
- 15. Acharya C, Sahingur SE, Bajaj JS. Microbiota, cirrhosis, and the emerging oral‐gut‐liver axis. JCI Insight. 2017;2(19):e94416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alakhali MS, Al‐Maweri SA, Al‐Shamiri HM, Al‐Haddad K, Halboub E. The potential association between periodontitis and non‐alcoholic fatty liver disease: a systematic review. Clin Oral Investig. 2018;22:2965‐2974. [DOI] [PubMed] [Google Scholar]
- 17. Bajaj JS, Betrapally NS, Hylemon PB, et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology. 2015;62:1260‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qin N, Yang F, Li A, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59‐64. [DOI] [PubMed] [Google Scholar]
- 19. Strnad P, Tacke F, Koch A, Trautwein C. Liver ‐ guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14:55‐66. [DOI] [PubMed] [Google Scholar]
- 20. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27:R1147‐R1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanwar S, Rhodes F, Srivastava A, Trembling PM, Rosenberg WM. Inflammation and fibrosis in chronic liver diseases including non‐alcoholic fatty liver disease and hepatitis C. World J Gastroenterol. 2020;26:109‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. 2018;36:247‐277. [DOI] [PubMed] [Google Scholar]
- 23. Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol. 2003;200:504‐515. [DOI] [PubMed] [Google Scholar]
- 24. Senoo H, Yoshikawa K, Morii M, Miura M, Imai K, Mezaki Y. Hepatic stellate cell (vitamin A‐storing cell) and its relative–past, present and future. Cell Biol Int. 2010;34:1247‐1272. [DOI] [PubMed] [Google Scholar]
- 25. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 26. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306‐321. [DOI] [PubMed] [Google Scholar]
- 27. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70:151‐171. [DOI] [PubMed] [Google Scholar]
- 28. Moon AM, Singal AG, Tapper EB. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol. 2020;18:2650‐2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McGill MR. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016;15:817‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301‐1314. [DOI] [PubMed] [Google Scholar]
- 31. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wight TN, Potter‐Perigo S. The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol. 2011;301:G950‐G955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473‐1492. [DOI] [PubMed] [Google Scholar]
- 36. Boianov B, Popov K, Nachev N, Vasilev P. Vliianie na eksperimentalnoto uvrezhdane na cherniia drob vŭrkhu parodonta [Effect of experimental injury of the liver on the parodontium]. Stomatologiia (Sofiia). 1969;51:219‐225. [PubMed] [Google Scholar]
- 37. Saraval U, De Biasio B. Quadro sieroproteico e funzionalità epatica nelle parodontopatie. [Blood protein picture & hepatic function in periodontal diseases] Minerva Med. 1957;48:3457‐3463. [PubMed] [Google Scholar]
- 38. Brunt EM, Wong VW, Nobili V, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 2015;1:15080. [DOI] [PubMed] [Google Scholar]
- 39. Smith BW, Adams LA. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat Rev Endocrinol. 2011;7:456‐465. [DOI] [PubMed] [Google Scholar]
- 40. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non‐alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005‐2023. [DOI] [PubMed] [Google Scholar]
- 41. Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: Prevalence and impact on world health. Hepatology. 2016;64:19‐22. [DOI] [PubMed] [Google Scholar]
- 42. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 43. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 44. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koliaki C, Szendroedi J, Kaul K, et al. Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739‐746. [DOI] [PubMed] [Google Scholar]
- 46. D'Aiuto F, Nibali L, Parkar M, Patel K, Suvan J, Donos N. Oxidative stress, systemic inflammation, and severe periodontitis. J Dent Res. 2010;89:1241‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Akinkugbe AA, Slade GD, Barritt AS, et al. Periodontitis and Non‐alcoholic Fatty Liver Disease, a population‐based cohort investigation in the Study of Health in Pomerania. J Clin Periodontol. 2017;44:1077‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Akinkugbe AA, Avery CL, Barritt AS, et al. Do genetic markers of inflammation modify the relationship between periodontitis and nonalcoholic fatty liver disease? Findings from the SHIP study. J Dent Res. 2017;96:1392‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alazawi W, Bernabe E, Tai D, et al. Periodontitis is associated with significant hepatic fibrosis in patients with non‐ alcoholic fatty liver disease. PLoS One. 2017;12:e0185902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen YL, Lin JZ, Mo YQ, et al. Deleterious role of hepatitis B virus infection in therapeutic response among patients with rheumatoid arthritis in a clinical practice setting: a case‐control study. Arthritis Res Ther. 2018;20:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iwasaki T, Hirose A, Azuma T, et al. Correlation between ultrasound‐diagnosed non‐alcoholic fatty liver and periodontal condition in a cross‐sectional study in Japan. Sci Rep. 2018;8:7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim JY, Lee GN, Song HC, et al. Association between fatty liver index and periodontitis: the Korea national health and nutrition examination survey. Sci Rep. 2020;10:3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qiao F, Fu K, Zhang Q, et al. The association between missing teeth and non‐alcoholic fatty liver disease in adults. J Clin Periodontol. 2018;45:941‐951. [DOI] [PubMed] [Google Scholar]
- 54. Shin HS. Association between periodontal status and non‐alcoholic fatty liver disease in a Korean adult population: A nationwide cross‐sectional study. J Periodontol. 2020;91:524‐532. [DOI] [PubMed] [Google Scholar]
- 55. Kuroe K, Furuta M, Takeuchi K, et al. Association between periodontitis and fibrotic progression of non‐alcoholic fatty liver among Japanese adults. J Clin Periodontol. 2021;48:368‐377. [DOI] [PubMed] [Google Scholar]
- 56. Helenius‐Hietala J, Suominen AL, Ruokonen H, et al. Periodontitis is associated with incident chronic liver disease‐A population‐ based cohort study. Liver Int. 2019;39:583‐591. [DOI] [PubMed] [Google Scholar]
- 57. Komazaki R, Katagiri S, Takahashi H, et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non‐ alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci Rep. 2017;7:13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakahara T, Hyogo H, Ono A, et al. Involvement of Porphyromonas gingivalis in the progression of non‐alcoholic fatty liver disease. J Gastroenterol. 2018;53:269‐280. [DOI] [PubMed] [Google Scholar]
- 59. Furusho H, Miyauchi M, Hyogo H, et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet‐induced steatohepatitis in mice. J Gastroenterol. 2013;48:1259‐1270. [DOI] [PubMed] [Google Scholar]
- 60. Yoneda M, Naka S, Nakano K, et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non‐alcoholic fatty liver disease. BMC Gastroenterol. 2012;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamata Y, Kessoku T, Shimizu T, et al. Efficacy and safety of PERIOdontal treatment versus usual care for Nonalcoholic liver disease: protocol of the PERION multicenter, two‐arm, open‐label, randomized trial. Trials. 2020;21:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seto WK, Lo YR, Pawlotsky JM, Yuen MF. Chronic hepatitis B virus infection. Lancet. 2018;392:2313‐2324. [DOI] [PubMed] [Google Scholar]
- 63. Tang LSY, Covert E, Wilson E, Kottilil S. Chronic hepatitis B infection: A review. JAMA. 2018;319:1802‐1813. [DOI] [PubMed] [Google Scholar]
- 64. Pradat P, Virlogeux V, Trépo E. Epidemiology and elimination of HCV‐related liver disease. Viruses. 2018;10:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Van der Eijk AA, Niesters HG, Götz HM, et al. Paired measurements of quantitative hepatitis B virus DNA in saliva and serum of chronic hepatitis B patients: implications for saliva as infectious agent. J Clin Virol. 2004;29:92‐94. [DOI] [PubMed] [Google Scholar]
- 66. De Waure C, Cefalo C, Chiaradia G, et al. Intrafamilial transmission of hepatitis C virus in Italy: a systematic review. J Epidemiol Community Health. 2010;64:843‐848. [DOI] [PubMed] [Google Scholar]
- 67. Ferreiro MC, Dios PD, Scully C. Transmission of hepatitis C virus by saliva? Oral Dis. 2005;11:230‐235. [DOI] [PubMed] [Google Scholar]
- 68. Lins L, Almeida H, Vitvisk L, Carmo T, Paraná R, Reis MG. Detection of hepatitis C virus RNA in saliva is not related to oral health status or viral load. J Med Virol. 2005;77:216‐220. [DOI] [PubMed] [Google Scholar]
- 69. Maticic M, Poljak M, Kramar B, et al. Detection of hepatitis C virus RNA from gingival crevicular fluid and its relation to virus presence in saliva. J Periodontol. 2001;72:11‐16. [DOI] [PubMed] [Google Scholar]
- 70. Roy KM, Bagg J, McCarron B, Good T, Cameron S, Pithie A. Predominance of HCV type 2a in saliva from intravenous drug users. J Med Virol. 1998;54:271‐275. [DOI] [PubMed] [Google Scholar]
- 71. Sosa‐Jurado F, Hernández‐Galindo VL, Meléndez‐Mena D, et al. Detection of hepatitis C virus RNA in saliva of patients with active infection not associated with periodontal or liver disease severity. BMC Infect Dis. 2014;14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Azatyan V, Yessayan L, Shmavonyan M, Melik‐Andreasyan G, Perikhanyan A, Porkshenyan K. Evaluation of IL‐2, IL‐10, IL‐4 and ɤ‐interferon levels in the oral fluids of patients with hepatitis C, B and HIV. J Infect Dev Ctries. 2019;13(5.1):69S‐74S. [DOI] [PubMed] [Google Scholar]
- 73. Henderson L, Muir M, Mills PR, et al. Oral health of patients with hepatitis C virus infection: a pilot study. Oral Dis. 2001;7:271‐275. [DOI] [PubMed] [Google Scholar]
- 74. Kazankov K, Jørgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16:145‐159. [DOI] [PubMed] [Google Scholar]
- 75. Coates EA, Brennan D, Logan RM, et al. Hepatitis C infection and associated oral health problems. Aust Dent J. 2000;45:108‐114. [DOI] [PubMed] [Google Scholar]
- 76. Surlin P, Gheorghe DN, Popescu DM, et al. Interleukin‐1α and ‐1β assessment in the gingival crevicular fluid of periodontal patients with chronic hepatitis C. Exp Ther Med. 2020;20:2381‐2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kitamoto S, Nagao‐Kitamoto H, Hein R, Schmidt TM, Kamada N. The Bacterial connection between the oral cavity and the gut diseases. J Dent Res. 2020;99:1021‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Șurlin P, Camen A, Stratul SI, et al. Optical coherence tomography assessment of gingival epithelium inflammatory status in periodontal ‐ Systemic affected patients. Ann Anat. 2018;219:51‐56. [DOI] [PubMed] [Google Scholar]
- 79. Marrone A, Lasserre J, Bercy P, Brecx MC. Prevalence and risk factors for peri‐implant disease in Belgian adults. Clin Oral Implants Res. 2013;24:934‐940. [DOI] [PubMed] [Google Scholar]
- 80. Cadwell K. The virome in host health and disease. Immunity. 2015;42:805‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen Y, Yang YC, Zhu BL, Wu CC, Lin RF, Zhang X. Association between periodontal disease, tooth loss and liver diseases risk. J Clin Periodontol. 2020;47:1053‐1063. [DOI] [PubMed] [Google Scholar]
- 82. Mahfouz M, Martin P, Carrion AF. Hepatic complications of inflammatory bowel disease. Clin Liver Dis. 2019;23:191‐208. [DOI] [PubMed] [Google Scholar]
- 83. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095‐2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Movin S. Relationship between periodontal disease and cirrhosis of the liver in humans. J Clin Periodontol. 1981;8:450‐458. [DOI] [PubMed] [Google Scholar]
- 85. Novacek G, Plachetzky U, Pötzi R, et al. Dental and periodontal disease in patients with cirrhosis–role of etiology of liver disease. J Hepatol. 1995;22:576‐582. [DOI] [PubMed] [Google Scholar]
- 86. Ladegaard Grønkjær L, Holmstrup P, Schou S, Jepsen P, Vilstrup H. Severe periodontitis and higher cirrhosis mortality. United European Gastroenterol J. 2018;6:73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Costa FO, Lages EJP, Lages EMB, Cota LOM. Periodontitis in individuals with liver cirrhosis: A case‐control study. J Clin Periodontol. 2019;46:991‐998. [DOI] [PubMed] [Google Scholar]
- 88. Jaiswal G, Deo V, Bhongade M, Jaiswal S. Serum alkaline phosphatase: a potential marker in the progression of periodontal disease in cirrhosis patients. Quintessence Int. 2011;42:345‐348. [PubMed] [Google Scholar]
- 89. Lins L, Bittencourt PL, Evangelista MA, et al. Oral health profile of cirrhotic patients awaiting liver transplantation in the Brazilian Northeast. Transplant Proc. 2011;43:1319‐1321. [DOI] [PubMed] [Google Scholar]
- 90. Jensen A, Ladegaard Grønkjær L, Holmstrup P, Vilstrup H, Kilian M. Unique subgingival microbiota associated with periodontitis in cirrhosis patients. Sci Rep. 2018;8:10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ghapanchi J, Bazargani A, Khorshidi H, et al. Isolation and identification of non‐ commensal pathogenic bacteria in the saliva of patients candidate for liver transplant: A cross sectional study in Shiraz. South of Iran. J Dent (Shiraz). 2020;21:81‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kakiyama G, Hylemon PB, Zhou H, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2014;306:G929‐G937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bajaj JS, Heuman DM, Hylemon PB, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60:940‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen Y, Yang F, Lu H, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562‐572. [DOI] [PubMed] [Google Scholar]
- 95. Wang J, Wang Y, Zhang X, et al. Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic hepatitis B patients. Front Microbiol. 2017;8:2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dubinkina VB, Tyakht AV, Odintsova VY, et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome. 2017;5:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kapil S, Duseja A, Sharma BK, et al. Small intestinal bacterial overgrowth and toll‐like receptor signaling in patients with non‐alcoholic fatty liver disease. J Gastroenterol Hepatol. 2016;31:213‐221. [DOI] [PubMed] [Google Scholar]
- 98. Garrido A, Djouder N. Cirrhosis: a questioned risk factor for hepatocellular carcinoma. Trends Cancer. 2021;7:29‐36. [DOI] [PubMed] [Google Scholar]
- 99. Michaud DS, Fu Z, Shi J, Chung M. Periodontal disease, tooth loss, and cancer risk. Epidemiol Rev. 2017;39:49‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ahn J, Segers S, Hayes RB. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 2012;33:1055‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yang B, Petrick JL, Abnet CC, et al. Tooth loss and liver cancer incidence in a Finnish cohort. Cancer Causes Control. 2017;28:899‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tamaki N, Takaki A, Tomofuji T, et al. Stage of hepatocellular carcinoma is associated with periodontitis. J Clin Periodontol. 2011;38:1015‐1020. [DOI] [PubMed] [Google Scholar]
- 103. Kwong A, Kim WR, Lake JR, et al. OPTN/SRTR 2018 Annual Data Report: Liver. Am J Transplant. 2020;20 (Suppl S1):193‐299. [DOI] [PubMed] [Google Scholar]
- 104. Goldberg D, Ditah IC, Saeian K, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Moreno R, Berenguer M. Post‐liver transplantation medical complications. Ann Hepatol. 2006;5:77‐85. [PubMed] [Google Scholar]
- 106. Di Profio B, Inoue G, Marui VC, et al. Periodontal status of liver transplant candidates and healthy controls. J Periodontol. 2018;89:1383‐1389. [DOI] [PubMed] [Google Scholar]
- 107. Guggenheimer J, Eghtesad B, Close JM, Shay C, Fung JJ. Dental health status of liver transplant candidates. Liver Transpl. 2007;13:280‐286. [DOI] [PubMed] [Google Scholar]
- 108. Martin P, DiMartini A, Feng S, Brown R Jr, Fallon M. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology. 2014;59:1144‐1165. [DOI] [PubMed] [Google Scholar]
- 109. Machtei EE, Falah M, Oettinger‐Barak O, Baruch Y, Horwitz J. Periodontal status in post‐ liver transplantation patients: 10 years of follow‐up. Quintessence Int. 2012;43:879‐885. [PubMed] [Google Scholar]
- 110. Aimetti M, Romano F, Marsico A, Navone R. Non‐surgical periodontal treatment of cyclosporin A‐induced gingival overgrowth: immunohistochemical results. Oral Dis. 2008;14:244‐250. [DOI] [PubMed] [Google Scholar]
- 111. Kauffels A, Schmalz G, Kollmar O, et al. Oral findings and dental behaviour before and after liver transplantation ‐ a single‐centre cross‐ sectional study. Int Dent J. 2017;67:244‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mouzaki M, Comelli EM, Arendt BM, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120‐127. [DOI] [PubMed] [Google Scholar]
- 113. Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079‐1089. [DOI] [PubMed] [Google Scholar]
- 114. Albillos A, de Gottardi A, Rescigno M. The gut‐liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. 2020;72:558‐577. [DOI] [PubMed] [Google Scholar]
- 115. Chassaing B, Etienne‐Mesmin L, Gewirtz AT. Microbiota‐liver axis in hepatic disease. Hepatology. 2014;59:328‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tripathi A, Debelius J, Brenner DA, et al. The gut‐ liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41‐50. [DOI] [PubMed] [Google Scholar]
- 118. Isayama F, Hines IN, Kremer M, et al. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318‐G1328. [DOI] [PubMed] [Google Scholar]
- 119. Arimatsu K, Yamada H, Miyazawa H, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. 2014;4:4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hyvärinen K, Tuomainen AM, Laitinen S, et al. Chlamydial and periodontal pathogens induce hepatic inflammation and fatty acid imbalance in apolipoprotein E‐deficient mice. Infect Immun. 2009;77:3442‐3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nagasaki A, Sakamoto S, Chea C, et al. Odontogenic infection by Porphyromonas gingivalis exacerbates fibrosis in NASH via hepatic stellate cell activation. Sci Rep. 2020;10:4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sasaki N, Katagiri S, Komazaki R, et al. Endotoxemia by Porphyromonas gingivalis injection aggravates non‐alcoholic fatty liver disease, disrupts glucose/lipid metabolism, and alters gut microbiota in mice. Front Microbiol. 2018;9:2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Seyama M, Yoshida K, Yoshida K, et al. Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165731. [DOI] [PubMed] [Google Scholar]
- 124. Varela‐López A, Bullón P, Ramírez‐Tortosa CL, et al. A diet rich in saturated fat and cholesterol aggravates the effect of bacterial lipopolysaccharide on alveolar bone loss in a rabbit model of periodontal disease. Nutrients. 2020;12:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fujita M, Kuraji R, Ito H, et al. Histological effects and pharmacokinetics of lipopolysaccharide derived from Porphyromonas gingivalis on rat maxilla and liver concerning with progression into non‐alcoholic steatohepatitis. J Periodontol. 2018;89:1101‐1111. [DOI] [PubMed] [Google Scholar]
- 126. Ding LY, Liang LZ, Zhao YX, et al. Porphyromonas gingivalis‐ derived lipopolysaccharide causes excessive hepatic lipid accumulation via activating NF‐ κB and JNK signaling pathways. Oral Dis. 2019;25:1789‐1797. [DOI] [PubMed] [Google Scholar]
- 127. Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin‐3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. 2006;103:5060‐5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zaitsu Y, Iwatake M, Sato K, Tsukuba T. Lipid droplets affect elimination of Porphyromonas gingivalis in HepG2 cells by altering the autophagy‐lysosome system. Microbes Infect. 2016;18:565‐571. [DOI] [PubMed] [Google Scholar]
- 129. Ishikawa M, Yoshida K, Okamura H, et al. Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the Akt/GSK‐3β signaling pathway. Biochim Biophys Acta. 2013;1832:2035‐2043. [DOI] [PubMed] [Google Scholar]
- 130. Zhang Z, Liu D, Liu S, Zhang S, Pan Y. The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol. 2021;10:585917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Olsen I, Yilmaz Ö. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol. 2019;11:1563410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yamazaki K, Sato K, Tsuzuno T, et al. Orally administered pathobionts and commensals have comparable and innocuous systemic effects on germ‐free mice. Microb Pathog. 2020;140:103962. [DOI] [PubMed] [Google Scholar]
- 133. Amado PPP, Kawamoto D, Albuquerque‐Souza E, et al. Oral and fecal microbiome in molar‐incisor pattern periodontitis. Front Cell Infect Microbiol. 2020;10:583761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lourenςo TGB, Spencer SJ, Alm EJ, Colombo APV. Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. J Oral Microbiol. 2018;10:1487741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Aly AM, Adel A, El‐Gendy AO, Essam TM, Aziz RK. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 2016;8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bajaj JS, Fagan A, Sikaroodi M, et al. Liver transplant modulates gut microbial dysbiosis and cognitive function in cirrhosis. Liver Transpl. 2017;23:907‐914. [DOI] [PubMed] [Google Scholar]
- 137. Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in Nonalcoholic Fatty Liver Disease. Hepatology. 2019;69:107‐120. [DOI] [PubMed] [Google Scholar]
- 139. Loos BG, Van Dyke TE. The role of inflammation and genetics in periodontal disease. Periodontol 2000. 2020;83(1):26‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rau M, Schilling AK, Meertens J, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. 2016;196:97‐105. [DOI] [PubMed] [Google Scholar]
- 141. Widita E, Yoshihara A, Hanindriyo L, Miyazaki H. Relationship between clinical periodontal parameters and changes in liver enzymes levels over an 8‐year period in an elderly Japanese population. J Clin Periodontol. 2018;45:311‐321. [DOI] [PubMed] [Google Scholar]
- 142. Mester A, Ciobanu L, Taulescu M, et al. Periodontal disease may induce liver fibrosis in an experimental study on Wistar rats. J Periodontol. 2019;90:911‐919. [DOI] [PubMed] [Google Scholar]
- 143. Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS One. 2011;6:e24422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper‐reactivity in chronic periodontitis. Innate Immun. 2015;21:714‐725. [DOI] [PubMed] [Google Scholar]
- 145. Wang CW, Colas RA, Dalli J, et al. Maresin 1 biosynthesis and proresolving anti‐infective functions with human‐localized aggressive periodontitis leukocytes. Infect Immun. 2015;84:658‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gustafsson A, Ito H, Asman B, Bergström K. Hyper‐reactive mononuclear cells and neutrophils in chronic periodontitis. J Clin Periodontol. 2006;33:126‐129. [DOI] [PubMed] [Google Scholar]
- 147. Ling MR, Chapple IL, Matthews JB. Neutrophil superoxide release and plasma C‐reactive protein levels pre‐ and post‐periodontal therapy. J Clin Periodontol. 2016;43:652‐658. [DOI] [PubMed] [Google Scholar]
- 148. Roberts HM, Ling MR, Insall R, et al. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol. 2015;42:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bohm T, Berger H, Nejabat M, et al. Food‐derived peroxidized fatty acids may trigger hepatic inflammation: a novel hypothesis to explain steatohepatitis. J Hepatol. 2013;59:563‐570. [DOI] [PubMed] [Google Scholar]
- 150. Cannito S, Morello E, Bocca C, et al. Microvesicles released from fat‐laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro‐inflammatory link between lipotoxicity and non‐alcoholic steatohepatitis. PLoS One. 2017;12:e0172575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Morinaga H, Mayoral R, Heinrichsdorff J, et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes. 2015;64:1120‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gaudilliere DK, Culos A, Djebali K, et al. Systemic immunologic consequences of chronic periodontitis. J Dent Res. 2019;98:985‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Liu P, Chen L, Zhang H. Natural killer cells in liver disease and hepatocellular carcinoma and the NK cell‐based immunotherapy. J Immunol Res. 2018;2018:1206737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Edlich B, Ahlenstiel G, Zabaleta Azpiroz A, et al. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon‐based therapy of hepatitis C patients. Hepatology. 2012;55:39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Miyagi T, Takehara T, Nishio K, et al. Altered interferon‐alpha‐signaling in natural killer cells from patients with chronic hepatitis C virus infection. J Hepatol. 2010;53:424‐430. [DOI] [PubMed] [Google Scholar]
- 156. Peppa D, Gill US, Reynolds G, et al. Up‐regulation of a death receptor renders antiviral T cells susceptible to NK cell‐mediated deletion. J Exp Med. 2013;210:99‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Cheng WC, van Asten SD, Burns LA, et al. Periodontitis‐associated pathogens P gingivalis and A. actinomycetemcomitans activate human CD14(+) monocytes leading to enhanced Th17/IL‐17 responses. Eur J Immunol. 2016;46:2211‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Paquissi FC. Immunity and fibrogenesis: The role of Th17/IL‐17 Axis in HBV and HCV‐induced chronic hepatitis and progression to cirrhosis. Front Immunol. 2017;8:1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Medara N, Lenzo JC, Walsh KA, et al. Peripheral memory T‐cell profile is modified in patients undergoing periodontal management. J Clin Periodontol. 2021;48:249‐262. [DOI] [PubMed] [Google Scholar]
- 160. Cifcibasi E, Ciblak M, Kiran B, et al. The role of activated cytotoxic T cells in etiopathogenesis of periodontal disease: does it harm or does it heal? Sci Rep. 2015;5:9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Medara N, Lenzo JC, Walsh KA, O'Brien‐Simpson NM, Reynolds EC, Darby IB. Peripheral T helper cell profiles during management of periodontitis. J Clin Periodontol. 2021;48:76‐90. [DOI] [PubMed] [Google Scholar]
- 162. Shin EC, Sung PS, Park SH. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat Rev Immunol. 2016;16:509‐523. [DOI] [PubMed] [Google Scholar]
- 163. Wei C, Ni C, Song T, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158‐1168. [DOI] [PubMed] [Google Scholar]
- 164. Gonçalves Fernandes J, Morford LA, Harrison PL, et al. Dysregulation of genes and microRNAs in localized aggressive periodontitis. J Clin Periodontol. 2020;47:1317‐1325. [DOI] [PubMed] [Google Scholar]
- 165. Yoneda T, Tomofuji T, Ekuni D, et al. Serum microRNAs and chronic periodontitis: A case‐control study. Arch Oral Biol. 2019;101:57‐63. [DOI] [PubMed] [Google Scholar]
- 166. Pirola CJ, Fernández Gianotti T, Castaño GO, et al. Circulating microRNA signature in non‐alcoholic fatty liver disease: from serum non‐coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64:800‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Waldron PR, Holodniy M. MicroRNA and hepatitis C virus–challenges in investigation and translation: a review of the literature. Diagn Microbiol Infect Dis. 2014;80:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wong CM, Tsang FH, Ng IO. Non‐coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 2018;15:137‐151. [DOI] [PubMed] [Google Scholar]