

Abstract
Background and purpose
This study was undertaken to determine the diagnostic and prognostic value of a panel of serum biomarkers and to correlate their concentrations with several clinical parameters in a large cohort of patients with amyotrophic lateral sclerosis (ALS).
Methods
One hundred forty‐three consecutive patients with ALS and a control cohort consisting of 70 patients with other neurodegenerative disorders (DEG), 70 patients with ALS mimic disorders (ALSmd), and 45 healthy controls (HC) were included. Serum neurofilament light chain (NfL), ubiquitin carboxyl‐terminal hydrolase isozyme L1 (UCHL1), glial fibrillary acidic protein (GFAP), and total tau protein levels were measured using ultrasensitive single molecule array.
Results
NfL correlated with disease progression rate (p < 0.001) and with the measures of upper motor neuron burden (p < 0.001). NfL was higher in the ALS patients with classic and pyramidal phenotype. GFAP was raised in ALS with cognitive–behavioral impairment compared with ALS with normal cognition. NfL displayed the best diagnostic performance in discriminating ALS from HC (area under the curve [AUC] = 0.990), DEG (AUC = 0.946), and ALSmd (AUC = 0.850). UCHL1 performed well in distinguishing ALS from HC (AUC = 0.761), whereas it was not helpful in differentiating ALS from DEG and ALSmd. In multivariate analysis, NfL (p < 0.001) and UCHL1 (p = 0.038) were independent prognostic factors. Survival analysis combining NfL and UCHL1 effectively stratified patients with lower NfL levels (p < 0.001).
Conclusions
NfL is a useful biomarker for the diagnosis of ALS and the strongest predictor of survival. UCHL1 is an independent prognostic factor helpful in stratifying survival in patients with low NfL levels, likely to have slowly progressive disease. GFAP reflects extramotor involvement, namely cognitive impairment or frontotemporal dementia.
Keywords: frontotemporal dementia, glial fibrillary acidic protein, neurofilament proteins, UCHL1 protein
Neurofilament light chain (NfL) is a useful biomarker for the diagnosis of amyotrophic lateral sclerosis and the strongest predictor of survival. Ubiquitin carboxyl‐terminal hydrolase isozyme L1 is an independent prognostic factor and may be helpful in further stratifying the prognosis of patients showing low NfL concentrations. Glial fibrillary acidic protein reflects extramotor involvement, namely, cognitive impairment or frontotemporal dementia.

INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease caused by a relentless motor neuron degeneration, leading to progressive muscle weakness, dysphagia, respiratory failure, and ultimately death [1]. ALS belongs to the broader spectrum of motor neuron disease, a heterogeneous group of disorders marked by the involvement of both upper and lower motor neurons [2, 3].
As of today, the diagnosis of ALS is based on clinical judgment, given a combination of progressive upper and lower motor neuron involvement without an alternative explanation for the presenting symptoms and signs [4]. The diagnosis of ALS can be challenging when its first manifestations overlap with those of ALS mimic disorders, and the lack of specific diagnostic tests prevents an early diagnosis [5]. Definition of prognosis in ALS is hampered by the heterogeneity of its clinical features, with variability in survival being the most salient feature [6, 7]. An accurate prediction of the individual outcome is crucial to establish early interventions as well as in clinical trial design [8, 9].
Despite extensive research efforts, only a few biomarkers have been validated for ALS, including neuroimaging, electrophysiological, and biofluid biomarkers [10]. The latter, frequently referred to as “wet biomarkers,” have gained increasing interest in recent years, in consideration of their advantages in terms of lower economic costs, better tolerability, and greater ease to acquire, process, and harmonize. Therefore, wet biomarkers are needed to aid clinical decision and achieve early diagnosis, track disease progression, and better define disease trajectories.
Consistent evidence has supported neurofilament light chain (NfL), a marker of axonal injury, as one of the most promising biomarkers for ALS. NfL is broadly considered a reliable prognostic biomarker for ALS [11, 12, 13] whereas the applicability of NfL as a diagnostic biomarker is still debated. Previous studies have shown that NfL yields a high diagnostic performance in discriminating ALS from healthy controls (HC) in both serum and cerebrospinal fluid (CSF) [11, 12, 13, 14, 15]. Conversely, studies aimed at investigating the applicability of CSF and serum NfL in distinguishing ALS from its mimic disorders achieved mixed results, possibly due to the heterogeneity of ALS and the lack of specificity of NfL when tested across neurodegenerative disorders [11, 16]. To overcome this limitation, the use of a panel of biomarkers, as already employed for the diagnosis of Alzheimer disease (AD), might be a promising strategy to improve the diagnostic performance in differentiating ALS from its mimic disorders [21].
An ideal biomarker panel in ALS should embrace markers that may already be tested in neurodegenerative disorders and that are easily measurable in serum. An example is ubiquitin carboxyl‐terminal hydrolase isozyme L1 (UCHL1), a multifunctional protein expressed in the cytoplasm of neurons [22, 23, 24]. It is elevated in both CSF and serum from sporadic ALS patients and from those carrying the C9orf72 hexanucleotide repeat expansion (C9‐ALS) [25, 26, 27]. Glial fibrillary acidic protein (GFAP) is an established brain marker of astrogliosis [28]. GFAP has been found elevated in both CSF and serum from patients with frontotemporal dementia (FTD), whereas it has not yet been fully investigated in ALS [29, 30, 31]. Total tau protein (tTAU) is a protein reflecting an ongoing neuronal injury process, and it is significantly raised in the CSF of AD patients [32, 33]. Several studies have attempted to investigate the potential of CSF and serum tTAU in ALS, achieving controversial results [34, 35, 36, 37].
Against this background, this study aims to investigate UCHL1, tTAU, and GFAP, addressing neuronal injury, neurodegeneration, and astroglial activation in a large cohort of ALS patients. The diagnostic and prognostic performance of these biomarkers were benchmarked against NfL, a marker of axonal injury.
MATERIALS AND METHODS
Participants and clinical characterization
A total of 143 consecutive ALS patients, diagnosed according to the revised El Escorial criteria, were recruited at the Department of Neurology of San Raffaele Scientific Institute, Milan, Italy, from March 2016 to March 2021 [4, 38, 39]. A control cohort consisting of 70 patients with other neurodegenerative disorders (DEG), 70 patients with ALS mimic disorders (ALSmd), and 45 HC was included in the study. All the patients enrolled in the study were recruited at our center. ALSmd were patients showing signs and symptoms resembling ALS, a diagnosis that was excluded after a throughout diagnostic workup. HC were unrelated to ALS patients. Baseline serum samples for biomarker assays were collected during the diagnostic workup. Additionally, a longitudinal biomarker evaluation was performed in subgroup of ALS patients.
At the collection time, demographics and clinical history information were registered and a neurological evaluation was performed collecting the following data: Medical Research Council (MRC) scale (12 muscles for each side; score = 0–120 points) [40]; MRC progression rate, calculated as (120 − MRC score) / disease duration; ALS Functional Rating Scale–Revised (ALSFRS‐R); disease progression rate, calculated as (48 − ALSFRS‐R) / disease duration; and upper motor neuron (UMN) score, calculated by totaling the number of pathological UMN signs at examination (score = 0–16) [40]. Patients were staged according to King's clinical staging system, and classified into eight different ALS phenotypes, in accordance with previously published criteria (Appendix S1) [41]. The presence of C9orf72 repeat expansion was screened in the whole ALS cohort, as previously described (Appendix S1) [42]. A neuropsychological assessment, performed as recommended by the Diagnostic Criteria for the Behavioural Variant of FTD and the ALSFTD Consensus Criteria, was available for 74 ALS patients who were assessed in a time period of ±2 months from serum sampling [43, 44, 45]. Patients were categorized into five different cognitive phenotypes, consistently with the neuropsychological assessment, as previously described [46]. Routine transcranial magnetic stimulation, performed for diagnostic purpose, was available from 90 patients, in a time period of ±2 months from serum sampling. Mean motor evoked potential/compound muscle action potential (MEP/cMAP) at the four limbs was calculated, as previously described [46].
Disease duration was defined as the time period between symptoms onset and date of sampling. Survival was defined as time from sampling to death/tracheostomy. Patients were followed up with periodical phone calls, and survival status was updated in April 2021.
Sample collection and biomarker analysis
Serum samples from all the patients included in the study were collected at the first evaluation performed at our center. Furthermore, serum samples from 34 patients with ALS were collected for longitudinal biomarker evaluation 6 months after the first venipuncture. Serum samples were processed within 1 h of blood collection and were stored at −80°C prior to analysis. Single‐protein array technology (Simoa, Quanterix) was used to quantify serum GFAP, UCHL‐1, NfL, and tTAU levels (pg/ml). The analysis was performed with the fully automated instrument HD‐1 Analyzer (Quanterix). Samples were run with appropriate standards and controls, and the technician performing the assays was blinded to clinical data. For the longitudinal assay, samples were assessed on the same plate for each ALS patient to reduce batch effects. The interassay coefficient of variation was <15%.
Statistical methods
Normality data distribution was explored with the Shapiro–Wilk test. Continuous variables are reported as median and interquartile range (IQR), and categorical variables as number and relative frequencies. We applied two‐tailed unpaired Mann–Whitney U test and Kruskal–Wallis test with Bonferroni post hoc comparison to verify differences among two and more than two groups, respectively. Correlation between parameters was calculated by Pearson correlation r at a 5% significance. Wilcoxon matched‐pairs signed‐rank test was performed to compare pairs of basal and follow‐up serum biomarker levels. Clinical variables were categorized accordingly with their tertile values to assess differences among groups.
Receiver operating characteristic (ROC) curve analysis was carried out to investigate diagnostic sensitivity, specificity, and positive and negative predictive values (PPV and NPV), and to calculate the area under the curve (AUC) of serum biomarkers, with corresponding 95% confidence interval (CI). The highest Youden index was used to calculate the optimal cutoff of each biomarker on an ROC analysis.
Kaplan–Meier (KM) univariate analysis was carried out to estimate the effect of biomarkers on survival. Patients were clustered according to biomarker tertile values. Log‐rank test (Mantel–Cox) was used to test for significant differences among groups. Patients who were alive at last follow‐up were censored. Multivariate analysis with Cox proportional hazards model (enter method) was performed to estimate the proportional hazard ratios of biomarkers on survival. Cox regression was adjusted for factors that negatively influenced ALS survival [6, 46].
All statistical tests were carried out using SPSS 26.0 software (IBM). Statistical significance was set at p < 0.05.
Patient consent for publication
The study was performed according to the Helsinki Declaration and approved by the ethics committee of our institute (Protocollo Genotipo‐Fenotipo, reference number DSAN 855‐A‐OS/3). All patients gave informed written consent to participate in the study.
RESULTS
Patients' clinical characteristics and correlations with serum biomarkers
Demographics and clinical characteristics of the ALS and control cohorts are given in Table 1 and Tables S1, S2, and S3. All groups were age‐ (p = 0.27) and sex‐matched (p = 0.89).
TABLE 1.
ALS patients' demographics and clinical characteristics
| Characteristic | ALS, n = 143 |
|---|---|
| Gender, M/F | 87/56 |
| Age at venipuncture, years | 64.0 (55.0–72.0) |
| Disease duration at venipuncture, months | 9.0 (7.0–12.0) |
| Diagnostic delay, months | 7.0 (5.0–10.0) |
| ALSFRS‐R | 41.0 (36.0–44.0) |
| ΔALSFRS‐R, points/month | 0.9 (0.5–1.5) |
| MRC total score | 106.0 (94.8–114.0) |
| ΔMRC, points/month | 1.7 (0.7–2.7) |
| UMN score | 8.0 (4.0–11.0) |
| ECAS ALS‐specific | 71.0 (56.0–84.0) |
| ECAS ALS‐nonspecific | 23.0 (18.0–27.0) |
| Total ECAS score | 93.0 (76.0–109.0) |
| Mean MEP/cMAP at four limbs | 0.2 (0.1–0.3) |
Given are median values and interquartile range.
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale–Revised; ECAS, Edinburgh Cognitive and Behavioural ALS Screen; F, female; M, male; MEP/cMAP, motor evoked potential/compound muscle action potential; MRC, Medical Research Council Scale; UMN, upper motor neuron; ΔALSFRS‐R, ALSFRS‐R progression rate; ΔMRC, MRC progression rate.
Serum NfL was significantly higher in ALS compared with HC, DEG, and ALSmd groups (p < 0.001; Figure 1, Table 2). UCHL1 was significantly higher in ALS compared with HC (p < 0.001). GFAP and tTAU were significantly lower in ALS compared with DEG (p < 0.001; Figure 1, Table 2). Serum biomarker median levels and IQRs of the ALS and control cohorts are summarized in Table 2.
FIGURE 1.

(a) Neurofilament light chain (NfL), (b) ubiquitin carboxyl‐terminal hydrolase isozyme L1 (UCHL1), (c) glial fibrillary acidic protein (GFAP), and (d) total tau protein (tTAU) serum concentration among different study groups. Boxes are median concentration and interquartile range. Whiskers are lowest and highest values. Biomarker levels are plotted on a 10‐logarithmic scale, whereas tTAU levels are plotted on a linear scale. *p < 0.05, **p < 0.01. ALS, amyotrophic lateral sclerosis; ALS‐md, ALS mimic diseases; DEG, neurodegenerative diseases; HC, healthy controls
TABLE 2.
Demographic characteristics and serum levels in the study groups
| Study groups | ||||
|---|---|---|---|---|
| ALS, n = 143 | HC, n = 45 | DEG, n = 70 | ALSmd, n = 70 | |
| Age, years | 64.5 (54.8–72.2) | 62.0 (58.0–70.0) | 66.0 (61.8–72.3) | 68.0 (54.8–77.0) |
| Sex, M/F | 87/56 | 25/20 | 42/28 | 44/26 |
| NfL | 112.1 (70.8–184.9) | 14.1 (10.9–19.2)** | 24.5 (15.8–37.4)**; # | 27.6 (16.9–53.0)**;## |
| UCHL1 | 41.9 (28.0–62.8) | 25.8 (17.2–36.2)** | 49.7 (26.3–62.9)## | 44.5 (24.5−67.4)## |
| GFAP | 131.4 (92.6–173.4) | 125.9 (84.2–156.5) | 206.2 (118.5–277.2)**;## | 149.8 (96.0–212.3) |
| tTAU | 0.8 (0.5–1.3) | 1.1 (0.7–2.0)** | 1.2 (0.4–1.8) | 0.6 (0.3–1.1)##; §§; |
Median is given with interquartile range in parentheses.
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSmd, ALS mimic disorders; DEG, neurodegenerative disorders; F, female; GFAP, glial fibrillary acidic protein; HC, healthy controls; M, male; NfL, neurofilament light chain; tTAU, total tau protein; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1.
*p < 0.05 vs ALS; **p < 0.01 vs ALS; # p < 0.05 vs HC; ## p < 0.01 vs HC; § p < 0.05 vs DEG; §§ p < 0.01 vs DEG.
Univariate Pearson pairwise analysis identified a weak correlation between NfL and tTAU (r = 0.19, p = 0.02), and a moderate correlation between NfL and GFAP (r = 0.39; p < 0.001). UCHL1 showed a weak correlation with GFAP (r = 0.18, p = 0.03) and tTAU (r = 0.18, p = 0.03).
When serum biomarker concentrations were correlated with ALS clinical characteristics and functional parameters of disease progression, we observed a correlation between GFAP and age at sampling (r = 0.42, p < 0.001). Disease progression rate was moderately correlated with NfL (r = 0.47, p < 0.001) and weakly correlated with GFAP (r = 0.27, p = 0.01). NfL (r = 0.34, p < 0.001), UCHL1 (r = 0.20, p = 0.02), and GFAP (r = 0.21, p = 0.02) showed a positive correlation with the UMN burden measured by UMN score. Furthermore, we observed a negative correlation between the mean MEP/cMAP values at the four limbs and NfL (r = −0.33, p = 0.01).
We explored the serum biomarker concentrations across the ALS phenotypes. Higher NfL levels were detected in pyramidal, classic, and bulbar patients compared with atypical or restricted phenotypes such as flail arm (FA), flail leg (FL), pure lower motor neuron (PLMN), and pure UMN (PUMN; Figure 2 and Table S1). Conversely, UCHL1, GFAP, and tTAU showed homogenous levels regardless of the ALS phenotypes (Figure S1 and Table S1). We investigated serum biomarker concentrations across ALS cognitive phenotypes, evidencing that GFAP was significantly higher in ALS with concomitant cognitive and/or behavioral impairment or FTD, compared with pure motor ALS (Figure 2 and Table S1). This result was confirmed also after correction for the age at sampling. NfL, UCHL1, and tTAU did not differ among cognitive phenotypes (Figure S2 and Table S1). In our ALS cohort, 20 patients were C9‐ALS. C9‐ALS and C9‐negative ALS groups had similar serum biomarker concentrations (Table S1). Thirty‐four patients underwent longitudinal serum biomarker evaluation. The median time occurring between baseline (T0) and the second sampling (T1) was 6 months (range = 5–7 months). Demographics and clinical characteristics of the longitudinal ALS cohort are given in the Table S4. A Wilcoxon matched‐pairs signed‐rank test to evaluate significant changes of repeated measures of biomarker concentrations showed no difference for NfL (p = 0.197), UCHL1 (p = 0.939), and GFAP (p = 0.109). Conversely, tTau levels were significantly increased at the second time point (p = 0.005; Figure S3).
FIGURE 2.
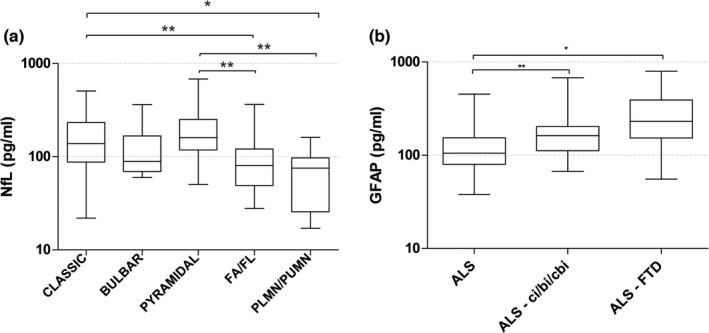
(a) Serum neurofilament light chain (NfL) levels in patients with amyotrophic lateral sclerosis (ALS) grouped according to previously published criteria [41] (b) Serum glial fibrillary acidic protein (GFAP) levels in ALS patients classified according to cognitive impairment [43] Boxes are median and interquartile range. Whiskers are highest and lowest values. Biomarker levels are plotted on a 10‐logarithmic scale. *p < 0.05, **p < 0.01. ci/bi/cbi, cognitive impairment/behavioral impairment/both; FA, flail arm; FL, flail leg; FTD, frontotemporal dementia; PLMN, pure lower motor neuron; PUMN, pure upper motor neuron
Biomarker diagnostic performance
Serum NfL displayed the best diagnostic performance among biomarkers when discriminating ALS from HC (AUC = 0.990, 95% CI = 0.978–1.00). UCHL1 exhibited good diagnostic performance (AUC = 0.761, 95% CI = 0.765–0.837); conversely, GFAP and tTAU had lower performance in differentiating ALS from HC (Figures S4, S5, and S6). NfL also had the highest diagnostic yield in distinguishing ALS from DEG (AUC = 0.946, 95% CI = 0.916–0.976); UCHL1, GFAP, and tTAU were not helpful in discriminating ALS from DEG (Figures S4, S5, and S6). When distinguishing ALS from ALSmd, the highest AUC value was observed for NfL (AUC = 0.850, 95% CI = 0.785–0.914), whereas UCHL1, GFAP, and tTAU showed lower AUC values (Figures S4, S5, and S6). The optimal cutoff, sensitivity, specificity, PPV, NPV, and AUC of each biomarker are given in Table S5.
Survival analysis
In a univariate analysis, higher NfL, UCHL1, and GFAP negatively affected prognosis. First, KM survival curves were obtained with ALS patients stratified according to serum NfL concentration tertile (Mantel–Cox; χ 2 = 42.3, p < 0.001). Survival estimations for NfL concentration tertile were as follows: first tertile (lower values), 40.3 months (95% CI = 25.1–57.0); second tertile, 21.0 months (95% CI = 14.2–25.8); third tertile, 9.0 months (95% CI = 5.9–12.1 months; Figure 3). Survival estimations for serum UCHL1 concentration tertile (Mantel–Cox; χ 2 = 11.3, p = 0.004) were as follows: first tertile (lower values), 30.0 months (95% CI = 22.7–37.3); second tertile, 18.0 months (95% CI = 13.9–22.1); third tertile, 15.0 months (95% CI = 10.7–19.2 months; Figure 3). For GFAP concentrations (Mantel–Cox; χ 2 = 7.6, p = 0.02), survival estimations were as follows: first tertile (lower values), 36.0 months (95% CI = 16.1–55.8); second tertile, 18.0 months (95% CI = 12.3–23.6); third tertile, 13.0 months (95% CI = 4.0–22.0 months; Figure 3). These results were confirmed when KM curves were adjusted by age at onset and disease duration at serum sample (data not shown). Conversely, tTAU concentration did not show any effect on survival.
FIGURE 3.
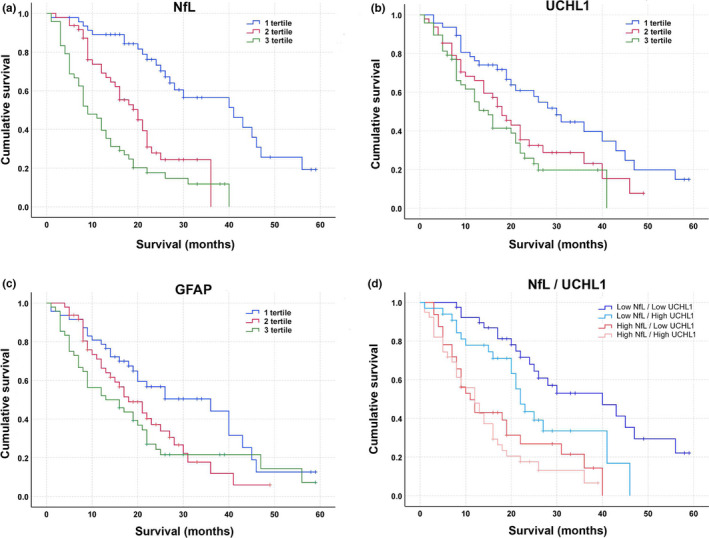
Kaplan–Meier (KM) curves estimate amyotrophic lateral sclerosis (ALS) cumulative survival according to (a) neurofilament light chain (NfL), (b) ubiquitin carboxyl‐terminal hydrolase isozyme L1 (UCHL1), and (c) glial fibrillary acidic protein (GFAP) serum levels. (d) KM survival curves in ALS patients combining NfL and UCHL1 levels: below median values for both biomarkers (Low NfL/Low UCHL1); NfL below median values and UCHL1 above median values (Low NfL/High UCHL1); NfL above median values and UCHL1 below median values (High NfL/Low UCHL1); above median values for both biomarkers (High NfL/High UCHL1). Survival was defined as the time from blood sample to death or tracheostomy [Colour figure can be viewed at wileyonlinelibrary.com]
Multivariate Cox regression models confirmed that higher levels of NfL and UCHL1 are independently associated with reduced survival in ALS, whereas GFAP did not reach any statistical significance. All the variables included in the analysis with respective p value, hazard ratio, and 95% CI are reported in Table 3.
TABLE 3.
Cox proportional hazards regression multivariate analysis on survival
| Covariates | Survival [from serum sample to death or tracheostomy] | |
|---|---|---|
| HR (95% CI) | p | |
| NfL levels, pg/ml | <0.001 a | |
| 1st tertile | 1 | |
| 2nd tertile | 3.24 (1.67–6.30) | <0.001 a |
| 3rd tertile | 5.62 (2.93–10.77) | <0.001 a |
| GFAP levels, pg/ml | 0.649 | |
| 1st tertile | 1 | |
| 2nd tertile | 1.15 (0.66–2.0) | 0.621 |
| 3rd tertile | 0.89 (0.48–1.65) | 0.716 |
| UCHL1 levels, pg/ml | 0.038 a | |
| 1st tertile | 1 | |
| 2nd tertile | 1.04 (0.59–1.85) | 0.80 |
| 3rd tertile | 1.88 (1.06–3.33) | 0.032 a |
| Age at venipuncture | 1.02 (0.99–1.04) | 0.18 |
| Diagnostic delay | 1.09 (1.01–1.17) | 0.03 a |
| Disease progression rate | 1.86 (1.49–2.33) | <0.001 a |
| C9orf72 expansion | ||
| No | 1 | |
| Yes | 1.75 (0.91–3.36) | 0.09 |
| Phenotype | ||
| Spinal | 1 | |
| Bulbar | 1.49 (0.86–2.59) | 0.15 |
Variables included in the model: NfL divided in tertiles, GFAP divided in tertiles, UCHL1 divided in tertiles, age at venipuncture, diagnostic delay, disease progression rate, C9orf72 expansion dichotomized as yes or no, ALS phenotype subdivided into spinal and bulbar.
Abbreviations: CI, confidence interval; GFAP, glial fibrillary acidic protein; HR, hazard ratio; NfL, neurofilament light chain; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1.
Statistically significant.
To further explore the prognostic role of UCHL1, we performed a combined NfL and UCHL1 KM analysis (Figure 3). We clustered patients according to serum median NfL and UCHL1 levels in four different groups. The KM survival analysis produced a significant result (Mantel–Cox; χ 2 = 35.4, p < 0.001). Patients with above median concentrations of NfL had similar prognosis regardless of UCHL1 concentrations (NfL‐high/UCHL1‐low and NfL‐high/UCHL1‐high median survival were 12.0 and 11.0 months, respectively); in contrast, with below median NfL levels, UCHL1 concentrations allowed an effective stratification of patients. In this NfL range, the median survival for ALS patients with below median UCHL1 concentrations (NfL‐low/UCHL1‐low) was 40.0 months (95% CI = 21.7–58.3), whereas it was 22.0 months (95% CI = 18.8–25.1) for ALS patients with UCHL1 concentration above the median (NfL‐low/UCHL1).
DISCUSSION
Our study aimed at screening serum UCHL1, tTAU, and GFAP, surrogates of neuronal injury, neurodegeneration, and astroglial activation, respectively, as potential novel candidate biomarkers for ALS. Their diagnostic and prognostic performance were benchmarked against NfL. In our large ALS cohort, we observed that NfL was raised in ALS phenotypes characterized by a fast disease progression and prominent UMN involvement, whereas GFAP was higher in ALS with concomitant cognitive impairment. Our findings showed the lower diagnostic performance of UCHL1, GFAP, and tTAU compared with NfL in discriminating ALS from HC, DEG, and ALSmd. Conversely, along with NfL, UCHL1 emerged as an independent prognostic factor for survival, proving itself a strong predictor of survival in patients with low NfL concentrations in serum.
NfL is pivotal in maintaining the cytoskeletal structure of neurons and is considered a reliable marker of axonal injury [8]. Despite axonal degeneration being a nonspecific process, shared by several neurological diseases, elevated NfL levels in biofluids have been consistently associated with ALS, supporting their introduction as a promising biomarker for this neurodegenerative disorder. In line with previous reports, NfL was higher in ALS compared with every other control group and showed a good correlation with the disease progression rate [11, 16, 17, 47]. We also demonstrated that NfL correlated with the UMN burden assessed by the UMN score and MEP/cMAP. Although our understanding of axonal degeneration at the system level is limited, our results indicate corticospinal tract degeneration is likely to be the main contributor to NfL outflow into biological fluid. In support of this hypothesis is the observation that NfL was heterogeneously distributed across ALS motor phenotypes; higher NfL levels were seen in ALS patients with prominent UMN burden compared with a restricted or predominantly lower motor neuron phenotypes such as PLMN, FA, and FL. These results are consistent with a previous study by our group, focusing on phosphorylated neurofilament heavy chain levels [46]. In the biomarker panel under investigation, UCHL1 appears to add to the estimation of prognosis and to the clinical stratification of ALS. UCHL1 is an enzyme selectively expressed in the cytoplasm of neurons and involved in the ubiquitin–proteasome system [48, 49]. It has been found upregulated in the CSF of ALS and C9‐ALS subjects, and significantly raised in both CSF and serum of ALS subjects compared with ALSmd [25, 27, 49]. In our study, UCHL1 concentration was higher in ALS compared with HC, but, contrary to a previous study [27] we did not detect any statistical difference between ALS and ALSmd. The larger sample size of our study and the intrinsic heterogeneity of the ALSmd group might explain these diverging results. UCHL1 was homogenous across the ALS motor and cognitive phenotypes. Similarly, no differences were observed between the ALS and DEG groups. Importantly, UCHL1 serum concentrations provide an additional tool for stratification and prognosis in those ALS individuals with low NfL serum concentrations (Figure 3). As lower NfL concentrations in ALS have been linked to slower disease progression, the use of UCHL1 may be informative on prognosis in this subgroup of patients, as UCHL1 levels were able to identify two groups of patients with extremely different prognostic trajectories (as highlighted by the 18‐month difference in median survival) among those with NfL levels below the median. Low serum NfL levels in asymptomatic ALS have been reported to increase in the prodromal and symptomatic phase of the disease and to remain stable throughout disease progression [50]. UCHL1 serum concentration should therefore be tested in longitudinal studies targeting the asymptomatic or early stage of the disease.
GFAP is a specific brain protein and established marker of astrogliosis [28]. The abnormal proliferation of astrocytes, a consequence of the neuronal damage, has been observed to be increased in frontal cortical tissue in FTD, and elevated in both CSF and serum of patients with symptomatic FTD [29, 30, 31, 51]. Consistent with this, we found that GFAP was raised in the DEG group compared to the ALS and HC groups and correlated with the age at symptom onset. Interestingly, we observed that GFAP was different among cognitive phenotypes; that is, ALS with concomitant cognitive and/or behavioral impairment or FTD had higher levels compared with ALS with normal cognition. In light of this result, higher GFAP levels might suggest a wider spreading of the neurodegenerative process, extended to the frontotemporal regions, as typically occurs in patients with ALS and concomitant cognitive impairment. Monitoring GFAP along with the neuropsychological status might be instrumental in tracking the occurrence and the evolution of cognitive decline in ALS. Further longitudinal studies are needed to better address this hypothesis.
Among the investigated biomarkers, NfL reached the best diagnostic performance for ALS. NfL showed almost optimal diagnostic yield in distinguishing ALS from HC (AUC = 0.990) and DEG (AUC = 0.946), with excellent sensitivity and specificity values (Table S5). Conversely, the diagnostic yield of NfL in discriminating ALS from ALSmd was lower (AUC = 0.850), with specificity decreasing to 78.0% (Table S5). Our results, consistent with previous findings, confirmed the potential of NfL as a diagnostic biomarker for ALS [11]. A previous study measured UCHL1, demonstrating good diagnostic performance in both CSF and serum and suggesting it as a promising biomarker for ALS [27]. Conversely, in our study, UCHL1 showed good diagnostic yield in discriminating ALS from HC (AUC = 0.761), but unsatisfactory performance in distinguishing ALS from DEG and ALSmd (Table S5). GFAP and tTAU were not helpful in differentiating ALS from any of the control cohorts.
In univariate analysis, NfL, UCHL1, and GFAP were significant in stratifying ALS survival as shown by the KM curves. Among them, NfL was confirmed to be the best predictor of outcome (Figure 3). However, only NfL and UCHL1 were significant in multivariate analysis. Therefore, we performed a survival analysis combining both biomarkers to improve the prognostic evaluation of NfL when measured alone. Our results demonstrated that UCHL1 is helpful in stratifying ALS prognosis in patients showing lower levels of NfL (below median value). Conversely, patients with high NfL have similar survival independently from UCHL1 level. Therefore, we demonstrated that UCHL1 is an independent prognostic factor for ALS, and the combined evaluation of both biomarkers might be useful in better defining a patient's prognosis. Hence, UCHL1 adds to the growing list of potentially useful prognostic wet biomarkers in ALS, together with serum C‐reactive protein and miR‐181, as well as plasmatic markers of ferroptosis [52, 53, 54].
Our study is not exempt from limitations. Although performed on a large ALS cohort, the lack of a validation cohort represents a limitation; further investigations are required to confirm UCHL1 as an independent prognostic factor, and a larger cohort of positive and negative control groups is needed to confirm our findings. Although a previous study demonstrated a strong correlation between CSF and serum concentration [27] we were not able to assess the diagnostic and prognostic performance of UCHL1 in CSF due to the lack of serum and CSF matched samples in our cohort. Despite that, a blood‐based biomarker would be preferable to a CSF one, due to easier collection practices.
Lastly, the longitudinal evaluation had a short follow‐up period of only six months reducing our ability to calculate a reliable slope for any biomarker in the disease progression using linear mixed models.
In conclusion, we confirmed NfL as a potential diagnostic and prognostic biomarker for ALS. The upcoming introduction of NfL in clinical practice needs the definition of standardized cutoff values, to provide consistency and allow comparisons between measurements from different laboratories. Although we confirm NfL as the strongest predictor of survival, UCHL1 is an independent prognostic factor for ALS and may be helpful in stratifying survival of patients with low NfL. Finally, GFAP might be useful to detect extramotor impairment, namely, cognitive impairment or FTD, in ALS. Future investigations should address finding novel, more specific biomarkers, to improve the diagnostic specificity when combined with NfL in differentiating ALSmd.
CONFLICT OF INTEREST
All the authors report no disclosures or conflicts of interest related to this article. M.F. is Editor‐in‐Chief of the Journal of Neurology, Associate Editor of Human Brain Mapping, Associate Editor of Radiology, and Associate Editor of Neurological Sciences; has received compensation for consulting services and/or speaking activities from Alexion, Almirall, Bayer, Biogen, Celgene, Eli Lilly, Genzyme, Merck‐Serono, Novartis, Roche, Sanofi, Takeda, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Merck‐Serono, Novartis, Roche, Teva Pharmaceutical Industries, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, and Fondazione Italiana di Ricerca per la SLA (ARiSLA). F.A. is Associate Editor of NeuroImage: Clinical; has received compensation for consulting services and/or speaking activities from Novartis, Biogen Idec, Philips, and Roche; and receives or has received research support from the Italian Ministry of Health, AriSLA, and the European Research Council. None of the other authors has any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
Yuri Matteo Falzone: Data curation (lead), formal analysis (lead), investigation (lead), methodology (equal), writing–original draft (lead). Teuta Domi: Data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), writing–original draft (lead). Alessandra Mandelli: Data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), writing–review & editing (equal). Laura Pozzi: Data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), writing–review & editing (equal). Paride Schito: Data curation (equal), investigation (equal), writing–review & editing (equal). Tommaso Russo: Data curation (equal), investigation (equal), writing–review & editing (equal). Alessandra Barbieri: Data curation (equal), investigation (equal), writing–review & editing (equal). Raffaella Fazio: Data curation (equal), writing–review & editing (equal). Maria Antonietta Volontè: Data curation (equal), writing–review & editing (equal). Giuseppe Magnani: Data curation (equal), writing–review & editing (equal). Ubaldo Del Carro: Data curation (equal), writing–review & editing (equal). Paola Carrera: Data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), writing–review & editing (equal). Andrea Malaspina: Writing–review & editing (equal). Federica Agosta: Data curation (equal), funding acquisition (equal), writing–review & editing (equal). Angelo Quattrini: Conceptualization (lead), funding acquisition (lead), writing–review & editing (lead). Roberto Furlan: Formal analysis (equal), funding acquisition (equal), methodology (equal), writing–review & editing (equal). Massimo Filippi: Conceptualization (equal), funding acquisition (lead), resources (lead), supervision (lead), writing–review & editing (equal). Nilo Riva: Conceptualization (lead), data curation (equal), formal analysis (lead), funding acquisition (equal), investigation (equal), supervision (lead), writing–original draft (equal), writing–review & editing (lead).
Supporting information
Falzone YM, Domi T, Mandelli A, et al. Integrated evaluation of a panel of neurochemical biomarkers to optimize diagnosis and prognosis in amyotrophic lateral sclerosis. Eur J Neurol. 2022;29:1930–1939. doi: 10.1111/ene.15321
Massimo Filippi and Nilo Riva share senior authorship.
Funding information
This work was supported by the Giovanni Marazzina Foundation and European Research Council (StG‐2016_714388_NeuroTRACK)
See commentary by P. Bede and J. Lope on page 1867
DATA AVAILABILITY STATEMENT
The anonymized data that support the findings of our study are available from the corresponding author (N.R.) on reasonable request and approval by the relevant boards of the corresponding institutions.
REFERENCES
- 1. Hardiman O, Al‐Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primer. 2017;3(1):17071. doi: 10.1038/nrdp.2017.71 [DOI] [PubMed] [Google Scholar]
- 2. Gentile F, Scarlino S, Falzone YM, et al. The peripheral nervous system in amyotrophic lateral sclerosis: opportunities for translational research. Front Neurosci. 2019;13:601. doi: 10.3389/fnins.2019.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riva N, Gentile F, Cerri F, et al. Phosphorylated TDP‐43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain. 2022;145(1):276‐284. doi: 10.1093/brain/awab285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 5. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population‐based study. Arch Neurol. 2000;57(1):109. doi: 10.1001/archneur.57.1.109 [DOI] [PubMed] [Google Scholar]
- 6. Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17(5):423‐433. doi: 10.1016/S1474-4422(18)30089-9 [DOI] [PubMed] [Google Scholar]
- 7. Beghi E, Chiò A, Couratier P, et al. The epidemiology and treatment of ALS: Focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Lateral Scler. 2011;12(1):1‐10. doi: 10.3109/17482968.2010.502940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falzone Y, Russo T, Domi T, et al. Current application of neurofilaments in amyotrophic lateral sclerosis and future perspectives. Neural Regen Res. 2021;16(10):1985. doi: 10.4103/1673-5374.308072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blasco H, Patin F, Descat A, et al. A pharmaco‐metabolomics approach in a clinical trial of ALS: Identification of predictive markers of progression. Guillemin GJ, Ed. PLoS One. 2018;13(6):e0198116. doi: 10.1371/journal.pone.0198116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verber NS, Shepheard SR, Sassani M, et al. Biomarkers in motor neuron disease: a state of the art review. Front Neurol. 2019;10:291. doi: 10.3389/fneur.2019.00291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verde F, Steinacker P, Weishaupt JH, et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(2):157‐164. doi: 10.1136/jnnp-2018-318704 [DOI] [PubMed] [Google Scholar]
- 12. Wilke C, Preische O, Deuschle C, et al. Neurofilament light chain in FTD is elevated not only in cerebrospinal fluid, but also in serum. J Neurol Neurosurg Psychiatry. 2016;87(11):1270‐1272. doi: 10.1136/jnnp-2015-312972 [DOI] [PubMed] [Google Scholar]
- 13. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. Reindl M, ed. PLoS One. 2013;8(9):e75091. doi: 10.1371/journal.pone.0075091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu CH, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247‐2257. doi: 10.1212/WNL.0000000000001642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gagliardi D, Faravelli I, Meneri M, et al. Diagnostic and prognostic value of CSF neurofilaments in a cohort of patients with motor neuron disease: a cross‐sectional study. J Cell Mol Med. 2021;25(8):3765‐3771. doi: 10.1111/jcmm.16240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feneberg E, Oeckl P, Steinacker P, et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology. 2018;90(1):e22‐e30. doi: 10.1212/WNL.0000000000004761 [DOI] [PubMed] [Google Scholar]
- 17. Gille B, De Schaepdryver M, Goossens J, et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2019;45(3):291‐304. doi: 10.1111/nan.12511 [DOI] [PubMed] [Google Scholar]
- 18. Rossi D, Volanti P, Brambilla L, Colletti T, Spataro R, La Bella V. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J Neurol. 2018;265(3):510‐521. doi: 10.1007/s00415-017-8730-6 [DOI] [PubMed] [Google Scholar]
- 19. Brodovitch A, Boucraut J, Delmont E, et al. Combination of serum and CSF neurofilament‐light and neuroinflammatory biomarkers to evaluate ALS. Sci Rep. 2021;11(1):703. doi: 10.1038/s41598-020-80370-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88(24):2302‐2309. doi: 10.1212/WNL.0000000000004029 [DOI] [PubMed] [Google Scholar]
- 21. Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med. 2018;284(6):643‐663. doi: 10.1111/joim.12816 [DOI] [PubMed] [Google Scholar]
- 22. Day INM, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurogibol. 2010;90(3):327‐362. doi: 10.1016/j.pneurobio.2009.10.020 [DOI] [PubMed] [Google Scholar]
- 23. Liu H, Povysheva N, Rose ME, et al. Role of UCHL1 in axonal injury and functional recovery after cerebral ischemia. Proc Natl Acad Sci. 2019;116(10):4643‐4650. doi: 10.1073/pnas.1821282116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT. The UCH‐L1 gene encodes two opposing enzymatic activities that affect α‐synuclein degradation and Parkinson’s disease susceptibility. Cell. 2002;111(2):209‐218. doi: 10.1016/S0092-8674(02)01012-7 [DOI] [PubMed] [Google Scholar]
- 25. Barschke P, Oeckl P, Steinacker P, et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J Neurol Neurosurg Psychiatry. 2020;91(5):503‐511. doi: 10.1136/jnnp-2019-322476 [DOI] [PubMed] [Google Scholar]
- 26. Zhu S, Wuolikainen A, Wu J, et al. Targeted multiple reaction monitoring analysis of CSF identifies UCHL1 and GPNMB as candidate biomarkers for ALS. J Mol Neurosci. 2019;69(4):643‐657. doi: 10.1007/s12031-019-01411-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li R, Wang J, Xie W, Liu J, Wang C. UCHL1 from serum and CSF is a candidate biomarker for amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(8):1420‐1428. doi: 10.1002/acn3.51141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(3):263‐270. doi: 10.1136/jnnp-2019-321954 [DOI] [PubMed] [Google Scholar]
- 29. Oeckl P, Weydt P, Steinacker P, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4‐10. doi: 10.1136/jnnp-2018-318868 [DOI] [PubMed] [Google Scholar]
- 30. Consortium for Frontotemporal Lobar Degeneration German , Oeckl P, Halbgebauer S, et al. Glial fibrillary acidic protein in serum is increased in alzheimer’s disease and correlates with cognitive impairment. J Alzheimers Dis. 2019;67(2):481‐488. doi: 10.3233/JAD-180325 [DOI] [PubMed] [Google Scholar]
- 31. Ishiki A, Kamada M, Kawamura Y, et al. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J Neurochem. 2016;136(2):258‐261. doi: 10.1111/jnc.13399 [DOI] [PubMed] [Google Scholar]
- 32. Sunderland T, Linker G, Mirza N, et al. Decreased β‐amyloid 1–42 and increased tau levels in cerebrospinal fluid of patients with alzheimer disease. JAMA. 2003;289(16):2094‐2103. doi: 10.1001/jama.289.16.2094 [DOI] [PubMed] [Google Scholar]
- 33. Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993;16(11):460‐465. doi: 10.1016/0166-2236(93)90078-Z [DOI] [PubMed] [Google Scholar]
- 34. Brettschneider J, Petzold A, Sussmuth SD, Ludolph AC, Tumani H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology. 2006;66(6):852‐856. doi: 10.1212/01.wnl.0000203120.85850.54 [DOI] [PubMed] [Google Scholar]
- 35. Grossman M, Elman L, McCluskey L, et al. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(4):442. doi: 10.1001/jamaneurol.2013.6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilke C, Deuschle C, Rattay TW, Maetzler W, Synofzik M. Total tau is increased, but phosphorylated tau not decreased, in cerebrospinal fluid in amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36(2):1072‐1074. doi: 10.1016/j.neurobiolaging.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 37. Paladino P, Valentino F, Piccoli T, Piccoli F, La Bella V. Cerebrospinal fluid tau protein is not a biological marker in amyotrophic lateral sclerosis. Eur J Neurol. 2009;16(2):257‐261. doi: 10.1111/j.1468-1331.2008.02405.x [DOI] [PubMed] [Google Scholar]
- 38. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497‐503. doi: 10.1016/j.clinph.2007.09.143 [DOI] [PubMed] [Google Scholar]
- 39. Schrooten M, Smetcoren C, Robberecht W, Van Damme P. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: a prospective study. Ann Neurol. 2011;70(1):79‐83. doi: 10.1002/ana.22380 [DOI] [PubMed] [Google Scholar]
- 40. Riva N, Mora G, Sorarù G, et al. Safety and efficacy of nabiximols on spasticity symptoms in patients with motor neuron disease (CANALS): a multicentre, double‐blind, randomised, placebo‐controlled, phase 2 trial. Lancet Neurol. 2019;18(2):155‐164. doi: 10.1016/S1474-4422(18)30406-X [DOI] [PubMed] [Google Scholar]
- 41. Chio A, Calvo A, Moglia C, Mazzini L, Mora G. PARALS study group. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2011;82(7):740‐746. doi: 10.1136/jnnp.2010.235952 [DOI] [PubMed] [Google Scholar]
- 42. Agosta F, Ferraro PM, Riva N, et al. Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol Aging. 2017;57:206‐219. doi: 10.1016/j.neurobiolaging.2017.05.024 [DOI] [PubMed] [Google Scholar]
- 43. Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis ‐ frontotemporal spectrum disorder (ALS‐FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Front Degener. 2017;18(3–4):153‐174. doi: 10.1080/21678421.2016.1267768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456‐2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Poletti B, Solca F, Carelli L, et al. The validation of the Italian Edinburgh Cognitive and Behavioural ALS Screen (ECAS). Amyotroph Lateral Scler Front Degener. 2016;17(7–8):489‐498. doi: 10.1080/21678421.2016.1183679 [DOI] [PubMed] [Google Scholar]
- 46. Falzone YM, Domi T, Agosta F, et al. Serum phosphorylated neurofilament heavy‐chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J Neurol. 2020;267(8):2272‐2280. doi: 10.1007/s00415-020-09838-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59‐e69. doi: 10.1212/WNL.0000000000009559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bishop P, Rocca D, Henley JM. Ubiquitin C‐terminal hydrolase L1 (UCH‐L1): structure, distribution and roles in brain function and dysfunction. Biochem J. 2016;473(16):2453‐2462. doi: 10.1042/BCJ20160082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oeckl P, Weydt P, Thal DR, Weishaupt JH, Ludolph AC, Otto M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020;139(1):119‐134. doi: 10.1007/s00401-019-02093-x [DOI] [PubMed] [Google Scholar]
- 50. Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion: Neurofilament Light in Presymptomatic ALS. Ann Neurol. 2018;84(1):130‐139. doi: 10.1002/ana.25276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Umoh ME, Dammer EB, Dai J, et al. A proteomic network approach across the als ‐ ftd disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol Med. 2018;10(1):48‐62. doi: 10.15252/emmm.201708202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lunetta C, Lizio A, Maestri E, et al. Serum C‐reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. 2017;74(6):660. doi: 10.1001/jamaneurol.2016.6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Devos D, Moreau C, Kyheng M, et al. A ferroptosis–based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci Rep. 2019;9(1):2918. doi: 10.1038/s41598-019-39739-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Magen I, Yacovzada NS, Yanowski E, et al. Circulating miR‐181 is a prognostic biomarker for amyotrophic lateral sclerosis. Nat Neurosci. 2021;24(11):1534‐1541. doi: 10.1038/s41593-021-00936-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The anonymized data that support the findings of our study are available from the corresponding author (N.R.) on reasonable request and approval by the relevant boards of the corresponding institutions.