

Abstract
Background and purpose
Mutations in DNAJB2 are associated with autosomal recessive hereditary motor neuropathies/ Charcot‐Marie‐Tooth disease type 2 (CMT2). We describe an Italian family with CMT2 due to a homozygous DNAJB2 mutation and provide insight into the pathomechanisms.
Methods
Patients with DNAJB2 mutations were characterized clinically, electrophysiologically and by means of skin biopsy. mRNA and protein levels were studied in lymphoblastoid cells (LCLs) from patients and controls.
Results
Three affected siblings were found to carry a homozygous DNAJB2 null mutation segregating with the disease. The disease manifested in the second to third decade of life. Clinical examination showed severe weakness of the thigh muscles and complete loss of movement in the foot and leg muscles. Sensation was reduced in the lower limbs. All patients had severe hearing loss and the proband also had Parkinson’s disease (PD). Nerve conduction studies showed an axonal motor and sensory length‐dependent polyneuropathy. DNAJB2 expression studies revealed reduced mRNA levels and the absence of the protein in the homozygous subject in both LCLs and skin biopsy. Interestingly, we detected phospho‐alpha‐synuclein deposits in the proband, as already seen in PD patients, and demonstrated TDP‐43 accumulation in patients’ skin.
Conclusions
Our results broaden the clinical spectrum of DNAJB2‐related neuropathies and provide evidence that DNAJB2 mutations should be taken into account as another causative gene of CMT2 with hearing loss and parkinsonism. The mutation likely acts through a loss‐of‐function mechanism, leading to toxic protein aggregation such as TDP‐43. The associated parkinsonism resembles the classic PD form with the addition of abnormal accumulation of phospho‐alpha‐synuclein.
Keywords: genetic and inherited disorders, neuropathology, Parkinson's disease, peripheral neuropathies
We report a family with Charcot‐Marie‐Tooth disease type 2 (CMT2), with hearing loss and parkinsonism, who harbor a novel homozygous DNAJB2 mutation, expanding the phenotype associated with the disease and providing new insights into its pathomechanism.

INTRODUCTION
Charcot‐Marie‐Tooth disease (CMT) is the most common degenerative disorder of the peripheral nervous system. It is associated with wide clinical and genetic heterogeneity and is classified into different forms according to inheritance pattern and neurophysiology [1]. Distal hereditary motor neuropathies (dHMN), also called distal spinal muscular atrophy (dSMA), are a related group of genetic neuropathies characterized by progressive muscle weakness and atrophy, predominating at the distal part of the limbs [2].
Recessive DnaJ homolog subfamily B member 2 (DNAJB2) gene mutations (first described as heat shock protein J1 [HSJ1] gene) have been identified as a so‐far uncommon cause of autosomal recessive peripheral neuropathies, which may present as either dSMA (dSMA5‐AR) or CMT type 2 (AR‐CMT2). Six different missense mutations and a large deletion have been identified in different families to date [3, 4, 5, 6, 7, 8, 9]. Symptoms involving the central nervous system have been described in some patients with DNAJB2 mutations, with early‐onset parkinsonism being reported in at least three patients from as many different families, expanding the phenotypic spectrum of the disease [6, 7, 9].
DNAJB2 is a key component in neuronal protein quality control and acts as a molecular chaperone. Specifically, it primarily directs the client proteins to the ubiquitin‐proteasome system through two ubiquitin interaction motifs that mediate binding to polyubiquitinated proteins and to the proteasome [10]. Supporting its role in protein aggregation prevention, DNAJB2 has recently been shown to bind insoluble TAR‐DNA binding protein‐43 (TDP‐43) and to be highly effective in preventing its aggregation by allowing it to interact with the chaperone protein HSP70 [11].
Alternative splicing produces two isoforms of the protein that differ in their C‐termini and show distinct subcellular localization: DNAJB2a (HSJ1a; 277 amino acids, 31 kDa) localizes to the cytosol and nucleus, whereas DNAJB2b (HSJ1b; 324 amino acids, 36 kDa) is associated with the cytoplasmic face of the endoplasmic reticulum through the C‐terminal [10]. DNAJB2 is predominantly expressed in neurons and the clearly predominant isoform in neuronal tissues is DNAJB2b. Moreover, colocalization of DNAJB2 and neurofilaments (NFs) was demonstrated in the isolated sciatic nerve of mice. A low level of the protein has been detected in other cells and tissues (e.g., cardiac and skeletal muscles), as well as in fibroblast cultures [12].
We report a CMT2 family harboring a novel homozygous DNAJB2 mutation, expanding the phenotype associated with the disease and providing new insights into its pathomechanism.
METHODS
Patients
We evaluated three affected individuals from a family with a clinical diagnosis of CMT2 (Figure 1a). Clinical impairment was evaluated using the validated CMT Examination Score, version 2 (CMTESv2) [13]. All patients underwent nerve conduction studies according to standard procedures as well as skin biopsies that were processed for either immunohistochemistry (IHC) or Western blot (WB) analysis. A healthy control population was included for the skin biopsy analysis. The index case also underwent muscle computed tomography (CT) and a DaT‐Scan test. In 1991, sural nerve biopsy was performed in patient II.1, at the age of 36 years, and processed according to the same methodology as that employed in a previous study [14].
FIGURE 1.
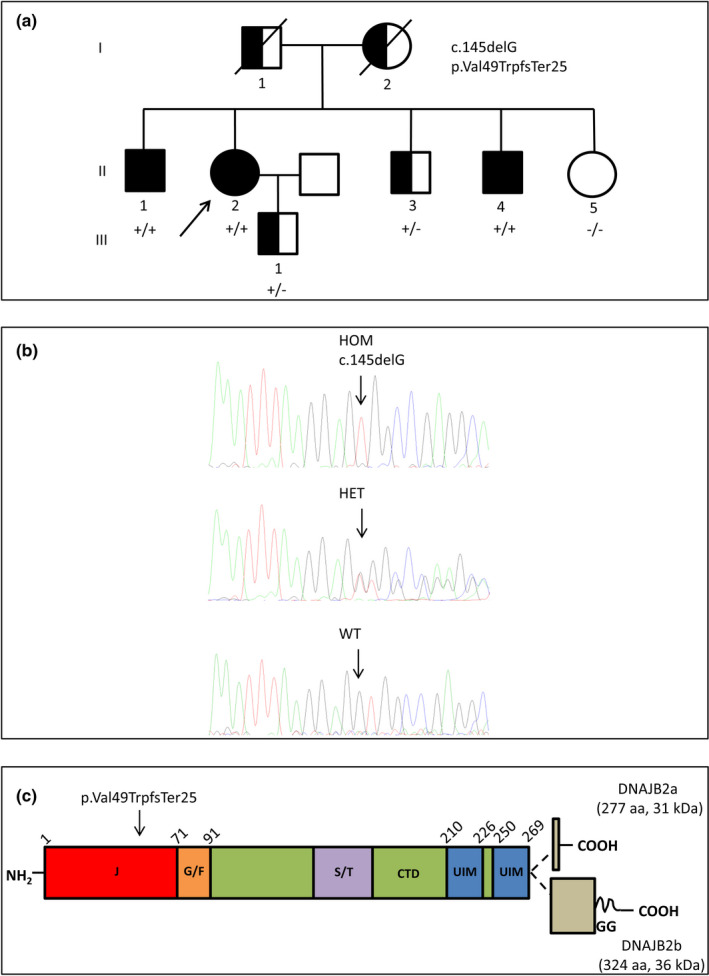
Pedigree, DNA sequencing and schematic representation of DNAJB2 protein domain structure. (a) Family pedigree. (b) Sanger chromatograms of patient II.4 with the homozygous (HOM) c.145delG variant in the DNAJB2 gene, patient II.3 (heterozygous [HET]) and II.5 (wild‐type [WT]). (c) DNAJB2 protein domains, the alternatively spliced C‐terminal parts of the two isoforms and the residue harboring the disease mutation (p. Val49TrpfsTer25) are indicated. The J, G/F common region, and C‐terminal domains (CTD), the Ser/Thr‐rich region, two ubiquitin interacting motifs (UIM) and the C‐terminal geranylgeranyl (GG) anchor of DNAJB2b are shown. Partly modified from Sarparanta et al., Int J Mol Sci. 2020; 21: 1409
The Institutional Ethics Committee of the Fondazione IRCCS Istituto Neurologico Carlo Besta approved the study and written informed consent was obtained from all participants in the study.
Genetic analysis
The proband (II.2) DNA was analyzed via a next‐generation sequencing (NGS) multigene panel for CMT and related disorders including 94 genes (Nextera Rapid Capture Custom Kit I; Illumina Inc.). Sequencing was performed using the Illimina MiSeq NGS sequencer (Illumina Inc.). The entire DNAJB2 coding region (10 coding exons and 20 bp of flanking introns) was sequenced by NGS, with a depth of coverage >50× (mean coverage 243×). The variant was confirmed in all patients by a Sanger sequencing method. An NGS multigene panel of Parkinson disease (PD)‐associated genes was performed in patient II.2. Copy number variations of alpha‐synuclein (SNCA) and parkin (PARK2) genes were analyzed using multiplex ligation‐dependent probe amplification according to the manufacturer's protocol (kit P051; MRC Holland). For bioinformatic analysis, reads were aligned to the reference human genome (hg19) and the identified variants were annotated and filtered, focusing on rare variants (≤1% in public databases: dbSNP137, gnomAD v2.1.1, NHLBI Exome Sequencing Project 6500, and 1000 Genomes project). Filtering passing variants were classified and prioritized according to the American College of Medical Genetics Standards and Guidelines [15]. Variant confirmation and segregation analysis were performed by Sanger sequencing.
Patient‐derived cell lines and mRNA analysis
Epstein–Barr‐virus‐stabilized lymphoblastoid cell lines (LCLs) from an affected patient (II.4), an unaffected carrier (III.1) and two unrelated control subjects were established and cultured as previously described [16].
Total RNA was extracted using the Maxwell® 16 LEV simplyRNA Cells Kit (Promega, Madison, WI, USA). cDNA synthesis was carried out using Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer's instructions, with oligo(dT)s and random primers. DNAJB2 mRNA level were evaluated with specific primer by quantitative PCR using a 7300 Real‐Time PCR System (Applied Biosystems) with FastStart Universal SYBR® Green Master (Sigma). Ct values were normalized to GAPDH mRNA level. Relative expression of DNAJB2 was determined by the 2‐∆∆Ct method. Data are presented as mean ± standard error of three experimental replicates.
Skin biopsy: Immunohistochemistry and immunoblot analysis
Skin samples for IHC or WB analysis were obtained by a 3‐mm punch from the proximal phalanx of the II finger (patient II.4) or from the shoulder (patients II.1 and II.2). Samples for IHC were fixed by immersion in 2% paraformaldehyde‐lysine‐periodate for 24 h at 4°C, washed, cryoprotected overnight and serially cut (50 μm) with a standard cryostat (Bio‐Optica). Assays were performed following standard free‐floating protocol. Skin sections were incubated with a panel of primary antibodies at room temperature overnight, washed and reacted with secondary antibodies for 1 h the following day, washed, dried and mounted for examination. The following primary antibodies were employed: anti‐DNAJB2 (1:85, 10838‐1‐AP; Proteintech), anti‐myelin protein zero (1:80, ab39375; Abcam, Cambridge, UK), anti‐neurofilament 200 (1:400, N0142, Sigma‐Aldrich), anti‐PGP9.5 (1:400, E3344; Spring Bioscience Corp.), anti‐phospho‐alpha‐synuclein (1:400, 825701; BioLegend). We used Alexa Fluor‐conjugated (Jackson ImmunoResearch) secondary antibodies. Fluorescent images were acquired with a Leica TSC SP8 confocal microscope (Leica Microsystems). Labeled fibers were chosen randomly from three non‐adjacent sections. Image acquisition was performed by the same investigator, blinded to the experimental condition.
Skin samples for WB analysis were immediately frozen in liquid nitrogen. Frozen skin biopsies were pulverized, dissolved in lysis buffer (RIPA buffer, R0278 [Sigma] and protease inhibitors) and sonicated. Samples were subsequently kept on ice for at least 30 min, and centrifuged at 14,000 g for 10 min at 4°C. The protein content of the supernatant was determined using a bovine serum albumin standard curve. Equal protein amounts were loaded onto SDS‐PAGE. Separated proteins were transferred onto PVDF membrane (Immobilon; Millipore). The membrane was blocked in 1% milk in tris‐buffered saline (TBS) 1× with 0.1% Tween20 for 1 h at room temperature, incubated with the appropriate primary antibody, washed in TBS 1× with 0.1% Tween20 and then incubated with a secondary antibody conjugated with horseradish peroxidase (HRP). The primary antibodies used were: DNAJB2 (1:500, 10838‐1‐AP; Proteintech) and phospho‐TDP43 (1:500, 22309‐1‐AP; Proteintech). HRP‐conjugated secondary antibodies (1:10000; GE HealthCare) were detected using enhanced chemiluminescence reagents (LiteAblot Extend Long Lasting Chemiluminescent Substrate; EuroClone Life Sciences). Densitometry analysis was performed using the Gene Tools from Syngene and normalized to appropriate loading controls signal intensity. Results were normalized for beta‐actin (1:5000, ab8224; Abcam).
Lymphoblasts: Immunoblot analysis
Lymphoblasts from patient II.4, III.1 and control were removed from each flask and transferred into 15‐ml tubes. Cells were centrifuged at 1800 g for 10 min. Supernatants were discarded and cell pellets were washed in PBS. The resulting cell pellets were lysed and processed as reported above for WB analysis of skin biopsies. Anti‐DNAJB2 primary antibody (1:500, 10838‐1‐AP; Proteintech) was employed (overnight 4°C).
Statistical analysis
Student's t‐test for unpaired data was used to compare mRNA and protein levels from patients and controls. A p value of <0.01 was taken to indicate statistical significance.
Data sharing and data accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
RESULTS
Patients
The disease onset occurred in the second or third decade of life, with gait difficulty and progressive weakness in the lower limbs, first distally and then proximally. Clinical examination showed mild‐to‐moderate distal paresis in the upper limbs, a severe weakness of the thigh muscles and complete loss of movement in the foot and leg muscles. Joint position, vibration and superficial sensation were reduced in the lower limbs. The disease severity, as determined by the CMTESv2, ranged from moderate to severe impairment (scores between 17 and 20). All patients were wheelchair‐bound from their fifth to sixth decade of life. Additionally, all patients had deafness, starting in the third or fourth decade of life. The proband developed typical PD at the age of 50 years, with asymmetric resting tremor, rigidity and hypokinesia responsive to L‐DOPA treatment. Individual CMTESv2 scores, along with detailed clinical information, are provided in Table 1.
TABLE 1.
Clinical features
| Patients | II.1 | II.2 | II.4 |
|---|---|---|---|
| Age at examination, years/sex | 61/M | 59/F | 52/M |
| Onset age, years/symptoms | 28/Gait difficulty | 28/Gait difficulty | 15/Gait difficulty |
| Motor symptoms legs | Wheelchair | Wheelchair | Wheelchair |
| Proximal/distal weakness UL | −/+ (4+; 4+) | −/+ (4+; 4−) | + (5; 4; 5)/+ (3; 3) |
| Proximal/distal weakness LL | + (4−; 2)/+ (0; 0) | + (2; 2)/+ (0; 0) | + (1; 1)/+ (0; 0) |
| Ankle deep tendon reflexes | Absent | Absent | Absent |
| Joint position sense UL/LL | Normal/toes | Normal/ankle | Normal/toes |
| Vibration sense UL/LL | Normal/knee | Normal/ankle | Normal/knee |
| Pinprick UL/LL | Normal/toes | Normal/ankle | Normal/toes |
| Light touch UL/LL | Normal/toes | Normal/ankle | Normal/toes |
| Pes cavus | N | N | Y (mild) |
| Additional features | Hearing loss | Hearing loss/Parkinson disease | Hearing loss |
| CMTESv2 (disease severity) | 13 (moderate) | 20 (severe) | 17 (moderate‐severe) |
Motor weakness based on Medical Research Council Scale: upper limb distal weakness assessed by first dorsal interosseous and abductor pollicis brevis; upper limb proximal weakness assessed by deltoids, biceps brachii and triceps. Lower limb distal weakness assessed by anterior tibialis and gastrocnemius; lower limb proximal weakness assessed by iliopsoas and quadriceps. Numbers are based on the side that had the worst score. For sensory examination, the level given is the highest level where a deficit was detected.
Abbreviations: −, weakness absent; +, weakness present; CMTESv2, Charcot‐Marie‐Tooth Examination Score, version 2; F, female; LL, lower limbs; M, male; N, not; UL, upper limbs; Y, yes.
Electrophysiological findings revealed a severe length‐dependent axonal motor‐sensory polyneuropathy in all affected patients (Table 2). Audiometry and brainstem auditory evoked potentials were consistent with bilateral sensorineural hearing loss. In the proband, muscle CT showed severe fatty replacement of both the anterior and posterior compartments of the leg more than the thigh muscles (Figure 2a), while DaT‐Scan disclosed a severe, bilateral but asymmetric reduction of tracer uptake indicative of presynaptic dopaminergic terminal degeneration (Figure 2b). Sural nerve biopsy in patient II.1, at the age of 36 years, disclosed a moderate reduction of nerve fiber density, mainly involving the large‐caliber fibers. A few fibers had thin myelin in relation to the axon diameter and were surrounded by one or two lamellae of Schwann cell cytoplasm. There were some acute axonal degeneration figures and rare regeneration clusters (Figure 2c).
TABLE 2.
Electrophysiological findings
| Age at examination |
Patient II.1 43 years |
Patient II.2 53 years |
Patient II.4 27 years |
|---|---|---|---|
| Motor conduction | |||
| L median nerve | |||
| DML (≤4.1 ms) | 3.3 | 3.8 | 3.2 |
| MNCV (≥50 m/s) | 52.3 | 54.3 | 51.5 |
| dCMAP (≥6 mV) | 11.5 | 13.4 | 8 |
| L ulnar nerve | |||
| DML (≤3.3 ms) | 3.2 | 2.5 | |
| MNCV (≥55 m/s) | 56.3 | 59.5 | |
| dCMAP (≥7 mV) | 11.9 | 8 | |
| R peroneal nerve | |||
| DML (≤5 ms) | NA | NA | NA |
| MNCV (≥43 m/s) | NA | NA | NA |
| dCMAP (≥5 mV) | 0 | 0 | 0 |
| R tibial nerve | |||
| DML (≤5 ms) | NA | NA | 9 |
| MNCV (≥44 m/s) | NA | NA | 30 |
| dCMAP (≥5 mV) | 0 | 0 | 0.1 |
| Sensory conduction | |||
| L median nerve | |||
| SNCV (≥50 m/s) | 44 | NA | |
| SNAP (≥10 μV) | 5 | 0 | |
| R radial nerve | |||
| SNCV (≥48 m/s) | 50 | ||
| SNAP (≥5 μV) | 2.3 | ||
| L ulnar nerve | |||
| SNCV (≥46 m/s) | 48 | 37 | |
| SNAP (≥6 μV) | 7 | 6 | |
| R sural nerve | |||
| SNCV (≥46 m/s) | NA | NA | |
| SNAP (≥6 μV) | 0 | 0 | |
| R superficial peroneal nerve | |||
| SNCV (≥43 m/s) | NA | ||
| SNAP (≥5 μV) | 0 |
Normal values between parentheses and abnormal values in bold.
Abbreviations: dCMAP, distal compound muscular action potential amplitude; DML, distal motor latency; L, left; MNCV/SNCV, motor/sensory nerve conduction velocity; NA, not applicable; R, right; SNAP, sensory nerve action potential amplitude.
FIGURE 2.
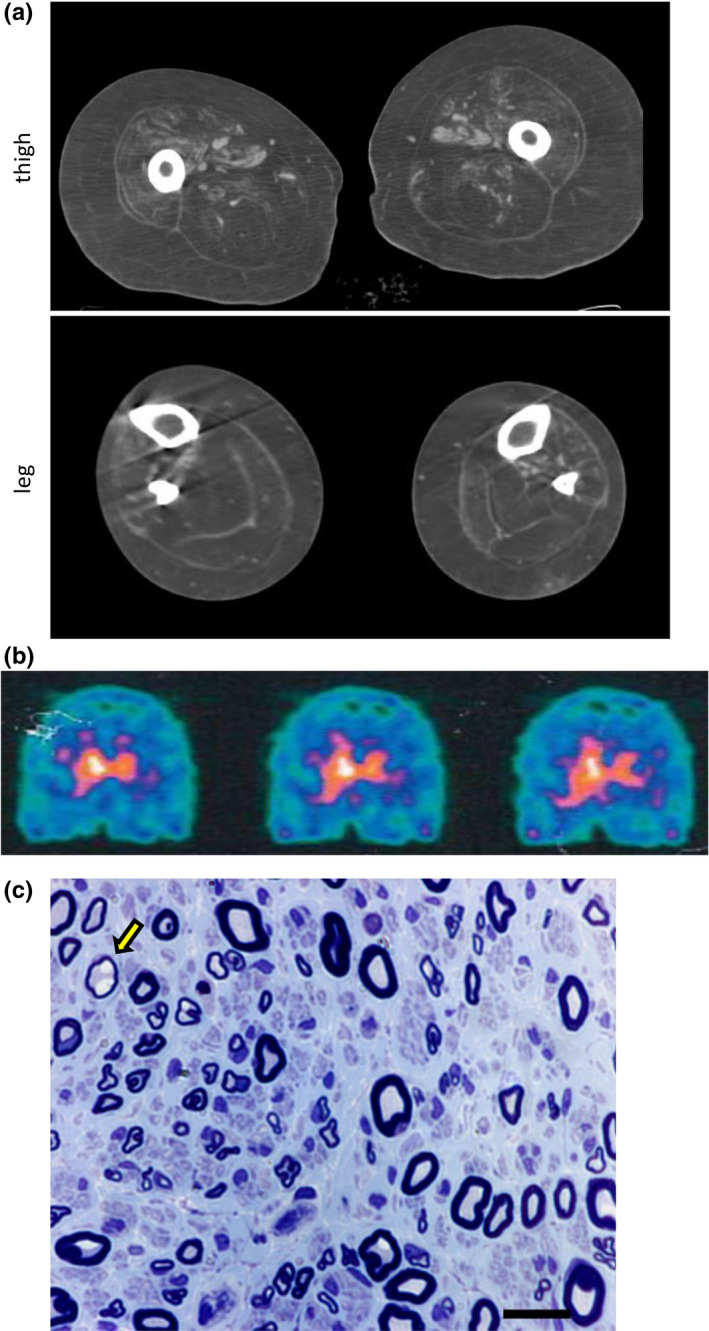
Dat‐Scan, muscle computed tomography (CT) and sural nerve biopsy. (a) Muscle CT of patient II.2 showing severe fatty replacement of thigh and leg muscles, in both the anterior and posterior compartments. (b) Dat‐Scan of patient II.2 showing a severe, bilateral but asymmetric reduction of tracer uptake indicative of presynaptic dopaminergic terminal degeneration. (c) Sural nerve biopsy of patient II.1, taken at age 36 years. Semithin sections, stained with toluidine blue, disclosing a mild‐to‐moderate reduction of nerve fiber density, mainly involving the fibers of large caliber. A few fibers had thin myelin in relation to the axon diameter (arrow). Some fibers showed evidence of acute axonal degeneration and there were rare regeneration clusters. Scale bar = 15 μm. CTRL, control
Genetic analysis
The CMT gene panel analysis in the proband (II.2) revealed the novel homozygous pathogenic variant c.145delG in the DNAJB2 gene (NM_006736.6), resulting in the frameshift p. Val49TrpfsTer25. The mutation segregated with the disease as it was present in homozygosity in two affected siblings (II.1, II.4) and absent in a healthy sister (II.5; Figure 1a–b).
The unaffected deceased parents were apparently not consanguineous but came from a small village in Northern Italy. One of the brothers (II.3) and the unaffected son of the proband (III.1) were found to be heterozygous for the mutation (Figure 1a–b). Sequence analysis of PD‐associated genes did not detect pathogenic or likely pathogenic variants in the proband, while copy number analysis excluded the presence of duplication/triplication of SNCA and deletion/rearrangements of PARK2 genes. The c.145delG DNAJB2 variant, absent in general population databases, is a null mutation which alters the amino acid sequence starting from Val49 in the J domain and causes a premature termination codon after 25 amino acids (Figure 1c).
mRNA and protein level in lymphoblastoid cells
To study the effects of the variant we investigated its impact on DNAJB2 mRNA and protein levels in both homozygous and heterozygous LCLs derived from patient II.4, an unaffected carrier (III.1) and controls.
Quantitative mRNA analysis revealed a significant reduction in DNAJB2 mRNA levels in homozygous and heterozygous subjects as compared to the controls (Figure 3a). The reduction was more severe in the homozygous subject, with a residual mRNA level of 33%, compared to the heterozygous subject who had a residual mRNA level of 65%. Consistently, WB analysis showed an absence of DNAJB2 protein in the homozygous patient and a significant reduction of protein level in the heterozygous carrier (40% of the control's protein level; Figure 3a–b).
FIGURE 3.
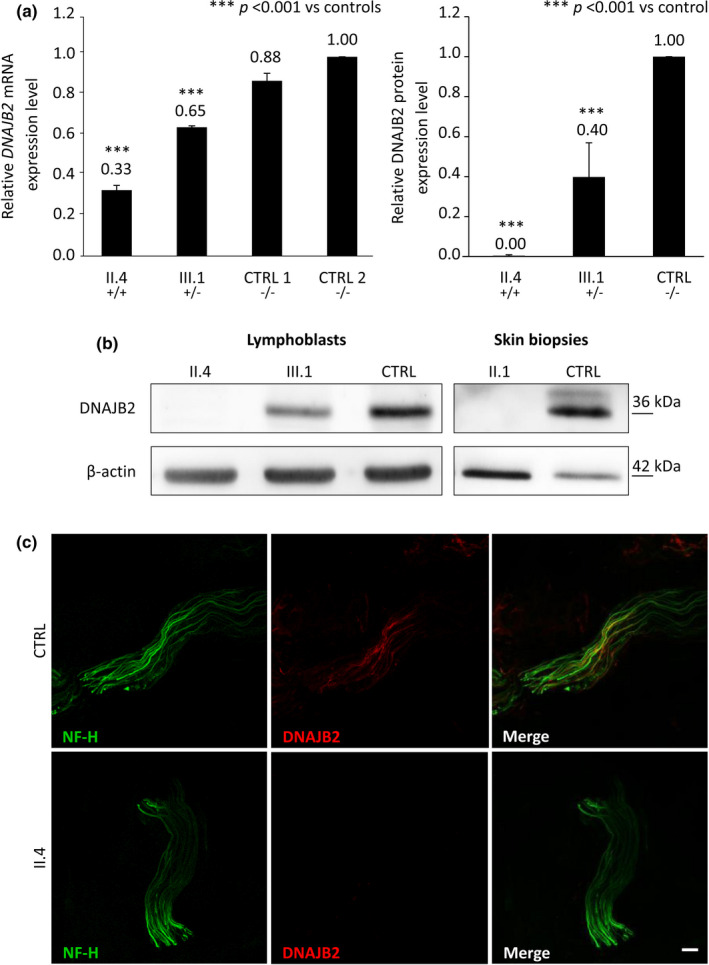
DNAJB2 mRNA and protein levels, expression of DNAJB2 protein in skin. (a) DNAJB2 mRNA and protein expression analysis in lymphoblastoid cell lines (LCLs) from an affected patient (II.4), an unaffected carrier (III.1) and control subjects. It shows in the homozygous subject residual mRNA level of 33% and the absence of the protein, and in the heterozygous one 65% of mRNA level and of 40% of the protein. DNAJB2 mRNA level was normalized to GAPDH mRNA level. Bars and vertical lines indicate mean and ±1 SD of three independent experiments, respectively. Asterisks indicate a statistically significant (***p ≤ 0.001) difference from controls, as determined by Student's t‐test. (b) Immunoblot analysis from LCLs and skin. DNAJB2 protein expression was missing in both LCLs and skin from the homozygous patients (II.1 and II.4) and significantly reduced in the LCLs from the heterozygous carrier (III.1). Beta‐actin expression was normal in all the samples. (c) Immunohistochemical staining on skin biopsy sections using antibodies direct to neurofilament‐200 (in green) and DNAJB2 protein (in red) in both patient II.4 and control. Sections from control (upper panel) demonstrated many axons labeled by the axonal marker Neurofilament heavy chain (NF‐H) and DNAJB2 antibodies, while in the patient (lower panel) axons were labeled with antibodies to NF‐H but not by DNAJB2 antibodies. Scale bar = 10 μm. CTRL, control
Skin biopsy
Cutaneous nerve fibers show loss of DNAJB2 expression and normal NF expression
We performed IHC on dermal nerves from patients and controls. To investigate whether DNAJB2 loss could lead to NF impairment and to explain the axonal degeneration mechanism, we marked dermal fibers with anti‐NF‐H antibodies. Sections from controls demonstrated many axons labeled by the axonal marker NF‐H and DNAJB2 antibodies (Figure 3c). By contrast, axons from patients were labeled with antibodies to NF‐H but not by anti‐DNAJB2 antibodies (Figure 3c).
Results from WB analysis of skin samples confirmed these data, showing the complete absence of the expected DNAJB2 band in patient II.1, whereas beta‐actin expression was normal (Figure 3b).
TDP‐43 accumulation in patients’ skin
As DNAJB2a was recently shown to prevent TDP‐43 aggregation [11], we assessed phospho‐TDP‐43 expression by immunoblot analysis of samples from skin biopsies and demonstrated the presence of TDP‐43 in patients II.1 and II.2 but not in the control (Figure 4a).
FIGURE 4.
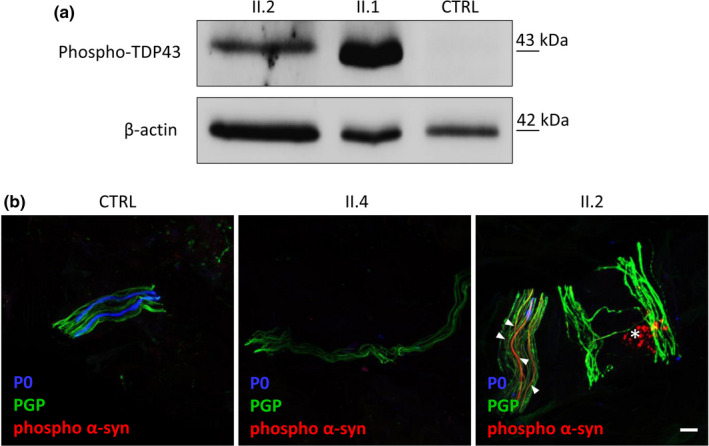
TDP‐43 expression and phospho‐alpha‐synuclein aggregates in patients’ skin. (a) Western blot analysis of phospho‐TDP43 in the skin tissue of patients II.1, II.2 and control. TDP‐43 is expressed in patients’ skin and absent in the control. Beta‐actin was used as loading control. (b) Immunohistochemical staining on skin biopsy sections using antibodies direct to phospho‐alpha‐synuclein (p‐alpha‐syn; in red), PGP 9.5 (pan‐axonal marker; in green) and P0 (myelinated fibers; in blue) in patient II.4, patient II.2, who had also Parkinson disease, and control. The analysis showed in patient II.2 aggregates of p‐alpha‐syn which were absent in both patient II.4 and control. In the true intraneural deposition, the p‐alpha‐syn and PGP 9.5 signals are colocalized (arrowhead) and continuous along the nerve fiber; otherwise, p‐alpha‐syn signal without colocalized PGP 9.5 staining (asterisk) is a non‐specifc signal due to fuorophore precipitates that often presents a dot‐like staining distribution. Scale bar = 10 μm
Phospho‐alpha‐synuclein deposits were present in skin nerves
Since the associated parkinsonism of our patient resembled classic PD, we performed IHC for phospho‐alpha‐synuclein (p‐alpha‐syn) to determine if the pathological phenotype was consistent with the classic form. Notably, IHC study of the proband's skin biopsy showed abnormal accumulation of p‐alpha‐syn along the nerve fiber, which was absent in the control (Figure 4b).
To study whether the abnormal accumulation of p‐alpha‐syn might precede the clinical onset of PD, we also analyzed the skin of patient II.4 showing no signs of parkinsonism. As in the control, no p‐alpha‐syn deposits were detected in his skin nerves (Figure 4b).
DISCUSSION
We report an Italian family with AR‐CMT2 due to a novel DNAJB2 mutation and characterize its multifaceted phenotype and possible pathomechanisms. Mutations in DNAJB2 have been reported in families with autosomal recessive dSMA/CMT2. The presenting phenotype in our patients was CMT2 as sensory abnormalities were detected at both clinical and electrophysiological examination. The clinical picture was quite severe as all patients had been wheelchair‐bound from their fifth to sixth decade. Muscle CT demonstrated extensive fatty infiltration in the thigh muscles as well as leg muscles; the sural nerve biopsy, obtained at an early disease stage (at age 36 years), showed a mild‐to‐moderate reduction of large myelinated nerve fibers. Additionally, all patients developed a severe deafness in early adulthood, never reported before for DNAJB2 mutations, broadening the clinical spectrum of the disease and providing evidence that DNAJB2 is another causative gene of axonal CMT with hearing loss. Auditory function is often impaired in both demyelinating and axonal forms of CMT [17]. In particular, sensorineural hearing loss has been reported in patients with CMT caused by mutations in different genes, including PMP22, MPZ, GJB1, SH3TC2, NDRG1, NEFL, PRPS1 and MORC2. Physiological studies in both CMT1 and CMT2 have localized the disorder to the auditory nerve, providing evidence of normal cochlear activity with disrupted auditory brainstem potentials [17]. We hypothesize the same mechanism in our patients and suggest to include DNAJB2 gene testing in cases of CMT2/dSMA and deafness.
The reported DNAJB2 mutation is a null mutation which alters the amino acid sequence starting from Val49 and causes a premature termination codon after 25 amino acids. It is predicted to act through a loss‐of‐function mechanism. Indeed, DNAJB2 mRNA and protein analysis revealed a significant reduction of DNAJB2 mRNA level in lymphoblasts, more severe in the homozygous than in the heterozygous subject, and absence of the protein in the homozygous patient and a significant reduction in the heterozygous carrier, in both lymphoblasts and skin. Consistent with these data, skin biopsy analysis showed a complete loss of DNAJB2 expression in patients that was not associated with an impairment of NF, leaving the pathomechanism of the axonal loss still unclear.
DNAJB2 is involved in the prevention of protein aggregation, and isoform A has been shown to bind insoluble TDP‐43 and prevent its accumulation in the cells [11]. Consistent with these data, we demonstrated TDP‐43 accumulation in patients’ skin but not in the control, supporting the hypothesis that in our patients the loss of DNAJB2 impairs the removal of toxic protein aggregates.
Mislocalization and aggregation of TDP‐43 is a common pathological hallmark of several neurodegenerative diseases. Mutations in Dynactin‐1 gene (DCTN1) cause two distinct neurodegenerative diseases: dHMN7B and Perry syndrome. Particularly, the latter shows some clinical and pathological similarities with those related to the DNAJB2 phenotype. It is characterized clinically by early‐onset parkinsonism, depression/apathy and central hypoventilation, and pathologically by degeneration of dopaminergic neurons and TDP‐43 proteinopathy [18].
Other evidence of the role of DNAJB2 in hindering protein aggregate formation is provided by the finding that DNAJB2a reduces the aggregation of mutated Parkin, whose corresponding gene is related to juvenile parkinsonism [19], and DNAJB2 mRNA transcripts are increased in the PD brain [20]. Thus far, PD has been reported in at least three patients with DNAJB2 mutations from as many different families [6, 7, 9]. All patients had early‐onset PD with resting tremor and rigidity beginning at 16 years in one, and left‐sided symptoms at 40 years in another. Interestingly, one patient in our family (patient II.2) was affected by PD with onset at the age of 50 years and no evidence of mutations in PD‐associated genes nor abnormal gene dosage of SNCA or PARK2. Therefore, our family further confirms that PD is indeed part of the phenotypic spectrum of the disease. In addition, we demonstrated p‐alpha‐syn accumulation in the skin nerves of the patient with associated PD, as recently shown in patients with classic PD [21]. The evidence of abnormal accumulation of p‐alpha‐syn and TDP‐43 provides clues as to the pathomechanism of the disease, suggesting an impaired function of DNAJB2 in preventing aggregate formation in mutation‐carrying patients, leading to a dysregulation of the protein quality control mechanisms. The lack of p‐alpha‐syn deposits in the patient without PD signs allows us neither to exclude nor to predict that he will develop PD in the future. The presence of abnormal deposits would have been evidence of a role for alpha‐synuclein as an early preclinical biomarker of PD in these patients. Why some patients with DNAJB2 mutations develop parkinsonism and others do not is still unclear. With respect to the ability of DNAJB2a to reduce mutated Parkin aggregates and rescue its function [20], we did not find any rare variant/polymorphism in the PARK2 gene as a possible predisposing factor for PD in our patient.
In conclusion, our study extends the clinical spectrum and the pathological knowledge of DNAJB2‐associated CMT, provides new insight into its pathomechanisms, confirming the role of skin biopsy to investigate disease mechanisms in neuropathies, and indicates the clearance of aggregated TDP‐43 as a possible therapeutic intervention.
CONFLICT OF INTEREST
The authors have nothing to report.
AUTHOR CONTRIBUTIONS
Paola Saveri: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Methodology (equal); Supervision (equal); Writing – original draft (equal); Writing – review and editing (equal). Stefania Magri: Data curation (equal); Formal analysis (equal); Methodology (equal); Writing – original draft (equal); Writing – review and editing (equal). Emanuela Maderna: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Francesca Balistreri: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Raffaella Lombardi: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Claudia Ciano: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Fabio Moda: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Barbara Garavaglia: Methodology (supporting); Supervision (supporting); Writing – review and editing (supporting). Chiara Reale: Data curation (supporting); Formal analysis (supporting); Methodology (supporting); Writing – review and editing (supporting). Giuseppe Lauria: Writing – review and editing (supporting). Franco Taroni: Methodology (equal); Supervision (equal); Writing – review and editing (equal). Davide Pareyson: Conceptualization (equal); Data curation (supporting); Formal analysis (supporting); Methodology (equal); Supervision (equal); Writing – original draft (supporting); Writing – review and editing (equal). Chiara Pisciotta: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Methodology (equal); Supervision (equal); Writing – original draft (equal); Writing – review and editing (equal).
ACKNOWLEDGEMENT
We thank Mike Cheetham for the DNAJB2 antibodies and Daniele Cartelli for technical support. P.S., S.M., F.T., D.P. and C.P. are members of the Inherited Neuropathy Consortium RDCRN. P.S., S.M., R.L., C.C., B.G., G.L.P., F.T., D.P. and C.P. are members of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO‐NMD).
Saveri P, Magri S, Maderna E, et al. DNAJB2‐related Charcot‐Marie‐Tooth disease type 2: Pathomechanism insights and phenotypic spectrum widening. Eur J Neurol. 2022;29:2056–2065. doi: 10.1111/ene.15326
Paola Saveri and Stefania Magri contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Pisciotta C, Shy ME. Neuropathy. Handb Clin Neurol. 2018;148:653‐665. [DOI] [PubMed] [Google Scholar]
- 2. Previtali SC, Zhao E, Lazarevic D, et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2019;90:1171‐1179. [DOI] [PubMed] [Google Scholar]
- 3. Blumen SC, Astord S, Robin V, et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol. 2012;71:509‐519. [DOI] [PubMed] [Google Scholar]
- 4. Gess B, Auer‐Grumbach M, Schirmacher A, et al. HSJ1‐related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology. 2014;83:1726‐1732. [DOI] [PubMed] [Google Scholar]
- 5. Gonzaga‐Jauregui C, Harel T, Gambin T, et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 2015;12:1169‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frasquet M, Chumillas MJ, Vílchez JJ, et al. Phenotype and natural history of inherited neuropathies caused by HSJ1 c.352+1G>A mutation. J Neurol Neurosurg Psychiatry. 2016;87:1265‐1268. [DOI] [PubMed] [Google Scholar]
- 7. Sanchez E, Darvish H, Mesias R, et al. Identification of a Large DNAJB2 deletion in a family with spinal muscular atrophy and parkinsonism. Hum Mutat. 2016;37:1180‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lupo V, García‐García F, Sancho P, et al. Assessment of targeted next‐generation sequencing as a tool for the diagnosis of Charcot‐Marie‐tooth disease and hereditary motor neuropathy. J Mol Diagn. 2016;18:225‐234. [DOI] [PubMed] [Google Scholar]
- 9. Frasquet M, Rojas‐García R, Argente‐Escrig H, et al. Distal hereditary motor neuropathies: mutation spectrum and genotype‐phenotype correlation. Eur J Neurol. 2021;28:1334‐1343. [DOI] [PubMed] [Google Scholar]
- 10. Sarparanta J, Jonson PH, Kawan S, Udd B. Neuromuscular diseases due to chaperone mutations: a review and some new results. Int J Mol Sci. 2020;21:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen HJ, Mitchell JC, Novoselov S, et al. The heat shock response plays an important role in TDP‐43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain. 2016;139:1417‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Claeys KG, Sozanska M, Martin JJ, et al. DNAJB2 expression in normal and diseased human and mouse skeletal muscle. Am J Pathol. 2010;176:2901‐2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot‐Marie‐Tooth disease. J Peripher Nerv Syst. 2011;16:191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sommer CL, Brandner S, Dyck PJ, et al. Peripheral Nerve Society Guideline on processing and evaluation of nerve biopsies. J Peripher Nerv Syst. 2010;15:164‐175. [DOI] [PubMed] [Google Scholar]
- 15. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gellera C, Castellotti B, Mariotti C, et al. Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics. 2007;8:289‐299. [DOI] [PubMed] [Google Scholar]
- 17. Rance G, Ryan MM, Bayliss K, et al. Auditory function in children with Charcot‐Marie‐Tooth disease. Brain. 2012;135:1412‐1422. [DOI] [PubMed] [Google Scholar]
- 18. Deshimaru M, Kinoshita‐Kawada M, Kubota K, et al. DCTN1 binds to TDP‐43 and regulates TDP‐43 aggregation. Int J Mol Sci. 2021;22:3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605‐608. [DOI] [PubMed] [Google Scholar]
- 20. Rose JM, Novoselov SS, Robinson PA, Cheetham ME. Molecular chaperone‐mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum Mol Genet. 2011;20:16‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donadio V. Skin nerve α‐synuclein deposits in Parkinson's disease and other synucleinopathies: a review. Clin Auton Res. 2019;29:577‐585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The data that support the findings of this study are available from the corresponding author upon reasonable request.