

Abstract
Aim
This phase I study investigated talazoparib pharmacokinetics (PK) and safety in patients with advanced solid tumours and varying degrees of hepatic function.
Methods
Patients with advanced solid tumours and normal hepatic function or varying degrees of hepatic impairment (mild, moderate or severe, based on National Cancer Institute Organ Dysfunction Working Group classification) received talazoparib 0.5 mg once daily for 22 calendar days. Plasma and urine samples after single and multiple doses were collected and analysed for talazoparib using validated assays. Plasma PK data from all patients were analysed using the population PK method. Plasma and urine PK parameters in PK‐evaluable patients were calculated using noncompartmental analysis (NCA). Safety was monitored in all enrolled patients.
Results
Thirty‐eight patients were enrolled; 37 had ≥1 PK concentration, among which 17 were evaluable for NCA. Population PK analysis (n = 37) indicated no significant impact of hepatic function on apparent clearance (CL/F) of talazoparib. Baseline creatinine clearance was the only significant covariate on CL/F (α = 0.05). NCA of data (n = 17) showed no clear trend for increase in exposure on day 22 with worsening hepatic function. Talazoparib protein binding was comparable in patients with varying hepatic function. Talazoparib was generally well tolerated, and the safety profile observed in this study was consistent with the known safety profile of the drug.
Conclusions
Hepatic impairment (mild, moderate or severe) has no impact on the PK of talazoparib. No dose modification is recommended for patients with advanced solid tumours and various degrees of hepatic impairment, and this labelling language has been approved by the US Food and Drug Administration and the European Medicines Agency.
Keywords: hepatic impairment, pharmacokinetics, phase I, PK modelling, talazoparib
1. What is already known about this subject
Although renal excretion is the primary route of talazoparib elimination, the potential impact of hepatic impairment on talazoparib clearance cannot be ruled out.
A previous population pharmacokinetic analysis indicated that there was no effect of mild hepatic impairment on talazoparib exposure. The effects of moderate or severe hepatic impairment are unknown.
What this study adds
Varying degrees of hepatic impairment have no significant impact on talazoparib exposure or clearance, and thus no dose modification is recommended in patients with advanced solid tumours and varying degrees of hepatic impairment.
1. INTRODUCTION
Talazoparib is a poly(ADP‐ribose) polymerase (PARP) inhibitor that has been approved for use as a monotherapy in the United States (2018) and the European Union (2019) for the treatment of germline BRCA‐mutated human epidermal growth factor receptor 2 (HER2)‐negative locally advanced/metastatic breast cancer. 1 , 2 Treatment with talazoparib was generally well tolerated, with anaemia, fatigue and nausea being the most common adverse events (AEs) observed in patients with advanced breast cancer. 3 , 4 Grade 3‐4 AEs were primarily hematologic and occurred in 55% of patients on talazoparib. 3
Following repeated once‐daily (QD) dosing of talazoparib 1 mg in patients with advanced tumours, talazoparib was rapidly absorbed with median time to maximum concentration (T max) ranging from 1.0 to 2.0 hours. 5 , 6 The mean half‐life of talazoparib is 89.8 hours following administration of a single dose in patients with advanced cancer, 7 and steady state was reached after 2‐3 weeks of daily dosing. 6 , 8 On the basis of phase I mass balance study in vitro and in vivo data, talazoparib underwent minimal hepatic metabolism and was predominantly excreted unchanged via the renal route (54.6% of the administered dose), while unchanged talazoparib recovered in the faeces accounted for 13.6%. 7 A previous population pharmacokinetic (PK) analysis using pooled data from four clinical studies that included 490 patients indicated that talazoparib exposure was affected by Asian race, renal impairment and concomitant administration with strong P‐glycoprotein (P‐gp) inhibitors. 8 The impact of strong P‐gp inhibitors and renal impairment was confirmed by two specific phase I studies. 9 , 10
Hepatic impairment may affect the PK of a drug through multiple mechanisms, such as altering the metabolism, biliary secretion and protein binding of drugs, or shunting of blood past the liver. 11 Although hepatic elimination plays a much lesser role in the clearance of talazoparib than renal excretion, the potential impact of moderate or severe hepatic impairment on talazoparib PK cannot be ruled out. 8 A previous population PK analysis indicated that there was no effect of mild hepatic impairment on talazoparib exposure. 8 In addition, AE profiles were generally comparable between patients with hepatic impairment and those with normal hepatic function. 12 In the phase III EMBRACA study (NCT01945775), for patients receiving talazoparib 1 mg QD, the biggest difference in AE incidence between hepatic function groups was for thrombocytopenia (27.8% in the mild hepatic impairment group vs 12.1% in the normal hepatic function group). Thrombocytopenia was also reported at a higher incidence in patients with hepatic impairment in the physician's choice treatment arm (11.1% vs 3.3%). 12 However, talazoparib had not been studied in patients with moderate or severe hepatic impairment. To address these knowledge gaps, this study was conducted to evaluate the PK and safety of talazoparib following daily administration in patients with advanced solid tumours and varying degrees of hepatic impairment compared with patients with normal hepatic function. This study aimed to support dose recommendations for talazoparib in patients with varying degrees of hepatic impairment.
2. METHODS
2.1. Study design
In this open‐label, nonrandomized, multicenter, phase I study (NCT02997176), eligible patients were assigned to one of four groups based on hepatic function per National Cancer Institute Organ Dysfunction Working Group (NCI‐ODWG) classification at study enrolment (day −1): normal hepatic function (Group A), mild hepatic impairment (Group B), moderate hepatic impairment (Group C) and severe hepatic impairment (Group D). See Supporting Information Table S1 for NCI‐ODWG classification.
Patients received oral talazoparib 0.5 mg QD for 22 calendar days. Patients were enrolled in parallel, and the enrolment was to be continued until at least six patients were evaluable for noncompartmental analysis (NCA) in each of the four study groups. Patients were considered evaluable for NCA (ie, PK‐evaluable) if they met the following criteria: completed the day 22 visit and missed ≤5 consecutive doses of talazoparib, received at least 10 consecutive days of talazoparib 0.5 mg without dosing interruption prior to day 22 PK sample collection, completed at least 85% of total plasma PK sample collection, and had not vomited the drug on day 1 and/or the final day of dosing. Treatment compliance was assessed based on patients’ used/unused study drug containers and their study drug diary.
Following evaluation by the principal investigator and approval of the sponsor, eligible patients could continue talazoparib treatment in a separate open‐label extension (OLE) study (NCT02921919).
The study was concluded before reaching six PK‐evaluable patients in the moderate and severe hepatic impairment groups due to extreme difficulties in enrolment and a low PK‐evaluable rate of enrolled patients. Results from an analysis of data from 34 patients collected by November 8, 2019, demonstrated that current data were sufficient to address the objective of this study by using population PK analysis in addition to NCA. Therefore, the sponsor and the US Food and Drug Administration (FDA) agreed to terminate the study.
2.2. Patient eligibility
Eligible patients were male or female and ≥18 years of age with histologically or cytologically confirmed advanced solid tumours with no available standard treatment options as per investigator assessment. Patients were included on the basis of an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, life expectancy of ≥3 months and hepatic function at screening/enrolment classified by NCI‐ODWG criteria as either normal function or mild, moderate or severe impairment. Patients were required to have adequate haematology and renal function at screening and enrolment.
The main exclusion criteria were prior treatment with any systemic anticancer therapy or investigational drug within 14 days or five half‐lives (whichever was longer) prior to enrolment, or use of a P‐gp inhibitor or inducer, or an inhibitor of breast cancer resistance protein within 7 days or five half‐lives (whichever was longer) prior to day 1. Additional eligibility criteria and a full list of excluded concomitant medications (Table S2) are detailed in Supporting Information.
2.3. Analysis sets
The PK concentration set was defined as patients who received at least one dose of talazoparib and had at least one reportable talazoparib concentration; this set was used for the population PK analysis. The PK‐evaluable analysis set included patients who met the PK‐evaluable criteria for NCA (defined under ‘Study design’) and had at least one defined talazoparib PK parameter of primary interest at the day 22 visit. The safety analysis set included all patients who received any amount of talazoparib.
2.4. PK assessment
On day 1 (single dose) and day 22 (multiple dose), serial plasma PK samples were taken at predose and 0.5, 1, 2, 4, 6, 8‐12 and 24 hours postdose. On days 8 and 15, predose plasma PK samples were collected. A single void urine sample was collected predose on day 1, and all urine voided postdose on days 1 and 22 was collected at intervals of 0‐12 hours and 12‐24 hours. In addition, plasma samples were collected at 2 hours postdose on days 1 and 22 for protein binding measurement. If plasma protein binding is comparable across hepatic function groups, total plasma concentration will be used for PK analysis. See the Supporting Information for a description of bioanalytical methods and assay performance. 10
2.5. Software
The software used for NCA and population PK analysis is described in the Supporting Information.
2.6. NCA and statistical analysis
Primary endpoints included area under the concentration‐time curve from 0 to 24 hours (AUC0−24), maximum observed plasma concentration (C max), unbound AUC0−24 (AUC0−24u) and unbound C max (C maxu) at steady state (day 22). Secondary PK parameters were the talazoparib plasma T max, predose concentration, apparent clearance from plasma after oral administration (CL/F), accumulation ratio (R ac), fraction of unbound drug in plasma (f u), unbound CL/F on day 22, amount of drug excreted in urine from time 0 to 24 hours (Ae0−24), percentage of dose excreted in urine (Ae0−24%) and renal clearance on day 22, as well as AUC0−24, C max, T max, f u, AUC0−24u, C maxu, Ae0−24 and Ae0−24% on day 1.
PK parameters obtained from NCA were summarized descriptively by hepatic function. PK parameters (AUC0−24, C max, AUC0−24u and C maxu on day 22 from the PK‐evaluable analysis set) were natural log‐transformed and analysed using an analysis of variance (ANOVA) model with group as a fixed effect to compare each hepatic impairment group (mild, moderate; Test) with the normal hepatic function group (Reference). Group D was not included in the ANOVA analysis due to the limited sample size (N = 2).
2.7. Population PK analysis
2.7.1. Data for analysis
All total talazoparib plasma concentration data from the PK concentration set were used for the population PK analysis, except that below limit of quantification (BLQ) data were omitted from the analysis, since the percentage of postdose BLQ data among all of the PK observations was low (1%).
2.7.2. Base model development
Talazoparib PK was described using a two‐compartmental model with first‐order absorption described earlier 8 and defined by the following parameters: CL/F, apparent volume of distribution of central compartment (V2/F), apparent volume of distribution of peripheral compartment (V3/F), intercompartmental clearance (Q/F), first‐order absorption rate constant (k a) and lag time (TLAG). To account for the large intrapatient PK variability as evidenced by the different shape of PK profiles between day 1 and day 22 and to obtain the best fit of the data, models that contain different population typical values for V2/F, V3/F and Q/F for day 22 (time‐varying parameters) were explored. The equation below describes the typical value of V2/F on day 22 in relation to those prior to day 22 (ie, days 1, 8 and 15).
where V2DAY22 represents the fold change of V2/F on day 22 compared to those on days prior to day 22. Similar equations apply to V3/F and Q/F, while population typical values for CL/F, k a and TLAG were kept the same for all the visit days.
2.7.3. Inclusion of covariates
Statistical testing of relevant covariates was performed by a stepwise covariate model building procedure (SCM) with statistical criteria of α = 0.05 for forward inclusion and α = 0.001 for backward elimination. The following covariates were tested in the SCM on CL/F and V2/F, respectively: baseline body weight (BWT), race and age. Since the primary objective of this analysis is to assess the impact of hepatic impairment on CL/F and baseline albumin, baseline aspartate aminotransferase and baseline total bilirubin were correlated with liver function. These covariates were not included in the covariate analysis to maximize the potential to see hepatic function as a covariate. Baseline creatinine clearance (BCCL) was not included in the initial SCM since BCCL was calculated using the Cockcroft‐Gault equation and thus correlated with demographic factors BWT and age. After the elimination algorithm of the SCM, BCCL was tested as a covariate on CL/F. Next, hepatic function group based on NCI‐ODWG (LIVER) was tested as a categorical covariate on CL/F.
2.7.4. Final model and assessment of final model
If the inclusion of BCCL and/or LIVER led to a drop in objective function value (OFV) of >3.84 (ie, reaching statistical difference at α = 0.05), they would be included in the final model.
Outliers in both the base and final models were identified using the criteria conditional weighted residuals with interaction (CWRESI) of >6. The outliers were considered influential if the key parameter estimates differed by more than 20% with and without the outliers.
Goodness‐of‐fit of different models was evaluated using standard methods. Model validation was conducted by using visual predictive check (VPC) and prediction‐corrected visual predictive check (pcVPC) based on the parameter estimates from the final model. The software used in this analysis is described in the Supporting Information.
2.8. Safety assessment and statistical analysis
Safety was evaluated at screening, at enrolment and during the treatment period by incidence of AEs, including AEs of special interest and serious AEs (SAEs), physical examination and vital signs, ECOG performance status, electrocardiogram and clinical laboratory tests. Results were reported descriptively as all‐causality treatment‐emergent adverse events (TEAEs) and treatment‐related TEAEs. Safety analyses were performed using the safety population and were summarized descriptively by hepatic function group.
3. RESULTS
3.1. Patient population
Since November 2019, when the interim results were presented to the FDA, four additional patients have been enrolled, none of whom were evaluable for NCA. In total, 38 patients were enrolled and received talazoparib between September 2016 and February 2020, and PK samples were collected for 37 patients. Only 17 patients (44.7%) were evaluable for NCA (n = 6, 6, 3 and 2 for groups A, B, C and D, respectively). Compared with the limited PK data evaluable for NCA, PK data from all 37 patients were utilized in the population PK analysis to evaluate the effect of hepatic impairment on talazoparib PK. More than half of patients (21/38 [55.3%]) enrolled in the OLE study and continued talazoparib treatment. Demographic information and disease characteristics of all enrolled patients are presented in Table 1.
TABLE 1.
Patient demographics and disease characteristics: safety analysis set
| Normal hepatic function (N = 7) | Mild hepatic impairment (N = 10) | Moderate hepatic impairment (N = 5) | Severe hepatic impairment (N = 16) | All patients (N = 38) | |
|---|---|---|---|---|---|
| Age (y) | |||||
| Mean (SD) | 60.3 (7.61) | 56.6 (17.08) | 60.4 (6.31) | 52.7 (11.98) | 56.1 (12.40) |
| Median (range) | 61.0 (52‐73) | 57.0 (33‐84) | 61.0 (53‐67) | 50.5 (37‐75) | 55.0 (33‐84) |
| Gender, n (%) | |||||
| Male | 2 (28.6) | 3 (30.0) | 0 | 7 (43.8) | 12 (31.6) |
| Female | 5 (71.4) | 7 (70.0) | 5 (100) | 9 (56.3) | 26 (68.4) |
| Race, n (%) | |||||
| White | 5 (71.4) | 8 (80.0) | 5 (100) | 13 (81.3) | 31 (81.6) |
| Asian | 1 (14.3) | 2 (20.0) | 0 | 1 (6.3) | 4 (10.5) |
| Other | 1 (14.3) | 0 | 0 | 2 (12.5) | 3 (7.9) |
| Weight (kg) | |||||
| Median (range) | 79.70 (63.3‐105.2) | 61.80 (46.6‐83.5) | 67.40 (65.7‐105.5) | 68.10 (41.7‐86.8) | 67.25 (41.7‐105.5) |
| Primary cancer site, n (%) | |||||
| Breast | 3 (42.9) | 2 (20.0) | 3 (60.0) | 4 (25.0) | 12 (31.6) |
| Colorectal | 0 | 2 (20.0) | 0 | 5 (31.3) | 7 (18.4) |
| Liver | 0 | 1 (10.0) | 0 | 3 (18.8) | 4 (10.5) |
| Pancreas | 0 | 2 (20.0) | 0 | 1 (6.3) | 3 (7.9) |
| Bile duct | 0 | 1 (10.0) | 0 | 1 (6.3) | 2 (5.3) |
| Other a | 4 (57.1) | 2 (20.0) | 2 (40.0) | 2 (12.5) | 10 (26.3) |
| ECOG performance status, n (%) | |||||
| 0 | 2 (28.6) | 0 | 0 | 0 | 2 (5.3) |
| 1 | 5 (71.4) | 9 (90.0) | 3 (60.0) | 11 (68.8) | 28 (73.7) |
| 2 | 0 | 1 (10.0) | 2 (40.0) | 5 (31.3) | 8 (21.1) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; N, number of patients in the group; n (%), number and percent of patients used in the calculation; SD, standard deviation.
ECOG performance status on the last assessment prior to study drug treatment is used.
Other includes primary cancer sites that contain <2 patients and unknown primary sites.
3.2. NCA
Summary statistics of PK parameters on day 22 are presented by hepatic function group in Table 2. Summary statistics for group D were not available due to the limited number of patients who met the PK‐evaluable criteria (N = 2). The median T max values on day 22 visits were similar across different hepatic function groups ranging from 1.05 to 2.13 hours. Large variability (coefficient of variation [CV%]) was observed for AUC0−24 and C max in groups A, B and C, with the largest variability observed in group B. No clear trend for increasing exposure or decreasing CL/F with worsening hepatic function was observed on day 22. Similarly, R ac was comparable among groups. The results of the statistical comparisons of AUC0−24 and C max on day 22 from the final statistical model (ANOVA analysis) are summarized in Supporting Information Table S3. The 90% confidence intervals (CIs) of the geometric mean ratio for AUC0−24 and C max in group B/group A and group C/group A encompassed 100%, indicating no statistically significant effect of hepatic function on AUC0−24 and C max. However, no conclusion can be made with regard to the impact of hepatic impairment on talazoparib PK based on NCA results because of large variability and small sample size (three and two PK‐evaluable participants for groups C and D, respectively).
TABLE 2.
Descriptive summary of plasma and urine talazoparib PK parameters following multiple oral 0.5‐mg doses of talazoparib by hepatic function (day 22): PK‐evaluable analysis set
| Parameter summary statistics a by hepatic function group | ||||
|---|---|---|---|---|
| Normal hepatic function | Mild hepatic impairment | Moderate hepatic impairment | Severe hepatic impairment | |
| Plasma PK | ||||
| N, n | 6, 6 | 6, 6 b | 3, 3 | 2, 2 c |
| AUC0−24 (ng•h/mL) | 111.8 (30) | 159.0 (99) | 123.6 (30) | 243.0 (184, 302) |
| C max (ng/mL) | 10.30 (23) | 11.30 (65) | 13.56 (23) | 17.15 (11.6, 22.7) |
| T max (h) | 1.50 (0.50, 2.13) | 2.13 (1.05, 4.00) | 1.05 (1.00, 2.75) | 4.000 (2.08, 5.92) |
| CL/F (L/h) | 4.471 (30) | 3.144 (99) | 4.044 (30) | 2.187 (1.66, 2.72) |
| R ac | 5.070 (24) | 5.134 (68) | 4.771 (31) | 22.83 (22.8, 22.8) |
| Ctrough (ng/mL) d | 2.624 (28) | 3.699 (197) | 3.553 (8) | 6.225 (2.05, 10.4) |
| f u (%) e | 26.98 (23) | 27.71 (18) | 27.10 (9) | 34.72 (23.1, 45.4) |
| AUC0−24u (ng•h/mL) | 30.17 (11) | 45.08 (84) | 33.50 (35) | 64.97 (60.2, 69.8) |
| C maxu (ng/mL) | 2.778 (27) | 3.204 (56) | 3.675 (28) | 4.518 (3.79, 5.24) |
| CLu/F (L/h) | 16.57 (11) | 11.09 (84) | 14.92 (35) | 7.739 (7.17, 8.31) |
| Urine PK | ||||
| N, n | 6, 6 | 6, 5 | 3, 3 | 2, 2 |
| Ae0−24 (mg) | 0.2229 (30) | 0.1819 (34) | 0.1867 (32) | 0.1475 (0.104, 0.191) |
| Ae0−24 (%) | 44.58 (30) | 36.36 (34) | 37.40 (31) | 29.50 (20.8, 38.2) |
| CLr (L/h) | 1.993 (57) | 1.449 (92) | 1.510 (39) | 0.5986 (0.565, 0.632) |
Abbreviations: Ae0−24, amount of drug excreted in urine from time 0 to 24 hours post‐dose; AUC0−24, area under the concentration‐time curve from 0 to 24 hours; AUC0−24u, unbound AUC0−24; CL/F, apparent clearance from plasma after oral administration; CLr, renal clearance; CLu/F, unbound CL/F; Cmax , maximum observed plasma concentration; C maxu, unbound C max; C trough, predose plasma drug concentration; f u, fraction of unbound drug in plasma; N, number of PK‐evaluable patients in each group; n, number of patients contributing to summary statistics (except for R ac, C trough and f u); NA, not available as n is less than 3; PK, pharmacokinetics; R ac, accumulation ratio; T max, time to C max.
Geometric mean (geometric coefficient of variation [CV%]) for all except median (range) for T max and arithmetic mean (min, max) for group D due to n = 2;
n = 3 for C trough, n = 8 for f u and n = 5 for R ac;
n = 5 for f u and n = 1 for R ac;
For C trough to be included in this summary, the plasma sample needs to be drawn within 24 ± 2 hours of the previous dose and not more than +10 min after the drug administration on the PK collection day;
fu data from all patients including PK non‐evaluable were included in the summary.
No trend was observed in the geometric mean f u of talazoparib in plasma with worsening hepatic function. Therefore, the conclusions of this study were based on the results obtained from the analyses of talazoparib total plasma PK parameters, and the population PK analysis was based on total concentration data.
3.3. Population PK analysis
Supporting Information Table S4 shows the baseline characteristics of patients in the PK concentration set, which included 485 observations from 37 patients (seven, nine, five and 16 patients in groups A, B, C and D, respectively). The majority of patients (84%) were White (Asian, n = 3) and had a normal renal function (23/37 [62%]), with a median BCCL of 106.3 mL/min. On average, there were 13.1 post‐treatment PK samples per patient with a range of seven to 17 PK samples per patient; one patient had three post‐dose PK samples.
Talazoparib PK was well characterized using a two‐compartmental PK model with first‐order absorption and TLAG. Different sets of V2/F, V3/F and Q/F values on day 22 were used to account for the different shapes of PK profiles on different days. The PK parameter estimates for the base model (model #2) are shown in Table 3. Since there was no correlation between BCCL and CL/F when BCCL was >90 mL/min in the base model (Supporting Information Figure S1), BCCL was capped at 90 mL/min for patients with normal renal function (defined as BCCL >90 mL/min) in the covariate analysis. Covariate analysis showed that BCCL was a significant covariate on CL/F at the α = 0.05 level but not at α = 0.001. BCCL was included as a covariate in model #2 before testing LIVER as a categorical covariate on CL/F in model #3. LIVER was found not to be significant at the α = 0.05 level. The estimated PK parameter values and associated statistics for model #3 are shown in Table 3. Numerically, it was estimated that patients with mild, moderate and severe hepatic impairment had 14.4%, 26.7% and 18.5% lower CL/F than patients with normal hepatic function based on model #3. However, the addition of LIVER as a covariate on CL/F (model #3) resulted in ∆OFV of −1.309 compared with model #2 (BCCL alone as a covariate on CL/F), which was not statistically significant. Interindividual variance of CL/F in model #3 (44.7%; Table 3) was similar to that of model #2 (45.4%), indicating that inclusion of hepatic function did not help explain the interpatient variability of CL/F. Furthermore, the 95% CI for the estimated effect of hepatic impairment on CL/F encompassed zero (Table 3); therefore, hepatic function had no statistically significant impact on CL/F and model #2 was the final model.
TABLE 3.
Parameter estimates for models with or without hepatic function as a covariate
| Model #2 (BCCL as covariate) | Model #3 (BCCL and LIVER as covariates) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Estimate | SE | RSE (%) | 95% CI lower, upper | Shrinkage (%) | Estimate | SE | RSE (%) | 95% CI lower, upper | Shrinkage (%) |
| θCL/F (L/h) | 5.155 | 0.644 | 12.492 | 3.893, 6.417 | – | 5.998 | 0.635 | 10.587 | 4.753, 7.242 | – |
| θV2/F (L) | 188.816 | 21.760 | 11.525 | 146.166, 231.466 | – | 188.961 | 21.543 | 11.401 | 146.736, 231.186 | – |
| θka (h−1) | 5.351 | 2.709 | 50.618 | 0.042, 10.660 | – | 5.364 | 2.084 | 38.852 | 1.279, 9.449 | – |
| θQ/F (L/h) | 52.897 | 11.023 | 20.839 | 31.291, 74.503 | – | 52.801 | 8.154 | 15.443 | 36.819, 68.783 | – |
| θV3/F (L) | 274.221 | 24.512 | 8.939 | 226.178, 322.264 | – | 275.323 | 23.428 | 8.509 | 229.405, 321.241 | – |
| θTLAG (h) | 0.422 | 0.019 | 4.574 | 0.385, 0.460 | – | 0.422 | 0.020 | 4.648 | 0.384, 0.461 | – |
| θV2DAY22 | −0.488 | 0.108 | −22.135 | −0.700, −0.277 | – | −0.489 | 0.107 | −21.969 | −0.699, −0.278 | – |
| θQDAY22 | −0.914 | 0.025 | −2.752 | −0.963, −0.865 | – | −0.914 | 0.025 | −2.696 | −0.962, −0.866 | – |
| θV3DAY22 | −0.538 | 0.150 | −27.950 | −0.833, −0.243 | – | −0.537 | 0.154 | −28.752 | −0.840, −0.234 | – |
| θBCCL on CL/F | 1.184 | 0.659 | 55.698 | −0.109, 2.476 | – | 1.130 | 0.667 | 58.966 | −0.176, 2.437 | – |
| θLIVER‐B on CL/F | – | – | – | – | – | −0.144 | 0.237 | −164.291 | −0.609, 0.320 | – |
| θLIVER‐C on CL/F | – | – | – | – | – | −0.267 | 0.179 | −67.120 | −0.618, 0.084 | – |
| θLIVER‐D on CL/F | – | – | – | – | – | −0.185 | 0.137 | −74.140 | −0.454, 0.084 | – |
| θσ | 0.386 | 0.032 | 8.245 | 0.323, 0.448 | – | 0.386 | 0.032 | 8.195 | 0.324, 0.448 | – |
| (CV%) | 0.206 (45.4) | 0.074 | 36.153 | 0.060, 0.352 | 13.626 | 0.200 (44.7) | 0.076 | 37.992 | 0.051, 0.348 | 13.894 |
| (CV%) | 0.389 (62.4) | 0.119 | 30.551 | 0.156, 0.622 | 11.003 | 0.389 (62.3) | 0.116 | 29.902 | 0.161, 0.616 | 10.990 |
| (CV%) | 4.750 (217.9) | 1.400 | 29.475 | 2.006, 7.494 | 18.250 | 4.765 (218.3) | 1.338 | 28.075 | 2.143, 7.387 | 18.291 |
| OFV | −204.035 | NA | – | NA, NA | – | −205.343 | NA | NA, NA | – | |
Abbreviations: σ, variance‐covariance matrix of the intra‐individual (residual) random effects in the measurements; BCCL, baseline creatinine clearance; CI, confidence interval; CL/F, apparent clearance; CV, coefficient of variation; F, bioavailability and was fixed at 1; k a, first‐order absorption rate constant; LIVER‐B on CL/F, fold change in CL/F in group B compared with control; LIVER‐C on CL/F, fold change in CL/F in group C compared with control; LIVER‐D on CL/F, fold change in CL/F in group D compared with control; NA, not applicable; OFV, objective function value; PK, pharmacokinetics; QDAY22, fold change in Q/F on day 22 compared with those on days prior to day 22; Q/F, intercompartmental clearance; RSE, relative standard error; SE, standard error; θ, typical population value; TLAG, lag time; V2/F, apparent volume of distribution of central compartment; V2DAY22, fold change in V2/F on day 22 compared with those on days prior to day 22; V3/F, apparent volume of distribution of peripheral compartment; V3DAY22, fold change in V3/F on day 22 compared with those on days prior to day 22; ω, variance of inter‐patient random effects in the PK parameters.
Overall, the final PK model described the observed data reasonably well, as shown by the diagnostic plots (Figure 1), and the predictive capability of the final PK model was validated through VPC and pcVPC (Figure 2).
FIGURE 1.
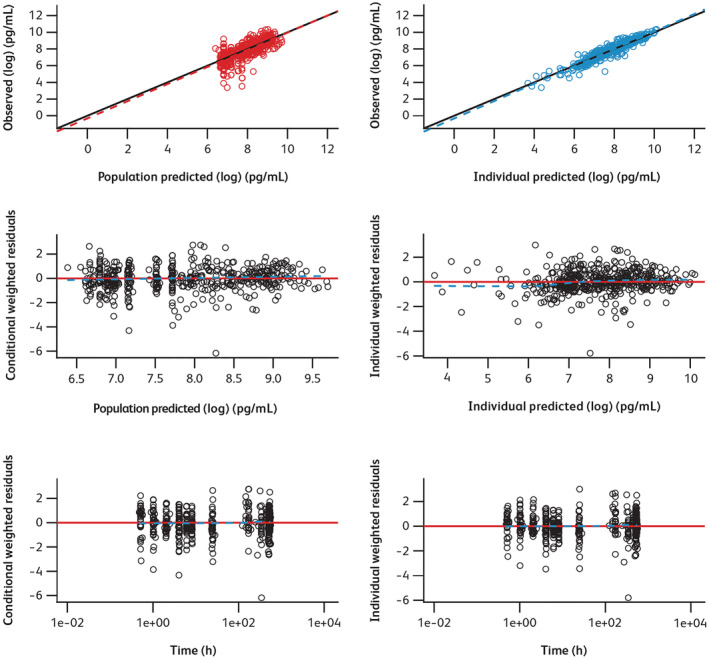
Diagnostic plots for final model. Time, time after the first dose. In the plots of log‐transformed observed values versus log‐transformed predicted values, circle points represent individual data points; the solid line and dotted line show the reference line (diagonal line) and linear regression line based on the individual data points, respectively. In the scatter plots of residuals, black circle points represent individual data points; the red solid line and blue dotted line show the reference line (y = 0) and the smooth line using locally weighted polynomial regression, respectively
FIGURE 2.
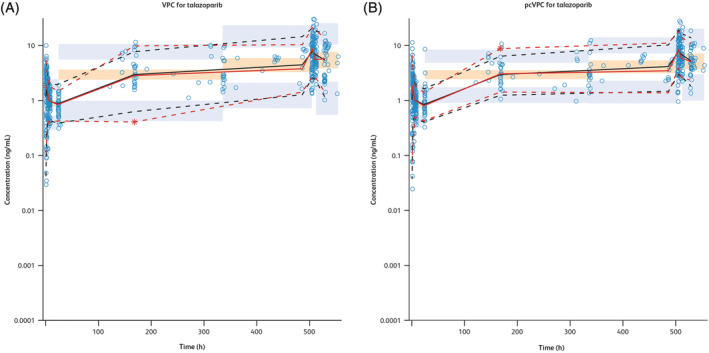
(A) VPC for the final PK model. (B) Prediction‐ and variance‐corrected VPC for the final PK model. pcVPC, prediction‐corrected visual predictive check; PK, pharmacokinetics; Time, time after first dose; VPC, visual predictive check. Observed concentration data points, represented by blue scatter points, are shown here. The red lines represent the median (solid line), 5th percentile (lower dash line) and 95th percentile (upper dash line) of the observed data. The median, 5th percentile and 95th percentile of simulated concentration values are represented by black lines. 95% confidence intervals for simulated median and each percentile are shown by an orange shaded area for median and a light‐blue shaded area for the 5th and 95th percentiles
One outlier was identified in the model development. When this outlier was excluded from the analysis, estimates for the key parameters were similar (<10% difference) to those when the outlier was included in both the base and final models. SCM results were comparable when the outlier was included; therefore, the final model was based on the entire dataset, including the outlier.
3.4. Safety
Overall, 31 patients (81.6%) experienced ≥1 all‐causality TEAE, of which 19 patients (50%) reported ≥1 TEAE of grade ≥3. Hyponatremia (five patients, 13.2%), hyperbilirubinemia (five patients, 13.2%) and disease progression (four patients, 10.5%) were the most commonly reported all‐causality grade ≥3 TEAEs (Table 4). The most frequently reported SAE was disease progression (n = 4), all of which occurred in the severe hepatic impairment group. However, none of the SAEs reported were considered related to talazoparib by investigators. Nine patients experienced ≥1 treatment‐related TEAE, with fatigue the most commonly reported, observed in four patients with normal hepatic function, one patient with mild hepatic impairment and one with severe hepatic impairment (n = 6, 15.8%; Table 5). Thrombocytopenia was the only ≥grade 3 TEAE considered related to talazoparib reported in one patient with severe hepatic impairment (2.6%) and led to permanent discontinuation of treatment. Two patients with moderate hepatic impairment and 10 patients with severe hepatic impairment permanently discontinued treatment because of AEs (12/38); however, none was considered treatment related by investigators. Similarly, of the eight patients (21.1%) that died (one patient with moderate and seven patients with severe hepatic impairment), three (7.9%) died because of progressive disease and five (13.2%) died because of AEs not associated with talazoparib treatment by investigator assessment.
TABLE 4.
All‐causality TEAEs that were grade 3 or higher by preferred term occurring in ≥5% of patients: safety analysis set
| Preferred term | Normal hepatic function (N = 7) n (%) | Mild hepatic impairment (N = 10) n (%) | Moderate hepatic impairment (N = 5) n (%) | Severe hepatic impairment (N = 16) n (%) | All patients (N = 38) n (%) |
|---|---|---|---|---|---|
| Any grade 3 or higher TEAE | 1 (14.3) | 4 (40.0) | 2 (40.0) | 12 (75.0) | 19 (50.0) |
| Blood and lymphatic system disorders | 0 | 1 (10.0) | 0 | 4 (25.0) | 5 (13.2) |
| Thrombocytopenia | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Anaemia | 0 | 1 (10.0) | 0 | 2 (12.5) | 3 (7.9) |
| Coagulopathy | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Gastrointestinal disorders | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Abdominal pain | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Nausea | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Vomiting | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Intestinal obstruction | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Respiratory, thoracic and mediastinal disorders | 0 | 1 (10.0) | 0 | 1 (6.3) | 2 (5.3) |
| Pleural effusion | 0 | 1 (10.0) | 0 | 0 | 1 (2.6) |
| Hypoxia | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Reproductive system and breast disorders | 1 (14.3) | 0 | 0 | 0 | 1 (2.6) |
| Breast pain | 1 (14.3) | 0 | 0 | 0 | 1 (2.6) |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 1 (14.3) | 0 | 1 (20.0) | 2 (12.5) | 4 (10.5) |
| Malignant pleural effusion | 1 (14.3) | 0 | 0 | 0 | 1 (2.6) |
| Neoplasm progression | 0 | 0 | 1 (20.0) | 2 (12.5) | 3 (7.9) |
| Cardiac disorders | 0 | 1 (10.0) | 0 | 1 (6.3) | 2 (5.3) |
| Coronary artery occlusion | 0 | 1 (10.0) | 0 | 0 | 1 (2.6) |
| Cardio‐respiratory arrest | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| Hepatobiliary disorders | 0 | 1 (10.0) | 1 (20.0) | 5 (31.3) | 7 (18.4) |
| Hyperbilirubinemia | 0 | 0 | 1 (20.0) | 4 (25.0) | 5 (13.2) |
| Cholangitis | 0 | 1 (10.0) | 0 | 1 (6.3) | 2 (5.3) |
| Metabolism and nutrition disorders | 0 | 0 | 1 (20.0) | 5 (31.3) | 6 (15.8) |
| Hyponatremia | 0 | 0 | 1 (20.0) | 4 (25.0) | 5 (13.2) |
| Hyperkalaemia | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Hypokalaemia | 0 | 0 | 0 | 1 (6.3) | 1 (2.6) |
| General disorders and administration site conditions | 0 | 0 | 0 | 4 (25.0) | 4 (10.5) |
| Disease progression | 0 | 0 | 0 | 4 (25.0) | 4 (10.5) |
| Nervous system disorders | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Hepatic encephalopathy | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Infections and infestations | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
| Sepsis | 0 | 0 | 0 | 2 (12.5) | 2 (5.3) |
Abbreviations: MedDRA, Medical Dictionary for Regulatory Activities; n (%), number (percentage) of patients with reported grade 3 or higher TEAEs; N, number of patients in the group; TEAE, treatment‐emergent adverse event.
MedDRA version 23.0 was used to code AEs. Patients were counted once for each preferred term. Preferred terms were sorted in descending frequency and then alphabetically.
TABLE 5.
Treatment‐related TEAEs that occurred in ≥5% of patients by preferred term: safety analysis set
| Preferred term | Normal hepatic function (N = 7) n (%) | Mild hepatic impairment (N = 10) n (%) | Moderate hepatic impairment (N = 5) n (%) | Severe hepatic impairment (N = 16) n (%) | All patients (N = 38) n (%) |
|---|---|---|---|---|---|
| Patients with at least one treatment‐related TEAE | 4 (57.1) | 3 (30.0) | 0 | 2 (12.5) | 9 (23.7) |
| Fatigue | 4 (57.1) | 1 (10.0) | 0 | 1 (6.3) | 6 (15.8) |
| Thrombocytopenia | 0 | 2 (20.0) | 0 | 2 (12.5) | 4 (10.5) |
| Constipation | 2 (28.6) | 0 | 0 | 0 | 2 (5.3) |
| Diarrhea | 2 (28.6) | 0 | 0 | 0 | 2 (5.3) |
| Nausea | 2 (28.6) | 0 | 0 | 0 | 2 (5.3) |
Abbreviations: MedDRA, Medical Dictionary for Regulatory Activities; n (%), number (percentage) of patients used in the calculation; N, number of patients in the group; TEAE, treatment‐emergent adverse event.
MedDRA version 23.0 was used to code AEs.
Patients were counted once for each preferred term. Preferred terms were sorted in descending frequency and then alphabetically.
4. DISCUSSION
In this study, the effect of hepatic impairment on the PK and tolerability of talazoparib after daily administration was evaluated in patients with advanced solid tumours. Because of a limited number of PK‐evaluable patients with only three moderate and two severe hepatic impairments evaluable for NCA, the conclusion for the PK assessment was based on the population PK analysis using plasma concentration data from 37 patients in the PK concentration analysis set. NCA using data from the 17 PK‐evaluable patients was included for completeness. Safety conclusions were based on the safety analysis set (38 patients).
The population PK analysis indicated that varying degrees of hepatic impairment (including mild, moderate and severe) had no significant impact on the clearance of talazoparib. This finding is corroborated by the NCA of data from 17 PK‐evaluable patients, which showed no clear trend for increase in plasma talazoparib AUC0−24 and C max on day 22 with worsening hepatic function. This finding is also consistent with renal excretion as the primary elimination pathway for talazoparib and is aligned with the previous population PK analysis that showed no significant association of mild hepatic impairment on talazoparib PK. 8
Using different sets of population typical values for V2/F, V3/F and Q/F on day 22 as the base model was an empirical approach to better fit the individual PK profiles on both day 1 and day 22. Single CL/F was used for both day 1 and day 22, and was able to describe the CL/F on both days well, which enabled the assessment of the effect of hepatic function on CL/F. The OFV of the base model was significantly lower than that of a model with time‐constant parameters (Supporting Information Table S5). A sensitivity analysis was performed to assess the effect of hepatic impairment on CL/F with time‐constant parameters for V2/F, V3/F and Q, and showed no difference in findings, as described in the Supporting Information.
In a previous population PK analysis, Asian race, renal impairment/BCCL and concomitant administration with strong P‐gp inhibitors were found to be significant covariates on CL/F. 8 As this study is a dedicated hepatic impairment study, major factors known to affect the PK of talazoparib were controlled, including prohibition of concomitant administration with strong P‐gp inhibitors. Talazoparib was taken under fasting conditions at day 1 and day 22 visits, and the majority of patients had normal renal function. Only three of the 37 patients in the population PK dataset were Asian; therefore, this current study includes a less diverse patient population than previous population PK analyses, and some previously identified significant covariates were not applicable in this analysis. The identification of BCCL as a significant covariate on CL/F is consistent with the previous findings. 8
Talazoparib was generally tolerated at a dose of 0.5 mg QD for 22 days in patients with advanced solid tumours and varying degrees of hepatic impairment, and results from this study were consistent with the known safety profile of the drug. Similarly, there were no unexpected safety findings identified, and reported TEAEs were considered consistent with the disease under study and the established safety profile of talazoparib. 4 The frequency of all‐causality SAEs was higher in patients with severe hepatic impairment; however, none of the SAEs reported in the study were considered related to talazoparib treatment by the investigator. Of the eight deaths that occurred in this study, seven of which were in patients with severe hepatic impairment, none were considered related to talazoparib. However, since patients in the severe hepatic impairment group had more advanced underlying disease, were possibly more heavily pre‐treated and were in worse general health condition, these factors provide a likely explanation for the greater number of treatment discontinuations, AEs and the low PK‐evaluable rate in the severe hepatic impairment group compared with the other groups.
This study showcased how a model‐based approach was used to address the enrolment and logistical challenges of assessing steady‐state PK in patients with cancer with hepatic impairment. Among the patients nonevaluable for NCA, 81% of them were due to missing >5 doses consecutively. The reason for the high dose interruption/discontinuation rate in patients with moderate and severe hepatic impairment was due to AEs not related to talazoparib (eg, disease progression) and likely due to the patients' underlying condition. If study enrolment had continued to achieve the planned number of evaluable patients for NCA, it could likely take much longer to complete the study and thereby delay the availability of dosing information for this population.
In conclusion, this study, based on the results of the population PK analysis and totality of the data, suggests that talazoparib can be administered to patients with varying degrees of hepatic impairment with no dose modification. This labelling language has been approved by the US FDA and the European Medicines Agency.
CONFLICT OF INTEREST
C.G., Y.Y., A.P., H.S., J.C., C.D. and D.D.W. are employees of Pfizer Inc. and own stocks. S.A.P.‐P. reports clinical trial research/grant funding (received through the institution): AbbVie, ABM Therapeutics, Acepodia, Alkermes, Aminex Therapeutics, Amphivena Therapeutics, BioMarin, Boehringer Ingelheim, Bristol Myers Squibb, Cerulean Pharma, Chugai Pharmaceutical, Curis, Daiichi Sankyo, Eli Lilly, ENB Therapeutics, Five Prime Therapeutics, Gene Quantum, Genmab A/S, GlaxoSmithKline, Helix BioPharma, Incyte, Jacobio Pharmaceuticals, MedImmune, Medivation, Merck Sharp & Dohme Corp., Novartis Pharmaceuticals, Pieris Pharmaceuticals, Pfizer, Principia Biopharma, Puma Biotechnology, RAPT Therapeutics, Seattle Genetics, Silverback Therapeutics, Taiho Oncology, Tesaro, TransThera Biosciences, NCI/NIH, and P30CA016672 – Core Grant (CCSG Shared Resources). R.M. reports honoraria from Genentech, Immunomedics, Lilly, Novartis, Pfizer and Seattle Genetics. Z.A.W. reports consulting fees from Array, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Lilly, Merck and Novartis.
CONTRIBUTORS
Contributions to the study conception or design: C.G. and D.W.W. All authors: contribution to the acquisition, analysis, or interpretation of data; drafting of the manuscript or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
PATIENT CONSENT STATEMENT
Written informed consent was obtained for each patient prior to entering the patient into the study, in accordance with the Declaration of Helsinki and other applicable local regulations.
Supporting information
Table S1Definition of assigned groups based on hepatic function as per NCI‐ODWG classification at study enrolment
Table S2 Prohibited concomitant therapies
Table S3 ANOVA results for day 22 parameters
Table S4 Summary of baseline demographics by hepatic function group
Table S5 Summary of attempted models
Table S6 Model #3a (time‐constant parameters with LIVER and baseline creatinine clearance as covariates on CL/F) parameter estimates
Figure S1 ETAs on CL/F versus covariates in base model. BALB, baseline albumin; BAST, baseline aspartate aminotransferase; BBIL, total baseline bilirubin; BCCL, baseline creatinine clearance; BWT, baseline body weight; CL/F, apparent clearance; ETA, empirical Bayes prediction of the interindividual random effect; RACEN = 1 and 2 represent non‐Asian and Asian patients; Renal = 1, 2, and 3 represent patients with normal renal function, mild renal impairment and moderate renal impairment; LIVER = 1, 2, 3 and 4 represent patients with normal hepatic function, mild hepatic impairment, moderate hepatic impairment and severe hepatic impairment. The black circles represent the estimates from individual patients. In the top six panels, the red solid line and blue dotted line show the reference line (y = 0) and local polynomial regression fitting line, respectively. In the bottom three panels, the mean and median for each group are represented by the blue diamond symbols and the black solid line; the red dotted line shows the reference line (y = 0)
ACKNOWLEDGMENTS
This study was sponsored by Medivation, which was acquired by Pfizer Inc. in September 2016. We acknowledge the contribution from Dr Ravindranath Patel to the enrolment of this study. Medical writing support was provided by Maddie Higgins, MBiolSci, of CMC AFFINITY, McCann Health Medical Communications, and funded by Pfizer. This study was sponsored by Medivation, which was acquired by Pfizer in September 2016.
Guo C, Yu Y, Chakrabarti J, et al. Evaluation of pharmacokinetics and safety of talazoparib in patients with advanced cancer and varying degrees of hepatic impairment. Br J Clin Pharmacol. 2022;88(7):3392-3403. doi: 10.1111/bcp.15294
The authors confirm that the PI for this study is Dr Wainberg and that he had direct clinical responsibility for patients.
Clinical trials registration: NCT02997176.
Funding information Medivation; Pfizer Inc.
DATA AVAILABILITY STATEMENT
Pfizer will provide access to individual de‐identified participant data and related study documents (eg, protocol, statistical analysis plan, clinical study report) on request from qualified researchers and subject to certain criteria, conditions and exceptions.
REFERENCES
- 1. European Medicines Agency TALZENNA® (talazoparib) Summary of Product Characteristics. December 2021. https://www.ema.europa.eu/en/documents/product-information/talzenna-epar-product-information_en.pdf. Accessed 10 March 2022.
- 2. Pfizer Inc TALZENNA® (talazoparib) prescribing information. September 2021. http://labeling.pfizer.com/ShowLabeling.aspx?id=11046. Accessed 10 March 2022.
- 3. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753‐763. doi: 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hurvitz SA, Gonçalves A, Rugo HS, et al. Talazoparib in patients with a germline BRCA‐mutated advanced breast cancer: Detailed safety analyses from the phase III EMBRACA trial. Oncologist. 2020;25(3):e439‐e450. doi: 10.1634/theoncologist.2019-0493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoffman J, Chakrabarti J, Plotka A, et al. Talazoparib has no clinically relevant effect on QTc interval in patients with advanced solid tumors. Anticancer Drugs. 2019;30(5):523‐532. doi: 10.1097/CAD.0000000000000772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose‐escalation, two‐part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7(6):620‐629. doi: 10.1158/2159-8290.CD-16-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu Y, Chung CH, Plotka A, et al. A phase 1 mass balance study of (14) C‐labeled talazoparib in patients with advanced solid tumors. J Clin Pharmacol. 2019;59(9):1195‐1203. doi: 10.1002/jcph.1415 [DOI] [PubMed] [Google Scholar]
- 8. Yu Y, Durairaj C, Shi H, Wang DD. Population pharmacokinetics of talazoparib in patients with advanced cancer. J Clin Pharmacol. 2020;60(2):218‐228. doi: 10.1002/jcph.1520 [DOI] [PubMed] [Google Scholar]
- 9. Elmeliegy M, Láng I, Smolyarchuk EA, et al. Evaluation of the effect of P‐glycoprotein inhibition and induction on talazoparib disposition in patients with advanced solid tumours. Br J Clin Pharmacol. 2020;86(4):771‐778. doi: 10.1111/bcp.14178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Durairaj C, Chakrabarti J, Ferrario C, et al. The effect of renal impairment on the pharmacokinetics and safety of talazoparib in patients with advanced solid tumors. Clin Pharmacokinet. 2021;60(7):921‐930. doi: 10.1007/s40262-020-00983-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. European Medicines Agency Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. 17 February 2005. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function_en.pdf. Accessed 10 March 2022.
- 12. European Medicines Agency Assessment report: Talzenna [Procedure No. EMEA/H/C/004674/0000]. 26 April 2019. https://www.ema.europa.eu/en/documents/assessment‐report/talzenna‐epar‐public‐assessment‐report_en.pdf. Accessed 10 March 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1Definition of assigned groups based on hepatic function as per NCI‐ODWG classification at study enrolment
Table S2 Prohibited concomitant therapies
Table S3 ANOVA results for day 22 parameters
Table S4 Summary of baseline demographics by hepatic function group
Table S5 Summary of attempted models
Table S6 Model #3a (time‐constant parameters with LIVER and baseline creatinine clearance as covariates on CL/F) parameter estimates
Figure S1 ETAs on CL/F versus covariates in base model. BALB, baseline albumin; BAST, baseline aspartate aminotransferase; BBIL, total baseline bilirubin; BCCL, baseline creatinine clearance; BWT, baseline body weight; CL/F, apparent clearance; ETA, empirical Bayes prediction of the interindividual random effect; RACEN = 1 and 2 represent non‐Asian and Asian patients; Renal = 1, 2, and 3 represent patients with normal renal function, mild renal impairment and moderate renal impairment; LIVER = 1, 2, 3 and 4 represent patients with normal hepatic function, mild hepatic impairment, moderate hepatic impairment and severe hepatic impairment. The black circles represent the estimates from individual patients. In the top six panels, the red solid line and blue dotted line show the reference line (y = 0) and local polynomial regression fitting line, respectively. In the bottom three panels, the mean and median for each group are represented by the blue diamond symbols and the black solid line; the red dotted line shows the reference line (y = 0)
Data Availability Statement
Pfizer will provide access to individual de‐identified participant data and related study documents (eg, protocol, statistical analysis plan, clinical study report) on request from qualified researchers and subject to certain criteria, conditions and exceptions.