

Abstract
Primary ciliary dyskinesia (PCD) is a heterogeneous disease, with impaired mucociliary clearance causing respiratory tract infections. A founding CCDC114 mutation has led to a relatively homogeneous and large Dutch PCD population in Volendam. Our aim was to describe their phenotype. Therefore, all Volendam PCD patients seen at the Amsterdam UMC were included in this study. Data were collected on lung function, microbiology, radiology, and ear‐nose‐throat (ENT) symptoms. A mixed effects model estimated lung function decline in %point per year (95% confidence interval [CI]). Thirty‐three (60%) out of approximately 56 Volendam PCD patients were treated at our center and included in this study. Only 30% of patients had situs inversus. FEV1 declined in children (−1.43%/year, CI: −1.80/−1.05), but not in adults (0.01%/year, CI: −0.36/0.38). Pseudomonas aeruginosa was cultured in 21% of children and 60% of adults, respectively. Patients who have been infected at some point with P. aeruginosa had a steeper decline in FEV1 as compared to patients that have never been infected. Neonatal symptoms (79%) and ENT problems (94%) were common; fertility issues however, were not (11%) common. Compared to other PCD cohorts, the Volendam/CCDC114 patients have a moderately severe phenotype with lung function decline predominantly occurring in childhood.
Keywords: CCDC114, lung function, phenotype, primary ciliary dyskinesia
1. INTRODUCTION
Primary ciliary dyskinesia (PCD) is a genetic ciliopathy, in which the mucociliary clearance is impaired due to abnormal function of the cilia lining the upper and lower respiratory tract surface, or in some cases, due to a severe reduction in multiple motile cilia (RGMC). Due to recurrent respiratory tract infections, lung damage and bronchiectasis ensue. PCD is generally assumed to have a relatively benign disease course compared to cystic fibrosis (CF), with an average lung function decline of approximately 0.5–0.8%/year in PCD (Marthin, Petersen, Skovgaard, & Nielsen, 2010; Shah et al., 2016) compared to 1–3%/year in CF, respectively (Caley, Smith, White, & Peckham, 2021; VandenBranden et al., 2012). However, PCD patients have the same level of structural lung disease and lung function decline as pancreatic sufficient CF patients (Cohen‐Cymberknoh et al., 2014). In addition, as was shown in a large international PCD (iPCD) cohort study, lung function is considerably lower, compared to reference values from a healthy population. In addition, quality of life is low and disease burden is high, especially in adults (Behan, Rubbo, Lucas, & Dunn Galvin, 2017; Pifferi et al., 2010).
PCD is a heterogeneous disease, both genetically and phenotypically. To date, over 50 genes have been described, with usually an autosomal recessive mode of inheritance (Lucas, Davis, Omran, & Shoemark, 2020). However, x‐chromosomal recessive (PIH1D3, ODF1, RPGR) (Höben et al., 2018; Paff et al., 2017; Vervoort & Wright, 2002) and autosomal dominant (FOXJ1) (Wallmeier et al., 2019) modes of inheritance have also been identified. With increasing knowledge about the genetic background of PCD, the genotype–phenotype correlations are becoming more apparent. Given the rare nature of PCD, with an estimated prevalence of at least 1:7,500 (Hannah et al., 2022), most knowledge on genotype–phenotype correlations has been derived from small case series. Mutations in multiciliogenesis genes, like CCNO and MCIDAS, have been associated with poor clinical outcome. Wallmeijer and colleagues found that in two out of 16 patients with a CCNO mutation, lung transplantation was necessary at the age of 34, due to respiratory failure (Wallmeier et al., 2019). Interestingly, situs inversus is not found in these patients, as the nodal cilia are not affected. More recently, a genotype–phenotype study was published by Shoemark and colleagues, using topological data‐analysis in 396 patients (Shoemark et al., 2021). They found that patients with mutations in CCDC39 (n = 35) and DNAH11 (n = 48) had a distinct respiratory phenotype, with the former having relatively worse and the latter relatively better lung function, compared to the rest of the study population. Still, many genotypes‐phenotypes have not been described yet.
In 2013, Onoufriadis and colleagues sought to identify the genetic defect in a population of PCD patients from Volendam (Onoufriadis et al., 2013). Volendam is a fishermen's village, founded in 1462, by 20 families in the neighborhood of Edam in Noord‐Holland. Due to its geographic nature and religious differences, the population remained relatively isolated. Consequently, the mutation c.742G>A; p.(Ala248 Thrfs52*) (ref. seq. NM_144577.4) (in the CCDC114/ODAD1 gene) carried within the founding ancestors could spread throughout a relatively inbred population. In a population of 22,461 inhabitants (January 2021), the carrier frequency is 1:10 with an estimated incidence of 1:400. This mutation causes loss of a splice donor site and 100% use of a cryptic splice donor site at c.742+79, leading to a frameshift. The clinical picture of these patients appears to be classical, with recurrent upper and lower respiratory tract infections and situs inversus in about half the patients, though fertility issues have not been reported (Onoufriadis et al., 2013).
In this article, we aim to give a phenotypical description of all PCD patients from Volendam treated at our center (approximately 2/3 of all Volendam patients). This relatively large and genetically homogeneous population provides us with the unique possibility to describe the natural disease course in CCDC114 c.742G>A patients, including lung function, microbiology, and clinical features.
2. METHODS
2.1. Study design and subjects
The present study is a cohort study using data entered in the international PCD registry (Werner et al., 2016). Written informed consent was obtained from all patients and/or representatives and forms were kept by the local institution. This is in accordance with the protocols approved by the institutional ethics review boards. For this study, we included all PCD‐patients from Volendam treated at the Amsterdam University Medical Centers (33 patients, out of 121 PCD‐patients treated at our center, are from Volendam). Diagnosis was based on high‐speed video microscopy (HSVM), transmission electron microscopy (TEM), genetics, and nasal nitric oxide. A diagnosis was considered definite if (a) confirmed by TEM and/or (b) confirmed by two copies of the CCDC114/ODAD1 mutation. Both adult and pediatric patients were included in this study.
2.2. Data extraction
Registry data were extracted on diagnosis, family history, lung function analysis, microbiology, ear‐nose‐throat symptoms, and medication. For both ENT symptoms and medication, data on the last 4 years (2017–2021) were used. All retrospectively available lung function data were analyzed. Forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) z‐scores and %predicted values were calculated using the Global Lung function Initiative (GLI) 2012 reference value (Quanjer et al., 2012). Lung function data from children aged <6 years were excluded to ensure the quality of the measurements and comparability with previously published lung function data of the iPCD cohort (Halbeisen et al., 2018). For this comparison, we used data published in 2018 of the iPCD cohort, which analyzed lung function of 991 PCD patients (Halbeisen et al., 2018). Several patients have been included in both the iPCD and Volendam cohorts; we were unable to exclude these from the data in the iPCD cohort, due to data being anonymized. We analyzed our lung function data in the same way, including the same age stratification, to allow for a direct comparison.
Microbiology test results were extracted from the year 2019, to minimize the effect of the COVID‐19 pandemic. Data from the previous year were also extracted to provide a full year of culture history per patient. Furthermore, we extracted if and at which age Pseudomonas aeruginosa (P. aeruginosa) was cultured for the first time in each patient. For the year 2019, patients were categorized as: never infected, free of infection, intermittent infection, or chronic infection; based on the Leeds criteria (Lee, Brownlee, Conway, Denton, & Littlewood, 2003). We did not adhere to the recommended minimum of four samples per patient, as data were not sufficiently available.
2.3. Statistical analysis
All analyses were performed using Rstudio version 3.6.2 (SAS Institute, Inc.). Demographic data were presented as number (%), median (interquartile range), or mean ± SD where appropriate. If data were collected at multiple time points, the most recent available data were used for the descriptive statistics. The Χ2‐test was used to compare the Volendam and iPCD cohorts. An exact binomial test has been performed to determine the situs solitus versus situs inversus distribution. Data were stratified between children and adults, or P. aeruginosa status, where applicable. Fischer's exact test was used to analyze differences between adults and children. Linear mixed effect models, with individual patients as random effect, were constructed to estimate the decline per year in FEV1 and FVC, for both %predicted and z‐scores. This was presented as slope per year, including 95% confidence interval (CI).
3. RESULTS
3.1. Population demographics and clinical features
Our Volendam cohort included 33 PCD patients out of the approximately 56 (1:400 inhabitants) PCD patients living in Volendam (Onoufriadis et al., 2013). The patients ranged from 1 to 71 years of age and included 14 (45%) males and 19 (55%) females. Frequency of follow‐up ranged from once a year at the expert center, with additional visits at a local center, to four times a year. This depended on several factors including age, children were seen more frequently, and disease severity. All patients received a definite PCD diagnosis based on TEM and/or genetic screening (only genetic testing: n = 8, genetics and TEM: n = 25), all TEM results showed an ODA defect, in keeping with the genetic mutation. The median age at PCD diagnosis was 2.8 years (inter quartile range: 0.4–10.3). For those with nasal nitric oxide (NO) data available (n = 16), all showed low NO levels of under 77 nl/min. Demographics of the Volendam cohort compared to the iPCD cohort can be found in Table 1.
TABLE 1.
Demographic data of the Volendam and iPCD cohort
| Volendam cohort (n = 33) | iPCD cohort (n = 991) | ||||
|---|---|---|---|---|---|
| n/mean/median | (%)/±SD /[IQR] | n/mean /median | (%)/±SD/[IQR] | p‐value | |
| Gender | .450 | ||||
| Male | 14 | (45%) | 487 | (49%) | |
| Female | 19 | (55%) | 504 | (51%) | |
| Time period of birth | .078 | ||||
| Before 1976 | 7 | (21%) | 151 | (15%) | |
| 1977–1996 | 6 | (18%) | 363 | (37%) | |
| 1997–2015 | 20 | (61%) | 477 | (48%) | |
| Affected family members a | NA | ||||
| Brother | 6 | (18%) | |||
| Sister | 5 | (15%) | |||
| Mother | 1 | (3%) | |||
| Father | 4 | (12%) | |||
| Cousin | 3 | (9%) | |||
| Other | 3 | (9%) | |||
| None | 12 | (36%) | |||
| Unknown | 4 | (12%) | |||
| Fertility (n = 9) | |||||
| Fertile (female) | 6 | (67%) | |||
| Less fertile (female) | 1 | (11%) | |||
| Fertile (male) | 2 | (22%) | |||
| Less fertile (male) | 0 | ‐ | |||
| Too young to investigate | 22 | ‐ | |||
| Unknown | 2 | ‐ | |||
| Diagnostic information | <.001 | ||||
| Definite PCD diagnosis b | 33 | (100%) | 611 | (62%) | |
| Only genetics | 8 | (24%) | |||
| Genetics + TEM | 25 | (76%) | |||
| Probable PCD diagnosis c | ‐ | ‐ | 207 | (21%) | |
| Clinical diagnosis only | ‐ | ‐ | 173 | (17%) | |
| Age at diagnosis (years) | 2.8 | [0.4–10.3] | |||
| Neonatal symptoms (n = 24) a | .027 | ||||
| Respiratory distress syndrome | 7 | (29%) | 469 | (40%) | |
| Pneumonia | 8 | (33%) | NA | ||
| Other | 5 | (21%) | NA | ||
| Wet lung | 1 | (3%) | |||
| Tachypnea | 1 | (3%) | |||
| Aspiration pneumonia | 1 | (3%) | |||
| Meconium aspiration | 1 | (3%) | |||
| Need for oxygen | 1 | (3%) | |||
| None | 5 | (21%) | 357 | (34%) | |
| Nasal NO (n = 16) | NA | NA | |||
| nl/min | 10.1 | ±8.0 | |||
| ≤77 nl/min | 16 | (100%) | |||
| >77 nl/min | 0 | (0%) | |||
| Not available | 17 | ‐ | |||
| Organ laterality | .002 | ||||
| Situs solitus | 23 | (70%) | 499 | (51%) | |
| Situs inversus totalis | 9 | (27%) | 348 | (35%) | |
| Heterotaxia | 1 | (3%) | 13 | (1%) | |
| Situs status not reported | ‐ | ‐ | 131 | (13%) | |
| Findings chest imaging a | NA | NA | |||
| Atelectasis | 12 | (36%) | |||
| Infiltration | 2 | (6%) | |||
| Bronchiectasis | 16 | (48%) | |||
| Other | 14 | (42%) | |||
| Normal | 4 | (12%) | |||
| BMI | .123 | ||||
| kg/m2 | 21.1 | ±3.8 | NA | ||
| z‐score d | 0.05 | ±0.38 | 0.21 | ±0.06 | |
| Underweight | 0 | (0%) | 59 | (6%) | |
| Normal | 26 | (88%) | 752 | (81%) | |
| Overweight | 1 | (3%) | 116 | (13%) | |
| Unknown | 3 | (9%) | |||
| FEV1 | NA | NA | |||
| L | 2.9 | ±1.0 | |||
| %predicted | 83.0 | ±15.1 | |||
| z‐score | −1.3 | ±1.1 | |||
| FVC | NA | NA | |||
| L | 4.0 | ±1.3 | |||
| %predicted | 97.1 | ±14.6 | |||
| z‐score | −0.2 | ±1.1 | |||
Note: Data are presented as n(%), mean ± SD, or median [IQR].
Abbreviations: BMI, body mass index; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; NA, not available; NO, nitric oxide.
Patients can be part of multiple subgroups.
Based on hallmark PCD electron microscopy findings and/or gene mutation identified.
Abnormal light or high frequency video microscopy finding and/or low nasal nitric oxide value.
z‐scores based on reference values from the World Health Organization, adjusted for age and sex.
In eight patients, fertility appeared to be normal (one or more offspring). In one female patient, fertility issues were reported (IVF treatment). Twenty‐two patients were either children (n = 10) or young adults (<25 years of age), who have not yet expressed any desire to have children (n = 12). There were no data available for two patients. Most (79%) of the patients showed signs of respiratory distress during the neonatal period, classified as either neonatal respiratory distress syndrome (NRDS), pneumonia, wet lung, or other (Table 1). In our cohort, the probability of having either situs inversus totalis or heterotaxia is 0.303 (95% CI: 0.156–0.487), this is significantly different from a 50/50 situs inversus/situs solitus distribution (p = .035). Patients frequently showed atelectasis (36%) or bronchiectasis (48%) on chest imaging (chest x‐ray and/or high resolution CT‐scan). Clinical features of both adults and children can be found in Table 2.
TABLE 2.
Clinical features of adults and children in this cohort
| Adults | Children | |||
|---|---|---|---|---|
| n/mean | (%)/±SD | n/mean | (%)/±SD | |
| Findings chest imaging a | N = 16 | N = 16 | ||
| Atelectasis | 5 | (31%) | 7 | (44%) |
| Infiltration | 0 | (0%) | 2 | (13%) |
| Bronchiectasis | 13 | (81%) | 3 | (19%) |
| Other | 2 | (13%) | 12 | (75%) |
| Normal | 2 | (13%) | 2 | (13%) |
| BMI | N = 15 | N = 13 | ||
| kg/m2 | 22.7 | ±3.8 | 19.4 | ±3.2 |
| z‐score b | 0.20 | ±1.1 | −0.14 | ±0.9 |
| Underweight | 0 | (0%) | 0 | (0%) |
| Normal | 14 | (93%) | 13 | (100%) |
| Overweight | 1 | (7%) | 0 | (0%) |
| FEV1 | N = 15 | N = 13 | ||
| L | 2.8 | ±0.9 | 3.1 | ±1.1 |
| %predicted | 77.9 | ±14.8 | 88.8 | ±13.8 |
| z‐score | −1.7 | ±1.0 | −0.9 | ±1.1 |
| FVC | N = 15 | N = 13 | ||
| L | 3.8 | ±1.0 | 4.3 | ±1.5 |
| %predicted | 90.7 | ±15.0 | 105.2 | ±9.3 |
| z‐score | −0.7 | ±1.1 | 0.4 | ±0.8 |
Note: Data are presented as n(%) or mean ± SD.
Abbreviations: BMI, body mass index; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity.
Patients can be part of multiple subgroups.
z‐scores based on reference values from the World Health Organization, adjusted for age and sex.
3.2. Microbiology
Of the 33 patients being treated at our center, 24 (10 adults and 14 children) had data available on sputum (n = 27) or cough swab cultures (n = 14) in the year 2019 (Figure 1 and Table 3). Of these cultures, 34% was positive for P. aeruginosa, accounting for nine patients (38%). P. aeruginosa showed a trend to be less prevalent in cultures from children compared to adults (21% vs. 60%, p = .09), corresponding to respectively three and six patients. Similar results were shown for Haemophilus influenzae, with 31% positive cultures overall accounting for 11 (46%) patients: 25% positive cultures in children (n = 5, 36%), compared to 46% positive cultures in adults (n = 6, 60%). This difference was not statistically significant (p = .40). Staphylococcus aureus bacteria were less commonly cultured: only 17% of all cultures, accounting for six patients (25%), with no significant differences between children (29%) and adults (20%).
FIGURE 1.
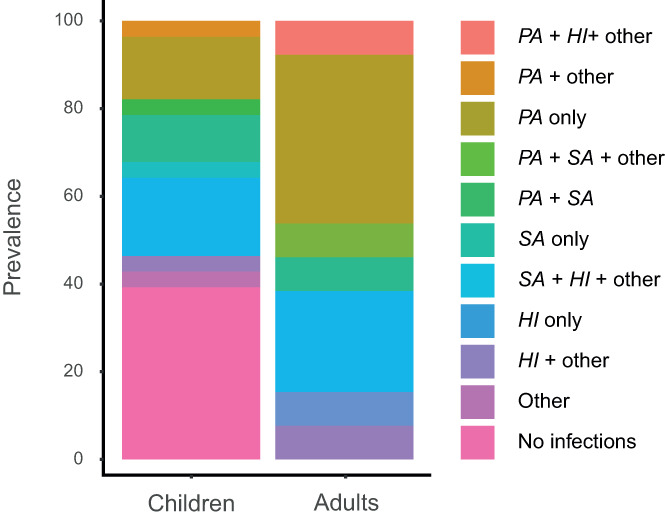
Microbes cultured in the year 2019. Stratified by children and adults. HI: H. Influenzae, PA: P. aeruginosa, SA: S. aureus
TABLE 3.
Patients [n (%)] who had a single or multiple positive cultures for microorganisms in 2019, stratified between adults and children
| Microorganisms | Overall (n = 24) | Adults (n = 10) | Children (n = 14) |
|---|---|---|---|
| Aspergillus fumigatus | 1 (4%) | 1 (10%) | 0 |
| Enterobacter species | 1 (4%) | 0 | 1 (7%) |
| Haemophilus influenzae | 11 (46%) | 6 (60%) | 5 (36%) |
| Pseudomonas aeruginosa | 9 (36%) | 6 (60%) | 3 (21%) |
| Staphylococcus aureus | 6 (25%) | 2 (20%) | 4 (29%) |
| Streptococcus pneumonia | 3 (13%) | 1 (10%) | 2 (14%) |
| Other | 1 (4%) | 0 | 1 (7%) |
Including data from the previous year, to provide a full year of culture history per patient, 53 samples were available. Of the 24 patients, 41% had one sample, 33% had two samples, 8% had three samples, and 17% had four or more samples available in 1 year. Based on the set criteria, 29% had never been infected with P. aeruginosa by 2019, 29% were free of infection in 2019, 29% had one or more positive cultures in 2019, and 13% had a chronic P. aeruginosa infection during 2019 (Figure 2a). More children (38%) than adults (25%) were never infected before. The median age of first isolation of P. aeruginosa was 6 years (inter quartile range: 2–9; Figure 2b).
FIGURE 2.
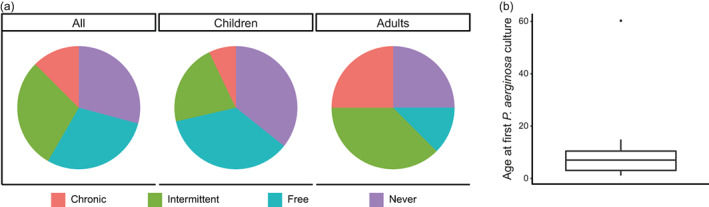
P. aeruginosa status in 2019. (a) Status based on Leeds criteria (Lee et al., 2003), data presented for all patients and stratified for children and adults. (b) Boxplot showing the median age at which patients first acquired a P. aeruginosa culture
3.3. Lung function
Lung function data were retrieved from as early as 1998 up to 2021, and data were available for 28 patients, including 346 data points (Figures S1 and S2). This accumulated to 171 patient years in the children population and 69 patient years in the adult population. The comparison of FEV1 and FVC z‐scores between the Volendam and iPCD cohort is shown in Figures 3a and 4a. The Volendam cohort showed a significant decline in both FEV1 (−0.97%/year, CI: −1.22/−0.71) and FVC (−0.81%/year, CI: −1.05/−0.56; Table 4). For FEV1, this was an absolute decline of 0.97% per year, or a drop of 0.07 in z‐score per year; for FVC, this was an absolute decline of 0.81% per year, or a drop of 0.07 in z‐score per year (Figure 3b). Stratification showed that the FEV1 decline predominately occurred in children (−1.43% FEV1/year, CI: −1.80/−1.05) as compared to adults, which showed no significant decline (0.01% FEV1/year, CI: −0.36/0.38). The same holds true for FVC, where children showed an absolute decrease of 1.46% per year (CI: −1.78/−1.14), and adults showed an absolute increase of 0.35% per year (CI: 0.01/0.69; Figure 4b). Analysis showed that patients who have been infected at some point with P. aeruginosa had a steeper decline in FEV1 (−1.13%/year, CI: −1.41/−0.85) but not in FVC (−0.89%/year, CI: −1.16/−0.62), as compared to patients that have never been infected (−0.55% FEV1/year, CI: −1.30/−0.56, and −0.91% FVC/year, CI: −1.56/−0.24; Figure 5).
FIGURE 3.
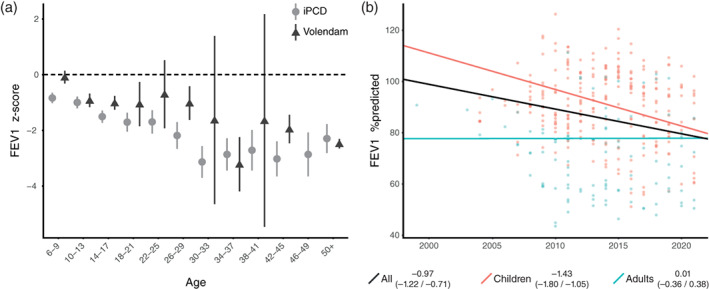
Forced expiratory volume in 1 s (FEV1). (a) Comparison of FEV1 z‐scores between the Volendam (▲) and IPCD (●) cohort. (b) The estimated FEV1 in %predicted as established by the mixed effects model, displayed for all patients (black), children (red), adults (blue). The slope with 95% confidence interval is stated in the legend
FIGURE 4.
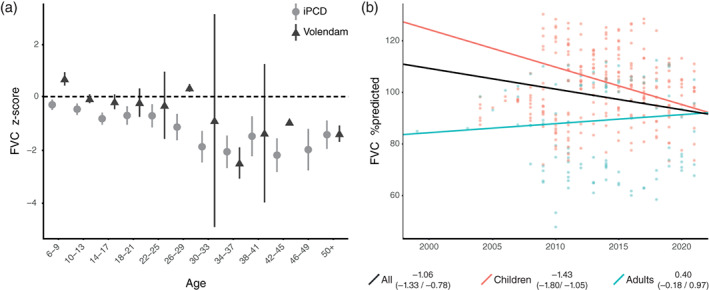
Forced vital capacity (FVC). (a) Comparison of FVC z‐scores between the Volendam (▲) and IPCD (●) cohort. (b) The estimated FVC in %predicted as established by the mixed effects model, displayed for all patients (black), children (red), adults (blue). The slope with 95% confidence interval is stated in the legend
TABLE 4.
The slope in %predicted per year and z‐score per year for both the forced expiratory volume in 1 s (FEV1) and the forced vital capacity (FVC), including 95% confidence interval (CI)
| %predicted | z‐score | ||||||
|---|---|---|---|---|---|---|---|
| 95% CI | 95% CI | ||||||
| Slope | Lower | Upper | Slope | Lower | Upper | ||
| FEV1 | All | −0.97 | −1.22 | −0.71 | −0.07 | −0.10 | −0.05 |
| Children | −1.43 | −1.80 | −1.05 | −0.12 | −0.15 | −0.09 | |
| Adults | 0.01 | −0.36 | 0.38 | 0.02 | 0.00 | 0.05 | |
| FVC | All | −0.81 | −1.05 | −0.56 | −0.07 | −0.10 | −0.05 |
| Children | −1.46 | −1.78 | −1.14 | −0.12 | −0.15 | −0.09 | |
| Adults | 0.35 | 0.01 | 0.69 | 0.04 | 0.01 | 0.06 | |
FIGURE 5.
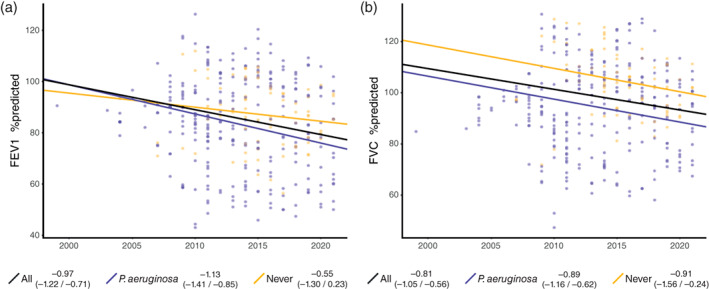
Lung function decline in relation to P. aeruginosa status. (a) The estimated FEV1 in %predicted as established by the mixed effects model. (b) The estimated FVC in %predicted as established by the mixed effects model. Results are displayed for all patients (black), patients who at some point required P. aeruginosa (purple), and patients that never acquired it (yellow). The slope with 95% confidence interval is stated in the legend. FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity
3.4. Ear‐nose‐throat
Ear‐nose‐throat (ENT) data were available from all patients; during the data collection timeframe, 12 (36%) patients were children, six (18%) transitioned from children to adults, and 15 (45%) were adults. Out of the 33 patients, 31 (94%) had ENT involvement (Table 5 and Figure 6). In these patients, the most common problems were rhinosinusitis (87%), hearing impairment (58%), and otitis media with effusion (OME) (42%). Severity of hearing impairment was not assessed as data about the original hearing tests were not always available. Therefore, these data were excluded from the study.
TABLE 5.
Ear‐nose‐throat (ENT) involvement in the Volendam cohort
| N | % | |
|---|---|---|
| ENT involvement | 31 | 94 |
| Hearing impairment | 18 | 58 |
| Hearing aids | 6 | 19 |
| Rhinosinusitis | 27 | 87 |
| Nasal polyps | 4 | 13 |
| Eardrum perforation | 7 | 23 |
| OM acuta/otorrea | 6 | 19 |
| OM with effusion | 13 | 42 |
| ENT surgery | 8 | 24 |
| Tympanostomy tube | 6 | 75 |
| Adenotomy | 1 | 13 |
| Sinus surgery | 1 | 13 |
| Mastoidectomy | 1 | 13 |
| Nasal therapy | 17 | 55 |
| Decongestant | 3 | 6 |
| Nasal corticosteroids | 13 | 82 |
| Hypertonic NaCl | 10 | 59 |
| Other | 1 | 17 |
FIGURE 6.
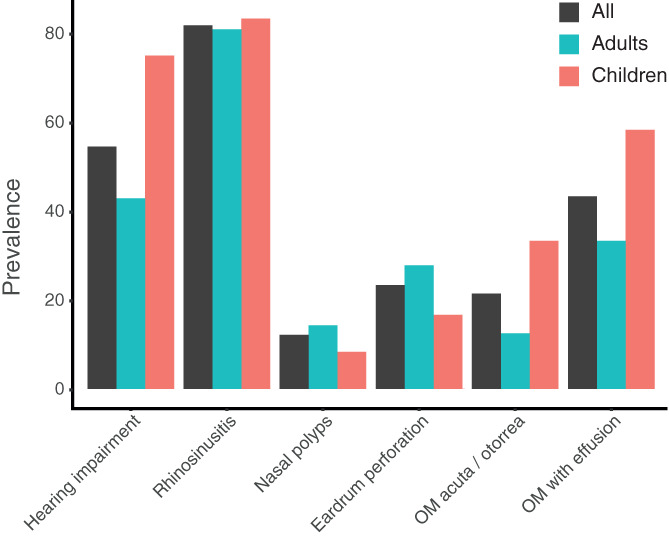
Ear‐nose‐throat involvement in the Volendam cohort (black), stratified for adults (blue) and children (red). OM: otitis media
3.4.1. Rhinosinusitis
Twenty‐seven patients suffered from symptoms of rhinosinusitis, of which four also known had nasal polyps. Seventeen (48%) of these 27 patients used local nasal therapy, with topical corticosteroids (n = 13) as the most frequently used medication. Additionally, 10 patients rinsed their noses with a saline solution, being the first treatment of choice for rhinosinusitis. One patient needed functional endoscopic sinus surgery, and one patient underwent adenoidectomy.
3.4.2. Hearing impairment
In 58% of the patients (n = 18), hearing was impaired. In all cases, this was caused by conductive hearing loss. Most often, conductive hearing loss was caused by OME (n = 12; 67% of cases). Other causes included: a perforation of the eardrum (n = 3), otitis media acuta/otorrea (n = 2), and previous sanation surgery of the ear for severe chronic otorrhoea (n = 1). Restoration with a hearing aid was necessary in six out of 18 patients (five caused by OME and one by sanation surgery). In six patients, ventilation tubes were inserted at least once to resolve the conductive hearing loss caused by OME.
3.4.3. Otitis media with effusion
Thirteen (42%) patients had at least one period of OME in the last 4 years. As described above, in six of these patients ventilation tubes were inserted at least once. In five other patients, hearing aids were chosen for restoration of the hearing loss. In two patients, the hearing impairment caused by OME did not require therapy. There were more children (n = 7, 58%) than adults (n = 6, 33%) with OME; however, this difference was not statistically significant (p = .26).
4. DISCUSSION
In this study, we describe the clinical phenotype of a cohort of PCD patients from Volendam (with CCDC114 c.742G>A mutation). This study shows a relatively low prevalence of situs inversus, a high prevalence of P. aeruginosa, a lung function decline during childhood followed by stabilization during adulthood, and frequent ENT involvement. Fertility issues were rarely reported. Overall, the CCDC114 c.742G>A genotype leads to a moderately severe phenotype.
The decline in lung function for the Volendam cohort (−0.97% FEV1/year) was steeper as compared to other published PCD cohorts (−0.5–0.8% FEV1/year) (Marthin et al., 2010; Shah et al., 2016). Shaw, Davis, and colleagues reported a lung function decline in patients with similar ultrastructural defects (outer dynein arm defects or combined outer and inner dynein defects) of 0.5–0.6% FEV1/year (Davis et al., 2019; Shah et al., 2016). The difference between our cohort and other cohorts with outer dynein arm mutations can probably be attributed to the genetic heterogeneity in the latter cohort and the large proportion of patients with mutations in the DNAH5 gene. These patients show a diverse phenotype, possibly reflecting the effects of different kinds of mutations in a large gene (Shoemark et al., 2021). In contrast, lung function decline in CCDC39 an CCDC40 mutated patients is generally steeper (−1.11% FEV1/year) (Davis et al., 2019), compared to our cohort and other general PCD cohorts. Overall, the founder CCDC114 c.742G>A mutation in our cohort seems to be associated with a moderately severe pulmonary outcome.
Interestingly, our data show that lung function decline mainly occurred during childhood in our center. In pediatric patients, the frequency of checkups is higher and the threshold for starting antibiotic treatment lower compared to adults (personal communication). However, we have no data on treatment compliance in our center. Nonetheless, the lung function decline during childhood is in line with a 2016 report on CF patients, where the steepest decline occurs between 11 and 15 years of age (Harun, Wainwright, Klein, & Hennig, 2016). Larger studies are necessary to confirm our findings and validate this in PCD patients.
Our study suggests a steeper lung function decline in PCD patients infected with P. aeruginosa. In previous studies, baseline FEV1 was lower in patients colonized with P. aeruginosa, while lung function decline was similar to non‐colonized PCD patients (Cohen‐Cymberknoh et al., 2017; Roden et al., 2019; Shah et al., 2016). It was hypothesized that PCD patients with a lower FEV1 are more vulnerable to infection with P. aeruginosa, but that infection does not necessarily increase the rate of lung function decline (Cohen‐Cymberknoh et al., 2017). Given the relatively long history of lung function data in our cohort, one can question this hypothesis and argue that infections with P. aeruginosa accelerate the progression of lung damage; further research is needed to validate this.
In our cohort, 21% of the children had a positive culture for P. aeruginosa, and 60% of the adults had a positive culture. Comparison of P. aeruginosa infection rates with previous studies is hampered by differences in characteristics (children vs. adults vs. both) and follow‐up (cross‐sectional vs. longitudinal). In general, the prevalence of positive cultures is assumed to be around 15–35% (Cohen‐Cymberknoh et al., 2017; Davis et al., 2019; Roden et al., 2019). Lower frequencies are reported for children (9–25%) than for adults (26–51%). Our cohort had a relatively high prevalence of P. aeruginosa positive cultures. One can speculate about possible causes including specific genotype‐associated factors, household transmission, frequency of antibiotic use, and the extent of lung damage. To explore these associations, further research is necessary. The relative high prevalence of P. aeruginosa is likely still an underestimation of the real‐life prevalence, due to the use of cough swabs that have a higher rate of false negatives compared to other culturing methods. Furthermore, the percentage of patients with chronic infection in our cohort was relatively low: 13%. Previous studies show that 14–32% of PCD patients are chronically colonized with P. aeruginosa (Cohen‐Cymberknoh et al., 2017; Piatti et al., 2020). There is a underestimation of chronic infection in our cohort; many patients had only one culture available and thus could not be classified as chronic infection (Lee et al., 2003).
Surprisingly, we found a relatively low prevalence of situs inversus (27%). This contrasts with findings in literature, which suggest organ laterality to be random in most PCD patients (Nonaka et al., 1998; Noone et al., 1999; Supp, Witte, Potter, & Brueckner, 1997), including a study by our group in 16 patients with the CCDC114 c.742G>A founder mutation (Onoufriadis et al., 2013). If left–right lateralization were truly random, one would expect at least half of the patients to have a situs inversus or heterotaxia, as situs abnormalities raise the clinical suspicion of PCD. The exact reason for this skewed distribution is unclear.
Other aspects of the phenotype of this cohort of CCDC114 c.742G>A patients are in line with results published in other cohorts: 79% of patients had neonatal symptoms (reported values of 65–87% (Goutaki et al., 2018; Noone et al., 2004) and in all patients nasal NO levels were below the cut‐off value of 77 nl/min. In contrast, fertility issues were rarely reported, similar to the study of Onoufriadis and colleagues (Onoufriadis et al., 2013). This relatively fertile phenotype partly enables the high prevalence of PCD within families; unfortunately, we had no family tree data available. There are two theories to explain the fertility; either over‐expression of CCDC63 partly replaces CCDC114‐function in sperm cells (Bastian et al., 2021), or CCDC114 might be less relevant in the ODA type 3 present in both sperm flagella and in the cilia of the efferent duct (Aprea et al., 2021; Whitfield et al., 2019). Additional studies are required to identify the exact mechanism of fertility in these patients and may provide pointers on restoration of ciliary function in the airways.
In our Volendam cohort, a high prevalence of sinonasal disease was observed (94%), for which nasal corticosteroids are most commonly prescribed. The prevalence is in the higher range when compared to other studies, with an incidence of 13–85% (Bhatt, Muhonen, Meier, Sagel, & Chan, 2019; Sommer et al., 2011; Wolter, Dell, James, & Campisi, 2012). The use of nasal steroids is not in line with the standard of care for rhinosinusitis, which suggests the use of saline solution as primary treatment. This is most likely due to the burden patients experience from daily rinsing.
We found that OME caused hearing impairment in 39% of the patients, with a similar prevalence in adults compared to children. Current literature suggests that OME becomes less prevalent over the age of 12, but can be persistent into adulthood (Hadfield, Rowe‐Jones, Bush, & Mackay, 1997; Majithia, Fong, Hariri, & Harcourt, 2005). Children suffering of persistent OME, causing conductive hearing loss, are prone for impaired speech language development, and therefore need regular otolaryngologic care. Management of persistent OME focuses on hearing improvement, and in non‐PCD‐patients placement of ventilation tubes is standard of care. The question remains if children with PCD suffering from hearing loss due to OME benefit most from conservative treatment or insertion of ventilation tubes. Insertion of ventilation tubes has been shown to be effective in preventing bilateral hearing loss caused by OME (Wolter et al., 2012). However, other studies do not report a clear effect of tube placement on hearing loss in PCD patients (Ghedia, Ahmed, Navaratnam, & Harcourt, 2018; Kreicher, Schopper, Naik, Hatch, & Meyer, 2018). Moreover, chronic problematic otorrhoea (up to 50%) following insertion of ventilation tubes in PCD patients has been reported, adversely affecting both hearing and quality of life (Andersen, Alanin, von Buchwald, & Nielsen, 2016; Campbell, Birman, & Morgan, 2009; El‐Sayed, Al‐Sarhani, & Al‐Essa, 1997; Wolter et al., 2012). The best treatment of OME and also recurrent OMA in patients with PCD is still debatable (Campbell et al., 2009; Sagel, Davis, Campisi, & Dell, 2011). Existing guidelines and recommendations advice to be restrictive concerning placement of ventilation tubes (Barbato et al., 2009; Hadfield et al., 1997). The ERS Consensus on clinical care of children with PCD recommended against placement of ventilation tubes and advised to watch for spontaneous resolution of OME often occurring in teenage years (Lucas et al., 2017). In case education or speech development is impaired, hearing aids may be required. In this cohort, 33% of patients with ventilation tubes suffered from otorrhoea postoperatively. This might be an underestimation because the ventilation tubes were sometimes placed in another hospital, and therefore information might be incomplete. Future research on potential predictive factors associated with postoperative otorrhoea could aid clinical decision‐making.
One of the strengths of this study was that it was conducted in a PCD Expertise Center, acknowledged by The Netherlands Federation of University Medical Centers (NFU) and the European Reference Network for the Lung (ERN‐Lung). In addition, due to the collaboration with the international PCD registry, we were able to collect and extract a wide range of data, to encompass as much clinical features in the phenotype description as possible. Furthermore, the setting of Volendam and the increased prevalence of one mutation in a small population has provided us with a unique cohort. Nonetheless, this study was limited by several factors. First, despite including ~60% of the patients from Volendam, the number of patients is limited. Therefore, the age groups used in the iPCD lung function analysis resulted in such small subgroups in our cohort that statistical analysis was not possible. Furthermore, we acknowledge possible selection bias because of only including patients at a University Medical Center. Therefore, the patients included in this study might have a more severe phenotype.
This study describes the phenotypical characteristics of a PCD‐population with a founder mutation in the CCDC114 gene (c.742G>A; p.(Ala248 Thrfs52*). This phenotype is not only relevant for the Volendam population as the c.742G>A mutation is also carried by other families of mainly European ancestry (Knowles et al., 2013). For patients with this phenotype, a relatively steep lung function decline is reported. Our results strengthen the notion in the field that there is a need for improvement of PCD treatment by preventing and treating lung function decline, especially in childhood. This substantiates the work of Strippoli and colleagues whom found substantial heterogeneity in management of PCD in children among different European countries and bargain for standardized disease management (Strippoli et al., 2012). Current treatment is based on CF‐guidelines, but these may not be sufficient. Since P. aeruginosa was associated with a steeper lung function decline, early detection is pivotal. However, detection of P. aeruginosa is especially challenging in children who often cannot expectorate sputum. Additional tools for detecting organisms such as P. aeruginosa may improve treatment strategies (Fenn et al., 2021). Laryngeal aspirates could potentially play a part in this; however, these are not routinely performed in many centers (Crowley, Holgersen, & Nielsen, 2019; Hanshew, Jetté, Tadayon, & Thibeault, 2017). Furthermore, comparison of P. aeruginosa management strategies across European centers shows that there is a large variation in both monitoring and management of P. aeruginosa infection, which may influence eradication success (Crowley et al., 2019). Therefore, studies on P. aeruginosa eradication, including the effect of eradication on lung function decline, are necessary. Finally, PCD‐specific treatments, like mRNA transcript therapy, are currently being developed to restore normal ciliary activity and consequently mucociliary clearance.
In conclusion, Volendam PCD patients with a CCDC114 founder mutation have a moderately severe phenotype. In these patients, P. aeruginosa is prevalent and lung function decline occurs predominantly during childhood. These findings stress the importance of and the need for the development of PCD‐specific treatments that should be initiated at an early age.
CONFLICT OF INTEREST
EGH is part of the BEAT‐PCD network that was initially funded by COST Action BM 1407, and now by European Respiratory Society Clinical Research Collaboration. In addition, he participates in the European Reference Network for Rare Respiratory Diseases (ERN‐LUNG) project identification number 739546. AHM has received research grants outside the submitted work from GSK, Boehringer Íngelheim and Vertex, she is the PI of a P4O2 (Precision Medicine for more Oxygen) public private partnership sponsored by Health Holland involving many private partners that contribute in cash and/or in kind (Boehringer Ingelheim, Breathomix, Fluidda, Ortec Logiqcare, Philips, Quantib‐U, Roche, Smartfish, SODAQ, Thirona, TopMD and Novartis), and she has served in advisory boards for AstraZeneca, GSK and Boehringer Ingelheim with money paid to her institution.
AUTHOR CONTRIBUTIONS
Renate Kos, Joël Israëls, Eric G Haarman: conception or design of the work; Renate Kos, Joël Israëls, Christine DL van Gogh, Josje Altenburg, Sandra Diepenhorst, Elles MJ Boon, Eric G Haarman: the acquisition and analysis; Renate Kos, Joël Israëls, Christine DL van Gogh, Josje Altenburg, Tamara Paff, Gerard Pals, Anne H Neerincx, Anke H Maitland‐van der Zee, Eric G Haarman: interpretation of data for the work; All authors were involved in drafting the work or revising it critically for important intellectual content; All authors approved the final version to be published; all authors were responsible for the agreement to be accountable.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
The authors would like to acknowledge all patients and parents for their interest in scientific research and for giving their consent. Furthermore, the authors would like to thank Jaap Schipper, Hans (JWM) Niessen, and Hedy Meijer for their help with the data collection, and Hans Gille for his contribution to the genotyping results.
Kos, R. , Israëls, J. , van Gogh, C. D. L. , Altenburg, J. , Diepenhorst, S. , Paff, T. , Boon, E. M. J. , Micha, D. , Pals, G. , Neerincx, A. H. , Maitland‐van der Zee, A. H. , Haarman, E. G. , & Amsterdam Mucociliary Clearance Disease (AMCD) Research Group (2022). Primary ciliary dyskinesia in Volendam: Diagnostic and phenotypic features in patients with a CCDC114 mutation. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:89–101. 10.1002/ajmg.c.31968
Renate Kos and Joël Israëls should be considered joint first author.
Contributor Information
Renate Kos, Email: r.kos@amsterdamumc.nl.
Eric G. Haarman, Email: eg.haarman@amsterdamumc.nl.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Andersen, T. N. , Alanin, M. C. , von Buchwald, C. , & Nielsen, L. H. (2016). A longitudinal evaluation of hearing and ventilation tube insertion in patients with primary ciliary dyskinesia. International Journal of Pediatric Otorhinolaryngology, 89, 164–168. 10.1016/j.ijporl.2016.08.011 [DOI] [PubMed] [Google Scholar]
- Aprea, I. , Raidt, J. , Höben, I. M. , Loges, N. T. , Nöthe‐Menchen, T. , Pennekamp, P. , … Omran, H. (2021). Defects in the cytoplasmic assembly of axonemal dynein arms cause morphological abnormalities and dysmotility in sperm cells leading to male infertility. PLoS Genetics, 17(2), e1009306. 10.1371/journal.pgen.1009306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato, A. , Frischer, T. , Kuehni, C. E. , Snijders, D. , Azevedo, I. , Baktai, G. , … Bush, A. (2009). Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. European Respiratory Journal, 34(6), 1264–1276. 10.1183/09031936.00176608 [DOI] [PubMed] [Google Scholar]
- Bastian, F. B. , Roux, J. , Niknejad, A. , Comte, A. , Fonseca Costa, S. S. , de Farias, T. M. , … Robinson‐Rechavi, M. (2021). The Bgee suite: Integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Research, 49(D1), D831–D847. 10.1093/nar/gkaa793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan, L. , Rubbo, B. , Lucas, J. S. , & Dunn Galvin, A. (2017). The patient's experience of primary ciliary dyskinesia: A systematic review. Quality of Life Research, 26(9), 2265–2285. 10.1007/s11136-017-1564-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt, J. M. , Muhonen, E. G. , Meier, M. , Sagel, S. D. , & Chan, K. H. (2019). Rhinosinusitis in pediatric primary ciliary dyskinesia: Impact of disease. Otolaryngology–Head and Neck Surgery, 161(5), 877–880. 10.1177/0194599819874842 [DOI] [PubMed] [Google Scholar]
- Caley, L. , Smith, L. , White, H. , & Peckham, D. G. (2021). Average rate of lung function decline in adults with cystic fibrosis in the United Kingdom: Data from the UKCF registry. Journal of Cystic Fibrosis, 20(1), 86–90. 10.1016/j.jcf.2020.04.008 [DOI] [PubMed] [Google Scholar]
- Campbell, R. G. , Birman, C. S. , & Morgan, L. (2009). Management of otitis media with effusion in children with primary ciliary dyskinesia: A literature review. International Journal of Pediatric Otorhinolaryngology, 73(12), 1630–1638. 10.1016/j.ijporl.2009.08.024 [DOI] [PubMed] [Google Scholar]
- Cohen‐Cymberknoh, M. , Simanovsky, N. , Hiller, N. , Hillel, A. G. , Shoseyov, D. , & Kerem, E. (2014). Differences in disease expression between primary ciliary dyskinesia and cystic fibrosis with and without pancreatic insufficiency. Chest, 145(4), 738–744. 10.1378/chest.13-1162 [DOI] [PubMed] [Google Scholar]
- Cohen‐Cymberknoh, M. , Weigert, N. , Gileles‐Hillel, A. , Breuer, O. , Simanovsky, N. , Boon, M. , … Kerem, E. (2017). Clinical impact of Pseudomonas aeruginosa colonization in patients with primary ciliary dyskinesia. Respiratory Medicine, 131, 241–246. 10.1016/j.rmed.2017.08.028 [DOI] [PubMed] [Google Scholar]
- Crowley, S. , Holgersen, M. G. , & Nielsen, K. G. (2019). Variation in treatment strategies for the eradication of Pseudomonas aeruginosa in primary ciliary dyskinesia across European centers. Chronic Respiratory Disease, 16, 147997231878791. 10.1177/1479972318787919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Rosenfeld, M. , Lee, H.‐S. , Ferkol, T. W. , Sagel, S. D. , Dell, S. D. , … Leigh, M. W. (2019). Primary ciliary dyskinesia: Longitudinal study of lung disease by ultrastructure defect and genotype. American Journal of Respiratory and Critical Care Medicine, 199(2), 190–198. 10.1164/rccm.201803-0548OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Sayed, Y. , Al‐Sarhani, A. , & Al‐Essa, A.‐R. (1997). Otological manifestations of primary ciliary dyskinesia. Clinical Otolaryngology and Allied Sciences, 22(3), 266–270. 10.1046/j.1365-2273.1997.00895.x [DOI] [PubMed] [Google Scholar]
- Fenn, D. , Abdel‐Aziz, M. I. , Brinkman, P. , Kos, R. , Neerincx, A. H. , Altenburg, J. , … Bos, L. D. J. (2021). Comparison of microbial composition of cough swabs and sputum for pathogen detection in patients with cystic fibrosis. Journal of Cystic Fibrosis., 21, 52–60. 10.1016/j.jcf.2021.08.031 [DOI] [PubMed] [Google Scholar]
- Ghedia, R. , Ahmed, J. , Navaratnam, A. , & Harcourt, J. (2018). No evidence of cholesteatoma in untreated otitis media with effusion in children with primary ciliary dyskinesia. International Journal of Pediatric Otorhinolaryngology, 105, 176–180. 10.1016/j.ijporl.2017.12.015 [DOI] [PubMed] [Google Scholar]
- Goutaki, M. , Halbeisen, F. S. , Wisse, A. C. , Amirav, I. , Barbato, A. , Casaulta, C. , … Kuehni, C. E. (2018). Neonatal manifestations in Primary Ciliary Dyskinesia: a multinational cohort study. European Respiratory Journal, 52(Suppl 62), PA5021. 10.1183/13993003.congress-2018.PA5021 [DOI] [Google Scholar]
- Hadfield, P. J. , Rowe‐Jones, J. M. , Bush, A. , & Mackay, I. S. (1997). Treatment of otitis media with effusion in children with primary ciliary dyskinesia. Clinical Otolaryngology and Allied Sciences, 22(4), 302–306. 10.1046/j.1365-2273.1997.00020.x [DOI] [PubMed] [Google Scholar]
- Halbeisen, F. S. , Goutaki, M. , Spycher, B. D. , Amirav, I. , Behan, L. , Boon, M. , … Kuehni, C. E. (2018). Lung function in patients with primary ciliary dyskinesia: An iPCD cohort study. European Respiratory Journal, 52(2), 1801040. 10.1183/13993003.01040-2018 [DOI] [PubMed] [Google Scholar]
- Hannah, W. B. , Seifert, B. A. , Truty, R. , Zariwala, M. A. , Ameel, K. , Zhao, Y. , … Gaston, B. (2022). The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: A genetic database analysis. The Lancet Respiratory Medicine. 10.1016/S2213-2600(21)00453-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanshew, A. S. , Jetté, M. E. , Tadayon, S. , & Thibeault, S. L. (2017). A comparison of sampling methods for examining the laryngeal microbiome. PLoS One, 12(3), e0174765. 10.1371/journal.pone.0174765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harun, S. N. , Wainwright, C. , Klein, K. , & Hennig, S. (2016). A systematic review of studies examining the rate of lung function decline in patients with cystic fibrosis. Paediatric Respiratory Reviews, 20, 55–66. 10.1016/j.prrv.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Höben, I. M. , Hjeij, R. , Olbrich, H. , Dougherty, G. W. , Nöthe‐Menchen, T. , Aprea, I. , … Omran, H. (2018). Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. The American Journal of Human Genetics, 102(5), 973–984. 10.1016/j.ajhg.2018.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, M. R. , Leigh, M. W. , Ostrowski, L. E. , Huang, L. , Carson, J. L. , Hazucha, M. J. , … Zariwala, M. A. (2013). Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. The American Journal of Human Genetics, 92(1), 99–106. 10.1016/j.ajhg.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreicher, K. L. , Schopper, H. K. , Naik, A. N. , Hatch, J. L. , & Meyer, T. A. (2018). Hearing loss in children with primary ciliary dyskinesia. International Journal of Pediatric Otorhinolaryngology, 104, 161–165. 10.1016/j.ijporl.2017.11.005 [DOI] [PubMed] [Google Scholar]
- Lee, T. W. R. , Brownlee, K. G. , Conway, S. P. , Denton, M. , & Littlewood, J. M. (2003). Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis, 2(1), 29–34. 10.1016/S1569-1993(02)00141-8 [DOI] [PubMed] [Google Scholar]
- Lucas, J. S. , Alanin, M. C. , Collins, S. , Harris, A. , Johansen, H. K. , Nielsen, K. G. , … Walker, W. T. (2017). Clinical care of children with primary ciliary dyskinesia. Expert Review of Respiratory Medicine, 11(10), 779–790. 10.1080/17476348.2017.1360770 [DOI] [PubMed] [Google Scholar]
- Lucas, J. S. , Davis, S. D. , Omran, H. , & Shoemark, A. (2020). Primary ciliary dyskinesia in the genomics age. The Lancet Respiratory Medicine, 8(2), 202–216. 10.1016/S2213-2600(19)30374-1 [DOI] [PubMed] [Google Scholar]
- Majithia, A. , Fong, J. , Hariri, M. , & Harcourt, J. (2005). Hearing outcomes in children with primary ciliary dyskinesia—A longitudinal study. International Journal of Pediatric Otorhinolaryngology, 69(8), 1061–1064. 10.1016/j.ijporl.2005.02.013 [DOI] [PubMed] [Google Scholar]
- Marthin, J. K. , Petersen, N. , Skovgaard, L. T. , & Nielsen, K. G. (2010). Lung function in patients with primary ciliary dyskinesia. American Journal of Respiratory and Critical Care Medicine, 181(11), 1262–1268. 10.1164/rccm.200811-1731OC [DOI] [PubMed] [Google Scholar]
- Nonaka, S. , Tanaka, Y. , Okada, Y. , Takeda, S. , Harada, A. , Kanai, Y. , … Hirokawa, N. (1998). Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell, 95(6), 829–837. 10.1016/S0092-8674(00)81705-5 [DOI] [PubMed] [Google Scholar]
- Noone, P. G. , Bali, D. , Carson, J. L. , Sannuti, A. , Gipson, C. L. , Ostrowski, L. E. , … Knowles, M. R. (1999). Discordant organ laterality in monozygotic twins with primary ciliary dyskinesia. American Journal of Medical Genetics, 82(2), 155–160. [DOI] [PubMed] [Google Scholar]
- Noone, P. G. , Leigh, M. W. , Sannuti, A. , Minnix, S. L. , Carson, J. L. , Hazucha, M. , … Knowles, M. R. (2004). Primary ciliary dyskinesia. American Journal of Respiratory and Critical Care Medicine, 169(4), 459–467. 10.1164/rccm.200303-365OC [DOI] [PubMed] [Google Scholar]
- Onoufriadis, A. , Paff, T. , Antony, D. , Shoemark, A. , Micha, D. , Kuyt, B. , … Mitchison, H. M. (2013). Splice‐site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. The American Journal of Human Genetics, 92(1), 88–98. 10.1016/j.ajhg.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paff, T. , Loges, N. T. , Aprea, I. , Wu, K. , Bakey, Z. , Haarman, E. G. , … Micha, D. (2017). Mutations in PIH1D3 cause X‐linked primary ciliary dyskinesia with outer and inner dynein arm defects. The American Journal of Human Genetics, 100(1), 160–168. 10.1016/j.ajhg.2016.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti, G. , De Santi, M. M. , Farolfi, A. , Zuccotti, G. V. , D'Auria, E. , Patria, M. F. , … Ambrosetti, U. (2020). Exacerbations and Pseudomonas aeruginosa colonization are associated with altered lung structure and function in primary ciliary dyskinesia. BMC Pediatrics, 20(1), 158. 10.1186/s12887-020-02062-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifferi, M. , Bush, A. , Di Cicco, M. , Pradal, U. , Ragazzo, V. , Macchia, P. , & Boner, A. L. (2010). Health‐related quality of life and unmet needs in patients with primary ciliary dyskinesia. European Respiratory Journal, 35(4), 787–794. 10.1183/09031936.00051509 [DOI] [PubMed] [Google Scholar]
- Quanjer, P. H. , Stanojevic, S. , Cole, T. J. , Baur, X. , Hall, G. L. , Culver, B. H. , … Stocks, J. (2012). Multi‐ethnic reference values for spirometry for the 3–95‐yr age range: The global lung function 2012 equations. European Respiratory Journal, 40(6), 1324–1343. 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden, L. , Görlich, D. , Omran, H. , Peters, G. , Große‐Onnebrink, J. , & Kahl, B. C. (2019). A retrospective analysis of the pathogens in the airways of patients with primary ciliary dyskinesia. Respiratory Medicine, 156, 69–77. 10.1016/j.rmed.2019.08.009 [DOI] [PubMed] [Google Scholar]
- Sagel, S. D. , Davis, S. D. , Campisi, P. , & Dell, S. D. (2011). Update of respiratory tract disease in children with primary ciliary dyskinesia. Proceedings of the American Thoracic Society, 8(5), 438–443. 10.1513/pats.201103-024SD [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, A. , Shoemark, A. , MacNeill, S. J. , Bhaludin, B. , Rogers, A. , Bilton, D. , … Loebinger, M. R. (2016). A longitudinal study characterising a large adult primary ciliary dyskinesia population. European Respiratory Journal, 48(2), 441–450. 10.1183/13993003.00209-2016 [DOI] [PubMed] [Google Scholar]
- Shoemark, A. , Rubbo, B. , Legendre, M. , Fassad, M. R. , Haarman, E. G. , Best, S. , … Lucas, J. S. (2021). Topological data analysis reveals genotype–phenotype relationships in primary ciliary dyskinesia. European Respiratory Journal, 58(2), 2002359. 10.1183/13993003.02359-2020 [DOI] [PubMed] [Google Scholar]
- Sommer, J. U. , Schäfer, K. , Omran, H. , Olbrich, H. , Wallmeier, J. , Blum, A. , … Stuck, B. A. (2011). ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co‐morbidities. European Archives of Oto‐Rhino‐Laryngology, 268(3), 383–388. 10.1007/s00405-010-1341-9 [DOI] [PubMed] [Google Scholar]
- Strippoli, M.‐P. F. , Frischer, T. , Barbato, A. , Snijders, D. , Maurer, E. , Lucas, J. S. A. , … Kuehni, C. E. (2012). Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. European Respiratory Journal, 39(6), 1482–1491. 10.1183/09031936.00073911 [DOI] [PubMed] [Google Scholar]
- Supp, D. M. , Witte, D. P. , Potter, S. S. , & Brueckner, M. (1997). Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature, 389(6654), 963–966. 10.1038/40140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenBranden, S. L. , McMullen, A. , Schechter, M. S. , Pasta, D. J. , Michaelis, R. L. , Konstan, M. W. , … McColley, S. A. (2012). Lung function decline from adolescence to young adulthood in cystic fibrosis. Pediatric Pulmonology, 47(2), 135–143. 10.1002/ppul.21526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoort, R. , & Wright, A. F. (2002). Mutations ofRPGR in X‐linked retinitis pigmentosa (RP3). Human Mutation, 19(5), 486–500. 10.1002/humu.10057 [DOI] [PubMed] [Google Scholar]
- Wallmeier, J. , Frank, D. , Shoemark, A. , Nöthe‐Menchen, T. , Cindric, S. , Olbrich, H. , … Omran, H. (2019). De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. The American Journal of Human Genetics, 105(5), 1030–1039. 10.1016/j.ajhg.2019.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, C. , Lablans, M. , Ataian, M. , Raidt, J. , Wallmeier, J. , Große‐Onnebrink, J. , … Omran, H. (2016). An international registry for primary ciliary dyskinesia. European Respiratory Journal, 47(3), 849–859. 10.1183/13993003.00776-2015 [DOI] [PubMed] [Google Scholar]
- Whitfield, M. , Thomas, L. , Bequignon, E. , Schmitt, A. , Stouvenel, L. , Montantin, G. , … Legendre, M. (2019). Mutations in DNAH17, encoding a sperm‐specific axonemal outer dynein arm heavy chain, cause isolated male infertility due to Asthenozoospermia. The American Journal of Human Genetics, 105(1), 198–212. 10.1016/j.ajhg.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter, N. E. , Dell, S. D. , James, A. L. , & Campisi, P. (2012). Middle ear ventilation in children with primary ciliary dyskinesia. International Journal of Pediatric Otorhinolaryngology, 76(11), 1565–1568. 10.1016/j.ijporl.2012.07.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.