

Summary
An essential step in the analysis of single‐cell RNA sequencing data is to classify cells into specific cell types using marker genes. In this study, we have developed a machine learning pipeline called single‐cell predictive marker (SPmarker) to identify novel cell‐type marker genes in the Arabidopsis root.
Unlike traditional approaches, our method uses interpretable machine learning models to select marker genes. We have demonstrated that our method can: assign cell types based on cells that were labelled using published methods; project cell types identified by trajectory analysis from one data set to other data sets; and assign cell types based on internal GFP markers.
Using SPmarker, we have identified hundreds of new marker genes that were not identified before. As compared to known marker genes, the new marker genes have more orthologous genes identifiable in the corresponding rice single‐cell clusters. The new root hair marker genes also include 172 genes with orthologs expressed in root hair cells in five non‐Arabidopsis species, which expands the number of marker genes for this cell type by 35–154%.
Our results represent a new approach to identifying cell‐type marker genes from scRNA‐seq data and pave the way for cross‐species mapping of scRNA‐seq data in plants.
Keywords: cell marker genes, gene expression, machine learning, root development, single‐cell genomics, single‐cell sequencing
Introduction
Single‐cell RNA sequencing (scRNA‐seq) has recently emerged as a powerful approach to investigate gene expression in multicellular organisms. Compared with bulk RNA‐seq, scRNA‐seq can identify rare cell populations and reveal transitions of cell states at different developmental stages, which are difficult to capture using traditional methods (Trapnell, 2015; Wang & Navin, 2015; Butler et al., 2018). As a transformative technology, scRNA‐seq is particularly important for plant research because traditional methods for determining gene expression in individual cell types rely on transgenic lines expressing cell type‐specific fluorescent markers, which are not available in most nonmodel species. Because of the advantages of using scRNA‐seq in plants, this approach has been applied in a number of studies to profile transcriptomes of Arabidopsis, rice (Oryza sativa), tomato (Solanum lycopersicum) and maize (Zea mays) (Jean‐Baptiste et al., 2019; Shulse et al., 2019; Satterlee et al., 2020; Bezrutczyk et al., 2021; Liu et al., 2021; Roszak et al., 2021). Among the published scRNA‐seq data in plants, the majority of data are from the Arabidopsis root, which is an ideal system to address important questions in plant biology, including the analysis of the expression patterns of rare cell types (Denyer et al., 2019; Ryu et al., 2019), determination of developmental trajectories of root cells (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Ryu et al., 2019; T‐Q. Zhang et al., 2019) and characterization of stress‐responsive genes at the single‐cell level (Jean‐Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019).
Determining cell types is a key step in the analysis and interpretation of scRNA‐seq data (Luecken & Theis, 2019). Currently, approaches to define Arabidopsis root cell types fall into three major categories, (1) Index of cell identity (ICI) method. This approach uses selected marker genes based on information theoretic scores from the published cell expression profiles (Efroni et al., 2015; Shulse et al., 2019; Turco et al., 2019). (2) Definition of cluster‐marker genes. This approach generates clusters of cells with unsupervised dimension reduction methods and assigns cell types by visualizing expression patterns using known marker genes (Jean‐Baptiste et al., 2019; Ryu et al., 2019; T‐Q. Zhang et al., 2019). (3) Correlation methods. These methods compute correlation coefficients between single cells and published gene expression data (Jean‐Baptiste et al., 2019; Shulse et al., 2019). All of these strategies rely on the knowledge of genes expressed in specific cell types (also known as cell marker genes), and each of these three approaches has limitations. The ICI method was developed using microarray data and only included 15 cell types with c. 20 marker genes per cell type. This method did not use more recent data from bulk RNA‐seq or single‐cell experiments. The cluster‐marker gene methods used marker genes that vary from publication to publication. There has not been a standardized statistical test to determine how many marker genes are optimal. The correlation methods require expression profiles of all expressed genes from all known cell types, and these methods have difficulty assigning cell types between highly diverged species due to the loss of orthologous marker genes in nonmodel species (Liu et al., 2021). In Arabidopsis, > 1500 cell‐type marker genes have been determined from fluorescent‐activated cell sorting (FACS)‐based gene expression data in the root cells of Arabidopsis (Birnbaum et al., 2005; Brady et al., 2007; Bruex et al., 2012; Efroni et al., 2015; Li et al., 2016). Several computational approaches have been developed to identify novel marker genes from scRNA‐seq data in nonplant systems (Butler et al., 2018; Wang et al., 2019; X. Zhang et al., 2019; Boufea et al., 2020); however, none of these approaches have been applied to plant systems. The aim of this work was to develop and compare machine learning (ML)‐based approaches in order to identify new cell marker genes from plant scRNA‐seq data.
ML has been widely applied to solve classification problems in genomics (Libbrecht & Noble, 2015). With regard to scRNA‐seq data, supervised ML algorithms have been used to build cell type classifiers, which have outperformed traditional correlation‐based approaches (Alquicira‐Hernandez et al., 2019; Pliner et al., 2019; A. W. Zhang et al., 2019). However, none of these ML methods addresses the question of selecting marker genes in scRNA‐seq data (see Supporting Information Table S1 for a comparison of 16 state‐of‐the‐art ML methods for single cell type assignment). Feature selection refers to a class of techniques that assign scores to the input features (genes) to indicate how much each feature contributes to the performance of a predictive ML model (Cai et al., 2018). Feature selection is a key component of modern ML methods because it provides interpretability to the ML models (Azodi et al., 2020). For scRNA‐seq analysis, a support vector machine (SVM)‐based recursive feature elimination was used to identify marker genes to differentiate developing neocortical cells from neural progenitor cells (Hu et al., 2016). These novel marker genes not only performed better than traditional gene sets, but also uncovered hidden regulatory networks with novel interactions (Hu et al., 2016). We have also developed a feature selection‐based approach to determine key regulators of transcription regulatory networks with single‐cell data (Song et al., 2020).
In this study, we have integrated five published scRNA‐seq data sets from the Arabidopsis root containing over 25 000 cells and 17 cell clusters (Fig. 1). Using the SPmarker pipeline, we have compared seven ML and conventional methods for the classification of 10 different root cell types in Arabidopsis. We selected the best performing methods, random forest (RF) and SVM, to use for the identification of marker genes. For RF, we used a novel feature selection method called shapley additive explanations (SHAP) method (Lundberg et al., 2020). By comparison, we used the method suggested by Hu et al. (2016) to identify SVM method‐based marker genes (SVMM). The SHAP and SVMM markers were compared with other sets of marker genes, including those that have been published (KNOW), selected using correlation (CORR), from bulk RNA‐seq (BULR) and those used in the index of cell identity model (ICIM). When tested with the two newly published data sets that were not used in the training of the models, the SPmarker method and the SHAP markers successfully assigned cells to respective cell types.
Fig. 1.
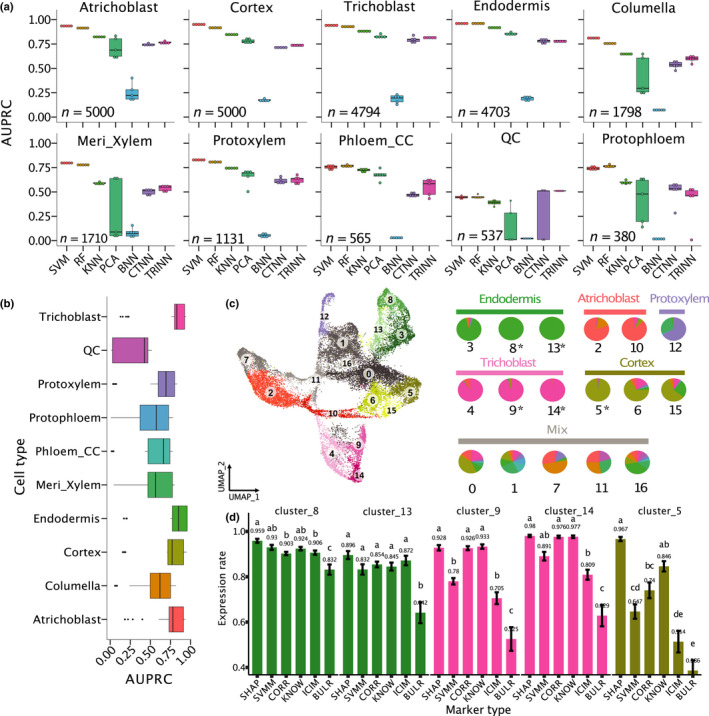
Classification performance of 10 root cell types of Arabidopsis. (a) Comparison between seven machine learning models on cell type classification. AUPRC, area under precision‐recall curve; SVM, support vector machine; RF, random forest; KNN, K‐nearest neighbours; PCA, principal component analysis; and BNN, CTNN and TRINN, baseline, contrastive and triplet neural networks. In these boxplots, the mid‐horizontal line represents the median and dots represent data points. (b) Comparison of classification performance of all the 10 cell types. Dots represent outliers. Colour coding for cell types in (b, c, d) is the same. (c) A UMAP plot where cells were clustered into 17 clusters. If one cell type is represented in more than one cluster, each cluster has a slightly different colour to distinguish the clusters. For example, green is used to represent endodermis, and clusters 3, 8 and 13 in the UMAP are all coloured in green. The right pie plot indicates cell composition in each cluster (Supporting Information Table S5). If over 50% of cells in a cluster belong to the same cell type, this type is defined as the dominant cell type. The labels above the pies are names of the dominant cell types of the clusters. Otherwise, the label for the clusters is ‘Mix’. *, clusters with > 95% of cells belong to the same cell type. (d) Comparisons of proportion of expressed cells among the six marker types. All pairwise comparisons are statistically significant as indicated by different letters. If two bars have the same letter, then they are not significantly different from each other. Error bars represent ±SE.
We further demonstrated the power of ML‐based marker selection is not dependent on any specific cell type assignment approach. For example, we trained SPmarker on cells that were labelled by a WEREWOLF (WER)‐GFP promoter line and identified new WER expressed cells. We also used SPmarker to determine annotations from additional markers that specify cell developmental stages in the root hair and epidermal cell types (Ryu et al., 2019). These new cell types could not be defined using traditional methods such as the ICI approach. We found that the majority of new cell marker genes identified by SPmarker were not identified before. Finally, we found that orthologous genes of SPmarkers showed a significant overlap with single‐cell marker genes found in rice, and in root hairs in five plant species, suggesting our approach can facilitate cell type identification in scRNA‐seq data from diverse plant species.
Materials and Methods
Data preprocessing
The scRNA‐seq data of root cells from five publications were downloaded from the NCBI GEO website (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019; T‐Q. Zhang et al., 2019). For each data set, raw counts were used as input data, and any samples from mutant background or under a treatment were removed. A gene was retained if it was expressed in more than three cells, and each cell was required to have at least 200 but not more than 5000 expressed genes. The cells that have over 5% mitochondrial counts were removed. A global‐scaling normalization method and multicanonical correlation analysis (seurat v.3.1) were used to normalize the expression data and to remove batch effects (Butler et al., 2018). Scrublet tool (Wolock et al., 2019) was used to predict doublet cells in this data set. The normalized expression values in this merged data set (57 333 cells and 25 092 genes) were used for the downstream analysis. In the processing of scRNA‐seq data from a GFP‐tagged line (Ryu et al., 2019), raw reads were mapped to the TAIR10 reference genome using Cell Ranger pipeline (v.2.1.1) with default settings (Zheng et al., 2017) to generate an expression matrix of 17 687 cells.
Cell type annotation and training data preparation
To assign cell types to cells collected from the previous five data sets, index of cell identity (ICI) score was computed (Efroni et al. (2015) for 15 root cell types including trichoblast, cortex, lateral root meristem (LRM), late phloem‐pole pericycle (Late_PPP), protophloem, meristematic xylem (Meri_Xylem), phloem_CC, protoxylem, phloem, pericycle, endodermis, atrichoblast, columella, quiescent centre (QC) and xylem‐pole pericycle (Late_XPP). A cell type with the highest ICI score was assigned to the cell as the cell type label. To compare the performance of our methods by using different ICI thresholds, we set two cut‐offs: ICI > 0.5 and > 0.9. There are 6662 cells with ICI scores higher than 0.9 and only seven cell types that have > 100 cells per cell type were retained for analysis at this ICI threshold. In addition to the ICI method, we also used two other methods to assign cell types: (1) by using reads mapped to an internal GFP marker genes and (2) by using manually identified developmental stage‐related markers from a trajectory analysis (Ryu et al., 2019).
Before the training process, two steps were used to balance the number of cells for each cell type: (1) five cell types with small number of cells (< 300) were removed; and (2) 5000 cells from atrichoblast and cortex were randomly selected to reduce the size of the training sets. Finally, 25 618 cells from 10 cell types were used for our analysis (Table S1). For GFP‐tagged cells, we labelled 955 cells as ‘positive’ because these cells contain reads that mapped to the GFP gene, and other randomly selected 955 cells without GFP reads as ‘negative’ examples. Same analysis was performed using WER‐AT (cells with reads mapped to AT5G14750, which is the ID for the WER gene) to label 1970 cells as ‘positive’ and 1970 cells as ‘negative’ examples. To train the models, the data sets were divided into a training data set of 90% cells and an independent testing data set of 10% cells. The training data sets (90% of cells) were separated into subtraining (80%) and validation (20%) sets for five‐fold cross‐validation. The independent testing data set was used to compare the performance of the ML methods.
Classification methods
ML approaches evaluated in this work include SVM, KNN, RF, baseline NN, triplet NN and contrastive NN methods that were implemented using sklearn v.0.23.1 and Keras v.2.2.4 (Pedregosa et al., 2011; Chollet, 2015). The implementations of neural networks were modified based on a published study (Alavi et al., 2018). Although a few published methods were able to classify cell types, we did not find methods that can provide the flexibility to select marker genes using all ML methods tested in our work (see Table S1 for a discussion of a list of published methods). Therefore, instead of trying to compare with other stand‐alone methods that implemented specialized methods for selecting marker genes, we used a generic ML package (sklearn) such that the methods are more directly comparable. The details for each ML approach are briefly described in Methods S1. Source code for our SPmarker pipeline is available at GitHub (https://github.com/LiLabAtVT/SPMarker).
Results
Comparison between different ML models for cell type classification
The SPmarker pipeline includes two major steps (Fig. S1). In the first step, the expression data of cells from different data sets were normalized and integrated (Fig. S2) using an established approach (Butler et al., 2018). The identities of these cells were assigned by three approaches: (1) using the ICI method (Efroni et al., 2015), (2) an internal GFP marker gene and (3) by manually identified developmental stage‐related markers from a trajectory analysis (Ryu et al., 2019). In the second step, several ML methods were trained and compared to determine the methods that best predict cell types.
To test different ML methods, we first used the ICI method to label the cell type for each cell (Efroni et al., 2015). The ICI score (0 ≤ score ≤ 1) of each cell represents a similarity of each cell to one of the 15 known cell types in Arabidopsis root, and only 56% of all cells were able to be assigned by ICI (ICI > 0.5) to a single‐cell type. These 56% of cells were used for comparing the performance of seven ML methods (Fig. 1). These methods were selected because they represent approaches where each is based on distinct underlying mechanisms (Fig. S1C). The area under precision‐recall curve (AUPRC) values are shown for all the methods (Fig. 1a), while other evaluation metrics were also calculated and compared between methods (Figs S3, S4). The SVM and RF had highest AUPRC among the seven models (Fig. 1a). For the deep learning‐based methods, the contrast NN and triplet NN had similar performance but had higher AUPRC than the baseline NN. In general, non‐neural network models showed relatively higher AUPRC than the NN‐based models. This is not unexpected, and further optimization of hyperparameters for NN models might improve the performance of NN‐based approaches. Performance comparisons were also obtained using seven other metrics (Figs S3–S5). Regardless of the evaluation metrics used, the performances of SVM and RF are better than other methods tested. Interestingly, the ML models performed better for some cell types (e.g. trichoblast and atrichoblast) than other cell types (QC cells, Fig. 1b). This is not entirely due to the low number of cells in QC (537 cells, see Table S2) because other cell types such as phloem companion cell (phloem_CC) and protophloem had similar or fewer cells (565 and 380 cells, respectively) as compared to QC, but the ML model performances on these cells are higher than QC (Fig. 1b). Due to their better performances, RF and SVM were used for downstream analyses to select marker genes using feature selection.
Next, we compared the marker genes identified by ML with marker genes identified by other methods by comparing their expressions in different cell clusters. To make these genes comparable, we selected only the top 20 markers for each cell type (200 markers for all cell types) for SHAP, SVMM and CORR, respectively. We also selected 180 BULR, 161 KNOW and 232 ICIM markers. We cannot select exactly 200 markers for these published types of markers because they were predetermined by previous publications (see Tables S3, S4 for the list of these marker genes). From the 17 clusters from the integrated single‐cell data set, we focused on five clusters (5, 8, 9, 13 and 14) with a dominant cell type that accounts for over 90% cells in each cluster (Fig. 1c, see Fig. S6 for a comparison of all cell clusters). These clusters were selected because they are the most homogenous clusters, making the results easier to interpret. For each marker gene in each cluster, we calculated the proportion of cells in which this marker gene is expressed, and we calculated the average ‘fraction of expressed cells’ for all 20 markers in each cluster. For example, given the 20 SHAP markers for cluster 8, on average each SHAP marker was detected in 95.9% of cells (Fig. 1d, Cluster 8, Endodermis). In these homogenous cell clusters, the SHAP markers achieved a higher or similar proportion of the expressed cells as compared to all the other markers. In cluster 5 that represents the cortex, the SHAP markers had significantly (P < 0.05) higher expression rate (96.7%) than all other marker types (< 85%) (Fig. 1d). The BULR markers were detected in the lowest number of cells and followed by the ICIM markers. Because the ICI method does not require all ICIM to be detected in a given cell, it is not surprising that ICIM was only found in 50% of cells in some clusters. On average, the percentage of cells expressing the SHAP markers was 29% more than the ICIM markers and 67% more than the BULR markers. Because both BULR and ICIM were not determined using the data from scRNA‐seq experiments, these results might suggest BULR and ICIM include cell type‐specific, but relatively low‐expressed genes that cannot be detected by scRNA‐seq.
Using newly developed markers to assign cell identity
Because only 20 marker genes were selected for each marker type per cell type, we compared the expression patterns for these six types of markers using heat maps (Figs S7–S12). Markers identified by SHAP, CORR and ICIM had stronger cell type‐specific expressions than those by KNOW, SVMM and BULR. To further quantify the specificity of the SHAP markers and other marker types, we calculated their cumulative correlation with specific cell types in the atrichoblast, trichoblast and endodermis (Fig. 2a–c). These three cell types were selected because all of these cell types have a higher number of cells than other cell types, and these cell types are consistently identified by the majority of marker types. The cumulative correlation rates for the SHAP markers were among the top three in all cell types, suggesting stronger preferential expression for this marker type. The SHAP markers had similar performance as compared to the CORR markers in atrichoblast and had higher performance than the CORR markers in two other cell types. The ICIM markers were among the top three in atrichoblast and endodermis but ranked fourth in trichoblast. This is consistent with the observation that not all the ICIM markers were detected in all cells.
Fig. 2.
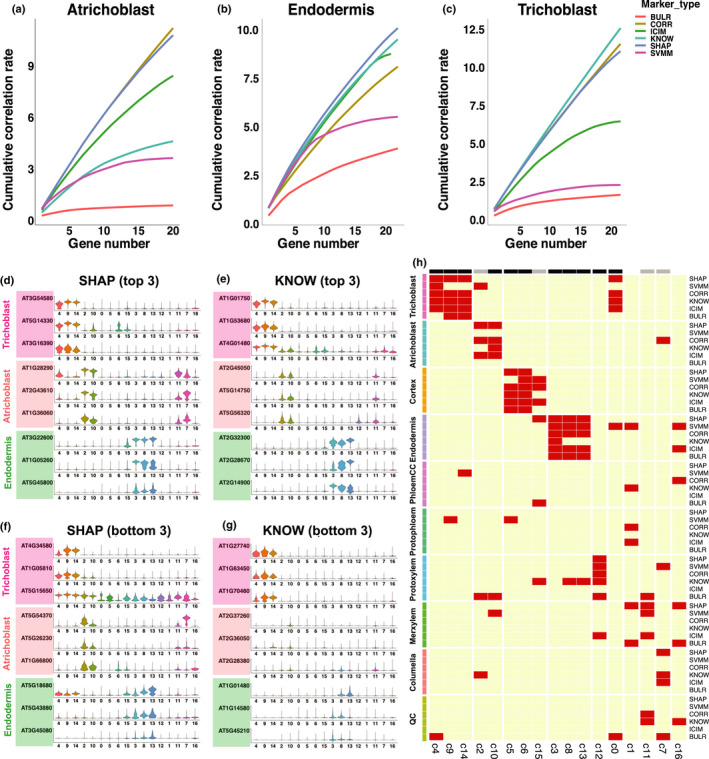
Assignments of cell identity using newly developed Arabidopsis markers. (a–c) Cumulative correlation plot for top 20 markers for six types of markers. (d–g) Violin plots that show the expression of top three markers (d, e) and bottom three markers (f, g) in three cell types, across all clusters. Only distributions of the marker gene expression are shown in (d–g). (h) Heat map of cell types assigned to each cluster by different markers. On top of the heat map, black bars show clusters assigned consistently by four or more methods, and grey bars show clusters assigned consistently by three out of six methods.
To demonstrate the specificity for the SHAP markers, we plotted the top three most specific markers from the 20 selected SHAP and KNOW markers (Fig. 2d,e). We also plotted the expression of the bottom 3 markers (ranked 18, 19 and 20 by marker specificity) from the 20 markers (Fig. 2f,g). We found that the SHAP markers showed high cell type specificity in both cases, whereas the specificity of the KNOW markers was lower in at least seven cases for those ranked at 18–20. One interesting observation is that most of the SVMM markers were highly expressed in multiple clusters (Fig. S13), suggesting SVM provides a different approach to detect cell types.
One potential limitation of the ML model selected markers is that other model parameters such as the decision thresholds for RF and feature weights for SVM associated with each group of markers has to be evaluated using model‐dependent algorithms. Because of the high correlation of the SHAP markers with specific cell types, we developed a voting procedure to simplify the process of assigning cell identities using the newly developed markers and the existing marker genes (Fig. 2h). Applying this method to the 17 clusters, we found 15 clusters were assigned consistently to the same cell types by three or more marker types (black and grey colour marked the clusters in Fig. 2h). These results show that cluster assignments are largely consistent between the existing markers and the new marker genes.
Identification of marker genes with different training labels
The SPmarker method is more flexible than traditional approaches because our method can be trained on different cell labels and select different sets of marker genes to classify cell types. To demonstrate this, we tested three additional scenarios: (1) label cells with a different ICI threshold; (2) label cells in the same lineage under different developmental stages; and (3) label cells with an internal GFP marker.
We first compared the performance of SPmarker with other conventional approaches using two different ICI thresholds, 0.5 and 0.9 (Fig. 3a,b). Marker genes were selected by RF and SVM models, and only the top 20 marker genes were used for the analysis to match the number of genes in ICIM and other marker types (CORR, KNOW and BULR). Random forest models were trained using these marker genes separately, and performances were compared using AUPRC. We have only five cell types with enough cells or enough marker genes from all methods for comparison with ICI > 0.9 (see Fig. S14 for all five cell types). ICIM performed best in both thresholds in five cell types tested, which is expected because the ICIM was used to label cells. Interestingly, we found that the performance of the SHAP and SVMM markers increased significantly for prediction cells with ICI > 0.9 as compared to cells with ICI > 0.5. These results may suggest that ICI > 0.9 cells are more specific as compared to cells with ICI > 0.5 and they are easier to be classified using different sets of markers. By contrast, the CORR, KNOW and BULR markers did not show an improvement in performance, partly because these marker genes were determined not based on cells labelled by training samples, thus are less flexible than marker genes determined by SPmarker.
Fig. 3.
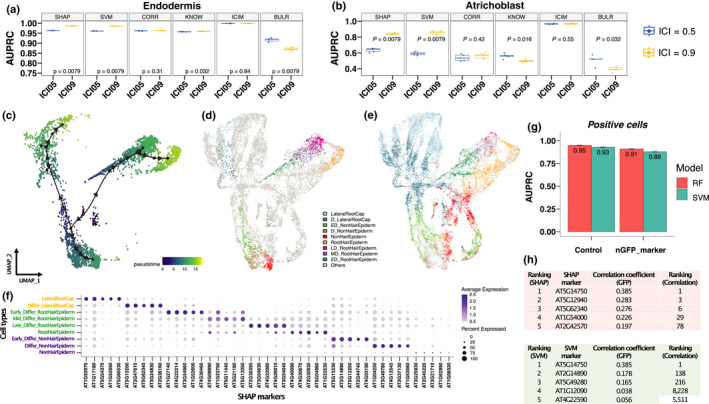
Identification of marker genes with three different ways to label cells in Arabidopsis root. (a, b) Comparison of classification performance based on index of cell identity (ICI) labelling method between 0.5 and 0.9 thresholds in (a) endodermis and (b) atrichoblast cells. P value < 0.05 indicates significant differences between ICI05 and ICI09 groups. (c–f) Label cells under different developmental stages. (c) Trajectory analysis of cells from root hair, nonroot hair and lateral root caps from our published single‐cell data. (d) A UMAP (Uniform Manifold Approximation and Projection) of nine developmental stages derived from these three cell types. The label ‘Others’ indicates cells from other publications including four studies with trichoblast and atrichoblast cells and with WEREWOLF (WER) cells generated from a WER‐promoter‐GFP line (Ryu et al., 2019). D, fully differentiated; ED, early differentiated; MD, middle differentiated; LD, late differentiated. (e) A UMAP shows predictions of cell identities from the other publications. The label ‘Others’ indicates cells from Ryu et al.’s study (2019). (f) Identification of the Shapley additive explanations (SHAP) markers in the nine developmental stages. (g–h) Identification of markers with labelling cells using internal green fluorescent protein (GFP) marker. (g) Comparison of classification performance on GFP‐labelled WER cells (positive cells) between using all genes (control) and genes without GFP marker (nGFP_marker) for both random forest (RF) and support vector machine (SVM) models. Error bars represent ±SE. (h) Ranking of best SHAP and SVMM markers to predict WER‐GFP‐positive cells. The third column indicates expression correlation between GFP and other genes. The fourth column indicates ranking based on correlation values.
We next tested whether we can use ML to transfer labels from one experiment to another (Fig. 3c–f). First, we used our published single‐cell data (Ryu et al., 2019) and selected cells from root hair, nonroot hair and lateral root caps. These cell types were selected because they are located at the outmost layer of roots and represent distinct cellular functions. These cells were further classified into nine different developmental stages based on a trajectory analysis (Ryu et al., 2019). SPmarker was trained on these data to select the SHAP and SVMM markers, and predictions were made on the other four data sets in the integrated root cell data set (Fig. 1c). In the UMAP plots, we found that cell types from Ryu et al. (2019) follow three separate trajectories that corresponded to the three selected cell types (Fig. 3c). Most importantly, the labelled cells (Fig. 3d) were overlapped strongly with the cells from other publications and form similar trajectories. For example, from the Ryu et al. (2019) data (Fig. 3c,d), we found that the ‘differentiating lateral root cap’ was located close to the centre of the UMAP (dark blue), whereas the mature lateral root cap cells were located towards the outskirt of the UMAP plot (light blue). Cells from other four publications showed similar distribution (Fig. 3e, dark and light blue cells). This is also observed for the root hair lineages (dark green, purple and dark pink cells), and for the nonhair lineages (light green, brown and red cells). We also identified new marker genes from these cell types at different developmental stages, which showed stage‐specific expression patterns (Fig. 3f). These results show that SPmarker can identify marker genes with fine‐grained resolution for cell types at different developmental stages.
Interestingly, some cells were not classified to the same type using RF or SVM (Fig. S15). In these cells, RF predicted these cells as nonhair epidermal, whereas SVM predicted these cells as lateral root caps. Upon further investigation, we found that these cells expressed the marker gene WER (Lee & Schiefelbein, 1999), which was highly expressed in root cap, and moderately expressed in nonhair cells. The prediction actually reflects the biological similarity of these cells and intrinsic cell identities of these cells. Following this observation, we tested our method using an internal control gene to label cells (Fig. 3g,h). In our published scRNA‐seq data (Ryu et al., 2019), we used a WER‐promoter‐GFP line to generate scRNA‐seq data. Therefore, in the scRNA‐seq data, we were able to identify reads that mapped to both GFP (WER‐GFP) and the WER (WER‐AT, AT5G14750) genes. We labelled cells using reads mapped to WER‐GFP first and trained the model to predict WER‐GFP‐positive cells. WER‐GFP was removed from the training data such that this gene will not be used as a marker. The same analysis was performed using the WER‐AT gene. As expected, when we labelled cells by WER‐GFP and selected marker genes using either RF or SVM, the best markers to predict WER‐GFP‐positive cells were WER‐AT genes in both methods (Fig. 3h). More interestingly, when we removed both WER‐GFP and WER‐AT from the gene expression matrix, we can still predict WER‐positive and WER‐negative cells with high AUPRC (Fig. 3g, nGFP_marker), suggesting that other marker genes can also provide predictions to the WER‐positive cells. We further compared the correlation between the SHAP and SVMM markers to the expression of the internal WER‐GFP tag. We found that the correlation was low (r = 0.385), and most importantly, the top ranked genes by SHAP or SVMM were not top ranked genes by correlation. When we labelled with WER‐AT instead of WER‐GFP, we observed similar results but obtained a different list of high‐ranking genes (Fig. S16). Since WER‐labelled cells were not included in the original ICI cell types, these results demonstrate that our ML method is applicable in identifying new cell types with alternative methods of cell labelling.
Most ML‐derived markers are new markers
Because SHAP and SVMM are very different from correlation markers in the WER‐GFP analysis, we sought to understand how many new markers can be identified in other cell types. We identified the SHAP marker genes that were unique to each cell type using ICI > 0.5. We found 1840 and 1460 unique marker genes for cortex and atrichoblast, respectively, while 63 and 37 unique marker genes were found for the QC and protophloem (Fig. 5a). In other words, there were almost 30 times more unique SHAP marker genes in cortex as compared to QC cells. By overlapping the cumulative SHAP values from genes with the unique SHAP marker, we found fewer than 50 unique genes accounted for 50% of the total SHAP values in each cell type (Fig. S17). This suggested that QC had a small number (Fig. 5a, 63 genes) of the unique SHAP markers and more markers (Fig. S17, 475 genes) were shared with other cell types, whereas cortex or atrichoblast had large numbers of the unique SHAP markers, but only a fraction of these markers carried the most weight.
Fig. 5.
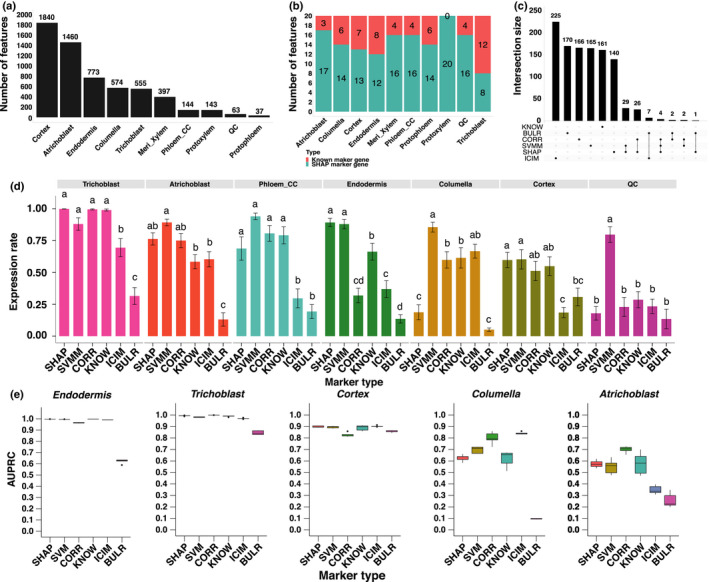
Testing of new markers with independently labelled cell types in Arabidopsis. (a) Number of Shapley additive explanations (SHAP) markers identified in each cell type. (b) Comparison of number of SHAP and known markers in the 20 genes with the highest SHAP value in each cell type. (c) Summary of gene counts from six marker types. Set size means gene count of different marker types. The dots under the bars mean the genes specifically exist in the relative marker type. The line connected between two or more dots under the bars mean genes exist in two or more marker types. If two or more marker types do not have a connection, it means these groups do not have shared genes. (d) Comparisons of proportion of expressed cells between the six marker types. All pairwise comparisons are statistically significant as indicated by different letters. If two bars have the same letter, then they are not significantly different from each other. Error bars represent ±SE. (e) Comparison between six marker types on cell type classification. The mid‐horizontal line represents the median, and dots represent outliers.
To study the identity of the SHAP marker genes, we focused on the top 20 genes in each cell type with the highest SHAP value. There were 146 genes out of 200 SHAP marker genes (73%) that were not identified before in a collection of 1813 marker genes from other publications (Table S6). In particular, in the protoxylem, all SHAP markers are new (Fig. 5b) and 80% of the SHAP markers from the atrichoblast and phloem_CC are new markers (Fig. S18). The same results were observed for SVMM, where the majority of new markers were specifically found by the SVM method but not by other methods (Fig. S19). Interestingly, there was little overlap between these marker genes such that 93.5% or 1027 marker genes were unique to a single method. When we compared the top 200 marker genes identified by six different methods, we found that most markers were found by a single method, whereas only 71 markers were found by two methods (Fig. 5c) and there was no single marker ranked as top 20 by more than three methods.
We also found unique biological functions for these newly identified marker genes (Fig. 4; Tables S7–S9). Among the three new marker types, we are most interested in the function of the SHAP markers and we studied the gene ontology (GO) annotation of the SHAP markers. These annotations were compared to the KNOW and ICIM markers which represent the majorities of published markers (Table S6). Nearly one third (30.5%; 61/200) of the SHAP markers were involved in the responsiveness of stimulus in the biological process. By contrast, nearly half of the terms (46.2%; 18/39) associated with the KNOW markers were related to root vegetative and reproduction development (Fig. 4a,c). In the ICIM markers, only two specific GO terms were identified. One possible reason that the KNOW markers were enriched in root developmental processes is that these KNOW marker genes might have been used to define GO annotations. The GO enrichment analysis also showed 19 SHAP markers were mainly enriched in water transport (GO:0006833), response to water (GO:0009415) and to water deprivation (GO:0009414) processes and participate in water transmembrane transporter (GO:0005372) and water channel activity (GO:0015250) (Fig. 4d; Table S7). Among these markers, more than half of them (11/19) were under cortex, endodermis, protoxylem and meri_xylem cells (Table S7). These four cell types are essential for water transportation and minerals assimilation (Steudle & Peterson, 1998; Qiao & Libault, 2013). Eighteen SHAP marker genes were also enriched in the cell wall biosynthesis (Fig. 4d). One third (6/18) of these SHAP markers were found for the protoxylem and meri_xylem cell types (Table S7) that are known to be important for cell wall formation (Oda & Fukuda, 2012). In summary, the SHAP markers are enriched with environmental response functions, especially water responsiveness and cell wall formation, which suggests that the SHAP marker genes can not only serve as cell identity markers but also may play important cell‐specific biological functions in roots.
Fig. 4.
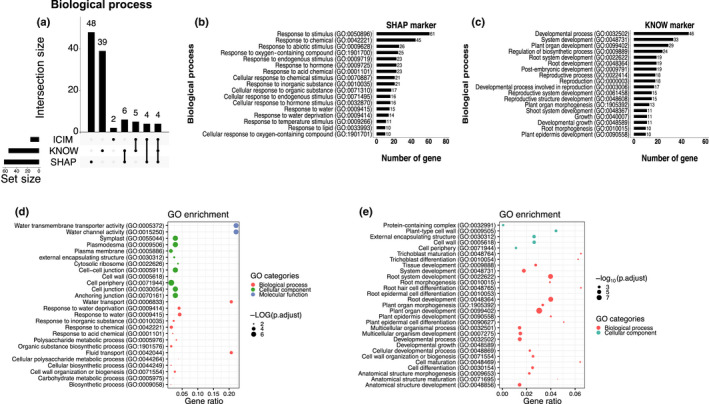
Biological function comparison between the Shapley additive explanations (SHAP) markers and the KNOW markers in Arabidopsis. (a) Number of unique gene ontology (GO) and biological processes identified in marker genes. The dots under the bars indicate the GO categories specifically exist in the relative marker type. The line connected between two or more dots under the bars mean GO categories exist in two or more marker types. If two or more marker types do not have connection, it means these groups do not have shared GO categories. (b, c) The number of markers annotated in the specific GO terms for the SHAP and KNOW markers. (d, e) The GO enrichment tests for the SHAP and KNOW markers.
To validate these newly identified marker genes, we searched literature for published experimental evidence for cell type specificity of these genes. We have identified 11 cases of published wet‐bench data supporting our new markers, and all the cases were not found by traditional methods for single‐cell data analysis (Table S10; Methods S1). More importantly, these 11 cases were generated by independent publications and thus are unbiased validation of our newly identified markers.
Testing new marker genes with independently labelled cell types and new single‐cell data
When we first implemented SPMarker, only five single‐cell data sets were available from Arabidopsis roots. To test whether SPmarker can predict cell types in data that were not used in training, we evaluated the model performance using two newly generated single‐cell data sets for Arabidopsis roots (Figs 5d,e, S20). In both data sets, different approaches were used to label cell clusters. In one paper, the top variable genes from each single‐cell cluster were selected and correlated with published bulk‐RNA‐seq data (Wendrich et al., 2020). In the other paper, three different methods were used to assign cell types and the consensus of three methods was to assign cell types (Shahan et al., 2022). To evaluate the new marker genes identified in our approach, we calculated how many cells in each cell type where these marker genes expressed (Figs 5d, S20). For example, for cells identified as trichoblast in one paper (Wendrich et al., 2020), > 99% of cells also expressed marker genes from SHAP, CORR and KNOW categories. About 88.1% of cells expressed the SVMM markers, and fewer than 70% of cells expressed the ICIM or BULR markers. This result shows that if only ICIM were used to assign cell types, > 30% of cells might not be assigned due to the lack of marker genes. This also shows that the SHAP, CORR and KNOW markers, and to a lesser extent, SVMM, work well to define trichoblast cells determined in this publication. Not all ML‐based markers performed well; for example, the SHAP markers have low expression rate in columella and QC cells (Fig. 5d). Interestingly, the BULR markers showed lower performance in all cell types. One possible reason is that the BULR markers are lowly expressed and cannot be detected in single‐cell data, but they are highly specific to individual cell types. These results are also observed when evaluating these marker genes in Shahan et al. (2022) (Fig. S20).
Finally, we tested the performance of different marker genes when we used them with a RF model to predict cell types (Fig. 5e). These models were trained using an integrated data set (Fig. 1c) that did not include the new data, thus serving as a completely independent validation. We found that AUPRC was close to 1.0 for five marker types in endodermis and trichoblast, and close to 0.9 for cortex, representing high performance of the model to transfer annotation to a different data set. By contrast, AUPRC for all types of markers was more variable and lower for columella and atrichoblast, suggesting additional methods would be needed to determine the best way to assign cell types in these cases. CORR had better performances in two cell types where the overall AUPRC was lower than 0.9, suggesting when cells are harder to classify, CORR might be better markers to use.
Newly identified marker genes can be used to assign cell types in other species
As scRNA‐seq experiments expand to other plant species, it becomes challenging to accurately determine cell types, because some marker genes in Arabidopsis may have altered their functions in other species or may be absent from genomes of other species. A major usage of new marker genes is to expand the list of candidate marker genes in other species. To this end, we compared the marker genes identified from a single‐cell sequencing data from rice roots (Fig. 6a; Table 1) (Liu et al., 2021). There are three clusters that have the same cell type names in both the Arabidopsis and rice data; therefore, we can only compare marker genes in these three clusters. We found very small number of Arabidopsis marker genes whose orthologous genes were also found in the corresponding cell types in rice. For example, for cortex, there are no marker genes from the ICIM and BULR that were also found in rice cortex cluster (Table 1). There are only 10 KNOW markers that were also found in rice cortex cluster, and such overlapping is not statistically significant. Although the ICIM and KNOW marker showed a significant overlapping in endodermis and trichoblast, respectively, the absolute number of overlapping markers is small. These results are consistent with the observations from the rice paper in that there is a limited number of marker genes in Arabidopsis that are also marker genes in rice (Liu et al., 2021). When we compared the SHAP and SVMM markers, we found a substantial increase of overlapping markers and such overlapping are statistically significant in five out of six cases (Table 1). By contrast, the overlapping of the CORR markers is only significant for trichoblast (P < 0.01) but not for other two cell types. When we analysed how many markers were also detected in the three cell types (Fig. 6a), we found c. 60% of cortex and endodermis cells and > 75% of trichoblast cells from rice also had orthologous genes of SHAP or SVMM markers. The CORR markers showed lower detection rates than one or both markers in the two cell types. The ICIM and KNOW markers were missing from one cell type and showed similar or lower detection rates than the SHAP and SVMM markers.
Fig. 6.
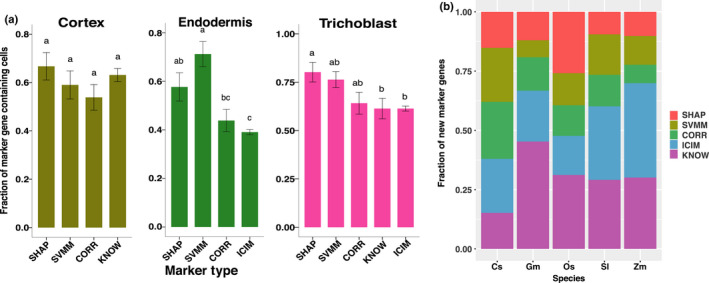
Testing of newly identified markers in other species. (a) Comparisons of proportion of expressed cells between the four marker types in a published rice scRNA‐seq data (Liu et al., 2021). All pairwise comparisons are statistically significant as indicated by different letters. If two bars have the same letter, then they are not significantly different from each other. Error bars represent ±SE. (b) Number of root hair marker genes identified using five marker types in five species including Cucumis sativus (Cs), Glycine max (Gm), Oryza sativa (Os), Solanum lycopersicum (Sl) and Zea mays (Zm). SHAP, Shapley additive explanations; SVMM, SVM method‐based marker genes; ICIM, index of cell identity model.
Table 1.
Comparison of number of overlapped rice markers in three cell types between six marker types.
| Marker type | Cell type | Arabidopsis marker number | Rice marker number | Overlap marker number | Overlap fraction | Binomial test P‐value |
|---|---|---|---|---|---|---|
| SHAP | Cortex | 2118 | 674 | 93 | 0.138 | 5.74E−11 |
| SVMM | Cortex | 1286 | 674 | 54 | 0.080 | 4.91E−06 |
| CORR | Cortex | 894 | 674 | 28 | 0.041 | 0.0320 |
| ICIM | Cortex | 42 | 674 | 0 | 0.000 | 1 |
| KNOW | Cortex | 304 | 674 | 10 | 0.014 | 0.1285 |
| BULR | Cortex | 62 | 674 | 0 | 0.000 | 1 |
| SHAP | Endodermis | 696 | 1295 | 25 | 0.019 | 0.7292 |
| SVMM | Endodermis | 479 | 1295 | 41 | 0.031 | 7.75E−06 |
| CORR | Endodermis | 4538 | 1295 | 107 | 0.082 | 1 |
| ICIM | Endodermis | 25 | 1295 | 6 | 0.004 | 0.0008 |
| KNOW | Endodermis | 66 | 1295 | 1 | 0.000 | 0.9205 |
| BULR | Endodermis | 81 | 1295 | 1 | 0.000 | 0.9664 |
| SHAP | Trichoblast | 511 | 1387 | 46 | 0.033 | 3.40E−12 |
| SVMM | Trichoblast | 1231 | 1387 | 58 | 0.041 | 2.64E−05 |
| CORR | Trichoblast | 4237 | 1387 | 237 | 0.170 | 5.43E−28 |
| ICIM | Trichoblast | 39 | 1387 | 3 | 0.002 | 0.1056 |
| KNOW | Trichoblast | 610 | 1387 | 29 | 0.020 | 0.0025 |
| BULR | Trichoblast | 29 | 1387 | 1 | 0.0007 | 0.5035 |
Arabidopsis marker number is the number of orthologous genes of Arabidopsis markers in the rice genome according to phytozome annotation. Overlap ratio is calculated as overlap marker number divided by rice marker number. Binomial test P‐values were calculated using 100 random draws of the same number of marker genes and compared these random overlap ratios with the observed overlap ratio. SHAP, Shapley additive explanations; SVMM, SVM method‐based marker genes; CORR, correlation; ICIM, index of cell identity model; BULR, bulk RNA‐seq.
Because we found a substantial increase in marker genes in the comparison between Arabidopsis and rice data, we asked whether such comparison could be expanded to other species. To address this question, we analysed root hair cell expression from five plant species including cucumber, soybean, rice, tomato and maize (Huang et al., 2017). To consider the most specific markers, we tested the top 20 markers from each marker type from single‐cell data. In these five species, we found a total of 172 genes were significantly differentially expressed in root hair cells that were also orthologous genes to the six types of marker genes (Fig. 6b). The SHAP and SVMM markers accounted for 26.1–60.7% of these root hair genes, and these new marker genes increased the number of candidate marker genes by 35.3–154.5% in these five species (Fig. 6b).
Discussion
The scRNA‐seq technology provides a novel platform to analyse the transcriptomic profile of individual cells to characterize heterogeneous cell populations. In plants, this process has been heavily reliant on the use of marker genes that are preferentially expressed in specific cell types (Ryu et al., 2021). Here, we introduce a ML‐based approach, SPmarker, to identify marker genes by analysing their feature importance. ML methods provide a number of principled approaches to evaluate marker performance including cross‐validation, leave‐out testing sets and evaluation metrics such as auROC (area under the receiver operating characteristic), auPRC (area under the precision‐recall curve) and F1 scores. These evaluation methods allow us to compare different marker genes in a more rigorous and unbiased fashion. In addition, the seurat package is a standard method that needs prior knowledge of cell clustering to identify marker genes. We have compared marker genes identified by ML methods with those identified by the FindAllMarkers function (with default parameters). We have found there is no statistically significant difference in the expression rate for the SHAP markers as compared to Seurat‐identified markers (Fig. S21). More importantly, the majority of the SHAP and SVMM markers are different from the Seurat markers (Fig. S22). We have shown that these new markers can yield high performance using ML‐based evaluation metrics. More importantly, because these machine learning derived markers are not based on the prior knowledge of gene functions, these markers may have new biological functions that have not been characterized before.
In the evaluation of different ML methods, the SVM and RF methods outperformed the three deep learning models (Fig. 2). One possible reason is SVM and RF are effective for relatively small data sets or fewer outliers (Ben‐Hur & Weston, 2010; Ali et al., 2012). The deep learning algorithms (Fig. S23) usually require a relatively large data set to work well and achieve good performance for solving more complex problems (Zou et al., 2019). A previous study utilized the contrastive NN and triplet NN to successfully classify cells in mouse by using > 100 000 cells to train these two models (Alavi et al., 2018), while our study used < 30 000 cells. If more cells with accurate cell identity were available in Arabidopsis, the performance of the deep learning model in our study may be improved (Eraslan et al., 2019). Based on the computational evaluation, SHAP and SVM performed consistently better than other methods. However, based on heat map analysis (Figs S7–S12), correlation analysis (Fig. 2a–c) and literature search (Table S10), SHAP and CORR markers have better correlation with expression patterns and literature support. One possible reason is that SVM is searching for ‘support vectors’, which are cases that separate different clusters of data, and thus, genes considered important by SVM are closer to the boundaries of cell types.
Cell populations of Arabidopsis roots are characterized by a high level of heterogeneity. Results from animal systems have demonstrated that, even within a cell population, the cells are not homogeneous because subpopulations may exist (Liu & Trapnell, 2016). Furthermore, it is not clear whether all cell types have been discovered for the Arabidopsis root (T‐Q. Zhang et al., 2019), in particular, for cells in a transition stage or regulated by periodical signals (Voß et al., 2015). This highlights the importance of identifying new marker genes, which may be expressed at different levels in subpopulations as compared to traditional marker genes.
The identification of new marker genes is particularly important for the plant biology research community because cell type markers are largely unknown from nonmodel species. We have demonstrated that our ML‐based approaches can substantially expand the number of known root hair marker genes and that orthologs of these marker genes can also be found in other plant species. One future direction is to define root cell types in nonmodel species from cross‐species mapping of marker genes and their expression pattern in roots.
Author contributions
HY, JL, QS and SL designed the experiments and performed the computational analysis. BZ and QL contributed to experimental validation of candidate genes. HY, JS and SL wrote the manuscript.
Supporting information
Fig. S1 Summary of the data processing pipeline and machine learning methods used.
Fig. S2 Integration of five datasets.
Fig. S3 Classification performance (AUROC) of 10 root cell types of Arabidopsis.
Fig. S4 Performance comparison among seven machine learning methods on six different evaluation scores.
Fig. S5 Performance comparisons among five cross validation random forest models.
Fig. S6 Comparisons of the proportion of expressed cells for different marker genes.
Fig. S7 Heat map of top 20 SHAP markers.
Fig. S8 Heat map of top 20 SVMM markers.
Fig. S9 Heat map of top 20 CORR markers.
Fig. S10 Heat map of all ICIM markers.
Fig. S11 Heat map of top 20 KNOW markers.
Fig. S12 Heat map of top 20 BULR markers.
Fig. S13 Violin map of top 20 SVMM markers of endodermis cell type.
Fig. S14 Comparison of classification performance based on ICI 0.5 and 0.9 thresholds in five cell types.
Fig. S15 A UMAP for SVM predicted cell types.
Fig. S16 Classification and predicted top markers.
Fig. S17 Cumulative SHAP values for all SHAP markers for each of the 10 cell types.
Fig. S18 Comparison of proportion of cumulative SHAP values.
Fig. S19 Summary of the SVMM markers.
Fig. S20 Expression rate of SHAP marker genes in Shahan et al. (2022).
Fig. S21 Expression rate comparison for five clusters between different marker types.
Fig. S22 Number of marker genes uniquely and commonly identified by different methods.
Fig. S23 Schematic demonstration of architecture for each type of neural network.
Methods S1 Additional materials and methods for this study.
Table S1 A summary of automatic cell classification and feature selection methods for scRNA‐seq data.
Table S2 The distribution of 25 618 cells from 10 cell types used to train models.
Table S3 The 1532 KNOW markers.
Table S4 The markers used in comparisons of expression patterns across clusters and cell‐type classifications.
Table S5 Assignment of cell type to cluster.
Table S6 The 1813 known marker genes, including those genes in Table S3 and other published markers.
Table S7 GO annotation of SHAP markers.
Table S8 Specific GO terms for ICIM, KNOW, and SHAP marker types.
Table S9 The cell types of SHAP marker genes enriched in GO terms.
Table S10 Additional wet‐bench experimental support for newly identified markers.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We would like to acknowledge Jeffress Trust Awards Program in Interdisciplinary Research (to JL and SL); United States Department of Energy Funding (DE‐SC0020358) to Sl and JS; and Hatch Programs from United States Department of Agriculture (to SL).
Data availability
These data were derived from the GEO and SRA repositories at NCBI with the following IDs: GSE123013, GSE121619, GSE122687 and GSE123818, and PRJNA517021.
References
- Alavi A, Ruffalo M, Parvangada A, Huang Z, Bar‐Joseph Z. 2018. A web server for comparative analysis of single‐cell RNA‐seq data. Nature Communications 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali J, Khan R, Ahmad N, Maqsood I. 2012. Random forests and decision trees. International Journal of Computer Science Issues 9: 272. [Google Scholar]
- Alquicira‐Hernandez J, Sathe A, Ji HP, Nguyen Q, Powell JE. 2019. scPred: accurate supervised method for cell‐type classification from single‐cell RNA‐seq data. Genome Biology 20: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azodi CB, Tang J, Shiu SH. 2020. Opening the black box: interpretable machine learning for geneticists. Trends in Genetics 36: 442–455. [DOI] [PubMed] [Google Scholar]
- Ben‐Hur A, Weston J. 2010. A user's guide to support vector machines. Methods in Molecular Biology 609: 223–239. [DOI] [PubMed] [Google Scholar]
- Bezrutczyk M, Zollner NR, Kruse CPS, Hartwig T, Lautwein T, Kohrer K, Frommer WB, Kim JY. 2021. Evidence for phloem loading via the abaxial bundle sheath cells in maize leaves. Plant Cell 33: 531–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum K, Jung JW, Wang JY, Lambert GM, Hirst JA, Galbraith DW, Benfey PN. 2005. Cell type–specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nature Methods 2: 615–619. [DOI] [PubMed] [Google Scholar]
- Boufea K, Seth S, Batada NN. 2020. scID uses discriminant analysis to identify transcriptionally equivalent cell types across single‐cell RNA‐seq data with batch effect. iScience 23: 100914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Orlando DA, Lee J‐Y, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN. 2007. A high‐resolution root spatiotemporal map reveals dominant expression patterns. Science 318: 801–806. [DOI] [PubMed] [Google Scholar]
- Bruex A, Kainkaryam RM, Wieckowski Y, Kang YH, Bernhardt C, Xia Y, Zheng X, Wang JY, Lee MM, Benfey P et al. 2012. A gene regulatory network for root epidermis cell differentiation in Arabidopsis. PLoS Genetics 8: e1002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. 2018. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Luo J, Wang S, Yang S. 2018. Feature selection in machine learning: a new perspective. Neurocomputing 300: 70–79. [Google Scholar]
- Chollet F. 2015. Keras: Deep learning library for theano and tensorflow. [WWW document] URL https://keras.io/about/ [accessed 1 December 2021]. [Google Scholar]
- Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, Timmermans MC. 2019. Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high‐throughput single‐cell RNA sequencing. Developmental Cell 48: 840–852.e5. [DOI] [PubMed] [Google Scholar]
- Efroni I, Ip P‐L, Nawy T, Mello A, Birnbaum KD. 2015. Quantification of cell identity from single‐cell gene expression profiles. Genome Biology 16: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G, Avsec Ž, Gagneur J, Theis FJ. 2019. Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics 20: 389–403. [DOI] [PubMed] [Google Scholar]
- Hu Y, Hase T, Li HP, Prabhakar S, Kitano H, Ng SK, Ghosh S, Wee LJK. 2016. A machine learning approach for the identification of key markers involved in brain development from single‐cell transcriptomic data. BMC Genomics 17: 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Shi X, Wang W, Ryu KH, Schiefelbein J. 2017. Diversification of root hair development genes in vascular plants. Plant Physiology 174: 1697–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean‐Baptiste K, McFaline‐Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C, Cuperus JT. 2019. Dynamics of gene expression in single root cells of Arabidopsis thaliana . Plant Cell 31: 993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MM, Schiefelbein J. 1999. WEREWOLF, a MYB‐related protein in Arabidopsis, is a position‐dependent regulator of epidermal cell patterning. Cell 99: 473–483. [DOI] [PubMed] [Google Scholar]
- Li S, Yamada M, Han X, Ohler U, Benfey PN. 2016. High‐resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Developmental Cell 39: 508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbrecht MW, Noble WS. 2015. Machine learning applications in genetics and genomics. Nature Reviews Genetics 16: 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Liang Z, Feng D, Jiang S, Wang Y, Du Z, Li R, Hu G, Zhang P, Ma Y et al. 2021. Transcriptional landscape of rice roots at the single‐cell resolution. Molecular Plant 14: 384–394. [DOI] [PubMed] [Google Scholar]
- Liu S, Trapnell C. 2016. Single‐cell transcriptome sequencing: recent advances and remaining challenges. F1000Research 5: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecken MD, Theis FJ. 2019. Current best practices in single‐cell RNA‐seq analysis: a tutorial. Molecular Systems Biology 15: e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg SM, Erion G, Chen H, DeGrave A, Prutkin JM, Nair B, Katz R, Himmelfarb J, Bansal N, Lee S‐I. 2020. From local explanations to global understanding with explainable AI for trees. Nature Machine Intelligence 2: 2522–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Fukuda H. 2012. Secondary cell wall patterning during xylem differentiation. Current Opinion in Plant Biology 15: 38–44. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V. 2011. Scikit‐learn: machine learning in Python. The Journal of Machine Learning Research 12: 2825–2830. [Google Scholar]
- Pliner HA, Shendure J, Trapnell C. 2019. Supervised classification enables rapid annotation of cell atlases. Nature Methods 16: 983–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Z, Libault M. 2013. Unleashing the potential of the root hair cell as a single plant cell type model in root systems biology. Frontiers in Plant Science 4: 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszak P, Heo J‐O, Blob B, Toyokura K, Sugiyama Y, de Luis Balaguer MA, Lau WWY, Hamey F, Cirrone J, Madej E et al. 2021. Cell‐by‐cell dissection of phloem development links a maturation gradient to cell specialization. Science 374: eaba5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, Schiefelbein J. 2019. Single‐cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiology 179: 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Zhu Y, Schiefelbein J. 2021. Plant cell identity in the era of single‐cell transcriptomics. Annual Review of Genetics 55: 479–496. [DOI] [PubMed] [Google Scholar]
- Satterlee JW, Strable J, Scanlon MJ. 2020. Plant stem‐cell organization and differentiation at single‐cell resolution. Proceedings of the National Academy of Sciences, USA 117: 33689–33699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahan R, Hsu C‐W, Nolan TM, Cole BJ, Taylor IW, Greenstreet L, Zhang S, Afanassiev A, Vlot AHC, Schiebinger G et al. 2022. A single‐cell Arabidopsis root atlas reveals developmental trajectories in wild‐type and cell identity mutants. Developmental Cell 57: 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulse CN, Cole BJ, Ciobanu D, Lin J, Yoshinaga Y, Gouran M, Turco GM, Zhu Y, O’Malley RC, Brady SM et al. 2019. High‐throughput single‐cell transcriptome profiling of plant cell types. Cell Reports 27: e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Lee J, Akter S, Rogers M, Grene R, Li S. 2020. Prediction of condition‐specific regulatory genes using machine learning. Nucleic Acids Research 48: e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steudle E, Peterson CA. 1998. How does water get through roots? Journal of Experimental Botany 49: 775–788. [Google Scholar]
- Trapnell C. 2015. Defining cell types and states with single‐cell genomics. Genome Research 25: 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco GM, Rodriguez‐Medina J, Siebert S, Han D, Valderrama‐Gómez MÁ, Vahldick H, Shulse CN, Cole BJ, Juliano CE, Dickel DE et al. 2019. Molecular mechanisms driving switch behavior in xylem cell differentiation. Cell Reports 28: 342–351.e4. [DOI] [PubMed] [Google Scholar]
- Voß U, Wilson MH, Kenobi K, Gould PD, Robertson FC, Peer WA, Lucas M, Swarup K, Casimiro I, Holman TJ et al. 2015. The circadian clock rephases during lateral root organ initiation in Arabidopsis thaliana . Nature Communications 6: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Liang S, Kumar T, Navin N, Chen K. 2019. SCMarker: a b initio marker selection for single cell transcriptome profiling. PLoS Computational Biology 15: e1007445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Navin NE. 2015. Advances and applications of single‐cell sequencing technologies. Molecular Cell 58: 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendrich JR, Yang B, Vandamme N, Verstaen K, Smet W, Van de Velde C, Minne M, Wybouw B, Mor E, Arents HE et al. 2020. Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science 370: eaay4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, Klein AM. 2019. Scrublet: computational identification of cell doublets in single‐cell transcriptomic data. Cell Systems 8: 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B et al. 2019. Probabilistic cell‐type assignment of single‐cell RNA‐seq for tumor microenvironment profiling. Nature Methods 16: 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T‐Q, Xu Z‐G, Shang G‐D, Wang J‐W. 2019. A single‐cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Molecular Plant 12: 648–660. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M et al. 2019. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Research 47: D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J et al. 2017. Massively parallel digital transcriptional profiling of single cells. Nature Communications 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. 2019. A primer on deep learning in genomics. Nature Genetics 51: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Summary of the data processing pipeline and machine learning methods used.
Fig. S2 Integration of five datasets.
Fig. S3 Classification performance (AUROC) of 10 root cell types of Arabidopsis.
Fig. S4 Performance comparison among seven machine learning methods on six different evaluation scores.
Fig. S5 Performance comparisons among five cross validation random forest models.
Fig. S6 Comparisons of the proportion of expressed cells for different marker genes.
Fig. S7 Heat map of top 20 SHAP markers.
Fig. S8 Heat map of top 20 SVMM markers.
Fig. S9 Heat map of top 20 CORR markers.
Fig. S10 Heat map of all ICIM markers.
Fig. S11 Heat map of top 20 KNOW markers.
Fig. S12 Heat map of top 20 BULR markers.
Fig. S13 Violin map of top 20 SVMM markers of endodermis cell type.
Fig. S14 Comparison of classification performance based on ICI 0.5 and 0.9 thresholds in five cell types.
Fig. S15 A UMAP for SVM predicted cell types.
Fig. S16 Classification and predicted top markers.
Fig. S17 Cumulative SHAP values for all SHAP markers for each of the 10 cell types.
Fig. S18 Comparison of proportion of cumulative SHAP values.
Fig. S19 Summary of the SVMM markers.
Fig. S20 Expression rate of SHAP marker genes in Shahan et al. (2022).
Fig. S21 Expression rate comparison for five clusters between different marker types.
Fig. S22 Number of marker genes uniquely and commonly identified by different methods.
Fig. S23 Schematic demonstration of architecture for each type of neural network.
Methods S1 Additional materials and methods for this study.
Table S1 A summary of automatic cell classification and feature selection methods for scRNA‐seq data.
Table S2 The distribution of 25 618 cells from 10 cell types used to train models.
Table S3 The 1532 KNOW markers.
Table S4 The markers used in comparisons of expression patterns across clusters and cell‐type classifications.
Table S5 Assignment of cell type to cluster.
Table S6 The 1813 known marker genes, including those genes in Table S3 and other published markers.
Table S7 GO annotation of SHAP markers.
Table S8 Specific GO terms for ICIM, KNOW, and SHAP marker types.
Table S9 The cell types of SHAP marker genes enriched in GO terms.
Table S10 Additional wet‐bench experimental support for newly identified markers.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
These data were derived from the GEO and SRA repositories at NCBI with the following IDs: GSE123013, GSE121619, GSE122687 and GSE123818, and PRJNA517021.