

Abstract
Nonalcoholic steatohepatitis (NASH) is a widely prevalent disease, but approved pharmaceutical treatments are not available. As such, there is great activity within the pharmaceutical industry to accelerate drug development in this area and improve the quality of life and reduce mortality for NASH patients. The use of quantitative systems pharmacology (QSP) can help make this overall process more efficient. This mechanism-based mathematical modeling approach describes both the pathophysiology of a disease and how pharmacological interventions can modify pathophysiologic mechanisms. Multiple capabilities are provided by QSP modeling, including the use of model predictions to optimize clinical studies. The use of this approach has grown over the last 20 years, motivating discussions between modelers and regulators to agree upon methodologic standards. These include model transparency, documentation, and inclusion of clinical pharmacodynamic biomarkers. Several QSP models have been developed that describe NASH pathophysiology to varying extents. One specific application of NAFLDsym, a QSP model of NASH, is described in this manuscript. Simulations were performed to help understand if patient behaviors could help explain the relatively high rate of fibrosis stage reductions in placebo cohorts. Simulated food intake and body weight fluctuated periodically over time. The relatively slow turnover of liver collagen allowed persistent reductions in predicted fibrosis stage despite return to baseline for liver fat, plasma ALT, and the NAFLD activity score. Mechanistic insights such as this that have been derived from QSP models can help expedite the development of safe and effective treatments for NASH patients.
KEY WORDS: fibrosis, modeling, NASH, QSP
Manuscript
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are diseases of the liver that are largely the result of excessive lipid accumulation and partitioning (1, 2). The incidence of NAFLD and NASH have grown substantially over the last 20 years, with estimates of 20–40% of populations in various locations throughout the world having NAFLD or NASH (3). As such, the interest in understanding and treating this disease has increased substantially. NASH is reasonably well-understood, with the overall understanding growing every year. The pathophysiology includes contributions from alterations in lipid partitioning, lipotoxicity, inflammation, and fibrosis (1, 2). Data from clinical studies with the numerous potential treatments have helped increase the overall understanding of the disease in addition to focused clinical studies.
NASH drug development efforts have increased substantially in the last 10 years. A considerable number of potential treatments are being developed across the pharmaceutical industry (4, 5). In addition to monotherapies, several compounds are being developed as combination treatments; complimentary mechanisms of action may provide additional clinical benefit. Unfortunately, the development of several compounds has been terminated (6). This has primarily been due to an insufficient ability to demonstrate favorable responses in treatment cohorts as compared with placebo cohorts.
Regulators want to ensure that NASH drugs being developed are both safe and effective. The United States’ Food and Drug Administration has provided definitions of endpoints that have the greatest chance of providing health benefits of patients (7). Data are not yet available identifying ideal surrogate endpoints for preventing adverse clinical outcomes. Measures derived from liver biopsies are currently employed to characterize resolution of NASH (based on specific histologic endpoints related to pathophysiologic components) as well as improvements in hepatic fibrosis (8, 9).
Efficient drug development will benefit NASH patients by minimizing the period of time for them to gain access to safe and effective medicines. Application of mathematical modeling practices such as quantitative systems pharmacology (QSP) can expedite clinical drug development (10). QSP modeling is a mechanism-based mathematical modeling approach that describes not only the pathophysiology of a disease, but also how pharmacological interventions can modify the mechanisms of pathophysiology (11–15). Simulated populations (aka virtual populations) provide a useful approach for capturing the pathophysiology of the disease inasmuch as they provide the ability to make predictions that account for inter-patient variability in both disease pathophysiology and clinical status (13, 14, 16). Predicted pharmacologic effects on simulated patients result from predicting compound exposure and pharmacodynamics (PD). Compound exposure at the site of the target can be predicted by the use of physiologically-based, pharmacokinetic (PBPK) modeling; this is particularly valuable when simulating pharmacologic intervention of intracellular targets. The PD and/or mechanism of action (MoA) of compounds can be simulated by translating laboratory and/or clinical data into equations that describe how existing pathophysiologic processes are altered by the actions of the compound.
QSP is a relatively new approach that has historical origins (17). Technological advances in computer chip design provided the ability for simulations results to be generated in much shorter periods of time, enabling this modeling approach to be applied to pharmaceutical drug development. The use of QSP modeling continues to grow (18, 19). Capabilities provided by QSP modeling include predicting efficacy, identifying responsive patient types, determining appropriate trial duration and measurement timing, delineating placebo response from pharmacologic response, understanding the link between mechanisms and biomarker responses, and predicting the efficacy potential for combination treatments (12, 20, 21). Simulation results of these sorts can inform clinical trial protocol design.
NAFLD and NASH provide some unique considerations with respect to QSP modeling. The pathophysiology includes several discrete, yet interactive areas: steatosis, lipotoxicity, inflammation, and fibrosis. QSP models of NAFLD and NASH should include representations of each of these areas, as the response to treatments usually invoke responses from each. Within the steatosis area, the uptake and/or de novo synthesis of fatty acids as well as esterification into triacylglycerol (TG) are primary components. Lipotoxicity is characterized by an excess of lipids that ultimately leads to hepatocellular apoptosis. Inflammation in NASH includes the recruitment of additional immune cells to the liver as well as increased production of key immune mediators. Fibrosis is characterized both by the presence of excessive amounts of collagen as well as the activated hepatic stellate cells that produce the extracellular matrix components. QSP models of NASH should include equations that describe these pathophysiologic mechanisms in addition to the primary biomarkers (e.g., plasma ALT) and histologic outputs that are used to determine efficacy.
QSP models of NASH also need to represent the various patient types that are recruited into NASH clinical studies. This includes overweight or obese individuals that have not had a diagnosis of NASH for Phase I studies; these patients may or may not have steatosis, and are unlikely to have much lipotoxicity, inflammation, and/or fibrosis. Phase IIa studies typically include patients with NASH and varying degrees of fibrosis. Phase IIb and Phase III studies typically recruit patients with stage 3 or 4 fibrosis in addition to NASH, as documented by liver histology. Simulated cohorts with each of these patient types will enable a NASH QSP model to support development throughout the lifecycle of the compound.
The awareness of QSP modeling within regulatory agencies such as the FDA has grown as the modeling approach has been increasingly utilized. Currently, the FDA has not issued any direct guidance about NASH QSP modeling, but the agency has indicated that there are certain recommendations that should be applied to all QSP modeling (18, 22). Model transparency enables reviewers to have access to all equations and parameters used to generate simulation results. Not only will it enable reviewers to recapitulate simulation results, model transparency will also allow reviewers to interpret simulation results. Similarly, documentation of the modeling rationale facilitates this process. Another recommendation from FDA about QSP modeling is for model developers to submit a minimum model. Reducing the complexity of the model further enables reviewers to better interpret simulation results. Similarly, prespecified quantitative or statistical criteria helps reviewers determine the validity of the predicted results. Finally, the FDA has recommended that there be maximal inclusion of clinical PD markers. Applying this recommendation to NASH indicates that the ability for the QSP model to predict changes to the histological measurements, NAFLD Activity Score (NAS) and fibrosis stage, is crucial; these are the currently acceptable endpoints for efficacy for NASH treatments (7). Additional biomarkers such as liver fat, plasma ALT, and serum Pro-C3 biomarkers have been routinely employed in NASH clinical studies; their inclusion in a NASH QSP model further enhances its utility.
An additional utility of a NASH QSP model would be the ability to reproduce the key results of clinical studies. This includes not only the response to the investigational treatment but also the response of the placebo group. The lack of separation between the treatment and placebo cohorts has been a significant reason for the termination of a number of promising potential treatments for NASH (6). In particular the fraction of patients within the placebo cohorts that show improvement in fibrosis stage over 26, 48, or 72 week studies is surprisingly high (20–30%, (23)). There is some indication that operator variability in the reading of the histological liver biopsy samples may play a significant role (24), and there may be contributions from other factors as well.
Several useful QSP models of hepatic steatosis, hepatic fibrosis, and NAFLD/NASH have been reported. Ashworth et al. developed a QSP model of lipid partitioning and steatosis, including zonal differences across the hepatic acinus (25). Liao et al. extended this model to include the effects of fructose on lipid partitioning and hepatic steatosis (26). Neither of these QSP models included representations of lipotoxicity, inflammation, fibrosis, or NASH biomarkers or the primary histologic endpoints of NAS and stage of fibrosis. Holzhutter and Berndt also developed a QSP model of zonal influences on hepatic steatosis (27); this model also includes hepatocellular death as a consequence of lipid accumulation. It does not, however, include representations of inflammation, fibrosis, or NASH biomarkers. Dutta-Moscato et al. developed an agent-based QSP model of hepatic fibrosis, but it is not specific to NASH (28).
A more comprehensive QSP model of NAFLD and NASH is NAFLDsym (29–33). This QSP model includes interactive sub-models of steatosis, lipotoxicity, inflammation, and fibrosis. Several important sub-models, such a hepatocyte death and proliferation, meal administration and post-prandial metabolites, mechanistic representations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) amongst others, were replicated from the quantitative systems toxicology (QST) model, DILIsym (34, 35). Also replicated from DILIsym are discrete representations of hepatocytes within the periportal, midlobular, and centrilobular sections of the hepatic acinus. NAFLDsym also includes representations of a variety of useful, NASH-specific biomarkers, including histologic NAS and fibrosis stage outputs as well as serum pro-C3. Moreover, there are more than 1700 simulated patients, enabling focused exploration of different subsets of NAFLD and NASH patients (e.g., fibrosis stage 3). These simulated patients have been validated by simultaneous comparison to many clinical data measurements that focus on specific aspects of the pathophysiology. A variety of treatments in clinical development have been simulated with NAFLDsym, including cenicriviroc and anti-FGFR1/KLB bispecific antibody (36, 37). Predictions of improvement with weight loss as well as disease progression with weight gain have also been reported with NAFLDsym (30), providing further validation of the model.
There are four sub-models within NAFLDsym that particularly address the major elements of NASH pathophysiology: steatosis, lipotoxicity, inflammation, and fibrosis. Concise descriptions of each submodel are given below. Figure 1 also displays several simulation results that characterize some of the key outputs of these sub-models.
Fig. 1.

Simulation results and clinical data enabling characterization of sub-model behavior and several important aspects of NASH pathophysiology as simulated by NAFLDsym. The relationship between liver fat and plasma ALT in the simulated patients is quite similar to the measured data from Maximos et al. (a); the simulated population retains a distribution of BMI that is comparable to the clinical data reported by Dudekala et al. (b); the relationship between fat mass and adipose fatty acid (FA) release rates is comparable between the simulated patients and the clinical data reported by Mittendorfer et al. Note that there are few simulated patients with adipose FA release rates in excess of 50 mmol/h (c); the ranges of de novo lipogenesis (DNL) and liver fat are comparable between the simulated patients and the clinical data reported by Lambert et al. and Smith et al. (d); the number of lobular macrophages in NAFLD and NASH patients is consistent with the clinical data reported by Tajiri et al. Note that there are minimal differences between patients below or above NAS = 4. (e); the range of TGF-beta levels in simulated patients and clinical cohorts with varying plasma ALT levels, as reported by Dal et al. (f); synthesis rates and quantities of hepatic collagen type I in clinical and simulated patients across a range of fibrosis scores. Clinical data were reported by Decaris et al. and Masugi et al. Clinical patients with fibrosis stage = 4 were excluded from figure, as data for only two patients were reported. (g,h). In each figure, black or grey symbols represent clinical data sets while red symbols represent simulated patients. In figures a, c-h, individual simulated patients are displayed.
The steatosis sub-model within NAFLDsym includes multiple processes essential for the trafficking of fatty acids, diacylglycerols, and TG within hepatocytes. One of these key processes included in NAFLDsym is the uptake of circulating fatty acids from the circulation in a concentration-dependent manner following the release of fatty acids into the circulation by adipocytes (38, 39). Hepatic de novo lipogenesis of fatty acids is also represented within NAFLDsym, including contributions in the overnight-fasted as well as the post-prandial states (40–42); hepatocytes in NAFLDsym take up glucose from the circulation in the post-prandial period, when plasma glucose levels are elevated (43, 44). In NAFLDsym, hepatocytes can also use either intracellular fatty acids or pyruvate to as fuels support the production of ATP to meet bioenergetic demands. The representation of ATP production is quite similar to what has previously been modeled in DILIsym (34, 35). Fatty acid oxidation is regulated by both ATP levels as well as the relative availability of both fatty acids and pyruvate (45). Fatty acids can be esterifed to TG in NAFLDsym; lipolysis of hepatocellular TG is also represented (46). Finally, the export of intracellular TG as part of very-low-density lipoprotein (VLDL-TG) is also represented (47, 48).
Lipotoxicity in NAFLDsym is primarily represented based on the mechanistic interactions between saturated fatty acids (SFA), oxidative stress, and hepatocellular apoptosis (49–51). A variety of clinical data were used to guide the optimization of parameter values within these processes (52–55). Both liver SFA and unsaturated fatty acids (UFA) are separately tracked within NAFLDsym. The accumulation of hepatocellular SFA elicits an increase in reactive oxygen species (49–51). The resultant cellular oxidative stress, in sufficient quantities, yields hepatocellular apoptosis; necrosis can occur at very high levels of oxidative stress. However, circulating biomarkers such as cleaved cytokeratin 19 (cK18) and histological ballooning indicate that apoptosis predominates (55, 56). The magnitude of hepatocyte loss due to lipotoxicity can be estimated by computational methods (57), providing validation of the steady state numbers of viable hepatocytes in the simulated NASH patients. Apoptotic hepatocytes release vesicles that contain cellular fragments. These vesicles and the fragments within them can interact with Kupffer cells, macrophages, neutrophils, and other immune cells to provide a degree of activation of the immune system (58–60).
The inflammation sub-model in NAFLDsym includes resident Kupffer cells as well as recruited macrophages and neutrophils. The numbers of each cell type in the simulated patients have been calibrated to be consistent with clinical data (61, 62). Interestingly, the numbers of macrophages and neutrophils do not vary much between NASH patients of differing severities (61, 62); the simulated patients within NAFLDsym are consistent with these reported data (Fig. 1). The Kupffer cells, macrophages and neutrophils generate a variety of mediators, depending on the various queues presented to them. Within NAFLDsym, these cells can produce TNF-alpha, IL-10, TGF-beta, PDGF, MMP, CCL3, and TIMP. Total mediator production is regulated by the number of viable cells, the ability of each cell type to make each mediator, as well as the cross-regulatory influence of mediators on each other’s production. The levels of each mediator across a range of disease severity in the simulated patients of NAFLDsym are consistent with a variety of different clinical data sets (63–68). Several mediators act on hepatocytes in addition to immune cells, while others participate in regulating fibrotic processes.
The fibrosis sub-model includes the activation and turnover of hepatic stellate cells (HSC) as well as the synthesis and breakdown of collagen. Early in the disease sequelae, the loss of hepatocytes to lipotoxic influences initiates collagen synthesis as part of the wound healing response. Chronic cell loss and ongoing activation of HSC lead to a fibrotic state in NASH patients, which is captured in a subset of the simulated patients of NAFLDsym. HSC are activated via stimulus from TGF-beta, transforming them to a myofibroblast-type state (69–72). The mediator PDGF encourages proliferation of HSC, regulating the number of activated HSC (73). Procollagen synthesis and collagen release by activated HSC are included in NAFLDsym as well as the release of the Pro-C3 fragment that can be used as a circulating biomarker of collagen synthesis rates (74, 75). Similar to the representation in the inflammation sub-model, the total amount of collagen synthesis reflects both the number of activated HSC as well as the propensity for HSC to produce collagen. Collagen synthesis rates are relatively slow (74, 76), and the simulated patients with fibrosis in NAFLDsym are consistent with this observation (Fig. 1). Also participating in the turnover of collagen in the simulated patients with fibrosis are MMP. This group of mediators are responsible for degrading collagen in a multi-step process (77). In addition to being released by macrophages and neutrophils, MMP are also released by activated HSC in the simulated patients (77).
NAFLDsym simulation outputs include representations of the currently-accepted histologic biomarkers of efficacy for NASH; the NAFLD activity score (NAS) and fibrosis stage (7). The representation of NAS includes the individual components, including steatosis, ballooning, and inflammation histologic (78, 79). These outputs have been validated by comparing with various types of clinical data (56, 80–85) (Fig. 2). Fibrosis stage is also simulated in NAFLDsym; this representation accounts for both the location (centrilobular, midlobular, peripoportal) and quantity of collagen within the liver and is consistent with the histologic scoring paradigm for NASH patients (79, 86). Additional simulation outputs that align with commonly-measured NASH biomarkers include the relative amount of liver fat (consistent with MRI-PDFF measurements), body weight, BMI, plasma ALT, plasma AST, and plasma triglycerides. As mentioned above, the circulating biomarker Pro-C3 is also simulated as an indicator of collagen biosynthesis. It should additionally be pointed out that NAFLDsym does not include several additional combination NASH biomarkers that continue to undergo qualification such as FIB-4, NFS, and ELF (87). These biomarkers could be added in the future as they become qualified.
Fig. 2.
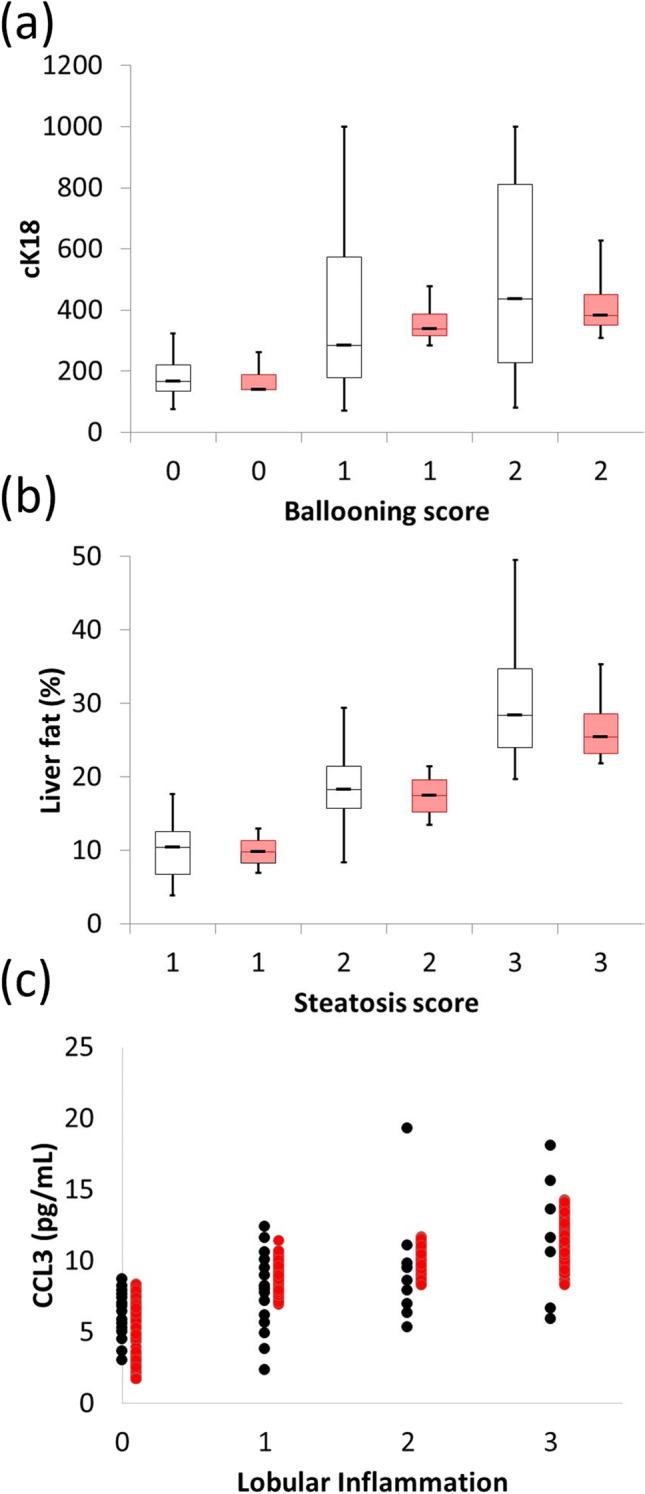
Simulation results and clinical data describing the correspondence between the simulated histologic components of NAS with related outputs. The range of cytokeratin-cleaved K18 (cK18) and histologic ballooning (a), liver fat measured by MRI-PDFF and histologic steatosis (b), andCCL3 and histologic lobular inflammation scores. Red bars or symbols denote results from simulated patients while black symbols and black bars denote clinical data from Aida et al. (a), Middleton et al. (b), and du Plessis et al. (c).
The substantial degree of inter-patient variability amongst NASH and NAFLD patients highlights the utility of the inclusion of > 1700 simulated patients within the NAFLD-NASH SimPops in NAFLDsym. This SimPops includes both mechanistic and clinical between-patient variability. Mechanistic variability is imposed within key aspects of each submodel, including parameters from the following categories across NASH pathophysiology: body weight (55, 88, 89), adipose fatty acid release rates (39), hepatic de novo lipogenesis (40, 41, 90, 91), hepatic VLDL-TG export (47, 48), hepatocellular mitochondrial function (92), hepatic glucose uptake(43, 44), hepatic and extrahepatic plasma TG clearance (55, 89, 93–97), hepatic antioxidant status (52, 98, 99), hepatocellular apoptotic sensitivity to oxidative stress (100), hepatocellular regeneration rates (101, 102), hepatocyte extracellular vesicle release rates (60), macrophage and neutrophil recruitment (80, 85), immune mediator production rates, hepatic stellate cell activation and proliferation (70–72), collagen synthesis and proteolysis rates (32, 33, 36). Parameters and references supporting the selection of these parameters are listed in Table I. Each simulated patient within the SimPops is validated by simultaneous comparison with clinical data across each of these axes. Simulated patients that produce simulation results within the reported ranges of these clinical data boundaries is considered acceptable. Some examples are given in Fig. 1. Inter-patient variability in key clinical measurements emerges from the parametrically-imposed mechanistic variability. Figure 3 illustrates the wide range of variability across several key clinical outputs, including fibrosis stage, NAS, plasma ALT, liver fat, and BMI. The SimPops includes patients with a wide range of NASH patient characteristics (no NASH to severe NASH, no fibrosis to substantial fibrosis, no steatosis to high liver fat, normal plasma ALT to above upper limit of normal). Simulated cohorts (SimCohorts) that include simulated patients with certain clinical characteristics can be selected from the larger SimPops to support simulations of clinical studies at various points in the clinical development pipeline (103).
Table I.
Summary of Parameters Included in NAFLD SimPops
| Parameter Name in NAFLDsym | Data Source for Distribution |
|---|---|
| Vmax for aHSC proliferation | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from Abdeen 2009, El Gendi 2012, Washington 2000 |
| ATP decrement necrosis Vmax | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with outcome data |
| Basal fasting glucose | Browning 2004, Maximos 2015, Copaci 2015, Dudekula 2014, Wong 2013, Stepnova 2010, Tanaka 2013, Zein 2012 |
| Basal plasma triglycerides concentration | Yki-Jarvinen 2014, Maximos 2015 |
| Basal value of mito ETC flux | Perez-Carreras 2003 |
| Rate constant for FFA release from Peripheral storage | Based on relationship between fat mass and adipose fatty acid release described by Mittendorfer 2009 |
| Basal liver triglycerides | Yki-Jarvinen 2014, Maximos 2015, Browning 2004 |
| Body Mass | Yki-Jarvinen 2014, Maximos 2015, Browning 2005 |
| Caspase-mediated apoptosis scaling constant | Bantel 2001 |
| Liver macrophage CCL3 production Vmax | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from DuPlessis 2015, DuPlessis 2016 |
| CL activated HSC apoptosis scalar | El-Gendi 2012, Abdeen 2012, Carpino 2004 (to provide steady state aHSC in accordance with the effects of the CL_aHSC crowding_scalar on aHSC proliferation) |
| CL activated HSC crowding scalar | El-Gendi 2012, Abdeen 2012, Carpino 2004 |
| CL fibrosis hepatocyte displacement scalar | Carpino 2004, D’Ambrosio 2012 |
| Collagen 1 baseline formation rate | Decaris 2017, Masugi 2018 |
| Collagen 1 formation rate | Decaris 2017, Masugi 2018 |
| Collagen 3 baseline formation rate | Decaris 2017, Masugi 2018 |
| Collagen 3 formation rate | Decaris 2017, Masugi 2018 |
| Extracellular vesicle release from apoptotic cells | Povero 2016 |
| Maximum LSEC HGF production rate per liver LSEC | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with outcome data |
| Maximum macrophage HGF production rate per macrophage | Dominguez-Perez 2016, Balaban 2006, Agrawal 2013 |
| Maximum neutrophil HGF production rate per liver neutrophil | Dominguez-Perez 2016, Balaban 2006, Agrawal 2013 |
| HGF mediated regeneration Vmax | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with outcome data |
| Vmax for HSC activation | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from Abdeen 2009, El Gendi 2012, Washington 2000 |
| Rate constant for DNL precursor production | Lambert 2014, Donnely 2005, Lee 2015, Diraison 2003 |
| Rate constant for lactate contribution to DNL | Lambert 2014, Donnely 2005, Lee 2015, Diraison 2003 |
| Conversion of mature to labile collagen rate constant | Arima 2004, D’Ambrosio 2012 |
| Rate constant for hepatic Chylo-TG uptake | Tushuizen 2010, McQuaid 2011 |
| Rate constant for hepatic glucose uptake | McMahon 1989, Cersosimo 2011 |
| Rate constant for hepatic VLDL-TG uptake | Yki-Jarvinen 2014, Maximos 2015, Mittendorfer 2003, Sane 1988, Beil 1982 |
| Km for FFA2DAG | Required to have appropriate dynamics with TG Lipolysis mechanism activated |
| Km for triglyceride lipolysis | Variability in this parameter provides variability in the liver TG-ALT relationship described by Yki-Jarvinen 2014, Maximos 2015, Browning 2004 |
| Rate constant for Chylo-TG uptake by peripheral tissues | Tushuizen 2010, McQuaid 2011 |
| Rate constant for VLDL-TG uptake by peripheral tissues | Yki-Jarvinen 2014, Maximos 2015, Mittendorfer 2003, Sane 1988, Beil 1982 |
| Vmax for LOX | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from Mesarwi 2015 |
| ML fibrosis hepatocyte displacement scalar | Carpino 2004, D’Ambrosio 2012 |
| Vmax for MMP | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D |
| Vmax for MMP (fragments) | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D |
| PP fibrosis hepatocyte displacement scalar | Carpino 2004, D’Ambrosio 2012 |
| Half-life for plasma Pro-C3 | Assumed standard deviation of ± 50% and parameter range of 2.5 times the S.D. and validated with data reported by Levin 2017 (abstract) |
| Procollagen 1 production rate | Decaris 2017, Masugi 2018 |
| Procollagen 1 baseline production rate | Decaris 2017, Masugi 2018 |
| Procollagen 3 production rate | Decaris 2017, Masugi 2018 |
| Procollagen 3 baseline production rate | Decaris 2017, Masugi 2018 |
| Scaling coeff. representing reserve mitochondria function | Perez-Carreras 2003 |
| Liver RNS/ROS baseline clearance Vmax | Hardwick 2010, Videla 2004, Tanaka 2013 |
| Serum adiponectin initial value | Adiels 2006 |
| Prior (weight) for liver TG % to steatosis Grade 0 model | Randomized distribution of histologic steatosis to provide variability between the four grades |
| Prior (weight) for liver TG % to steatosis Grade 1 model | Randomized distribution of histologic steatosis to provide variability between the four grades |
| Prior (weight) for liver TG % to steatosis Grade 2 model | Randomized distribution of histologic steatosis to provide variability between the four grades |
| Prior (weight) for liver TG % to steatosis Grade 3 model | Randomized distribution of histologic steatosis to provide variability between the four grades |
| Triglyceride lipolysis switch | Required to ensure TG Lipolysis mechanism activated |
| Liver macrophage TGF-beta production Vmax | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from Das 2011 |
| Maximum inhibition of MMP by TIMPs | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D |
| Liver macrophage TIMP production Vmax | Ando 2018, Miele 2009 |
| Liver macrophage TNF-alpha production Vmax | Assumed standard deviation of ± 20% and parameter range of 2.5 times the S.D. and validated with data from Das 2011, Zahran 2013, Hui 2004, Paredes-Turrubiarte 2016 |
| Vmax for FFA2DAG | Required to ensure appropriate hepatocyte fatty acid and DAG dynamics |
| VLDL-triglyceride secretion rate Vmax | Fabbrini 2008, Adiels 2006 |
Fig. 3.

Simulation results for untreated simulated patients within SimPops in NAFLDsym, including liver fat (a, units are %), plasma ALT (b, units are U/L), NAS (c), fibrosis stage (d), and BMI (e, units are kg/m2). Note that each simulated patient retains the same position on each radial plot.
While there has been substantial activity towards developing pharmaceutical treatments for NASH patients, none are currently available. As such, the standard of care for NASH treatment remains weight loss (104–107). Weight loss has been shown to be an efficacious treatment approach, although patient compliance is challenging (107). Consistent with current QSP methodologies, we applied weight loss to our SimPops to further validate the simulated patients (13, 14, 16). Included as a submodel within NAFLDsym is the QSP model of body weight developed by Hall et al. (108). As such, NAFLDsym is capable of accurately predicting weight loss and changes in body composition with reduced caloric intake. This submodel mechanistically interacts with the other NAFLDsym sub-models in the following ways: Reduced food intake acta to diminish substrate availability for hepatic de novo lipogenesis and adipose fatty acid release rates are reduced as fat mass is decreased (38, 42, 109). These mechanisms combine to reduce the lipid burden upon the liver, leading to further improvements in the downstream pathophysiology of NASH as well. Figure 4 illustrates the simulation results for SimCohorts selected to align with clinical cohorts for the given studies. Of note is the appropriate simulated reductions in liver fat, NAS (including individual components), and fibrosis stage with 5–10% weight loss via reduced caloric intake over 6–12 months. NAFLDsym has additionally been used to simulate several potential treatments for NASH patients (e.g., anti-FGFR1/KLB bispecific antibody, cenicriviroc), including those that elicited improvements in NASH and others that did not (36, 37). The accurate prediction of the responses to these treatments further validates NAFLDsym and the SimPops.
Fig. 4.

Simulation results and clinical data illustrating the appropriate degree of relief in simulated NASH patients in response to 5–10% weight loss achieved via restriction of caloric intake. Liver fat before and after six months of 10% weight loss, as compared with clinical data from Smith et al. Mean responses and individual simulated and clinical patient results displayed (a); absolute change in overall NAS and respective components after 5–7% weight loss over 12 months, as compared with clinical data from Vilar-Gomez et al. and Hameed et al. Note that a negative value indicates reduction relative to initial values (b); fraction of patients with worsened, stabilized, or regressed fibrosis stage after 12 months of 5–7% weight loss, as compared with clinical data from Vilar-Gomez et al. (c). Clinical results are summarized in figure on left, while simulation results are in figure on right Red bars or symbols denote results from SimCohorts while black or gray symbols denote clinical data.
NAFLDsym has recently been used to identify potential mechanistic contributors to the relatively high response rate in improved fibrosis stage within placebo cohorts in clinical studies (23). Specifically, NAFLDsym was used to simulate subtle adjustments in food intake over the course of a simulated 52 week study that led to periodic weight loss and weight gain; body weight was not predicted to change appreciably by the end of the simulated study (Fig. 5a, 5b). A simulated cohort of 90 simulated NASH patients was subjected to this so-called yo-yo dieting protocol. The baseline characteristics of the simulated cohort are given in Table II. Food intake was adjusted every four weeks to enable 1% weight gain and weight loss in repeated order. Figure 5 illustrates not only the small changes in body weight over the simulated study, but also the consequent predicted changes in liver fat, plasma ALT, and liver collagen. Both liver fat and plasma ALT are predicted to decline with the reduced food intake (and associated weight loss) and increase with the periodic rebound in body weight. The absolute amounts of body weight, liver fat, and plasma ALT at the conclusion of the 52 week simulated study are quite similar to the values at the beginning of the study. However, liver collagen levels did not follow the same pattern as the other biomarkers. Rather liver collagen was predicted to decline, leading to a predicted reduction in fibrosis stage in 10% of the simulated patients within the cohort (Table III). The notably slow turnover rate of liver collagen (74, 76) prevents liver collagen from having the same periodicity that body weight, liver fat, and plasma ALT do in response to the cyclical adjustments in simulated food intake. These simulation results suggest that, within clinical studies, some proportion of NASH patients in placebo cohorts with reduced fibrosis stage could be experiencing the yo-yo dieting behavioral pattern. Confirmation of these simulation results with additional clinical studies and/or data collection during existing clinical studies could impact the interpretation of data from the placebo cohorts in these studies.
Fig. 5.

Predicted relative changes (left) in and absolute levels (right) of body weight (a, b), liver fat (c, d), plasma ALT (e, f), and liver collagen (g,h) in NASH SimCohorts over time due to yo-yo dieting. Mean ± standard deviation plotted for all figures.
Table II.
Baseline Simulated Cohort Characteristics
| Body weight (kg) |
Liver fat (%) |
Plasma ALT (U/L) |
NAS (score) |
Fibrosis score = 2 |
Fibrosis score = 3 |
|---|---|---|---|---|---|
| 89.1 ± 19.4 | 17 ± 5 | 50 ± 12 | 5.6 ± 3.2 | 39% | 61% |
Table III.
Proportion of Simulated NASH Patients with Predicted Histologic Reductions Over time with Yo-Yo Dieting
| 13 weeks | 26 weeks | 39 weeks | 52 weeks | |
|---|---|---|---|---|
| Fibrosis | 6% | 10% | 12% | 10% |
| NAS | 4% | 0% | 0% | 0% |
Conclusion
The increasing prevalence of NAFLD and NASH accentuates the need for available treatments in this patient populations. Application of QSP modeling to the development of NASH drug development can help accelerate this process. Moreover, such NASH QSP models should adhere to recommendations put forth by regulatory agencies such as the FDA. These include model transparency and documentation, minimizing complexity of the QSP model, utilizing predefined, quantitative criteria for model validation, as well as the inclusion of useful NASH clinical biomarkers such as NAS and fibrosis stage. Several useful QSP models are currently available to help support NASH drug development to varying extents, with NAFLDsym, in particular, providing the capability of simulating numerous useful aspects of NASH. Ultimately, the partnership between clinical studies and QSP modeling should help provide safe and effective medicines for NASH patients in short order.
ACKNOWLEDGMENTS AND DISCLOSURES
The author would like to acknowledge the contributions of several individuals who contributed to the manuscript via numerous useful discussions. This list includes Lisl Shoda, Grant Generaux, Christina Battista, Zack Kenz, Kyunghee Yang, Jeff Woodhead, Brett Howell, Fulya Akpinar-Singh, Richard Allen, and Theodore Rieger. The author contributed to the development of the NASH QSP model, NAFLDsym, and works for the owner of the model, Simulations Plus.
Funding
This work was supported by Simulations Plus, as the author is an employee of the organization.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the american association for the study of liver diseases, american college of gastroenterology, and the american gastroenterological association. Am J Gastroenterol. 2012;107(6):811–826. doi: 10.1038/ajg.2012.128. [DOI] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Therapeut. 2011;34(3):274–85. [DOI] [PubMed]
- 3.Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. 2019;71(4):793–801. doi: 10.1016/j.jhep.2019.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Brown E, Hydes T, Hamid A, Cuthbertson DJ. Emerging and established therapeutic approaches for nonalcoholic fatty liver disease. Clin Ther. 2021;43(9):1476–1504. doi: 10.1016/j.clinthera.2021.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Satiya J, Snyder HS, Singh SP, Satapathy SK. Narrative review of current and emerging pharmacological therapies for nonalcoholic steatohepatitis. Transl Gastroenterol Hepatol. 2021;6:60–60. doi: 10.21037/tgh-20-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drenth JPH, Schattenberg JM. The nonalcoholic steatohepatitis (NASH) drug development graveyard: established hurdles and planning for future success. Expert Opin Investig Drugs. 2020;29(12):1365–1375. doi: 10.1080/13543784.2020.1839888. [DOI] [PubMed] [Google Scholar]
- 7.FDA-CDER. Noncirrhotic Nonalcoholic Steatohepatitis with Liver Fibrosis: Developing Drugs for Treatment: Guidance for Industry. https://www.fdagov/media/119044/download. 2018;
- 8.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24(1):3–20. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 9.Brunt EM, Neuschwander-Tetri BA, Oliver D, Wehmeier KR, Bacon BR. Nonalcoholic steatohepatitis: Histologic features and clinical correlations with 30 blinded biopsy specimens. Hum Pathol. 2004;35(9):1070–1082. doi: 10.1016/j.humpath.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Fediuk DJ, Nucci G, Dawra VK, Callegari E, Zhou S, Musante CJ, et al. End-to-end application of model-informed drug development for ertugliflozin, a novel sodium-glucose cotransporter 2 inhibitor. CPT Pharmacometrics Syst Pharmacol. 2021;10(6):529–542. doi: 10.1002/psp4.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, Califano A, et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. An NIH White Paper by the QSP Workshop Group. 2011.
- 12.Musante C, Ramanujan S, Schmidt B, Ghobrial O, Lu J, Heatherington A. Quantitative systems pharmacology: a case for disease models. Clin Pharmacol Ther. 2017;101(1):24–27. doi: 10.1002/cpt.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt BJ, Casey FP, Paterson T, Chan JR. Alternate virtual populations elucidate the type I interferon signature predictive of the response to rituximab in rheumatoid arthritis. BMC Bioinformatics. 2013;14(1):221. doi: 10.1186/1471-2105-14-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbiati RA, Pourdehnad M, Carrancio S, Pierce DW, Kasibhatla S, McConnell M, et al. Quantitative systems pharmacology modeling of avadomide-induced neutropenia enables virtual clinical dose and schedule finding studies. AAPS J. 2021;23(5):103. doi: 10.1208/s12248-021-00623-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rieger TR, Musante CJ. Benefits and challenges of a QSP approach through case study: Evaluation of a hypothetical GLP-1/GIP dual agonist therapy. Eur J Pharm Sci. 2016;30(94):15–19. doi: 10.1016/j.ejps.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Allen RJ, Rieger TR, Musante CJ. Efficient generation and selection of virtual populations in quantitative systems pharmacology models. CPT Pharmacometrics Syst Pharmacol. 2016;5(3):140–146. doi: 10.1002/psp4.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guyton AC, Coleman TG, Granger HJ. Circulation: overall regulation. Annu Rev Physiol. 1972;34:13–46. doi: 10.1146/annurev.ph.34.030172.000305. [DOI] [PubMed] [Google Scholar]
- 18.Bai JPF, Earp JC, Florian J, Madabushi R, Strauss DG, Wang Y, et al. Quantitative systems pharmacology: Landscape analysis of regulatory submissions to the US Food and Drug Administration. CPT Pharmacom & Syst Pharma. 2021;10(12):1479–1484. doi: 10.1002/psp4.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azer K, Kaddi CD, Barrett JS, Bai JPF, McQuade ST, Merrill NJ, et al. History and future perspectives on the discipline of quantitative systems pharmacology modeling and its applications. Front Physiol. 2021;25(12):637999. doi: 10.3389/fphys.2021.637999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leil TA, Bertz R. Quantitative systems pharmacology can reduce attrition and improve productivity in pharmaceutical research and development. Front Pharmacol. 2014;5:247. doi: 10.3389/fphar.2014.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ermakov S, Schmidt BJ, Musante CJ, Thalhauser CJ. A survey of software tool utilization and capabilities for quantitative systems pharmacology: what we have and what we need. CPT Pharmacometrics Syst Pharmacol. 2019;8(2):62–76. doi: 10.1002/psp4.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai JPF, Earp JC, Strauss DG, Zhu H. A perspective on quantitative systems pharmacology applications to clinical drug development. CPT Pharmacometrics Syst Pharmacol. 2020;9(12):675–677. doi: 10.1002/psp4.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong X fei, Wang Q yi, Zhao X yan, Sun Y meng, Wu X ning, Yang L ling, et al. Histological assessment based on liver biopsy: the value and challenges in NASH drug development. Acta Pharmacol Sin [Internet]. 2022 Feb 14 [cited 2022 Mar 17]; Available from: https://www.nature.com/articles/s41401-022-00874-x [DOI] [PMC free article] [PubMed]
- 24.Rowe IA, Parker R. The placebo response in randomized trials in nonalcoholic steatohepatitis simply explained. Clin Gastroenterol Hepatol. 2022;20(3):e564–e572. doi: 10.1016/j.cgh.2021.05.059. [DOI] [PubMed] [Google Scholar]
- 25.Ashworth WB, Davies NA, Bogle IDL. A computational model of hepatic energy metabolism: understanding zonated damage and steatosis in NAFLD. Beard DA, editor. PLoS Comput Biol. 2016 Sep 15;12(9):e1005105. [DOI] [PMC free article] [PubMed]
- 26.Liao Y, Davies NA, Bogle IDL. Computational modeling of fructose metabolism and development in NAFLD. Front Bioeng Biotechnol. 2020;22(8):762. doi: 10.3389/fbioe.2020.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holzhütter HG, Berndt N. Computational hypothesis: how intra-hepatic functional heterogeneity may influence the cascading progression of free fatty acid-induced non-alcoholic fatty liver disease (NAFLD) Cells. 2021;10(3):578. doi: 10.3390/cells10030578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta-Moscato J, Solovyev A, Mi Q, Nishikawa T, Soto-Gutierrez A, Fox IJ, et al. A multiscale agent-based in silico model of liver fibrosis progression. frontiers in bioengineering and biotechnology [Internet]. 2014 May 30 [cited 2018 Dec 18];2. Available from: http://journal.frontiersin.org/article/10.3389/fbioe.2014.00018/abstract [DOI] [PMC free article] [PubMed]
- 29.Generaux G. QSP modeling of liver AMPK activation using NAFLDsym is predicted to reduce steatosis in NAFLD patients. Clin Pharmacol Ther. 2017;101(S1):S65. [Google Scholar]
- 30.Siler SQ. Using systems pharmacology modeling to understand the pathophysiology of NAFLD and response to dietary intervention in a simulated population. Clin Pharmacol Ther. 2017;101(S1):S88. [Google Scholar]
- 31.Siler SQ. Using quantitative systems pharmacology modeling to understand the effects of acetyl CoA carboxylase inhibition on liver and plasma triglycerides in a simulated population. Hepatology. 2018;68(S1):448A. [Google Scholar]
- 32.Longo D. Zonal hepatic stellate cell activation in nonalcoholic steatohepatitis characterized by a mathematical model. Hepatology. 2018;68(s1):625A. [Google Scholar]
- 33.Generaux G. Zonal Extracellular Matrix (ECM) accumulation in nonalcoholic steatohepatitis (NASH) characterized by a mathematical model of fibrosis. J Pharmacokinet Pharmacodyn. 2018;45:s17. [Google Scholar]
- 34.Shoda LKM, Woodhead JL, Siler SQ, Watkins PB, Howell BA. Linking physiology to toxicity using DILIsym(®), a mechanistic mathematical model of drug-induced liver injury. Biopharm Drug Dispos. 2014;35(1):33–49. doi: 10.1002/bdd.1878. [DOI] [PubMed] [Google Scholar]
- 35.Woodhead JL, Howell BA, Yang Y, Harrill AH, Clewell HJ, 3rd, Andersen ME, et al. An analysis of N-acetylcysteine treatment for acetaminophen overdose using a systems model of drug-induced liver injury. J Pharmacol Exp Ther. 2012;342(2):529–540. doi: 10.1124/jpet.112.192930. [DOI] [PubMed] [Google Scholar]
- 36.Kenz Z, Howell BA, Dash A, Wong C, Yeh F, Chinn L, et al. Mathematical Modeling with NAFLDsym Supports the Role of Adiponectin in the Reduction of Steatosis by the Anti-FGFR1/KLB Bispecific Antibody. In 2020.
- 37.Battista C, Shoda LKM, Akpinar F, Siler SQ, Generaux G. Quantitative systems pharmacology (QSP) model predicts lack of efficacy for cenicriviroc, a CCR2/5 antagonist, in NAFLD/NASH patients. Hepatology. 2021;74(1S):1149A. [Google Scholar]
- 38.Klein S, Luu K, Gasic S, Green A. Effect of weight lipid metabolism loss on whole body and cellular in severely obese humans. American Journal of Physiology - Endocrinology And Metabolism. 1996;33:739–745. doi: 10.1152/ajpendo.1996.270.5.E739. [DOI] [PubMed] [Google Scholar]
- 39.Mittendorfer B, Magkos F, Fabbrini E, Mohammed BS, Klein S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity (Silver Spring) 2009;17(10):1872–1877. doi: 10.1038/oby.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130(3):1453–1460. doi: 10.1172/JCI134165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cersosimo E, Gastaldelli a, Cervera a, Wajcberg E, Sriwijilkamol a, Fernandez M, et al. Effect of exenatide on splanchnic and peripheral glucose metabolism in type 2 diabetic subjects. The Journal of clinical endocrinology and metabolism. 2011 Jun;96(6):1763–70. [DOI] [PubMed]
- 44.McMahon M, Marsh HM, Rizza R a. Effects of basal insulin supplementation on disposition of mixed meal in obese patients with NIDDM. Diabetes. 1989 Mar;38(3):291–303. [DOI] [PubMed]
- 45.Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14(4):263–283. doi: 10.1002/(SICI)1099-0895(199812)14:4<263::AID-DMR233>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 46.Quiroga AD, Lehner R. Pharmacological intervention of liver triacylglycerol lipolysis: The good, the bad and the ugly. Biochem Pharmacol. 2018;155:233–241. doi: 10.1016/j.bcp.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):424–431. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49(4):755–765. doi: 10.1007/s00125-005-0125-z. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284(22):14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noguchi Y, Young JD, Aleman JO, Hansen ME, Kelleher JK, Stephanopoulos G. Effect of anaplerotic fluxes and amino acid availability on hepatic lipoapoptosis. J Biol Chem. 2009;284(48):33425–33436. doi: 10.1074/jbc.M109.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Sack MN. SIRT3 is regulated by nutrient excess and modulates suscptibility to lipotoxicity. Free Radical Biol Med. 2010;49(7):1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. Differences in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2010;38:2293–2301. doi: 10.1124/dmd.110.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356(2):148–156. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 54.Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126(19):2283–2292. doi: 10.1161/CIRCULATIONAHA.112.104125. [DOI] [PubMed] [Google Scholar]
- 55.Maximos M, Bril F, Portillo Sanchez P, Lomonaco R, Orsak B, Biernacki D, et al. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology (Baltimore, MD) 2015;61(1):153–160. doi: 10.1002/hep.27395. [DOI] [PubMed] [Google Scholar]
- 56.Aida Y, Abe H, Tomita Y, Nagano T, Seki N, Sugita T, et al. Serum cytokeratin 18 fragment level as a noninvasive biomarker for non-alcoholic fatty liver disease. Int J Clin Exp Med. 2014;7(11):4191–4198. [PMC free article] [PubMed] [Google Scholar]
- 57.Longo DM, Generaux GT, Howell BA, Siler SQ, Antoine DJ, Button D, et al. Refining liver safety risk assessment: application of mechanistic modeling and serum biomarkers to cimaglermin alfa (GGF2) clinical trials. Clin Pharmacol Ther. 2017;102(6):961–969. doi: 10.1002/cpt.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150(4):956–967. doi: 10.1053/j.gastro.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. CMGH Cellular and Molecular Gastroenterology and Hepatology. 2015;1(1):17–27. doi: 10.1016/j.jcmgh.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, et al. Circulating extracellular vesicles with specific proteome and Liver MicroRNAs are potential biomarkers for liver injury in experimental fatty liver disease. Sookoian SC, editor. PLoS ONE. 2014 Dec 3;9(12):e113651. [DOI] [PMC free article] [PubMed]
- 61.Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology [Internet]. 2017 Sep 21 [cited 2017 Nov 10]; Available from: http://doi.wiley.com/10.1002/hep.29544 [DOI] [PubMed]
- 62.Tajiri K, Shimizu Y, Tsuneyama K, Sugiyama T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease: European Journal of Gastroenterology & Hepatology. 2009 Jun;21(6):673–80. [DOI] [PubMed]
- 63.Zahran WE, Salah El-Dien KA, Kamel PG, El-Sawaby AS. Efficacy of tumor necrosis factor and interleukin-10 analysis in the follow-up of nonalcoholic fatty liver disease progression. Indian J Clin Biochem. 2013;28(2):141–146. doi: 10.1007/s12291-012-0236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das SK, Balakrishnan V. Role of cytokines in the pathogenesis of non-alcoholic fatty liver disease. Indian J Clin Biochem. 2011;26(2):202–209. doi: 10.1007/s12291-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology (Baltimore, MD) 2004;40(1):46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY, et al. Extrahepatic platelet-derived growth factor-β, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014;147(6):1378–1392. doi: 10.1053/j.gastro.2014.08.038. [DOI] [PubMed] [Google Scholar]
- 67.Miele L, Forgione A, La Torre G, Vero V, Cefalo C, Racco S, et al. Serum levels of hyaluronic acid and tissue metalloproteinase inhibitor-1 combined with age predict the presence of nonalcoholic steatohepatitis in a pilot cohort of subjects with nonalcoholic fatty liver disease. Transl Res. 2009;154(4):194–201. doi: 10.1016/j.trsl.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 68.Ando W, Yokomori H, Tsutsui N, Yamanouchi E, Suzuki Y, Oda M, et al. Serum matrix metalloproteinase-1 level represents disease activity as opposed to fibrosis in patients with histologically proven nonalcoholic steatohepatitis. Clin Mol Hepatol. 2018;24(1):61–76. doi: 10.3350/cmh.2017.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao XK, Yu L, Cheng ML, Che P, Lu YY, Zhang Q, et al. Focal Adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci Rep. 2017;7(1):4032. doi: 10.1038/s41598-017-04317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Washington K, Wright K, Shyr Y, Hunter EB, Olson S, Raiford DS. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol. 2000;31(7):822–828. doi: 10.1053/hupa.2000.8440. [DOI] [PubMed] [Google Scholar]
- 71.Abdeen SM, Olusi SO, Askar HA, Thalib L, Al-Azemi A, George S. The predictive value of CD38 positive hepatic stellate cell count for assessing disease activity and fibrosis in patients with chronic hepatitis. Acta Histochem. 2009;111(6):520–530. doi: 10.1016/j.acthis.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 72.El-Gendi SM, Shaaban AAA, Ali ES. CD38+ hepatic stellate cell count as a predictor of disease activity and fibrosis in chronic hepatitis C virus patients: Egyptian Journal of Pathology. 2012 Jul;32(1):59–67.
- 73.Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Caligiuri A, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;178(3):785–800. [PMC free article] [PubMed]
- 74.Decaris ML, Li KW, Emson CL, Gatmaitan M, Liu S, Wang Y, et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood: Decaris et al. Hepatology. 2017 Jan;65(1):78–88. [DOI] [PMC free article] [PubMed]
- 75.Boyle M, Tiniakos D, Schattenberg JM, Ratziu V, Bugianessi E, Petta S, et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. 2019;1(3):188–198. doi: 10.1016/j.jhepr.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Decaris ML, Emson CL, Li K, Gatmaitan M, Luo F, Cattin J, et al. Turnover rates of hepatic collagen and circulating collagen-associated proteins in humans with chronic liver disease. PLoS ONE. 2015;10(4):e0123311. doi: 10.1371/journal.pone.0123311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geervliet E, Bansal R. Matrix metalloproteinases as potential biomarkers and therapeutic targets in liver diseases. Cells. 2020 May 13;9(5). [DOI] [PMC free article] [PubMed]
- 78.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 79.Brunt EM, Neuschwander-Tetri BA. Nonalcoholic Steatohepatitis: A Proposal for Grading and Staging the Histological Lesions. 1999;94(9):8. [DOI] [PubMed]
- 80.du Plessis J, Korf H, van Pelt J, Windmolders P, Vander Elst I, Verrijken A, et al. Pro-inflammatory cytokines but not endotoxin-related parameters associate with disease severity in patients with NAFLD. Strnad P, editor. PLOS ONE. 2016 Dec 19;11(12):e0166048. [DOI] [PMC free article] [PubMed]
- 81.Middleton MS, Heba ER, Hooker CA, Bashir MR, Fowler KJ, Sandrasegaran K, et al. Agreement between magnetic resonance imaging proton density fat fraction measurements and pathologist-assigned steatosis grades of liver biopsies from adults with nonalcoholic steatohepatitis. Gastroenterology. 2017;153(3):753–761. doi: 10.1053/j.gastro.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Permutt Z, Le TA, Peterson MR, Seki E, Brenner DA, Sirlin C, et al. Correlation between liver histology and novel magnetic resonance imaging in adult patients with non-alcoholic fatty liver disease - MRI accurately quantifies hepatic steatosis in NAFLD. Aliment Pharmacol Ther. 2012;36(1):22–29. doi: 10.1111/j.1365-2036.2012.05121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwimmer JB, Middleton MS, Behling C, Newton KP, Awai HI, Paiz MN, et al. Magnetic resonance imaging and liver histology as biomarkers of hepatic steatosis in children with nonalcoholic fatty liver disease. Hepatology. 2015;61(6):1887–1895. doi: 10.1002/hep.27666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feldstein A, Kleiner D, Kravetz D, Buck M. Severe hepatocellular injury with apoptosis induced by a hepatitis C polymerase inhibitor. J Clin Gastroenterol. 2009;43(4):374–381. doi: 10.1097/MCG.0b013e318178d91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology. 2015;149(3):635–648.e14. doi: 10.1053/j.gastro.2015.05.044. [DOI] [PubMed] [Google Scholar]
- 86.Albhaisi S, Sanyal AJ. Applying non-invasive fibrosis measurements in NAFLD/NASH: progress to date. Pharmaceut Med. 2019;33(6):451–463. doi: 10.1007/s40290-019-00305-z. [DOI] [PubMed] [Google Scholar]
- 87.Alkhouri N, Tincopa M, Loomba R, Harrison SA. What does the future hold for patients with nonalcoholic steatohepatitis: diagnostic strategies and treatment options in 2021 and beyond? Hepatol Commun. 2021;5(11):1810–1823. doi: 10.1002/hep4.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology. 2004;40(6):1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 89.Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2(11):901–910. doi: 10.1016/S2213-8587(14)70032-4. [DOI] [PubMed] [Google Scholar]
- 90.Lee JJ, Lambert JE, Hovhannisyan Y, Ramos-Roman MA, Trombold JR, Wagner DA, et al. Palmitoleic acid is elevated in fatty liver disease and reflects hepatic lipogenesis. Am J Clin Nutr. 2015;101(1):34–43. doi: 10.3945/ajcn.114.092262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29(5):478–485. doi: 10.1016/S1262-3636(07)70061-7. [DOI] [PubMed] [Google Scholar]
- 92.Pérez-Carreras M, Del Hoyo P, Martín M a, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md). 2003 Oct;38(4):999–1007. [DOI] [PubMed]
- 93.Tushuizen ME, Pouwels PJ, Bontemps S, Rustemeijer C, Matikainen N, Heine RJ, et al. Postprandial lipid and apolipoprotein responses following three consecutive meals associate with liver fat content in type 2 diabetes and the metabolic syndrome. Atherosclerosis. 2010;211(1):308–314. doi: 10.1016/j.atherosclerosis.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 94.McQuaid SE, Hodson L, Neville MJ, Dennis AL, Cheeseman J, Humphreys SM, et al. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes. 2011;60(1):47–55. doi: 10.2337/db10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mittendorfer B, Patterson BW, Klein S. Effect of sex and obesity on basal VLDL-triacylglycerol kinetics. Am J Clin Nutr. 2003;77(3):573–579. doi: 10.1093/ajcn/77.3.573. [DOI] [PubMed] [Google Scholar]
- 96.Sane T, Nikkilä EA. Very low density lipoprotein triglyceride metabolism in relatives of hypertriglyceridemic probands. Evidence for genetic control of triglyceride removal. Arteriosclerosis. 1988 Jun;8(3):217–26. [DOI] [PubMed]
- 97.Beil U, Grundy SM, Crouse JR, Zech L. Triglyceride and cholesterol metabolism in primary hypertriglyceridemia. Arteriosclerosis. 1982;2(1):44–57. doi: 10.1161/01.ATV.2.1.44. [DOI] [PubMed] [Google Scholar]
- 98.Videla L a, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clinical science (London, England : 1979). 2004;106(3):261–8. [DOI] [PubMed]
- 99.Tanaka S, Miyanishi K, Kobune M, Kawano Y, Hoki T, Kubo T, et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J Gastroenterol. 2013;48(11):1249–1258. doi: 10.1007/s00535-012-0739-0. [DOI] [PubMed] [Google Scholar]
- 100.Bantel H, Lügering A, Heidemann J, Volkmann X, Poremba C, Strassburg CP, et al. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology (Baltimore, MD) 2004;40(5):1078–1087. doi: 10.1002/hep.20411. [DOI] [PubMed] [Google Scholar]
- 101.Balaban YH. Metabolic syndrome, non-alcoholic steatohepatitis (NASH), and hepatocyte growth factor (HGF) Ann Hepatol. 2006;5(2):109–14. doi: 10.1016/S1665-2681(19)32027-7. [DOI] [PubMed] [Google Scholar]
- 102.Agrawal R, Sheroan V, Ola V, Sulemani A, Singh N, Sirohi P, et al. Hepatocyte growth factor, adiponectin and hepatic histopathology in non-alcoholic steatohepatitis. J Assoc Physicians India. 2013;61:4. [PubMed] [Google Scholar]
- 103.Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: Current and emerging. J Hepatol. 2018;68(2):362–375. doi: 10.1016/j.jhep.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 104.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology. 2015 Aug;149(2):367–78.e5; quiz e14–15. [DOI] [PubMed]
- 105.Sevastianova K, Santos A, Kotronen A, Hakkarainen A, Makkonen J, Silander K, et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am J Clin Nutr. 2012;96(4):727–734. doi: 10.3945/ajcn.112.038695. [DOI] [PubMed] [Google Scholar]
- 106.Browning JD, Baker J a, Rogers T, Davis J, Satapati S, Burgess SC. Short-term weight loss and hepatic triglyceride reduction : evidence of a metabolic advantage with dietary carbohydrate restriction 1 – 3. American Journal of Clinical Nutrition. 2011;93:1048–52. [DOI] [PMC free article] [PubMed]
- 107.Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology (Baltimore, MD) 2010;51(1):121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hall KD. Predicting metabolic adaptation, body weight change, and energy intake in humans. AJP: Endocrinology and Metabolism. 2010 Mar 1;298(3):E449–66. [DOI] [PMC free article] [PubMed]
- 109.McMurray RG, Ben-Ezra V, Forsythe WA, Smith AT. Responses of endurance-trained subjects to caloric deficits induced by diet or exercise. Med Sci Sports Exerc. 1985;17(5):574–579. doi: 10.1249/00005768-198510000-00010. [DOI] [PubMed] [Google Scholar]