

Abstract
Extracellular vesicles (EVs) have shown promise as potential therapeutics for the treatment of various diseases. However, their rapid clearance after administration could be a limitation in certain therapeutic settings. To solve this, an engineering strategy is employed to decorate albumin onto the surface of the EVs through surface display of albumin binding domains (ABDs). ABDs were either included in the extracellular loops of select EV‐enriched tetraspanins (CD63, CD9 and CD81) or directly fused to the extracellular terminal of single transmembrane EV‐sorting domains, such as Lamp2B. These engineered EVs exert robust binding capacity to human serum albumins (HSA) in vitro and mouse serum albumins (MSA) after injection in mice. By binding to MSA, circulating time of EVs dramatically increases after different routes of injection in different strains of mice. Moreover, these engineered EVs show considerable lymph node (LN) and solid tumour accumulation, which can be utilized when using EVs for immunomodulation, cancer‐ and/or immunotherapy. The increased circulation time of EVs may also be important when combined with tissue‐specific targeting ligands and could provide significant benefit for their therapeutic use in a variety of disease indications.
Keywords: albumin binding domains, circulation time, extracellular vesicles, lymph node accumulation, tetraspanins
![]()
1. INTRODUCTION
Extracellular vesicles (EVs) are lipid‐bilayer membrane particles released from almost all types of mammalian cells (Van Niel et al., 2018). Apart from the established role in cell‐cell or inter‐organ communications, an increasing number of studies are unravelling the capacity of EVs to functionally deliver biological cargos, such as proteins and nucleic acids (Kamerkar et al., 2017; Keller et al., 2020; Lathwal et al., 2021; Poggio et al., 2019; Zhang et al., 2019). They have also been successfully loaded with various small molecules and clinically used drugs. EVs have been studied for vaccine development, and dendritic cell‐(DC) derived EVs have been shown to modulate inflammation and be exploited as immunotherapy (Lindenbergh et al., 2020; Mehanny et al., 2021; Zitvogel et al., 1998). These characteristics highlight the potential therapeutic application of EVs (Fuhrmann et al., 2015; Vader et al., 2016). However, the half‐life of EVs after injection constitutes a potential barrier for certain clinical applications (Matsumoto et al., 2020). This rapid clearance in bloodstream is attributed to the considerable scavenging by monocyte/macrophage or reticuloendothelial system (RES) (Imai et al., 2015; Willekens et al., 2005). The half‐life of mouse plasma‐derived small EVs was revealed to be only about 7 min and tumour‐derived exosomes have been shown to be cleared from the circulation even faster, with a half‐life as short as 2 min (Matsumoto et al., 2020; Takahashi et al., 2013). To circumvent this hurdle and extend the circulation time of EVs, many strategies are being developed (Lathwal et al., 2021; Watson et al., 2016).
As an example, surface display of CD47 was used to protect EVs from phagocytosis by monocytes and macrophages and thereby enhanced retention of EVs in the circulation (Belhadj et al., 2020; Kamerkar et al., 2017; Wei et al., 2021). However, CD47 is expressed by normal red blood cells and platelets to prevent phagocytosis by binding to the signal‐regulatory protein alpha (SIRPα) on the surface of macrophages, eliciting “don't eat me” signals (Catani et al., 2011; Ishikawa‐Sekigami et al., 2006; Oldenborg, 2004). If the EV surface CD47 occupied the SIRPα sites, the normal red blood cells could be phagocytosed by macrophages, holding potential to decrease red blood cell count (Buatois et al., 2018; Chao et al., 2010). Another strategy used to extend circulation time of EVs is to use polyethylene glycol (PEG) conjugation or hydrogel encapsulation (Gómez‐Cid et al., 2021; Mardpour et al., 2019). PEGylated EVs were shown to have enhanced circulation time and could be detected 60 min post‐injection compared to only 10 min for the unmodified EVs (Kooijmans et al., 2016). However, there are concerns about the toxicity and immunogenicity related to PEGylated‐micelle carrier system, associated with the development of anti‐PEG antibodies (Kawaguchi et al., 2009; Shiraishi & Yokoyama, 2019). There is thus an unmet need to develop new strategies to extend the circulation time of EVs without significant adverse effects.
Here, we proposed to decorate EVs with albumin to extend the circulation time of EVs. The half‐life of human albumin is as long as 3 weeks (Figure 1A) (Andersen et al., 2013) and conjugation of albumin binding domains (ABDs) or albumin decoration have been widely used to extend circulation time of therapeutic proteins or synthetic nanoparticles (Ishihara et al., 2021; Ma et al., 2019; Mehta et al., 2020; Nessler et al., 2020; Wei et al., 2018; Yousefpour et al., 2018). To promote binding of albumin to EVs, we inserted engineered ABDs (Jonsson et al., 2008; Nilvebrant & Hober, 2013b) into the extracellular loops of tetraspanins or directly fused ABD with single‐transmembrane EV‐sorting domains (Figure 1B). The engineered ABD‐EVs bound to human serum albumin (HSA) with high affinity in vitro and bound to mouse serum albumins (MSA) after injection in mice (Figure 1A). The MSA‐bound EVs substantially enhanced the circulation time and accumulated in lymph‐nodes and solid tumours (Figure 1A), similar to the findings demonstrated by other studies using ABD‐conjugation (Liu et al., 2014; Nessler et al., 2020; Rivoltini et al., 2016; Wang et al., 2018, 2015). In summary, we have developed a novel approach to extend the circulation time of EVs and enrich EVs in lymph‐nodes, thus overcoming one of the main hurdles in utilizing EVs for therapeutic applications.
FIGURE 1.
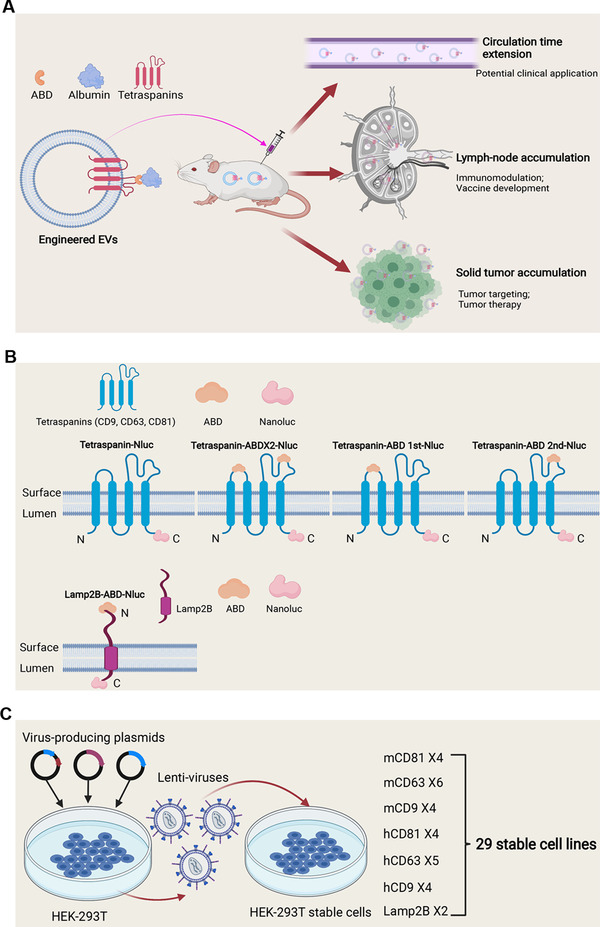
Schematic illustration of the aims, engineering strategy and generation of stable cell lines for the study. (A) Schematic graph of the hypothesis. (B) Strategies to engineer EVs with ABDs. (C) Generation of stable cells for EV production by lentiviral transduction
2. RESULTS
2.1. Strategy to engineer EVs with albumin binding capacity
To achieve binding of engineered EVs with albumin, we exploited the widely used engineered ABDs with affinity to albumin from different species (Johansson et al., 2002; Jonsson et al., 2008), which were inserted into the extracellular loops of tetraspanins (Figure 1B). Mouse and human CD9, CD63, and CD81 were either engineered with ABDs in both loops (ABD×2), or a single ABD in each respective loop, designated as ABD 1st and ABD 2nd (Figure 1B) and fused to the reporter protein nanoluc‐luciferase (Nluc) for high‐sensitivity detection. For the production of EVs, a total of 29 stable cell lines derived from HEK‐293T cells were generated by lenti‐viral transduction, as shown in Figure 1C. To validate the expression of the transgenes in the stable cells, as well as in the corresponding isolated EVs, protein expression was measured by western blotting (Figure 2A‐F). The expression of Nluc in the stable cells did not always correspond to the same expression profile in their respective EVs, demonstrated most distinctly by the faint bands of Nluc in the cells engineered with mouse and human CD81 construct, where the EVs demonstrated higher levels. Interestingly, constructs in which the ABD was inserted into loop 1 expressed at lower level in EVs, compared to constructs containing ABD in the second or both loops. (Figure 2A and D). Both stable cells and isolated EVs expressed CD9 transgenes well as demonstrated in Figure 2C and E, with expression detected even in ABD 1st as well as the other groups. The stable cells with mouse CD63 (mCD63) were positive for Nluc, but no expression could be observed when the ABD was in the first loop or the ABDs included in both loops (Figure 2B), suggesting potential structural change that led to failure of loading. This was, however, not further investigated. In contrast, all the hCD63 transgenes were significantly expressed in the isolated EVs (Figure 2E), indicating that the tetraspanins respond differently to loop‐engineering, despite the high species conservation between human and mouse (Tomlinson, 2009).
FIGURE 2.
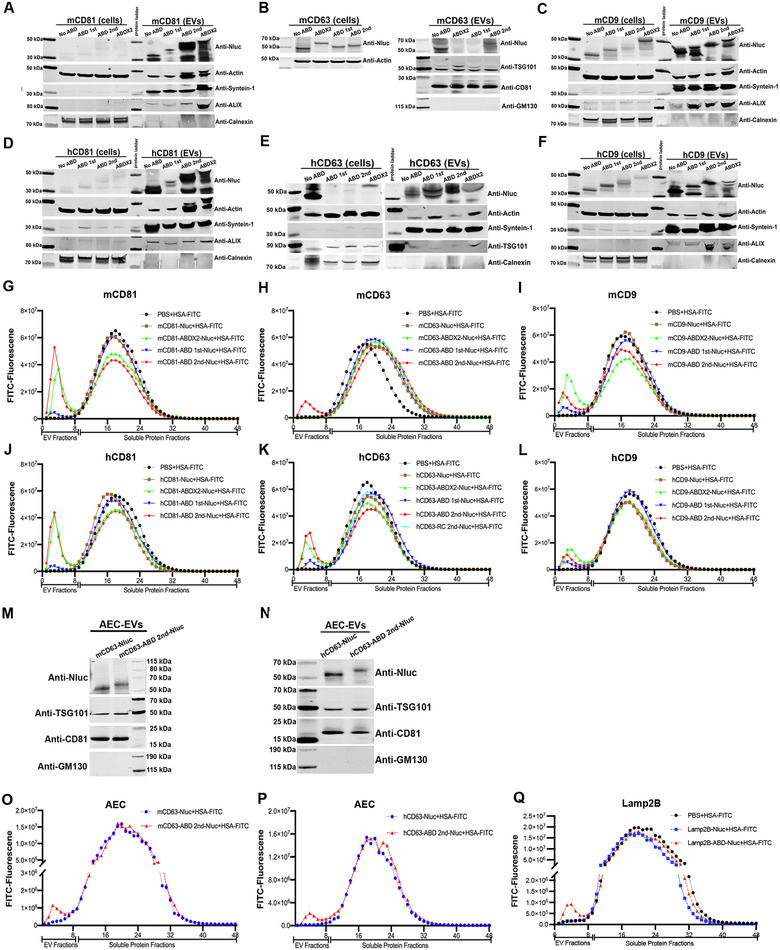
Engineering strategies to generate EVs with albumin binding capacity. (A‐F) Protein expression of transgenes in EV‐producing cells and corresponding isolated EVs (A: mCD81, B: mCD63, C: mCD9, D: hCD81, E: hCD63 and F: hCD9). Cell lysate from 5×105 cells and 1×1010 EVs were used for Western blotting (WB) assay, respectively. (G‐L). The binding of ABD‐engineered EVs to HSA‐FITC (G: mCD81, H: mCD63, I: mCD9, J: hCD81, K: hCD63 and L: hCD9). (M). Protein expression of mCD63 2nd ABD construct in AEC‐derived EVs. (N). Protein expression of hCD63 2nd ABD construct in AEC‐derived EVs. (O) The binding of mCD63 ABD‐engineered EVs derived from AEC cells with HSA‐FITC. (P) The binding of hCD63 ABD‐engineered EVs derived from AEC cells with HSA‐FITC. (Q) The binding of Lamp2B ABD‐engineered EVs derived from HEK‐293T cells to HSA‐FITC
Next, we evaluated the binding of the ABD‐engineered EVs to commercially available FITC‐labelled human serum albumin (HSA‐FITC). The isolated EVs were incubated with HSA‐FITC followed by size exclusion chromatography (SEC). During elution, 48 fractions were collected from each sample, with 300 μl per fraction, as previously described (Gupta et al., 2020). The first nine fractions are considered to contain EVs (EV fractions), corroborated by luciferase intensity (Figure S1A‐F), indicating the successful loading of fused proteins by ABD engineering. In contrast, fraction 10–48 mainly contain soluble proteins (soluble protein fractions). The fractions were subsequently examined for fluorescence and a peak in the EV fractions thus indicative of colocalization and was considered as confirmation of binding of EVs to albumin. As shown in Figure 2G‐L, almost all the ABD‐engineered EVs bound to HSA‐FITC except the mCD63‐ABD 1st and mCD63‐ABD×2 groups. These results were consistent with the protein expression assay (Figure 2A‐F).
In most of the groups, ABD in the second loop showed the best binding capacity to albumin, while ABD in the first loop had the lowest albumin binding, thus indicating that the second tetraspanin loop is preferred for ABD engineering. To our knowledge, this is the first report showing differences in functionality in regards to which extracellular loop of tetraspanins that is being engineered, providing useful information when conducting tetraspanin loop‐engineering.
To extend these findings, which were obtained using HEK‐293T cells, the ABD‐engineering was also conducted in another therapeutically relevant cell type, amniotic epithelial cells (AEC). Following transient transfection, the isolated EVs were used for protein expression assay and the expression of the transgenes was confirmed (Figure 2 M and N). In addition, and as expected, the luciferase intensities were found only in the EV fractions (Figure S1G‐H). The albumin binding assay was conducted as described above, showing successful albumin binding to AEC derived ABD‐engineered EVs (Figure 2O and P), indicating that the albumin binding capacity of ABD‐engineered EVs is not cell source‐dependent. Following this, Lamp2B, a single‐pass transmembrane EV‐sorting domain, commonly used for EV surface display of targeting ligands, was assessed as a comparison to the tetraspanins. Following successful engineering (Figure S1I), the binding of Lamp2B‐ABD‐EVs with albumin was confirmed (Figure 2Q), indicating the versatility of the ABD engineering strategy. Theoretically, any EV sorting domain with ABD on the surface of EVs would bind to albumin.
The engineered EVs from both HEK‐293T and AEC cells were characterized by Nanoparticle tracking analysis (NTA) and the size distribution of EVs was not affected by the introduction of ABD on the surface (Figure S2A‐D). Moreover, transmission electron microscope (TEM) was used to assess the integrity and shape of the ABD‐engineered vesicles. As shown in Figure S2E, the ABD‐engineered EVs remained intact and spherically shaped, similar to the EVs from control group. However, after incubating with HSA, the ABD‐engineered EVs exhibited coating of proteins on the surface (Figure S2E), further confirming HSA binding. Taken together, ABD‐engineering of EVs impart albumin binding capacity without changing the EV characteristics.
2.2. ABD‐engineered EVs bind to HSA in vitro
To further confirm the binding of ABD‐EVs to albumin, widefield imaging was performed on EVs isolated from engineered HEK‐293T cells and the procedure of this experiment was shown in Figure 3SA. The EVs were incubated with Alexa Fluor 488‐conjugated HSA and a mixture of fluorescent antibodies against CD9/CD63/CD81 (Alexa Fluor 647‐conjugated), to visualize all EVs. Optiprep gradient (density range of 14–36%) centrifugation was used to separate vesicular components from unbound HSA and fluorescent antibodies. The fractions of EVs were then collected and placed into 18‐well glass bottom chambers to allow EVs to adhere to the glass and subsequently imaged with Nanoimager (Figure 3SA). The fractions of EVs used for imaging were all from Optiprep gradient ultracentrifugation, which were different from that got from SEC used for fluorescent intensity and Nluc activity evaluation. Both mCD63 and hCD63 ABD‐engineered EVs co‐localized with HSA, except for the mCD63‐ABD 1st and mCD63‐ABD×2 groups (Figure 3A and B), corroborating the expression profile previously shown (Figure 2B and H). Of note, the imaging data were well‐matched to the data of the fluorescent intensity assay. For instance, mCD9 ABD×2 and hCD9 ABD×2 groups showed the best binding while ABD in the second loop exhibited the best binding for other groups (Figure 2G‐L; Figure 3A and B; Figure S3‐S6). Furthermore, super‐resolution (dSTORM) microscopy was performed, showing single vesicles bound to a high amount of albumin, confirming the decoration of albumin on the surface of ABD‐engineered EVs (Figure 3C).
FIGURE 3.
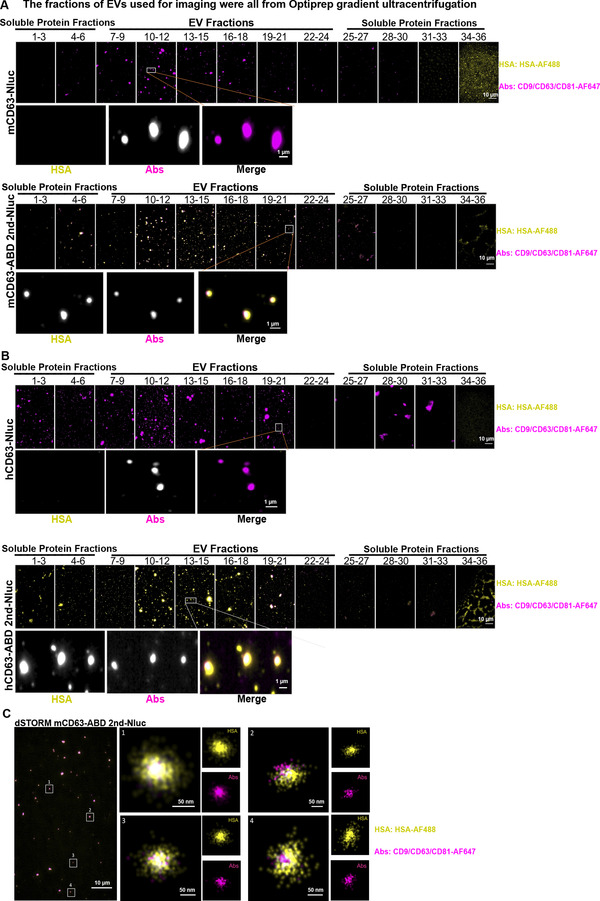
ABD‐engineered EVs bind to HSA in vitro. (A) mCD63‐ABD EVs were bound by HSA as detected by widefield imaging of single vesicles. The upper and lower panels indicated the binding of mCD63‐Nluc EVs and mCD63‐ABD 2nd‐Nluc EVs to HSA, respectively. AF647‐labelled CD9/CD63/CD81 antibodies cocktail was exploited to detect EVs while AF488‐labelled HSA was used to show the co‐localization of EVs with HSA. (B) hCD63‐ABD EVs were bound by HSA detected by widefield imaging of single vesicles. The binding of hCD63‐Nluc EVs and hCD63‐ABD 2nd‐Nluc EVs with HSA was indicated in the upper and lower panels, respectively. (C) ABD EVs bound to HSA as detected by super‐resolution imaging (dSTORM) of single vesicles
2.3. ABD‐engineered EVs bind to MSA in vivo after injection
Since the ABD‐engineered EVs exhibited robust albumin binding capacity in vitro, we next sought to assess their albumin binding capacities in vivo. For this, plasma was drawn from mice 10 min after injection of ABD‐engineered (mCD63‐ABD 2nd) EVs into NMRI mice (Figure 4A). The plasma was then incubated with FITC‐labelled mouse albumin antibody (MSA Ab‐FITC). Subsequently, the plasma was separated by SEC and 48 fractions were collected for fluorescence and luciferase detection, as previously described (Figure 2G‐L; Figure S1A‐F). Even though the components of plasma were more complex than the isolated EVs, a fluorescence peak in the EV fractions could be detected (Figure 4B), indicating the binding of MSA with injected ABD‐EVs. Most of the Nluc luciferases were found in the EV fractions (Figure 4C), implying that intact EVs remained in the plasma. Similarly, the widefield imaging was used to examine the binding of the injected ABD‐EVs to MSA and almost all the detected EVs were decorated with MSA in vivo (Figure 4D). These results provided a strong basis for evaluating the potential circulation time extension effects of the ABD‐EVs considering MSA has a half‐life as long as 35 h.
FIGURE 4.
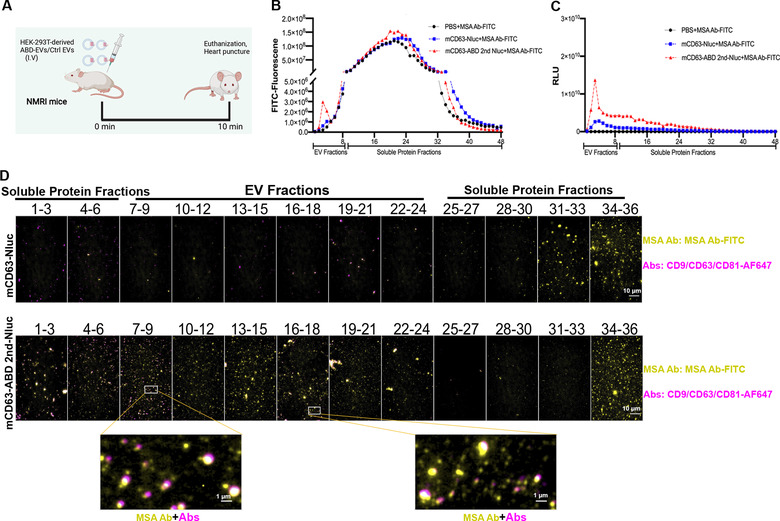
ABD‐engineered EVs bind to MSA in vivo after injection. (A) Schematic illustration of the animal experimental method. Plasma was harvested 10 min after I.V injection. (B) The binding assay for the injected ABD‐EVs with MSA after injection. FITC‐labelled MSA antibody was used to incubate with the plasma and SEC was performed to get different fractions. (C) Nluc luciferase activity of the injected EVs. (D) Single vesicles bound to MSA after injection detected by widefield imaging. AF647‐labelled CD9/CD63/CD81 antibodies cocktail was exploited to detect EVs whereas FITC‐labelled MSA antibody was used to confirm the co‐localization of EVs with MSA
2.4. Albumin‐decoration by displaying ABD on the surface of EVs extended circulation time in vivo
In order to evaluate the circulation time in vivo, a small screen of the tetraspanin ABD 2nd‐enginnered EVs was conducted according to the experimental plan presented in Figure 5A. As shown in Figure 5B, mCD63 and hCD63 engineered EVs exhibited the most significant fold‐increase (> 10‐fold) compared to the corresponding control groups in terms of circulating concentrations at 4.5 h after injection of the EVs, pointing to a retention in plasma by the binding to MSA. The mCD81 and hCD81‐ABD EVs showed the lowest fold‐increase (Figure 5B). Engineered EVs from hCD9 had around 10‐fold increase of circulation concentration while the mCD9 EVs had a slightly lower increase (about 6‐fold) (Figure 5B). Based on this, ABD‐EVs from mCD63 and hCD63 groups were chosen for further investigation. Different injection routes (intravenously (I.V), intraperitoneally (I.P) and subcutaneously (S.C)) were assessed for mCD63 ABD‐EVs derived from HEK‐293T cells. Regardless of the route of administration, it was found that ABD‐EVs were present at a higher concentration in plasma at the investigated time points, as compared to control EVs, with a significant overall fold increase (Figure 5C‐E). Similarly, ABD‐EVs from hCD63 and Lamp2B groups showed significant circulation time extension effects at different time points after I.V injection (Figure 5F and G). To further confirm the results, ABD‐EVs derived from AEC cells were intravenously injected into NMRI mice and the extended circulation time was again observed (Figure 5H‐J), though to a lower extent. We next tried another mouse strain, C57BL/6, and similar circulation time extension effects were achieved by using mCD63 engineered ABD‐EVs from both HEK‐293T and AEC cells after 2 h of I.V injection (Figure 5K‐M).
FIGURE 5.
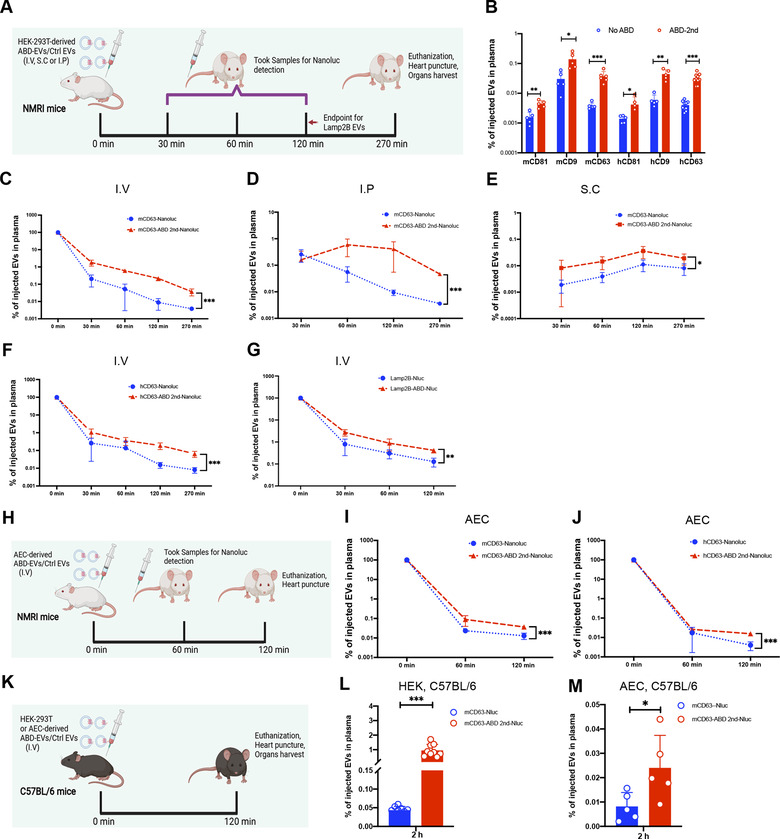
Albumin‐decoration by displaying ABD on the surface of EVs extends circulation time in vivo. (A) Illustration of the animal experimental set‐up with indicated injection routes and time points for sample collection. (B) Screen of best tetraspanin candidates for engineering to extend circulation time of EVs, N = 5 for mCD81, mCD9, hCD81 and hCD9; N = 6 for mCD63; N = 8 for hCD63. The endpoint of this experiment was 270 min after I.V injection. (C‐E) Circulation time extension by mCD63‐ABD engineered EVs after different routes of injections (C: I.V, N = 6, D: I.P, N = 5, and E: S.C, N = 5), endpoint = 270 min. (F) Extension of circulation time by hCD63‐ABD engineered EVs after I.V injection, N = 9, endpoint = 270 min. (G) Lamp2B‐ABD engineered EVs extended their circulation time after I.V injection, N = 5, endpoint = 120 min. (H) Illustration of the animal experimental set‐up for the injection of AEC derived EVs. (I) mCD63‐ABD engineered EVs derived from AEC cells exert circulation time extension after I.V injection, N = 5, endpoint = 120 min. (J) Circulation time extension by hCD63‐ABD engineered EVs derived from AEC cells after I.V injection, N = 5, endpoint = 120 min. (K) Illustration of the animal experimental outline. (L) Circulation amounts of injected mCD63‐ABD engineered EVs derived from HEK‐293 cells after I.V injection in C57BL/6 mice, N = 9, endpoint = 120 min. (M) Plasma concentration of injected mCD63‐ABD engineered EVs derived from AEC cells after I.V injection in C57BL/6 mice, N = 5, endpoint = 120 min. N: number of mice used. * P < 0.05; ** P < 0.01; *** P < 0.001
2.5. Albumin‐decorated EVs accumulated in LNs and tumours
Published literature clearly demonstrate that ABD‐conjugation or albumin decorated nanoparticles accumulate in LNs and solid tumours (Liu et al., 2014; Nessler et al., 2020; Rivoltini et al., 2016; Wang et al., 2018, 2015). Therefore, we harvested the inguinal lymph nodes (ILNs) or solid tumours after ABD‐EVs injection for luciferase activity detection. The HEK‐293T‐derived ABD‐EVs from both mCD63 and hCD63 groups showed significant increase of Nluc signals (RLU/mg) in the ILNs of NMRI mice as well as in C57BL/6 mice following I.V injection (Figure 6A‐C). Similar increase was found when using AEC‐derived ABD‐engineered EVs from both mCD63 and hCD63 groups (Figure 6D and E). In addition, I.P injection of HEK‐293T‐derived mCD63 ABD‐EVs also resulted in increased accumulation of EVs in ILNs as compared to control EVs (Figure 6F). To confirm solid tumour accumulation of EVs, we performed in vitro uptake of the ABD‐EVs in B16F10 cells. As shown in Figure S7A, the uptake of ABD‐engineered EVs significantly increased at investigated time points. And then we subcutaneously inoculated B16F10 cells in C57BL/6 mice and subsequentially injected the ABD‐engineered EVs after tumour formation. Compared to control EVs, both HEK‐293T and AEC‐derived mCD63 ABD‐EVs accumulated more in tumoural tissues (Figure 6G and H). The property of ABD‐engineered EVs accumulating in ILNs may potentially be harnessed for immunomodulation where the EVs are equipped with immune‐regulation moieties. Their ability to accumulate in tumoural tissue allows for the delivery of a more concentrated dose of tumour‐killing chemical drugs when the ABD‐engineered EVs are packaged with bioactive payloads, possibly leading to the eradication of solid tumours. In addition, ABD‐engineering combined with tumour targeting moieties could enhance the penetration of EVs inside tumours and thus hold promise to develop efficient tumour‐therapeutic EVs.
FIGURE 6.
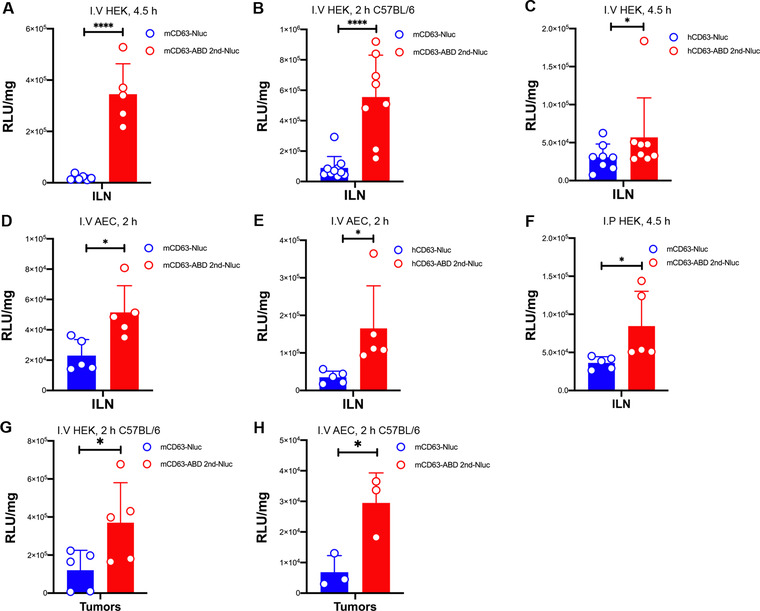
Albumin‐binding EVs accumulate in ILNs and tumours. All graphs depict Nluc luciferase activity in RLU/mg of harvested ILNs or tumours. (A) Nluc activity in ILNs at 4.5 h after I.V injection of mCD63‐ABD EVs derived from HEK‐293T cells in NMRI mice, N = 5. (B) 2 h after I.V injection of mCD63‐ABD EVs derived from HEK‐293T cells in C57BL/6 mice, luciferase activity was evaluated in ILNs, N = 9 for control group and N = 8 for ABD group. (C) ILNs were harvested at 4.5‐h time point after I.V injection of hCD63‐ABD EVs derived from HEK‐293T cells in NMRI mice and then Nluc activity was evaluated, N = 8. (D) Luciferase activity determined in ILNs after 2 h of I.V injection of mCD63‐ABD EVs derived from AEC cells in NMRI mice, N = 5. (E) hCD63‐ABD EVs derived from AEC cells were injected (I.V) into NMRI mice for 2 h, and then Nluc activity in ILNs was measured, N = 5. (F) Harvested ILNs in NMRI mice were subjected to luciferase activity measurements after 4.5 h of I.P injection of mCD63‐ABD EVs derived from HEK‐293T cells, N = 5. (G) Nluc activity in B16F10 melanoma after 2 h of I.V injection of mCD63‐ABD EVs derived from HEK‐293T cells in C57BL/6 mice, N = 5. (H) B16F10 melanomas were harvested after 2 h of I.V injection of mCD63‐ABD EVs derived from AEC cells in C57BL/6 mice and then Nluc luciferase activity was evaluated, N = 3. N: number of mice used. * P < 0.05; **** P < 0.0001
2.6. Albumin‐decorated EVs accumulate across organs
The biodistribution of the engineered ABD‐EVs in vivo was measured by checking Nluc signals from blood and other harvested organs. In contrast to control EVs, HEK‐293T‐derived ABD‐EVs accumulated more in liver, spleen and kidney after different routes of administration (I.V, I.P and S.C), but remained unchanged in lung, in NMRI mice (Figure 7A‐C). I.V injection of HEK‐293T‐derived mCD63 ABD‐EVs showed similarly significant increase of Nluc signals in C57BL/6 mice (Figure 7D). When calculating the percentage of injected EVs in plasma or other organs, the plasma enriched most of the injected ABD‐EVs as expected since they bound to MSA (Figure 7E‐H). Compared to the control EVs, the biodistribution of ABD‐EVs increased in liver, spleen and kidney, but remained unchanged in lung in most of the cases (Figure 7E‐H), similar to the RLU/mg data (Figure 7A‐C). We hypothesized that if the circulation time of engineered EVs was extended, the chance to distribute to other organs increased. In addition, the albumin‐decoration may protect the EVs from fast clearance by circumventing the RES system.
FIGURE 7.
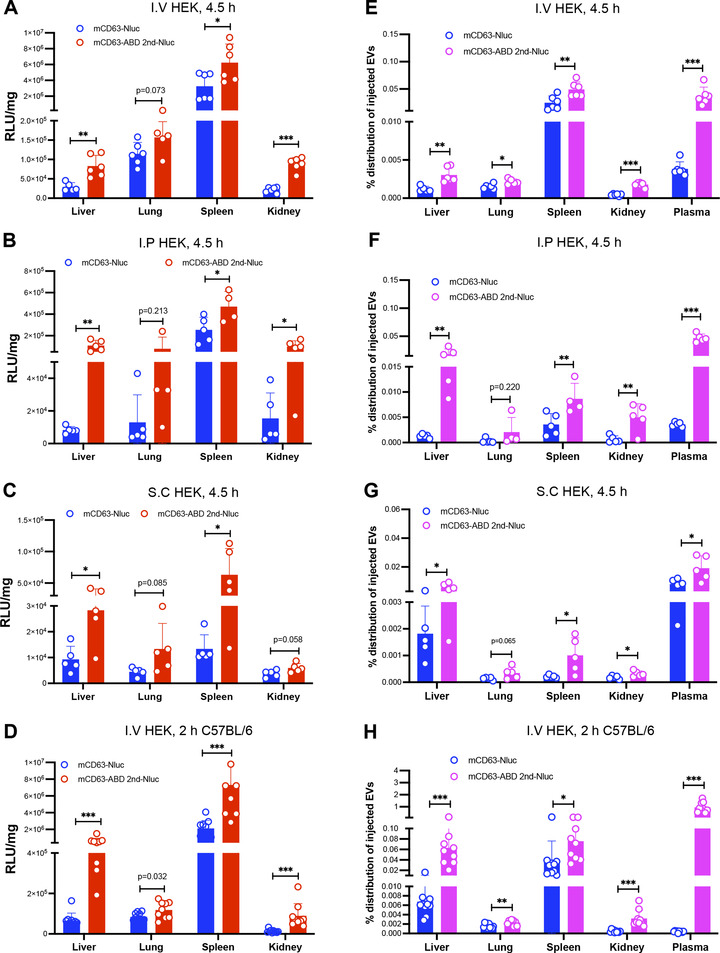
Albumin‐decorated EVs accumulate across organs. (A) Biodistribution of mCD63‐ABD EVs derived from HEK‐293T cells at 4.5‐h time point after I.V injection in terms of RLU/mg in NMRI mice, N = 6. (B) Different organ‐distribution of the mCD63‐ABD EVs derived from HEK‐293T cells after 4.5 h of I.P injection in terms of RLU/mg in NMRI mice, N = 5. (C) Nluc signals from a variety of organs after 4.5 h of S.C injection of mCD63‐ABD EVs derived from HEK‐293T cells in terms of RLU/mg in NMRI mice, N = 5. (D) I.V injection of mCD63‐ABD EVs derived from HEK‐293T cells for 2 h and then biodistribution was evaluated in terms of RLU/mg in C57BL/6 mice, N = 9 for control group and N = 8 for ABD group. (E) Organ and plasma distribution of the mCD63‐ABD EVs derived from HEK‐293T cells after 4.5 h of I.V injection in terms of percentage of injected EVs in NMRI mice, N = 6. (F) After 4.5 h of I.P injection of the mCD63‐ABD EVs derived from HEK‐293T cells, biodistribution of EVs was evaluated in terms of percentage of injected EVs in NMRI mice, N = 5. (G) Biodistribution of the mCD63‐ABD EVs derived from HEK‐293T cells after 4.5 h of S.C injection in terms of percentage of injected EVs in NMRI mice, N = 5. (H) mCD63‐ABD EVs derived from HEK‐293T cells were injected (I.V) into C57BL/6 mice for 2 h, and then biodistribution of EVs was demonstrated in terms of percentage of injected EVs in, N = 9 for control group and N = 8 for ABD group. N: number of mice used. * P < 0.05; ** P < 0.01; *** P < 0.001
2.7. Live imaging demonstrated delayed clearance of albumin‐decorated EVs
Finally, live imaging was performed to show the dynamic change of the injected engineered ABD‐EVs. For this, we used mCD63‐2nd ABD fused to thermoluciferase (Tluc) (mCD63‐ABD 2nd‐Tluc), which we recently demonstrated to exhibit higher tissue penetration compared to Nluc (Gupta et al., 2020). The success of engineering (Figure S8A) and albumin binding capacity of these ABD‐engineered Tluc EVs were confirmed (Figure 8SB‐D) prior to in vivo experiments. The EVs with the same amount of relative light units (RLU) from both ABD‐engineered group and control group were injected into NMRI mice and the luciferase signal was assessed between 10 and 30 min post injection. The clearance of the signals in the ABD‐EVs group was much slower than that in the control group (Figure 8A), indicating retention of ABD‐EVs in vivo. These results were also replicated in another strain of white mice, BALB/c, showing similar delayed clearance of the ABD‐engineered EVs (Figure 8B). In addition, live imaging combined with computed tomography (CT) scanning was performed to investigate the 3D biodistribution of injected EVs. The results show that the ABD‐engineered EVs distributed to the main organs, such as liver, spleen, lung and kidney after 10 min of I.V injection (Figure 8C). These results are in line with our previous publications on EV biodistribution (Gupta et al., 2020; Wiklander et al., 2015).
FIGURE 8.
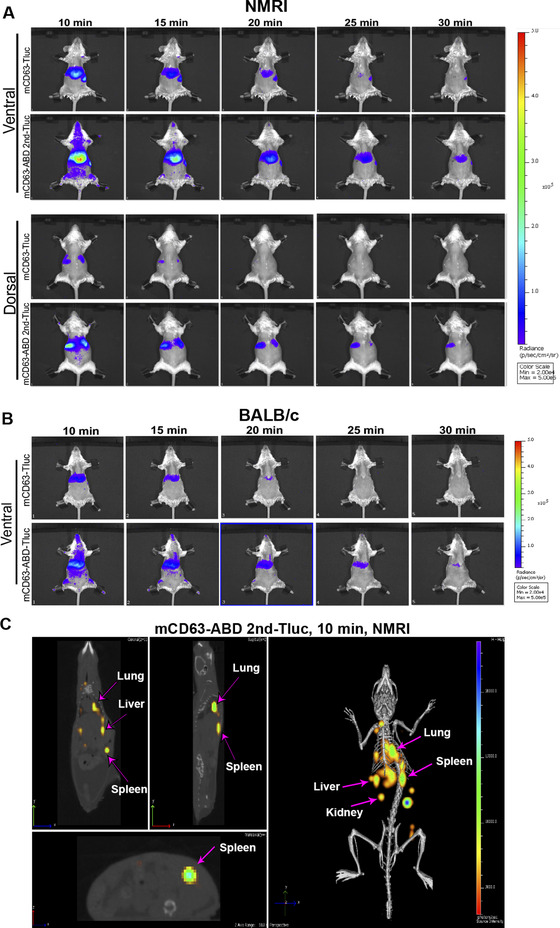
Delayed clearance of albumin‐decorated EVs demonstrated by live imaging based on Tluc activity. (A) Dynamic change of luciferase activities in mice injected into mCD63‐ABD‐Tluc EVs in NMRI mice. The upper and lower panels indicated the dorsal and ventral dynamic images, respectively. The monitoring of the luciferase activity started from 10 min after injection and recorded every 5 min. (B) luciferase activity dynamic change in mice injected with mCD63‐ABD‐Tluc EVs in BALB/c mice. Ventral position images were shown. (C) 3D imaging showing the biodistribution of the mCD63‐ABD‐Tluc EVs at 10 min after injection in NMRI mice. Live imaging combined with computed tomography (CT) scanning was used to demonstrate the luciferase signals in different organs
3. DISCUSSION
To our knowledge, this is the first report utilizing ABD to engineer EVs to achieve similar effects as observed in synthetic nanoparticles (Nessler et al., 2020; J. Wei et al., 2018). We achieved albumin decoration on EVs by inserting ABDs into the extracellular loops of tetraspanins or directly fusing the ABD to single‐transmembrane EV‐sorting domain. Albumin‐bound EVs significantly extended the circulation time in vivo after different routes of administration in different strains of mice. The delayed clearance of ABD‐engineered EVs was further corroborated by IVIS live imaging (Figure 8). The extension in circulation time may be favourable from a clinical point of view as it would allow reduced frequency of EV dosing (Chen & Liu, 2016; Herrmann et al., 2021).
The engineering strategy we developed could be adapted easily since we showed that the display of ABDs on the surface of EVs can be achieved either by using tetraspanins or single‐pass transmembrane EV‐sorting domain. A potential concern in regards to using ABDs pertains to their bacterial origin (Johansson et al., 2002), which holds potential immunogenicity in vivo. In a natural state, bacteria utilize ABD to bind to plasma albumin and thereby camouflage themselves with bound albumin to evade the immune system (Egesten et al., 2011; Nilvebrant & Hober, 2013a). Similarly, the ABDs on the surface of the engineered EVs were bound by serum albumin and therefore likely shielded from the recognition of the immune system, which may lead to reduced immunogenicity. Another concern about ABD engineering is the potential change of internalization of albumin‐coating EVs in recipient cells. To test this, we did uptake experiments of the ABD‐EVs in mouse cell lines and the albumin coating significantly increased EV‐uptake (Figure S7). Although not explored in our study, one interesting point would be to understand how the ABD‐engineered EVs will be internalized in vivo after administration. Soluble albumin binds to its receptor, such as neonatal Fc receptor (FcRn), and thus gets recycled through transcytosis in epithelial cells, prolonging its lifespan (Chaudhury et al., 2003). Albumin‐bound nanoparticles, such as the FDA‐approved 130‐nanometer albumin‐bound paclitaxel (nab‐paclitaxel; Abraxane) were found to bind to albumin receptor (gp60) (similar to the interaction between soluble albumin and recipient cells) on the surface of endothelial cells of tumour vessels, leading to transcytosis across endothium and accumulation of albumin‐nanoparticles within the tumour interstitium (Hawkins et al., 2008; Loureiro et al., 2016). Since EVs are natural cell‐derived nanoparticles, we assume the fate of ABD‐EVs may be similar to soluble albumin (get recycled to extend the circulation time) and synthetic nanoparticles (tumour accumulation) in vivo after injection.
EV engineering of tetraspanins often involves the insertion of targeting moieties into the extracellular loops for surface display. This approach might result in functional EVs depending on the intended aims; however, the engineering is performed without any regard to which loop favourably allows the display of the protein of interest (Duong et al., 2019; Lozano‐Andrés et al., 2019). Our data clearly showed that the second loop of the tetraspanins was more compatible for engineering while the first loop was more sensitive to inserting polypeptide of interest (Figure 2). The characterized structure of large extracellular loop (LEL, also the second loop) of CD81 was well defined (Charrin et al., 2009; Kitadokoro et al., 2001; Zimmerman et al., 2016), and the stabilization of the CD81 LEL by de novo disulphide bond was found to enhance its amenability for engineering into a more versatile protein scaffold (Vogt et al., 2018), which echoed our finding for ABD engineering. The LEL is larger than the first loop and is protruded from the membrane, allowing interaction with a variety of other protein partners (Ivanusic et al., 2021; Susa et al., 2020). The protrusion of LEL may allow engineering with different functional moieties, which is not delineated in our study, but is warranted for further investigation.
Due to the constitutive movement of albumin from circulation to lymphatic system, albumin decorated drugs or nanoparticles accumulate in lymph nodes (Ishihara et al., 2021; Liu et al., 2014; Wang et al., 2015). As expected, the ABD‐engineered EVs used in this study were found to similarly accumulate in the ILNs. This implies the potential utility of the technology for immunomodulation if the ABD‐engineered EVs were further equipped with immunomodulatory moieties, such as DC‐targeting ligands and tumour antigens. By this method, the EVs would be efficiently taken up and processed inside of DCs, facilitating antigen presentation and the priming of the immune system. Therefore, this type of engineered EVs may be used as tumour vaccines to prevent cancer development. In addition, the ABD‐engineered EVs were found to accumulate in tumoural tissues as a result of the potential disrupted lymphatic drainage inside of the tumours, similar to the findings reported when using ABD‐conjugated synthetic nanoparticles (Jonsson et al., 2008; Wei et al., 2018). This may be exploited to fight against established tumours if the ABD‐engineered EVs were loaded with chemotherapeutic drugs. Taken together, our results show that engineered EVs displaying ABD have extended circulation time, as well as differential biodistribution characteristics that unlock improved therapeutic potential for EVs.
4. MATERIALS AND METHODS
4.1. Constructs generation
All the transgenes used in this study were codon optimized and bought from IDT (Integrated DNA Technologies, USA). The transgenes were first cloned into pLEX vector by using EcoRI and XhoI sites and then were transferred to lenti‐viral vector by using the same restriction sites.
4.2. Cell culture
HEK‐293T cells were maintained in DMEM medium supplemented with 10% foetal bovine serum (FBS, Gibco, USA) and 1% Anti‐anti (Gibco, USA). Cells were cultured at 37°C in a humidified air atmosphere containing 5% CO2. All the cells used were in low passages (p < 10) and of high viability (> 90%). The AEC cells were cultured in PEM medium supplemented with 1% Anti‐anti (Gibco, USA) in a shaking system.
4.3. Viruses production
The lenti‐viral vectors containing corresponding transgenes were co‐transfected into HEK‐293T cells with pCD/NL‐BH (Helper plasmid) and pcoPE01(Envelope plasmid) and incubated overnight. The second day morning, the medium was changed to full DMEM medium (plus 10% FBS and 1% Anti‐anti) supplemented with 10 mM sodium butyrate (Sigma‐Aldrich) and incubated for 6–8 h. And then the medium was changed back to full DMEM medium. Around 22–24 h late, the medium containing virus particles was filtered with 0.45 μm syringe filter (VWR) and put into the 50 ml tubes specific for virus collection. The medium was centrifuged at 25,000 × g for 90 min at 4°C. And then the pellets were resuspended with freshly prepared medium (IMDM with 20%FBS) and aliquoted into cryo‐tubes and put in ‐80°C freezer for long‐time storage.
4.4. Stable cell generation
HEK‐293T cells were seeded into 6‐well plate with 0.8 million cells/well and incubated overnight. The corresponding virus particles, either freshly prepared or stored in ‐80°C freezer, were added into the cells directly. Three doses of virus particles were used for the brief titration, 2 μl, 10 μl, and 50 μl. 48 h after virus transduction, the cells were washed with PBS and trypsinized to cell suspensions. And then the cells were transferred to T25 flasks and suffered from selection by puromycin (4 μg/ml). After selection for 48 h, the medium was changed to fresh medium with puromycin and the colonies survived during selection formed the following week. Expanded the selected cells and verified the transgene expression either by Western blot or by flow cytometry.
4.5. EV production and isolation
To produce the EVs for in vivo injection, the corresponding stable cells were seeded in 15‐cm dishes with 10 million cells/ dish with full DMEM medium. Two days late, the medium was changed to Opti‐MEM medium (Gibco, USA) with 1% Anti‐anti. And then the conditional medium (CM) was harvested 48 h late, serially centrifuged at 700 × g for 5 min and 2000× g at 4°C for 10 min. The CM was filtered with bottle top filters system (Corning, low protein binding) with cellulose acetate membrane. This system contains a 0.22 μm pore size within the membrane and can remove the remaining larger particles. The EVs in the filtered CM were isolated with Tangential flow filtration (TFF, MicroKross, 20 cm2, Spectrum labs). The cut‐off of TFF is 300 kDa and the particles bigger than 300 kDa are remained in the system and concentrated. And then the concentrated particles were further concentrated by Amicon Ultra‐15 100 kDa (Millipore) spin filter, centrifuged at 4000× g for 30 min to several hours at 4°C depending on the amount EVs in the samples. At last, the concentrated EVs were collected in maxirecovery 1.5 ml Eppendorf tubes (Axygene, USA) and the concentrations were detected by Nanoparticle Tracking Analysis (NTA).
4.6. Nanoparticle Tracking Analysis (NTA)
Before checking the particle sizes and concentrations of the samples with a NanoSight NS500 instrument, the samples were diluted with freshly 0.22 μm‐filtered PBS. The cameral levels of the instrument were set up to 13 or 14, and at least five videos longer than 30 s were recorded in light scatter mode. The NTA 2.3 analytical software equipped in the machine was used to analyse the gained data, with the constant setting (screen gain 10, detection threshold 7) for all measurements. The number of EVs used for Western blot, Luminescence detection and animal injection was according to the concentrations measured by NTA.
4.7. HSA binding in vitro
The ABD‐engineered EVs (1×1011 dose) and the corresponding control EVs were incubated with FITC labelled HSA (HSA‐FITC, ab8030, Abcam) and the final concentration of HSA‐FITC was 150 μg/ml at 37°C for 120 min. And then the EVs were subjected to size exclusions by using the IZON qEV columns (IZON, qEVoriginal/70 nm). 48 fractions were collected from each sample with 300 μl per fraction. 150 μl of each fraction was taken in 96‐well plate for fluorescence intensity detection by using SpectraMax i3x (MOLECULAR DEVICES, USA). 30 μl of each fraction was taken for luciferases (Nanoluc or Thermoluc) activities assay by using GloMax 96 Microplate Luminometer machine (Promega, USA).
4.8. Western blot
The total proteins were isolated from HEK‐293T cells with RIPA lysis buffer. The cell pellets containing 2 million cells were resuspended with 100 μl of RIPA buffer and the cells were lysed for 25 min on ice. And then the cell lysates were centrifuged at 14,000 × rpm for 15 min at 4°C. The supernatants were collected to new clean tubes. 24 μl of cell lysate or 1E10 EVs were mixed with the 4x loading buffer (the resume is 0.4 M sodium carbonate, 10% glycerol, 0.5 M dithiothreitol, and 8% SDS) for each sample. The mixed samples were heated at 70°C for 5 min. NuPAGE Novex 4–12% Bis‐Tris Protein Gels (Invitrogen, Thermo Fisher Scientific) were used to load the heated samples. The gels were run in NuPAGE MES SDS running buffer (Invitrogen, Thermo Fisher Scientific) at 120 V for 2 h or longer depending on the targeting protein sizes. iBlot system was used to transfer the proteins inside of the gels to iBlot nitrocellulose membrane (Invitrogen, Thermo Fisher Scientific). Before incubating with the corresponding primary antibodies overnight at 4°C, the membranes were blocked with Odyssey blocking buffer (LI‐COR) for 50 min at RT. One day late, the primary antibodies were collected and can be re‐used for future and the membranes were washed with PBS‐T (PBS with 0.1% Tween‐20, Sigma) for 3 times, 10 min for each time. And then the corresponding secondary antibodies (LI‐COR) were added and with the membranes were incubated for 50 min at RT. After this, the membranes were washed with PBS‐T for 3 times, 10 min for each time. During the last wash, PBS was used and the washing time is 5 min. Finally, the Odyssey infrared imaging system (LI‐COR) was used to visualize the images. The primary antibodies used in this study are the following: anti‐NanoLuc (Promega, 1:1000 dilution); anti‐Alix (ab117600, Abcam, 1:1000 dilution); anti‐CD81 (Santa Crus, 1:200 dilution); and anti‐actin (Sigma 1:20000 dilution); anti‐TSG101 (ab125011, Abcam, 1:1000); anti‐Calnexin (PA5‐19169, ThermoFisher, 1:1000). The secondary antibodies used in this study are the following: goat anti‐mouse IRDye800CW or 680LT (1:8000) or goat/anti‐rabbit IRDye800CW or 680LT (1:8000).
4.9. Luminescence (Nluc and Tluc) detection
Determined amount of EVs (such as 1×109) or 10 μl of tissue lysate from mice internal organs were used for Nanoluc detection. The EVs or tissue lysate were added into white‐walled 96‐well plates. 25 μl diluted Nano‐Glo substrates (Nano‐Glo Luciferase Assay System: Promega), were added into each well by injection mode of the GloMax 96 Microplate Luminometer machine (Promega, USA). The background wells without any nanoluc proteins were also included for future calculations.
4.10. Transmission electron microscope
Engineered HEK293T cell derived‐EVs were spotted on a glow‐discharged formvar‐carbon type B coated grid (Ted Pella Inc.) and stained with 2% uranyl acetate solution (TAAB). The grids were washed with distilled water and imaged with a FEI Tecnai 10 transmission electron microscope at an accelerating voltage of 100 kV.
4.11. In vivo animal work
NMRI, C57BL/6 and BALB/c mice were used for the animal work. The female mice were bought and adapted for more than 1 week at KI animal facility. And then the mice were randomly divided into different control and experimental groups. 1×1011 EVs were injected into the mice by different injection routes dependent on the requirement of the experiments. Blood were sampled at different time points (30 min, 60 min, 120 min, and 450 min for NMRI mice. 120 min for C57BL/six mice). The sampled blood was collected into the 0.5 M EDTA‐treated tubes and centrifuged at 1000× g for 10 min to get the plasma for nanoluc detection. At 450 min time or 120 min time points, the mice were sacrificed and internal organs (brain, liver, lung, spleen, kidney, tumours and lymph nodes) were harvested. The organs were collected into 2 ml tubes filled with 1 ml lysis buffer (PBS supplemented with 0.1% TritonX‐100) and were lysed in the TissueLyser machine. And then the tissue lysates were used for nanoluc luciferase activities detection.
4.12. In vivo bioluminescence imaging/computed tomography (CT)
The distribution of EVs was observed by an IVIS Spectrum (PerkinElmer, Waltham, MA, USA). Quantum FX (Perkin Elmer, Waltham, MA, USA) was also used to co‐register optical signals with anatomical μCT. NMRI mice were I.P administered with 150 mg/kg D‐luciferin and after 5 min EVs in 100 μl PBS were injected I.V(tail vein). The 2D/3D bioluminescence imaging and μCT scans were performed at different time points post injection. The 2D bioluminescence images were acquired with open filter, and 3D bioluminescence images were acquired at wavelengths 600, 620, and 640 nm. The mouse in the Mouse Imaging Shuttle (MIS, 25 mm high, PerkinElmer) was then transferred to the Quantum FX‐μCT and subjected to a dynamic CT scan with an X‐ray source current of 200 μA, voltage of 70 kV, FOV (field of view) 60 mm x 60 mm, scan time 17 s. All images were analysed using Living Image 4.3.1 (PerkinElmer, Waltham, MA, USA).
4.13. Widefield imaging and super‐resolution (dSTORM) imaging
100 μl HEK EVs (mCD63‐Nluc – concentration 1.37×1012 particles/ml, mCD63‐ABDx2‐Nluc – concentration 5.00×1011 particles/ml, mCD63‐ABD‐1st loop Nluc – concentration 1.17×1012 particles/ml, mCD63‐ABD‐2nd loop Nluc – concentration 6.5×1011 particles/ml) were incubated with 40 ug/ml Alexa Fluor 488 conjugated Human Serum Albumin (Jackson ImmunoResearch cat.no. 009‐540‐051) and a mix of CD9/CD63/CD81 fluorescent antibodies (R&D System mouse anti‐hCD63 Alexa Fluor 647 cat.no. FAB4615R and mouse anti‐hCD9 Alexa Fluor 647 cat.no. A6208T100; Biolegend mouse anti‐hCD63 Alexa Fluor 647 cat.no. 353016) for 2 h at room temperature. EVs were then ultracentrifuged (with ultracentrifuge Thermo Scientific Sorvall WX) for 16 h at 4°C on an Optiprep (Sigma Aldrich cat.no. D1556) 14–36% step gradient to separate vesicular components from unbound Human Serum Albumin and fluorescent antibodies. After ultracentrifugation, 300 μl were collected and 50 μl from each three fractions were pulled into a poly‐lysine coated Ibidi 18‐well glass bottom chambers (Ibidi cat. no. 81817) and incubated overnight to allow EVs to adhere to the glass. The next day, the supernatant from the chambers was removed and PBS was added prior to imaging.
Imaging: wide‐field and super‐resolution (dSTORM) microscopy was performed by using Nanoimager (Oxford Nanoimaging) equipped with a 100x Nikon objective, numerical aperture 1.4. Image processing was performed by using ImageJ (for wide‐field microscopy): background from images was removed by using build‐in ImageJ function “subtract background” and same brightness and contrast settings were applied. For super‐resolution microscopy (dSTORM), image processing was performed by using Oxford Nanoimaging software.
4.14. EV uptake
B16F10 and TCMK‐1 cells were seeded in 24‐well plates and incubated at 37°C overnight. 1×109 EVs from different groups were added directly into the cells the second day. After 2 h, 4 h and 8 h incubation of the EVs with recipient cells, the cell supernatants were removed and washed with 1 ml PBS twice and then the cells were lysed with 100 μl 0.1% TritonX‐100, 15 min in a shaking machine (900 rmp/min). 30 μl of the cell lysates were took for Nluc activity assay by using GloMax 96 Microplate Luminometer machine (Promega, USA).
4.15. Statistical analysis
The data were analysed and plotted by the GraphPad Prism 8 (GraphPad Prism Software, USA). Two‐tailed Student's t test in two groups or 1‐way ANOVA for analysis of multiple groups was used for the statistical calculations. Statistical significance was set at P < 0.05 (** P < 0.01; *** P < 0.001; **** P < 0.0001).
CONFLICT OF INTEREST
Oscar P. B. Wiklander, Joel Z. Nordin, Dhanu Gupta, Samir E. L. Andaloussi are consultants and stakeholders in Evox Therapeutics Limited, Oxford, United Kingdom. Valentina Galli, Nathalie Howe, Christopher Davies, Justin Hean, Eleni Kyriakopoulou are employees of Evox Therapeutics Limited, Oxford, United Kingdom. Other authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Xiuming Liang: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resourses; Validation; Writing‐original draft; Funding acquisition. Zheyu Niu: Data curation; Formal analysis; Investigation; Methodology. Valentina Galli: Data curation; Methodology; Resourses. Nathalie Howe: Data curation; Methodology; Resourses. Ying Zhao: Investigation; Methodology. Oscar P.B. Wiklander: Methodology; Funding acquisition; Writing‐review and editing. Wenyi Zheng: Investigation; Methodology; Writing‐review and editing. Rim Jawad: Methodology; Writing‐review and editing. Giulia Corso: Methodology. Christopher Davies: Methodology. Justin Hean: Methodology; Resourses. Eleni Kyriakopoulou: Methodology. Doste R. Mamand: Methodology. Risul Amin: Methodology. Joel Z. Nordin: Formal analysis; Investigation; Funding acquisition; Writing‐review and editing. Dhanu Gupta: Conceptualization; Data curation; Investigation; Methodology; Writing‐review and editing. Samir EL Andaloussi: Conceptualization; Investigation; Supervision; Funding acquisition; Resources; Writing‐review and editing.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGEMENTS
This study was supported by the Swedish Research Council (VR‐Med, Dnr 2020‐01322), Swedish Foundation of Strategic research (SSF‐IRC), ERC‐CoG DELIVER, Evox Therapeutics and Center for Medical Innovation (CIMED, FoUI‐963452).
Liang, X. , Niu, Z. , Galli, V. , Howe, N. , Zhao, Y. , Wiklander, O. P. B. , Zheng, W. , Wiklander, R. J. , Corso, G. , Davies, C. , Hean, J. , Kyriakopoulou, E. , Mamand, D. R. , Amin, R. , Nordin, J. Z. , Gupta, D. , & Andaloussi, S. EL. (2022). Extracellular vesicles engineered to bind albumin demonstrate extended circulation time and lymph node accumulation in mouse models. Journal of Extracellular Vesicles, 11, e12248. 10.1002/jev2.12248
Contributor Information
Xiuming Liang, Email: xiuming.liang@ki.se.
Samir EL Andaloussi, Email: samir.el-andaloussi@ki.se.
DATA AVAILABILITY STATEMENT
The data supporting the main findings of this study are available from the corresponding authors.
REFERENCES
- Andersen, J. T. , Cameron, J. , Plumridge, A. , Evans, L. , Sleep, D. , & Sandlie, I. (2013). Single‐chain variable fragment albumin fusions bind the neonatal Fc receptor (FcRn) in a species‐dependent manner: Implications for in vivo half‐life evaluation of albumin fusion therapeutics. Journal of Biological Chemistry, 288(33), 24277–24285. 10.1074/jbc.M113.463000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhadj, Z. , He, B. , Deng, H. , Song, S. , Zhang, H. , Wang, X. , Dai, W. , & Zhang, Q. (2020). A combined “eat me/don't eat me” strategy based on extracellular vesicles for anticancer nanomedicine. Journal of Extracellular Vesicles, 9(1), 1806444. 10.1080/20013078.2020.1806444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buatois, V. , Johnson, Z. , Salgado‐Pires, S. , Papaioannou, A. , Hatterer, E. , Chauchet, X. , Richard, F. , Barba, L. , Daubeuf, B. , Cons, L. , Broyer, L. , D'Asaro, M. , Matthes, T. , LeGallou, S. , Fest, T. , Tarte, K. , Clarke Hinojosa, R. K. , Ferrer, E. G. , Ribera, J. M. , …, & Ferlin, W. G. (2018). Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B‐cell lymphoma and leukemia. Molecular Cancer Therapeutics, 17(8), 1739–1751. 10.1158/1535-7163.MCT-17-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catani, L. , Sollazzo, D. , Ricci, F. , Polverelli, N. , Palandri, F. , Baccarani, M. , Vianelli, N. , & Lemoli, R. M. (2011). The CD47 pathway is deregulated in human immune thrombocytopenia. Experimental Hematology, 39(4), 486–494. 10.1016/j.exphem.2010.12.011 [DOI] [PubMed] [Google Scholar]
- Chao, M. P. , Jaiswal, S. , Weissman‐Tsukamoto, R. , Alizadeh, A. A. , Gentles, A. J. , Volkmer, J. , Weiskopf, K. , Willingham, S. B. , Raveh, T. , Park, C. Y. , Majeti, R. , & Weissman, I. L. (2010). Calreticulin is the dominant pro‐phagocytic signal on multiple human cancers and is counterbalanced by CD47. Science Translational Medicine, 2(63), 63ra94. 10.1126/scitranslmed.3001375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrin, S. , Le Naour, F. , Silvie, O. , Milhiet, P. E. , Boucheix, C. , & Rubinstein, E. (2009). Lateral organization of membrane proteins: Tetraspanins spin their web. Biochemical Journal, 420(2), 133–154. 10.1042/BJ20082422 [DOI] [PubMed] [Google Scholar]
- Chaudhury, C. , Mehnaz, S. , Robinson, J. M. , Hayton, W. L. , Pearl, D. K. , Roopenian, D. C. , & Anderson, C. L. (2003). The major histocompatibility complex‐related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. Journal of Experimental Medicine, 197(3), 315–322. 10.1084/jem.20021829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , & Liu, Z. (2016). Albumin carriers for cancer theranostics: A conventional platform with new promise. Advanced Materials, 28(47), 10557–10566. 10.1002/adma.201600038 [DOI] [PubMed] [Google Scholar]
- Duong, N. , Curley, K. , Brown, A. , Campanelli, A. , Do, M. A. , Levy, D. , Tantry, A. , Marriott, G. , & Lu, B. (2019). Decoy exosomes as a novel biologic reagent to antagonize inflammation. International Journal of Nanomedicine, 14, 3413–3425. 10.2147/IJN.S196975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egesten, A. , Frick, I. M. , Mörgelin, M. , Olin, A. I. , & Björck, L. (2011). Binding of albumin promotes bacterial survival at the epithelial surface. Journal of Biological Chemistry, 286(4), 2469–2476. 10.1074/jbc.M110.148171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann, G. , Herrmann, I. K. , & Stevens, M. M. (2015). Cell‐derived vesicles for drug therapy and diagnostics: Opportunities and challenges. Nano Today, 10(3), 397–409. 10.1016/j.nantod.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Cid, L. , López‐Donaire, M. L. , Velasco, D. , Marín, V. , González, M. I. , Salinas, B. , Cussó, L. , García, Á. , Bravo, S. B. , Fernández‐Santos, M. E. , Elvira, C. , Sierra, J. , Arroba, E. , Bañares, R. , Grigorian‐Shamagian, L. , & Fernández‐Avilés, F. (2021). Cardiac extracellular matrix hydrogel enriched with polyethylene glycol presents improved gelation time and increased on‐target site retention of extracellular vesicles. International Journal of Molecular Sciences, 22(17), 9226. 10.3390/ijms22179226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, D. , Liang, X. , Pavlova, S. , Wiklander, O. P. B. , Corso, G. , Zhao, Y. , Saher, O. , Bost, J. , Zickler, A. M. , Piffko, A. , Maire, C. L. , Ricklefs, F. L. , Gustafsson, O. , Llorente, V. C. , Gustafsson, M. O. , Bostancioglu, R. B. , Mamand, D. R. , Hagey, D. W. , Görgens, A. , …, & EL Andaloussi, S. (2020). Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. Journal of Extracellular Vesicles, 9(1), 1800222. 10.1080/20013078.2020.1800222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, M. J. , Soon‐Shiong, P. , & Desai, N. (2008). Protein nanoparticles as drug carriers in clinical medicine. Advanced Drug Delivery Reviews, 60(8), 876–885. 10.1016/j.addr.2007.08.044 [DOI] [PubMed] [Google Scholar]
- Herrmann, I. K. , Wood, M. J. A. , & Fuhrmann, G. (2021). Extracellular vesicles as a next‐generation drug delivery platform. Nature Nanotechnology, 16(7), 748–759. 10.1038/s41565-021-00931-2 [DOI] [PubMed] [Google Scholar]
- Imai, T. , Takahashi, Y. , Nishikawa, M. , Kato, K. , Morishita, M. , Yamashita, T. , Matsumoto, A. , Charoenviriyakul, C. , & Takakura, Y. (2015). Macrophage‐dependent clearance of systemically administered B16BL6‐derived exosomes from the blood circulation in mice. Journal of Extracellular Vesicles, 4(2015), 1–8. 10.3402/jev.v4.26238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara, A. , Ishihara, J. , Watkins, E. A. , Tremain, A. C. , Nguyen, M. , Solanki, A. , Katsumata, K. , Mansurov, A. , Budina, E. , Alpar, A. T. , Hosseinchi, P. , Maillat, L. , Reda, J. W. , Kageyama, T. , Swartz, M. A. , Yuba, E. , & Hubbell, J. A. (2021). Prolonged residence of an albumin–IL‐4 fusion protein in secondary lymphoid organs ameliorates experimental autoimmune encephalomyelitis. Nature Biomedical Engineering, 5(5), 387–398. 10.1038/s41551-020-00627-3 [DOI] [PubMed] [Google Scholar]
- Ishikawa‐Sekigami, T. , Kaneko, Y. , Saito, Y. , Murata, Y. , Okazawa, H. , Ohnishi, H. , Oldenborg, P. A. , Nojima, Y. , & Matozaki, T. (2006). Enhanced phagocytosis of CD47‐deficient red blood cells by splenic macrophages requires SHPS‐1. Biochemical and Biophysical Research Communications, 343(4), 1197–1200. 10.1016/j.bbrc.2006.03.094 [DOI] [PubMed] [Google Scholar]
- Ivanusic, D. , Madela, K. , Bannert, N. , & Denner, J. (2021). The large extracellular loop of CD63 interacts with gp41 of HIV‐1 and is essential for establishing the virological synapse. Scientific Reports, 11(1), 1–14. 10.1038/s41598-021-89523-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, M. U. , Frick, I. M. , Nilsson, H. , Kraulis, P. J. , Hober, S. , Jonasson, P. , Linhult, M. , Nygren, P. Å. , Uhlén, M. , Björck, L. , Drakenberg, T. , Forsén, S. , & Wikström, M. (2002). Structure, specificity, and mode of interaction for bacterial albumin‐binding modules. Journal of Biological Chemistry, 277(10), 8114–8120. 10.1074/jbc.M109943200 [DOI] [PubMed] [Google Scholar]
- Jonsson, A. , Dogan, J. , Herne, N. , Abrahmsén, L. , & Nygren, P. Å. (2008). Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Engineering, Design and Selection, 21(8), 515–527. 10.1093/protein/gzn028 [DOI] [PubMed] [Google Scholar]
- Kamerkar, S. , Lebleu, V. S. , Sugimoto, H. , Yang, S. , Ruivo, C. F. , Melo, S. A. , Lee, J. J. , & Kalluri, R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature, 546(7659), 498–503. 10.1038/nature22341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, T. , Honda, T. , Nishihara, M. , Yamamoto, T. , & Yokoyama, M. (2009). Histological study on side effects and tumor targeting of a block copolymer micelle on rats. Journal of Controlled Release, 136(3), 240–246. 10.1016/j.jconrel.2009.02.011 [DOI] [PubMed] [Google Scholar]
- Keller, M. D. , Ching, K. L. , Liang, F. X. , Dhabaria, A. , Tam, K. , Ueberheide, B. M. , Unutmaz, D. , Torres, V. J. , & Cadwell, K. (2020). Decoy exosomes provide protection against bacterial toxins. Nature, 579(7798), 260–264. 10.1038/s41586-020-2066-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitadokoro, K. , Bordo, D. , Galli, G. , Petracca, R. , Falugi, F. , Abrignani, S. , Grandi, G. , & Bolognesi, M. (2001). CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO Journal, 20(1–2), 12–18. 10.1093/emboj/20.1.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Fliervoet, L. A. L. , Van Der Meel, R. , Fens, M. H. A. M. , Heijnen, H. F. G. , Van Bergen En Henegouwen, P. M. P. , Vader, P. , & Schiffelers, R. M. (2016). PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. Journal of Controlled Release, 224, 77–85. 10.1016/j.jconrel.2016.01.009 [DOI] [PubMed] [Google Scholar]
- Lathwal, S. , Yerneni, S. S. , Boye, S. , Muza, U. L. , Takahashi, S. , Sugimoto, N. , Lederer, A. , Das, S. R. , Campbell, P. G. , & Matyjaszewski, K. (2021). Engineering exosome polymer hybrids by atom transfer radical polymerization. Proceedings of the National Academy of Sciences of the United States of America, 118(2), 1–11. 10.1073/pnas.2020241118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbergh, M. F. S. , Wubbolts, R. , Borg, E. G. F. , van ’T Veld, E. M. , Boes, M. , & Stoorvogel, W. (2020). Dendritic cells release exosomes together with phagocytosed pathogen; potential implications for the role of exosomes in antigen presentation. Journal of Extracellular Vesicles, 9(1), 1798606. 10.1080/20013078.2020.1798606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Moynihan, K. D. , Zheng, Y. , Szeto, G. L. , Li, A. V. , Huang, B. , Van Egeren, D. S. , Park, C. , & Irvine, D. J. (2014). Structure‐based programming of lymph‐node targeting in molecular vaccines. Nature, 507(7493), 519–522. 10.1038/nature12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro, A. , Azoia, N. G. , Gomes, A. C. , & Cavaco‐Paulo, A. (2016). Albumin‐based nanodevices as drug carriers. Current Pharmaceutical Design, 22(10), 1371–1390. 10.2174/1381612822666160125114900 [DOI] [PubMed] [Google Scholar]
- Lozano‐Andrés, E. , Libregts, S. F. , Toribio, V. , Royo, F. , Morales, S. , López‐Martín, S. , Valés‐Gómez, M. , Reyburn, H. T. , Falcón‐Pérez, J. M. , Wauben, M. H. , Soto, M. , & Yáñez‐Mó, M. (2019). Tetraspanin‐decorated extracellular vesicle‐mimetics as a novel adaptable reference material. Journal of Extracellular Vesicles, 8(1), 1573052. 10.1080/20013078.2019.1573052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. , Dichwalkar, T. , Chang, J. Y. H. , Cossette, B. , Garafola, D. , Zhang, A. Q. , Fichter, M. , Wang, C. , Liang, S. , Silva, M. , Kumari, S. , Mehta, N. K. , Abraham, W. , Thai, N. , Li, N. , Dane Wittrup, K. , & Irvine, D. J. (2019). Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science, 365(6449), 162–168. 10.1126/science.aav8692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardpour, S. , Ghanian, M. H. , Sadeghi‐Abandansari, H. , Mardpour, S. , Nazari, A. , Shekari, F. , & Baharvand, H. (2019). Hydrogel‐mediated sustained systemic delivery of mesenchymal stem cell‐derived extracellular vesicles improves hepatic regeneration in chronic liver failure. ACS Applied Materials and Interfaces, 11(41), 37421–37433. 10.1021/acsami.9b10126 [DOI] [PubMed] [Google Scholar]
- Matsumoto, A. , Takahashi, Y. , Chang, H. Y. , Wu, Y. W. , Yamamoto, A. , Ishihama, Y. , & Takakura, Y. (2020). Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. Journal of Extracellular Vesicles, 9(1), 1696517. 10.1080/20013078.2019.1696517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehanny, M. , Lehr, C. M. , & Fuhrmann, G. (2021). Extracellular vesicles as antigen carriers for novel vaccination avenues. Advanced Drug Delivery Reviews, 173, 164–180. 10.1016/j.addr.2021.03.016 [DOI] [PubMed] [Google Scholar]
- Mehta, N. K. , Pradhan, R. V. , Soleimany, A. P. , Moynihan, K. D. , Rothschilds, A. M. , Momin, N. , Rakhra, K. , Mata‐Fink, J. , Bhatia, S. N. , Wittrup, K. D. , & Irvine, D. J. (2020). Pharmacokinetic tuning of protein–antigen fusions enhances the immunogenicity of T‐cell vaccines. Nature Biomedical Engineering, 4(6), 636–648. 10.1038/s41551-020-0563-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nessler, I. , Khera, E. , Vance, S. , Kopp, A. , Qiu, Q. , Keating, T. A. , Abu‐Yousif, A. O. , Sandal, T. , Legg, J. , Thompson, L. , Goodwin, N. , & Thurber, G. M. (2020). Increased tumor penetration of single‐domain antibody‐drug conjugates improves in vivo efficacy in prostate cancer models. Cancer Research, 80(6), 1268–1278. 10.1158/0008-5472.CAN-19-2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilvebrant, J. , & Hober, S. (2013a). The albumin‐binding domain as a scaffold for protein engineering. Computational and Structural Biotechnology Journal, 6(7), e201303009. 10.5936/csbj.201303009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilvebrant, J. , & Hober, S. (2013b). The albumin‐binding domain as a scaffold for protein engineering Abstract : The albumin‐binding domain is a small, three‐helical protein domain found in various surface proteins expressed by gram‐positive bacteria. Albumin binding is important in bacter. Pone.0103094.Pdf, March.
- Oldenborg, P. A. (2004). Role of CD47 in erythroid cells and in autoimmunity. Leukemia and Lymphoma, 45(7), 1319–1327. 10.1080/1042819042000201989 [DOI] [PubMed] [Google Scholar]
- Poggio, M. , Hu, T. , Pai, C. C. , Chu, B. , Belair, C. D. , Chang, A. , Montabana, E. , Lang, U. E. , Fu, Q. , Fong, L. , & Blelloch, R. (2019). Suppression of exosomal PD‐L1 induces systemic anti‐tumor immunity and memory. Cell, 177(2), 414–427.e13. 10.1016/j.cell.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivoltini, L. , Chiodoni, C. , Squarcina, P. , Tortoreto, M. , Villa, A. , Vergani, B. , Bürdek, M. , Botti, L. , Arioli, I. , Cova, A. , Mauri, G. , Vergani, E. , Bianchi, B. , Della Mina, P. , Cantone, L. , Bollati, V. , Zaffaroni, N. , Gianni, A. M. , Colombo, M. P. , & Huber, V. (2016). TNF‐related apoptosis‐inducing ligand (trail)‐armed exosomes deliver proapoptotic signals to tumor site. Clinical Cancer Research, 22(14), 3499–3512. 10.1158/1078-0432.CCR-15-2170 [DOI] [PubMed] [Google Scholar]
- Shiraishi, K. , & Yokoyama, M. (2019). Toxicity and immunogenicity concerns related to PEGylated‐micelle carrier systems: a review. Science and Technology of Advanced Materials, 20(1), 324–336. 10.1080/14686996.2019.1590126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susa, K. J. , Seegar, T. C. M. , Blacklow, S. C. , & Kruse, A. C. (2020). A dynamic interaction between cd19 and the tetraspanin CD81 controls B cell co‐receptor trafficking. ELife, 9, 1–20. 10.7554/eLife.52337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, Y. , Nishikawa, M. , Shinotsuka, H. , Matsui, Y. , Ohara, S. , Imai, T. , & Takakura, Y. (2013). Visualization and in vivo tracking of the exosomes of murine melanoma B16‐BL6 cells in mice after intravenous injection. Journal of Biotechnology, 165(2), 77–84. 10.1016/j.jbiotec.2013.03.013 [DOI] [PubMed] [Google Scholar]
- Tomlinson, M. G. (2009). Platelet tetraspanins: Small but interesting. Journal of Thrombosis and Haemostasis, 7(12), 2070–2073. 10.1111/j.1538-7836.2009.03613.x [DOI] [PubMed] [Google Scholar]
- Vader, P. , Mol, E. A. , Pasterkamp, G. , & Schiffelers, R. M. (2016). Extracellular vesicles for drug delivery. Advanced Drug Delivery Reviews, 106, 148–156. 10.1016/j.addr.2016.02.006 [DOI] [PubMed] [Google Scholar]
- Van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. In Nature Reviews Molecular Cell Biology, 19(4), 213–228. 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- Vogt, S. , Stadlmayr, G. , Stadlbauer, K. , Sádio, F. , Andorfer, P. , Grillari, J. , Rüker, F. , & Wozniak‐Knopp, G. (2018). Stabilization of the CD81 large extracellular loop with De Novo disulfide bonds improves its amenability for peptide grafting. Pharmaceutics, 10(3), 138. 10.3390/pharmaceutics10030138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Zhao, P. , Dong, S. , Xu, T. , He, X. , & Chen, M. (2018). An albumin‐binding polypeptide both targets cytotoxic T lymphocyte vaccines to lymph nodes and boosts vaccine presentation by dendritic cells. Theranostics, 8(1), 223–236. 10.7150/thno.21691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Lang, L. , Huang, P. , Wang, Z. , Jacobson, O. , Kiesewetter, D. O. , Ali, I. U. , Teng, G. , Niu, G. , & Chen, X. (2015). In vivo albumin labeling and lymphatic imaging. Proceedings of the National Academy of Sciences of the United States of America, 112(1), 208–213. 10.1073/pnas.1414821112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, D. C. , Bayik, D. , Srivatsan, A. , Bergamaschi, C. , Valentin, A. , Niu, G. , Bear, J. , Monninger, M. , Sun, M. , Morales‐Kastresana, A. , Jones, J. C. , Felber, B. K. , Chen, X. , Gursel, I. , & Pavlakis, G. N. (2016). Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials, 105, 195–205. 10.1016/j.biomaterials.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, J. , Bera, T. K. , Liu, X. F. , Zhou, Q. , Onda, M. , Ho, M. , Tai, C. H. , & Pastan, I. (2018). Recombinant immunotoxins with albumin‐binding domains have long half‐lives and high antitumor activity. Proceedings of the National Academy of Sciences of the United States of America, 115(15), E3501–E3508. 10.1073/pnas.1721780115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Z. , Chen, Z. , Zhao, Y. , Fan, F. , Xiong, W. , Song, S. , Yin, Y. , Hu, J. , Yang, K. , Yang, L. , Xu, B. , & Ge, J. (2021). Mononuclear phagocyte system blockade using extracellular vesicles modified with CD47 on membrane surface for myocardial infarction reperfusion injury treatment. Biomaterials, 275(May), 121000. 10.1016/j.biomaterials.2021.121000 [DOI] [PubMed] [Google Scholar]
- Wiklander, O. P. B. , Nordin, J. Z. , O'Loughlin, A. , Gustafsson, Y. , Corso, G. , Mäger, I. , Vader, P. , Lee, Y. , Sork, H. , Seow, Y. , Heldring, N. , Alvarez‐Erviti, L. , Edvard Smith, C. I. , Le Blanc, K. , Macchiarini, P. , Jungebluth, P. , Wood, M. J. A. , & El Andaloussi, S. (2015). Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. Journal of Extracellular Vesicles, 4(2015), 1–13. 10.3402/jev.v4.26316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willekens, F. L. A. , Werre, J. M. , Kruijt, J. K. , Roerdinkholder‐Stoelwinder, B. , Groenen‐Döpp, Y. A. M. , Van Den Bos, A. G. , Bosman, G. J. C. G. M. , & Van Berkel, T. J. C. (2005). Liver Kupffer cells rapidly remove red blood cell‐derived vesicles from the circulation by scavenger receptors. Blood, 105(5), 2141–2145. 10.1182/blood-2004-04-1578 [DOI] [PubMed] [Google Scholar]
- Yousefpour, P. , McDaniel, J. R. , Prasad, V. , Ahn, L. , Li, X. , Subrahmanyan, R. , Weitzhandler, I. , Suter, S. , & Chilkoti, A. (2018). Genetically encoding albumin binding into chemotherapeutic‐loaded polypeptide nanoparticles enhances their antitumor efficacy [rapid‐communication]. Nano Letters, 18(12), 7784–7793. 10.1021/acs.nanolett.8b03558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Higginbotham, J. N. , Jeppesen, D. K. , Yang, Y. P. , Li, W. , McKinley, E. T. , Graves‐Deal, R. , Ping, J. , Britain, C. M. , Dorsett, K. A. , Hartman, C. L. , Ford, D. A. , Allen, R. M. , Vickers, K. C. , Liu, Q. , Franklin, J. L. , Bellis, S. L. , & Coffey, R. J. (2019). Transfer of functional cargo in exomeres. Cell Reports, 27(3), 940–954.e6. 10.1016/j.celrep.2019.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, B. , Kelly, B. , McMillan, B. J. , Seegar, T. C. M. , Dror, R. O. , Kruse, A. C. , & Blacklow, S. C. (2016). Crystal structure of a full‐length human tetraspanin reveals a cholesterol‐binding pocket. Cell, 167(4), 1041–1051.e11. 10.1016/j.cell.2016.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel, L. , Regnault, A. , Lozier, A. , Wolfers, J. , Flament, C. , Tenza, D. , Ricciardi‐Castagnoli, P. , Raposo, G. A. S. , Zitvogel, L. , Regnault, A. , Lozier, A. , Wolfers, J. , Flament, C. , Tenza, D. , Ricciardi‐Castagnoli, P. , Raposo, G. , & Amigorena, S. (1998). Eradication of established murine tumors using a novel cell‐free vaccine: dendritic cell‐derived exosomes. Nature Medicine, 4(5), 594–600. http://www.ncbi.nlm.nih.gov/pubmed/9585234%0Ahttp://www.nature.com/naturemedicine [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data supporting the main findings of this study are available from the corresponding authors.