

Abstract
Background
Mim8 is a novel, next‐generation factor VIIIa mimetic in development for subcutaneous prophylactic treatment of patients with hemophilia A with and without inhibitors. In vitro and in vivo models indicate that Mim8 has a distinct hemostatic potential.
Objectives
To test the nonclinical safety and pharmacodynamics of Mim8.
Methods
The Mim8 nonclinical safety program in cynomolgus monkeys consisted of three studies of 4–26 weeks in duration with Mim8 doses ranging from 0.3–60 mg/kg/week intravenously or subcutaneously. After sacrifice, macroscopic and microscopic pathological examinations were performed.
Results
Mim8 was well tolerated with no noteworthy clinical observations. No signs of excessive coagulation or pathological macroscopic or microscopic findings were observed at doses 0.3–3 mg/kg/week subcutaneous. Thrombosis‐related findings were detected during histopathological examination in a small proportion of animals (16%) receiving doses ranging 6–20 mg/kg/week. Dose‐dependent increases in factor X (FX) and factor IX (FIX) concentrations were observed. Shortening of activated partial thromboplastin time (APTT) and increased thrombin generation under ex vivo hemophilia A‐like conditions were observed at all Mim8 dose levels.
Conclusions
Thrombosis‐related findings observed at doses above 6 mg/kg/week Mim8 may have been exaggerated pharmacological reactions to a procoagulant compound in normocoagulant animals. Increases in FX and FIX concentrations could be because of a half‐life prolongation due to binding to Mim8, but were limited at clinically relevant exposure levels. Subcutaneous administration of up to 3 mg/kg/week (several fold greater than expected clinical exposure) for 26 weeks resulted in relevant pharmacodynamic effects, observed in thrombin generation and APTT, with no signs of thrombi or excessive coagulation activation.
Keywords: antibodies, bispecific, drug evaluation, factor VIII, hemophilia A, nonclinical, safety
Essentials.
Mim8, a novel factor VIIIa mimetic, shows in vitro and in vivo hemostatic potential.
Nonclinical safety studies in monkeys confirmed the hemostatic potential of Mim8 ex vivo.
No thrombi or adverse effects were seen at dose levels up to 3 mg/kg/week for 26 weeks.
The data support clinical evaluation in hemophilia A patients with or without inhibitors.
1. INTRODUCTION
For decades, prophylactic factor VIII (FVIII) replacement therapy has been the standard of care for hemophilia A (HA), whereby patients receive intravenous (IV) injections two to three times per week to maintain sufficient FVIII activity to prevent spontaneous bleeding. 1 Successful management of HA with FVIII replacement therapy may be hampered by the development of neutralizing alloantibodies (inhibitors) against FVIII. It is estimated that 15–40% of previously untreated patients with HA develop inhibitors. 2 , 3 , 4 , 5
Hemophilia patients with inhibitors require on‐demand treatment with bypassing agents (e.g., activated recombinant factor VII [FVIIa] or activated prothrombin complex concentrate [APCC]) to resolve bleeding episodes. 1 However, on‐demand treatment is an inadequate alternative, and high annualized bleeding rates and associated joint and musculoskeletal comorbidities may result if patients do not receive appropriate prophylactic therapy. 1 , 6 Inhibitors can be overcome by immune tolerance induction (ITI), but it is costly, time consuming, often a burden for the patients. ITI is not always successful and is often met with diminished success in patients with high inhibitor titers. 7 To overcome inhibitors, and the barrier to treatment they represent, several novel drugs are in clinical development. The recently approved FVIIIa mimetic, emicizumab (Hemlibra®), which is a bispecific antibody that bridges activated factor IX (FIXa) and factor X (FX), is currently used for treatment of patients with HA with and without inhibitors in many countries. 8 , 9 , 10
Furthermore, the injection frequency of FVIII replacement therapy can represent a treatment burden for patients with HA. Antibodies, in general, have a high bioavailability after subcutaneous (SC) administration and long circulatory half‐life, and they can offer less invasive administration, longer time between dosing intervals, and a steady level of bleed protection. Overall, this shows great promise in terms of treatment convenience, a potential for improved compliance, and steady levels of effective drug. 11
Mim8 is a novel, next‐generation FVIIIa mimetic designed for the subcutaneous prophylactic treatment of patients with HA with and without inhibitors. Mim8 is a fully human, bispecific antibody that mimics FVIIIa function by bridging FIXa and FX on the phospholipid surface of activated platelets, enhancing the proteolytic activity of FIXa, and thus facilitating effective FX activation. Data from studies using in vitro HA‐like human blood, as well as in vivo HA mouse models, indicate that Mim8 is ~15‐fold more potent than a sequence identical analogue (SIA) of the FVIII mimetic emicizumab. 12 The nonclinical characterization of Mim8 has demonstrated that it potently increases thrombin generation triggered by both tissue factor and FXIa. 12 This increased potency is primarily due to the monovalent anti‐FIXa arm’s strong stimulation of FIXa’s proteolytic activity. 12 In animal models of hemophilia in both mice and cynomolgus monkeys (Macaca fascicularis), Mim8 was able to effectively reduce bleeding. 12 , 13 These studies support the potent pharmacological activity of Mim8. 13 Mim8 is currently being evaluated in a phase I/II clinical trial (NCT04204408).
A high potency may lead to exaggerated pharmacology at high dose levels, which for a procoagulant compound could present as excessive coagulation such as the development of thrombi. 12 , 14 Cynomolgus monkeys are the only nonclinical model species shown to have cross‐reactivity to Mim8. Thus, to support safe administration of Mim8 to humans in clinical trials, we tested the safety and pharmacodynamics of various dose levels of Mim8 in cynomolgus monkeys in three nonclinical safety studies.
2. MATERIALS AND METHODS
2.1. General study design
Cynomolgus monkeys were selected for these studies because these are the only nonclinical model species shown to have cross‐reactivity to Mim8, and we have previously demonstrated that Mim8 binds cynomolgus monkey and human FIX and FX epitopes with similar affinities. 12 Furthermore, cynomolgus monkeys have comparable plasma FIX and FX concentrations and high homology compared to human FIX and FX.
This nonclinical program in cynomolgus monkeys consisted of three studies: (1) a 4‐week dose range finding (DRF) study, (2) a 13‐week toxicity study (with a 4‐week interim sacrifice and 13‐week recovery period), and (3) a 26‐week toxicity study.
In the 4‐week DRF study, 10 animals (one male and one female per dose) received 1, 4, 20, or 60 mg/kg Mim8 SC or 20 mg/kg IV once weekly for 4 weeks. All animals were sacrificed 1 day after the last dose, and a full necropsy was performed (Figure S1A in supporting information).
In the 13‐week study, three animals per sex received 0.3 mg/kg/week Mim8 SC and six animals per sex received Mim8 in doses of 6 or 12 mg/kg/week SC or vehicle control. Three animals per sex received 1 mg/kg/week Mim8 IV. Three animals per sex dosed with 6 or 12 mg/kg/week Mim8 SC or vehicle control were sacrificed after 4 weeks of dosing (interim sacrifice) and the remaining animals dosed with SC Mim8 were sacrificed after 13 weeks of dosing. Animals administered IV Mim8 were sacrificed after 8 weeks of dosing. Finally, two animals per sex were dosed with 12 mg/kg/week SC Mim8 for 13 weeks and sacrificed after a 13‐week recovery period in which no doses of Mim8 were administered, to examine if any adverse effects potentially seen after 13 weeks of dosing were reversible (Figure S1B). At the end of the study, a full necropsy was performed.
In the 26‐week study, four sexually mature animals per sex received 0.3, 1, or 3 mg/kg/week SC Mim8 or vehicle control. All animals were sacrificed, and a full necropsy was performed after 26 weeks of dosing (Figure S1C).
2.2. Animal housing and handling
For all experiments, cynomolgus monkeys were purchased from Noveprim. On arrival, all animals underwent clinical inspection and were tested for tuberculosis. Monkeys were acclimated for at least 2 weeks before dosing. Body weights were recorded twice weekly or weekly during the predose, dosing, and recovery period and prior to necropsy. At the beginning of experiments, animals were between 118–123 weeks old (4‐week study); 101–119 weeks, (13‐week study), or 62–102 months (26‐week study).
2.3. Exposure and anti‐drug antibodies
To measure exposure in animals dosed SC, blood samples were taken in 3.8% trisodium citrate at 0, 8, 24, 48, 96, and 168 h postdose in the penultimate or last dosing of Mim8. In the animals dosed IV in the 4‐week and 13‐week studies, blood was sampled at 0.25, 4, 8, 24, 72, and 168 h post‐dose. A non‐validated fit‐for‐purpose assay electrochemiluminescent assay specific for human IgG4 was used in the 4‐week DRF study and a validated immunoassay specific for Mim8 was used in the 13‐ and 26‐week studies. Non‐compartmental analysis of plasma concentration data was performed using Phoenix WinNonlin version 8.1 (Certara). For evaluation of anti‐drug antibodies (ADAs), blood was sampled in 3.8% trisodium citrate pretreatment and predose on days 85 and 176 in the 26‐week study. For the 4‐week and 13‐weeks studies, samples for ADA measurement were only taken pretreatment and predose on the penultimate or last day of dosing. The blood was processed for plasma and analyzed by a fit‐for‐purpose enzyme‐linked immunosorbent assay (ELISA) using mouse anti‐human IgG as a capture antibody and horseradish peroxidase labelled anti‐monkey IgG to measure ADAs. Neutralizing effects of ADAs were determined by decreases in exposure or thrombin generation and/or prolongation of activated partial thromboplastin time (APTT). Samples from control animals were not analyzed for the presence of ADAs.
2.4. Prothrombin time and activated partial thromboplastin time
Plasma separated from citrate stabilized blood samples taken at predetermined time points were used for measurements of prothrombin time (PT; PT‐fibrinogen HS PLUS [Instrumentation Laboratory]) and APTT (SynthASil® [Instrumentation Laboratory]). In the 26‐week study, PT and APTT were measured pretreatment; after 3 days; and at 13, 20, and 26 weeks of dosing.
2.5. Thrombin‐antithrombin and D‐dimer evaluation
In all studies, D‐dimers were measured using the Immunoclone D‐dimer ELISA kit (Sekisui Diagnostics), and analysis for thrombin‐antithrombin (TAT) was performed using the Enzygnost TAT Micro ELISA kit (Siemens Healthineers). In the 26‐week study, TAT and D‐dimers were evaluated on pretreatment and after 3, 43, 85, 134, and 183 days of dosing.
2.6. FIX and FX evaluation
For FIX and FX antigen evaluation, blood was sampled in 3.8% trisodium citrate prior to the first dose and at predetermined time points during the dosing and recovery periods in all three studies. In the 26‐week study, FIX and FX was assessed pretreatment and after 3, 43, 85, 134, and 183 days of dosing. Plasma was separated and analyzed for FX in an assay based on a commercial ELISA for human FX (Zymutest Factor X; HYPHEN BioMed) with an in‐house cynomolgus monkey FX standard. FIX was measured using LOCI with one commercial polyclonal biotinylated antibody (LS‐B7226; LSBio) and one in‐house monoclonal FIX antibody.
2.7. Thrombin generation
Plasma was prepared from blood samples collected in 3.8% trisodium citrate prior to the first dose and at predetermined time points during the dosing and recovery periods in all three studies. For the 26‐week study, samples were obtained pretreatment and after 3, 43, 85, 134, and 183 days of dosing.
Pretreatment samples were analyzed directly to measure the normal thrombin generation capacity in the animals and with an anti‐FVIII polyclonal antibody (Hematological Technologies Inc.) added (0.26 mg/ml) to induce hemophilia A‐like conditions. All postdose samples were analyzed with the anti‐FVIII antibody added. The thrombin generation assay (TGA) was performed by adding activated factor XI (FXIa; Hematological Technologies Inc.) diluted in phospholipid reagent (MP reagent, Thrombinoscope, Stago) to a final FXIa plasma concentration of 50 mU/ml and FluCa reagent (Thrombinoscope). The thrombin generation was followed for 90 min using a Fluoroskan Ascent plate reader (Thermo Fisher). The fluorescent signal was converted to thrombin activity using Thrombinoscope’s thrombin calibrator and version 5.0.0 software (Synapse B.V.).
2.8. Blood sampling for clinical pathology
For the 26‐week nonclinical toxicity study, blood sampling was performed once during the pretreatment phase, and after 3 days and 13, 20, and 26 weeks of the dosing phase, as well as on the day before necropsy. Similarly, blood was sampled for clinical pathology at predetermined time points during the dosing and recovery periods in the 4‐week and 13‐week studies. Blood samples stabilized with ethylenediaminetetraacetic acid were evaluated for hematological parameters, including platelet counts. Lists of examined hematological parameters are included in the supplemental material (Tables S1–S2 in supporting information).
2.9. Necropsy
Animals were administered a lethal IV overdose of sodium pentobarbitone. Once death was confirmed, animals were exsanguinated by severing major blood vessels. A full macroscopic examination was performed under the supervision of a pathologist. Organs were weighed and histological samples were prepared from selected target organs in the 4‐week DRF study, and from all major organ systems in the 13‐week and 26‐week studies. Up to 54 tissues from all animals were fixed, sectioned, stained, and examined microscopically. For a list of examined tissues, please see Table S3 in supporting information.
The histopathologic evaluation was performed by an experienced and qualified pathologist. Histopathological alterations were evaluated in a semiquantitative manner, based on a 5‐point grading system: Grade 1—minimal, Grade 2—slight, Grade 3—moderate, Grade 4—marked, and Grade 5—severe. 15
2.10. Animal handling ethics statement
All animal studies were performed at Covance (Harrogate, UK or Münster, Germany), and conformed with the requirements of internal ethical committees at both Novo Nordisk A/S and Covance, national legislation, and Directive 2010/63/EU of the European Parliament and Council of 22 September 2010 on the protection of animals used for scientific purposes.
2.11. Data analysis and statistics
For the 4‐week and 13‐week study, only data from pretreatment and the final sampling point are presented for pharmacodynamic effects (TGA, APTT) and biomarkers (PT, FIX, FX).
Data from animals that developed ADAs and in which reduced exposure was observed were omitted from the pharmacokinetic, pharmacodynamic, and biomarker analysis from the time of ADA development. For the pharmacokinetic data, APTT and thrombin generation, the data for ADA‐positive animals are included (see Tables 1 and 3), but no statistical analyses were performed on these data. Tests for significance between time points for pharmacodynamic effects (TGA, APTT) and biomarkers (PT, FIX, FX) were performed using a one‐way analysis of variance comparing the vehicle group with the different dose groups corrected for multiple comparisons. In the case of variance heterogeneity or lack of normal distribution, a nonparametric test (Kruskall‐Wallis with Dunnet’s post‐test) was performed. P < .05 was considered statistically significant. No statistical comparisons were made on the data from the 4‐week study due to low number of animals and lack of control group.
TABLE 1.
Exposure to Mim8 in cynomolgus monkeys after weekly administration
| Study | Dose (mg/kg/week) | Administration route | Neutralizing ADA status (n) | Cmax, nmol/L a | Cmax/dose b | AUC0–168 h, h × nmol/L a | AUC0–168 h/dose b |
|---|---|---|---|---|---|---|---|
| 4‐week | 1 | SC | ‐‐ (2) | 150 (11.3) | 150 | 21,700 (9.56) | 21,700 |
| 4 | SC | ‐‐ (2) | 441 (26.9) | 110 | 68,600 (29.5) | 17,200 | |
| 20 | SC | ‐‐ (2) | 1960 (2.16) | 98 | 276,000 (0.574) | 13,800 | |
| 60 | SC | ‐‐ (2) | 7500 (3.77) | 125 | 974,000 (6.81) | 16,200 | |
| 20 | IV | ‐‐ (2) | 4460 (2.85) | 223 | 480,000 (7.17) | 24,000 | |
| 13‐week | 0.3 | SC | ‐‐ (6) | 58.2 (15.3) | 194 | 8840 (16.4) | 29,500 |
| 6 | SC | ‐‐ (4) | 750 (12.1) | 125 | 107,000 (13.9) | 17,800 | |
| + (2) | 30.5 (119) | 5.08 | 1640 (128) | 273 | |||
| 12 c | SC | ‐‐ (7) | 2320 (13.5) | 193 | 352,000 (13.3) | 29,400 | |
| + (3) | 564 (38.6) | 47 | 54,600 (41.5) | 4550 | |||
| 1 | IV | ‐‐ (1) | 306 | 306 | 37,100 | 37,100 | |
| + (5) | 61.7 (90.9) | 61.7 | 1580 (149) | 1580 | |||
| 26‐week | 0.3 | SC | ‐‐ (4) | 81.6 (30.2) | 272 | 12,700 (33.0) | 42,300 |
| + (4) | N/A d | N/A d | N/A d | N/A d | |||
| 1 | SC | ‐‐ (5) | 241 (13.0) | 241 | 36,500 (10.5) | 36,500 | |
| + (3) | N/A d | N/A d | N/A d | N/A d | |||
| 3 | SC | ‐‐ (4) | 730 (3.53) | 243 | 109,000 (2.04) | 36,400 | |
| + (4) | N/A d | N/A d | N/A d | N/A d |
Abbreviations: ADA, anti‐drug antibodies; AUC, area under the curve; Cmax, maximum plasma concentration; h, hours; IV, intravenous; SC, subcutaneous.
Cmax and AUC values are presented as the geometric mean (CV%).
Normalized to 1 mg/kg.
Including exposure after 13 weeks in the 4 recovery animals dosed for 13 weeks followed by a 13‐week recovery phase.
In the 26‐week study all ADA‐positive animals had no exposure to Mim8 at the end of the study.
TABLE 3.
Overview of activated partial thromboplastin time (APTT) and thrombin peak in cynomolgus monkeys following exposure to Mim8
| Study | Dose (mg/kg/week) | Admininistration route | ADA status a |
APTT predose, s (n) |
APTT before final dose, s (n) |
Thrombin peak predose, nM (n) |
Thrombin peak before final dose, nM (n) |
|---|---|---|---|---|---|---|---|
| 4‐week | 1 | SC | ‐ | 20.9 (1) | 13.6 ± 0.2 (2) | N/A | 363 ± 111 (2) |
| 4 | SC | ‐ | 21.9 ± 0.5 (2) | 12.1 ± 0.2 (2) | 4.7 ± 1.1 (2) | 443 ± 23 (2) | |
| 20 | SC | ‐ | ‐ | 11.5 ± 0.1 (2) | 7.9 (1) | 331 (1) | |
| 60 | SC | ‐ | 26.6 ± 4.4 (2) | 13.1 ± 0.7 (2) | 15.7 ± 8.5 (2) | 331 ± 105 (2) | |
| 20 | IV | ‐ | 20.5 ± 0.8 (2) | 11.5 ± 0.2 (2) | 5.8 (1) | 379 ± 29 (2) | |
| 13‐week | 0 | SC | NA | 20.0 ± 0.5 (12) b | 20.0 ± 0.3 (6) | 4.9 ± 1.3 (12) b | 3.5 ± 1.4 (6) |
| 0.3 | SC | ‐ | 20.0 ± 0.3 (6) | 14.9 ± 0.2 (6) | 5.3 ± 1.8 (6) | 131 ± 8.0 (6) | |
| 6 | SC | ‐ | 19.8 ± 0.2 (12) b | 12.7 ± 0.9 (4)** | 6.5 ± 1.7 (12) b | 237 ± 17 (4)* | |
| + | ‐ | 20.6 ± 0.5 (2) | ‐ | 4.7±0.7 (2) | |||
| 12 | SC | ‐ | 20.2 ± 0.3 (16) c | 11.7 ± 0.2 (7)*** | 6.2 ± 2.2 (16) c | 292 ± 9.0 (7)*** | |
| + | ‐ | 19.0 ± 1.0 (2) | ‐ | 15.3 ± 3.5 (3) | |||
| 12 d | SC | ‐ | ‐ | 14.5 ± 1.5 (3) | ‐ | 125 ± 32 (3) | |
| + | ‐ | 18.7 (1) | ‐ | 4.4 (1) | |||
| 1 | IV | ‐ | 20.3 ± 0.3 (6) | 14 (1) | 9.7 ± 3.1 (6) | 234 (1) | |
| + | ‐ | 19.7 ± 1.1(5) | ‐ | 44.8 ± 38.8 (5) | |||
| 26‐week | 0 | SC | NA | 20.9 ± 0.7 (8) | 20.4 ± 0.4 (8) | 4.6 ± 1.1 (8) | 4.8 ± 1.0 (8) |
| 0.3 | SC | ‐ | 20.8 ± 0.5 (8) | 12.6 ± 0.6 (4)*** | 5.4 ± 0.7 (8) | 208 ± 19 (4) | |
| + | 20.5 ± 0.7 (4) | ‐ | 5.8 ± 1.4(4) | ||||
| 1 | SC | ‐ | 20.7 ± 0.5 (8) | 12.7 ± 0.6 (5)*** | 4.5 ± 0.7 (8) | 249 ± 20 (5)* | |
| + | ‐ | 21.4 ± 1.3 (3) | ‐ | 4.5 ± 0.6 (3) | |||
| 3 | SC | ‐ | 21.5 ± 0.7 (8) | 11.3 ± 0.1 (4)*** | 4.8 ± 0.9 (8) | 410 ± 57 (4)*** | |
| + | ‐ | 22.1 ± 1.2 (4) | ‐ | 6.4 ± 2.1(4) |
Abbreviations: IV, intravenous; N/A, not available; s, seconds; SC, subcutaneous.
*P < .05, **P < .01, ***P < .001. Data are mean ± standard error of the mean (SEM). Data are from three separate nonclinical studies. Thrombin peak was measured by thrombin generation assay. All thrombin peaks were measured are after adding a neutralizing FVIII antibody ex vivo to induce HA‐like conditions.
Status for neutralizing anti‐drug‐antibody development (ADA) at the end of the study: ‘+’: animals with ADA’s and affected exposure; ‘‐’: Animals with expected exposure; most of these animals were ADA negative, however some animals in the 4‐week and 13‐week study were ADA positive, without affected exposure.
Including data from interim animals (n = 6) taken down after 4 weeks;
Including data from interim animals sacrificed after 4 weeks (n = 6) and recovery animals (n = 4).
Animals dosed 13 weeks followed by a 13‐week recovery phase.
3. RESULTS
3.1. Exposure and anti‐drug antibodies
During all three studies, Mim8 exposure increased approximately dose‐proportionally, with the highest exposure (maximum plasma concentration [Cmax]: 7500 nmol/L; AUC0‐168h: 974 000 h × nmol/L) after 4 weeks at 60 mg/kg/week SC in the 4‐week study (Table 1). ADAs were detected in three animals at ≥20 mg/kg/week in the 4‐week study, with no apparent effect on exposure (Table 2). In the 13‐week study, ADAs were detected in most animals with doses ≥1 mg/kg/week. ADAs affected Mim8 exposure in 4/12 (33.3%), 3/16 (18.8%), and 5/6 (83.3%) animals in the 6 mg/kg/week SC, 12 mg/kg/week SC, and 1 mg/kg/week IV groups, respectively. In the 26‐week toxicity study, approximately half of the animals in all dose groups developed ADAs and all animals with ADA had reduced Mim8 exposure (Table 2). A clear effect of the ADAs in the form of reduced exposure (Table 1), as well as lack of Mim8 effect in APTT and TGA was observed both in the 13‐week and the 26‐week studies (Table 3).
TABLE 2.
Overview of toxicology studies with Mim8 in cynomolgus monkeys
| Study | Dose (mg/kg/week) | Administration route | n a | ADAs (neutralizing ADAs) b | Noteworthy adverse findings in single animals |
|---|---|---|---|---|---|
| 4‐week | 1 | SC | 2 | 0/2 (0/2) | ‐ |
| 4 | SC | 2 | 0/2 (0/2) | ‐ | |
| 20 | SC | 2 | 1/2 (0/2) | One microscopic pulmonary thrombus | |
| 60 | SC | 2 | 1/2 (0/2) | ‐ | |
| 20 | IV | 2 | 1/2 (0/2) | ‐ | |
| 13‐week | 0 c | SC | 6 | ‐ | ‐ |
| 6 c | SC | 6 | 5/6 (2/6) | Three microscopic pulmonary thrombi | |
| 12 c | SC | 6 | 3/6 (0/6) | Necrosis adrenal medulla | |
| 0 | SC | 6 | ‐ | ‐ | |
| 0.3 | SC | 6 | 0/6 (0/6) | ‐ | |
| 6 | SC | 6 | 6/6 (2/6) | Necrosis axial lymph node | |
| 12 | SC | 6 | 4/6 (2/6) | One microscopic thrombus | |
| 12 d | SC | 4 | 3/4 (1/4) | ‐ | |
| 1 e | IV | 6 | 5/6 (5/6) | ‐ | |
| 26‐week | 0 | SC | 8 | ‐ | ‐ |
| 0.3 | SC | 8 | 4/8 (4/8) | ||
| 1 | SC | 8 | 3/8 (3/8) | ‐ | |
| 3 | SC | 8 | 4/8 (4/8) | ‐ |
Abbreviations: ADAs, anti‐drug antibodies; IV, intravenous; SC, subcutaneous.
Equal numbers of male and female animals were used.
Neutralizing based on reduced exposure and reduced effect in thrombin generation assay/activated partial thromboplastin time at the end of the study.
Interim animals sacrificed after 4 weeks of dosing.
Animals dosed for 13 weeks followed by a 13‐week recovery phase.
Animals from IV group dosed for 8 weeks.
[Correction added on March 17, 2022, after first online publication: The heading for Table 2 has been updated.]
3.2. Tolerability, safety, and pathological evaluation
Across all three studies, Mim8 was well tolerated, and no noteworthy clinical observations were made during the exposure period. During the post‐mortem examinations, no noteworthy macroscopic pathological findings were made.
In the 4‐week study, the histopathological examination of tissues revealed a minimal, non‐occlusive microscopic thrombus in the left anterior lung lobe of one monkey administered 20 mg/kg/week Mim8 SC (Table 2). This thrombus was not associated with any pathology involving the parenchyma.
At the interim (4‐week) sacrifice of the 13‐week study, the histopathological examination revealed moderate adrenal necrosis in a single animal administered 12 mg/kg/week Mim8 SC and minimal microscopic non‐occlusive pulmonary thrombi in three medium‐sized veins of a single animal administered 6 mg/kg/week Mim8 SC. During the terminal (13‐week) sacrifice, a minimal microscopic non‐occlusive pulmonary thrombus was observed in one animal administered 12 mg/kg/week Mim8 SC and moderate necrosis of the axillary lymph node adjacent to the SC injection site (draining lymph node) was observed in an animal administered 6 mg/kg/week Mim8 SC. The observed minimal microscopic non‐occlusive lung thrombi were not associated with inflammatory changes or tissue damage in the surrounding parenchyma of the lung.
No treatment‐related findings were observed for animals administered 12 mg/kg/week Mim8 SC after a 13‐week recovery phase, and no findings were observed for animals administered Mim8 1 mg/kg/week Mim8 IV or 0.3 mg/kg/week Mim8 SC.
In the 26‐week toxicity study, no pathological findings were made. There were no macroscopic or microscopic observations of thrombi or other signs of excessive coagulation at dose levels up to 3 mg/kg/week SC (Table 2).
Local tolerance was assessed in the 13‐ and 26‐week studies. The histopathological findings recorded at the SC injection sites after 13 weeks were of similar incidence and severity in treated groups and vehicle control. In the 26‐week study, the histopathological findings at the SC injection sites consisted of mononuclear cell/mixed inflammatory cell infiltrates, some with edema/hemorrhage in 13 out of 32 animals. The findings primarily occurred in the animals with detected ADAs (9 of 13 animals).
3.3. Pharmacodynamic effects (APTT and thrombin generation)
In accordance with the Mim8 mode of action, a pharmacodynamic effect in the form of a shortening of APTT was observed in all dose groups across all studies, reaching statistical significance compared to controls at 6 mg/kg/week and above in the 13‐week study (P < .01 or .001) and at all dose levels in the 26‐week study (P < .001). There were limited differences between the dose levels, except for the time point 3 days after the first dose, where the effect on APTT tended to be lower in the low dose group compared to the other groups (Table 3; Figure 1A). Consistent with this, APTT was shortened compared to predose values in all groups in the 4‐week study, with no apparent differences between dose levels. In animals with ADAs and affected exposure, the APTT returned to pretreatment levels (Table 3).
FIGURE 1.
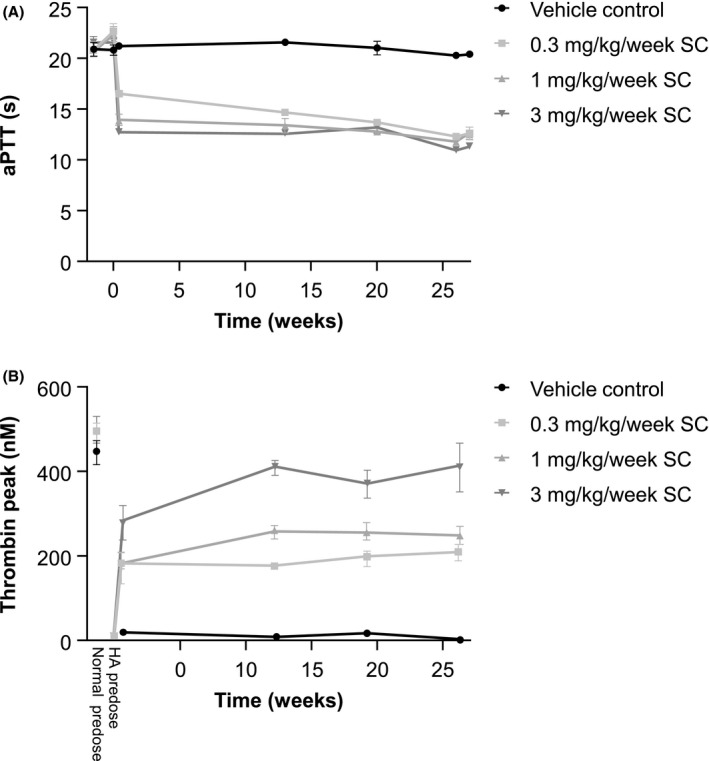
Activated partial thromboplastin time (APTT) and thrombin peak in cynomolgus monkeys administered 0, 0.3, 1, or 3 mg/kg/week subcutaneous Mim8 at predose and after 3 days and 13, 20, and 26 weeks exposure to Mim8. A, APTT of plasma from monkeys administered Mim8. B, Thrombin generation was measured prior to and after addition of anti–factor VIII (FVIII) antibody to mimic hemophilia A‐like conditions. In addition, predose samples were analyzed without anti‐FVIII antibody. Data from samples positive for anti‐drug antibodies were omitted. Data are mean ± standard error of the mean (SEM); 4–8 animals were measured at each time point
Ex vivo addition of anti‐FVIII antibody to plasma taken from monkeys pre‐Mim8 dosing rendered it HA‐like and resulted in very low thrombin generation (Table 3; Figure 1B). A dose‐dependent increase in thrombin generation was observed in HA‐like plasma from monkeys dosed with Mim8, reaching statistical significance compared to predose HA‐like controls at 6 and 12 mg/kg/week SC dosing levels in the 13‐week study (P < .05 or .001, respectively), and at 1 and 3 mg/kg/week SC in the 26‐week study (P < .05 or .001, respectively). The level of thrombin generation of samples from monkeys dosed with Mim8 from week 13 and onward in the 26‐week study was stable, indicating steady‐state exposure (Table 3; Figure 1B). Furthermore, thrombin generation approached that of normal predose blood in the 3 mg/kg/week dose group (Figure 1B). In the 4‐week study, the thrombin generation of samples from monkeys dosed with Mim8 was numerically higher than predose levels of HA‐like plasma and generally comparable across all dose levels up to 60 mg/kg/week. In animals with ADAs and affected exposure, the thrombin generation returned to pretreatment levels (Table 3).
3.4. Biomarkers
Across all studies, no noteworthy changes in platelet counts or fibrinogen were observed at any time point (data not shown). In the 13‐week study, a statistically significant prolongation of PT compared to controls was observed at 6 and 12 mg/kg/week (P < .05 and .001, respectively). Similarly, the PT was numerically longer compared to the predose values in the 4‐week study at dose levels of 20 mg/kg/week or above (Table 4). In the 26‐week study, no significant changes in PT were observed when testing dose levels went up to 3 mg/kg/week (Figure 2A).
TABLE 4.
Overview of FIX and FX concentrations and prothrombin time (PT) in cynomolgus monkeys following exposure to Mim8
| Study | Dose (mg/kg/week) | Admin route |
FIX Predose, ng/ml (n) |
FIX Before final dose, ng/ml (n) |
FX Predose, ng/ml (n) |
FX Before final dose, ng/ml (n) |
PT Predose, s (n) |
PT Before final dose, s (n) |
|---|---|---|---|---|---|---|---|---|
| 4‐week | 1 | SC | 3005 ± 135 (2) | 3835 ± 525(2) | 5580 ± 860 (2) | 7020 ± 1140 (2) | 14.1 (1) | 13.1 ± 0.5 (2) |
| 4 | SC | 3900 ± 180 (2) | 5700 ± 420 (2) | 5120 ± 40 (2) | 13 750 ± 250 (2) | 13.4 ± 0 (2) | 14.3 ± 0.8 (2) | |
| 20 | SC | 3440 ± 80 (2) | 4345 ± 855 (2) | 4560 ± 200 (2) | 15 650 ± 450 (2) | ‐ | 16.2 ± 0.1 (2) | |
| 60 | SC | 3600 ± 520 (2) | 5760 ± 1120 (2) | 4620 ± 420 (2) | 20 500 ± 100 (2) | 16.2 ± 3.5 (2) | 20.2 ± 0.1 (2) | |
| 20 | IV | 3935 ± 425 (2) | 7520 ± 1320 (2) | 4820 ± 540 (2) | 18 750 ± 150 (2) | 13.6 ± 0.5 (2) | 17.7 ± 0.6 (2) | |
| 13‐week | 0 | SC | 4991 ± 417 (12) a | 4743 ± 322 (6) | 3742 ± 84 (12) a | 4013 ± 194 (6) | 13.0 ± 0.2 (12) a | 13.5 ± 0.2 (6) |
| 0.3 | SC | 5087 ± 624 (6) | 5102 ± 265 (6) | 4007 ± 215 (6) | 5022 ± 344 (6) | 13.2 ± 0.2 (6) | 13.8 ± 0.3 (6) | |
| 6 | SC | 5186 ± 216 (12) a | 6513 ± 712(4) | 3841 ± 226 (12) a | 8473 ± 1008 (4) | 13.2 ± 0.2 (12) a | 14.8 ± 0.2 (4)* | |
| 12 | SC | 4274 ± 189 (16) b | 7461 ± 576 (7)** | 3794 ± 136 (16) b | 15 429 ± 719 (7)*** | 13.1 ± 0.1 (16) b | 16.6 ± 0.4 (7)*** | |
| 12 c | SC | ‐ | 3800 ± 230 (3) | ‐ | 4740 ± 210 (3) | ‐ | 13.3 ± 0.2 (3) | |
| 1 | IV | 3810 ± 208 (6) | 5090 (1) | 3703 ± 207 (6) | 6600 (1) | 13.1 ± 0.2 (6) | 14.1 (1) | |
| 26‐week | 0 | SC | 6158 ± 288 (8) | 5628 ± 394 (8) | 4043 ± 195 (8) | 4505 ± 170 (8) | 14.9 ± 0.3 (8) | 14.8 ± 0.3 (8) |
| 0.3 | SC | 5908 ± 500 (8) | 5825 ± 394 (4) | 4463 ± 176 (8) | 5785 ± 585 (4) | 14.5 ± 0.3 (8) | 13.7 ± 0.2 (4) | |
| 1 | SC | 6074 ± 327 (8) | 6054 ± 367 (5) | 3974 ± 217 (8) | 6306 ± 578 (5)* | 14.7 ± 0.2 (8) | 15.3 ± 0.1 (5) | |
| 3 | SC | 5448 ± 242 (8) | 6823 ± 618 (4) | 4025 ± 131 (8) | 10 670 ± 768 (4)*** | 14.4 ± 0.2 (8) | 14.9 ± 0.3 (4) |
Abbreviations: FIX, factor IX; FX, factor X; IV, intravenous; s, seconds; SC, subcutaneous
*P < .05, **P < .01, ***P < .001. Data are mean ± standard error of the mean (SEM). Data are from three separate nonclinical studies. Data from samples positive for anti‐drug antibodies that affected exposure were omitted.
Including data from interim animals (n = 6) taken down after 4 weeks.
Including data from interim animals sacrificed after 4 weeks (n = 6) and recovery animals (n = 4)
Animals dosed 13 weeks followed by a 13‐week recovery phase.
FIGURE 2.
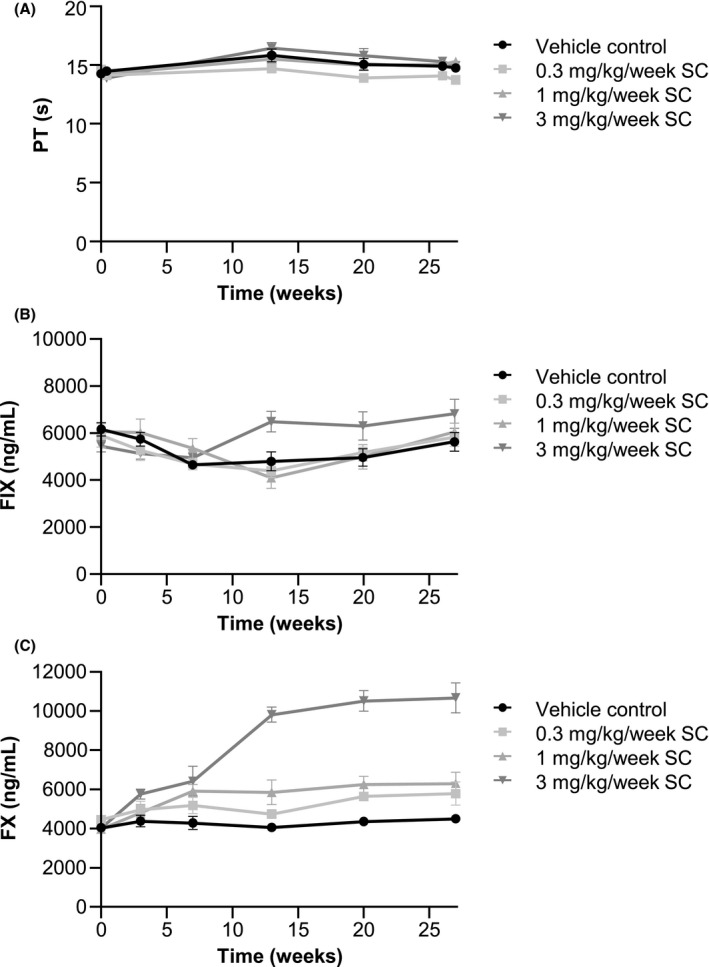
Prothrombin time (PT) and FX and FIX concentrations in cynomolgus monkeys during weekly subcutaneous (SC) Mim8 administration for 26 weeks. (A) PT, (B) FX concentration, and (C) FIX concentrations in blood from monkeys treated with 0, 0.3, 1, or 3 mg/kg/week SC Mim8. Data from samples positive for anti‐drug antibodies were omitted. Data are mean ± SEM. FIX, factor IX; FX, factor X; SEM, standard error of the mean; 4–8 animals were measured at each time point
No significant changes in FIX concentrations were observed in the 26‐week study, testing dose levels up to 3 mg/kg/week, but a numerical increase compared to control animals was observed at the highest dose level from week 13 onward (Figure 2B). Only when testing higher dose levels in the 13‐week study, a dose‐dependent increase in FIX concentrations was observed, reaching statistical significance compared to control animals at 12 mg/kg/week (P < .01). In the 4‐week study testing dose levels up to 60 mg/kg/week, the FIX concentrations also tended to be higher than predose levels in dosing groups ≥4 mg/kg/week (Table 4).
Plasma concentration of FX tended to increase dose‐dependently in all three studies, reaching a numerical increase of up to 4.5‐fold of predose levels at 60 mg/kg/week in the 4‐week study, and almost 4‐fold higher levels compared to control at 12 mg/kg/week in the 13‐week study (P < .001) (Table 4). Accordingly, statistically significant increases in FX were observed at 1 and 3 mg/kg/week in the 26‐week study (P < .05 and .001, respectively), with average concentration of up to 2.4‐fold of control levels at 3 mg/kg/week (Figure 2C).
TAT and D‐dimers (DDM) were measured at predetermined time points before and after dose start in all three studies. High inter‐ and intra‐individual variability was observed, and the levels after dosing were generally comparable to the levels in controls and predose samples. No trends for increased TAT or DDM concentrations were seen in monkeys with observations of minimal microscopic thrombi (Figure S2A & B in supporting information).
4. DISCUSSION
To support administration of the next‐generation FVIIIa mimetic, Mim8, in clinical trials, the safety and pharmacodynamics of Mim8 were tested in three separate, subsequent nonclinical toxicology studies in cynomolgus monkeys; a 4‐week DRF study, a 13‐week toxicity study (with a 4‐week interim sacrifice and 13‐week recovery), and a 26‐week toxicity study.
In all three studies, Mim8 exposure increased approximately dose‐proportionally. Some variation in the dose‐corrected area under the curve (AUC) was observed, but there were no systematic trends toward non‐linearity. However, for the 6 mg/kg/week group in the 13‐week study, lower than expected exposure was observed and the dose‐corrected AUC for this group was considerably lower at the end of the study than for the other groups in this study. As most of the animals in this group had ADAs at the end of the study, this could potentially be explained by an impact of the ADA on the Mim8 concentration in the plasma at this time point.
Relevant pharmacodynamic effects were seen at all dose levels of Mim8 in all studies. Thus, thrombin generation increased at all dose levels in plasma from monkeys dosed with Mim8 and made hemophilia A‐like by ex vivo addition of anti‐FVIII antibodies. At the highest Mim8 dose used in the 26‐week study, the thrombin generation approached the thrombin generation of normal blood obtained before dosing, indicating a good response with no over‐normalization of the coagulation system. Furthermore, administration of Mim8 shortened APTT in plasma at all dose levels, adding further proof‐of‐concept to evidence the effect of Mim8 observed in animal bleeding models. 12
In safety studies with procoagulant compounds performed in normocoagulant animals, high dose levels are used to search for potential adverse findings related to thrombosis. Thus, in the 4‐ and 13‐week studies, which tested very high dose levels up to 60 mg/kg/week of Mim8, leading to plasma concentrations up to 7500 nmol/L, adverse findings, considered potentially related to thrombosis, were observed in a small proportion of the animals. These included minimal non‐occlusive microscopic lung thrombi, as well as necroses in the adrenal medulla and the lymph node draining the SC injection site, seen as sequelae to a previous thrombosis. The reported thrombi were only observed in a few animals (1/10 Mim8 dosed animals in the 4‐week study and 4/40 Mim8 dosed animals in the 13‐week study; overall 16% of animals dosed between 6 and 20 mg/kg/week, but no thrombi were seen at lower dose levels [≤3 mg/kg/week]) and only seen during the histopathological examination. The lung thrombi were non‐occluding and were considered non‐adverse due to the minimal severity of the microscopic change, the absence of occlusion of the blood vessel, the absence of any inflammatory changes or tissue damage in the surrounding parenchyma of the lung, and the absence of any clinical observations and clinical pathology changes for these animals.
As mentioned, these were expected findings of exaggerated pharmacology in animals with a normal coagulation system using concentrations of Mim8 that are considerably above the expected dosing concentration in patients with HA in the clinical setting (plasma exposures ranging from 6–180 nmol/L are currently being investigated in clinical phase I/II trials). The use of monkeys with normal coagulation systems is according to toxicology guidelines, and no well‐characterized HA models exist for use in toxicology studies.
The more clinically relevant doses of 0.3, 1, or 3 mg/kg/week Mim8 SC were then chosen for the 26‐week study, and were well tolerated in the cynomolgus monkeys with normal coagulation. The no‐observed adverse effects level in the 26‐week study, based on clinical observations and microscopic evaluation, was the highest concentration used (3 mg/kg/week), resulting in exposure levels around 730 nmol/L, which is several fold higher than the likely clinical exposure. Moreover, there were no noteworthy changes in other hematological parameters, for example platelet counts, in any of the safety studies.
It should be noted that for molecules like Mim8, a bell‐shaped dose‐response curve would be expected both regarding efficacy and safety due to the prozone (or hook) effect. This has been shown in vitro for Mim8 12 and has also been previously described for emicizumab. 16 The data from the presented toxicity studies are in agreement with this, as thrombosis‐related findings were seen in a few animals at 6, 12, and 20 mg/kg/week SC, whereas no thrombi were seen at 60 mg/kg/week SC or 20 mg/kg/week IV, both doses leading to considerably higher exposures, and at dose levels of 3 mg/kg/week SC or lower. A prolongation of PT in animals was observed in the 4‐week and 13‐week studies, employing dose levels at or above 6 mg/kg/week SC or IV. This may be an assay artifact caused by slight inhibition of FX in the assay due to the high concentrations of Mim8. Similar observations were seen with emicizumab, for which authors speculated PT prolongation was due to steric interference of emicizumab‐FX/FXa binding in the assay. 17 Furthermore, in global assays performed on samples taken from monkeys dosed with Mim8, a clear procoagulant effect of Mim8 was observed on thrombin generation, indicating that the prolongation of PT is either an assay effect (only relevant in the in vitro conditions of the PT assays) or that this effect is outweighed by the procoagulant effect of Mim8.
In the 26‐week study, in which no pathological findings were made, the FX concentration was up to 2.4‐fold higher at the highest Mim8 dose level (3 mg/kg/week SC, with a maximum Mim8 plasma exposure of 730 nmol/L) compared to the control level. Mim8 binds FX in the second epidermal growth factor and serine protease domain not interfering with the active site, and the increase in FX levels could be mediated by prolongation of FX half‐life through (1) protection of FX from scavenger receptors/molecules by steric hindrance and/or (2) rescue of internalized FX through FcRn‐mediated recycling of Mim8. These effects are potentially exaggerated in this study, because Mim8 binds cynomolgus monkey FX with approximately 2‐fold higher affinity than human FX. The Mim8 affinity for FIX is considerably lower than for FX, 12 and congruently, the effect of Mim8 on FIX levels is smaller compared to the effect on FX levels. Thus, increases in FIX concentrations were only observed at the highest dose levels in the 4‐ and 13‐week studies, with maximum Mim8 plasma concentrations of 4460 and 2320 nmol/L, respectively. Similarly, increases in FIX and FX have been reported in toxicity studies with emicizumab. 18 The changes in FIX and FX antigen levels were observed at Mim8 concentrations several fold higher than the expected clinical exposures (the currently ongoing phase I/II clinical trial is investigating Mim8 plasma concentrations up to 180 nmol/L). At exposure levels closer to the expected clinical range in the 26‐week study, the FIX concentration was comparable to the control level, and a less than 30% increase in FX concentration was observed.
Previous studies have found that the half‐life of Mim8 in cynomolgus monkey is approximately 2 weeks, and steady‐state exposure is achieved after ~5 half‐lives (i.e., 10 weeks). 12 APTT, thrombin generation, and concentrations of FIX and FX remained stable from week 13 onwards in the 26‐week study, indicating a steady‐state concentration of Mim8 was achieved after 13 weeks of dosing.
During the 26‐week study, ADAs developed in approximately half of the animals, and in 11 of 24 monkeys, these ADAs caused reductions in exposure and neutralization of pharmacodynamic effects with both APTT and thrombin generation reverting toward predose levels. This was anticipated following dosing with a foreign‐species antibody and is not predictive of immunogenicity in humans. An assessment of 27 monoclonal antibodies approved in the EU between 1988 and 2010 showed that the ADA incidence in non‐human primates was high (>45%) for approximately 25% (7/27) of the monoclonal antibodies, of which 6/7 showed negligible (<2%) or tolerable (<15%) immunogenicity clinically, whereas 11/27 monoclonal antibodies had intermediate ADA incidence (6–45%) in non‐human primates, of which all had negligible or tolerable immunogenicity clinically. 19 Furthermore, in nonclinical studies with emicizumab, 30% of animals dosed for 26 weeks developed ADAs. 18 Subsequently, the overall neutralizing ADA incidence across the pivotal phase III emicizumab studies was 2.7%. 20
Local injection site observations in animals following administration of Mim8 in this study were considered related to SC dosing of Mim8 and were most likely a local immunogenic effect related to administration of a species‐foreign protein to immunocompetent animals as the findings primarily occurred in the animals with detected ADAs.
Taken together, pathological findings at high Mim8 doses in the 4‐ and 13‐week studies were considered related to an expected exaggerated pharmacological effect. In accordance, all changes in pharmacodynamic safety biomarkers were considered related to the pharmacological activation of the coagulation system or Mim8’s interaction with coagulation factors. Data show relevant effects on thrombin generation and no safety concerns for the weekly subcutaneous administration of 0.3, 1, or 3 mg/kg/week Mim8 SC to monkeys with normal coagulation systems, which at the highest led to exposure levels that were several fold greater than those expected to be relevant for patients with HA in the clinical setting. These data support further development of Mim8, which is currently being tested in a clinical phase I/II trial.
CONFLICTS OF INTEREST
At the time of this study, all authors were employees at Novo Nordisk. Stine Kjellev is currently employed by Y‐mAbs Therapeutics, Inc., Hørsholm, Denmark. Kevin A. Keane is currently employed at Blueprint Medicines in Cambridge, MA, USA.
AUTHOR CONTRIBUTIONS
B. Lauritzen planned and performed experiments and interpreted data. Stine Kjellev planned experiments and interpreted data. M. Bjelke, O. Björkdahl, E. Bloem, K. Keane, M. Kjalke, M. Rossen, S. L. Lippert, K. N. Weldingh, and M. Skydsgaard performed experiments and interpreted data. All authors critically reviewed and participated in the writing of this manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Novo Nordisk A/S (Måløv, Denmark). Safety study in cynomolgus monkeys was performed and analyzed by Covance (Harrogate, UK or Münster, Germany). The authors thank Kasper Jensen, Charlotte Gandsø Fjordager, and Thomas Nygaard Jensen for expert technical assistance. Writing and editorial support was provided by Ashfield MedComms GmbH (Mannheim, Germany), funded by Novo Nordisk A/S.
Lauritzen B, Bjelke M, Björkdahl O, et al. A novel next‐generation FVIIIa mimetic, Mim8, has a favorable safety profile and displays potent pharmacodynamic effects: Results from safety studies in cynomolgus monkeys. J Thromb Haemost. 2022;20:1312–1324. doi: 10.1111/jth.15682
Manuscript handled by: Joost Meijers
Final decision: Joost Meijers, 18 February 2021
REFERENCES
- 1. Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma‐derived or recombinant factor VIII concentrates: a systematic review. J Thromb Haemost. 2010;8(6):1256‐1265. [DOI] [PubMed] [Google Scholar]
- 3. Fischer K, Lassila R, Peyvandi F, et al. Inhibitor development in haemophilia according to concentrate. Four‐year results from the European HAemophilia Safety Surveillance (EUHASS) project. Thromb Haemost. 2015;113(5):968‐975. [DOI] [PubMed] [Google Scholar]
- 4. Calvez T, Chambost H, d’Oiron R, et al. Analyses of the FranceCoag cohort support differences in immunogenicity among one plasma‐derived and two recombinant factor VIII brands in boys with severe hemophilia A. Haematologica. 2018;103(1):179‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054‐2064. [DOI] [PubMed] [Google Scholar]
- 6. Leissinger C, Gringeri A, Antmen B, et al. Anti‐inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N Engl J Med. 2011;365(18):1684‐1692. [DOI] [PubMed] [Google Scholar]
- 7. Dimichele DM, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recommendations. Haemophilia. 2007;13(Suppl 1):1‐22. [DOI] [PubMed] [Google Scholar]
- 8. HEMLIBRA® (emicizumab): EPAR – Product information ‐ Summary of product characteristics. 2018. Accessed January 21, 2021.
- 9. HEMLIBRA® (emicizumab): US prescribing information (Oct 2018), 1 DNA Way, CA 94080‐4990, Genentech, Inc. 2020. Accessed January 20, 2021.
- 10. Shima M, Hanabusa H, Taki M, et al. Factor VIII‐mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044‐2053. [DOI] [PubMed] [Google Scholar]
- 11. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open‐label, non‐randomised phase 3 study. Lancet Haematol. 2019;6(6):e295‐e305. [DOI] [PubMed] [Google Scholar]
- 12. Østergaard H, Lund J, Greisen PJ, et al. A factor VIIIa‐mimetic bispecific antibody, Mim8, ameliorates bleeding upon severe vascular challenge in hemophilia A mice. Blood. 2021;138(14):1258‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ley C, Kjalke M, Holm TL, et al. Improved effect of Mim8, a next‐generation FVIII mimetic, translates from human in vitro to humanized mouse and cynomolgus models. Haemophilia. 2020;26(S2):P028. [Google Scholar]
- 14. Hartmann R, Feenstra T, Valentino L, Dockal M, Scheiflinger F. In vitro studies show synergistic effects of a procoagulant bispecific antibody and bypassing agents. J Thromb Haemost. 2018;16(8):1580‐1591. [DOI] [PubMed] [Google Scholar]
- 15. Crissman JW, Goodman DG, Hildebrandt PK, et al. Best practices guideline: toxicologic histopathology. Toxicol Pathol. 2004;32(1):126‐131. [DOI] [PubMed] [Google Scholar]
- 16. Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa‐mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117(7):1348‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adamkewicz JI, Chen DC, Paz‐Priel I. Effects and interferences of emicizumab, a humanised bispecific antibody mimicking activated factor VIII cofactor function, on coagulation assays. Thromb Haemost. 2019;119(7):1084‐1093. [DOI] [PubMed] [Google Scholar]
- 18. Hemlibra®Assessment Report. https://www.ema.europa.eu/en/documents/assessment‐report/hemlibra‐epar‐public‐assessment‐report_en.pdf Accessed 2021, EMA/88475/2018.
- 19. van Meer PJ, Kooijman M, Brinks V, et al. Immunogenicity of mAbs in non‐human primates during nonclinical safety assessment. Mabs. 2013;5(5):810‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmitt C, Emrich T, Chebon S, et al. Low immunogenicity of emicizumab in persons with haemophilia A. Haemophilia. 2021;27(6):984‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material