

Abstract
Ether‐based electrolytes offer promising features such as high lithium‐ion solvation power and stable interface, yet their limited oxidation stability impedes application in high‐voltage Li‐metal batteries (LMBs). Whereas the fluorination of the ether backbone improves the oxidative stability, the resulting solvents lose their Li+‐solvation ability. Therefore, the rational molecular design of solvents is essential to combine high redox stability with good ionic conductivity. Here, we report the synthesis of a new high‐voltage fluorinated ether solvent through integrated ring‐chain molecular design, which can be used as a single solvent while retaining high‐voltage stability. The controlled Li+‐solvation environment even at low‐salt‐concentration (1 M or 2 M) enables a uniform and compact Li anode and an outstanding cycling stability in the Li|NCM811 full cell (20 μm Li foil, N/P ratio of 4). These results show the impact of molecular design of electrolytes towards the utilization of LMBs.
Keywords: Electrolyte Chemistry, Electrolyte Engineering, Lithium-Metal Battery, Solvation Structure, Solvent Design
An integrated fluorinated ring‐chain molecular approach was developed through structural modification of a low‐cost, commercial ether for a new solvent 2‐ethoxy‐4‐(trifluoromethyl)‐1,3‐dioxolane (cFTOF), which served as a single ether solvent for lithium‐metal batteries combining high‐voltage stability up to 6 V, and regulated Li+ solvation structure, increasing Li+ ion‐pairing with FSI anions in a low‐salt‐concentration electrolyte system.
Introduction
Conventional lithium‐ion batteries (LIBs) [1] bearing graphite anodes (372 mAh g−1), hardly satisfy the ever‐increasing demand with respect to the energy density for electric vehicles and consumer electronics. [2] Metallic lithium (Li) has been considered as the most promising anode material for the post‐LIBs due to its ultra‐high specific capacity (3860 mAh g−1) and the lowest reduction potential (−3.04 V vs. standard hydrogen electrode). [3] However, there are still significant challenges to be overcome for their practical application. These challenges include Li dendrite growth arising from uneven Li plating and high reactivity of metallic Li with the electrolyte that results in low Coulombic efficiency (CE) and poor cyclability. Li dendrite growth also destabilizes the solid electrolyte interphase (SEI) layer, which further aggravates dendrite growth. [4]
Among various strategies, [5] electrolyte engineering is one of the most effective approaches, as it can impart critical properties to the electrolyte such as controlled reactivity with Li metal, compatibility with high‐voltage cathodes, and adoptability in existing manufacturing schemes. [6] Commercial carbonate electrolytes are widely applied in high‐voltage LIBs. However, they usually form a Li2O/Li2CO3‐rich inhomogeneous SEI layer with high fragility at the anode interface, which induces the growth of mossy lithium with a porous and mechanically weak morphology. [7] Significant efforts have been devoted to improve the CE in commercial carbonate electrolytes for Li‐metal protection. [8] However, even with some additives such as fluoroethylene carbonate (FEC) [9] and vinylene carbonate (VC) that promote the formation of LiF or modify the SEI layer by reductive polymerization, [10] the CE values were still low for practical applications. High‐concentration electrolytes (HCEs) such as 7 M lithium bis(fluorosulfonyl)imide (LiFSI) in FEC [11] and 3.27 mol kg−1 LiN(SO2C2F5)2/PC [12] were also used to decrease the number of free solvent molecules that induce the inorganic‐rich SEI layer. These HCE approaches preferentially decompose Li salt to form a more robust SEI layer. Nevertheless, super concentrated carbonate electrolytes sharply increase the cell cost to the level far beyond the market price range and also cause insufficient wetting on the separator.
Ether electrolytes show better compatibility with the Li‐metal electrode compared to carbonates owing to their intrinsic reductive stability. [13] However, the oxidative stability of conventional ether electrolytes is poor (<4.0 V), limiting their practical value in high‐voltage LMBs. Some strategies based on localized high concentration electrolytes (LHCEs) have been reported to overcome these challenges [14] by introducing a non‐solvating diluent, such as 1,1,2,2‐tetrafluoroethyl‐2,2,3,3‐tetrafluoropropyl ether (TTE) [15] and tris(2,2,2‐trifluoroethyl)orthoformate (TFEO), [16] bis(2,2,2‐tri‐fluoroethyl) ether (BTFE) [17] and 1,2‐difluorobenzene (1,2‐dfBen). [7b] As the diluent molecules are compatible with Li metal, yet can hardly solvate Li+ ions by themselves, it is necessary to combine them with a small amount of strongly solvating solvent for the dissolution of salts, which still causes the low CE in the initial cycles due to the parasitic side reactions. Whereas electrolyte engineering has shown great improvements in LMBs in various aspects, it is desirable to develop ether‐based electrolyte chemistry that combines Li+ solvation ability with high oxidative stability at low salt concentrations. [18] There are, however, rather limited number of examples in the literature. For example, Bao and co‐workers [18] elegantly reported a new linear chain, ether‐based solvent by extending the distance between −O− atoms in an ether and introducing −CF2− groups in between. The resulting solvent solvates Li‐ions through simultaneous coordination of −O− and −F atoms while retaining high oxidative stability. Accordingly, further research in this area is critical in order to establish design principles for new solvents with the aforementioned properties. Herein, we present an integrated fluorinated ring‐chain molecular approach for a solvent design. The solvent, namely 2‐ethoxy‐4‐(trifluoromethyl)‐1,3‐dioxolane (cFTOF), synthesized in a facile, one‐pot reaction from a commercial ether (triethyl orthoformate; TOF), served as a single solvent for the state‐of‐the‐art high‐voltage LMBs. The asymmetric addition of −CF3 moiety to the ring component allowed us to regulate the solvation of Li+ ions by decreasing the electron density on the −O− atoms and increasing their binding with FSI anions, thus forming an inorganic LiF‐rich SEI and a uniform Li plating morphology. cFTOF showed a superior cycling performance when tested as a single solvent in Li|LiNi0.8Co0.1Mn0.1O2 (NCM811) full cells.
Results and Discussion
We identified TOF as the target commercial solvent due to its open‐chain structure, high Li+ solvation power and facile derivatization (Figure 1a). We reasoned that covalent attachment of fluorinated groups could increase the oxidative stability of TOF. As opposed to the direct attachment of fluorinated groups to −O− atoms, which would substantially decrease their solvation power, we opted for a cyclic structure bearing a −CF3 group, where an additional −CH2− moiety is present in between the −CF3 group and the −O− atom. This ring‐chain design enabled us to achieve high‐voltage stability, while retaining the solvation power of −O− atoms. Notably, the asymmetric addition of the −CF3 group also created a binding site for the Li+ ion located between the −O− atom of the ring and that of the chain component. The cFTOF was synthesized (Figure 1b) by reacting 1,1,1‐trifluoro‐2,3‐propanediol [19] (Figure S1) with TOF at 100 °C for 3 h using p‐toluenesulfonic acid as a catalyst in 56 % yield (Figure S2). The formation of cFTOF was verified (Figures S2–S4) by nuclear magnetic resonance (NMR) spectroscopy analysis.
Figure 1.
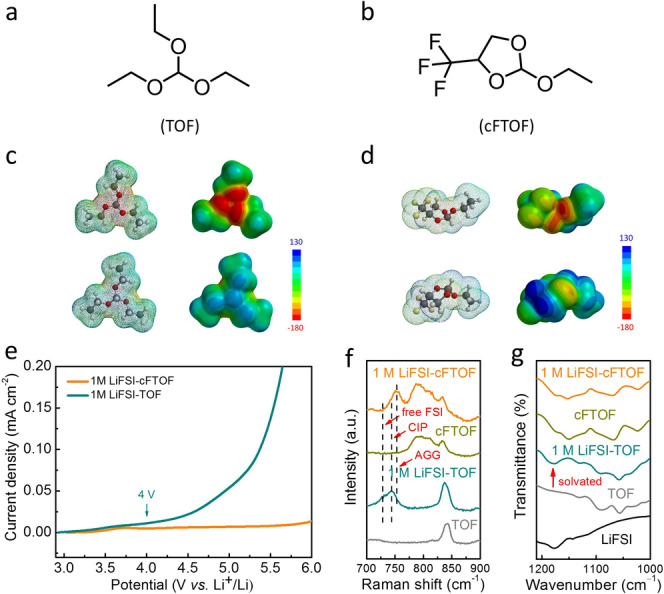
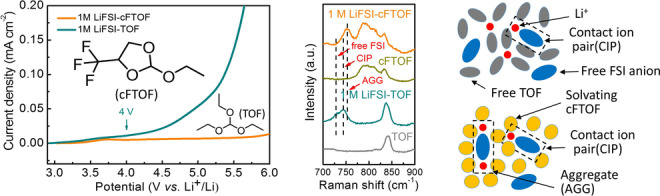
a, b) Molecular structures of solvents, TOF and cFTOF. c, d) Electrostatic potential map (ESP) comparisons of TOF and cFTOF with different views. The ESP scale bar is the electrostatic potential range (kJ mol−1). e) Oxidation stability of two electrolytes in Li|Al half cells tested by LSV. f) Raman spectra of 1 M LiFSI‐cFTOF and 1 M LiFSI‐TOF electrolytes. g) FT‐IR spectra of 1 M LiFSI‐cFTOF and 1 M LiFSI‐TOF electrolytes.
The cFTOF is a chiral molecule (Figures 1b and S2–S4) isolated as a racemic mixture, which maintains the ability to solvate Li salt as a transparent liquid solvent. We performed electrostatic potential (ESP) calculations to determine the charge densities of the atoms in the solvent and, particularly, the −O− atoms participating in the Li+ solvation. [18] Similar to the structurally symmetric solvents, which show same electron density for all −O− atoms, we observed high electron density for all the three oxygen atoms in the TOF (Figure 1c). By contrast, in the case of cFTOF solvent, due to the asymmetric incorporation of −CF3 group, the high electron density concentrates on the two oxygen atoms for Li+ coordination: one oxygen located on the ring (close to −CF3) and the other one on the chain (Figure 1d). In comparison to TOF, the decreased electron density of −O− atom located on the ring component leads to a decreased solvation power for the cFTOF solvent.
The 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes were obtained by dissolving 1 M LiFSI salt in the corresponding solvents. The comparative analysis of properties of 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF is presented in Table S1. Remarkably, an improved Li‐ion transference number (LTN) (0.78) was achieved for 1 M LiFSI‐cFTOF, compared to that of 1 M LiFSI‐TOF (0.64) (Figure S5), which are both significantly higher compared to 1,2‐dimethoxyethane (DME), 0.39. [18]
Linear sweep voltammetry (LSV) measurements were performed on Li|Al cells to probe the electrochemical stability windows of the 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes. 1 M LiFSI‐TOF electrolyte showed a slight oxidation starting at 4 V due to the low anodic stability, while the oxidative stability of the 1 M LiFSI‐cFTOF electrolyte extended above 6 V (Figure 1e), thus making it compatible with commercial high‐voltage cathode materials. By benchmarking a classic reference, [20] LSV was also conducted in a Li|LiMn2O4 (LMO) cell configuration to study the electrochemical stability windows of the electrolytes in practical batteries (Figure S6), where the 1 M LiFSI‐cFTOF electrolyte exhibited an oxidation stability up to 4.5 V. The highest occupied molecular orbital (HOMO) energy level was also utilized to assess the oxidation vulnerability at positive potentials to avoid overestimated electrolyte stability. [21] In agreement with the LSV result, cFTOF exhibited a lower HOMO energy level (−7.95 eV) compared to that of TOF (−7.5 eV) (Figure S7) due to the introduction of an electron‐withdrawing −CF3 group [22] to a ring‐chain molecular structure.
In order to study the solvation structure of the electrolytes, Raman spectroscopy analysis was conducted for the two electrolytes. In 1 M LiFSI‐TOF electrolyte, the majority of FSI− exists as a free anion (730 cm−1) and as a contact‐ion pair (CIP, an FSI anion coordinating to one Li+) (743 cm−1) due to the strong Li+ coordinating ability of TOF solvent. In the case of 1 M LiFSI‐cFTOF electrolyte, however, there were no free FSI anions present and almost all FSI anions remain in an aggregate (AGG, a FSI− anion coordinating to two or more Li+) (753 cm−1) state, suggesting the majority of cFTOF exists in a free state and more FSI− anions move to the anode along with Li+ to induce anion‐derived SEI composition (Figure 1f). [23] We also tested the effect of higher salt concentration, that is 2 M LiFSI (2 M LiFSI‐cFTOF), and the Raman band corresponding to AGG was remarkably enhanced through the intensive association between Li+ and FSI anions (Figure S8). The unusual solvation structure with predominantly AGG state even at low‐salt‐concentration is ascribed to the fluorinated ring‐chain molecular design of the solvent. These findings were also further verified by Fourier transform infrared spectroscopy (FTIR) analysis, where a solvated peak of the TOF solvent coordinating to Li+ appeared in the 1 M LiFSI‐TOF electrolyte, whereas it was significantly suppressed in the 1 M LiFSI‐cFTOF electrolyte (Figures 1g and S9) due to the weakened solvation power of cFTOF. [18]
Scanning electron microscopy (SEM) analysis was conducted to examine the morphologies of the deposited Li in different electrolytes. Firstly, we recorded the morphologies of Li deposits with a capacity of 4 mAh cm−2 at 1 mA cm−2 in coin‐type Li|Cu half cells. The deposited Li in 1 M LiFSI‐TOF electrolyte showed smaller and loosely packed Li grains, whereas more flat and larger Li grains were clearly observed for 1 M LiFSI‐cFTOF electrolyte (Figures 2a, b and S10), leading to a reduced surface area and decreased reactivity of Li metal towards the electrolyte. After 50 cycles, the plated Li in the case of 1 M LiFSI‐TOF electrolyte showed accumulated SEI on the Li anode surface, while the large Li grains were still observed for 1 M LiFSI‐cFTOF electrolyte (Figures 2c, d). The LiFSI‐TOF electrode showed a highly porous dark‐colored Li layer with an average thickness of 22 μm (Figure 2e), whereas the thickness of deposited Li with 1 M LiFSI‐cFTOF electrolyte was only 17 μm and the clear Li metal surface was observed (Figure 2f). This distinct morphology remarkably points to a homogeneous electrochemical stripping and plating process with a minimal dead Li through the fluorinated ring‐chain electrolyte structure.
Figure 2.

SEM images of 4 mAh cm−2 Li plating morphology at 1 mA cm−2 in a) 1 M LiFSI‐TOF and b) 1 M LiFSI‐cFTOF electrolytes after the first cycle. The morphology of Li surface after 50 cycles at 1 mA cm−2 with a cut‐off capacity of 1 mAh cm−2 in c) 1 M LiFSI‐TOF and d) 1 M LiFSI‐cFTOF electrolytes. Cross‐sectional SEM images of Li on Cu substrate and the optical images of the Li‐anode surface (shown in insets) in e) 1 M LiFSI‐TOF and f) 1 M LiFSI‐cFTOF electrolytes after 50 cycles at 1 mA cm−2 with 1 mAh cm−2.
Coulombic efficiencies (CEs) of Li|Cu cells with both electrolytes were measured to evaluate the reversibility of Li‐metal plating and stripping. As shown in Figure 3a, when cycled at a current density of 0.5 mA cm−2 with a capacity of 1 mAh cm−2, 1 M LiFSI‐TOF electrolyte presented an average CE of only 95.6 % for the initial 100 cycles and an abrupt decay after 135 cycles, mainly originating from the rough Li deposition morphology and the recurring breakdown/repair of the loose SEI layer. In stark contrast, the cell with 1 M LiFSI‐cFTOF electrolyte demonstrated a superior and stable average CE of 97.8 % for the first 100 cycles and 99.0 % for the following 200 cycles. When measured at an increased Li deposition current density of 1.0 mA cm−2 with the capacity of 1 mAh cm−2, CE of the cell with 1 M LiFSI‐TOF electrolyte was only 96.1 % for the initial 100 cycles and decayed rapidly to 74.5 % after 155 cycles, while an average efficiency of 97.9 % was obtained for 200 cycles in the case of 1 M LiFSI‐cFTOF electrolyte (Figure 3b). The corresponding voltage vs. capacity profiles are provided in Figure S11. At a current density of 1 mA cm−2 and a high areal capacity of 3 mAh cm−2, a stable and high CE of 98.7 % was achieved for 1 M LiFSI‐cFTOF electrolyte over 80 cycles compared with 97.2 % for 1 M LiFSI‐TOF electrolyte (Figure S12). Symmetric Li|Li cells were also assembled to compare the plating and stripping cycling performance of both electrolytes. The voltage polarization of the cell with 1 M LiFSI‐TOF electrolyte began to increase sharply after around only 8000 minutes at a current density of 1 mA cm−2 with an areal capacity of 1 mAh cm−2, while the voltage hysteresis of the cell with 1 M LiFSI‐cFTOF electrolyte remained stable over 15000 minutes with a slightly higher overpotential, which is attributed to the moderate ionic conductivity of cFTOF, 0.7 mS cm−1 (Figure S13). [18]
Figure 3.
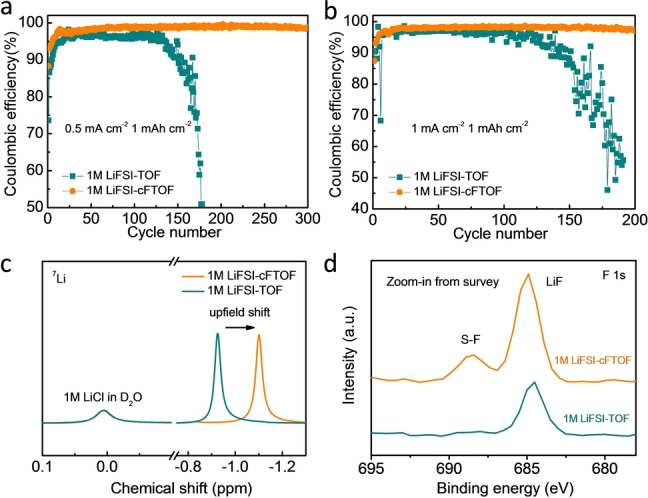
Comparison of CE of Li plating/stripping in 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes at a) 0.5 mA cm−2 current density of 1 mAh cm−2 Li and b) 1 mA cm−2 current density of 1 mAh cm−2 Li. c) 7Li NMR spectra of 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes. d) XPS zoom‐in spectra of F 1s in 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes after cycling.
The anode‐compatibility of electrolytes was examined by cyclic voltammetry (CV) in a Li|Cu half‐cell configuration. As shown in Figure S14, 1 M LiFSI‐cFTOF electrolyte showed high reversibility in Li plating/stripping in comparison to 1 M LiFSI‐TOF electrolyte. The electrochemical stability of both electrolytes was further studied by electrochemical impedance spectroscopy (EIS) after 1 cycle and 10 cycles at 1 mA cm−2 with 1 mAh cm−2 in Li|Cu half cells (Figure S15). After 1 cycle, the higher bulk resistance (R b) of 61 Ω for 1 M LiFSI‐cFTOF electrolyte than 1 M LiFSI‐TOF electrolyte of 7 Ω is attributed to its lower ionic conductivity. [18] Moreover, after 10 cycles, the SEI resistance (R SEI) for 1 M LiFSI‐cFTOF electrolyte decreased from 60 to 55 Ω while that of 1 M LiFSI‐TOF electrolyte increased from 45 to 69 Ω, indicating the formation of a stable SEI layer in the case of 1 M LiFSI‐cFTOF.
In order to understand the solvation structure of electrolytes, 7Li‐ and 19F NMR analyses were carried out (Figures 3c and S16) using 1 M LiCl in D2O as an internal standard in a coaxial NMR tube. In 1 M LiFSI‐cFTOF electrolyte, we observed an upfield shift in both 7Li (−0.92 to −1.1 ppm) and 19F spectra (52.5 to 52.3 ppm) compared with 1 M LiFSI‐TOF, a clear indication for the increased Li+ ion‐pairing with FSI anions [24] and the decreased solvation power of cFTOF.
X‐ray photoelectron spectroscopy (XPS) was conducted to further characterize the Li metal surface and corroborate on the proposed solvation structure. In the F 1s zoom‐in spectra of 1 M LiFSI‐cFTOF electrolyte, an obvious peak assigned to FSI anion at 688.6 eV [25] in S‐F orbital branch was observed after 20 cycles at 1 mA cm−2 with 1 mAh cm−2 in Li|Cu half cells. Moreover, the enhanced intensity of LiF at 685 eV [26] was largely associated with the reduction of FSI anion (Figures 3d and S18), while the other elements did not show any obvious difference between 1 M LiFSI‐TOF and 1 M LiFSI‐cFTOF electrolytes (Figure S17). These results collectively suggest that the solvation structure of LiFSI‐cFTOF is under AGG state even at low salt concentrations, which results in LiF‐rich SEI formation and the superior cycling performance.
Based on the improved compatibility with Li metal and high‐voltage stability of cFTOF‐based electrolytes, we further compared cycling performance of Li|NCM811 full cells with limited Li metal reservoir (20 μm Li foil, N/P ratio of 4) and different electrolytes at 0.5 C (1 C=200 mA g−1) shown in Figure 4. Carbonate‐based electrolyte (1 M LiPF6‐EC‐EMC) was taken as the benchmark, which exhibited severe capacity fading (only 77 % retention after 50 cycles) and low CE (below 90 %) during cycling due to the limited amount of Li anode (20 μm) in Li|NCM811 full cells. Moreover, the incompatibility of 1 M LiPF6‐EC‐EMC with Li metal was also proven by the low CE (below 85 %) in the Li|Cu half cell (Figure S19). Li|NCM811 full cells with TOF‐based electrolytes (both 1 M and 2 M) stabilized for only 14 cycles followed by a serious capacity fading, which was attributed to the low oxidation stability and continuous electrolyte decomposition (Figures 4a and c). In stark contrast, the full cell with 1 M LiFSI‐cFTOF electrolyte exhibited a good capacity retention of 94 % after 100 cycles. In order to further optimize the performance, the salt concentration was increased to 2 M. Li|NCM811 full cell with 2 M LiFSI‐cFTOF electrolyte demonstrated excellent capacity retention of 100 % after 112 cycles at 0.5 C (Figures 4b, c) with good reproducibility (Figure S20). Electrochemical performance comparison of electrolytes is summarized in Table S2, the full cell performance of 2 M LiFSI‐cFTOF electrolyte is found to be highly competitive based on a single‐ether solvent and a single‐salt formulation paired with NCM cathode.
Figure 4.
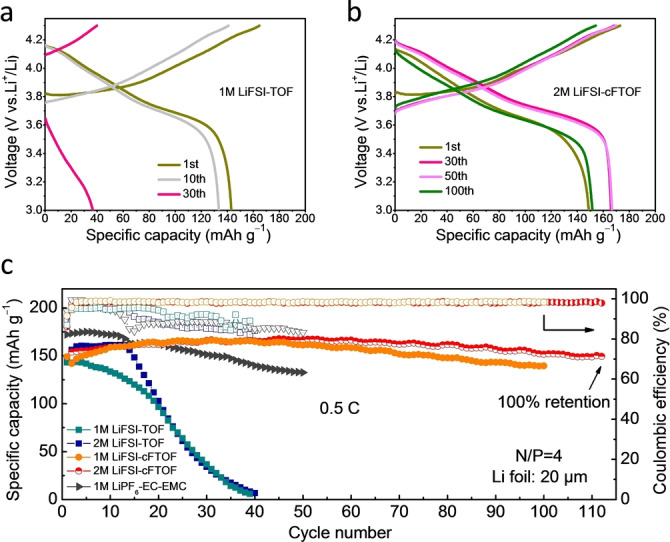
a, b) Galvanostatic charge‐discharge profiles of Li|NCM811 full cells in a) 1 M LiFSI‐TOF and b) 2 M LiFSI‐cFTOF electrolytes. c) Cycling performance and CE at 0.5 C of Li|NCM811 full cells with different electrolytes (1 C=200 mA g−1).
Considering the obvious temperature influence on electrochemical performance of Li metal anodes and electrolytes, [27] the full cell with 2 M LiFSI‐cFTOF electrolyte was evaluated at different temperatures (Figure S21). Al corrosion by electrolytes was also investigated (Figure S22). Compared with the severe corrosion and crack formation on Al foil in the case of 1 M LiFSI‐TOF electrolyte, cFTOF‐based electrolytes exhibited excellent Al anti‐corrosion properties at 5 V for 48 hours. The cathode electrolyte interphase (CEI) was characterized by XPS analysis to evaluate the compatibility between the electrolyte and high voltage cathode (Figures S23–S26). The formation of a LiF‐rich CEI layer was observed in the cFTOF‐based electrolytes, and this modified CEI layer plays a pivotal role in extending the cycle life. Cathode morphology changes in Li|NCM811 full cells with different electrolytes after 20 cycles at 0.5 C were also compared (Figure S27), where we observed no obvious difference between electrolytes. CEI of cathode with different electrolytes was also analyzed by transmission electron microscopy (TEM) after 100 cycles (Figure S28). cFTOF‐based electrolytes exhibited a thinner and more uniform CEI layer compared to 1 M LiFSI‐TOF electrolyte. The systematic analysis of cFTOF‐based electrolytes revealed that cFTOF can, on the anode side, promote anion decomposition to form LiF‐rich SEI layer through strong Li+‐FSI binding leading to stable Li plating, while facilitating the formation of stable, LiF‐rich and thin CEI layer on the cathode surface to realize stable cycling.
Conclusion
We demonstrated a unique integrated ring‐chain approach to tune the solvation power and solvent structure through molecular engineering of a low‐cost, commercial ether to a solvent with improved electrochemical performance for high‐voltage LMBs. Notably, this molecule can serve as a single ether‐based solvent for LMBs combining high anodic stability and regulated Li+ solvation power. The desirable solvation structure in the electrolyte promoted the formation of Li+ ion‐pairing with FSI anions in a low‐salt‐concentration system and led to a high transference number, enabling a uniform and compact, LiF‐rich SEI layer. This new class of electrolyte greatly expands the electrolyte possibilities through modification of electrolyte chemistry and introduces design principles for the development of new electrolytes.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
A.C. acknowledges the support from the Swiss National Science Foundation (SNF) for funding of this research (200021‐188572). J.W.C. acknowledges the support by the National Research Foundation of Korea (NRF) grants (NRF‐ 2021R1A2B5B03001956), the Technology Innovation Program (20012341) funded by the Ministry of Trade, Industry & Energy (MOTIE) of Korea and generous support from the Institute of Engineering Research (IOER) and Inter‐university Semiconductor Research Center (ISRC), and Research Institute of Advanced Materials (RIAM) at Seoul National University. We would like to thank Dr. Dominika Baster at PSI for XPS test and Timur Ashirov at Unifr for TEM test. Open access funding provided by Universite de Fribourg.
T. Zhou, Y. Zhao, M. El Kazzi, J. W. Choi, A. Coskun, Angew. Chem. Int. Ed. 2022, 61, e202115884; Angew. Chem. 2022, 134, e202115884.
Contributor Information
Prof. Jang Wook Choi, Email: jangwookchoi@snu.ac.kr.
Prof. Ali Coskun, Email: ali.coskun@unifr.ch.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Etacheri V., Marom R., Elazari R., Salitra G., Aurbach D., Energy Environ. Sci. 2011, 4, 3243. [Google Scholar]
- 2. Choi J. W., Aurbach D., Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar]
- 3. Liu B., Zhang J.-G., Xu W., Joule 2018, 2, 833–845. [Google Scholar]
- 4.
- 4a. Goodenough J. B., Kim Y., Chem. Mater. 2010, 22, 587–603; [Google Scholar]
- 4b. Wood K. N., Noked M., Dasgupta N. P., ACS Energy Lett. 2017, 2, 664–672; [Google Scholar]
- 4c. Fang C., Wang X., Meng Y. S., Trends Chem. 2019, 1, 152–158. [Google Scholar]
- 5. Tikekar M. D., Choudhury S., Tu Z., Archer L. A., Nat. Energy 2016, 1, 16114. [Google Scholar]
- 6. Zhang J. G., Xu W., Xiao J., Cao X., Liu J., Chem. Rev. 2020, 120, 13312–13348. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Leung K., Soto F., Hankins K., Balbuena P. B., Harrison K. L., J. Phys. Chem. C 2016, 120, 6302–6313; [Google Scholar]
- 7b. Yoo D. J., Yang S., Kim K. J., Choi J. W., Angew. Chem. Int. Ed. 2020, 59, 14869–14876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 14979–14986. [Google Scholar]
- 8. Xu K., Chem. Rev. 2004, 104, 4303–4417. [DOI] [PubMed] [Google Scholar]
- 9. Schroder K., Alvarado J., Yersak T. A., Li J., Dudney N., Webb L. J., Meng Y. S., Stevenson K. J., Chem. Mater. 2015, 27, 5531–5542. [Google Scholar]
- 10. Ota H., Shima K., Ue M., Yamaki J.-i., Electrochim. Acta 2004, 49, 565–572. [Google Scholar]
- 11. Suo L., Xue W., Gobet M., Greenbaum S. G., Wang C., Chen Y., Yang W., Li Y., Li J., Proc. Natl. Acad. Sci. USA 2018, 115, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeong S.-K., Seo H.-Y., Kim D.-H., Han H.-K., Kim J.-G., Lee Y. B., Iriyama Y., Abe T., Ogumi Z., Electrochem. Commun. 2008, 10, 635–638. [Google Scholar]
- 13. Jiao S., Ren X., Cao R., Engelhard M. H., Liu Y., Hu D., Mei D., Zheng J., Zhao W., Li Q., Nat. Energy 2018, 3, 739–746. [Google Scholar]
- 14. Wang J., Yamada Y., Sodeyama K., Chiang C. H., Tateyama Y., Yamada A., Nat. Commun. 2016, 7, 12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren X., Zou L., Cao X., Engelhard M. H., Liu W., Burton S. D., Lee H., Niu C., Matthews B. E., Zhu Z., Wang C., Arey B. W., Xiao J., Liu J., Zhang J.-G., Xu W., Joule 2019, 3, 1662–1676. [Google Scholar]
- 16. Cao X., Ren X., Zou L., Engelhard M. H., Huang W., Wang H., Matthews B. E., Lee H., Niu C., Arey B. W., Cui Y., Wang C., Xiao J., Liu J., Xu W., Zhang J.-G., Nat. Energy 2019, 4, 796–805. [Google Scholar]
- 17. Chen S., Zheng J., Mei D., Han K. S., Engelhard M. H., Zhao W., Xu W., Liu J., Zhang J. G., Adv. Mater. 2018, 30, 1706102. [DOI] [PubMed] [Google Scholar]
- 18. Yu Z., Wang H., Kong X., Huang W., Tsao Y., Mackanic D. G., Wang K., Wang X., Huang W., Choudhury S., Zheng Y., Amanchukwu C. V., Hung S. T., Ma Y., Lomeli E. G., Qin J., Cui Y., Bao Z., Nat. Energy 2020, 5, 526–533. [Google Scholar]
- 19.M. Y. Koh, Akiyoshi; Tanaka, Michiru; Saita, Tomohide, JP2008230970A, Japan, 2008.
- 20.
- 20a. Kasnatscheew J., Streipert B., Röser S., Wagner R., Laskovic I. C., Winter M., Phys. Chem. Chem. Phys. 2017, 19, 16078–16086; [DOI] [PubMed] [Google Scholar]
- 20b. Hilbig P., Ibing L., Streipert B., Wagner R., Winter M., Cekic-Laskovic I., Curr. Top. Electrochem 2018, 20, 1–13. [Google Scholar]
- 21. Peljo P., Girault H. H., Energy Environ. Sci. 2018, 11, 2306–2309. [Google Scholar]
- 22. von Aspern N., Roschenthaler G. V., Winter M., Cekic-Laskovic I., Angew. Chem. Int. Ed. 2019, 58, 15978–16000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16124–16147. [Google Scholar]
- 23. Xie J.-D., Patra J., Chandra Rath P., Liu W.-J., Su C.-Y., Lee S.-W., Tseng C.-J., Gandomi Y. A., Chang J.-K., J. Power Sources 2020, 450, 227657. [Google Scholar]
- 24. Amanchukwu C. V., Yu Z., Kong X., Qin J., Cui Y., Bao Z., J. Am. Chem. Soc. 2020, 142, 7393–7403. [DOI] [PubMed] [Google Scholar]
- 25. Miao R., Yang J., Xu Z., Wang J., Nuli Y., Sun L., Sci. Rep. 2016, 6, 21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Markevich E., Salitra G., Rosenman A., Talyosef Y., Chesneau F., Aurbach D., J. Mater. Chem. A 2015, 3, 19873–19883. [Google Scholar]
- 27. Yan K., Wang J., Zhao S., Zhou D., Sun B., Cui Y., Wang G., Angew. Chem. Int. Ed. 2019, 58, 11364–11368; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11486–11490. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.