

Abstract
Loss‐of‐function mutations in RNF43 induce activation of Wnt ligand‐dependent Wnt/β‐catenin signaling through stabilization of the Frizzled receptor, which is often found in microsatellite instability (MSI)‐type colorectal cancer (CRC) that develops from sessile serrated adenomas. However, the mechanism underlying how RNF43 mutations promote tumorigenesis remains poorly understood. In this study, we established nine human CRC‐derived organoids and found that three organoid lines carried RNF43 frameshift mutations associated with MSI‐high and BRAF V600E mutations, suggesting that these CRCs developed through the serrated pathway. RNF43 frameshift mutant organoids required both Wnt ligands and R‐spondin for proliferation, indicating that suppression of ZNRF3 and retained RNF43 function by R‐spondin are required to achieve an indispensable level of Wnt activation for tumorigenesis. However, active β‐catenin levels in RNF43‐mutant organoids were lower than those in APC two‐hit mutant CRC, suggesting a lower threshold for Wnt activation in CRC that developed through the serrated pathway. Interestingly, transplantation of RNF43‐mutant organoids with intestinal myofibroblasts accelerated the β‐catenin nuclear accumulation and proliferation of xenograft tumors, indicating a key role of stromal cells in the promotion of the malignant phenotype of RNF43‐mutant CRC cells. Sequencing of subcloned organoid cell‐expressed transcripts revealed that two organoid lines carried monoallelic RNF43 cis‐mutations, with two RNF43 frameshift mutations introduced in the same allele and the wild‐type RNF43 allele remaining, while the other organoid line carried two‐hit biallelic RNF43 trans‐mutations. These results suggest that heterozygous RNF43 frameshift mutations contribute to CRC development via the serrated pathway; however, a second‐hit RNF43 mutation may be advantageous in tumorigenesis compared with a single‐hit mutation through further activation of Wnt signaling. Finally, treatment with the PORCN inhibitor significantly suppressed RNF43‐mutant cell‐derived PDX tumor development. These results suggest a novel mechanism underlying RNF43 mutation‐associated CRC development and the therapeutic potential of Wnt ligand inhibition against RNF43‐mutant CRC. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: RNF43, serrated pathway, colorectal cancer, organoid, Wnt ligand, PORCN inhibitor
Introduction
The Wnt signaling pathway plays a role in the maintenance of the stem cells of adult tissues, which is regulated by the destruction complex, in which GSK3 phosphorylates β‐catenin, leading to the degradation of β‐catenin by the ubiquitin–proteasome pathway [1]. Wnt signaling is also regulated at the receptor level; namely, E3 ligases RNF43 and ZNRF3 downregulate Wnt signaling by ubiquitination of the Wnt receptor Frizzled (FZD), leading to its turnover. Mouse genetic studies have shown that the simultaneous disruption of Rnf43 and Znrf3 in the intestine results in Wnt ligand‐independent growth and development of intestinal tumors through Wnt signaling activation [2, 3, 4].
Consistent with this, genetic alterations in RNF43 are found in approximately 18% of colorectal cancer (CRC) cases [5]. RNF43 mutations were also identified in cancers in the ovary, stomach, and pancreas [6, 7, 8]. In hereditary serrated polyposis, germline RNF43 mutations were identified [9], and second‐hit inactivation via loss of heterozygosity (LOH) was also found in tumors [10]. In addition, loss of wild‐type RNF43 was also reported in ovarian cancer [6]. These results suggest a role for two‐hit RNF43 mutations in the promotion of tumorigenesis.
Serrated intestinal adenomas are considered precursors of CRC [11]. Notably, RNF43 mutations in sessile adenomas and serrated pathway‐type CRC were associated with the BRAF V600E mutation and CpG island methylator phenotype (CIMP), leading to the methylation of the MLH1 promoter, which further causes microsatellite instability (MSI) without DNA mismatch repair gene mutations [4, 10, 11, 12, 13, 14, 15]. In addition, RNF43 mutations were detected in MSI cancers in the stomach, esophagus, and uterine endometrium [16], suggesting that RNF43 mutations are major drivers of MSI‐type cancer. Furthermore, colitis‐associated colon tumor development was accelerated in Rnf43 −/− mice [17], and in vivo screening using the Sleeping Beauty transposon identified Rnf43 as a candidate driver of intestinal tumorigenesis [18]. These results, taken together, indicate that an increased level of FZD receptors caused by RNF43 dysfunction drives intestinal tumorigenesis via Wnt signaling activation, particularly in MSI‐CRC through the serrated pathway. Wnt is palmitoleated by porcupine O‐acyltransferase (PORCN), which is required for Wnt secretion [19], and a PORCN inhibitor suppressed RNF43‐mutant CRC cell growth [12, 20]. Consistent with this, translational research indicates that PORCN inhibition is an effective therapeutic strategy against Wnt ligand‐dependent cancer [19, 21].
Recent studies have identified a link between RNF43 mutation types and their tumorigenic potential [22, 23, 24]. However, the genetic information for RNF43 mutations is still not sufficient to understand the mechanism of CRC development. Furthermore, it has been shown that 37% of Wnt ligand‐dependent pancreatic cancer cells require exogenous Wnt ligands, while others utilize endogenous ones [25]. Such cell‐type differences have not been well characterized in CRC.
In the present study, we established human CRC‐derived organoids and found RNF43 frameshift mutations in CIMP‐high/MSI‐high CRC. Importantly, the RNF43‐mutant organoids were dependent on endogenous or exogenous Wnt ligands, and PORCN inhibitors significantly suppressed the organoid proliferation and xenograft tumorigenesis. Furthermore, sequencing of subcloned cell‐derived transcripts suggested that heterozygous monoallelic RNF43 mutations drive the development of CRC and that second‐hit mutations may be advantageous for tumorigenesis.
Materials and methods
Establishment of patient tumor‐derived organoids
Human primary CRC samples were obtained from 50 patients who underwent surgical resection at Ishikawa Prefectural Central Hospital, Japan. All specimens were used for the establishment of organoids and histological analyses. All experiments using human samples were approved by the Human Genome/Gene Analysis Research Ethics Committee of Kanazawa University (2016‐086‐433), and written informed consent was obtained from the patients.
Organoid culture experiments
The CRC specimens were incubated with 1% collagenase type I (#17100‐017; Life Technologies, Carlsbad, CA, USA) for 30 min, filtered with a 100‐μm‐pore filter, and embedded in growth factor‐reduced (GFR) Matrigel (#356231; Corning, Corning, NY, USA). The organoid culture medium is described in Supplementary materials and methods. Organoids were cultured with 50% of L‐WRN cell‐conditioned medium (CM) containing Wnt3a, R‐spondin, and Noggin [26]. In the absence of WRN‐CM (WRN 0%), 100 ng/ml Noggin (Peprotech, Cranbury, NJ, USA) was added to the medium. For Wnt‐ and R‐spondin dependency experiments, 30 ng/ml human Wnt3a (#5036; R&D Systems, Minneapolis, MN, USA) and/or 1 μg/ml human R‐spondin 3 (#3500; R&D Systems) were added. To examine the growth rate, luciferase activity was analyzed using a Cell Titer‐Glo 3D Cell Viability Assay (#G9682; Promega, Madison, WI, USA). To inhibit Wnt ligand signaling, organoids were cultured with PORCN inhibitor: 100 nm Wnt‐C59 (C59) (S7037; Selleckchem, Houston, TX, USA) or 10 μm IWP2 (a gift from Dr David Virshup). In this study, we established one normal colon tissue‐derived organoid (NC8).
Immortalized intestinal myofibroblasts (LmcMF) have been described previously [27]. LmcMF cells were cultured in 8 ml of DMEM for 48 h, and their conditioned medium was collected. Organoids were cultured with 66.7% LmcMF‐CM. To label organoid cells, Venus and tdTomato cDNAs were subcloned into a pPB‐CAG‐IP PiggyBac transposon vector (a gift from Dr Hitoshi Niwa) and co‐transfected with transposase expression vector using Lipofectamine (Thermo Fisher Scientific, Rockford, IL, USA). Transfected clones were selected by puromycin screening.
Patient‐derived xenograft (PDX) mice
All animal experiments were performed using the protocol approved by the Committee on Animal Experimentation of Kanazawa University.
Organoid cells were subcutaneously (s.c.) transplanted (1 × 105 cells per site) into immunodeficient SCID Hairless Outbred (SHO) mice (Crlj:SHO‐Prkdc scid Hr hr ; Charles River, Yokohama, Japan), and tumors were analyzed at 4 weeks after transplantation (n = 4 for each organoid line). For inhibitor treatment, mice were treated with the PORCN inhibitor ETC‐159 (S6616; Selleckchem) at 50 mg/kg per day (per oral) for 5–5.5 weeks after transplantation (n = 6–8 sites for each cell line). For co‐transplantation experiments, C45 cells were monocultured (2 × 105 cells per site) or co‐cultured with LmcMF (2 × 105 cells per site for both lines) for 1 day and transplanted s.c. into SHO mice (n = 4 for each condition).
Histology and immunohistochemistry
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4‐μm‐thick sections. The sections were used for H&E or immunohistochemistry. The antibodies used for immunohistochemistry are described in Supplementary materials and methods. Immunostaining signals were visualized using an ImmPACT DAB Substrate Kit (Vector Laboratories, Burlingame, CA, USA).
Immunofluorescence
Cell proliferation was examined by EdU labeling. Organoids were cultured with 10 μm EdU for 1 h and fixed in 4% PBS–formaldehyde and then permeabilized with 0.1% Triton X‐100. Antibodies are described in Supplementary materials and methods. Fluorescence images were obtained using a TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Immunoblotting
Organoids were lysed in TNE buffer with Complete Mini protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), and 10 μg of the protein samples was separated using 10% SDS‐polyacrylamide gel. Antibodies for immunoblotting are described in Supplementary materials and methods. Relative band intensities were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Whole‐exome sequencing
Whole‐exome sequencing was performed using original organoid cells (not subclones). Genomic DNA was extracted using the NucleoSpin Tissue XS (Macherey‐Nagel, Düren, Germany). Paired‐end libraries were prepared using a SureSelect Human All Exon V6 kit (Agilent, Fremont, CA, USA) and sequenced using an Illumina NovaSeq 6000 (outsourced to Takara Bio, Kusatsu, Japan). The generated fastq files were mapped onto human reference genome version GRCh37 (hg19) using the DRAGEN Bio‐IT Platform (Illumina, San Diego, CA, USA). Frameshift mutations, nonsense mutations, and reported oncogenic mutations were examined in APC, CTNNB1, RNF43, ZNRF3, KRAS, BRAF, TGFBR2, ACVR2A, and TP53.
CIMP analyses
Genomic DNA was modified with sodium bisulfite using an EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). Levels of DNA methylation of classic CIMP markers (MINT1, MINT2, MINT31, MLH1, and CDKN2A) and new CIMP markers (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1) were analyzed by bisulfite pyrosequencing as described previously [28, 29].
MSI analyses
Genomic DNA was extracted, and the MSI analysis was performed according to the Revised Bethesda Guidelines for two mononucleotides (BAT25 and BAT26) and two dinucleotide (D2S123 and D5S346) microsatellite markers (System Biotics, Sagamihara, Japan) [30]. If two or more of these markers showed instability, the sample was diagnosed as MSI‐high.
Organoid subcloning and RNF43 genotyping
Organoids were dissociated to single cells by trypsin treatment and single cells were seeded into 96‐well plates at 1 cell per well to obtain subclones. Total RNA was prepared from the subclones, and mRNA was reverse‐transcribed (RT) by SuperScript III (Thermo Fisher Scientific), amplified by polymerase chain reaction (PCR), and subcloned into the plasmid vector with Mighty Cloning Reagent Set (#6027; Takara Bio). PCR primer sequences are provided in Supplementary materials and methods. Subcloned cDNAs were sequenced using an Applied Biosystems 3500 genetic analyzer (Thermo Fisher Scientific).
Genotyping of RNF43 codon 370
Genotypes of RNF43 codon 370 were examined using the TaqMan SNP Genotyping Assay (Applied Biosystems, Waltham, MA, USA). The sequences of TaqMan primers and probes for wild‐type RNF43 codon 370 (VIC) and p.Rpo370fs (FAM) are provided in Supplementary materials and methods. The PCR results were analyzed by an allele discrimination/SNP system in Mx3000P (Stratagene, La Jolla, CA, USA). The custom RNF43 p.Pro370fs cDNAs were synthesized to obtain homozygous mutant controls.
Human gene expression database analyses
Human CRC expression data were downloaded from the Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/about‐nci/organization/ccg/research/structural‐genomics/tcga) [31]. The expression data of CRC carrying APC‐deep deletions (n = 19) or RNF43 frameshift mutations at an N‐ter of codon 659 (n = 12) were extracted, and upstream regulators were examined using Ingenuity Pathway Analysis (IPA) (Ingenuity Systems, Qiagen; www.ingenuity.com). A Z‐score of ≥2 and a P value of <0.05 were considered to indicate activation with statistical significance.
Transposon insertion site analyses
Transposon mutagenesis screening was previously performed in mice carrying sensitizing driver mutations of CRC [18]. Using the screening data, we analyzed the mutation frequency of Rnf43, Znrf3, and Apc in Trp53 R172H mouse intestinal tumors, as described in Supplementary materials and methods.
Statistical analyses
The data were analyzed using two‐sided unpaired t‐tests and are presented as the mean ± SD for in vitro experiments and the mean ± SEM for in vivo analyses. p < 0.05 was considered statistically significant.
Results
Establishment of human CRC‐derived organoids and PDXs
Primary CRC tissues were collected from 50 patients, and nine CRC‐derived organoids were successfully established. The clinicopathological characteristics of the nine patients are indicated in supplementary material, Table S1. Histologically, all CRCs, with the exception of C31, were diagnosed as tubular adenocarcinoma; C31 was diagnosed as mucinous adenocarcinoma. Ki67 immunostaining confirmed the increased proliferation of cancer cells in all tissues (Figure 1A). Nuclear accumulation of β‐catenin was detected in C3, C15, and C46 cancer cells, suggesting transcriptional activation of Wnt/β‐catenin signaling.
Figure 1.
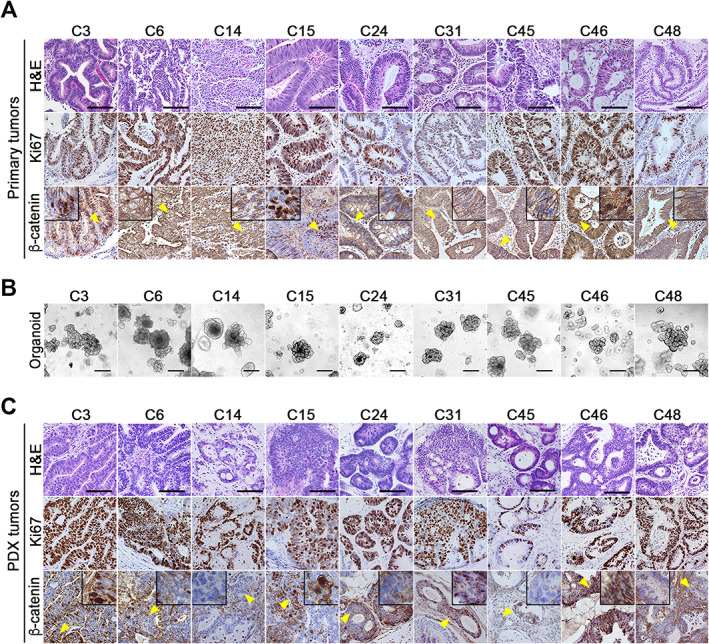
Establishment of human col orectal cancer (CRC)‐derived organoids and patient‐derived xenograft (PDX) models. (A) Representative photographs of histology (H&E) (top) and immunohistochemistry for Ki67 (middle) and β‐catenin (bottom) in the primary CRC tissues of nine patients. Insets are enlarged images of the regions indicated by yellow arrowheads. Bars: 100 μm. (B) Representative photographs of the respective CRC tissue‐derived organoids. Bars: 250 μm. The images are representative of n = 3 independent cultures. (C) Representative photographs of histology (H&E) (top) and immunohistochemistry for Ki67 (middle) and β‐catenin (bottom) of the organoid‐transplanted patient‐derived xenograft (PDX) tumors. Insets are enlarged images of the regions indicated by yellow arrowheads. Bars: 100 μm.
All established organoid lines showed tubular structures (Figure 1B). When these organoids were s.c. transplanted into SHO mice, all organoids developed tumors with continuous proliferation detected by Ki67 immunostaining (Figure 1C). Histologically, the tubular morphology of the PDX tumors resembled the original primary tumors. Furthermore, β‐catenin nuclear accumulation was detected in the C3, C15, and C46 PDX tumors, indicating that the morphology and Wnt signaling activity of the primary tumors remained in the organoids and PDX tumors.
RNF43 mutations in MSI‐type CRC organoids
We performed whole‐exome sequencing of all organoid lines, and mutations in the selected CRC driver genes were analyzed (Figure 2A and supplementary material, Table S2). Possible two‐hit mutations in APC were found in C6, C15, C46, and C48 organoids, while CTNNB1 mutations were not found in any lines. Thus, these CRCs were considered to develop through a conventional pathway (i.e. the adenoma–carcinoma sequence initiated by APC two‐hit mutations). Notably, missense mutations in RNF43 were found in all organoid lines. However, the p.Ile47Val, p.Arg117His, and p.Leu418Met mutations have been shown to maintain the RNF43 function, indicating that they do not contribute to Wnt activation [24]. Notably, C14, C24, and C45 carried frameshift mutations of RNF43, i.e. NM_017763.6:c.673del; p.(Arg225Alafs*194), NM_017763.6:c.1109del; p.(Pro370Hisfs*49), and NM_017763.6:c.1976del; p.(Gly659Valfs*41) (hereafter, p.Arg225fs, p.Pro370fs, and p.Gly659fs, respectively). Similar frameshift mutations of RNF43 were frequently found in serrated adenomas that were often accompanied by CIMP, MSI, and BRAF V600E mutations [10, 11, 12, 13, 14, 15]. Pyrosequencing of CIMP markers showed that all classic CIMP markers (MINT1, MINT2, MINT31, MLH1, and CDKN2A) and new CIMP markers (CACNA1G, IGF2, NEUROG1, RUNX3) except for SOCS1 were methylated >20% in C14, C24, and C45 cells, indicating that these cells were CIMP‐high (Figure 2B and supplementary material, Figure S1 and Table S2). Moreover, a microsatellite marker analysis revealed different patterns of BAT25 and BAT26 (mononucleotide repeats) in C14, C24, and C45 cells and D5S346 (dinucleotide repeats) in C14 and C24, indicating an MSI‐high status of these organoid cells (Figure 2C and supplementary material, Table S2). Furthermore, these cells carried BRAF V600E mutations and frameshift mutations in TGFBR2 and ACVR2A at polyadenine repeats, which have been reported in MSI CRC cells (Figure 2A) [32, 33]. Taken together, these results suggest that C14, C24, and C45 CRCs developed through the serrated pathway, with BRAF mutations as a possible initiation event and subsequent Wnt activation mutation causing a dysplastic phenotype [10, 11, 34]. Loss‐of‐function mutations in ZNRF3 were not found in any organoids. The molecular characteristics of RNF43 frameshift mutant organoids are summarized in Table 1.
Figure 2.
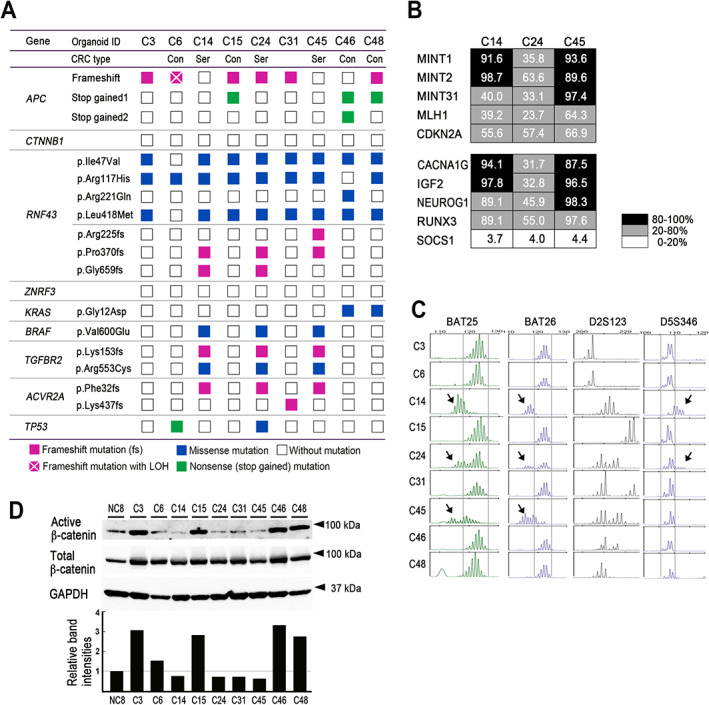
Genetic characterization and β‐catenin activation of CRC‐derived organoid lines. (A) Mutations of the CRC driver genes identified in the respective CRC‐derived organoid lines. The variants of mutations are indicated. Con, conventional pathway CRC; Ser, serrated pathway CRC. (B) The results of CIMP analyses for classic CIMP markers (top) and new CIMP markers (bottom). The methylation levels for each marker in C14, C24, and C45 cells are indicated as a heatmap. (C) The results of microsatellite instability (MSI) marker analyses in BAT25, BAT26, D2S123, and D5S346. Arrows indicate different patterns from other organoid lines. (D) Immunoblotting results for active β‐catenin and total β‐catenin in the respective organoid lines (top). GAPDH was used as an internal control. Normalized band intensities relative to the NC8 level are indicated in the bar graph (bottom).
Table 1.
Molecular characteristics of RNF43 frameshift mutant CRC organoids.
| Organoid lines | C14 | C24 | C45 | |
|---|---|---|---|---|
| Genetic/epigenetic status | RNF43 mutation | p.Pro370fs | p.Pro370fs | p.Arg225fs |
| p.Gly659fs | p.Gly659fs | p.Pro370fs | ||
| BRAF mutation | p.Val600Glu | p.Val600Glu | p.Val600Glu | |
| CIMP | High | High | High | |
| MSI | High | High | High | |
| Organoid growth | Wnt ligand dependency | + | + | + |
| Exogenous Wnt ligand | No need* | No need* | Required | |
| R‐spondin dependency | + | + | + | |
| Exogenous R‐spondin | Required | Required | Required |
C14 and C24 cells utilize endogenously expressed Wnt ligands.
Interestingly, the immunoblotting results showed significantly higher active β‐catenin levels in CRC organoids with APC two‐hit mutations (C15, C46, and C48) than in those with RNF43 frameshift mutations (C14, C24, and C45) (Figure 2D and supplementary material, Table S2), which is consistent with the nuclear β‐catenin accumulation noted in APC‐mutant cancer cells (Figure 1A). Furthermore, an IPA using a public database [29] indicated that Wnt/β‐catenin pathways, such as CTNNB1, WNT1, and lithium chloride, are significantly activated in CRC with APC homozygous deletions compared with CRC with RNF43 truncation mutations (supplementary material, Table S3). These results suggest that the Wnt activation level is lower in serrated pathway CRC cells than in conventional‐type CRC.
Wnt ligand and R‐spondin dependency of RNF43 frameshift mutant organoids
We next examined the Wnt ligand dependency of RNF43 frameshift mutant organoids by reducing the WRN‐conditioned medium (WRN‐CM), which included Wnt3a, R‐spondin, and Noggin. As expected, the number of proliferating cells was significantly decreased in C14, C24, and C45 organoids in the absence of WRN‐CM (Noggin was added to the medium), while other organoids continued proliferating (Figure 3A). Consistent with this, the EdU labeling efficiency was dramatically decreased in RNF43 frameshift mutant organoids in the WRN‐0% + Noggin condition (supplementary material, Figure S2). Supplementation of WRN‐0% cultures with both recombinant Wnt3a and R‐spondin rescued the proliferation of C14, C24, and C45 organoids to a similar degree as culture with WRN‐50% (Figure 3B,C). Notably, R‐spondin alone also rescued the C14 and C24 organoid proliferation in WRN‐0% + Noggin, while Wnt3a alone did not. Thus, C14 and C24 cells required exogenous R‐spondin but utilized endogenously expressed Wnt ligands (Figure 3D). In contrast, neither Wnt ligand nor R‐spondin rescued C45 cell proliferation, indicating that C45 cells required both exogenous Wnt3a and R‐spondin.
Figure 3.
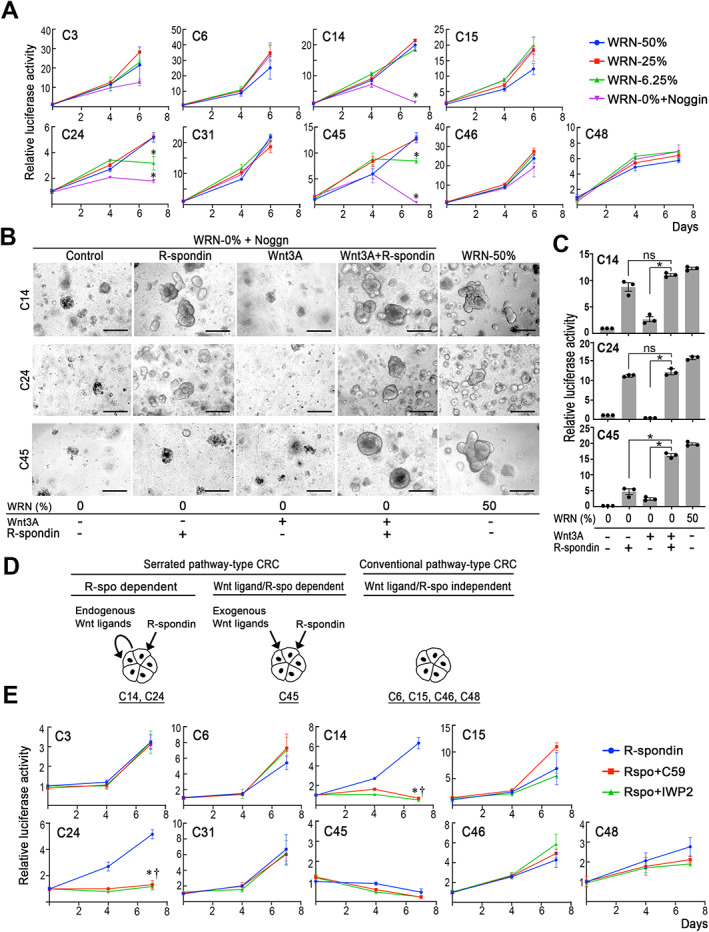
Wnt ligand and R‐spondin dependency of RNF43 frameshift mutant organoids. (A) Relative cell proliferation, as examined by the luciferase activity of the respective organoids cultured at various concentrations of WRN‐CM, is indicated by line graphs (mean ± SD). A two‐sided t‐test was used to calculate statistical significance. *p < 0.05 versus WRN‐50%. (B) Representative photographs of the respective organoids. Culture conditions are indicated below the photographs. Bars: 250 μm. The images are representative of n = 3 independent experiments. (C) Relative cell proliferations, as examined by the luciferase activity of organoids cultured under the indicated medium conditions relative to those at control (WRN‐0%), are shown in bar graphs (mean ± SD). Individual data are indicated with dots. A two‐sided t‐test was used to calculate statistical significance. *p < 0.05; ns, not significant. (D) Schematic drawing of Wnt ligand and R‐spondin dependency for R‐spondin and endogenous Wnt ligand‐dependent serrated pathway‐type CRC (C14 and C24) (left), R‐spondin and exogenous Wnt ligand‐dependent serrated pathway‐type CRC (C45) (center), and conventional pathway‐type CRC (C6, C15, C46, and C48) (right). (E) Relative cell proliferation, examined by the luciferase activity of the respective organoids cultured in the presence or absence of PORCN inhibitors (C59 or IWP2), is indicated by line graphs (mean ± SD). A two‐sided t‐test was used to calculate statistical significance. Asterisks (for R‐spo + C59) and daggers (for R‐spo + IWP2), p < 0.05 versus R‐spondin only (no inhibitor) control (blue lines).
To confirm the Wnt ligand dependency, we treated organoids with the PORCN inhibitors C59 and IWP2 in the presence of R‐spondin under WRN‐0% + Noggin conditions. As expected, both PORCN inhibitors significantly suppressed C14 and C24 cell growth, confirming the need for endogenously expressed Wnt ligand (Figure 3E). In this assay, C45 cells were unable to proliferate because of the absence of exogenous Wnt ligand.
Proliferation of RNF43 ‐mutant cells with support from the microenvironment
Lung cancer cells have two distinct subpopulations: one forms a niche providing a Wnt ligand to the other, activating Wnt signaling [35]. It is thus possible that Wnt ligand‐dependent CRC cells utilize the Wnt ligands secreted by other subpopulations. To assess this possibility, C14 and C45 organoid cells were differentially labeled with tdTomato and Venus, respectively, and cultured separately or mixed. We confirmed that R‐spondin rescued the proliferation of C14, but not C45, cells under WRN‐0% + Noggin conditions (Figure 4A). However, C45 cells proliferated under the same culture conditions when co‐cultured with C14 organoids, suggesting that C14‐secreting Wnt ligands activate the Wnt signaling of C45 cells to promote their proliferation.
Figure 4.
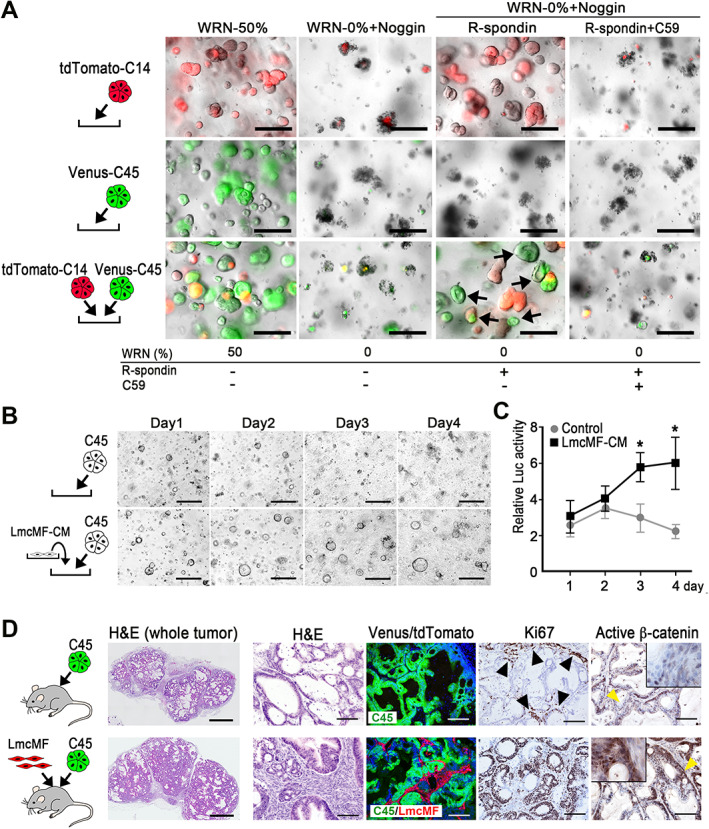
The survival and proliferation of RNF43 frameshift mutant organoid cells by support from cancer cells and stromal cells. (A) Representative photographs of tdTomato‐labeled C14 (top) and Venus‐labeled C45 (middle) monocultures, and co‐cultures of both organoids (bottom). Culture conditions are indicated below the photographs. Arrows indicate surviving Venus‐labeled C45 cells in a co‐culture with C14 cells under WRN‐0% conditions. Bars: 250 μm. The images are representative of n = 3 independent experiments. (B) Representative photographs of C45 organoids cultured under WRN‐0% conditions in the absence (top) or presence of LmcMF cell‐derived conditioned medium (CM) (bottom). Bars: 250 μm. The images are representative of n = 3 independent cultures. (C) Relative cell proliferation examined by the luciferase activities of organoids cultured with LmcMF‐CM or control, shown in B, is indicated as a line graph (mean ± SD). A two‐sided t‐test was used to calculate statistical significance. *p < 0.05 versus control. (D) Representative photographs of histology (H&E) and immunohistochemistry for Venus/tdTomato (to detect C45 and LmcMF, respectively), Ki67, and active β‐catenin (left to right) of C45 cell‐transplanted tumors (top) and C45 and LmcMF co‐transplanted tumors (bottom). Bars: 1 mm in H&E‐stained whole tumor (left) and 100 μm for other sections. Insets are enlarged images of the regions indicated by yellow arrowheads. Arrowheads indicate Ki67‐positive cells in the marginal zone of C45 tumor tissues. Images are representative of n = 4 biologically independent samples.
Stromal cells are also an important source of Wnt ligands and R‐spondin [36]. We therefore cultured C45 organoids in the presence of CM from intestinal myofibroblasts (LmcMF cells) [27] and found that the C45 cells proliferated under WRN‐0% + Noggin conditions, suggesting that LmcMF cells provided sufficient Wnt ligand and R‐spondin to C45 cells (Figure 4B,C). We next co‐transplanted C45 organoids and LmcMF cells into SHO mice s.c. (Figure 4D). Histological analyses indicated that a fibroblast‐rich microenvironment was generated by tdTomato‐labeled LmcMF cells in co‐transplanted tumors (Figure 4D). Ki67‐positive proliferating cells were found only in the marginal zone of C45‐alone tumors (Figure 4D, arrowheads), while proliferation was found in the whole tumor area of co‐transplanted tumors. Furthermore, clear nuclear accumulation of β‐catenin was detected in C45 tumor cells when co‐transplanted with LmcMF cells, which was rarely found in C45‐alone tumors. These results suggest that cancer‐associated fibroblasts contribute to malignant phenotypes of RNF43‐mutant CRC by expressing Wnt ligands and R‐spondin.
Characterization of RNF43 frameshift mutation in CRC cells
C14, C24, and C45 organoid cells carried two RNF43 frameshift mutations: p.Pro370fs and p.Gly659fs in C14 and C24, and p.Arg225fs and p.Pro370fs in C45. To examine whether these mutations were heterozygous (monoallelic) or biallelic – i.e. cis or trans mutations – we performed subcloning of the parental organoid lines and examined the mutation types in subclones via the amplification of RNF43 mRNA by RT‐PCR followed by cDNA sequencing (Figure 5A). PCR primers were designed to amplify cDNA fragments, including two mutation sites at both ends (Figure 5B). cDNA sequencing indicated a 1‐bp deletion in (C)5 for p.Arg225fs and p.Pro370fs and in (G)7 for p.Gly659fs, resulting in amino acid changes (Figure 5C).
Figure 5.
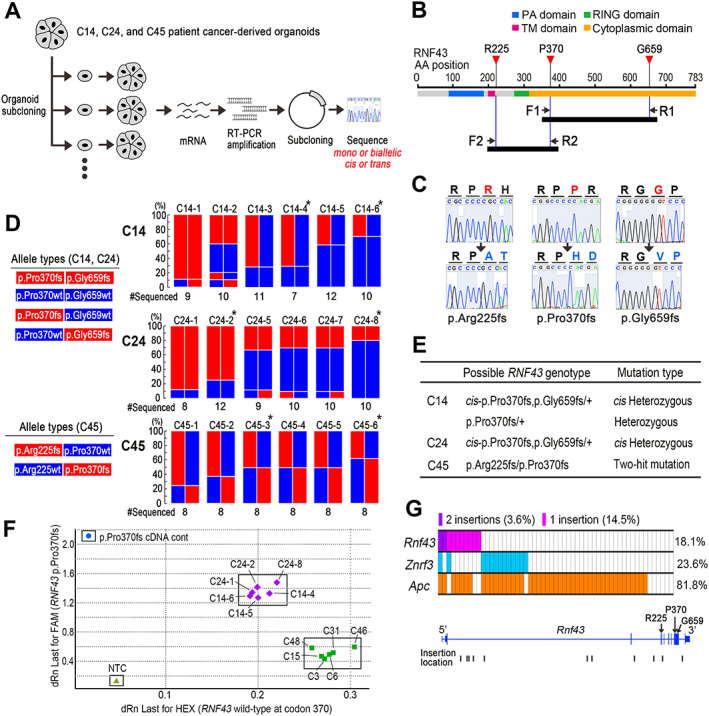
Characterization of RNF43 mutations in CRC cells. (A) Schematic drawing of the experimental strategy. C14, C24, and C45 RNF43 frameshift mutant organoids were subjected to subcloning (n = 6 subclones for each line); RNF43 mRNAs were amplified by RT‐PCR, and cDNAs were subcloned to the plasmid (n = 7–12 cDNAs for each subclone). RNF43 mutation patterns were then examined by sequencing. (B) The RT‐PCR strategy to examine cis or trans mutations of p.Pro370fs and p.Gly659fs by the primer F1 and R1, and p.Arg225fs and p.Pro370fs by the primer F2 and R2. Arrows indicate the locations of the respective primers. Closed bars indicate expected amplified cDNA fragments by the primers. (C) Representative sequencing results of wild‐type (top) and mutant RNF43 (bottom) for p.Arg225fs (left), p.Pro370fs (center), and p.Gly659fs (right) positions. Note that the mutations cause amino acid changes (blue). (D) Schematic drawing of RNF43 allele types identified by sequencing in the C14 and C24 subclones (left, top) and C45 subclones (left, bottom). The frequencies of the respective allele types in each subclone are indicated as color bars for C14 (right, top), C24 (right, middle), and C45 (right, bottom). Asterisks indicate subclones used for in vitro and xenograft drug dosing experiments (Figure 6 and supplementary material, Figures S3 and S4). The numbers indicated below the bar graphs indicate the total number of independently amplified and sequenced cDNAs. (E) List of possible RNF43 genotypes of the parental organoid lines. (F) The results of TaqMan SNP genotyping at RNF43 codon 370 (wild‐type: HEX versus p.Pro370fs: FAM) for C14 and C24 subclones (purple diamonds) and conventional pathway‐type CRC organoids (green squares) are shown as a scatter plot with RNF43 pPro370fs cDNA control (blue circle) and no template control (NTC) (light‐green triangle). (G) Top: the frequencies of transposon insertions in the Rnf43, Znrf3, and Apc genes in SB‐induced intestinal tumors. Bottom: the locations of transposon insertions in the Rnf43 gene. Exons including R225, P370, and G659 are indicated by arrows.
Before sequencing, we confirmed the Wnt ligand and R‐spondin dependency of the organoid subclones (supplementary material, Figure S3A). Furthermore, AXIN2 and ZNRF3 expression was significantly decreased in C14‐, C24‐, and C45‐derived subclones compared with APC‐mutated conventional‐type CRC organoids (supplementary material, Figure S3B,C), which was a characteristic of CIMP‐high/MSI‐high serrated CRC [12, 22, 37].
RNF43 mutations in the cDNA fragments were then examined by sequencing, and a majority of C24 subclones showed cis‐p.Pro370fs, p.Gly659fs mutations or wild‐type RNF43, indicating that the RNF43 genotype of the parental C24 organoids was an RNF43 cis‐p.Pro370fs,p.Gly659fs/+ heterozygous mutation (Figure 5D, middle; 5E). In contrast, C45 subclones showed either simple p.Arg225fs or p.Pro370fs mutations, indicating that the RNF43 genotype of parental C45 cells was RNF43 p.Arg225fs/p.Pro370fs, a two‐hit biallelic mutation (Figure 5D, bottom; 5E). In contrast, C14 subclones showed at least two different genotypes (i.e. RNF43 cis‐p.Pro370fs,p.Gly659fs/+ and RNF43 p.Pro370fs/+) (Figure 5D, top; 5E). It is possible that C14 parental organoids carried genetic heterogeneity and that different genotype cells were subcloned, although this point remains to be examined.
Importantly, allele‐specific genomic PCR indicated that C14 and C24 subclones possessed both RNF43 codon 370 wild‐type and mutant genes with the same copy number, while conventional pathway‐type CRC organoids had only wild‐type RNF43 (Figure 5F). These results exclude the possibility of loss of wild‐type RNF43 by LOH in C14 and C24 cells. Interestingly, the mutant allele frequency increased to approximately 90% in C14‐1 and C24‐1 subclones. The wild‐type RNF43 gene may have been lost by LOH during the subcloning process, allowing LOH cells to become dominant because of a possible growth advantage.
We further examined the data previously obtained by transposon mutagenesis screening in mouse intestine with the Trp53 R172H genetic background [18]. The frequencies of Rnf43, Znrf3, and Apc mutations by transposon insertions in intestinal tumors are indicated in Figure 5G (top). Single insertions in Rnf43 were found in 14.5% of tumors, and two cases did not carry mutations in Apc and Znrf3. Most transposon insertions were found in introns 2 and 3, resulting in truncation at the N‐terminal of RNF43 (Figure 5G, bottom). These results support the idea that a single‐hit RNF43 mutation contributes to intestinal tumorigenesis.
Suppression of RNF43 ‐mutant PDX tumors by a PORCN inhibitor
Finally, we examined whether or not a PORCN inhibitor suppresses the tumor development of RNF43 frameshift mutant cells in PDX models. C14, C24, and C45 parental and subclone organoids were transplanted s.c. into SHO mice, and mice were treated with the PORCN inhibitor ETC‐159 (Figure 6A). Importantly, ETC‐159 treatment nearly completely suppressed the tumor development in all PDX model mice (Figure 6B,C and supplementary material, Figure S4A). Histological analyses indicated that ETC‐159 treatment resulted in decreased branching and enlarged glands with fewer dysplastic features and significantly decreased proliferation (Figure 6D and supplementary material, Figure S4B). It is possible that cancer cells were differentiated by PORCN inhibition‐induced suppression of Wnt signaling, as previously reported [21].
Figure 6.
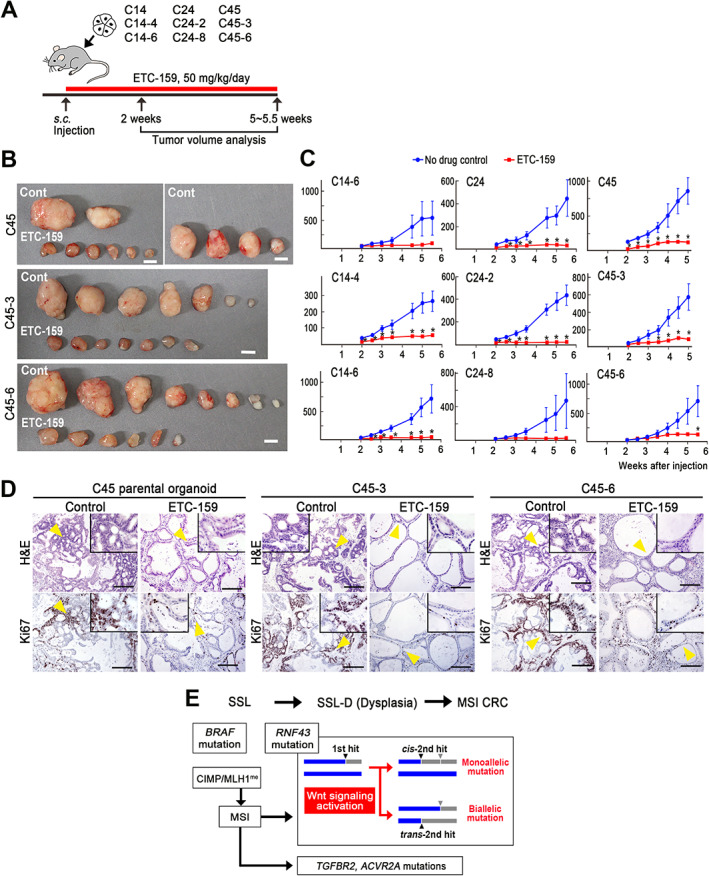
Suppression of RNF43 frameshift mutant PDX tumor development by a PORCN inhibitor. (A) Schematic drawing of the strategy for the drug dosing experiment. The red bar indicates the PORCN inhibitor (ETC‐159) treatment period. (B) Representative photographs of PDX tumors of C45 parental organoids and two subclones (C45‐3 and C45‐6) developed in no‐drug control mice (top) and ETC‐159‐treated mice (bottom). Bars: 5 mm. Photographs of PDX tumors of C14, C24, and subclones are shown in supplementary material, Figure S4A. (C) Tumor volume changes of the control (blue) and ETC‐159‐treated PDX mice (red) are indicated as line graphs (mean ± SEM). Transplanted organoid clone numbers are indicated. A two‐sided t‐test was used to calculate statistical significance.*p < 0.05 versus control. (D) Representative histology photographs of PDX tumors developed in the control (left) and ETC‐159‐treated mice (right) for C45 parental organoids and C45‐3 and C45‐6 subclones (left to right). H&E (top) and immunohistochemistry for Ki67 (bottom). Insets are enlarged images of the regions indicated by yellow arrowheads. Bars: 200 μm. The images are representative of n = 6–8 biologically independent experiments. Histology results for C14, C24, and subclones are shown in supplementary material, Figure S4B. (E) Schematic illustration of serrated pathway CRC development. SSL, sessile serrated lesion; SSL‐D, SSL with dysplasia; CIMP, CpG island methylator phenotype; MLH1me, MLH1 gene promoter hypermethylation.
Discussion
Serrated polyps with MSI are reportedly associated with a predisposition to progress to CRC [10, 11]. Moreover, RNF43 mutations were frequently found in sessile serrated lesions (SSLs) and MSI‐type CRC, suggesting that RNF43 mutation drives CRC development through the serrated pathway (Figure 6E). Consistently, we found RNF43 frameshift mutations in CIMP‐high and MSI‐high CRC‐derived organoids, which were associated with BRAF V600E mutations.
Interestingly, the level of active β‐catenin in RNF43‐mutant CRC was significantly lower than that in APC two‐hit CRC, and nuclear accumulation of β‐catenin was found only in APC‐mutant CRC. These results suggest that the threshold of Wnt activation for serrated pathway tumorigenesis is lower than that via the conventional pathway. Consistently, it has been reported that Wnt activation is greatly reduced in MSI‐high hypermutated CRCs relative to non‐hypermutated CRCs [37]. It is possible that a high level of Wnt activation is needed for the expansion of intestinal stem/progenitor cells to form adenomatous polyps as an initial event of the conventional pathway, as found in Apc Δ716 knockout mice [38]. Although adenomatous proliferation was found in Rnf43 −/− Znrf3 −/− compound mouse intestine [2, 3], it was not observed in Rnf43 −/− simple mutant mice [17]. These results suggest that the degree of Wnt activation induced by RNF43 mutation is not sufficient for the initiation of conventional‐type CRC but can contribute to serrated pathway tumorigenesis.
In the present study, all RNF43 frameshift mutant CRC cells proliferated in an R‐spondin‐dependent manner. It is possible that suppression of ZNRF3 function and retention of RNF43 function by R‐spondin is required to achieve the level of Wnt signaling necessary for tumorigenesis in RNF43‐mutant cells.
Loss of wild‐type RNF43 by LOH or second‐hit somatic mutations has been reported in ovarian tumors and serrated colon tumors [6, 10], suggesting the need for two‐hit inactivation of RNF43 for tumorigenesis. However, the present results showed heterozygous monoallelic RNF43 mutations in C14 and C24 cells, indicating that biallelic inactivation of RNF43 is not necessarily required for serrated pathway tumor development. In contrast, we also found two‐hit biallelic trans‐mutations in RNF43 in C45 cells, suggesting that two‐hit mutations of RNF43 are advantageous for CRC development, possibly through an increase in the Wnt activation level (Figure 6E). Furthermore, cis mutations in RNF43 for p.Pro370fs and p.Gly659fs were found in the same allele. A recent analysis indicated that the most common mutation of RNF43, p.Gly659fs, modestly compromises the RNF43 function [24], and N‐terminal truncation mutations are more effective at increasing Wnt signaling activity than C‐terminal mutations [23]. Therefore, the p.Gly659fs mutation may have been introduced at an early stage of tumorigenesis in C14 and C24 with a slight increase in Wnt signaling. An additional N‐terminal p.Pro370fs mutation was then introduced at the same allele, which may have facilitated tumor promotion by increasing the Wnt activity (Figure 6E).
Importantly, co‐transplantation of RNF43 frameshift mutant CRC cells with myofibroblasts increased the β‐catenin nuclear accumulation and cell proliferation of PDX tumors, indicating that the malignant phenotype of RNF43‐mutant cells is highly dependent on stromal cells. Several studies have demonstrated the role of Wnt signaling in metastasis; namely, Wnt activation promotes the epithelial–mesenchymal transition program by increasing the nuclear Smai2 expression [39], and liver metastasis of colon cancer cells is accelerated by the nuclear accumulation of β‐catenin and FOXO3 [40]. Accordingly, it is possible that metastatic niche cells play a key role in the proliferation of disseminated RNF43‐mutant CRC cells through activation of Wnt signaling. In this study, we demonstrated the significant suppression of RNF43‐mutant PDX tumors by ETC‐159 treatment. Thus, it is important to further examine whether or not the development of metastatic foci by RNF43‐mutant cells can be suppressed by PORCN inhibitors as a future clinical strategy.
In conclusion, we established RNF43 frameshift CRC‐derived organoids. The Wnt/β‐catenin activation level of RNF43‐mutant serrated pathway CRC was lower than that of conventional pathway‐type CRC. Furthermore, heterozygous RNF43 frameshift mutation may contribute to tumorigenesis through the serrated pathway. However, second‐hit RNF43 mutations may facilitate CRC development by increasing the Wnt activation level. Finally, PORCN inhibition significantly suppressed PDX tumor development of RNF43‐mutant CRC cells, suggesting that PORCN inhibitors are an effective therapeutic strategy against RNF43‐mutant CRC development and metastasis.
Author contributions statement
HO and MO designed the study and planned the experiments. DY, HO and KK were involved in organoid preparation and performed immunohistochemistry. DY, DW, XL, MN and KM were involved in in vitro and in vivo experiments. MT and HS were involved in CIMP analysis. HaT examined the transposon data. TO provided cancer‐associated fibroblasts. HiT, NI and MO supervised the project. All of the authors were involved in writing the paper and had final approval of the submitted version.
Supporting information
Supplementary materials and methods
Figure S1. The results of CIMP analysis for RNF43 frameshift mutant organoid cells
Figure S2. Wnt ligand and R‐spondin dependency of RNF43 frameshift mutant organoid cells
Figure S3. Biological characteristics of RNF43 frameshift mutant CRC cells
Figure S4. Suppression of RNF43 frameshift mutant tumor development by PORCN inhibitor
Table S1. Clinicopathological characteristics of CRC patients
Table S2. Mutation variants and characteristics of CRC organoids
Table S3. Activated upstream regulators in APC‐deleted CRCs compared with RNF43 truncation mutation CRCs (IPA)
Acknowledgements
We wish to thank Manami Watanabe, Ayako Tsuda, and Yoshie Jomen for their technical assistance. This work was supported by Grants‐in‐Aid for Scientific Research (A) (18H04030) (MO) and (B) (19H03498) (HO) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; AMED (19ck0106259h0003, 20ck0106541h0001) (MO) from the Japan Agency for Medical Research and Development, Japan; Takeda Science Foundation; and The Hokkoku Cancer Foundation.
No conflicts of interest were declared.
References
- 1. Nusse R, Clevers H. Wnt/β‐catenin signaling, disease, and emerging therapeutic modalities. Cell 2017; 169: 985–999. [DOI] [PubMed] [Google Scholar]
- 2. Koo BK, Spit M, Jordens I, et al. Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012; 488: 665–669. [DOI] [PubMed] [Google Scholar]
- 3. Koo BK, van Es JH, van den Born M, et al. Porcupine inhibitor suppresses paracrine Wnt‐driven growth of Rnf43;Znrf3‐mutant neoplasia. Proc Natl Acad Sci U S A 2015; 112: 7548–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lannagan TRM, Le YK, Wang T, et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019; 68: 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giannakis M, Hodis E, Mu XJ, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014; 46: 1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryland GL, Hunter SM, Doyle MA, et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J Pathol 2013; 229: 469–476. [DOI] [PubMed] [Google Scholar]
- 7. Wang K, Yuen ST, Xu J, et al. Whole‐genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 2014; 46: 573–582. [DOI] [PubMed] [Google Scholar]
- 8. Jiang X, Hao HX, Growney JD, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2013; 110: 12649–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taupin D, Lam W, Rangiah D, et al. A deleterious RNF43 germline mutation in a severely affected serrated polyposis kindred. Hum Genome Var 2015; 2: 15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan HHN, Lai JCW, Ho SL, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017; 66: 1645–1656. [DOI] [PubMed] [Google Scholar]
- 11. Crockett SD, Nagtegaal ID. Terminology, molecular features, epidemiology, and management of serrated colorectal neoplasia. Gastroenterology 2019; 157: 949–966.e4. [DOI] [PubMed] [Google Scholar]
- 12. Bond CE, McKeone DM, Kalimutho M, et al. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016; 7: 70589–70600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsai JH, Liau JY, Yuan CT, et al. RNF43 is an early and specific mutated gene in the serrated pathway, with increased frequency in traditional serrated adenoma and its associated malignancy. Am J Surg Pathol 2016; 40: 1352–1359. [DOI] [PubMed] [Google Scholar]
- 14. Sekine S, Yamashita S, Tanabe T, et al. Frequent PTPRK–RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol 2016; 239: 133–138. [DOI] [PubMed] [Google Scholar]
- 15. Kleeman SO, Koelzer VH, Jones HJ, et al. Exploiting differential Wnt target gene expression to generate a molecular biomarker for colorectal cancer stratification. Gut 2020; 69: 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018; 173: 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eto T, Miyake K, Nosho K, et al. Impact of loss‐of‐function mutations at the RNF43 locus on colorectal cancer development and progression. J Pathol 2018; 245: 445–455. [DOI] [PubMed] [Google Scholar]
- 18. Takeda H, Wei Z, Koso H, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet 2015; 47: 142–150. [DOI] [PubMed] [Google Scholar]
- 19. Zhong Z, Virshup DM. Wnt signaling and drug resistance in cancer. Mol Pharmacol 2020; 97: 72–89. [DOI] [PubMed] [Google Scholar]
- 20. van de Wetering M, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015; 161: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Madan B, Ke Z, Harmston N, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016; 35: 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tu J, Park S, Yu W, et al. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci Rep 2019; 9: 18557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li S, Lavrijsen M, Bakker A, et al. Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β‐catenin signaling and unlikely confer Wnt‐dependency onto colorectal cancers. Oncogene 2020; 39: 3458–3472. [DOI] [PubMed] [Google Scholar]
- 24. Yu J, Yusoff PAM, Woutersen DTJ, et al. The functional landscape of patient‐derived RNF43 mutations predicts sensitivity to Wnt inhibition. Cancer Res 2020; 80: 5619–5632. [DOI] [PubMed] [Google Scholar]
- 25. Seino T, Kawasaki S, Shimokawa M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 2018; 22: 454–467.e6. [DOI] [PubMed] [Google Scholar]
- 26. Miyoshi H, Ajima R, Luo CT, et al. Wnt5a potentiates TGF‐β signaling to promote colonic crypt regeneration after tissue injury. Science 2012; 338: 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawasaki H, Ohama T, Hori M, et al. Establishment of mouse intestinal myofibroblast cell lines. World J Gastroenterol 2013; 19: 2629–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamamoto E, Suzuki H, Yamano H, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol 2012; 181: 1847–1861. [DOI] [PubMed] [Google Scholar]
- 29. Niinuma T, Kitajima H, Kai M, et al. UHRF1 depletion and HDAC inhibition reactivate epigenetically silenced genes in colorectal cancer cells. Clin Epigenetics 2019; 11: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parsons R, Myeroff LL, Liu B, et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res 1995; 55: 5548–5550. [PubMed] [Google Scholar]
- 33. Hempen PM, Zhang L, Bansal RK, et al. Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res 2003; 63: 994–999. [PubMed] [Google Scholar]
- 34. Hashimoto T, Yamashita S, Yoshida H, et al. WNT pathway gene mutations are associated with the presence of dysplasia in colorectal sessile serrated adenoma/polyps. Am J Surg Pathol 2017; 41: 1188–1197. [DOI] [PubMed] [Google Scholar]
- 35. Tammela T, Sanchez‐Rivera FJ, Cetinbas NM, et al. A Wnt‐producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017; 545: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greicius G, Kabiri Z, Sigmundsson K, et al. PDGFRα + pericryptal stromal cells are the critical source of Wnts and RSPO3 for murine intestinal stem cells in vivo . Proc Natl Acad Sci U S A 2018; 115: E3173–E3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Donehower LA, Creighton CJ, Schultz N, et al. MLH1‐silenced and non‐silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. J Pathol 2013; 229: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oshima H, Oshima M, Kobayashi M, et al. Morphological and molecular processes of polyp formation in ApcΔ716 knockout mice. Cancer Res 1997; 57: 1644–1649. [PubMed] [Google Scholar]
- 39. Wu ZQ, Li XY, Hu CY, et al. Canonical Wnt signaling regulates Slug activity and links epithelial–mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A 2012; 109: 16654–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tenbaum SP, Ordóñez‐Morán P, Puig I, et al. β‐catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 2012; 18: 892–901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. The results of CIMP analysis for RNF43 frameshift mutant organoid cells
Figure S2. Wnt ligand and R‐spondin dependency of RNF43 frameshift mutant organoid cells
Figure S3. Biological characteristics of RNF43 frameshift mutant CRC cells
Figure S4. Suppression of RNF43 frameshift mutant tumor development by PORCN inhibitor
Table S1. Clinicopathological characteristics of CRC patients
Table S2. Mutation variants and characteristics of CRC organoids
Table S3. Activated upstream regulators in APC‐deleted CRCs compared with RNF43 truncation mutation CRCs (IPA)